

SUMMARY
While transcriptional control of innate immune gene expression is well characterized, almost nothing is known about how pre-mRNA splicing decisions influence, or are influenced by, macrophage activation. Here, we demonstrate that the splicing factor hnRNP M is a critical repressor of innate immune gene expression and that its function is regulated by pathogen sensing cascades. Loss of hnRNP M led to hyperinduction of a unique regulon of inflammatory and antimicrobial genes following diverse innate immune stimuli. While mutating specific serines on hnRNP M had little effect on its ability to control pre-mRNA splicing or transcript levels of housekeeping genes in resting macrophages, it greatly impacted the protein’s ability to dampen induction of specific innate immune transcripts following pathogen sensing. These data reveal a previously unappreciated role for pattern recognition receptor signaling in controlling splicing factor phosphorylation and establish pre-mRNA splicing as a critical regulatory node in defining innate immune outcomes.
In Brief
West et al. report that hnRNP M represses expression of a cohort of innate immune transcripts in infected macrophages. IL6 splicing repression is relieved when hnRNP M is phosphorylated at specific residues, demonstrating that post-translational modification of splicing factors downstream of pathogen sensing can control maturation of innate immune mRNAs.
Graphical Abstract
INTRODUCTION
When innate immune cells like macrophages sense pathogens, they undergo a massive reprogramming of gene expression. Although innate immune gene expression is mostly studied in the context of transcriptional activation, multiple lines of evidence support a crucial role for pre-mRNA splicing regulation in shaping the macrophage transcriptome. For example, when primary mouse macrophages are treated with a Toll-like receptor 4 (TLR4) agonist, individual transcripts show significant variation in the time it takes for them to be fully spliced, with some pre-mRNAs remaining unprocessed for hours after transcriptional activation (Bhatt et al., 2012; Pandya-Jones et al., 2013). Like-wise, computational analyses of human primary macrophages reveal a robust increase in mRNA isoform diversity and a global preference for exon inclusion following lipopolysaccharide (LPS) treatment or Salmonella enterica serovar Typhimurium infection (Pai et al., 2016). The production of functionally diverse protein isoforms via alternative splicing is also known to influence innate immune responses. Several important innate immune molecules that function downstream of pattern recognition receptors, such as the TLR adaptor protein MyD88 (Janssens et al., 2003), the interleukin-1 receptor associated kinase 1, IRAK1 (Rao et al., 2005), and even some of the TLRs themselves (TLR3, TLR4 co-receptor MD2) (Gray et al., 2010; Seo et al., 2015), are regulated through expression of truncated isoforms that auto-inhibit full-length protein function and dampen inflammatory responses. In the case of MyD88, splicing factors like SF3a1 have been directly implicated in generating the MyD88 short isoform (MyD88-S), which inhibits expression of pro-inflammatory cytokines like interleukin-6 (IL-6) following LPS treatment (De Arras and Alper, 2013; De Arras et al., 2013).
To date, only a handful of RNA-binding proteins (RBPs) have been studied in the context of the innate immune response. For example, TLR4 signaling via LPS treatment promotes the shuttling of hnRNP U (heterogeneous nuclear ribonucleoprotein particle U) from the nucleus to the cytosol, resulting in differential expression of several innate immune cytokines (TNF-α, IL-6, IL-1β) via hnRNP U-dependent stabilization of cytosolic mRNAs (Zhao et al., 2012). Tristetraprolin (TTP), human antigen R (HuR), T cell intracellular antigen 1-related protein (TIAR), and hnRNP K have also been implicated in controlling gene expression in LPS-activated macrophages, with TTP and HuR regulating mRNA decay and TIAR and hnRNP K causing translational repression (Chen et al., 2013; Liepelt et al., 2014; Ostareck and Ostareck-Lederer, 2019). Phosphorylation is generally thought to control subcellular localization and protein-protein interactions between these RBPs (Allemand et al., 2005; Cobianchi et al., 1993; Huang et al., 2004; Ostareck-Lederer et al., 2002; Shin et al., 2004; Stamm, 2008), but the kinases/phosphatases responsible for modifying them and the conditions under which these modifications are controlled remain poorly understood.
Two recent publications measured macrophage protein phosphorylation following infection with the intracellular pathogens Mycobacterium tuberculosis (Penn et al., 2018) and Cryptococcus neoformans (Pandey et al., 2017). Intriguingly, a substantial number of these differentially phosphorylated peptides were derived from splicing factors. In fact, “spliceosome” was the top over-represented phosphorylated pathway in C. neoformans-infected cells, suggesting that post-translational modification (PTM) of splicing factors is critical for controlling innate immune responses to pathogens. One of the proteins that was significante differentially phosphorylated in each of these datasets was hnRNP M. hnRNP M is a splicing factor and RBP that has been repeatedly implicated in cancer metastasis (Chen et al., 2014; Passacantilli et al., 2017; Thomas et al., 2011; Xu et al., 2014) and muscle differentiation (Chen et al., 2017). Its role in regulating innate immune gene expression in macrophages is unknown, although interestingly, it has also been found to influence dengue virus replication (Viktorovskaya et al., 2016), suggesting a role in antiviral responses.
Here, we demonstrate that abrogating hnRNP M expression in a macrophage cell line leads to hyperinduction of over 100 transcripts following distinct innate immune stimuli, including infection with the gram-negative bacteria Salmonella enterica serovar Typhimurium, treatment with TLR2 and TLR4 agonists, and transfection of cytosolic dsDNA. While our data reveal that hnRNP M co-transcriptionally represses gene expression by influencing both constitutive and alternative splicing decisions, regulation of hnRNP M’s function via phosphorylation at S574 specifically controls the protein’s ability to inhibit intron removal of innate immune-activated transcripts. Consistent with its role in downregulating macrophage activation, macrophages lacking hnRNP M are better able to control viral replication, emphasizing the importance of pre-mRNA splicing regulation in modulating the innate immune response to infection.
RESULTS
RNA Sequencing (RNA-Seq) Analysis Reveals Immune Response Genes Are Regulated by hnRNP M during Salmonella Infection
To investigate a role for hnRNP M in regulating the innate immune response, we first tested how loss of hnRNP M globally influenced macrophage gene expression. Stably selected, constitutive hnRNP M knockdown (hnRNP M KD) cell lines were generated by transducing RAW 264.7 mouse macrophages with lentiviral short hairpin RNA (shRNA) constructs designed to target hnRNP M or a control scramble (SCR) shRNA. Western blot and qRT-PCR analysis confirmed ~80% and 60% knockdown of hnRNP M using two different shRNA constructs (KD1 and KD2, respectively) (Figure 1A). Because several attempts to knock out hnRNP M in RAW 264.7 macrophages by CRISPR/Cas9 guide RNAs (gRNAs) resulted exclusively in clones with in-frame insertions or deletions (data not shown), we concluded that hnRNP M is essential in macrophages and continued our experiments using the viable knockdown cell lines.
