

SUMMARY
The widespread availability of energy-dense, rewarding foods is correlated with the increased incidence of obesity across the globe. Overeating during mealtimes and unscheduled snacking disrupts timed metabolic processes which further contribute to weight gain. The neuronal mechanism by which the consumption of energy-dense food restructures the timing of feeding is poorly understood. Here, we demonstrate that dopaminergic signaling within the suprachiasmatic nucleus (SCN), the central circadian pacemaker, disrupts the timing of feeding, resulting in overconsumption of food. D1 dopamine receptor (Drd1) null mice are resistant to diet-induced obesity, metabolic disease, and circadian disruption associated with energy-dense diets. Conversely, genetic rescue of Drd1 expression within the SCN restores diet-induced overconsumption, weight gain, and obesogenic symptoms. Access to rewarding food increases SCN dopamine turnover, and elevated Drd1-signaling decreases SCN neuronal activity, which we posit disinhibits downstream orexigenic responses. These findings define a connection between the reward and circadian pathways in the regulation of pathological calorie consumption.
Graphical Abstract

eTOC Blurb
Palatable diets rich in fat and sugar incentivize overeating which leads to obesity. Grippo and Tang et al. discover a unique role for dopamine signaling in the central circadian clock that promotes overconsumption outside of mealtimes.
INTRODUCTION
Obesity and its comorbid conditions including type 2 diabetes, cardiovascular disease, and metabolic syndrome are pandemics that significantly reduce lifespan [1–4]. Lifestyle interventions such as decreasing caloric intake and increasing exercise are the primary strategies used to combat obesity, yet they remain ineffective for long-term success [5]. Energy-rich, rewarding foods encourage snacking outside of regular mealtimes, lowering adherence to dietary interventions and therefore preventing sustained weight loss [6, 7]. Determining the neural mechanisms by which palatable foods alter feeding amount and meal-timing is a necessary step toward developing effective therapies against obesity.
Proper maintenance of energy homeostasis requires the synchronization of meals with daily metabolic rhythms [8]. Surprisingly, even during conditions of isocaloric energy intake, high-fat food consumption out of phase with rhythmic metabolic processes leads to obesity through altered energy utilization and increased energy storage in mice [9, 10]. Consistent with these observations, late-night eaters tend to be refractory to weight-loss therapy [11], while temporal restriction of meal timing improves the metabolic profiles of pre-diabetic men [12]. These findings emphasize the importance of both the amount and timing of calorie intake on metabolic health. While becoming recognized as critically important for well-being, the precise mechanisms by which energy-dense, rewarding foods alter food intake and feeding patterns remain unknown.
Dopamine (DA) is a good candidate for pathological regulation of feeding behavior because it is known for reward processing, and is required for sustained motivation and execution of goal-directed behaviors [13, 14]. Consistent with this, genetic ablation of DA production leads to hypophagia and eventual death by starvation in mice [15]. Modulation of DA-dependent neural activity and behavior is mediated by a group of G protein-coupled receptors expressed in anatomically distinct regions throughout the central and peripheral nervous systems. Both D1-like (Gs-coupled) and D2-like (Gi-coupled) receptors have been implicated in non-homeostatic consumption of palatable foods and obesity [16–18]. Selective activation of D1 dopamine receptor (Drd1) expressing neurons in the prefrontal cortex induces food intake [19], and prolonged access to a high-fat diet alters Drd1 expression within the reward centers of the brain [20, 21]. Additionally, treatment with a Drd1 antagonist reduces food intake in rodents, and causes weight loss in humans [22, 23]. Given the importance of DA signaling in food reward responding and feeding regulation, we sought to determine the role of this neuromodulatory system in diet-induced obesity.
In this study, we show that Drd1 expression is necessary for overconsumption of a high-fat, high-sugar (HFD) diet. Moreover, Drd1 signaling in response to HFD perturbs circadian feeding and activity rhythms. Unlike wild-type mice, Drd1-knockout mice provided ad libitum access to palatable foods are resistant to weight gain. Most strikingly, Drd1-knockout mice on a HFD maintain robust daily rhythms of food consumption, foraging, and fuel utilization, while wild-type mice demonstrate altered circadian regulation of metabolism. Selective restoration of Drd1 expression within the nucleus accumbens (NAc), the predominant reward processing center of the brain, fails to rescue the overconsumption of energy-rich food and attendant weight gain observed in wild-type mice. However, Drd1 rescue specifically within the central circadian clock (suprachiasmatic nucleus: SCN) of Drd1-knockout mice completely restores the obesogenic phenotype. The overall excitability of the SCN is reduced in response to Drd1 agonist treatment, likely due to the increased activity of GABAergic Drd1-expressing-SCN neurons inhibiting the local post-synaptic cells. Therefore, we posit that in response to rewarding foods, the increased dopaminergic tone on the circadian pacemaker dampens SCN neuronal activity, disinhibiting downstream orexigenic targets and promoting out-of-phase foraging.
RESULTS
Drd1-KO Mice are Protected from High-Fat Diet-Induced Obesity
Mice with ad libitum access to rewarding, calorically dense food rapidly develop obesity, diabetes, and metabolic disease [24, 25]. To explore the potential relationship between Drd1 signaling and obesity, we monitored response to dietary challenge using Drd1aCre/Cre (KO) mice, in which both Drd1a alleles were replaced by Cre recombinase (Cre) [26]. We monitored weekly changes in body weight (BW) between adult male wild-type (WT) and KO mice during ad libitum access to either standard chow diet (SCD; 19% fat, 0% sucrose) or high-fat, high-sugar diet (HFD; 45% fat, 17% sucrose) (Figures 1A and S1A–B). Percent and total BW change was indistinguishable between adult male WT and KO mice on SCD (WT-SCD: 5.0±2.6% increase; KO-SCD: 5.9±1.1% increase; Student’s two-tailed t-test, p=0.8). As expected, WT mice on HFD (WT-HFD) significantly increased BW (Figures 1B and S1C), however, KO mice fed the same diet (KO-HFD) were completely resistant to diet-induced weight gain (WT-HFD: 29.5±3.1% increase; KO-HFD: 5.4±1.6% increase; Student’s two-tailed t-test, p<0.001; Figures 1B–C, and S1C). We also observed resistance to obesity in a separate Drd1a null (Drd1−/−) mouse model [27], following twelve weeks access to a palatable high-fat, low sugar diet (60% fat, 10.7% sucrose) (Figure S1D). These data indicate that Drd1 signaling is necessary for weight gain on a variety of calorically dense food sources in a strain independent manner.
Figure 1. Drd1-KO Mice Are Protected from HFD-Induced Obesity.

(A) Food access paradigm. Diet switch from SCD to HFD (week 0).
(B) Percent BW change relative to week 0 for WT and KO mice on SCD or HFD. Repeated-measures three-way ANOVA with Bonferroni post-hoc comparison, n=7–13/group; Fgenotype(1,37)=4.8, p=0.03; Fdiet(1,37)=5.9, p=0.02. Arrow marks diet switch from SCD to HFD. Statistical significance between WT-HFD and KO-HFD is depicted.
(C) Representative images of WT or KO mice from 1A–B. Scale bar=1cm.
(D) Calorie intake during the 12-hour light- (Day) or dark-phases (Night) for WT and KO mice on SCD or HFD. Repeated-measures three-way ANOVA with Bonferroni post-hoc comparison, n=13/group; Ftime(1,96)=1,544, p<0.001; Fgenotype(1,96)=57.1, p<0.001; Fdiet(1,96)=12.6, p<0.001.
(E) Percent of total daily food intake during the Day for WT and KO mice on SCD or HFD. Two-way ANOVA with Bonferroni post-hoc comparison, n=13/group; Fgenotype(1,48)=203.2, p<0.001; Fdiet(1,48)=170.2, p<0.001.
(F) Latency to retrieve a HFD pellet, Day or Night, for WT and KO mice (See also Video S1–S2). Student’s two-tailed t-test, n=9–15/group.
Data are represented as mean±SEM. **p<0.01,***p<0.001, ns=not significant.
Paralleling their weight gain, daily HFD consumption of WT mice was significantly increased compared to all other groups (Figures S1E–F). Diets rich in fat not only increase food consumption, but also alter feeding patterns resulting in food intake that extends into the rest phase [9, 28]. This disruption in feeding rhythms induces weight gain independent of overconsumption, highlighting the importance of maintaining robust feeding rhythms for proper metabolic regulation [10]. While caloric intake of either diet was equivalent between genotypes during the active (Night) phase (Figure 1D), WT mice exhibited a significant increase in rest (Day) phase HFD consumption that was nearly absent in KO-HFD mice (Figures 1D–E, and S1E). Attenuated daytime consummatory behavior in KO animals was also present during access to a highly palatable liquid diet (Ensure), demonstrating that this phenotype is observed in response to a variety of hypercaloric food sources (Figure S1G).
The inhibitory effects of light on general activity and feeding (masking) have been well documented [29]. However, when placed into constant dark conditions (DD), KO mice still consumed significantly less during the subjective day (DD-day) compared to WT-HFD controls (WT-HFD: 42.8±2.9%; KO-HFD: 27.8±3.9%; Student’s two-tailed t-test, p=0.009; Figures S1H–I). Furthermore, KO mice remained resistant to weight gain following ten days of HFD access in DD, indicating that light exposure is not the primary determinant of reduced HFD consumption in KO mice (WT-HFD: 17.3±2.1%; KO-HFD: −0.4±1.8% BW change; Student’s two-tailed t-test, p<0.001; Figure S1J). Next, we sought to ascertain whether the reduced daytime food intake in KO mice was a result of diurnally regulated motivation for rewarding foods. Although progressive ratio breakpoint analysis is the standard test for motivation, because of the deficit in operant conditioning performance in KO mice during both phases of the day (Figures S1K–M) [30], we designed a simpler foraging assay, which more closely mimics ad libitum food access conditions. In this assay, habituated mice are placed into an arena with a HFD pellet buried under the bedding at one end. The latency for the mice to find and consume food is recorded. Consistent with reduced daytime food intake, KO mice exhibited an increased latency to forage during the day compared to WT controls (ZT8; WT: 21.9±4.2s; KO: 58.6±12.9s). However, no detectable differences in foraging latency were observed at night (ZT15; WT: 32.9±12.2s; KO: 38.4±8.5s), demonstrating that Drd1 is important for out-of-phase palatable food-seeking, and the observed daytime reduction of food intake in KO mice is not a generalized impairment in olfaction or locomotion (Figure 1F, and Video S1–S2) [31]. Taken together, these data suggest that Drd1-dependent overconsumption of palatable food, predominantly during the light phase, leads to obesity and metabolic disease. These findings not only reaffirm the importance of circadian regulation in energy balance [32], but also implicate an essential role for the Drd1-dependent DA-signaling in this process.
Drd1-KO Mice Are Resistant to Dampening of Metabolic Rhythms and Metabolic Disease
One consequence of ad libitum calorie-rich food consumption is the adaptation of metabolic processes, which switch from a high amplitude circadian oscillation between carbohydrate utilization (during the active phase) and lipid oxidation (during the rest phase), to a low amplitude oscillation reflective of metabolic shunting towards energy storage [33–35]. While WT-SCD and KO-SCD mice exhibited robust circadian rhythms of respiratory exchange ratio (RER) (Figures 2A–B) indicating efficient energy source cycling between predominantly carbohydrates (RER of ~1) or fats (RER of ~0.7), the circadian amplitude of RER in WT-HFD mice was significantly dampened (Figures 2D–E, and S2A). Interestingly, nighttime RER values were reduced in both WT-HFD and KO-HFD mice likely due to the elevated fat/carbohydrate ratio of the diet (Figures 2B and 2D). However, KO-HFD mice sustained higher-amplitude RER rhythms reflecting increased fat utilization during the rest phase (Figure 2D). Consistent with a lower RER value and adipocyte lipolysis, KO-HFD mice had elevated activated phosphorylated hormone sensitive lipase (pHSL) levels during the rest phase (ZT 6) (Figures S2B and S2C) [36, 37]. These data reveal that KO-HFD mice maintain SCD-like metabolic rhythms compared to WT-HFD animals, which is favorable for metabolic well-being and protection from weight gain.
Figure 2. Drd1-KO Mice are Resistant to HFD-Induced Metabolic Disease.
