

Abstract
With more than 795,000 cases occurring every year, stroke has become a major problem in the United States across all demographics. Stroke is the leading cause of long-term disability and is the fifth leading cause of death in the US. Ischemic stroke represents 87% of total strokes in the US, and is currently the main focus of stroke research. This literature review examines the risk factors associated with ischemic stroke, changes in cell morphology and signaling in the brain after stroke, and the advantages and disadvantages of in vivo and in vitro ischemic stroke models. Classification systems for stroke etiology are also discussed briefly, as well as current ischemic stroke therapies and new therapeutic strategies that focus on the potential of stem cells to promote stroke recovery.
Keywords: Ischemic stroke, photothrombotic model, MCAO, tissue plasminogen activator, regenerative therapy, dental pulp stem cells, hematopoietic stem cells
1. Introduction
Stroke has become a serious issue in the United States, as more than 795,000 people in the US have a stroke every year. About 610,000 of those are first time strokes, while 185,000 occur in patients who have had a stroke before. Stroke is also responsible for about 140,000 deaths in the US every year, which is about one out of every twenty deaths in the country. This makes it the fifth leading cause of death in America. When cost of health care services, medicine, and work days missed are taken into account, the total yearly cost of stroke in the US is about 34 billion dollars. It is also the leading cause of long-term disability in the United States [1].
Stroke is more likely to occur at an older age, but can happen at any time. In 2009, 34% of people who were hospitalized for a stroke were under 65 years old. Race and ethnicity contribute to differences in stroke trends: first stroke risks are twice as high in African Americans as in Caucasians. African Americans also suffer from the highest rate of deaths due to strokes in the US. However, death rates from stroke have declined among every race and ethnicity, excepting Hispanic populations, in whom death rates due to stroke have increased since 2013 [1]. Statistically, females also have a higher risk of experiencing a stroke-related fatality than males do. On average, females represent six out of every ten people that die due to both ischemic and hemorrhagic strokes [2]. There are multiple reasons for this higher risk. One of them is that females on average live longer than males, and the longer average lifespan increases the odds that they will have a stroke. Other unique risk factors that females face include high blood pressure during pregnancy and high blood pressure caused by certain forms of birth control medications. They also are more likely to experience depression and anxiety, and often report higher levels of stress than males do. All of these factors increase the overall risk of stroke in females [3–5]. The trends seen between races in both males and females are also observed when looking specifically at females. African American females are, on average, twice as likely to have a stroke as Caucasian females. According to the Centers for Disease Control and Prevention (CDC), this is due to higher prevalence of high blood pressure, obesity, and diabetes in African American females. These statistics show that across all demographics, stroke is an extremely prevalent medical development that needs to be addressed.
Two specific types of stroke make up the vast majority of stroke cases. Hemorrhagic strokes are caused by rupture of a blood vessel inside the brain, and ischemic strokes are caused by blockage of an artery in the brain; both conditions cause local hypoxia that damages brain tissue. Though both are serious and prevalent, ischemic strokes are more common, making up 87% of all strokes in the US. The blockages in ischemic stroke are usually caused by blood clots that become lodged in one of the arteries in the brain. Currently, tissue plasminogen activator (tPA), a thrombolytic drug that breaks down the clot, is the only FDA-approved therapeutic for the treatment of ischemic stroke. However, the patient experiencing a stroke must receive this treatment within 4.5 hours of the onset of stroke symptoms [6]. Treatment with tPA outside of this therapeutic time window can result in a hemorrhagic transformation, which can cause additional damage to the brain. If the patient does not make it to the hospital within the time window appropriate for treatment with tPA, there are other options for treatment if the clot does not clear itself, including a thrombectomy to physically remove the clot. Preventative treatments, such as anticoagulants and blood pressure lowering and cholesterol lowering drugs, may be administered as well, as there is an increased risk of having a second stroke immediately after the initial stroke [1]. Prompt administration of these treatments can help lessen the effects of disabilities that stroke can cause [7]. Common disabilities post-stroke include motor impairments such as hemiparesis (weakness of the left or right side of the body), hemiplegia (paralysis on the left or right side of the body), and central facial paresis [8]. Also common are language and speech disorders, such as global or mixed aphasia (impairment of language comprehension) and dysarthria (speech impairment) [9, 10]. Other disabilities include altered levels of consciousness, impaired vision, and decreased blood flow to parts of the brain [11]. All of these disabilities have drastic effects on the quality of life for stroke patients.
Overall, the available treatments for ischemic stroke are extremely limited, and more research must be done to not only explore treatment of the condition when the patient arrives at the hospital, but also possible avenues of recovery after the event. This review will examine more closely the risk factors associated with stroke, morphological changes and cell signaling post-stroke, experimental modeling, current therapies, and therapies under development for stroke patients.
2. Stroke Classification
In 2005, a study was performed to better identify and categorize the causes of ischemic stroke. A commonly used stroke cause classification system is named Trial of Org 10172 Acute Stroke Treatment (TOAST). The method proposed in the study was called Stop Stroke Study-TOAST (SSS-TOAST) classification. The study’s 50 participants underwent different imaging techniques to assign the likelihood that their conditions would lead to stroke. Likelihoods were listed as “evident, probable, or possible” for each condition’s propensity to cause a stroke, based on a parameter called positive likelihood ratio (PLR), which “was used to describe the strength of associations among clinical and imaging features and particular stroke mechanisms” [12]. Mathematically, PLR was defined as “the probability that a person with a given stroke subtype will have a particular clinical or imaging feature by the probability that a person with no such mechanism will have the same clinical or imaging features.” Statistically, there was no significant difference between the TOAST classification system and the SSS-TOAST classification system [12].
In 2011, a new method to assign causality of stroke was published that examined the reliability of the Causative Classification of Stroke (CCS) system. In the study, twenty members of the International Stroke Genetics Consortium independently assessed the same 50 stroke patients via abstracted case summaries [13]. When comparing Kappa values (which are used to measure inter-rater reliability [14]) obtained by CCS to those obtained by other classification systems, including TOAST, CCS was found to be the most reliable [13]. However, both CCS and TOAST are well-established and are still commonly used to classify stroke causes. They are not interchangeable, as they require different data and criteria, but the two systems have been found to be in agreement about 70% of the time [15].
Reliable classification of direct causes of ischemic stroke could lead to better treatment and medical outcomes of the condition. With more accurate assessments of stroke causality, therapies can be better tailored to either prevent or treat ischemic stroke and its aftermath.
3. Risk Factors
Many pathological and behavioral conditions have been shown to contribute to a higher risk of experiencing a stroke. These factors include, but are not limited to, diet, smoking habits, hypertension, and diabetes. Many of these risk factors involve a negative effect on the cardiovascular system, which is often the best way to assess stroke risk [16].
Relationships have been established between an increasing stroke rate in younger populations (under 50 years old) and traditional risk factors seen in patients over 50 years old, including conditions such as hypercholesterolemia, hypertension, and diabetes [17, 18]. These conditions and their relation to stroke in young patients have not been as widely investigated in literature. This is because in younger patients there are specific risk factors or underlying pathologies such as antiphospholipid syndrome (commonly found in females under 50 years old) that are a greater subject of research in this age group. An increased prevalence of traditional risk factors has been observed in young stroke victims. The most dramatic increases were seen in hypercholesterolemia and hypertension in ages between 35 and 44 years old for both males and females. A dramatic increase in the development of two stroke risk factors in the same age group has also been observed. However, it should be noted that one of the risk factors involved in those statistics was smoking, meaning that for smokers only one pathological risk factor had to manifest itself for there to be two risk factors present. There are excess mortalities in every age group across the board for young stroke victims, but, again, in individuals 35 to 50 years of age, there is a significant increase in excess mortality. It is difficult to establish any kind of direct causality from the information considered, but there is certainly a correlation between the increase of strokes in younger patients and the increased prevalence of particular risk factors at earlier ages that should not be ignored [17].