Figure 1. hnRNP M Regulates Expression of Innate Immune Genes during Salmonella Typhimurium Infection.

(A) Western blot analysis and qRT-PCR of hnRNP M in RAW 264.7 macrophages with β-actin as a loading control. Valúes are mean (SD) representative of 3 biological replicates.
(B) Volcano plot (t test) showing gene expression analysis of hnRNP M KD RNA-seq data from uninfected cells. x axis shows fold change of gene expression, and y axis shows statistical significance. Downregulated genes are plotted on the left, and upregulated genes are on the right.
(C) Gene expression analysis of hnRNP M KD cells compared to SCR control for uninfected cells. Each column represents a biological replicate. Red, genes downregulated in hnRNP M KD; blue, genes upregulated in hnRNP M KD.
(D) Gene expression analysis of hnRNP M KD cells compared to SCR control for Salmonella-infected cells. Each column represents a biological replicate.
(E) Ingenuity pathway analysis of gene expression changes in uninfected and Salmonella-infected cells.
(F) Manually annotated hnRNP M-dependent innate immune genes. Each column represents a biological replicate.
(G) qRT-PCR of Rnf26, Rnf128, and Slc6a4 in uninfected hnRNP M KD cells.
(H) qRT-PCR of mature IL6, Mx1, and Gbp5 transcripts in Salmonella-infected hnRNP M KD cells at 2 and 4 h post-infection.
(I) qRT-PCR of IL1b and Tnfa transcripts in Salmonella-infected hnRNP M KD cells at 4 h post-infection.
(G)-(I) represent 3 biological replicates ± SEM, n = 3. For all experiments in this study, statistical significance was determined using two-tailed Students’ t test. *p < 0.05, **p < 0.01, ***p < 0.001, n.s., not significant.
We next performed RNA-seq analysis on total poly(A)+ selected RNA collected from uninfected and Salmonella Typhimurium infected cells (MOI = 10) at the key innate immune time point of 4 h post-infection, at which time transcriptional activation downstream of both MyD88 and TRIF adapters would be expected (Yamamoto et al., 2003). Using CLC Genomics Workbench, we identified a number of genes that were differentially expressed in uninfected hnRNP M KD cells compared to SCR control cells, with 391 genes upregulated and 174 downregulated (Figure 1B). Looking specifically at transcripts with a fold change of > ±1.5 (p < 0.05), we observed similar numbers of impacted genes in uninfected hnRNP M KD and SCR macrophages (Figure 1C) and those infected with Salmonella (Figure 1D). The ratio of upregulated (blue) and downregulated (red) transcripts was also quite similar between the two conditions and consistent with previous reports of hnRNP M repressing pre-mRNA splicing (Hovhannisyan and Carstens, 2007; Marko et al., 2010). Interestingly, we observed only 25% overlap between genes that were differentially expressed in uninfected and Salmonella-in-fected macrophages, suggesting that hnRNP M has distinct modes of operation depending on the activation state of a macrophage (Figure S1A). Unbiased canonical pathways analysis revealed strong enrichment for differentially expressed genes in innate immune signaling pathways in Salmonella-infected hnRNP M KD cells (Figure 1E), and manual analysis of these lists revealed a number of important chemokines (e.g., Cxcl16, Ccl17, Ccl2, Ccl7), antiviral molecules (e.g., Isg15, Mx1, Rsad2), and pro-inflammatory cytokines (e.g., IL6, Mip1a [Ccl3], IL18) whose expression were dramatically affected by loss of hnRNP M (Figure 1F). Additional pathways enriched for hnRNP M-dependent genes can be found in Figures S1B and S1C, and a list of all impacted genes (±1.5-fold change) can be found in Table S1.
To validate the RNA-seq gene expression changes, we used qRT-PCR to measure transcript levels of genes from both lists (uninfected SCR versus hnRNP M KD and Salmonella-infected SCR versus hnRNP M KD). We confirmed overexpression of several genes in uninfected hnRNP M KD cells (Rnf26, Rnf128, Slc6a4; Figure 1G), as well as hyperinduction of transcripts in hnRNP M KD cells at 2 and 4 h post-Salmonella infection (IL6, Mx1, Gbp5, Adora2a, and Marcks) (Figures 1H and S1D). Importantly, induction of other pro-inflammatory mediators such as IL1b and Tnfa did not rely on hnRNP M (Figure 1I), suggesting that hnRNP M’s ability to regulate gene expression is conferred by specificity at the transcript level, rather than being common to a transcriptional regulon (e.g., nuclear factor κB [NF-κB], IRF3, STAT1). Together, these results reveal a previously unappreciated role for hnRNP M in repressing specific innate immune transcripts in macrophages.
hnRNP M Regulates a Specific Subset of Innate Immune Genes upon Treatment with Diverse Innate Immune Stimuli
Salmonella encodes several pathogen-associated molecular patterns (PAMPs) that serve as potent activators of pattern recognition receptors. Salmonella can also activate pro-inflammatory gene expression via its virulence-associated type III secretion system (Sun et al., 2018). To begin to determine the nature of the signal through which hnRNP M-dependent gene expression changes occur, we first tested whether LPS, a potent agonist of TLR4 (Poltorak et al., 1998) and component of the Salmonella outer membrane, was sufficient to hyperinduce IL6 expression in hnRNP M KD macrophages (Figure 2A). Similar to Salmonella infection, we observed a 3- to 4-fold hyperinduction of IL6 in hnRNP M KD cells treated with 100 ng/mL LPS (from E. coli) for 2 and 4 h, confirming that hnRNP M acts downstream of TLR4 activation (Figure 2B). Importantly, hyperinduction of IL6 mRNA in both LPS-treated and Salmonella-infected hnRNP M KD macrophages increased IL-6 protein levels 3- to 6-fold (Figure 2C), indicating that hnRNP M repression of IL6 mRNA processing impacts protein outputs in a biologically meaningful way. We believe hnRNP M mainly functions to repress IL6 expression early in macrophage activation, as we did not observe statistically significant differences in IL6 mRNA levels between SCR and hnRNP M KD at later time points (6 h) post-LPS treatment (Figure S2B), and this trend generally held for several other hnRNP M-dependent transcripts (Figure S2B). We did not observe any significant changes in hnRNP M protein expression over the same time course of LPS treatment (Figure S2C) nor did we observe significant differences in IκBα degradation over a course of LPS treatment in hnRNP M KD versus SCR control cells (Figure S2D), demonstrating that signaling downstream of TLR4 activation is intact in the absence of hnRNP M.
Figure 2. hnRNP M-Dependent Regulation of Innate Immune Gene Expression Occurs Downstream of Sensing Multiple Innate Immune Stimuli.

(A) Model of TLR4 and TLR2 signaling.