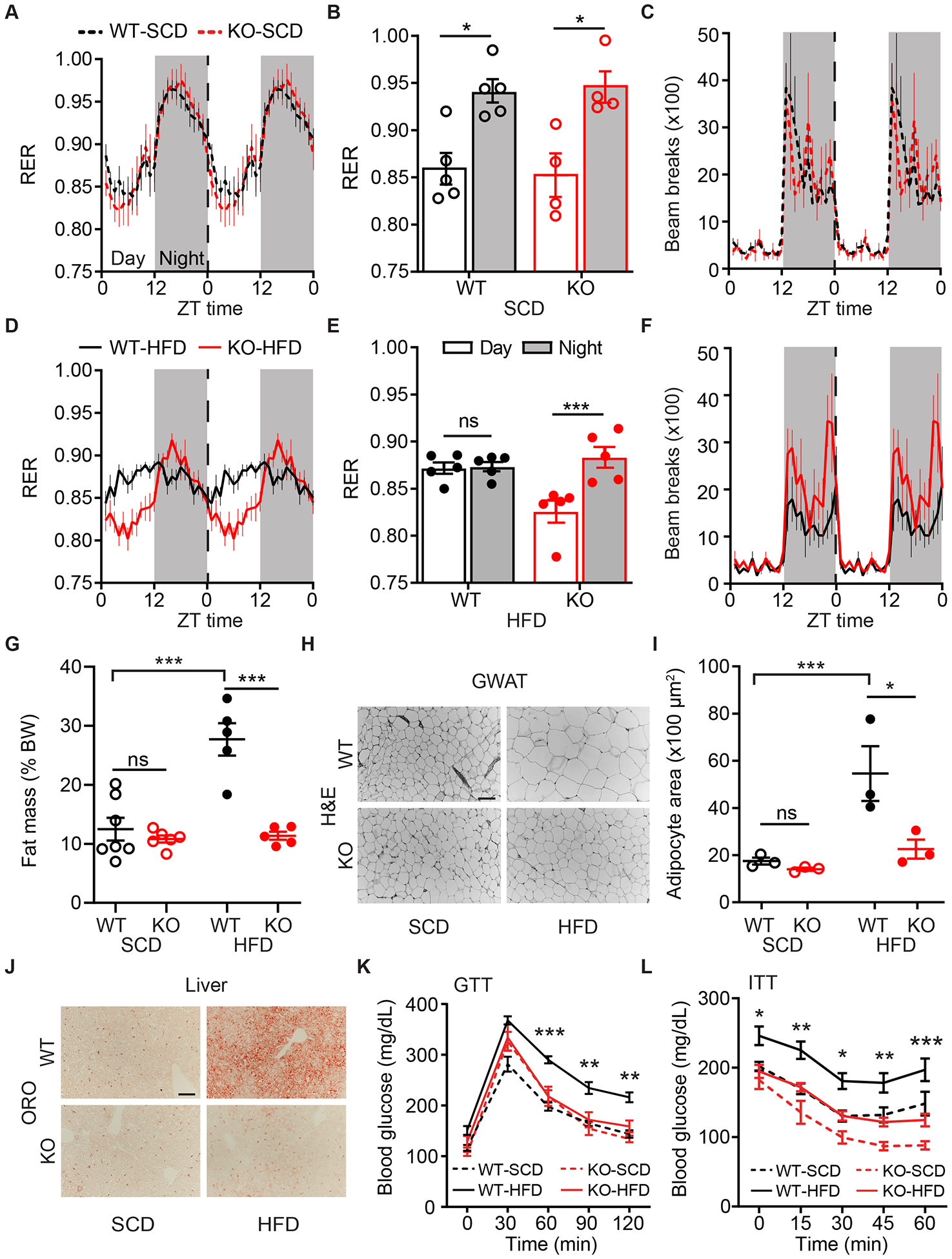
(A) RER for WT-SCD and KO-SCD mice. Repeated-measures two-way ANOVA, n=4–5/group. Time is represented as Zeitgeber time (ZT), where ZT0 indicates onset of Day and ZT12 indicates onset of Night (shaded grey). Data are plotted in 1-hour intervals. Vertical dotted line indicates the point at which twenty-four-hour data are double-plotted for clarity. Same representation applies to panels (C), (D) and (F).
(B) Average RER during the Day or Night from panel (A). Repeated-measures two-way ANOVA with Bonferroni post-hoc comparison, n=4–5/group; Ftime(1,7)=57.5, p<0.001.
(C) Double-plotted ambulatory infrared beam-break activity for WT-SCD and KO-SCD mice. Repeated-measures two-way ANOVA with Bonferroni post-hoc comparison, n=4–5/group.
(D) Double-plotted RER for WT-HFD and KO-HFD mice. Repeated-measures two-way ANOVA with Bonferroni post-hoc comparison, n=5/group.
(E) Average RER during the Day or Night from panel (D). Repeated-measures two-way ANOVA with Bonferroni post-hoc comparison, n=5/group; Ftime(1,8)=15.8, p=0.004.
(F) Double-plotted ambulatory infrared beam-break activity for WT-HFD and KO-HFD mice. Repeated-measures two-way ANOVA with Bonferroni post-hoc comparison, n=5/group; Fgenotype(1,8)=5.4, p=0.05.
(G) Fat mass as percent BW for WT and KO mice on SCD or HFD. Two-way ANOVA with Bonferroni post-hoc comparison, n=5–7/group; Fdiet(1,19)=20.3, p<0.001; Fgenotype(1,19)=26.5, p<0.001.
(H) Representative GWAT Hematoxylin and Eosin (H&E) stain for groups in (G). Scale bar=100μm.
(I) Quantification of GWAT adipocyte area. Two-way ANOVA with Bonferroni post-hoc comparison, n=3/group; Fdiet(1,8)=13.5, p=0.006; Fgenotype(1,8)=8.2, p=0.02.
(J) Representative liver Oil-red-O (ORO) histology for groups in (G), where hepatic lipid content stains as red. Scale bar=100μm.
(K) Blood glucose levels during glucose tolerance test (GTT). Repeated-measures three-way ANOVA with Bonferroni post-hoc comparison, n=8–12/group; Fgenotype(1,35)=5.5, p=0.03; Fdiet(1,35)=21.9, p<0.001. And (L) insulin tolerance test (ITT). Repeated-measures three-way ANOVA with Bonferroni post-hoc comparison; n=9–12/group; Fgenotype(1,36)=26.4, p<0.001; Fdiet(1,36)=18.4, p<0.001. Statistical significance between WT-HFD and KO-HFD is depicted.
Data are represented as mean±SEM. *p<0.05,**p<0.01,***p<0.001.
It has been reported that Drd1 expression within the nucleus accumbens (NAc) is involved in the maintenance of wakefulness [38], which could interfere with time spent foraging. Both WT and KO mice exhibited similar hourly putative sleep duration during the day on HFD (WT-HFD: 42.0±5.1 min/hour; KO-HFD: 48.5±1.8 min/hour; Student’s two-tailed t-test, p=0.4) suggesting that the observed feeding phenotype is unlikely to be caused by increased sleep bouts in KO mice. In fact, KO-HFD mice exhibit marginal hyperactivity at night, however, this does not translate to a difference in twenty-four hour energy expenditure (EE), resting metabolic rate (RMR) or excess energy lost to heat compared to WT-HFD mice (Figures 2C, 2F, and S2D–G) [39, 40]. Lastly, while there was no difference in fecal energy density between all groups (Figure S2H), KO-HFD mice show diminished daily energy assimilation and hence the reduced BW in these animals is not due to increased energy excretion (Figure S2I).
Obesity from HFD consumption also results in increased fat mass, adipocyte hypertrophy, hepatic steatosis, glucose intolerance, and insulin resistance [25, 41]. As expected, WT-HFD mice accumulated significant total body adiposity, while KO-HFD animals, maintained similar body fat composition to SCD controls (WT-SCD: 12.5±1.9%; KO-SCD: 10.8±0.6%; WT-HFD: 27.7±2.7%; KO-HFD: 11.4±0.7%;Figures 2G and S2L). Additionally, adipocyte cell hypertrophy in gonadal white adipose tissue (GWAT) and posterior subcutaneous adipose tissue (SCAT) were reduced, and hepatic lipid accumulation was diminished in KO-HFD mice compared to WT-HFD animals (Figures 2H–J, S2J–K, and Table S1). Most strikingly, KO-HFD mice were protected from glucose intolerance and insulin resistance, two hallmarks of obesity-induced metabolic disease (Figures 2K–L, and S2M–N) [42, 43]. Taken together, these data demonstrate that unlike WT mice, KO mice on HFD maintain high amplitude rhythms in activity, temperature, and energy source, as well as reduced adiposity and robust responsiveness to glucose and insulin fluctuations.
The Effect of HFD on KO Peripheral Circadian Rhythms
Following chronic nutrient overload, development of diet-induced insulin resistance has been associated with microbiome dysbiosis [44–46]. Therefore, we evaluated the fecal microbiome in WT-SCD, WT-HFD, and KO-HFD mice from samples collected every four hours across the day (Figure S3). Compared to SCD, HFD significantly reduced the alpha diversity of fecal microbiome in WT mice (Mann–Whitney U test, p<0.001; Figure 3B), and accounted for over 40% of the variation in microbial composition (PERMANOVA, R2=0.4; p=0.001; Figure 3A). While diet had a significant effect on species richness and composition of the microbiome, no substantial genotype differences in alpha diversity (Mann–Whitney U test, p=0.4; Figure 3B) or microbial composition (PERMANOVA, R2 0.04; p=0.2; n=18/group; Figure 3A) could be detected on HFD. However, when fecal samples were collected at ZT1 and ZT13 from the same animal, we observed that time of collection did have a significant effect (PERMANOVA, R2=0.2; p=0.001; n=8/group), but again no substantial differences between genotype could be distinguished (PERMANOVA, R2=0.04; p=0.05; n=8/group; Figure 3C). These data reinforce that the composition of the diet, and not the obese phenotype is primarily responsible for observed differences in the fecal microbiome.
Figure 3. HFD-Induced Peripheral Abnormalities are Only Partially Protected in Drd1-KO Mice.
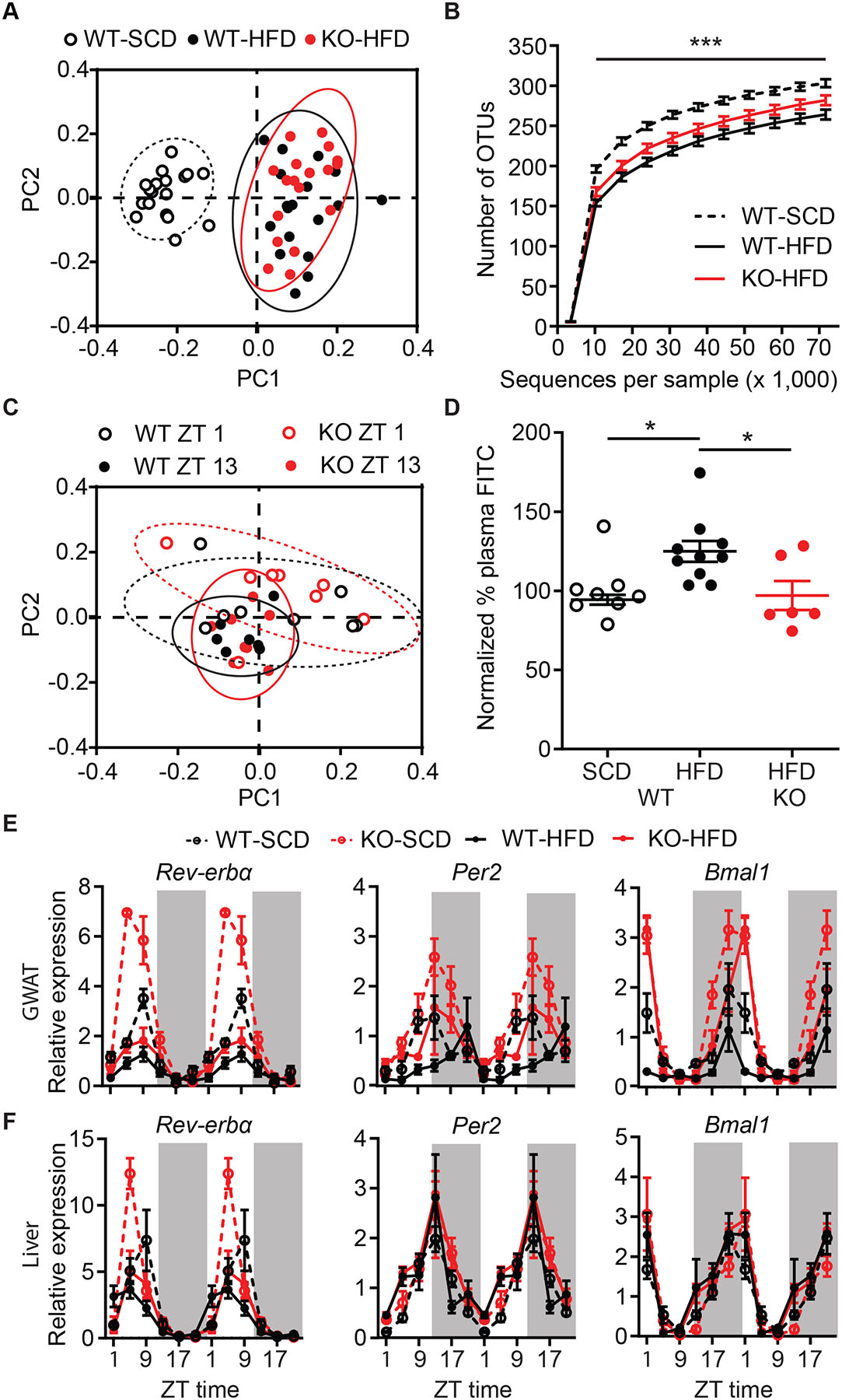
(A) Weighted UniFrac principal component analysis (PCA) of microbiome composition comparing WT-SCD, WT-HFD, and KO-HFD mice. 95% normal confidence ellipses are illustrated.
(B) Alpha diversity of fecal microbiome operational taxonomic units (OTUs). Repeated measures two-way ANOVA with Bonferroni post-hoc comparison, n=18/group; Ftreatment(2,51)=15.1, p<0.001. Statistical significance between WT-SCD and WT-HFD is depicted.
(C) Weighted UniFrac PCA analysis of fecal samples collected from the same mouse at ZT1 and ZT13 for WT-HFD and KO-HFD mice. 95% normal confidence ellipses are illustrated.
(D) Plasma fluorescence normalized to WT-SCD levels following oral gavage of FITC-Dextran. One-way ANOVA with Bonferroni post-hoc comparison, n=6–10/group; F(2,21)=4.9, p=0.02.