One study examining stroke pathology included information on risk factors for the stroke patients that were assessed. They examined 27 patients who were diagnosed with acute ischemic stroke, all aged between 65 and 75 years old. Out of the 27 patients, 22 had hypertension, which was the most common risk factor observed in the study. The next most common risk factor was atrial fibrillation, which was seen in 14 patients, followed by hypercholesterolemia in 10 patients, coronary artery disease in 9 patients, and diabetes in 8 patients. The study also examined the family history of cerebrovascular ischemic events for each patient’s family and found that 6 of the patients’ families had a history of these events. Mortalities were also recorded; 15 of the patients died within three days of the onset of ischemic stroke, 4 died four to seven days later, and 8 died eight or more days after ischemic stroke. Several conditions were termed as “predictors” for early mortality by the study, with coma being the most common of these predictors. Other factors included uncontrolled hypertension, diabetes, and acute coronary syndrome. As discussed above, it is difficult to assign any specific causality between the strokes that occurred and the risk factors involved, especially since many of the patients may have suffered from multiple risk factors at once. The study also suffers from a relatively small sample size, which means that some of the results may not be generalizable to a larger population [11]. However, the findings of the study are consistent with others that find a correlation between stroke and several of the risk factors assessed, such as hypertension, hypercholesterolemia, and diabetes [19–21].
A sedentary lifestyle may also contribute to higher stroke risks. Exercise has been shown to be an effective method of stroke prevention, as it decreases both cardiovascular and cerebrovascular risk [22, 23]. Exercise also upregulates expression of certain neuroprotective factors, such as endothelial nitric oxide synthase, brain derived neurotrophic factor, and insulin-like growth factor 1 (IGF-1) [24–26].
Overall, many of the risk factors associated with ischemic are manageable. Controlling factors such as diabetes and hypertension, as well as exercising regularly, could go a long way in prevention of ischemic stroke in higher risk populations.
4. Morphological Changes and Signaling Post-Stroke
Another important aspect of stroke research is assessment of the morphological changes in the brain after ischemic stroke occurs. Different types of cells in the brain undergo unique changes in morphology after ischemia occurs.
Ischemia observed in stroke patients causes injury in multiple cell types in the brain, including neurons, glial cells, and blood vessels. More specifically, neurons in the center of the ischemic area undergo liquefactive necrosis, a process that has been observed in other ischemic stroke studies in which the body and the axons of the neurons disappear [27, 28]. In large neurons, swelling, vacuolization of neuroplasm, and disappearance of the core and the nucleolus have been observed. Smaller neurons’ shapes get distorted, and the core of the cell condenses. These symptoms indicate severe damage to organelles including mitochondria, which no longer operate properly and cannot produce energy for the cell. These symptoms are also seen in glial cells, astrocytes, oligodendrocytes, and microglial cells [11]. The penumbra, which contains viable neurons surrounding the ischemic region, also features what are called “red neurons” or “ischemic neurons.” These neurons are characterized by several factors such as acidophilic cytoplasm, changes in neuronal proteins, and disintegration of endoplasmic ribosomes and Nissl bodies.
Lymphocytes, granulocytes, and “rare macrophages” have been found in both the ischemic and penumbral regions [29, 30]. This indicates that the processes involving cell apoptosis in these areas lead to the gathering of these cells and an immune response. The migration of these cells from blood vessels to the ischemic region has been seen in multiple studies and has been the starting point for further studies on the potential roles that monocytes play in ischemic conditions [31, 32]. Furthermore, increased permeability of both capillaries and the blood-brain barrier is observed in the penumbra, ischemic core, and in other areas, leading to perivascular and perineuronal edema. Changes in the blood-brain barrier occur quickly after an ischemic stroke. The development of pronounced extravasates and perivascular edema often occurs. The increased permeability of the blood-brain barrier leads to the influx of serum protein between four to six hours after stroke, and also leads to vasogenic edemas. Vasogenic edema peaks between one and two days after the stroke, and the water content of the tissue increased by over a hundred percent [11].
There is little reaction from blood-borne leukocytes within the first three days after the stroke. In this period, astrocytes undergo reactive gliosis (astrogliosis), which is a common occurrence when there is damage to the central nervous system [33]. In a healthy central nervous system, astrocytes are a diverse group of cells that show variability in both morphology and functionality, as they regulate several different physiological processes, including blood flow regulation, supplying of energy metabolites to neurons, and participation in synaptic function [34]. They also regulate ion and fluid balances, and direct the maintenance of transmitter molecules [35]. Upon damage to the central nervous system, the astrocytes undergo changes in both molecular expression and morphology. This reaction is not an “all or nothing” reaction, as it occurs in a series of steps that can be attenuated at different stages. The changes may result in altered gene expression or reversible changes to morphology, or they could be permanent changes, such as those that lead to scarring. The degree to which astrocytes react to central nervous system damage is mediated by both extracellular and intracellular signaling mechanisms that direct the reactions [36]. The purpose of the reaction is to protect neurons and to limit both inflammation and infection around the affected area. This process is necessary to support neuronal recovery in the penumbral region of the injury [37]. Unfortunately, this also comes with adverse effects that are often harmful to the patient. Astrogliosis has been observed to contribute to or cause further problems in the central nervous system, whether by gaining new, detrimental effects, or by the loss of existing functionality [35, 38].
Microglial cells, also called brain macrophages, also undergo transformations following ischemic stroke. They become swollen and release inflammatory proteins, such as cytokines, chemokines, proteases, cyclooxygenase-2, ROS, prostaglandins, HO-1, and other metabolites, which leads to the consumption of damaged tissue from the ischemic core [39]. However, these inflammatory factors can also be toxic to surrounding cells, which will be discussed in detail later in this section [11].
Brains of patients who die between four and seven days after suffering a stroke show signs of neural damage both inside and outside the ischemic region, demonstrating the potential for a stroke’s extended reach over the central nervous system. Within this same time period, neurons present in grey matter seem to be more susceptible to damage from hypoxia as compared to those in white matter [11]. In addition, many large macrophages have at that time already been actively phagocytosing dead tissue, which is widespread in both the ischemic and penumbral regions. Apoptosis of neuronal cells is most common in the first few days after the stroke occurs. By assessing the quantity of cells that express caspase proteins (caspase is a well-known marker for cells that are undergoing apoptosis), the number of cells undergoing apoptosis has been observed to be lower in patients who die after three days, suggesting that the majority of apoptosis occurs at early on after the stroke [11].
The inflammatory reaction of the brain to ischemia is another facet of stroke that is of great interest in research. This process includes cells that mediate the response as well as any factors, such as cytokines or proteins that are released, that could have an effect on the surrounding tissues [40].
Cells that originate from both the brain and blood play roles in the inflammatory response in the brain. Microglial cells are the primary immune cells in the central nervous system and can be activated within minutes of the damaging event [41]. They reach peak amounts of activity two to three days after the injury and linger for weeks afterward. The role these cells play in ischemia is not well understood, since there are multiple types of microglia that perform different functions. One example of this is that microglia are observed to release both neuroprotective and neurotoxic factors after damage occurs.
Astrocytes are also an integral part of brain function, however, like microglial cells, exhibit a duality in their function after ischemia. As mentioned above, astrocytes undergo astrogliosis, in which they exhibit glial fibrillary acidic protein (GFAP) and begin to secrete factors that both complicate and improve ischemic recovery [40].
Other cells that play inflammatory roles migrate to the brain from the blood. Leukocytes are the first to arrive at the ischemic site. These are responsible for the release of neurotoxic factors, including inducible nitric oxide synthase and matrix metalloproteinases (MMPs), which have been associated with both increased compromise of the blood brain barrier, as well as hemorrhagic transformation after an ischemic stroke [40, 42]. Studies suggest that neutrophils are more harmful than beneficial to recovery after ischemic stroke, as removal or exclusion of the neutrophils has been shown to decrease injury and improve recovery [40].