(B) qRT-PCR of IL6 mRNA levels in SCR control and hnRNP M KD cells treated with LPS for 2 and 4 h.
(C) IL6 ELISA with supernatants collected 4 h post-Salmonella infection or LPS treatment.
(D) qRT-PCR of IL1b transcripts in LPS-treated hnRNP M KD cells (4 h).
(E) qRT-PCR of Mx1 and Gbp5 mRNA levels in SCR control and hnRNP M KD cells treated with LPS (4 h).
(F) qRT-PCR and western analysis demonstrating effective depletion of hnRNP M in siRNA-transfected BMDMs. β-actin was used as a loading control.
(G) qRT-PCR of mature IL6 in negative control and hnRNP M siRNA BMDMs treated with 100 ng/mL of LPS for 1 and 2 h.
(H) qRT-PCR of mature IL6 in negative control and hnRNP M siRNA BMDMs treated with 10 ng/mL of LPS for 2 and 4 h.
(I) As in (H) but Adora2a.
(J) qRT-PCR of Gbp5 and Mx1 in negative control and hnRNP M siRNA BMDMs treated with 10 ng/mL of LPS for 4 and 6 h.
(K) qRT-PCR of mature IL6 in SCR control and hnRNP M KD cells treated with Pam3CSK4 for 4 h.
(L) Model of cGAS-mediated cytosolic DNA sensing and IFNAR signaling.
(M) qRT-PCR of Mx1 and Ifnb mRNA levels in SCR control and hnRNP M KD cells at 4 h following ISD transfection.
(N) qRT-PCR of ISGs (Ifit and Irf7) in SCR control and hnRNP M KD cells at 4 h following ISD transfection
(O) qRT-PCR of Mx1 transcript in SCR control and hnRNP M KD cells treated with recombinant IFN-β for 4 h.
All experiments represent 3 biological replicates where values are means ± SEM, n = 3.
Consistent with our RNA-seq and qRT-PCR data from Salmonella-infected cells, Mx1, Gbp5, and Marcks were hyperinduced in hnRNP M KD cells after LPS treatment at 2 and 4 h (Figure 2E; Figure S2A), while IL1b (Figure 2D) and Tnfa (Figure S2A) showed no changes in expression after LPS treatment in hnRNP M KD versus SCR control cells, despite both transcripts being tremendously upregulated. Rather than being activated by NF-κB, transcription of Mx1 and Gbp5 occurs via STAT1 downstream of interferon (IFN)-β signaling, following IFN-β expression via the TRIF/IRF3 axis (Figure 2L). These results hinted at a mechanism for hnRNP M-dependent repression that is independent of transcription factor specificity and is instead dependent on individual transcripts.
In order to confirm that hnRNP M’s ability to regulate innate immune gene expression was not unique to RAW 264.7 macrophages, we used siRNAs to knockdown hnRNP M in primary mouse bone marrow-derived macrophages (BMDMs) for 72 h, alongside negative control (designed to not target anything) and positive control (designed to target GAPDH) siRNAs (Figure 2F; Figure S2E) and treated these macrophages with LPS. Because BMDMs are incredibly responsive to innate immune agonists, we used two different concentrations of LPS, 100 ng/mL (same as in the RAW 264.7 experiments) and 10 ng/mL. In both cases, hnRNP M siRNA KD in BMDMs recapitulated the phenotype that we observed in the RAW 264.7 KD macrophages, i.e., hyperinduction of IL6 (Figures 2G, 100 ng/mL, and 2H, 10 ng/mL), albeit with slightly different kinetics and dose responses than we observed in the RAW 264.7 cell line. Importantly, we also measured hyperinduction of Gpb5, Mx1, and Adora2a and no change in Tnfa or IL1b (Figures 2I, 2J, and S2F) in hnRNP M siRNA KD cells, consistent with our results in the macrophage cell line. Together, these results argue for hnRNP M playing a crucial, conserved role in innate immune gene expression in both primary murine macrophages and murine macrophage cell lines.
To more directly test the idea that hnRNP M’s target specificity is at the level of the transcript itself, we tested whether genes like Mx1 and IL6 were hyperinduced in hnRNP M KD RAW 264.7 macrophages treated with a panel of innate immune agonists. Treatment with 100 ng/mLof theTLR2/1 agonist Pam3CSK4 hyperinduced IL6 expression in hnRNP M KD cells compared to SCR controls (Figure 2K; Figure S2G), while Tnfa and IL1b mRNA levels remained similar (Figure S2G). Likewise, transfection of hnRNP M KD cells with 1 μg/mL IFN stimulatory DNA (ISD), a potent agonist of cytosolic DNA sensing and IRF3-mediated transcription downstream of the cGAS/STING/TBK1 axis (Stetson and Medzhitov, 2006) (Figure 2L), led to hyperinduction of Mx1 in hnRNP M KD cells (Figure 2M). Ifnb and other IFN-stimulated genes (ISGs) regulated by IRF3 (Ifit1 and Irf7) were expressed at similar levels (Figures 2M and 2N). Direct engagement of the IFN receptor (IFNAR) with recombinant IFN-β also resulted in Mx1 hyperinduction in hnRNP M KD cells (Figure 2O). Collectively, these results bolster a model whereby hnRNP M represses mRNA expression of a specific subset of innate immune genes, regardless of how those genes are induced.
hnRNP M Influences Gene Expression Outcomes at the Level of Pre-mRNA Splicing
Because previous studies of hnRNP M have shown that it can enhance or silence splicing of alternatively spliced exons (Cho et al., 2014; Hovhannisyan and Carstens, 2007; Llères et al., 2010; Park et al., 2011), we first asked whether loss of hnRNP M could specifically influence constitutive intron removal and/or alternative splicing in LPS-activated macrophages. We chose IL6 as a model transcript because (1) it has a simple intron-exon architecture with four relatively short introns (165, 1,271, 3,059, and 1,226 nucleotides, respectively); (2) it was robustly hyperinduced by loss of hnRNP M (Figures 1F and 1H); and (3) it is a crucial component of the macrophage inflammatory response. Using qRT-PCR, we first measured the relative abundance of each IL6 intron-exon junction (Figure 3A) in SCR control cells to assess how intron removal proceeded on IL6 pre-mRNAs in cells containing hnRNP M. Primers were designed to only amplify introns that are still part of pre-mRNAs and not released intron lariats. At 2 h post-LPS treatment, most of the IL6 transcripts we detected were partially processed, with intron 1 and to some extent intron 4 being preferentially removed and introns 2 and 3 being retained (Figure 3B). We then compared the relative abundance of IL6 introns in SCR control cells to those in hnRNP M KD macrophages and observed a dramatic and specific decrease in intron 3-containing IL6 pre-mRNAs in the absence of hnRNP M. This decrease in IL6 intron 3 starkly contrasted other IL6 intron-exon and exon-exon junctions, which were overall more abundant in the absence of hnRNP M (Figure 3C). The fact that we observe vastly different amounts of intron 2 and 4-containing IL6 pre-mRNAs compared to intron-3-containing IL6 pre-mRNAs in hnRNP M KD macrophages speaks against hnRNP M impacting IL6 expression transcriptionally and instead argues strongly for the protein playing a role in IL6 pre-mRNA processing. These data demonstrate that IL6 pre-mRNAs accumulate in the absence of hnRNP M and suggest that IL6 intron 3 plays a privileged role in dictating the maturation of IL6 mRNAs. Specifically, we propose that retention of intron 3 serves as a rate-limiting step in IL6 pre-mRNA processing so that in the absence of hnRNP M, when IL6 intron 3 is removed more efficiently, higher levels of mature IL6 mRNA are made (Figures 1F, 1H, and 2B). Consistent with a role for hnRNP M in controlling splicing specifically, we did not observe any significant differences in the stability of IL6 mRNAs in hnRNP M KD macrophages compared to SCR controls following a time course of Actinomycin D treatment (Figure S3A).