(E–F) Double-plotted RNA expression level of circadian genes Rev-erbα, Per2 and Bmal1 in GWAT (E) and liver (F). Three-way ANOVA with Bonferroni post-hoc comparison, n=3/time point/group (See also Table S2). Time is represented in ZT.
Data are represented as mean±SEM. *p<0.05,***p<0.001.
Contrary to the reported associations between gut microbiota and metabolic state, KO-HFD mice remain protected from adipocyte hypertrophy and insulin resistance despite having a dysbiotic microbiome (Figures 2–3) [44, 45]. Diet-induced changes in intestinal permeability provide the proinflammatory bacterial products access to the peripheral tissues [47–49]. Indeed, we observed that WT-HFD mice had enhanced permeability compared to both WT-SCD and KO-HFD animals. (Figure 3D). The decreased intestinal permeability and the resultant attenuated microbial byproduct infiltration in KO mice could provide some of the protection against the HFD-induced metabolic dysfunction observed in WT-HFD mice [48, 50].
Prolonged access to HFD also dampens circadian clock gene rhythms in peripheral tissues (i.e. WAT and liver) [33]. Because KO-HFD animals maintain a predominantly nocturnal pattern of feeding and robust metabolic rhythms, we evaluated circadian clock gene (Rev-erbα, Per2, and Bmal1) expression in GWAT and liver every four hours across the twenty-four-hour day (Figures 3E–F, and Table S2). Similar to previous reports, GWAT circadian clock genes in WT-HFD mice exhibited a reduced oscillatory amplitude compared to WT-SCD controls [10, 33] as did the KO-HFD group compared to KO-SCD mice. Despite their lean phenotype, the daily oscillation of clock genes in KO-HFD mice were generally indistinguishable from the WT-HFD mice. Therefore, the self-imposed restricted food consumption pattern of KO-HFD animals is not sufficient to sustain robust peripheral circadian clock rhythmicity as has been observed in mice on eight-hour time restricted feeding (TRF) [10]. Unlike the TRF paradigm, the protection from metabolic disease in KO-HFD mice cannot be fully explained by a resilient amplitude of clock gene expression in the periphery.
Re-expression of Drd1 Within the NAc Increases Daytime HFD Consumption without Inducing Obesity.
Energy homeostasis is controlled by a variety of neuronal circuits that influence food intake and energy expenditure suggesting that the obesity resistant phenotype of KO mice is due to a reduced central drive to overconsume palatable foods [51–53]. Therefore, we limited Drd1 ablation to distinct populations of neurons by crossing floxed Drd1 (Drd1fl/fl) mice with different Cre driver lines. Drd1 is mostly expressed in GABAergic populations except for a select group of CaMKII-positive cells in the hippocampus [54, 55]. To distinguish the role of Drd1 between these populations during palatable food consumption, we genetically ablated Drd1 expression using either CaMKIICre/+ or VGATCre/+ driver lines, respectively [55–57]. On HFD, CaMKIICre/+;Drd1fl/fl mice gained an equivalent amount of weight compared to CaMKIICre/+;Drd1+/+ controls (Figure S4B). However, like germline Drd1-null animals (Figure S1D), select ablation of Drd1 expression in GABAergic neurons (VGATCre/+;Drd1fl/fl mice) completely protected animals from diet-induced obesity (Figures 4A–B, and S4A).
Figure 4. Selective Re-expression of Drd1 Within the NAc of Drd1-KO Mice Partially Rescues HFD-Induced Phenotypes Excluding Obesity.
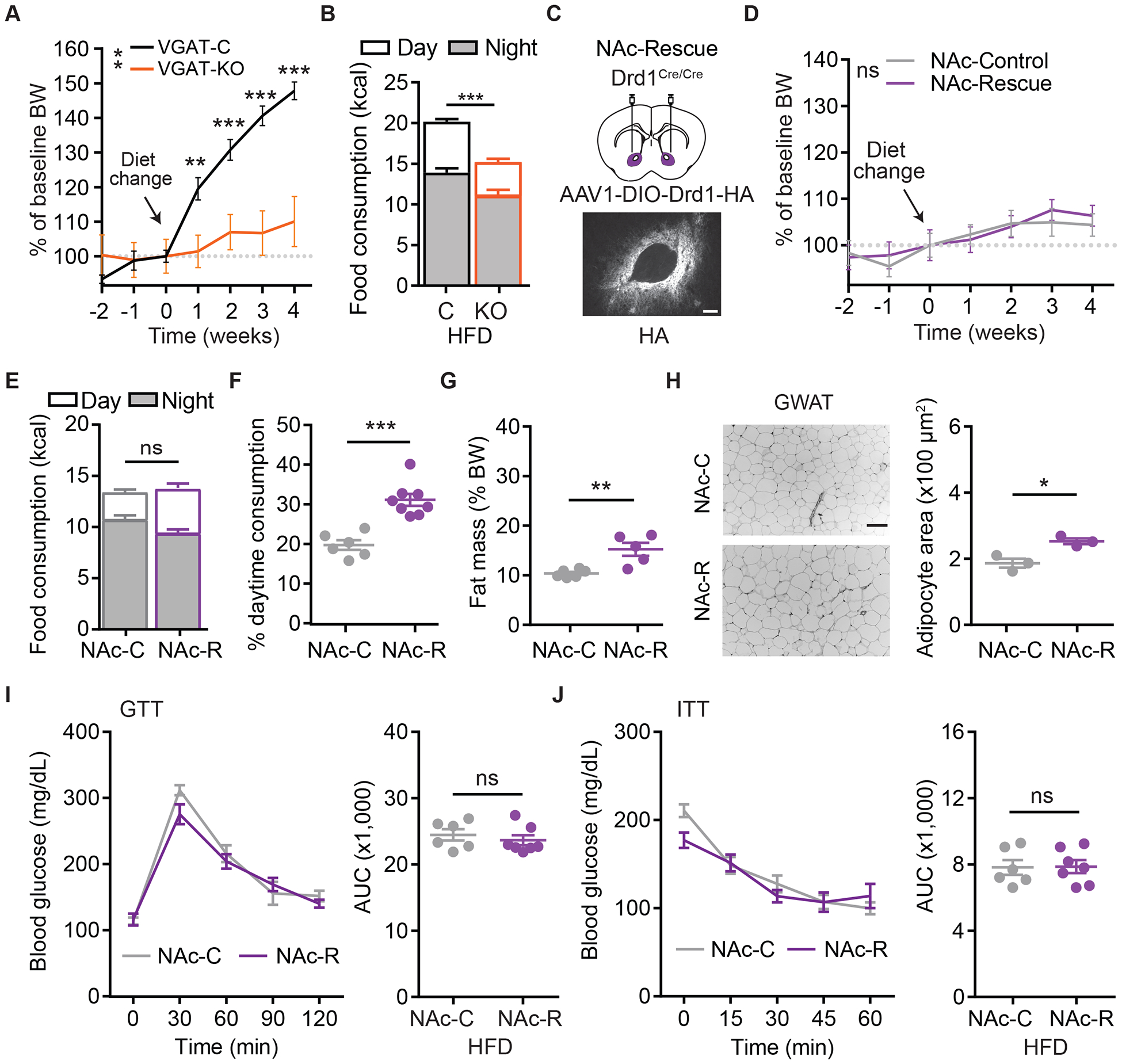
(A) Percent BW change relative to week 0 for VGAT+/+;Drd1fl/fl (VGAT-C), and VGATCre/+;Drd1fl/fl (VGAT-KO) mice following access to high fat/low sugar diet. Repeated-measures two-way ANOVA with Bonferroni post-hoc comparison, n=7–16/group; Fgenotype(1,21)=10.4, p=0.004.
(B) Daily total calorie consumption of VGAT-C and VGAT-KO mice on HFD. Day (white) and Night (grey) consumption are segregated for clarity. Student’s two-tailed t-test, n=7–16/group.
(C) Top: Schematic diagram illustrating NAc bilateral injection of AAV1-DIO-Drd1-HA or AAV1-ChR2-YFP in KO (Drd1cre/cre) mice. Bottom: Anti-HA immunohistochemical labeling within the NAc. Scale bar=100um.
(D) Percent BW change relative to week 0 for NAc-control (NAc-C) and NAc-Rescue (NAc-R) mice on HFD. Repeated-measures two-way ANOVA with Bonferroni post-hoc comparison, n=6–8/group.
(E) Daily total calorie consumption of NAc-control (NAc-C) and NAc-Rescue (NAc-R) mice on HFD. Student’s two-tailed t-test, n=6–8/group.
(F) Portion of Day consumption on HFD. Student’s two-tailed t-test, n=6–8/group.
(G) Fat mass as percent BW on HFD. Student’s two-tailed t-test, n=5–6/group.
(H) Representative GWAT histology with H&E stain (left) and quantification of GWAT adipocyte area (right). Scale bar=100μm. Student’s two-tailed t-test, n=3/group.
(I) and (J) Blood glucose levels over time (left; repeated-measures two-way ANOVA with Bonferroni post-hoc comparison, n=6–7/group) and area under the curve (AUC) quantification (right; Student’s two-tailed t-test, n=6–7/group) during GTT (I) or ITT (J).
Data are represented as mean±SEM. *p<0.05,**p<0.01,***p<0.001, ns=not significant.
Next, we restored Drd1 expression to the nucleus accumbens (NAc), a primary DA recipient region within the mesolimbic pathway, in Drd1-KO mice by bilaterally delivering an adeno-associated virus 1 (AAV1) containing a Cre recombinase dependent Drd1 transgene (Drd1-HA; NAc-Rescue). Because Cre is knocked-in to replace the endogenous Drd1a gene in these mice, Drd1-HA expression remains confined to the cells with active Drd1a promoter at the targeted site [30]. Successful targeting of Drd1-HA was determined by anti-HA immunohistochemistry (Figures 4C and S4E), and functional expression was evaluated following intraperitoneal injections of the Drd1 agonist SKF-81297 (i.p.; 7.5 mg/kg) (Figure S4D) [30]. Drd1-KO mice injected with AAV1 encoding the light-activated, membrane-localized cation channel Channelrhodopsin-2 (ChR2-eYFP) to the NAc were used as controls (NAc-Control). Surprisingly, re-expression of Drd1 within the NAc resulted in no appreciable increase in BW on HFD (Figures 4D, S4F, and S4I). Additionally, we observed no difference in twenty-four-hour consumption compared to controls on either diet, however, NAc-Rescue mice increased the portion of Day consumption on HFD (NAc-Control: 19.8±1.2%; NAc-Rescue: 31.1±1.5%; Figures 4E–F, and S4G–H). This elevated rest phase consumption resulted in a marginal, but statistically significant increase in percentage fat mass, GWAT adipocyte area, and hepatic steatosis (Figures 4G–H, and S4J–L). However, similar to germline KO mice, NAc-Rescue mice maintained glucose tolerance and insulin sensitivity (Figures 4I–J). Despite increasing the portion of rest-phase feeding, re-expression of Drd1 within the NAc is not sufficient to induce HFD overconsumption and obesity, indicating that another Drd1-expressing brain region governs this response.
HFD and Drd1-signaling Modulate Behavioral Circadian Period and SCN Neurophysiology
The observed temporal change of feeding pattern in WT-HFD suggests a disruption in circadian rhythmicity. In addition to alterations in peripheral circadian clock gene expression, access to HFD lengthens centrally regulated activity rhythms [33]. To determine whether HFD-induced disruption of circadian behavioral rhythms is dependent on Drd1 signaling, we monitored wheel running activity of WT and KO mice following four weeks of SCD or HFD access. As expected, WT-HFD mice exhibited a lengthened free-running period compared to WT-SCD animals that was reversible upon diet change. However, no difference between KO-HFD and SCD controls was observed (Figures 5A–C, and S5A–B). Resistance to HFD-induced period lengthening in KO mice suggests a role for Drd1 signaling in the disruption of central circadian rhythms during dietary challenge.
Figure 5. Elevated Drd1 Signaling Reduces Neuronal Firing Rate in the SCN.

(A) Representative double-plotted actogram of wheel running activity in constant darkness (DD). White and grey background indicates the light and dark phase of the LD cycle, respectively.
(B) Average daily activity onset time. Repeated-measures three-way ANOVA with Bonferroni post-hoc comparison, n=7–10/group; Fgenotype(1,28)=11.7, p=0.002, Fdiet(1,28)=15.5, p<0.001.
(C) Average DD free running period (Tau). Two-way ANOVA with Bonferroni post-hoc comparison, n=6–9/group; Fgenotype(1,27)=16.2, p<0.001, Fdiet(1,27)=14.01, p<0.001.
(D) Quantification of dopamine turnover (DOPAC/DA ratio) following one-hour SCD or HFD exposure in hypothalamic tissue punches containing the SCN. Student’s two-tailed t-test, n=7/group.
(E) Mean firing rate of cell attached SCN neuron recordings following SKF-81297 (5μM) incubation at ZT8–11 (Day) or ZT14–17 (Night). Two-way ANOVA, n=13–26/group; Ftime(1,70)=16.9, p<0.001, Ftreatment(1,70)=6.8, p=0.01.
(F) Schematic of bilateral AAV2-DIO-ChR2-YFP injection in the SCN of Drd1cre/+ mice.
(G) Representative traces from whole-cell current-clamp recordings. SCN neurons exhibit both depolarizing (top) and hyperpolarizing (bottom) responses to 20 Hz stimulation.
(H) ChR2 stimulation-induced average peak responses for depolarizing (Depol.), hyperpolarizing (Hyper.) and non-responsive (None) neurons. Distribution of recorded responses are depicted. (B.) indicates one cell exhibiting both responses (see Figure S5H).