Monocytes and macrophages also migrate to the brain through the bloodstream and, like microglial cells and astrocytes, have a complicated relationship with the inflammatory response to stroke [43, 44]. Blood monocytes fall into two categories when it comes to inflammatory responses: anti-and pro-inflammatory. Anti-inflammatory monocytes are so named because they exhibit anti-inflammatory cytokines like interleukin 10 (IL-10). Pro-inflammatory monocytes are so named because they release factors such as IL-1β and tumor necrosis factor (TNF-α), both of which have are known to have neurotoxic effects [45]. Monocytes, when they reach the brain, have been observed to differentiate into dendritic cells, or cells that are similar to microglia and macrophages. Blood macrophages fall into similar categories in regards to the inflammatory response; generally, M1 macrophages are considered pro-inflammatory and M2 macrophages are anti-inflammatory. Like monocytes, these cells release both the pro-inflammatory factors IL-1β and TNF-α as well as the anti-inflammatory factors IL-10 and transforming growth factor beta (TGF-β) according to their M1/M2 phenotype [40].
Lymphocytes also migrate to the brain after an ischemic stroke. T cells have been the emphasis of research, as many kinds have been associated with the response to ischemia. Of great interest are CD4+, CD8+, and γδT cells, which release cytokines such as IFN-γ and IL-17 that are detrimental to the brain. However, regulatory T cells produce cytokines with anti-inflammatory properties, such as IL-10, indicating that T cells, like blood monocytes and macrophages, have a complex relationship with the inflammatory response to ischemia that needs to be examined further [40].
The cytokines released by these cells have a pronounced impact on certain aspects of inflammation and stroke recovery. Some of the effects seen from these cytokines are positive, some are negative, and some are not entirely understood (Table–I). Beneficial cytokines include IL-10, TGF-β, TNF-α-inducible protein 8-like-2 (TIPE-2), and IGF-1, all of which have been seen to decrease the volume of damaged tissue after ischemia. Cytokines that have negative impacts on recovery and are associated with increased damage volume are more commonly studied. These include IL-1α, IL-1β, and E-selectin, adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1), and chemokines like monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-1α (MIP-1α), and fractalkine (CX3CL1) [40, 46, 47]. These cytokines mediate damage in various ways. Some examples are IL-1β’s contribution to blood-brain barrier disruption and vasogenic edema, promotion of inflammation by adhesion molecules that cause neutrophils to adhere to the vascular endothelium and infiltrate to the damaged site, and the ability of CX3CL1 to activate and attract microglia to the damaged region of the brain [48]. Other cytokines appear to play contradictory roles in response to ischemic stroke. For example, TNF-α is most often considered a pro-inflammatory factor, and plays a major role in glutamate excitotoxicity, as it blocks glutamate transporters on astrocytes and increases surface expression of α-amino-3-hydroxy-5-methyl-4-isxazolepropionic acid (AMPA) receptors at glutamatergic synapses [49, 50]. However, studies report that TNF-α has both pro-inflammatory and neuroprotective tendencies. One explanation for this duality that is being explored is the relationship between these responses and the two receptors to which TNF-α binds. However, TNF-α is not the only cytokine with conflicting effects. MMP-9 also has different effects based on the point in time at which it is released. It is associated with increases in blood-brain barrier permeability, edema, and hemorrhagic transformation in the early stages of post-stroke, but at later stages, it induces several growth factors that stimulate angiogenesis. The cytokine P-selectin also has dual roles in ischemia; blockage of P-selectin decreases brain injury but also decreases survivability of animals that undergo middle cerebral artery occlusion (MCAO) experiments. Given these examples, it is obvious that more research must be done in order to understand exactly how these cytokines affect the brain in relation to ischemic stroke.
Table I.
Effector function of secreted molecules after ischemic stroke.
| Molecules Affecting Infarct Volume in Ischemic Stroke | ||
|---|---|---|
| Increase Infarct Volume |
Decrease Infarct Volume |
Dual Roles |
| IL-1α | IL-10 | TNF-α |
| IL-1β | TGF-β | MMP-9 |
| E-Selectin | TIPE-2 | P-Selectin |
| ICAM-1 | IGF-1 | |
| VCAM-1 | ||
| MCP-1 | ||
| MIP-1α | ||
| CX3CL1 | ||
There are multiple pathways that are responsible for releasing these cytokines at the ischemic site. These include Toll-like receptor (TLR) pathways, mitogen-activated protein kinase (MAPK) pathways, and the nuclear factor-kappa B (NF-κB) pathway. TLRs have been closely associated with ischemic stroke in animal models. Inhibition of TLR2 and TLR4 specifically has been shown to decrease susceptibility to brain damage after ischemia in mice, showing smaller volumes of infarct tissue. However, other studies have shown that stimulation of TLRs prior to ischemic injury could have neuroprotective effects by inhibiting TNF-α, one of the final products of the TLR2 pathway [51, 52]. Earlier stimulation of this pathway actually inhibits NF-κB expression (the precursor to TNF-α), and enhances expression of IFN-β, which is a precursor for anti-inflammatory cytokines [53–55]. Unfortunately, there is little potential to use this as a preventative measure for ischemic stroke, as the alteration in the pathway must be present before the stroke takes place, which is difficult to predict ahead of time.
The MAPK pathways also have a close relationship with ischemic stroke, as all of these pathways are activated when ischemic stroke occurs. Their exact roles in ischemic stroke are unclear, but studies have shown that the pathways that include JNK and p38 contribute to ischemic injury [56], since inhibition of these pathways decreases the amount of damaged tissue [57]. These pathways contribute to the production of pro-inflammatory cytokines such as TNF-α and IL-1β. However, several of the MAPK pathways may also have neuroprotective qualities. Activation of the ERK1 or ERK2 may be beneficial as they increase expression of the protein Bcl-2, which prevents apoptosis. The ERK5 pathway could also act in a neuroprotective capacity when activated prior to an ischemic event [40].
Another set of pathways activated after an ischemic event are the NF-κB pathways, which are responsible for induction of pro-inflammatory factors such as MMPs [58]. Activation of both the p50 and p65 pathways have been shown to be harmful in regards to ischemic stroke. Studies also show that mice with the p50 gene knocked out had a smaller volume of damaged tissue in a permanent stroke model. However, results of some studies are again contradictory, as inhibition of NF-κB with diethyldithiocarbamate in rats caused DNA fragmentation and higher volumes of damaged tissue [40].
Identifying the changes that cells undergo post-ischemia, especially signaling changes in the brain, have been some the most challenging aspects of stroke research. Better understanding of the mechanisms that are heavily involved with post-stroke pathology could contribute substantially to development of therapies for ischemic stroke.
5. Experimental Models for Ischemic Stroke
5.1. In Vivo Models
Many of the experiments to examine mammalian responses to stroke have been performed in mainly in rodents such as mice and rats. In order to conduct these experiments, a situation that simulates a stroke in the animal must occur. The most common methods are Middle Cerebral Artery Occlusion (MCAO) procedures, and there are several different MCAO methods that are used in research.
The MCAO method used the most in research is the intraluminal suture model, where a filament is fed into the middle cerebral artery from either the external or internal carotid arteries to block blood flow for a set amount of time, resulting in transient occlusion [59, 60] (Fig. 1). This model causes a large volume of infarct tissue and has been instrumental to the progression of stroke research regarding issues such as blood-brain barrier disruption and inflammatory response to ischemia [61, 62].
Figure 1.
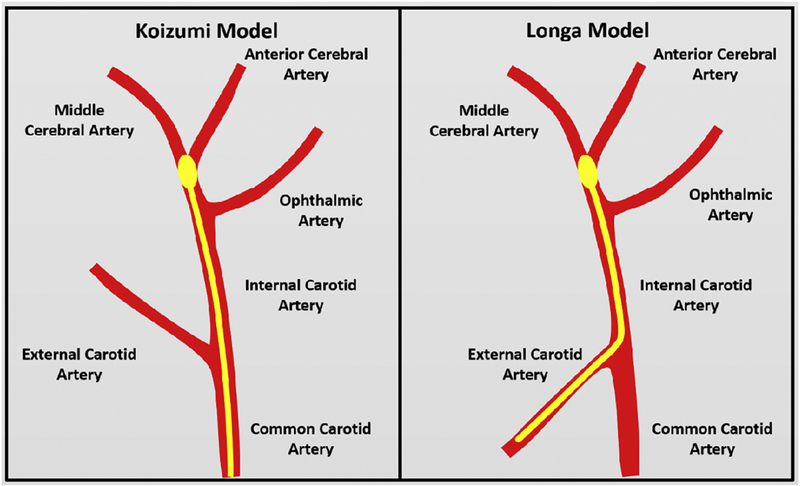
Two intraluminal filament methods can be used to achieve middle cerebral artery occlusion The Longa method (right) has been deemed more useful for stroke research than the Koizumi method (left). The Longa method sees a much lower mortality rate post-surgery than that of the Koizumi. In the Longa method, the filament is inserted in the external carotid artery and is guided through the internal carotid artery to the middle cerebral artery, as opposed to insertion from the common carotid artery as seen in the Koizumi method.