Figure 3. hnRNP M Influences Gene Expression Outcomes at the Level of Pre-mRNA Splicing.

(A) Diagram of IL6 pre-mRNA with introns (gray) and exons (blue).
(B) qRT-PCR of IL6 exon-exon and intron-exon junctions in SCR control macrophages at 2 h post-LPS treatment.
(C) qRT-PCR of IL6 exon-exon and intron-exon junctions in SCR versus hnRNP M KD1 and KD2 at 2 h post-LPS treatment.
(D) Categorization of alternative splicing events identified via MAJIQ in uninfected SCR versus hnRNP M KD1 samples and in Salmonella-infected SCR versus hnRNP M KD1 samples.
(E) Ingenuity Pathway Analysis of hnRNP M-dependent genes from MAJIQ analysis in uninfected and Salmonella-infected cells.
(F) VOILA output of Mx1 transcript model in SCR and hnRNP M KD1 cells infected with Salmonella.
(G) Violin plots depicting the delta PSI of hnRNP M-dependent local splicing variations in Mx1.
(H) As in (G) but for Commd8, alongside semiquantitative RT-PCR validation.
(I) As in (H) but for Nmt2.
(B) and (C) are representative of two independent experiments that showed the same result with values representing means (SD), n = 3.
We next wanted to explore whether loss of hnRNP M also influenced alternative splicing in uninfected and Salmonella-in-fected macrophages. To do so, we employed an algorithm for local splice variation (LSV) analysis called MAJIQ (Modeling Alternative Junction Inclusion Quantification) (Vaquero-Garcia et al., 2016). MAJIQ allows identification, quantification, and visualization of diverse LSVs, including alternative 5′ or 3′ splice site usage and exon skipping, across different experimental conditions. MAJIQ identified a total of 94 LSVs in uninfected SCR versus hnRNP M KD macrophages and 67 LSVs in Salmonella-infected SCR versus hnRNP M KD macrophages (probability [|delta PSI|, ≥20%], >95%) (Figure 3D). The vast majority of the LSVs identified in SCR versus hnRNP M KD cells were exon skipping events (Figure 3D). Subsequent visualization of these LSVs by Voila analysis revealed that loss of hnRNP M generally correlated with increased exon inclusion in both uninfected and Salmonella-infected macrophages. In other words, the presence of hnRNP M led to more exon skipping, which is consistent with a role for hnRNP M in splicing repression. We also conducted IPA pathway analysis of alternatively spliced transcripts to identify pathways enriched for hnRNP M-dependent changes. In contrast to our global gene expression IPA analysis, we observed no enrichment for genes in innate immune-related pathways in either uninfected or Salmonella-infected macrophages (Figure 3E). In fact, only 3 transcripts had both significant expression changes (via RNA-seq) and significant delta PSI changes (via MAJIQ), suggesting that hnRNP M’s role in influencing steady-state gene expression of innate immune transcripts is distinct from its role in controlling alternative splicing decisions (Figure S3B). Interestingly, the greatest number of splicing changes were induced by Salmonella infection itself (Figure S3C), consistent with previously published datasets (Kalam et al., 2017; Pai et al., 2016).
Mx1, an anti-viral GTPase, was one of the three transcripts significantly impacted by loss of hnRNP M at the levels of gene expression (Figures 1F and 1H) and alternative splicing (Figure 3F). Specifically, MAJIQ identified an exon inclusion event of Mx1 “exon 9” that was significantly more frequent in hnRNP M KD uninfected macrophages versus SCR control uninfected macrophages (delta PSI exon 8-exon 9 = 0.703 versus exon 8-exon 10 = 0.298) (Figure 3G). Inclusion of this exon 9 introduces a premature stop codon and exon-9-containing transcript isoforms of Mx1 are annotated as nonsense-mediated decay targets. Together with our RNA-seq analysis, these results suggest that the overall abundance of Mx1 protein may be regulated by hnRNP M at multiple post-transcriptional processing steps, i.e., bulk transcript abundance and proportion of functional protein-encoding transcripts. MAJIQ also reported increased exon inclusion events for Commd8, a putative transcriptional regulator, and Nmt2, an N-myristoyltransferase. We confirmed each of these LSVs by semi-quantitative RT-PCR (Figures 3H, 3I, and S3D). Collectively, these data illustrate that hnRNP M can repress splicing of both constitutive and alternative introns, leading to distinct transcript and protein expression outcomes in macrophages.
hnRNP M Is Enriched at the Level of Chromatin and at the IL6 Genomic Locus
To get a better understanding of how hnRNP M controls pre-mRNA splicing, we next asked where hnRNP M localizes in RAW 264.7 macrophages and whether its localization changed upon TLR4 activation. Other hnRNP family members have been found to translocate to the cytoplasm in response to several different types of stimuli including vesicular stomatitis virus (VSV) infection, osmotic shock, and inhibition of transcription (Allemand et al., 2005; Lichtenstein et al., 2001; Pettit Kneller et al., 2009), and hnRNP U has been shown to shuttle out of the nucleus following LPS treatment of macrophages (Zhao et al., 2012). Based on our data implicating hnRNP M in splicing, we predicted that it can function in the macrophage nucleus and indeed, several algorithms including nuclear localization signal (NLS) Mapper (Kosugi et al., 2009) and PredictProtein (Yachdav et al., 2014) predicted hnRNP M is a predominantly nuclear protein (NLS Mapper score 8.5/10; PredictProtein 98/100) (Figure 4A).
Figure 4. hnRNP M Is a Nuclear Protein that Associates with the IL6 Genomic Locus in an RNA-Dependent Fashion.

(A) Schematic diagram of hnRNP M, highlighting the nuclear localization signal (purple) and three RNA Recognition Motifs (green).
(B) Immunofluorescence images of uninfected RAW 264.7 macrophages immunostained with anti-hnRNP M (green).