(I) Response latency of each group in (H). Student’s two-tailed t-test, n=11/group.
(J) Representative traces from whole-cell current-clamp recordings for ChR2 stimulation-induced postsynaptic responses before and after 5 min 100μM PTX incubation.
(K) ChR2 stimulation-induced average peak responses before and after PTX incubation. Student’s two-tailed paired t-test, n=5/group.
Data are represented as mean±SEM. *p<0.05,**p<0.01,***p<0.001.
See also Figure S5.
Since the HFD-induced circadian period lengthening is Drd1-dependent, we sought to determine whether acute exposure to HFD changes DA tone in the SCN. To address this, we determined the DOPAC and DA content from hypothalamic tissue punches containing the SCN in WT animals following one-hour access to either SCD or HFD (Figure S5C). The DOPAC/DA ratio is an estimate of DA turnover reflecting active DA release and catabolism, which rapidly increases within the NAc in response to HFD [21, 58]. WT mice given one hour access to HFD during their inactive phase (ZT6) consumed significantly more food than SCD controls (SCD: 0.3±0.08kcal; HFD: 5.4±0.5kcal; Student’s two-tailed t-test, p<0.001). Accordingly, the hypothalamic DOPAC/DA ratio of animals fed HFD was elevated (SCD: 0.24±0.02; HFD: 0.35±0.02; Figure 5D). Previously, we have demonstrated that a fraction of the ventral tegmental area (VTA) DA neurons innervate the SCN [59], representing a potential source of DA to this region. By using a Cre-dependent rabies viral tracing strategy, we validated that the SCN-Drd1 neurons specifically receive VTA dopaminergic projections (Figure S5D). Thus, consumption of palatable foods increases DA turnover around the SCN region and therefore may modulate the function of the central circadian clock.
Rewarding foods have been shown to hasten a behavioral phase shift in response to photic stimulation and modulate circadian entrainment, as daily presentation of palatable food entrains circadian rhythms to the time of food availability [60–62]. Because KO mice fail to show HFD-induced circadian period lengthening, we evaluated the consequence of long-term HFD access on the central circadian clock by quantification of two rhythmic molecular markers within the SCN, c-Fos and Per2, at antiphasic time points (ZT1 and 13). We detected a significant time of day effect in both c-Fos and Per2 expression, however, we observed no diet or genotype effect (Figures S5E–F). These data suggest that while acute access to HFD elevates DA turnover within the SCN, prolonged access to HFD does not significantly disrupt the peak-to-trough expression pattern of circadian regulated proteins.
To reconcile the dichotomous observations that in response to HFD there is increased DA turnover in the SCN but no change in circadian gene expression patterns, we evaluated the impact of increased Drd1 signaling on neuronal activity within the SCN. To accomplish this, we incubated acute brain slices from WT mice with the selective Drd1 agonist (SKF-81279; 5μM) and performed loose cell attached recordings at ZT8–11 (Day) and ZT14–17 (Night). Incubation with SKF-81297 significantly decreased the firing rate of neurons within the SCN during both phases (Day: DMSO 4.3±0.4Hz; Day: SKF 3.2±0.4Hz; Night: DMSO 2.6±0.4Hz; Night: SKF 1.5±0.4Hz; Figure 5E). Since Drd1 is a Gs-coupled GPCR, and the majority of SCN neurons are GABAergic [63, 64] (Figure S5G), we hypothesized that stimulation of Drd1-expressing SCN neurons attenuates the overall firing rate of the local microcircuitry. Therefore, we performed whole cell current clamp recordings from the SCN of Drd1Cre/+ mice injected with an AAV2 that expresses ChR2 Cre-dependently (AAV2-DIO-ChR2-YFP; Figure 5F). Neurons were randomly selected during electrophysiological recordings independent of ChR2 expression within the anatomically defined region of the SCN. While 26% of recorded cells (8 cells) had no response to light stimulation, 35% of the cells depolarized within 1–2ms of the stimulation with time-locked optogenetic responses indicative of ChR2 expression in these cells (11 cells). Additionally, 35% of the SCN-neurons hyperpolarized following a minimum of 4 ms delay (11 cells; Figures 5G–I, and S5H), while one neuron displayed both light-induced action potentials and post-stimulation hyperpolarization (Figure S5I). Perfusion of a GABAA channel inhibitor, picrotoxin (PTX; 100 μM), diminished the amplitude of the inhibitory responses, confirming that they are ChR2-driven postsynaptic currents (Figures 5J–K, and S5J). These data identify a local inhibitory response to Drd1-SCN neuron activation, revealing that the HFD-induced elevated DA tone may directly modulate SCN firing rate.
Re-expression of Drd1 Within the SCN Restores Diet-induced Obesity
Although HFD does not significantly dampen circadian gene expression within the SCN (Figure S5), the increased DA turnover within the SCN in response to HFD and the dampened electrical firing rate in response to Drd1 agonist treatment (Figure 5) suggests that DA-dependent SCN-Drd1 signaling may play a prominent role in the increased daytime food intake and obesity observed in WT-HFD mice (Figures 1 and 5). To demonstrate whether Drd1 expression in the SCN is sufficient to establish HFD-induced obesity, we virally re-introduced Drd1 within the central circadian pacemaker by delivering Drd1-HA to the SCN of KO mice (SCN-Rescue) (Figure 6A) [59, 65]. Mice injected with AAV1 expressing ChR2-eYFP were used as controls (SCN-Control). Remarkably, HFD-induced obesity and the subsequent metabolic consequences were completely restored in SCN-Rescue animals as we observed a significant increase in BW, food consumption, adipose mass, adipocyte hypertrophy, hepatic steatosis, glucose intolerance, and insulin resistance relative to SCN-Controls (Figures 6B–J, and S6A–G). SCN-Rescue mice maintained a predominantly nocturnal pattern of food consumption on SCD, while on HFD they significantly increased daytime consumption (SCD: 20.3±3.1%; HFD: 37.4±2.3%; Student’s two-tailed t-test, p<0.001) (Figures 6D–E, and S6B–C). Most notably, nighttime consumption was not reduced on HFD, leading to an overall increase in food consumption and substantial weight gain (Figures 6B–E). Unlike restoration in the NAc, genetic reconstruction of Drd1 expression within the SCN of KO mice enabled out-of-phase overconsumption of HFD leading to all of the metabolic consequences observed in WT-HFD mice.
Figure 6. Re-expression of Drd1 Within the SCN of Drd1-KO Mice Restores HFD-Induced Obesity.

(A) Top: Schematic diagram illustrating AAV1-DIO-Drd1-HA SCN bilateral injection in the Drd1-KO (Drd1cre/cre) mice. Bottom: Anti-HA fluorescent immunohistochemistry staining within the SCN of SCN-Rescue mice. Scale bar=100μm.
(B) Percent BW change relative to week 0 for SCN-C and SCN-R mice on HFD. Repeated-measures two-way ANOVA with Bonferroni post-hoc comparison, n=10–12/group; Fgenotype(1,20)=4.5, p=0.05.
(C) Representative images of SCN-C and SCN-R mice (6A–B). Scale bar=1cm.
(D) Daily total calorie intake on HFD. Day and Night consumption are segregated for clarity. Student’s two-tailed t-test, n=10–11/group.
(E)Portion of food intake during the Day on HFD. Student’s two-tailed t-test, n=10–11/group.
(F) Fat mass as percent BW. Student’s two-tailed t-test, n=7/group.
(G) Left: representative GWAT H&E. Scale bar=100μm. Right: quantification of GWAT adipocyte area. Student’s two-tailed t-test, n=3/group.
(H) ORO of liver. Scale bar=100μm.
(I) GTT, repeated-measures two-way ANOVA with Bonferroni post-hoc comparison, n=8–9/group; Fgenotype(1,15)=27.1, p<0.001.
(J) ITT, repeated-measures two-way ANOVA with Bonferroni post-hoc comparison, n=6–8/group; Fgenotype(1,12)=11.2, p=0.006.
Data are represented as mean± SEM. *p<0.05,**p<0.01,***p<0.001.
DISCUSSION
Modern society places a significant burden on the primitive circadian machinery whereby the central pacemaker is under constant dysregulation by artificial lighting. Moreover, easy access to palatable foods contributes to maladaptive eating, obesity and metabolic disease in rodents and humans [8, 66, 67]. Diets rich in fat and sugar increase out-of-phase food consumption, however, the neuronal correlates of meal timing have remained elusive. Here, we identify a novel mechanism by which energy-rich foods dysregulate properly timed consumption by interfering with neural activity of the central circadian pacemaker within the SCN. This modulation results in increased foraging between meals and overconsumption leading to obesity. We observe that Drd1 expression is necessary for HFD-induced rest-phase hyperphagia, amplitude reduction in circadian oscillations of metabolic rhythms, lengthening of wheel running circadian period, and obesity. We also demonstrate that DA turnover within the SCN increases in response to HFD, while stimulation of the Drd1 signaling in the SCN dampens the overall activity of this nucleus. Lastly, Drd1 expression only within the SCN is sufficient to promote HFD overconsumption and metabolic disease. Therefore, we hypothesize that elevated DA during access to HFD increases out-of-phase foraging and overconsumption by Drd1 mediated reduction in SCN activity (Figure 7). In this view, DA, beyond providing hedonistic signaling via the mesolimbic circuit, also hinders anorexic output from the central circadian pacemaker between mealtimes contributing to excessive calorie intake.
Figure 7. High-energy diet drives ectopic dopaminergic tone in the SCN, resulting in out-of-phase feeding.

As nocturnal animals, mice eat 80% of their food during the night when chronically exposed to a healthy diet (left). In contrast, mice on high calorie diet eat roughly 40% of their food during the day. Rewarding high-energy foods induce dopamine release. We hypothesize that the increase in SCN dopaminergic tone attenuates overall SCN excitability via Drd1 signaling resulting in: 1. Increased out-of-phase daytime foraging, 2. Overconsumption of calories, 3. Reduced synchrony between central and peripheral clocks, 4. Obesity and metabolic disorder (right). Remarkably, Drd1 signaling in the SCN enables feeding and metabolic defects associated with chronic exposure to high calorie food.
Diet regimens that involve time restricted feeding are growing in popularity because they attenuate BW gain and significantly improve metabolic profiles in rodents and humans [10, 12, 68, 69]. Our findings demonstrating that the lack of Drd1-signaling imposes self-restricted, energy-rich food consumption predominantly during the active-phase, and provides timely insight to how rewarding, energy dense foods interfere with meal timing and diet adherence. Consumption of food beyond regular activity periods generates peripheral circadian desynchrony [10, 33]. Surprisingly, despite maintaining a lean phenotype, KO-HFD mice exhibit peripheral circadian gene expression profiles similar to the WT-HFD group (Figure 3 and Table S2). This suggests that peripheral clock gene expression in GWAT is mainly influenced by the dietary composition and the observed peripheral clock disruption likely precedes weight-gain. Similarly we observed that changes in the fecal microbial composition are primarily dependent on the diet type [70]. Our data suggest that, rather than the microbiome composition alone, an increased permeability of the intestinal barrier resulting from overconsumption contributes to the negative consequences of a dysbiotic microbiome [49]. Given that WT and KO animals on HFD maintain a similar microbiome and rhythmicity of peripheral clock gene expression, the obesity resistant phenotype of Drd1-ablated animals is primarily driven by the central processes that control the circadian phase and amount of food consumed.
Surprisingly, re-expression of Drd1 signaling within the NAc, a hub of reward processing, fails to increase daily HFD consumption and induce obesity in KO mice (Figure 4). These results parallel another study in which NAc-Rescue mice fail to perform at control levels in a progressive ratio task although they exhibit marked improvements in instrumental responding for reward [30]. Of note, NAc-Rescue animals increase their daytime HFD consumption, but compensate for the increase by reducing nighttime consumption, which leads to the maintenance of twenty-four hour HFD intake similar to the KO mice levels (Figure 1 and 6). Although this pattern of feeding in NAc-Rescue mice does not induce weight gain or alteration in glucose and insulin responsiveness, it leads to a moderate increase in adiposity (Figure 4). This increase might be the result of the metabolic disorganization caused by out of phase food consumption similar to what has been observed in day time TRF experiments [10, 34]. By contrast, re-expression of Drd1 within the SCN increases daytime foraging, without significantly disrupting feeding at night, resulting in daily overconsumption (Figure 6). We postulate that the Drd1-dependent DA signaling within the NAc supports foraging for palatable foods [71], but the increased out of phase energy intake is compensated for by a homeostatic response. Our observations reveal a critical role for the Drd1-SCN signaling in energy balance during HFD consumption and also suggests that increased daytime consumption without an increase in overall energy intake is insufficient for significant weight gain.
Activities that enhance dopaminergic signaling such as exercise, access to a running wheel, or energy-dense diets are known to influence circadian regulated behaviors [33, 72–74]. Most relevant to our studies, consumption of HFD increases the period length of general activity rhythms, which is driven by the period of the Drd1-positive SCN cells (Figure 5) [33, 74]. Accordingly, Drd1 agonist treatment lengthens the period of Per2 expression in SCN slices [75]. In line with these reports, the wheel running activity period of WT but not Drd1-KO mice rapidly and reversibly changes in response to HFD (Figures 5 and S5) suggesting that the diet composition governs behavioral circadian periods in a Drd1-signaling dependent manner.