Direct occlusion of the middle cerebral artery takes place via a craniectomy. In this method, occlusion is usually performed by pinching the artery closed when reperfusion is desired or by cauterization and transection of the artery if a permanent occlusion is needed. Craniectomy models can be used for many species, which makes it a popular model for simulating ischemic stroke [59].
The photothrombosis model is another method of occlusion that is frequently used in rats as well as in mice. Briefly, Rose Bengal, a photosensitive dye, is injected systemically into the animal. Following injection, a laser at a wavelength of 532 nanometers is illuminated directly onto the skull of the animal, causing the dye to react [63]. This produces reactive oxygen species (ROS), which damage the endothelial lining of the vessels (Fig. 2). The commonly proposed mechanism for clot formation is that the damage to the endothelial cells initiates the contact activation pathway of the coagulation cascade, and that an aggregation of platelets occludes the artery [64, 65] (Fig. 3). However, some have called into question the role of platelet activation in the formation of the clot, with some authors suggesting that the model is not suitable for stroke research, but only blood-brain barrier research [66, 67]. This model has many advantages, including reproducibility, ability to select which region of the brain sustains the occlusion, and low mortality in animals that undergo the procedure [59].
Figure 2.
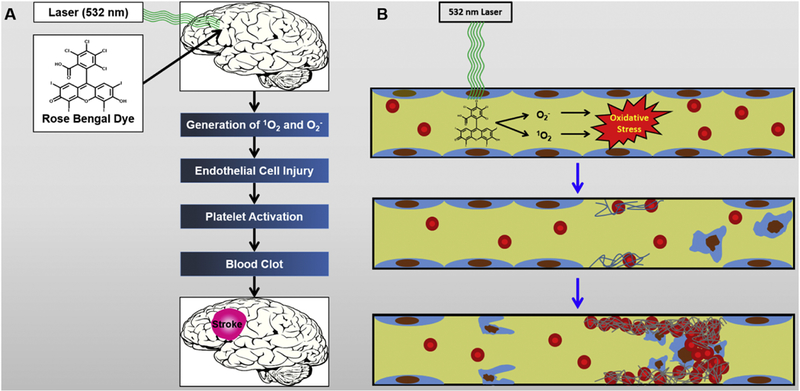
A. Summary of stroke generation using Rose Bengal Dye. B. Stepwise depiction of stroke generation at molecular and cellular levels.
Figure 3.
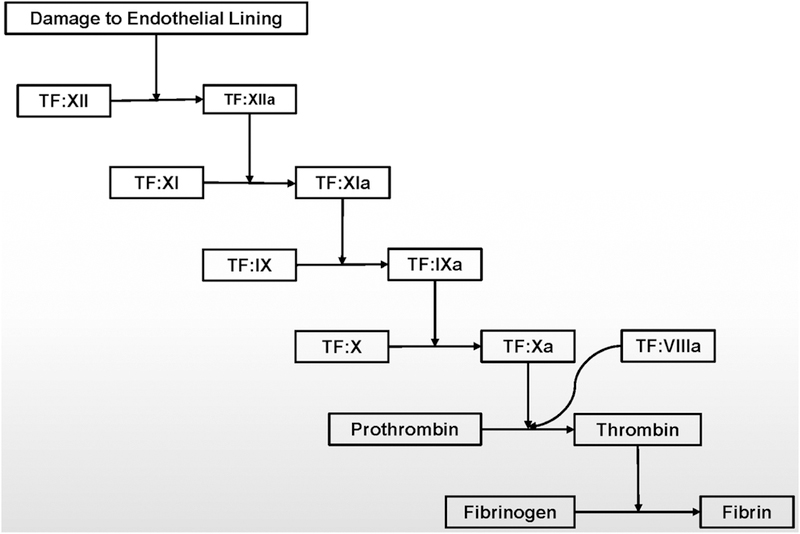
Stepwise summary of the intrinsic pathway of the coagulation cascade.
Another commonly used method for modeling stroke in vivo is by use of endothelin-1, a potent vasoconstrictor. The peptide can be applied in a number of ways, including directly onto an exposed vessel, on the brain’s surface, or via injection into the brain. Duration and level of the occlusion can be modified by changing the concentration of endothelin-1 that is used in the treatment. Endothelin-1 models are used in both mice and rats, and though they are commonly used for short-term experiments [68, 69], they are generally more useful for long time recovery studies [59].
While these models certainly have their advantages, the fact that hundreds of attempted stroke therapies have failed to translate to human treatments shows that there is still much work to do to accurately recreate ischemic conditions in experimental models. The above models each have advantages, but they all have significant shortcomings that undermine the potential for translation of stroke remedies to human treatments. For example, the intraluminal filament MCAO model often involves a very sudden reperfusion, not a gradual one as is seen in ischemic stroke in clinical settings. This causes different pathways to be activated, which more closely resemble global ischemia, making the translation of results to human studies extremely difficult. Other disadvantages include possible hemorrhaging of blood vessels, induced hyperthermia in the animal, and the possibility of an incomplete occlusion of the artery [59, 70].
The major drawback of direct occlusion models via craniectomy is the amount of trauma that must be introduced to the brain in order to gain access to the middle cerebral artery. Along with the skull trauma, there are possibilities of directly injuring the brain during the craniectomy procedure, as well as issues that could rise from exposing the brain to an open atmosphere. The possible change in the temperature of the exposed part of the brain, as well as the disruption of the intracranial pressure are extremely different from the occurrence of stroke in a human. In addition to these flaws in the model, it shares the same shortcoming of the intraluminal filament model in that reperfusion is sudden, causing a different set of pathways to be activated [59].
The photothrombotic model has only two big disadvantages in regards to recreating ischemic conditions, one of which is that there is a lack of a penumbral region in the area surrounding the ischemic region. This makes it extremely difficult to evaluate whether or not treatments have any effect on the volume of damaged tissue in the stroke model. However, there have been plenty of attempts to work around this flaw. One such attempt is a modified photothrombotic model published in 2016, in which the penumbra could be determined by a combination of perfusion-weighted imaging, diffusion-weighted imaging, and other MRI technologies [63]. The results were promising, and if the determination of a penumbra that is similar to that in humans becomes easy to achieve, then one of the major issues with this model will have been solved. The other issue in the model is that the photothrombotic model causes both vascular and cytotoxic edemas, while ischemic strokes in humans cause mainly cytotoxic edemas that do not immediately compromise the blood-brain barrier, meaning that even if the identification of the penumbra is achieved, there may still be difficulty in translation to human treatments [59].
The endothelin-1 model also has a number of flaws. The onset of ischemia is slow and there is very little edema formation. Endothelin-1 also affects astrocytes, neurons, and can cause axonal sprouting, all of which can cause inconsistencies when compared to strokes in humans [59, 70].
While there have been a few advances in stroke research using the above models, the vast lack of translation of these models to human strokes is a major reason that new, effective stroke treatments have remained elusive. There is much room for improvement in these areas of preclinical research, and until these models can more accurately illustrate strokes in humans, it may remain difficult to find new therapeutic strategies to combat ischemic stroke.