(C) Immunofluorescence images of RAW 264.7 macrophages stimulated with LPS for the respective time points and immunostained with anti-hnRNP M (green). Scale bar, 10 μM.
(D) Western blot analysis of cellular fractions with anti-hnRNP M and loading controls of cytoplasm (tubulin), nucleoplasm (hnRNP L) and chromatin (H3) fractions of uninfected and LPS stimulated RAW 264.7 macrophages.
(E) ChIP-qPCR primers designed to tile the IL-6 locus.
(F) qPCR of ChIP at the IL6 genomic locus with anti-hnRNP M in resting RAW 264.7 macrophages.
(G) As in (F) but using an anti-histone H3 antibody.
(H) Western blot analysis of nuclear and chromatin fractions with anti-hnRNP M and Histone H3 (control) with untreated and RNase-treated nuclear fractions.
(I) As in (F) but with 30 min incubation at 37° with RNase A.
(J) As in (F) but with macrophages treated with 100 ng/mL LPS for 1 h.
(K) Venn diagrams representing hnRNP M eCLIP (ENCODE) gene overlap with our MAJIQ, uninfected RNA-seq, and Salmonella-infected RNA-seq results.
(F) and (G) values are means ± SEM representative of 2 biological replicates, n = 2. (I) and (J) values are means ± SEM representative of 3 biological replicates, n = 3.
To examine hnRNP M localization, we performed immunofluorescence microscopy in uninfected macrophages using an anti-hnRNP M antibody and observed significant nuclear enrichment (Figure 4B) with no major changes over a 2 h time course of LPS treatment (Figure 4C). This was true for endogenous hnRNP M and a 3xFLAG-hnRNP M allele stably expressed in macrophages (Figure S4A). As a control, we monitored the translocation of hnRNP U upon LPS treatment and observed nuclear to cytoplasmic translocation, consistent with previous reports (Figure S4B). Based on these results, we concluded that hnRNP M is a nuclear protein in macrophages and that LPS treatment does not trigger translocation to another cellular compartment.
We next sought to understand more precisely where in the nucleus hnRNP M was enriched since intron recognition and removal can occur at the level of chromatin, while nascent transcripts are still tethered to RNA polymerase II (Bhatt et al., 2012; Moehle et al., 2014; Nojima et al., 2016; Pandya-Jones and Black, 2009). To this end, we performed a cellular fractionation experiment in RAW 264.7 macrophages over a time course of LPS treatment and visualized hnRNP M localization via western blot (Figure 4D). We observed hnRNP M in both the nucleoplasm and the chromatin over the course of LPS treatment, while no hnRNP M was detectable in the cytoplasmic fraction. Macrophages stably expressing 3xFLAG-hnRNP M showed a similar hnRNP M distribution between the nucleoplasm and chromatin (Figure S4C). We did not observe significant redistribution of either endogenous or 3xFLAG-hnRNP M between the nucleoplasm and chromatin fractions upon LPS treatment (Figures 4D and S4C). Residual hnRNP M protein expressed in KD cell lines was similarly distributed between the chromatin and nucleoplasm (Figure S4D). Together, fractionation and immunofluorescence experiments confirmed that a population of hnRNP M associates with chromatin, and the protein does not grossly redistribute in the cell upon LPS treatment.
hnRNP M’s Association with the IL6 Locus Is RNA Dependent and Controlled by TLR4 Signaling
We next wanted to determine whether hnRNP M’s association with chromatin was specific for the genomic loci of genes whose regulation was impacted by hnRNP M (Figure 1F). We hypothesized that, if hnRNP M repression of IL6 intron 3 removal occurs at the nascent transcript level, then hnRNP M may associate with the IL6 genomic locus. To test this, we performed chromatin immunoprecipitation (ChlP)-qPCR. ChIP has been used extensively in yeast and to some extent in mammals as a spatiotemporal read out of splicing factor recruitment to nascent transcripts (Bieberstein et al., 2014; Moehle et al., 2012; Neves et al., 2017; Nissen et al., 2017; Patrick et al., 2015). Endogenous hnRNP M was immunoprecipitated from untreated macrophages, and association with the IL6 locus (DNA) was determined using a series of tiling primers spaced approximately 500 bp apart (Figure 4E). We observed no enrichment of hnRNP M in the promoter region of IL6, consistent with it playing a mainly post-transcriptional role in IL6 processing (Figure 4F, primer set 1). We did, however, observe significant enrichment of hnRNP M at several primer sets in the IL6 gene, most notably over the intron 2-intron 3 region (Figure 4F, primer sets 3–5). Previously published cross-linking immunoprecipitation (CLIP)-seq experiments identified a GUGGUGG consensus site for hnRNP M (Huelga et al., 2012); such a site exists in intron 2 of IL6, and several similar motifs are found in IL6 intron 3 (Figure S4E). Indeed, of all the transcripts in Figure 1D, >75% of them contain at least one consensus hnRNP M motif in an intron (Table S2). ChIP-qPCR of histone H3, which showed clear depletion of nucleosomes around the IL6 transcription start site (primer sets 1 and 2), was performed to control for genomic DNA accessibility and/or primer set efficiency (Figure 4G). Together, these results reveal that hnRNP M can associate with the genomic locus of transcripts like IL6 whose splicing it represses, suggesting that it functions co-transcriptionally. Importantly, treatment with RNase A shifted hnRNP M from the chromatin into the nucleoplasm (Figure 4H). Likewise, RNase A treatment (Bieberstein et al., 2014) abolished hnRNP M enrichment at the IL6 genomic locus via ChIP-qPCR as well, confirming that its association with chromatin and the IL6 gene depends on RNA (Figure 4I).
If hnRNP M acts as a repressor of IL6 splicing by binding to nascent transcripts at the IL6 locus, we hypothesized that this repression might be relieved upon TLR4 activation, thus allowing a cell to robustly induce IL-6 expression following pathogen sensing. To test this, we performed ChIP-qPCR of hnRNP M at the IL6 locus in RAW 264.7 macrophages treated with LPS for 1 h. Remarkably, we observed a complete loss of hnRNP M enrichment at all primer sets along the IL6 gene body, including those over intron 2 and 3, following LPS treatment (Figure 4J). This result strongly links hnRNP M’s ability to repress IL6 with its presence at the IL6 genomic locus and suggests that TLR4 signaling controls hnRNP M’s repressor activity.