Drd1-DA neurotransmission outside the SCN has also been implicated in a variety of circadian behaviors [38, 76–80]. For instance, while NAc Drd1-activity regulates the transitions from sleep to wakefulness, Drd1 in the dorsal striatum plays a critical role in food anticipatory activity (FAA) following calorie restriction [78]. Ablation of these striatal dopamine signaling pathways in Drd1-KO mice could formally contribute to the observed circadian feeding patterns on HFD. However, in our studies, WT and KO mice demonstrate similar levels of rest phase locomotor activity (Figure 2) and hourly putative sleep duration (Figure S2) suggesting reduced roles for these pathways in the resistance of diet-induced obesity in KO-HFD mice. Additionally, reinstatement of WT-like HFD-induced metabolic and behavioral responses in SCN-Rescue animals reiterates that the circadian structure of rewarding food consumption and the associated weight gain is facilitated primarily through Drd1 signaling within the SCN. A complement to our current approach is selective SCN ablation of the Drd1 expression, which would reveal whether Drd1-signaling in this nucleus is required for diet-induced obesity. However, our efforts towards this goal could only accomplish partial Drd1 knock-down (Figures S6H–K), which was not sufficient to prevent diet-induced obesity. Therefore, future studies will focus on combinatorial transgenic ablation strategies to generate a complete SCN specific Drd1-KO line.
A vast majority of SCN neurons are GABAergic and exhibit synchronous neural activity that fluctuates across the day:night cycle [64]. Their activity is high during the day, producing an inhibitory tone within their projection sites throughout the hypothalamus (DMH and SPVZ), thalamus, and basal forebrain [81, 82]. In mice, this diurnal activity suppresses behavioral responses usually observed during the nighttime such as feeding and foraging [83]. Here, we demonstrated that increased SCN-Drd1 signaling, or optogenetic stimulation of Drd1-SCN neurons reduces neuronal activity within the SCN or inhibits postsynaptic SCN neurons, respectively (Figure 5). Therefore, we postulate that in response to rewarding foods, increased DA tone decreases the overall excitability within the SCN. This decrease in activity mimics the nighttime SCN neuronal activity, which is permissive for foraging, leading to overconsumption of available energy-rich foods. The precise downstream targets of the SCN-Drd1 circuit will be the focus of future investigations seeking to uncover how circadian rhythms integrate with the orexigenic signals at the neurocircuitry level.
LEAD CONTACT AND MATERIALS AVAILABILITY
This study did not generate new unique reagents or mouse lines. Further information and requests should be directed to and will be fulfilled by the Lead Contact, Ali D. Güler (aguler@virginia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal care experiments were conducted in concordance with University of Virginia Institutional Animal Care and Use Committee (IACUC). Animals were housed in a temperature and humidity controlled vivarium (22–24°C, ~40% humidity) until experimental use on a 12-hour:12-hour light:dark (LD) cycle and were provided with food and water ad libitum and cotton nesting material (Ancare, Bellmore, NY). All behavioral assays were conducted with male C57BL6/J mice which were 12–14 weeks old at the onset of food intake experiments. In addition to wild-type C57BL6/J mice, the following mouse lines were used: Drd1aCre/+ (Drd1-HET) [26], CamKIIa-creT29-1Stl/J [84], Drd1tm2.1Stl/J (Floxed-Drd1 or Drd1fl/fl) [55], Drd1tm1Jcd/J (Drd1 −/−) [27], Slc32a1tm2(cre)Lowl/J (VGAT-Cre) [57], and C57BL/6-Tg(Nms-icre)20Ywa/J (NMS-Cre) [85]. Drd1aCre/Cre (KO) mice and littermates were raised on Teklad 8664 (Envigo, United Kingdom) placed on the cage floor as Drd1-KO mice were more likely to consume food when it was readily accessible.
METHOD DETAILS
Mouse Diets
Standard chow diet (SCD):Teklad 8664 (Envigo, United Kingdom: 3.1 kcal/gram; 19% fat, 31% protein, 50% carbohydrates), Teklad 7912 (Envigo, United Kingdom: 3.1 kcal/gram; 17% fat, 25% protein, 58% carbohydrates) or PicoLab Rodent Diet 20 5053 (3.07 kcal/gram; 13% fat, 24% protein, 62% carbohydrates; 3.2% sucrose). High-fat, high-sugar diet (HFD): Open Source D12451 (4.73 kcal/gram; 45% fat, 20% protein, 35% carbohydrates; 17% sucrose). For Figure 4 and Figure S4 floxed animals, Normal chow diet (NCD), Teklad 2018 (3.1 kcal/gram; 18% fat, 24% protein, 58% carbohydrates). High-fat, low-sugar diet: BioServ F3282 (Flemington, New Jersey: 5.49 kcal/gram; 60% fat, 15% protein, 26% carbohydrates). Ensure original vanilla (24.3% fat, 16.2% protein, 59.5% carbohydrates).
Food Intake Measurements
Adult mice were housed individually in standard home cages with ad libitum access to SCD food and water throughout all experiments. For longitudinal feeding experiments, 12–14 week old mice were then randomly assigned into two different feeding regimens, SCD or HFD. All mice were maintained on SCD for an additional two weeks to measure baseline food intake and weight change. SCD animals remained on SCD for an additional four weeks at the time of the “diet switch” while the HFD group was given ad libitum access to HFD. Pre-weighed food pellets were placed on the cage floor and refreshed weekly. Body mass was measured weekly. For the daily feeding studies, animals were maintained on SCD or habituated to HFD for at least three days prior to the start of intake measurements. Pre-weighed food pellets were given on the cage floor at the start of the experiment, and body mass and food intake was measured at intervals of 12 h (ZT 0 and ZT 12) over a 72 h period starting at ZT 12. Food intake measurements were obtained by subtracting the mass of the residual food pellets from the total food given. Total caloric intake was calculated by multiplying the calories per gram of food and the mass of food consumed.
Constant Darkness Food Intake
Mice were individually housed in standard cages, entrained to a 12-hour:12-hour light:dark (LD) cycle, then released into constant darkness (DD) for at least seven days. Following release to DD, homecage general activity was monitored using infrared (IR) beam beaks with six minute internals. The initiation of subjective night and circadian free-running period (DD night; CT 12) was predicted for each animal using ClockLab collection and analysis system (Actimetrics, Wilmette, IL). Initiation of subjective day (DD day; CT 0) was calculated by the addition of half the free-running period length to CT 12 for each individual animal. The final collection point was determined by adding the duration of the endogenous free running period to the CT 12.
Fecal Bomb Calorimetry
Fecal output data was collected over a period of 72 hours. TheGl feces within the bedding was collected, freeze dried (Labconco) and weighed. For analysis of fecal calorie content, samples were collected, 2 mice/ n sample (~1 gram/sample) for fecal bomb calorimetry (Par 6200 isoperibol calorimeter, UT southwestern).
Histological analysis and imaging
Animals were deeply anesthetized (ketamine:xylazine, 280:80 mg/kg, i.p.) and perfused intracardially with ice cold 0.01 M phosphate buffer solution (PBS) followed by fixative solution (4% paraformaldehyde (PFA) in PBS at a pH of 7.4). After perfusion, brains were dissected and post-fixed overnight at 4°C in P FA. Freshly collected brains, after cervical dislocation were incubated in cold 4% PFA for 48 hours. Fixed brains were then rinsed in PBS, transferred into 30% sucrose in PBS for 24 hours, and then frozen on dry ice. Coronal sections (30 μm) were collected with a cryostat (Microm HM 505 E). Sections were permeabilized with 0.3% Triton X-100 in PBS (PBS-T) and blocked with 3% normal donkey serum (Jackson ImmunoResearch) in PBS-T (PBS-T DS) for 30 min at room temperature. Sections were then incubated overnight in primary antibodies diluted in PBS-T DS. For visualization, sections were washed with PBS-T and incubated with appropriate secondary antibodies diluted in the blocking solution for 2 hours at room temperature. Sections were washed three times with PBS and mounted using DAPI Fluoromount-G (Southern Biotech). Images were captured on a Zeiss Axioplan 2 Imaging microscope equipped with an AxioCam MRm camera using AxioVision 4.6 software (Zeiss). Confocal microscope imaging was performed in W.M. Keck Center for Cellular Imaging, University of Virginia, with Leica SP5 X imaging system. The following primary antibodies were used for fluorescent labelling: anti-Drd1 (rat, 1:500, Sigma D2944), anti-HA (rabbit, 1:500, Cell Signaling Technology C29F4), anti-c-Fos (rabbit, 1:1000, synaptic systems), anti-GAD65/67 (rabbit, 1:2,000, Abcam ab49832), anti-tyrosine hydroxylase (chicken, 1:500, Millipore AB9702), anti-GFP (rabbit, 1:1k, Invitrogen A-6455). The secondary antibodies (Jackson ImmunoResearch) used were Cy2- or Cy3-conjugated donkey anti-rat IgG (1:250), donkey anti-rabbit (1:250) and Alexa Fluor 488-conjugated goat anti-chicken IgY (1:250, Invitrogen A11039).
PER2-DAB
Brains sections were permeabilized and blocked via the process described above. anti-PER2 (rabbit; 1:1000 AlphaDiagnostic International, PER21-A) primary antibody was incubated at 4 °C for 20 hours. Secondary antibody and DAB staining was processed with a VECTASTAIN Elite ABC-HRP Kit following the company-recommended protocol.
Oil-Red-O
PFA fixed tissue was rinsed in 1% PBS, transferred to 30% sucrose overnight, then frozen in 2-Methylbutane chilled by dry ice. 8 μm thick sections were collected with a cryostat (Microm HM 505 E) and mounted onto gelatin coated slides. Slides were treated with Oil Red O (ORO) solution (3mg/ml in 60% isopropanol) for 18 hours. Sections were then imaged by Nikon ECLIPSE Ti microscope equipped with a Nikon DS-Ri2 color camera.
Hematoxylin & Eosin (H & E) staining
Bouin’s solution fixed tissues were washed 2 × 5 min in PBS and 3 × 5 min in 70% EtOH. Tissues were then sent to UVa Research Histology Core for paraffin embedding, sectioning (6μm) and H&E staining. Sections were imaged in the same way as ORO sections above.
Western Blot p-HSL
Samples of GWAT were collected from ad libitum fed mice at ZT 6 and flash frozen in liquid nitrogen. Adipose samples were homogenized in lysis buffer and centrifuged at 18,000g at 4°C for 20 minutes. The resultant supern atant was collected into a fresh eppendorf tube and protein levels in extracts were determined using the BCA method (Peirce). Protein was loaded equally (30 μg/lane) into lanes of a 4–15% gradient gel (BioRad) and separated by SDS-PAGE, and blotted onto 0.2 micron PVDF membranes. Blots were blocked for 1 hour at room temperature in a half-strength mixture of LiCOR Blocker (LiCOR Blocker (TBS): 1X TBS buffer). Primary antibody against HSL (Cell Signalling Technology 4107), and pHSL 660 (Cell Signalling Technology 4126) rabbit polyclonal used at 1:2000. The blots were washed in 1X TBS containing 0.01% Tween-20, and secondary antibody goat-anti-rabbit LiCOR 800CW was applied in LiCOR Blocker (TBS): 1X TBS buffer, used at 1:10,000 dilution for one hour at room temperature. Signal was normalized against tubulin using hFAB rhodamine-labelled anti-tubulin antibody fragment (BioRad 12004166), used at 1:2,000, and incubated along with LiCor 800 CW secondary. Finally, the blot was washed 6 times for 5–10 minutes each in 1X TBS + 0.01% Tween-20 followed by 2 final rinses for 5–10 minutes in 1X TBS alone. The blot was dried and imaged by fluorescence on a LiCor BioRad ChemiDoc MP station and bands quantified using BioRad ImageLab software. Protein concentrations of pHSL were determined by quantitative blot immunolabeling by a trained experimenter, blind to genotype and dietary conditions
Viral Expression and Stereotaxic Surgery
During surgery, animals were anesthetized with isoflurane (induction 5%, maintenance 2%–2.5%; Isothesia) and placed in a stereotaxic apparatus (Kopf). A heating pad was used for the duration of the surgery to maintain body temperature and ocular lubricant was applied to the eyes to prevent desiccation. A double-floxed inverted open reading frame (DIO) cassette containing recombinant AAV was used to express specific transgenes in Cre-expressing neurons. AAV was delivered using a 10 μl syringe (Hamilton) and 26-gauge needle (Hamilton) at a flow rate of 100 nl/min driven by a microsyringe pump controller (World Precision Instruments, model Micro 4). The syringe needle was left in place for 10 min and was completely withdrawn 20 min after viral delivery. Following surgery, mice were administered ketoprofen (3 mg/kg) subcutaneously as an analgesic. Animals were tested at least two weeks following virus injection to ensure optimal transgene expression. All surgical procedures were performed in sterile conditions and in accordance with University of Virginia IACUC guidelines.
Viral constructs
AAV2-hSyn-DIO-ChR2-YFP, AAV1-hSyn-ChR2-eYFP, AAV1-CAG-DIO-Drd1-HA (500 nl; 1.1 × 10^13 viral genomes/ul) were injected into the NAc (ML: + 1.15 mm, AP: + 0.98 mm, DV: − 5.75 mm), SCN (ML:±0.29 mm, AP: − 0.30 mm, DV: −5.75 mm). All coordinates are relative to bregma (George Paxinos and Keith B. J. Franklin).
Retrograde tracing
Rabies virus tracing: 500nl AAV1-synP-FLEX-splitTVA-EGFP-B19G was injected to SCN of Drd1-HET mice using the method described above. Three weeks after AAV injection, 120nl EnvA-dG-Rabies-H2B-mCherry was delivered to the same coordinates. After one week, animals were deeply anesthetized (ketamine:xylazine, 280:80 mg/kg, i.p.) and perfused intracardially with ice cold 0.01 M phosphate buffer solution (PBS) followed by fixative solution (4% paraformaldehyde (PFA) in PBS at a pH of 7.4). Tissue was then processed for antibody labeling as described above.