5.2. In Vitro Models
In vitro models are also commonly used in ischemic stroke. Many of these models include a co-culture of different types of cells, such as astrocytes and endothelial cells. Using co-cultures, it is possible to mimic certain properties of the blood-brain barrier and examine how different cell types interact with one another under ischemic conditions [71]. Other in vitro models use primary cultures taken from brain tissue of animals. These cultures can be useful for investigating effects of different treatments on specific aspects of the brain, such as neurons in the cerebral cortex [72]. Both types of in vitro models are used to examine specific cellular reactions to conditions comparable to ischemia, such as oxygen and glucose deprivation. These models have a number of advantages, as they allow researchers to study both animal and human cells directly [73]. Since species differentiation plays a key role in translating findings from bench to bedside, use of human-based in vitro models can aid in understanding differences between human and animal responses to stroke. However, in vitro studies, as important as they are, must be combined with in vivo studies to obtain the full picture of ischemia in humans [59].
6. Current Therapies for Ischemic Stroke
As mentioned above, tPA is the only one FDA-approved treatment for ischemic stroke. Endogenous tPA is a serine protease that plays an essential role in the fibrinolytic system of the body. Blood clots are formed by the aggregation of platelets onto fibrin meshes that form after an injury to a vessel. tPA initiates the process by which blood clots are dissolved via the activation of plasminogen to plasmin, which cleaves fibrin and dissolves the clots. tPA is regulated by binding to fibrin, which increases tPA’s catalytic ability [74]. The recombinant form of tPA was approved as a drug in 1996 after a clinical trial by the National Institute of Neurological Disorders and Stroke’s tPA Stroke Study Group was published a year earlier. The study showed a relative risk reduction of 30% for the chances of having no or little disability when compared to a placebo at 90 days [7]. The therapeutic time window for tPA is extremely narrow, as it must be administered within 4.5 hours of the onset of stroke, otherwise there is a high risk of a hemorrhagic transformation, which can cause other complications in treatment [6]. Unfortunately, some are hesitant to administer tPA even within the therapeutic time window, as there are other risks that are involved, including anaphylaxis and systemic bleeding [7], and there are still clinical trials being conducted to determine what risk factors should disqualify someone from receiving tPA treatment for an ischemic stroke [75].
Attempts have been made to extend the therapeutic time window of tPA [76–78]. If the extension of the window can be achieved, it would give patients more time to get to the hospital and be evaluated by a doctor for treatment with tPA. Combination therapies with tPA have been studied in preclinical settings to try to achieve this goal. One study assessed the possibility of using the Rho-kinase inhibitor Fasudil, which was loaded into liposomes and applied two hours before administration of tPA in rats (Fig. 4). The stroke model that was used was the photothrombotic occlusion of the middle cerebral artery. In the study, treatment with the Fasudil-loaded liposomes (Fasudil-Lip) and tPA was shown to decrease the amount of damaged tissue in the rat brains when compared to treatments with Fasudil alone, Fasudil-Lip alone, and tPA alone. With this treatment, the therapeutic time window of tPA treatment for rats was extended dramatically to 4.5 hours, more than double that of treatments with only tPA (the therapeutic time window for tPA administration in rats was found in the study to be just under 2 hours) [6]. As was mentioned earlier, most stroke patients don’t arrive at a hospital in time to receive tPA treatments. Further, patients who arrive at the hospital in a timely manner must still be evaluated to eliminate the possibility that they are suffering from a hemorrhagic stroke, which cannot be treated with tPA, since it would lead to further bleeding in the brain. As tPA is the only currently available drug to treat ischemic stroke, it is imperative to give patients as much time as possible to receive tPA at the hospital once they do arrive. Extending the therapeutic time window may save patients from severe disability or death, when they may not have arrived within the current therapeutic time window for tPA administration.
Figure 4.
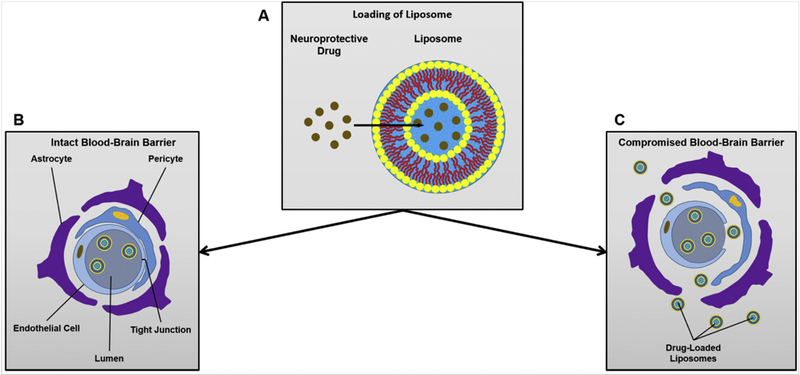
A. Liposomes can be loaded with drugs to be delivered to the brain. B. Delivery of drug-loaded liposomes is impossible when the blood-brain barrier is intact. C. When the blood-brain barrier is compromised (such as after a stroke), liposomes can be used to deliver neuroprotective drugs to the tissue surrounding the vessel.
A rapidly developing field of stroke recovery studies involves the use of micro-RNA (miRNA) to manipulate the expression of genes and protein in the post-stroke brain. It is estimated that as many as 30% of human protein coding genes could be regulated by miRNAs [79]. miRNAs are believed to play central roles in embryonic development, cell differentiation, and metabolism, and have been used in research to try to treat cancer and liver and heart diseases. In addition, one miRNA-based treatment for Hepatitis C has already advanced into Phase II clinical trials, demonstrating the great potential that this field of research possesses [79]. Briefly, mature miRNAs integrate into a multiprotein structure called the RNA-induced silencing complex (RISC) and bind to the 3’ untranslated region of a target mRNA. This process can block translation of or cleave the target mRNA, leading to lower expression of the corresponding protein [80]. miRNAs are believed to play instrumental roles in the progression of ischemic stroke pathology through changes in the expression of select miRNA, such as miR-15a, which contributes to ischemic damage through inhibition of BCL-2, an anti-apoptotic gene [80]. BCL-2 family genes have been one of the major focuses in the research of miRNA therapies for ischemic stroke, since they play central roles in cell death and are regulated by multiple miRNAs after ischemia, such as miR-29, miR-181, and the aforementioned miR-15 [79, 81]. A specific example of a potential therapeutic approach is the blocking of miR-181 activity after ischemic stroke in mice using an antagomir and engineered oligonucleotide that binds to the mRNA region targeted by the miRNA of interest, preventing the regulation of the desired mRNA by miRNA. A study reported that application of a miR-181a antagomir after MCAO yielded decreased expression of NF-B, increased levels of BCL-2, and a decreased infarct size. Long-term recovery of treated mice was also improved [82]. While these results are promising, miRNA research is still relatively new and comes with its own challenges. A single miRNA could target a number different mRNAs, and while this aspect has the potential benefit of silencing multiple detrimental mRNAs with one miRNA, there is also the possibility of unwanted side-effects through unintended targeting of beneficial mRNAs [79]. In addition to potential problems with target specificity, effective delivery and rapid degradation of miRNAs also pose potential problems for which solutions are still being investigated [80]. Yet, despite these obstacles, miRNAs represent a promising field of possible therapies that could have a substantial impact on ischemic stroke treatment.
7. Regenerative Therapies for Future Studies
The use of stem cell therapies has been explored in a wide range of conditions [83], including neurological diseases and in autoimmune diseases such as rheumatoid arthritis and inflammatory bowel diseases, in which inflammation plays key roles [84–86]. Transplantation of hematopoietic stem cells (HSCs) possessing CD34+, CD133+, and CXCR4 phenotypes, which are taken from human umbilical cords, has been investigated as a therapy for myocardial ischemia, and cardiac function was improved when analyzed using rat models [87]. However, for therapies that involve CD34+ HSC transplantation, millions of stem cells are needed, thus there has to be a way to rapidly produce stem cells ex vivo for these therapies to be plausible. A procedure for rapid expansion of CD34+ HSCs using aminated polyethersulfone (PES) nanofibers as scaffolds has already been established. Using this method, a 225-fold expansion of was achieved without inducing differentiation, showing that developing a required number of stem cells ex vivo should not be a hindrance in developing potential stem cell therapies further [88]. Expansion using this procedure produced a sufficient number of cells treated to overexpress proangiogenic factors for transplantation to treat injuries from limb ischemia by neovascularization [89, 90].