Having demonstrated that hnRNP M’s ability to associate with the IL6 chromatin locus relies on RNA, we were curious to see whether hnRNP M directly binds to transcripts whose expression were hnRNP M dependent (Figures 1C and 1D). To begin to answer this question, we leveraged previously published datasets of hnRNP M-bound transcripts in two human cell lines HepG2 (human liver carcinoma cells) and K562 (human chronic myelogenous leukemia cells) (Van Nostrand et al., 2016). Remarkably, we observed almost 60% overlap between our MAJIQ genes and the enhanced CLIP (eCLIP) datasets (Figure 4K; Table S3), suggesting that hnRNP M’salternative splicing targets are highly conserved between mouse and human and that the transcripts identified in our MAJIQ analysis are direct hnRNP M targets. While the overlap between the eCLIP hits and differentially expressed transcripts from our RNA-seq analysis (RNA-seq hits, Figures 1C and 1D) was lower (22% for Salmonella-infected transcripts, 21% for uninfected transcripts), this result is not altogether surprising, as HepG2 cells and K562 cells would not be expected to express many of the same transcripts as a macrophage. However, together these data reinforce the idea that RBPs like hnRNP M can play specialized roles in different cellular contexts, while also regulating a core set of conserved target transcripts.
Phosphorylation of hnRNP M at S574 Downstream of TLR4 Activation Controls Its Ability to Repress Expression of Innate Immune Transcripts
A recently published phosphoproteomics dataset identified a number of splicing factors that were differentially phosphorylated during infection with the intracellular bacterium Mycobacterium tuberculosis (Penn et al., 2018). Because it is not a gram-negative bacterium, M. tuberculosis does not activate TLR4 via LPS, but it does express the lipoglycan lipomannan (LM) and other lipoproteins, which are agonists of TLR2. Having confirmed hnRNP M-dependent regulation of IL6 following treatment with a TLR2 agonist (Pam3CSK4) (Figure 2K), we reasoned that TLR2 activation upon M. tuberculosis infection may lead to the same changes in hnRNP M phosphorylation as would TLR4 activation during Salmonella infection. We thus leveraged the M. tuberculosis global phosphoproteomics dataset from Penn et al. (2018), identified 5 differentially phosphorylated serine residues on hnRNP M (S85, S431, S480, S574, and S636) (Figure 5A), and generated 3xFLAG-hnRNP M constructs with phosphomimic (S→D) or phosphodead (S→A) mutationsat each of the serines and made stable RAW 264.7 macrophages expressing each of these alleles in wild-type RAW 264.7 macrophages that still contain a wild-type hnRNP M allele. Importantly, we did not observe any significant differences in the expression level of these mutant alleles compared to the wild-type 3xFLAG-hnRNP M either in resting macrophages or over a course of LPS treatment (Figure S5A).
Figure 5. Phosphorylation of hnRNP M at S574 Downstream of TLR4 Activation Controls Its Ability to Repress Expression of Innate Immune Transcripts.

(A) Protein diagram of hnRNP M indicating location of phosphorylation sites identified by SILAC/mass spectrometry (Penn et al., 2018) with nuclear localization signal shown in purple and RNA-recognition motifs (RRM) shown in in green.
(B) qRT-PCR of mature IL6 in wild-type (WT) hnRNP M-FLAG and phosphomutants in macrophages infected with Salmonella for 4 h.
(C) As in (B) but for Mx1.
(D) qRT-PCR of Rnf128 and Slc6a4 in uninfected, WT 3xFL-hnRNP M, and phosphomutants.
(E) Semiquantitative PCR of Commd8 alternative splicing in cells expressing SCR or hnRNP M KD constructs alongside phosphomutant-expressing alleles.
(F) ChIP-qPCR of hnRNP M-S574A/D alleles at the IL6 genomic locus.
(G) ChIP-qPCR of wild-type 3xFL-hnRNP M in the presence of 100 ng/mL LPS and various MAPK inhibitors (SB203580, SP600125, and U0126).
(H) RT-qPCR of IL6 intron-exon junctions and the exon 4–5 mature junction in 3xFL-hnRNP M and 3xFL-hnRNP M 574A and 574D phosphomutants, in macrophages infected with Salmonella for 4 h.
(B)-(D) are representative of 3 biological replicates with values indicating means ± SEM, n = 3. (F)-(H) are representative of 2 biological replicates values indicating means ± SEM, n = 2. (I) is representative of 2 independent experiments that showed the same result with values representing means (SD), n = 3.
Although the phosphoproteomics dataset predicts infectiondependent gain of phosphorylation at some sites and loss of phosphorylation at others, we were curious as to whether we could detect bulk hnRNP M phosphorylation changes via western blot analysis. While we were unable to detect any higher-or lower-molecular-weight species using antibodies against the endogenous protein, we consistently measured accumulation of a higher-molecular-weight species of the wild-type 3xFLAG-hnRNP M allele over the course of LPS treatment, with an initial increase in the species seen as early as 15 min post-treatment (Figure S5B), consistent with a population of hnRNP M being post-translationally modified upon pathogen sensing.
To determine how these individual serine residues contribute to hnRNP M activity during macrophage activation, we infected each of the phosphomutant/mimic-expressing cell lines, as well as a control expressing a wild-type allele, with Salmonella and measured IL6 and Mx1 expression at 4 h. Remarkably, expression of hnRNP M harboring a single serine mutation (hnRNP M 574D) caused dramatic hyperinduction of both IL6 and Mx1 compared to cells expressing the wild-type 3xFLAG-hnRNP M allele (Figures 5B and 5C). Several other phosphomutant alleles (S85D, red bar, S431A, light-blue bar, S480A/D, green bars) also affected IL6 and Mx1 induction but to a lesser extent (Figures 5B and 5C). Mutating S587, which is a repeat of the S574-containing sequence (MGANS(ph)LER), did not affect the regulation of IL6 or Mx1, suggesting the location of these serines is critical and that phosphorylation-dependent regulation of hnRNP M is specific for select serine residues (Figure S5C). Curiously, expression of the S574D allele in the hnRNP M KD cell lines did not recapitulate this derepression phenotype, suggesting the 574D allele disrupts the activity of wild-type hnRNP M itself—perhaps via interfering with hnRNP M protein oligomerization and/or higher-order complexes that form at innate immune targets (Figure S5D).
Having implicated hnRNP M phosphorylation at S574 in controlling IL6 and Mx1 expression, we next wanted to see how phosphorylation affected transcripts whose expression in uninfected cells was higher in the absence of hnRNP M (Figure 1G). While we again observed elevated expression of these transcripts in the absence of hnRNP M (hnRNP M KD, gray bars), expression of the phosphomutant alleles (S431A/D and S574A/D) had no effect on Rnf128, Rnf26, or Slc6a4 transcript levels (Figure 5D). Expression of these genes was similarly unaffected by the other hnRNP M phosphomutants (Figure S5E). Alternative splicing of Commd8 was also unaffected by any of the phophosmutants in either uninfected or Salmonella-infected cells (Figure 5E). Together, these data provide strong evidence that hnRNP M’s ability to regulate the expression of constitutively expressed genes and/or influence alternative splicing decisions does not rely on phosphorylation at serine 574, whereas its role in regulating innate immune transcripts induced during infection is specifically controlled by PTMs downstream of pathogen sensing.