Circadian Behavioral Analysis
To record the rhythm of locomotor activity, adult male mice were individually housed in activity wheel-equipped cages (Nalgene) in light-tight boxes under a 12h :12h LD cycle for at least 7 days. Fluorescent lights (100 mW/cm2) were used for behavioral experiments. Food and water were provided ad libitum. Wheel running rhythms were monitored and analyzed with ClockLab collection and analysis system (Actimetrics, Wilmette, IL). The free-running period was calculated according to the onset of activity across seven days in constant darkness. Activity onset was identified through ClockLab software as the first bin above a threshold of 5 counts preceded by at least 6 hours of inactivity and followed by at least 6 hours of activity. When necessary, onset and offset points were edited by eye. All data was analyzed by a trained scorer blind to genotype.
Putative sleep analysis by infrared chamber
WT-HFD and KO-HFD mice were individually housed in standard cages with ad libitum access to water and food. Assessment of activity/inactivity was recorded by Animal Activity Meter: Opto-M4 (Columbus Instruments, Columbus, OH). This system provides horizontal infrad-red beams that are 0.5 inches apart. The accompanying software provides counts of total and ambulatory activity of animals, in 10 second intervals. We examined total activity counts in the X-plane. Zero total beam-break activity for more than 40 consecutive seconds was determined to be putative sleep as described in [86].
SKF-81297 to NAc-Rescue Locomotor Assay
Mice were habituated to experimental housing conditions for 30 minutes. Following 20 minutes of baseline activity recordings, 7.5 mg/kg SKF-81297 (in saline) was administered by i.p. Injection. An additional 90 minutes of animal activity was recorded for analysis. Movement was tracked by EthoVision XT 11 (Noldus) and analyzed by EthoVision XT 11 (Noldus) and a custom Matlab script.
Glucose tolerance
Five weeks after the time of diet change, mice were fasted for 16 hours (ZT 10 - ZT 2), and fasted glucose was recorded using a Glucometer (One Touch Ultra) by tail bleeds [10]. Subsequently, mice received an i.p injection of glucose (1g/kg BW in saline), and blood glucose was measured in intervals of 30 minutes for 2 hours.
Insulin tolerance
Five weeks after the time of diet change, mice were fasted for 4 hours (ZT 1 - ZT 5), and fasted glucose was recorded using a Glucometer (One Touch Ultra) by tail bleeds. Subsequently, mice received an i.p injection of insulin (0.75units/kg of BW in saline), and blood glucose was measured in intervals of 15 minutes for 1 hour [87]. The time of collection (ZT 2 for GTT and ZT 5 for ITT) was chosen during peak response to glucose challenge which falls between ZT 2 and ZT 8 [88].
Body composition
Six weeks after time of diet change SCAT, GWAT, and liver were harvested, weighed, placed into a chilled 4% PFA solution or Bouin’s solution (fisher scientific) for fixation. Whole tissue was imaged (Canon EOS Rebel Xsi) before fixation. A ruler was used to scale each tissue.
Echo MRI
Body fat and lean mass four weeks after diet change were assessed using an Echo MRI (The EchoMRI™−100H) following manufacturer’s protocol.
Comprehensive Lab Animal Monitoring System (CLAMS)
Indirect calorimetry in a CLAMS system (Columbus Instruments) was used to evaluate whole-body states and ambulatory locomotor activity during ad libitum access to SCD or HFD. Four weeks after diet change, mice were acclimated to metabolic cages for 48 hours and then monitored for 72 hours following the manufacturer’s instructions. Energy expenditure (EE) in watts per kilogram of lean mass [W/kg] was calculated with the following formula described in [89].
The unit Watts was converted to kcal/hour by multiplying factor of 0.86 to report EE as kcal/hour/kg of lean mass. The resting metabolic rate (RMR) was quantified and defined as the average of the lowest 5 consecutive energy expenditure values (18 minute intervals) across a twenty-four hour day.
QPCR
Six weeks after the time of diet change, WT-SCD, KO-SCD, WT-HFD, and KO-HFD mice were sacrificed by cervical dislocation every four hours along the LD cycle (3 mice/timepoint: ZT 1,5,9,13,17,21). Tissue collected from mice during the Night phase were sacrificed in the dark under IR light with night-vision goggles. Dissected liver and GWAT tissue was flash frozen in liquid nitrogen. RNA was extracted with the RNeasy Lipid Tissue Mini Kit (QIAGEN). For liver and GWAT, 1100 ng of RNA was reverse-transcribed using a SuperScript™ IV First-Strand Synthesis System kit (Thermo Fisher). Quantitative PCR was performed using the iQ™ SYBR® Green Supermix system (BIO-RAD). Beta-actin was used as a housekeeping gene for the analysis of Bmal1, Per2, and Rev-erbα. The relative mRNA levels were calculated using the 2–ΔCt method. The ΔCt values were obtained by calculating the differences: Ct(gene of interest) – Ct(housekeeping gene) in each sample. All groups were normalized relative to WT-SCD median of 1.
FITC-Dextran
For the intestinal permeability assay, tracer fluorescein isothiocyanate (FITC) labeled dextran (4kDa; Sigma-Aldrich) was used to assess in vivo intestinal permeability. At ZT 2 mice were deprived of food 4 hours prior to and 4 hours following an oral gavage (ZT 6) using 150 μl of 80 mg/ml FITC-dextran. Blood (50μl) was collected from tail bleeds, and fluorescence intensity was measured on fluorescence plates using an excitation wavelength of 485 nm and an emission wavelength of 530 nm [90].
Foraging assay
Animals were habituated to experimental conditions for 4 days prior to testing. On the 1st and 2nd day 1 pellet of HFD was placed on the homecage floor for diet habituation. On the 3rd day mice were moved to a larger testing cage (144 in2; 18 (L) × 8 (W) × 8 (H)) with ad libitum access to SCD and 1 pellet of HFD on cage floor. 4th day: ad libitum access to SCD in home cage. After this habituation, the foraging assay was performed at either ZT 8 (Day) or ZT 15 (Night). Mice were place into the testing cage with 1~1.5cm deep bedding for 30 minutes. A pellet of HFD diet was buried beneath the bedding at one end of the testing cage. Mice were then placed onto the opposite end of the testing cage, video recorded, and tracked by EthoVision XT 11 (Noldus) (Day) or a night-vision sports camera (Night) for 3 minutes. The latency of the animals’ first intentional foraging of the food area scored by an investigator blind to genotype. The mice (1/15 WT and 2/10 KO) that failed to perform a foraging behavior within 2 minutes were counted as 120 seconds.
Progressive Ratio Test
Operant conditioning occurred in noise-controlled chambers under red lighting (Med-Associates, St. Alban, VT, USA). Prior to progressive ratio (PR) test, mice underwent fixed ratio (FR) training sessions for a minimum of three days. During training, a single nosepoke (FR1) in the center hole (of three possible entries) resulted in the delivery of a food reward (chocolate pellet) from an automatic dispenser. After earning 50 reinforcers, training advanced to a FR3 schedule of reinforcement, whereby three nosepokes in the center hole were required for the delivery of the food reward. After earning 50 reinforcers on this schedule, training advanced to FR5. For progressive ratio test, one reward pellet was delivered per completed trial in which the nosepoke requirement increased on the following schedule: 1, 3, 5, 10, 20, 30, 50, 70, 100, 130, 170, 210, 260, 310, and 370. Animals were allowed 30 minutes to complete each level of trial following the initial nosepoke. Mice were trained and tested during both the light and dark phase and the order of testing was counterbalanced. In between tests, animals were placed in a reversed light cycle for 7 days for entrainment. Training and testing began at ZT 0 (Day) or ZT 12 (Night).
Open Field Testing
Animals participating in the progressive ratio experiments underwent open field testing to measure locomotor behavior. Testing occurred in a closed, translucent plexiglass open field chamber (42×42 cm) at ZT 1 (Day) and ZT 13 (Night). Mice were placed on the edge of the open field and given five minutes to explore the area while distance travelled (cm) was recorded with EthoVision XT tracking software (Noldus, Leesburg, VA, USA).
HPLC
WT mice were habituated to HFD or SCD on cage floor on the day prior to tissue collection. Animals were given 1 hour access to either HFD or SCD and sacrificed by cervical dislocation. Fresh brains were dissected from adult wild-type male animals and 1 mm coronal slices were collected using a mouse brain matrix (Zivic Instruments). Hypothalamic samples containing the SCN were collected with a 1.5 mm in diameter tissue biopsy punch (Miltex), frozen in liquid nitrogen, and stored at 80°C until further processing. Tissue punches were homogenized by sonication in 50 μL of 0.04N perchloric acid solution. The homogenate was centrifuged at 13,200 rmp for 12 min at 4°C, followed by spin filtration (Sigma Aldrich) at 11,000 rpm for 4 min at 4°C. 15 μL of the resulting supernatant was loaded into an autosampler connected to a high-performance liquid chromatography instrument with an electrochemical detector (Decade, Antec Leyden B.V., Zoeterwoude, the Netherlands) to measure the levels DA and 3,4-Dihydroxyphenylacetic acid (DOPAC) and serotonin. Retention time for was determined through comparison with standards diluted in 0.04N perchloric acid. DOPAC tR range 4.56±0.015 min, DA: tR range 10.01±0.24 min, Serotonin: tR range: 22.165±0.035 min. Mobile phase: pH 3, 10% acetonitrile, 0.50mmol DSA. 0.8V 0.125 ml/min, 26 °C 24 minutes 0.5μM of DSA. Identification of serotonin within the chromatogram was used to confirm the SCN in collected tissue punch [91].
Microbiome
Six weeks after the diet change, fresh fecal samples from WT-SCD, WT-HFD and KO-HFD mice were collected, flash frozen in liquid nitrogen and stored at −80 until further processing. One or two fecal pellets weighing 10 to 100 mg from each sample were used for DNA extraction with the ZR-96 Fecal DNA Kit (Zymo Research). Bacterial 16S rRNA gene (V4 region) was amplified with barcodes from the extracted DNA, quantified and pooled for Illumina Miseq sequencing at the Genomic Core Facility of UVa. Demultiplexed sequence reads were trimmed based on quality score and checked for chimeric reads, and de novo operational taxonomic units (OTUs) were then picked at 97% similarity threshold using QIIME [92]. Mitochondrial and chloroplast OTUs and OTUs with no more than 100 sequences across all samples were filtered out. For each sample, sequences were rarefied to 64,455 sequences per sample to normalize the sequencing effort for diversity analysis. The weighted Unifrac metric was applied to calculate the dissimilarity between samples, and alpha diversity was measured as the observed number of OTUs. PERMANOVA test was applied to examine the effect of diet and genotype on the microbiome composition. The non-parametric JTK analysis that detects cycling elements was performed to determine whether a particular taxonomic group was cyclically fluctuating [93]. A taxonomic group was considered cyclical if both its adjusted permutation-based p-value (ADJ.P) and Benjamini-Hochberg q-values (BH.Q) were smaller than 0.05.
Temperature recordings
Body temperature were measured using a rectal probe attached to a digital thermometer (Physitemp Instruments, Clifton, NJ). Temperatures were collected at ZT 6 (Day) and ZT 18 (Night).
Slice electrophysiology
Slice visualization and data collection:
SCN cells were visualized with infrared DIC in an upright Slicescope 6000 microscope. The SCN was identified by the shape of the both 3rd ventricle and most inferior middle region of the slice, as well as the presence of the optic chiasm. Images of patched brain regions were taken using Scientifica SciPro camera and Ocular imaging software. A Multiclamp 700B amplifier and Digidata 1550B digitizer (Molecular Devices; San Jose, California) were used to perform all patch clamp experiments. All experiments were conducted using 2.5–6MΩ microelectrodes pulled with a Sutter P97 puller. All brain slice solutions were saturated with 95% O2 and 5% CO2 gas. SKF-81297 was used at 5μM concentration in all incubation experiments.
Cell attached recordings:
Male and female WT mice were individually housed in light-tight boxes under a 12 hr:12 hr LD cycle for at least 7 days. Mice between P31 and P55 were deeply anesthetized with isoflurane and brains were rapidly dissected and mounted for slicing in the compresstome slicer. Slices were taken with in ice cold HEPES based holding ACSF solution containing (in mM): 92 NaCl, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 2 CaCl2·4H2O and 2 MgSO4·7H2O with pH ranging from 7.3 to 7.4 and osmolarity ranging from 300 to 310 mOsm [94]. Slices were allowed to recover for ≤12 min at 34°C in the same HEPES based holding ACSF so lution, and then allowed to come to room temperature. ‘Day’ collections condition (sacrificed at ZT 5–6) were incubated for a minimum of 90 minutes, and data was collected between ZT 8 and 11. ‘Night’ collections condition (sacrificed at ZT 11–12) were incubated and data was collected at ZT 14 to 17. After a minimum of 90 minutes of incubation slices were transferred to the microscope recording bath and superfused with a continuous flow (1.5 – 2 ml/min) recording ACSF which consisted of (in mM): 124 NaCl, 2.5 KCl, 1.2 NaH2PO4, 24 NaHCO3, 5 HEPES, 10 glucose, 2 CaCl2·4H2O and 2 MgSO4·7H2O with pH ranging from 7.3 to 7.4 and osmolarity ranging from 300 to 310 mOsm, and included the vehicle or agonist. The resulting DMSO concentration in these buffers ranged from 0.01% to 0.12%. Vehicle control solutions contained the same amount of DMSO as the treatment conditions. Drug solutions were added directly to the bath after the recovery incubation. Cell attached recordings were made at 32 °C. For cell attached recordings the pipette was filled with ACSF, slight positive pressure applied when approaching the cell was released and the seal was allowed. Seal magnitude ranged from 5 to 50MΩ. In all experiments the pipette offset was <15mV, typically ranging from 1 to 11mV. Pipette offset was set just before initiating a recording, and the seal resistance was monitored closely. Recordings measuring spontaneous spiking lasted approximately one minute. Recordings were filtered offline at 1kHz and baseline was manually adjusted using ClampFit (Molecular devices) software. Action potential events were then extracted using ClampFit and analyzed using custom scripts in Matlab (Mathworks).