Neural differentiation of CD34+ HSCs has been examined in regards to treatment of neurological diseases such as Parkinson’s disease (PD) and the loss of dopaminergic neurons. In the study, mice underwent induction of a brain lesion by administration of 1 methyl-4-phenyl-1, 2, 3, 6-tetrehyropyrine (MPTP-mice). CD34+ HSCs were expanded using PES nanofibers as a scaffold. In total, 4.5 million cells were generated from an initial number of 20,000. They were then differentiated in vitro in media containing neural differentiating factors for three weeks. Successful differentiation was characterized by the presence of neural markers such as GFAP, MAP2, and oligo2, and nestin, none of which were expressed in undifferentiated cells that were used as a control. The differentiated cells also changed in morphology beginning on day 3, and became very similar in appearance to glial cells and neurons. After peripheral administration of the neural-differentiated HSCs, recovery of dopaminergic neurons, as well as angiogenesis were observed in both the caudate/putamen and substantia nigra pars compacta regions of the brains in MPTP-mice, showing great promise for stem cell therapies for neurological diseases [91]. These current advances in stem cell research and the exploration of the effect that stem cells have in PD models show that stem cell therapies have great potential for success in the treatment of ischemic stroke (Fig. 5).
Figure 5.
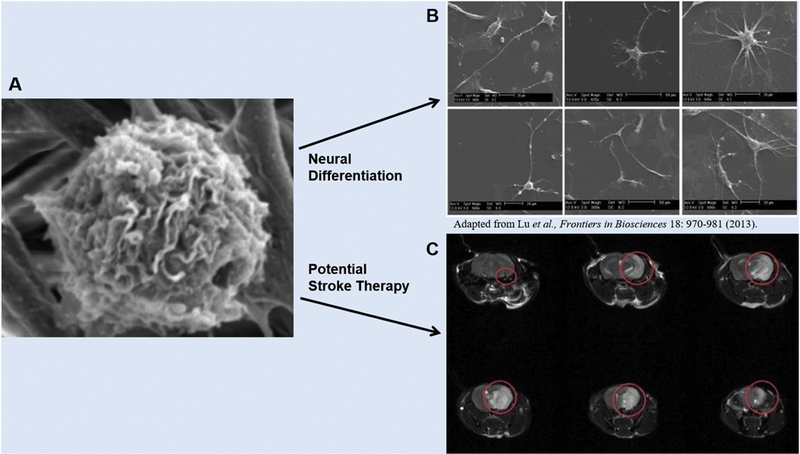
A. Scanning electron micrograph of nanofiber expanded CD34+ hematopoietic stem cells. B. Scanning electron micrograph of neural cells differentiated from nanofiber expanded CD34+ hematopoietic stem cells. C. Serial images of brain taken with magnetic resonance imaging machine after middle cerebral artery occlusion-mediated stroke generated in non-obese diabetic/severe combined immunodeficient mice.
Dental pulp stem cells (DPSCs) have also been of interest in the treatment of neurodegenerative conditions. More specifically, they show great potential to promote neuroregeneration after injuries such as ischemic stroke [92]. As they originate in the neural crest, they are prime candidates to replace neuronal cells lost in neurodegenerative conditions such as ischemic stroke In a study published in 2017, DPSCs were plated in matrigel with hippocampal slices taken from adult mice. When co-cultivated with the hippocampal slices, the DPSCs did differentiate along a neuronal lineage as expected, but they also secreted neurotrophic factors that supported the growth and preservation of the hippocampal tissue [94]. Another study carried out in 3-day-old rats demonstrated the ability of DPSCs to integrate into a damaged brain. Neural-predifferentiated DPSCs were transplanted intrathecally after an induced lesion by means of touching a metal stamp cooled to −60˚C to the skull above the forelimb motor complex. The tagged cells were localized in the neural progenitor zones in the rat brains, and, four weeks after injury, were also detected in the injured part of the brain. These cells also exhibited neuronal markers such as NeuN and GFAP as well as voltage-dependent ion channel activity [93]. While the study does not mimic the conditions of an ischemic stroke, the successful integration of pre-differentiated DPSCs into the injured rat brain is a promising result, and warrants further testing in a scenario that simulates ischemic stroke, which is already being explored [95–97].
With the success of advanced methods for expansion and differentiation of HSCs and DPSCs, stem cells represent a class of possible stroke treatments that is being explored extensively. Successful expansion, differentiation, and evidence of neuronal regeneration and angiogenesis with the CD34+ HSCs, as well as in experiments using DPSCs, show that stem cells have great potential for incorporation into stroke research.
While development of new therapies remains elusive in ischemic stroke research, there are still many avenues of treatments that can be explored more thoroughly. From combination therapies with tPA to stem cell therapies to manipulation of inflammatory factors, there are still many strategies that can be explored in the treatment of ischemic stroke.
8. Conclusion
Ischemic stroke is a condition that causes significant morbidity and mortality worldwide. While there have been several advances in experimental modeling and prospective treatments, research has left much to be desired when it comes to understanding the mechanisms and factors at play when a stroke occurs. It is not entirely understood how all of the various inflammatory factors affect the brain during and after ischemia. In vivo and in vitro models, though useful for recapitulating specific aspects of stroke-related pathological changes, still fall short in many respects when trying to simulate the human brain during an ischemic event. Because of this discrepancy between models and reality, ischemic stroke therapies are extremely difficult to produce despite the many different available strategies from which to approach the issue. While there is always room for improvement in the research of diseases, this is especially true for ischemic stroke research, and the development of new, more accurate models and more effective therapies still presents countless challenges.
Highlights.
This comprehensive review focuses on risk factors associated with ischemic stroke.
Advantages and disadvantages of in vivo and in vitro ischemic stroke models.
Current ischemic stroke therapies and new therapeutic strategies.
Focuses on potential of stem cells to promote stroke recovery.
Acknowledgments
This work was supported in part by National Institutes of Health grants, R01AR068279 (NIAMS) and STTR 1R41EY024217 (NEI). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stroke Facts | cdc.gov. 2017. 2017–09-06T08:10:23Z; Available from: https://www.cdc.gov/stroke/facts.htm.
- 2.Goldstein LB, et al. , Primary prevention of ischemic stroke: A Statement for Healthcare Professionals From the Stroke Council of the American Heart Association. Stroke, 2018. 32(1): p. 280–99. [DOI] [PubMed] [Google Scholar]
- 3.Jackson CA and Mishra GD, Depression and risk of stroke in midaged women: a prospective longitudinal study. Stroke, 2013. 44(6): p. 1555–60. [DOI] [PubMed] [Google Scholar]
- 4.Leffert LR, et al. , Hypertensive disorders and pregnancy-related stroke: frequency, trends, risk factors, and outcomes. Obstet Gynecol, 2015. 125(1): p. 124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sacco S, et al. , Hormonal contraceptives and risk of ischemic stroke in women with migraine: a consensus statement from the European Headache Federation (EHF) and the European Society of Contraception and Reproductive Health (ESC). J Headache Pain, 2017. 18(1): p. 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukuta T, et al. , Combination therapy with liposomal neuroprotectants and tissue plasminogen activator for treatment of ischemic stroke. FASEB J, 2017. 31(5): p. 1879–1890. [DOI] [PubMed] [Google Scholar]
- 7.Chapman SN, et al. , Current perspectives on the use of intravenous recombinant tissue plasminogen activator (tPA) for treatment of acute ischemic stroke. Vasc Health Risk Manag, 2014. 10: p. 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cramer SC and Chopp M, Recovery recapitulates ontogeny. Trends Neurosci, 2000. 23(6): p. 265–71. [DOI] [PubMed] [Google Scholar]
- 9.NIDCD. Aphasia 2015. 2015–08-18; Available from: https://www.ncbi.nlm.nih.gov/pubmed/.