Like wild-type hnRNP M, each hnRNP M phosphomutant was enriched in the chromatin in untreated cells (Figure S5F). However, in ChIP experiments looking specifically at the IL6 locus, the S574D phosphomimic allele displayed virtually no enrichment compared to the S574A phosphodead allele, whose enrichment profile was similarto that of wild-type hnRNP M (Figure 5F and Figure 4F). Indeed, hnRNP M 574D ChIPs more closely resembled those from RNase- or LPS-treated samples (Figures 4I and 4J). These data point to phosphorylation of residue S574 in controlling hnRNP M’s ability to co-transcriptionally repress processing of chromatin-associated IL6 pre-mRNAs.
We next sought to better understand how hnRNP M is phosphorylated at these key residues. TLR4 activation triggers a number of signaling cascades, including p38, MEK1/2 (ERK), and JNK MAP kinases. Previous reports have implicated each of these pathways in regulating IL6 expression downstream of innate immune stimuli (Costa-Pereira, 2014), but it is not known whether these cascades control splicing factor phosphorylation. To test the role of each cascade in hnRNP M-dependent repression of IL6, we performed ChIP experiments in the presence of LPS and specific inhibitors of p38 (SB203580), JNK (SP600125), or MEK (U0126). We again observed LPS-dependent loss of hnRNP M enrichment at IL6 (primer sets 4–6), and treatment with JNK and MEK inhibitors had no effect on hnRNP M release. However, in the presence of the p38 inhibitor, hnRNP M remained associated with the IL6 genomic locus after LPS treatment (Figure 5G), demonstrating that p38 signaling pro-motes release of hnRNP M from the IL-6 genomic locus.
Last, to interrogate the mechanism driving IL6 hyperinduction in hnRNP M S574D-expressing cells, we asked whether IL6 intron removal was affected by expression of the phosphomutant alleles. Using the same qRT-PCR approach used in Figure 3B, we detected an increase in IL6 pre-mRNAs containing introns 2 and 3 in macrophages overexpressing a wild-type hnRNP M allele, consistent with hnRNP M slowing IL6 intron removal. Conversely, these same introns were removed more efficiently in the presence of hnRNP M S574D, while no difference was observed in S574A-expressing cells (Figure 5H). These data strongly support a model whereby phosphorylation of hnRNP M at S574 relieves its ability to act as a splicing repressor, allowing for rapid removal of IL6 introns and upregulation of IL6 mRNA, and demonstrate a previously unappreciated role for constitutive intron removal in mediating IL6 expression in macrophages.
Loss of hnRNP M Enhances Macrophages’ Ability to Control Viral Infection
Because loss of hnRNP M resulted in hyperinduction of a variety of cell-intrinsic antimicrobial molecules and ISGs, we hypothesized that hnRNP M KD cells would be better at controlling viral replication. We infected SCR and hnRNP M KD RAW 264.7 macrophages with VSV, an enveloped RNA virus that can replicate and elicit robust gene expression changes in RAW 264.7 macrophages (Kandasamy et al., 2016). Viral replication (levels of VSV-G) was measured over an 8 h time course by qRT-PCR in cells infected with a viral MOI of 1 and 0.1. At both MOIs, loss of hnRNP M correlated with dramatic restriction of VSV replication, particularly at the 8 h time point (Figure 6A). As expected, infection with VSV, a potent activator of cytosolic RNA sensing via RIG-I/MAVS (Kandasamy et al., 2016), led to robust induction of Ifnb levels in an hnRNP M-independent fashion at both MOIs, as we previously observed in hnRNP M KD cells transfected with cytosolic dsDNA (Figures 6B, S6A, and 2M, respectively). Consistent with hnRNP M-dependent regulation occurring downstream of diverse immune stimuli (Figure 2), VSV-infected hnRNP M KD cells at MOI = 1 and MOI = 0.1 hyperinduced both Mx1 and IL6 (Figures 6C, 6D, S6B, and S6C).
Figure 6. Knockdown of hnRNP M Enhances a Macrophage’s Ability to Control Viral Infection.

(A) Viral replication in hnRNP M KD and SCR control RAW 264.7 macrophages infected with VSV (MOI = 1.0, MOI = 0.1, or Mock) at 2, 4, and 8 h post-infection.
(B) qRT-PCR of Ifnb mRNA levels in SCR control and hnRNP M KD cells at 2 and 4 h post-infection, MOI = 1.
(C) qRT-PCR of Mx1 transcript in VSV-infected SCR control and hnRNP M KD cells at 2, 4, and 8 h post-infection, MOI = 1.
(D) qRT-PCR of IL6 transcript in SCR control and hnRNP M KD cells at 2, 4, and 8 h post-infection MOI = 1.
(E) Differential gene expression in hnRNP M KD cells compared to SCR control macrophages from earlier RNA-seq analysis (Figure 1) highlighting known viral response genes.
(F) qRT-PCR of Ifit, Irf7, Rsad2, and Gbp5 mRNA levels in SCR control and hnRNP M KD cells at 4 h post-infection, MOI = 1.
(A)-(E) are representative of 2 biological replicates with values indicating means ± SEM, n = 2. (F) is representative of 2 independent experiments that showed the same result with values representing means (SD), n = 3.
While Mx1 itself is a well-known anti-viral GTPase, cell lines derived from inbred mouse strains like RAW 264.7 have been shown to carry non-functional Mx1 alleles (Shin et al., 2015). Therefore, to begin to predict what other hnRNP M-regulated genes may be responsible for enhanced VSV restriction, we manually examined hnRNP M-regulated transcripts in our RNA-seq data from uninfected and Salmonella-infected (i.e., TLR4-activated) macrophages and identified a number of genes known to be important for controlling RNA viral replication (Figure 6E). qRT-PCR confirmed hyperinduction of several antiviral ISGs in hnRNP M KD macrophages at 4 h post-VSV infection including Rsad2 (Viperin), Ifitl, Irf7, and Gbp5 (Figure 6F). Interestingly, neither Ifit1 nor Irf7 was identified as an hnRNP M-dependent transcript during Salmonella infection, even though both can be expressed downstream of TLR4 through IRF3/IFNAR/STAT1 signaling. This difference may simply reflect kinetic differences in transcript induction following RNA sensing versus TLR4 activation or may indicate that hnRNP M regulates an even broader set of transcripts in macrophages following RNA virus infection. We propose that inhibition of VSV replication in hnRNP M KD macrophages ultimately results from a combination of pro-viral gene downregulation (red genes, Figure 6E) and anti-viral gene upregulation (blue genes, Figure 6E and Figure 6F). Collectively, these data are consistent with hnRNP M playing a critical role in slowing innate immune gene expression and suggest that the presence of hnRNP M can actually blunt macrophage antiviral defenses at early time points following infection with VSV.