Optogenetic stimulation:
Heterozygous Drd1-HET mice minimum of 8 weeks of age received bilateral injections of AAV2-DIO-ChR2-YFP to the SCN as previously described. After a minimum of 5 weeks recovery animals were deeply anesthetized (ketamine:xylazine, 280:80 mg/kg, i.p.) and transcardially perfused with an NMDG recovery ACSF solution containing (in mM): 92 NMDG, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 0.5 CaCl2·4H2O and 10 MgSO4·7H2O, pH adjusted to 7.3 to 7.4, osmolarity 300 to 310 mOsm [94]. Slices were held in the NMDG recovery ACSF for ≤ 12 min at 34°C, and then transferred to recording ACSF (described above) and allowed to rest at room temperature for 45 minutes before being transferred to microscope bath for data collection. All recordings were made at room temperature and the same K-Glu intracellular solution was used as described above. SCN cells were randomly patched without regard for YFP expression. After formation of gigaohm seal (>2GΩ), cell membrane was ruptured and current clamp recordings acquired. A minimal current injection hold of 0 to −15pA was used to compensate for minor current leakage. Whole cell voltage measurements made as described for whole cell incubation recordings. Optogenetic stimulation consisted a 500ms 20 Hz square pulses of 488 nm light (10 pulses total, each pulse 10ms). Light was delivered at the beginning of each 10 second trace. For a subset of cells (5) which showed a hyperpolarized response to blue light stimulation, recording ACSF with 100μM Picrotoxin (PTX) was perfused for 6 minutes and cell responses to the optogenetic stimulation protocol were recorded. No further recordings were made on slices which were exposed to PTX. Response magnitudes and latencies were manually extracted in ClampFit, and consisted of an average of 5–6 traces.
QUANTIFICATION AND STATISTICAL ANALYSIS
Adipose area quantification
Samples of GWAT were collected and fixed for H & E staining as described above. Three digital images (20×) from non-overlapping fields were captured for each sample (Nikon ECLIPSE Ti microscope equipped with Nikon DS-Ri2 color camera). Quantitative analyses of adipocyte area were made using ImageJ (National Institutes of Health, Bethesda, MD). Results are expressed as mean±SE μm2 per cell. Exclusions: any cut off adipocytes on the image edge, cells that were artificially divided into multiple cells due to artifact, cells that were combined due to faintness of borders in the original image, and cells <350μm2 [95].
Statistical Analysis
When comparing two groups of normally distributed data, a Student’s two tailed t-test was used. To compare the effects of genotype and diet within 4 groups, two-way ANOVA test was used. When data was collected from the same animals across time, a three-way ANOVA test was performed to analyze time, genotype and diet effect. In experiments with single variable and more than two groups, a one-way ANOVA was performed. Following a significant effect in the ANOVA test, Bonferroni’s post hoc comparison was used to determine differences between individual data points. Permutational multivariate analysis of variance (PERMANOVA) was used to evaluate microbiome OTUs and conducted in R. Analyses were conducted using the GraphPad Prism 8 statistical software for Windows, or Prism 8 for Mac OS. All data are presented as means±standard error of the mean with p<0.05 considered statistically significant.
DATA AND SOFTWARE AVAILABILITY
The metagenomic sequence data were deposited at NCBI under Bioproject: PRJNA580025, available from: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA580025
Supplementary Material
Video S1: Representative video of the foraging assay for WT and Drd1-KO mice at Day (ZT8). Related to Figure 1. Video is accelerated to 4 times of speed. Real time is depicted at the bottom of the video.
Video S2: Representative video of the foraging assay for WT and Drd1-KO mice at Night (ZT15). Related to Figure 1. Video is accelerated to 4 times of speed. Real time is depicted at the bottom of the video.
Highlights.
Diet-induced obesity (DIO) requires dopamine (DA)-Drd1 signaling
DA-Drd1 signaling in the central circadian clock (SCN) instigates DIO
SCN DA-Drd1 signaling disinhibits hedonic food consumption in between meals
Diet drives dysbiosis and peripheral circadian desynchrony independent of Drd1
ACKNOWLEDGEMENTS
We are especially thankful for technical assistance from Aundrea Rainwater, Elena Tenore, Tarun Vippa, Charles Brennan, Sydney P. Williams, Charles Sheehan, and Yağmur Halezeroğlu. Jacqueline Parker and Alexander Duval (HPLC), Wenfan Ke and Eyleen O’Rourke (Oil-Red-O), Maisie Crook, Elise L. Savier, and Jianhua Cang (Rabies virus tracing). Additionally, we are grateful for assistance from the research histology core, UVA school of medicine (H & E histology), UVA department of biology genomics core facility (AnThu Ngyugen, 16s sequencing), and UT Southwestern metabolic core (fecal bomb calorimetry). Lastly, we are thanking Ignacio Provencio for manuscript revision, as well as Michael Menaker for experiment feedback. This work was supported by NIH National Institute of General Medicinal Sciences R01GM121937 (A.D.G.), University of Virginia (UVA) Faculty Start-up Funds (A.D.G.), UVA Brain Institute 2018 Seed Funding Award (A.D.G.), UVA Presidential Fellowship for Collaborative Neuroscience (Q.Z.), NIH5R01GM84128 (J.H.), NIH National Institute of General Medical Sciences unC3GM125570 (A.D.S.), Whitehall Foundation Research Grant (A.D.S.), 1R01MH116694-01A1 (M.M.S.), American Diabetes Association, Pathway to Stop Diabetes Award 1-18-INI-14 (J.N.C.), and lastly we acknowledge the Keck Center for Cellular Imaging for the usage of the Leica confocal microscopy system (RR025616).
Footnotes
DECLARATION OF INTERERESTS
The authors declare no competing interests.
REFERENCES
- 1.Guh DP, Zhang W, Bansback N, Amarsi Z, Birmingham CL, and Anis AH (2009). The incidence of co-morbidities related to obesity and overweight: a systematic review and meta-analysis. BMC Public Health 9, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hurt RT, Kulisek C, Buchanan LA, and McClave SA (2010). The obesity epidemic: challenges, health initiatives, and implications for gastroenterologists. Gastroenterol Hepatol (N Y) 6, 780–792. [PMC free article] [PubMed] [Google Scholar]
- 3.Eckel RH, Kahn SE, Ferrannini E, Goldfine AB, Nathan DM, Schwartz MW, Smith RJ, and Smith SR (2011). Obesity and type 2 diabetes: what can be unified and what needs to be individualized? J. Clin. Endocrinol. Metab 96, 1654–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Mutsert R, Sun Q, Willett WC, Hu FB, and van Dam RM (2014). Overweight in early adulthood, adult weight change, and risk of type 2 diabetes, cardiovascular diseases, and certain cancers in men: a cohort study. Am. J. Epidemiol 179, 1353–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montesi L, El Ghoch M, Brodosi L, Calugi S, Marchesini G, and Dalle Grave R (2016). Long-term weight loss maintenance for obesity: a multidisciplinary approach. Diabetes Metab. Syndr. Obes 9, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Webb VL, and Wadden TA (2017). Intensive Lifestyle Intervention for Obesity: Principles, Practices, and Results. Gastroenterology 152, 1752–1764. [DOI] [PubMed] [Google Scholar]
- 7.Del Corral P, Bryan DR, Garvey WT, Gower BA, and Hunter GR (2011). Dietary adherence during weight loss predicts weight regain. Obesity (Silver Spring) 19, 1177–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, et al. (2005). Obesity and metabolic syndrome in circadian Clock mutant mice. Science 308, 1043–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arble DM, Ramsey KM, Bass J, and Turek FW (2010). Circadian disruption and metabolic disease: findings from animal models. Best Pract. Res. Clin. Endocrinol. Metab 24, 785–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, Leblanc M, Chaix A, Joens M, Fitzpatrick JA, et al. (2012). Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab 15, 848–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garaulet M, Gomez-Abellan P, Alburquerque-Bejar JJ, Lee YC, Ordovas JM, and Scheer FA (2013). Timing of food intake predicts weight loss effectiveness. Int. J. Obes. (Lond.) 37, 604–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sutton EF, Beyl R, Early KS, Cefalu WT, Ravussin E, and Peterson CM (2018). Early Time-Restricted Feeding Improves Insulin Sensitivity, Blood Pressure, and Oxidative Stress Even without Weight Loss in Men with Prediabetes. Cell Metab. 27, 1212–1221 e1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wise RA (2004). Dopamine, learning and motivation. Nat. Rev. Neurosci 5, 483–494. [DOI] [PubMed] [Google Scholar]
- 14.Wise RA (2006). Role of brain dopamine in food reward and reinforcement. Philos. Trans. R. Soc. Lond. B Biol. Sci 361, 1149–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou QY, and Palmiter RD (1995). Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell 83, 1197–1209. [DOI] [PubMed] [Google Scholar]
- 16.Missale C, Nash SR, Robinson SW, Jaber M, and Caron MG (1998). Dopamine receptors: from structure to function. Physiol. Rev 78, 189–225. [DOI] [PubMed] [Google Scholar]
- 17.Beaulieu JM, and Gainetdinov RR (2011). The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev 63, 182–217. [DOI] [PubMed] [Google Scholar]
- 18.Johnson PM, and Kenny PJ (2010). Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat. Neurosci 13, 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Land BB, Narayanan NS, Liu RJ, Gianessi CA, Brayton CE, Grimaldi DM, Sarhan M, Guarnieri DJ, Deisseroth K, Aghajanian GK, et al. (2014). Medial prefrontal D1 dopamine neurons control food intake. Nat. Neurosci 17, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alsio J, Olszewski PK, Norback AH, Gunnarsson ZE, Levine AS, Pickering C, and Schioth HB (2010). Dopamine D1 receptor gene expression decreases in the nucleus accumbens upon long-term exposure to palatable food and differs depending on diet-induced obesity phenotype in rats. Neuroscience 171, 779–787. [DOI] [PubMed] [Google Scholar]
- 21.Carlin J, Hill-Smith TE, Lucki I, and Reyes TM (2013). Reversal of dopamine system dysfunction in response to high-fat diet. Obesity (Silver Spring) 21, 2513–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terry P, and Katz JL (1994). A comparison of the effects of the D1 receptor antagonists SCH 23390 and SCH 39166 on suppression of feeding behavior by the D1 agonist SKF38393. Psychopharmacology (Berl.) 113, 328–333. [DOI] [PubMed] [Google Scholar]
- 23.Astrup A, Greenway FL, Ling W, Pedicone L, Lachowicz J, Strader CD, Kwan R, and Ecopipam Obesity Study G (2007). Randomized controlled trials of the D1/D5 antagonist ecopipam for weight loss in obese subjects. Obesity (Silver Spring) 15, 1717–1731. [DOI] [PubMed] [Google Scholar]
- 24.Winzell MS, and Ahren B (2004). The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 53 Suppl 3, S215–219. [DOI] [PubMed] [Google Scholar]
- 25.Hariri N, and Thibault L (2010). High-fat diet-induced obesity in animal models. Nutr. Res. Rev 23, 270–299. [DOI] [PubMed] [Google Scholar]
- 26.Heusner CL, Beutler LR, Houser CR, and Palmiter RD (2008). Deletion of GAD67 in dopamine receptor-1 expressing cells causes specific motor deficits. Genesis 46, 357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drago J, Gerfen CR, Lachowicz JE, Steiner H, Hollon TR, Love PE, Ooi GT, Grinberg A, Lee EJ, Huang SP, et al. (1994). Altered striatal function in a mutant mouse lacking D1A dopamine receptors. Proc. Natl. Acad. Sci. U. S. A 91, 12564–12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Espinosa-Carrasco J, Burokas A, Fructuoso M, Erb I, Martin-Garcia E, Gutierrez-Martos M, Notredame C, Maldonado R, and Dierssen M (2018). Time-course and dynamics of obesity-related behavioral changes induced by energy-dense foods in mice. Addict. Biol 23, 531–543. [DOI] [PubMed] [Google Scholar]
- 29.Mrosovsky N (1999). Masking: history, definitions, and measurement. Chronobiol. Int 16, 415–429. [DOI] [PubMed] [Google Scholar]
- 30.Gore BB, and Zweifel LS (2013). Genetic reconstruction of dopamine D1 receptor signaling in the nucleus accumbens facilitates natural and drug reward responses. J. Neurosci 33, 8640–8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riera CE, Tsaousidou E, Halloran J, Follett P, Hahn O, Pereira MMA, Ruud LE, Alber J, Tharp K, Anderson CM, et al. (2017). The Sense of Smell Impacts Metabolic Health and Obesity. Cell Metab 26, 198–211 e195. [DOI] [PubMed] [Google Scholar]
- 32.Stenvers DJ, Scheer F, Schrauwen P, la Fleur SE, and Kalsbeek A (2019). Circadian clocks and insulin resistance. Nat. Rev. Endocrinol 15, 75–89. [DOI] [PubMed] [Google Scholar]
- 33.Kohsaka A, Laposky AD, Ramsey KM, Estrada C, Joshu C, Kobayashi Y, Turek FW, and Bass J (2007). High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab 6, 414–421. [DOI] [PubMed] [Google Scholar]