- 10.Rathore SS, et al. , Characterization of incident stroke signs and symptoms: findings from the atherosclerosis risk in communities study. Stroke, 2002. 33(11): p. 2718–21. [DOI] [PubMed] [Google Scholar]
- 11.Slujitoru AS, et al. , Clinical and morphological correlations in acute ischemic stroke. Rom J Morphol Embryol, 2012. 53(4): p. 917–26. [PubMed] [Google Scholar]
- 12.Ay H, et al. , An evidence-based causative classification system for acute ischemic stroke. Ann Neurol, 2005. 58(5): p. 688–97. [DOI] [PubMed] [Google Scholar]
- 13.Arsava EM, et al. , The Causative Classification of Stroke system: an international reliability and optimization study. Neurology, 2010. 75(14): p. 1277–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McHugh ML, Interrater reliability: the kappa statistic. Biochem Med (Zagreb), 2012. 22(3): p. 276–82. [PMC free article] [PubMed] [Google Scholar]
- 15.McArdle PF, et al. , Agreement between TOAST and CCS ischemic stroke classification: the NINDS SiGN study. Neurology, 2014. 83(18): p. 1653–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benjamin EJ, et al. , Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation, 2017. 135(10): p. e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maaijwee NA, et al. , Ischaemic stroke in young adults: risk factors and long-term consequences. Nat Rev Neurol, 2014. 10(6): p. 315–25. [DOI] [PubMed] [Google Scholar]
- 18.Vijayan M and Reddy PH, Stroke, Vascular Dementia, and Alzheimer’s Disease: Molecular Links. J Alzheimers Dis, 2016. 54(2): p. 427–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Collins VE, et al. , Hypertension and experimental stroke therapies. J Cereb Blood Flow Metab, 2013. 33(8): p. 1141–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larsson SC, Akesson A, and Wolk A, Primary prevention of stroke by a healthy lifestyle in a high-risk group. Neurology, 2015. 84(22): p. 2224–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khoury JC, et al. , Diabetes mellitus: a risk factor for ischemic stroke in a large biracial population. Stroke, 2013. 44(6): p. 1500–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sacco RL, et al. , Leisure-time physical activity and ischemic stroke risk: the Northern Manhattan Stroke Study. Stroke, 1998. 29(2): p. 380–7. [DOI] [PubMed] [Google Scholar]
- 23.Lee IM, et al. , Exercise and risk of stroke in male physicians. Stroke, 1999. 30(1): p. 1–6. [DOI] [PubMed] [Google Scholar]
- 24.Cotman CW, Berchtold NC, and Christie LA, Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci, 2007. 30(9): p. 464–72. [DOI] [PubMed] [Google Scholar]
- 25.Endres M, et al. , Mechanisms of stroke protection by physical activity. Ann Neurol, 2003. 54(5): p. 582–90. [DOI] [PubMed] [Google Scholar]
- 26.Rasmussen P, et al. , Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp Physiol, 2009. 94(10): p. 1062–9. [DOI] [PubMed] [Google Scholar]
- 27.Park JY, et al. , Neuroprotective effect of human placental extract on hypoxic-ischemic brain injury in neonatal rats. Brain Dev, 2013. 35(1): p. 68–74. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen TV, et al. , Multiplex immunoassay characterization and species comparison of inflammation in acute and non-acute ischemic infarcts in human and mouse brain tissue. Acta Neuropathol Commun, 2016. 4(1): p. 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, et al. , Accumulation of natural killer cells in ischemic brain tissues and the chemotactic effect of IP-10. J Neuroinflammation, 2014. 11: p. 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang ZK, et al. , Infiltration of invariant natural killer T cells occur and accelerate brain infarction in permanent ischemic stroke in mice. Neurosci Lett, 2016. 633: p. 62–68. [DOI] [PubMed] [Google Scholar]
- 31.Margaritescu O, et al. , Histopathological changes in acute ischemic stroke. Rom J Morphol Embryol, 2009. 50(3): p. 327–39. [PubMed] [Google Scholar]
- 32.ElAli A and Jean LeBlanc N, The Role of Monocytes in Ischemic Stroke Pathobiology: New Avenues to Explore. Front Aging Neurosci, 2016. 8: p. 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roy Choudhury G, et al. , Involvement of p38 MAPK in reactive astrogliosis induced by ischemic stroke. Brain Res, 2014. 1551: p. 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khakh BS and Sofroniew MV, Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci, 2015. 18(7): p. 942–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sofroniew MV, Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci, 2009. 32(12): p. 638–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koyama Y, Signaling molecules regulating phenotypic conversions of astrocytes and glial scar formation in damaged nerve tissues. Neurochem Int, 2014. 78: p. 35–42. [DOI] [PubMed] [Google Scholar]
- 37.Sims NR and Yew WP, Reactive astrogliosis in stroke: Contributions of astrocytes to recovery of neurological function. Neurochem Int, 2017. 107: p. 88–103. [DOI] [PubMed] [Google Scholar]
- 38.Huang L, et al. , Glial scar formation occurs in the human brain after ischemic stroke. Int J Med Sci, 2014. 11(4): p. 344–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yenari MA, Kauppinen TM, and Swanson RA, Microglial activation in stroke: therapeutic targets. Neurotherapeutics, 2010. 7(4): p. 378–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin R, et al. , Role of inflammation and its mediators in acute ischemic stroke. J Cardiovasc Transl Res, 2013. 6(5): p. 834–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor RA and Sansing LH, Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin Dev Immunol, 2013. 2013: p. 746068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lakhan SE, et al. , Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front Neurol, 2013. 4: p. 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ritzel RM, et al. , Functional differences between microglia and monocytes after ischemic stroke. J Neuroinflammation, 2015. 12: p. 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gronberg NV, et al. , Leukocyte infiltration in experimental stroke. J Neuroinflammation, 2013. 10: p. 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clausen BH, et al. , Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J Neuroinflammation, 2008. 5: p. 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonaventura A, et al. , Update on Inflammatory Biomarkers and Treatments in Ischemic Stroke. Int J Mol Sci, 2016. 17(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murray KN, Parry-Jones AR, and Allan SM, Interleukin-1 and acute brain injury. Front Cell Neurosci, 2015. 9: p. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amantea D, et al. , Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front Neurosci, 2015. 9: p. 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pickering M, Cumiskey D, and O’Connor JJ, Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol, 2005. 90(5): p. 663–70 [DOI] [PubMed] [Google Scholar]
- 50.Beattie EC, et al. , Control of synaptic strength by glial TNFalpha. Science, 2002. 295(5563): p. 2282–5. [DOI] [PubMed] [Google Scholar]
- 51.Stephenson D, et al. , Transcription factor nuclear factor-kappa B is activated in neurons after focal cerebral ischemia. J Cereb Blood Flow Metab, 2000. 20(3): p. 592–603. [DOI] [PubMed] [Google Scholar]
- 52.Schneider A, et al. , NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med, 1999. 5(5): p. 554–9. [DOI] [PubMed] [Google Scholar]
- 53.Veldhuis WB, et al. , Interferon-beta blocks infiltration of inflammatory cells and reduces infarct volume after ischemic stroke in the rat. J Cereb Blood Flow Metab, 2003. 23(9): p. 1029–39. [DOI] [PubMed] [Google Scholar]
- 54.Liu H, et al. , Interferon-beta administration confers a beneficial outcome in a rabbit model of thromboembolic cerebral ischemia. Neurosci Lett, 2002. 327(2): p. 146–8. [DOI] [PubMed] [Google Scholar]
- 55.Inacio AR, et al. , Endogenous IFN-beta signaling exerts anti-inflammatory actions in experimentally induced focal cerebral ischemia. J Neuroinflammation, 2015. 12: p. 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu X, et al. , Inhibition of thioredoxin-1 with siRNA exacerbates apoptosis by activating the ASK1-JNK/p38 pathway in brain of a stroke model rats. Brain Res, 2015. 1599: p. 20–31. [DOI] [PubMed] [Google Scholar]