- 34.Arble DM, Bass J, Laposky AD, Vitaterna MH, and Turek FW (2009). Circadian timing of food intake contributes to weight gain. Obesity (Silver Spring) 17, 2100–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pendergast JS, Branecky KL, Yang W, Ellacott KL, Niswender KD, and Yamazaki S (2013). High-fat diet acutely affects circadian organisation and eating behavior. Eur. J. Neurosci 37, 1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kraemer FB, and Shen WJ (2002). Hormone-sensitive lipase: control of intracellular tri-(di-)acylglycerol and cholesteryl ester hydrolysis. J. Lipid Res 43, 1585–1594. [DOI] [PubMed] [Google Scholar]
- 37.Akasheh RT, Pini M, Pang J, and Fantuzzi G (2013). Increased adiposity in annexin A1-deficient mice. PLoS One 8, e82608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo YJ, Li YD, Wang L, Yang SR, Yuan XS, Wang J, Cherasse Y, Lazarus M, Chen JF, Qu WM, et al. (2018). Nucleus accumbens controls wakefulness by a subpopulation of neurons expressing dopamine D1 receptors. Nat Commun 9, 1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mills EL, Pierce KA, Jedrychowski MP, Garrity R, Winther S, Vidoni S, Yoneshiro T, Spinelli JB, Lu GZ, Kazak L, et al. (2018). Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 560, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wan M, Easton RM, Gleason CE, Monks BR, Ueki K, Kahn CR, and Birnbaum MJ (2012). Loss of Akt1 in mice increases energy expenditure and protects against diet-induced obesity. Mol. Cell. Biol 32, 96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kopelman PG (2000). Obesity as a medical problem. Nature 404, 635–643. [DOI] [PubMed] [Google Scholar]
- 42.Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, and Lazar MA (2001). The hormone resistin links obesity to diabetes. Nature 409, 307–312. [DOI] [PubMed] [Google Scholar]
- 43.Kahn SE, Hull RL, and Utzschneider KM (2006). Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444, 840–846. [DOI] [PubMed] [Google Scholar]
- 44.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, et al. (2003). Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest 112, 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dandona P, Aljada A, and Bandyopadhyay A (2004). Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol. 25, 4–7. [DOI] [PubMed] [Google Scholar]
- 46.Tilg H, and Kaser A (2011). Gut microbiome, obesity, and metabolic dysfunction. J. Clin. Invest 121, 2126–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frazier TH, DiBaise JK, and McClain CJ (2011). Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. JPEN J. Parenter. Enteral Nutr 35, 14S–20S. [DOI] [PubMed] [Google Scholar]
- 48.Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palu G, and Martines D (2007). Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol 292, G518–525. [DOI] [PubMed] [Google Scholar]
- 49.Caricilli AM, and Saad MJ (2013). The role of gut microbiota on insulin resistance. Nutrients 5, 829–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang CJ, Lu CC, Lin CS, Martel J, Ko YF, Ojcius DM, Wu TR, Tsai YH, Yeh TS, Lu JJ, et al. (2018). Antrodia cinnamomea reduces obesity and modulates the gut microbiota in high-fat diet-fed mice. Int. J. Obes. (Lond.) 42, 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferrario CR, Labouebe G, Liu S, Nieh EH, Routh VH, Xu S, and O’Connor EC (2016). Homeostasis Meets Motivation in the Battle to Control Food Intake. J. Neurosci 36, 11469–11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stamatakis AM, Van Swieten M, Basiri ML, Blair GA, Kantak P, and Stuber GD (2016). Lateral Hypothalamic Area Glutamatergic Neurons and Their Projections to the Lateral Habenula Regulate Feeding and Reward. J. Neurosci 36, 302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rossi MA, and Stuber GD (2018). Overlapping Brain Circuits for Homeostatic and Hedonic Feeding. Cell Metab 27, 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerfen CR, and Surmeier DJ (2011). Modulation of striatal projection systems by dopamine. Annu. Rev. Neurosci 34, 441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarinana J, and Tonegawa S (2016). Differentiation of forebrain and hippocampal dopamine 1-class receptors, D1R and D5R, in spatial learning and memory. Hippocampus 26, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X, Zhang C, Szabo G, and Sun QQ (2013). Distribution of CaMKIIalpha expression in the brain in vivo, studied by CaMKIIalpha-GFP mice. Brain Res 1518, 9–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vong L, Ye C, Yang Z, Choi B, Chua S Jr., and Lowell BB (2011). Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71, 142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davis JF, Tracy AL, Schurdak JD, Tschop MH, Lipton JW, Clegg DJ, and Benoit SC (2008). Exposure to elevated levels of dietary fat attenuates psychostimulant reward and mesolimbic dopamine turnover in the rat. Behav. Neurosci 122, 1257–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grippo RM, Purohit AM, Zhang Q, Zweifel LS, and Guler AD (2017). Direct Midbrain Dopamine Input to the Suprachiasmatic Nucleus Accelerates Circadian Entrainment. Curr. Biol 27, 2465–2475 e2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mendoza J, Angeles-Castellanos M, and Escobar C (2005). A daily palatable meal without food deprivation entrains the suprachiasmatic nucleus of rats. Eur. J. Neurosci 22, 2855–2862. [DOI] [PubMed] [Google Scholar]
- 61.Mendoza J, Pevet P, and Challet E (2008). High-fat feeding alters the clock synchronization to light. J. Physiol 586, 5901–5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mendoza J, Clesse D, Pevet P, and Challet E (2010). Food-reward signalling in the suprachiasmatic clock. J. Neurochem 112, 1489–1499. [DOI] [PubMed] [Google Scholar]
- 63.Okamura H, Berod A, Julien JF, Geffard M, Kitahama K, Mallet J, and Bobillier P (1989). Demonstration of GABAergic cell bodies in the suprachiasmatic nucleus: in situ hybridization of glutamic acid decarboxylase (GAD) mRNA and immunocytochemistry of GAD and GABA. Neurosci. Lett 102, 131–136. [DOI] [PubMed] [Google Scholar]
- 64.Moore RY, and Speh JC (1993). GABA is the principal neurotransmitter of the circadian system. Neurosci. Lett 150, 112–116. [DOI] [PubMed] [Google Scholar]
- 65.McNulty S, Schurov IL, Sloper PJ, and Hastings MH (1998). Stimuli which entrain the circadian clock of the neonatal Syrian hamster in vivo regulate the phosphorylation of the transcription factor CREB in the suprachiasmatic nucleus in vitro. Eur. J. Neurosci 10, 1063–1072. [DOI] [PubMed] [Google Scholar]
- 66.Fonken LK, Aubrecht TG, Melendez-Fernandez OH, Weil ZM, and Nelson RJ (2013). Dim light at night disrupts molecular circadian rhythms and increases body weight. J. Biol. Rhythms 28, 262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gill S, and Panda S (2015). A Smartphone App Reveals Erratic Diurnal Eating Patterns in Humans that Can Be Modulated for Health Benefits. Cell Metab 22, 789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gabel K, Hoddy KK, Haggerty N, Song J, Kroeger CM, Trepanowski JF, Panda S, and Varady KA (2018). Effects of 8-hour time restricted feeding on body weight and metabolic disease risk factors in obese adults: A pilot study. Nutr Healthy Aging 4, 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chaix A, Manoogian ENC, Melkani GC, and Panda S (2019). Time-Restricted Eating to Prevent and Manage Chronic Metabolic Diseases. Annu. Rev. Nutr 39, 291–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, Chen YY, Knight R, Ahima RS, Bushman F, and Wu GD (2009). High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 137, 1716–1724 e1711–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Berridge KC, and Robinson TE (1998). What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res. Brain Res. Rev 28, 309–369. [DOI] [PubMed] [Google Scholar]
- 72.Gabriel BM, and Zierath JR (2019). Circadian rhythms and exercise - re-setting the clock in metabolic disease. Nat. Rev. Endocrinol 15, 197–206. [DOI] [PubMed] [Google Scholar]
- 73.Pendergast JS, Branecky KL, Huang R, Niswender KD, and Yamazaki S (2014). Wheel-running activity modulates circadian organization and the daily rhythm of eating behavior. Front. Psychol 5, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smyllie NJ, Chesham JE, Hamnett R, Maywood ES, and Hastings MH (2016). Temporally chimeric mice reveal flexibility of circadian period-setting in the suprachiasmatic nucleus. Proc. Natl. Acad. Sci. U. S. A 113, 3657–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Landgraf D, Joiner WJ, McCarthy MJ, Kiessling S, Barandas R, Young JW, Cermakian N, and Welsh DK (2016). The mood stabilizer valproic acid opposes the effects of dopamine on circadian rhythms. Neuropharmacology 107, 262–270. [DOI] [PubMed] [Google Scholar]
- 76.Wisor JP, Nishino S, Sora I, Uhl GH, Mignot E, and Edgar DM (2001). Dopaminergic role in stimulant-induced wakefulness. J. Neurosci 21, 1787–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eban-Rothschild A, Rothschild G, Giardino WJ, Jones JR, and de Lecea L (2016). VTA dopaminergic neurons regulate ethologically relevant sleep-wake behaviors. Nat. Neurosci 19, 1356–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gallardo CM, Darvas M, Oviatt M, Chang CH, Michalik M, Huddy TF, Meyer EE, Shuster SA, Aguayo A, Hill EM, et al. (2014). Dopamine receptor 1 neurons in the dorsal striatum regulate food anticipatory circadian activity rhythms in mice. Elife 3, e03781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mendoza J (2019). Eating Rewards the Gears of the Clock. Trends Endocrinol. Metab 30, 299–311. [DOI] [PubMed] [Google Scholar]
- 80.Grippo RM, and Guler AD (2019). Dopamine Signaling in Circadian Photoentrainment: Consequences of Desynchrony. Yale J. Biol. Med 92, 271–281. [PMC free article] [PubMed] [Google Scholar]
- 81.Kalsbeek A, Palm IF, La Fleur SE, Scheer FA, Perreau-Lenz S, Ruiter M, Kreier F, Cailotto C, and Buijs RM (2006). SCN outputs and the hypothalamic balance of life. J. Biol. Rhythms 21, 458–469. [DOI] [PubMed] [Google Scholar]
- 82.Moore RY (1995). Organization of the mammalian circadian system. Ciba Found. Symp 183, 88–99; discussion 100–106. [PubMed] [Google Scholar]
- 83.Schaap J, Pennartz CM, and Meijer JH (2003). Electrophysiology of the circadian pacemaker in mammals. Chronobiol. Int 20, 171–188. [DOI] [PubMed] [Google Scholar]
- 84.Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, and Tonegawa S (1996). Subregion- and cell type-restricted gene knockout in mouse brain. Cell 87, 1317–1326. [DOI] [PubMed] [Google Scholar]
- 85.Lee IT, Chang AS, Manandhar M, Shan Y, Fan J, Izumo M, Ikeda Y, Motoike T, Dixon S, Seinfeld JE, et al. (2015). Neuromedin s-producing neurons act as essential pacemakers in the suprachiasmatic nucleus to couple clock neurons and dictate circadian rhythms. Neuron 85, 1086–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pack AI, Galante RJ, Maislin G, Cater J, Metaxas D, Lu S, Zhang L, Von Smith R, Kay T, Lian J, et al. (2007). Novel method for high-throughput phenotyping of sleep in mice. Physiol. Genomics 28, 232–238. [DOI] [PubMed] [Google Scholar]
- 87.Vinue A, and Gonzalez-Navarro H (2015). Glucose and Insulin Tolerance Tests in the Mouse. Methods Mol. Biol 1339, 247–254. [DOI] [PubMed] [Google Scholar]
- 88.la Fleur SE, Kalsbeek A, Wortel J, Fekkes ML, and Buijs RM (2001). A daily rhythm in glucose tolerance: a role for the suprachiasmatic nucleus. Diabetes 50, 1237–1243. [DOI] [PubMed] [Google Scholar]
- 89.Fischer AW, Cannon B, and Nedergaard J (2018). Optimal housing temperatures for mice to mimic the thermal environment of humans: An experimental study. Mol Metab 7, 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, Loukov D, Schenck LP, Jury J, Foley KP, et al. (2017). Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell Host Microbe 21, 455–466 e454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morin LP (1999). Serotonin and the regulation of mammalian circadian rhythmicity. Ann. Med 31, 12–33. [DOI] [PubMed] [Google Scholar]
- 92.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hughes ME, Hogenesch JB, and Kornacker K (2010). JTK_CYCLE: an efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J. Biol. Rhythms 25, 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ting JT, Daigle TL, Chen Q, and Feng G (2014). Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol. Biol 1183, 221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Parlee SD, Lentz SI, Mori H, and MacDougald OA (2014). Quantifying size and number of adipocytes in adipose tissue. Methods Enzymol. 537, 93–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1: Representative video of the foraging assay for WT and Drd1-KO mice at Day (ZT8). Related to Figure 1. Video is accelerated to 4 times of speed. Real time is depicted at the bottom of the video.
Video S2: Representative video of the foraging assay for WT and Drd1-KO mice at Night (ZT15). Related to Figure 1. Video is accelerated to 4 times of speed. Real time is depicted at the bottom of the video.
Data Availability Statement
The metagenomic sequence data were deposited at NCBI under Bioproject: PRJNA580025, available from: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA580025