- 57.Wallace BK, Jelks KA, and O’Donnell ME, Ischemia-induced stimulation of cerebral microvascular endothelial cell Na-K-Cl cotransport involves p38 and JNK MAP kinases. Am J Physiol Cell Physiol, 2012. 302(3): p. C505–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harari OA and Liao JK, NF-kappaB and innate immunity in ischemic stroke. Ann N Y Acad Sci, 2010. 1207: p. 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sommer CJ, Ischemic stroke: experimental models and reality. Acta Neuropathol, 2017. 133(2): p. 245–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith HK, et al. , Critical differences between two classical surgical approaches for middle cerebral artery occlusion-induced stroke in mice. J Neurosci Methods, 2015. 249: p. 99–105. [DOI] [PubMed] [Google Scholar]
- 61.Knowland D, et al. , Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron, 2014. 82(3): p. 603–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang L, et al. , Human neural stem cells rapidly ameliorate symptomatic inflammation in early-stage ischemic-reperfusion cerebral injury. Stem Cell Res Ther, 2014. 5(6): p. 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qian C, et al. , Precise Characterization of the Penumbra Revealed by MRI: A Modified Photothrombotic Stroke Model Study. PLoS One, 2016. 11(4): p. e0153756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith SA, Travers RJ, and Morrissey JH, How it all starts: Initiation of the clotting cascade. Crit Rev Biochem Mol Biol, 2015. 50(4): p. 326–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Labat-gest V and Tomasi S, Photothrombotic ischemia: a minimally invasive and reproducible photochemical cortical lesion model for mouse stroke studies. J Vis Exp, 2013(76). [DOI] [PMC free article] [PubMed]
- 66.Kleinschnitz C, et al. , Blocking of platelets or intrinsic coagulation pathway-driven thrombosis does not prevent cerebral infarctions induced by photothrombosis. Stroke, 2008. 39(4): p. 1262–8. [DOI] [PubMed] [Google Scholar]
- 67.Frederix K, et al. , Platelet adhesion receptors do not modulate infarct volume after a photochemically induced stroke in mice. Brain Res, 2007. 1185: p. 239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yan H, et al. , Endothelin-1-induced focal cerebral ischemia in the growth hormone/IGF-1 deficient Lewis Dwarf rat. J Gerontol A Biol Sci Med Sci, 2014. 69(11): p. 1353–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wolinski P and Glabinski A, Chemokines and neurodegeneration in the early stage of experimental ischemic stroke. Mediators Inflamm, 2013. 2013: p. 727189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Durukan A and Tatlisumak T, Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav, 2007. 87(1): p. 179–97. [DOI] [PubMed] [Google Scholar]
- 71.Yang L, Shah KK, and Abbruscato TJ, An in vitro model of ischemic stroke. Methods Mol Biol, 2012. 814: p. 451–66. [DOI] [PubMed] [Google Scholar]
- 72.Baik SH, et al. , Pin1 promotes neuronal death in stroke by stabilizing Notch intracellular domain. Ann Neurol, 2015. 77(3): p. 504–16. [DOI] [PubMed] [Google Scholar]
- 73.Page S, Munsell A, and Al-Ahmad AJ, Cerebral hypoxia/ischemia selectively disrupts tight junctions complexes in stem cell-derived human brain microvascular endothelial cells. Fluids Barriers CNS, 2016. 13(1): p. 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gravanis I and Tsirka SE, Tissue-type plasminogen activator as a therapeutic target in stroke. Expert Opin Ther Targets, 2008. 12(2): p. 159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.ClinicalTrials.gov. 2018; Available from: https://www.clinicaltrials.gov/.
- 76.Tan Z, et al. , Bryostatin extends tPA time window to 6 h following middle cerebral artery occlusion in aged female rats. Eur J Pharmacol, 2015. 764: p. 404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Esmaeeli-Nadimi A, Kennedy D, and Allahtavakoli M, Opening the window: Ischemic postconditioning reduces the hyperemic response of delayed tissue plasminogen activator and extends its therapeutic time window in an embolic stroke model. Eur J Pharmacol, 2015. 764: p. 55–62. [DOI] [PubMed] [Google Scholar]
- 78.Culp WC, et al. , Dodecafluoropentane Emulsion Extends Window for tPA Therapy in a Rabbit Stroke Model. Mol Neurobiol, 2015. 52(2): p. 979–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ouyang YB, et al. , microRNAs: innovative targets for cerebral ischemia and stroke. Curr Drug Targets, 2013. 14(1): p. 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rink C and Khanna S, MicroRNA in ischemic stroke etiology and pathology. Physiol Genomics, 2011. 43(10): p. 521–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ouyang YB and Giffard RG, MicroRNAs affect BCL-2 family proteins in the setting of cerebral ischemia. Neurochem Int, 2014. 77: p. 2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu LJ, et al. , Post-stroke treatment with miR-181 antagomir reduces injury and improves long-term behavioral recovery in mice after focal cerebral ischemia. Exp Neurol, 2015. 264: p. 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Trounson A and McDonald C, Stem Cell Therapies in Clinical Trials: Progress and Challenges. Cell Stem Cell, 2015. 17(1): p. 11–22. [DOI] [PubMed] [Google Scholar]
- 84.Pluchino S and Cossetti C, How stem cells speak with host immune cells in inflammatory brain diseases. Glia, 2013. 61(9): p. 1379–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lopez-Santalla M, et al. , Human Adipose-Derived Mesenchymal Stem Cells Modulate Experimental Autoimmune Arthritis by Modifying Early Adaptive T Cell Responses. Stem Cells, 2015. 33(12): p. 3493–503. [DOI] [PubMed] [Google Scholar]
- 86.Martinez-Montiel Mdel P, Gomez-Gomez GJ, and Flores AI, Therapy with stem cells in inflammatory bowel disease. World J Gastroenterol, 2014. 20(5): p. 1211–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lu J, Pompili VJ, and Das H, Hematopoietic stem cells: ex-vivo expansion and therapeutic potential for myocardial ischemia. Stem Cells Cloning, 2010. 3: p. 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lu J, et al. , A novel technology for hematopoietic stem cell expansion using combination of nanofiber and growth factors. Recent Pat Nanotechnol, 2010. 4(2): p. 125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Das H, et al. , Ex vivo nanofiber expansion and genetic modification of human cord blood-derived progenitor/stem cells enhances vasculogenesis. Cell Transplant, 2009. 18(3): p. 305–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Joseph M, et al. , Retention of stemness and vasculogenic potential of human umbilical cord blood stem cells after repeated expansions on PES-nanofiber matrices. Biomaterials, 2014. 35(30): p. 8566–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lu J, et al. , Hematopoietic stem cells improve dopaminergic neuron in the MPTP-mice. Front Biosci (Landmark Ed), 2013. 18: p. 970–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Varga G and Gerber G, Mesenchymal stem cells of dental origin as promising tools for neuroregeneration. Stem Cell Res Ther, 2014. 5(2): p. 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kiraly M, et al. , Integration of neuronally predifferentiated human dental pulp stem cells into rat brain in vivo. Neurochem Int, 2011. 59(3): p. 371–81. [DOI] [PubMed] [Google Scholar]
- 94.Xiao L, et al. , Human Dental Pulp Cells Differentiate toward Neuronal Cells and Promote Neuroregeneration in Adult Organotypic Hippocampal Slices In Vitro. Int J Mol Sci, 2017. [DOI] [PMC free article] [PubMed]
- 95.Nagpal A, et al. , TOOTH (The Open study Of dental pulp stem cell Therapy in Humans): Study protocol for evaluating safety and feasibility of autologous human adult dental pulp stem cell therapy in patients with chronic disability after stroke. Int J Stroke, 2016. 11(5): p. 575–85. [DOI] [PubMed] [Google Scholar]
- 96.Leong WK, et al. , Human adult dental pulp stem cells enhance poststroke functional recovery through non-neural replacement mechanisms. Stem Cells Transl Med, 2012. 1(3): p. 177–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang X, et al. , Intravenous administration of DPSCs and BDNF improves neurological performance in rats with focal cerebral ischemia. Int J Mol Med, 2018. 41(6): p. 3185–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]