DISCUSSION
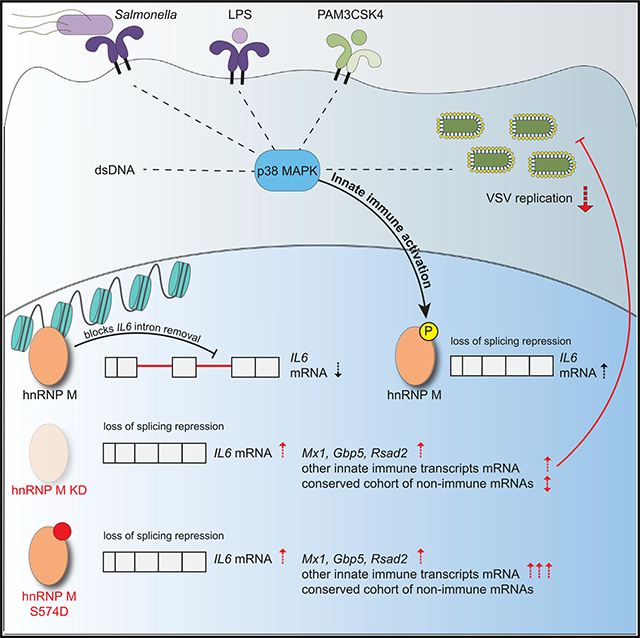
Despite the substantial impact pre-mRNA splicing has on gene expression outcomes, little is known about how components of the spliceosome are modified and regulated during cellular reprogramming events, such as macrophage pathogen sensing. Here, we demonstrate that the splicing factor hnRNP M is a critical repressor of a unique regulon of innate immune transcripts (see model in Figure 7). These transcripts were hyperinduced in hnRNP M KD macrophages downstream of a variety of innate immune stimuli (i.e., Salmonella infection, TLR4/TLR2 agonists, recombinant IFN-β, cytosolic dsDNA, RNA virus infection [VSV]) (Figure 2; Figure 6), and hyperinduction of this regulon correlated with enhanced capacity of hnRNP M KD macrophages to control VSV replication at early time points (Figure 6). We propose that in innate immune cells like macrophages repression of pre-mRNA splicing by hnRNP M serves as a safeguard, dampening the initial ramping up of innate immune gene expression and preventing spurious expression of potent pro-inflammatory molecules in situations where the cell has not fully engaged with a pathogen. The latter situation is supported by experiments in which low doses of LPS (10 and 50 ng/mL), were sufficient to hyperinduce IL6 in the absence of hnRNP M without inducing significant a change in the amount of IL6 mRNA expressed in SCR control cells (Figure S7A). These data support a role for hnRNP M in slowing IL6 processing in macrophages that are “sampling” PAMPsor that have just received an initial innate immune stimulus. The requirement for cells totightly control expression of potent inflammatory mediators like IL6 is evidenced by the fact that multiple transcriptional and post-transcriptional mechanisms exist to regulate IL6, including chromatin remodeling (Ramirez-Carrozzi et al., 2009), mRNA stability (Masuda et al., 2013), subcellular localization (Higa et al., 2018), and now, based on these data, pre-mRNA splicing. While hnRNP M’s role in regulating many alternative splicing decisions is conserved across diverse cell types in mice and humans (as evidenced by overlap in our MAJIQ hits and eCLIP datasets [Figure 4K]), its role in controlling innate immune transcripts is uniquely influenced by phosphorylation downstream of pathogen sensing. Based on these observations, we propose that hnRNP M and likely other splicing factors possess distinct capacities for interacting with RNAs and/or proteins depending on how they are post-translationally modified. In this way, innate immune sensing cascades may remodel splicing complexes, for example, by promoting release of hnRNP M from chromatin-associated RNA-proteins complexes via p38-MAPK cascades.
Figure 7. Proposed Model for hnRNP M-Dependent Repression for IL6 Expression in Resting, Early-, and Late-Activated Macrophages.

(Left) In resting macrophages, hnRNP M associates with chromatin, at the IL6 genomic locus, through interactions with RNA. These interactions may be direct with target transcripts expressed at lowlevels or spuriously or indirect via protein interactions with other RNA binding proteins or through interactions with other chromatin-associated RNAs, e.g., linc-RNAs. (Middle panel) When macrophages receive an innate immune stimulus, they transcriptionally activate genes like IL6. A population of “poised” hnRNP M can associate with chromatin-bound pre-mRNAs in these cells, slowing IL6 intron removal and preventing full maturation of IL6 nascent transcripts. (Right) As early macrophage activation proceeds, hnRNP M is phosphorylated at S574 in a p38-MAPK-dependent fashion. Phosphorylation of hnRNP M releases it from the IL6 genomic locus, relieves inhibition of IL6 splicing, and allows for full induction of IL6 gene expression. Figure generated using BioRender software.
While we do not fully understand the mechanisms driving hnRNP M’s target specificity, our RNA-seq data as well as other datasets (Bhatt et al., 2012) demonstrate the presence of cryptic exons in a number of hnRNP M-regulated transcripts (Figure S7). Previous work investigating the RNA-binding landscape of a panel of hnRNP proteins in a non-macrophage cell line (HEK293Ts, human embryonic kidney cells) revealed that hnRNP M has a strong preference for binding distal intronic regions (>2 kb from an exon-intron junction) (Huelga et al., 2012). Its binding profile was somewhat unique among the hnRNPs queried and was more reminiscent of another RBP, TDP-43. TDP-43 also binds UG-rich sites in distal introns and is crucial for repressing splicing of cryptic exons for a set of transcripts in the brain (Ling et al., 2016; 2015). We speculate that hnRNP M regulates splicing of macrophage transcripts through a similar mechanism where it binds to UG-rich regions downstream of cryptic exons and inhibits assembly of the spliceosome on these introns, thus slowing intron removal.
While hnRNP M’s ability to associate with the IL6 genomic locus via ChIP is RNA dependent, it is conceivable that hnRNP M controls innate immune gene expression through mechanisms that are independent of direct contacts between hnRNP M and regulated transcripts. Because a number of splicing factors have been shown to impact histone markers and chromatin remodeling, it is possible that hnRNP M promotes epigenetic changes at specific target transcripts (de Almeida et al., 2011; Kim et al., 2011; Martins et al., 2011; Saldi et al., 2016). hnRNP M may also interact with one or more long non-coding RNAs (lncRNAs), a number of which are regulated by TLR activation (Carpenter et al., 2013) and have been shown to control IL6 expression (Atianand et al., 2016; Carpenter et al., 2013). Experiments designed to identify hnRNP M-associated RNAs in uninfected and infected macrophages will provide important insights into how hnRNP M recognizes chromatin-associated target transcripts and help illuminate how pre-mRNA splicing decisions shape the innate immune transcriptome.
STAR⋆METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Robert Watson (robert.watson@tamu.edu). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
RAW 264.7 macrophages
RAW 264.7 macrophages (ATCC) (originally isolated from male BALB/c mice) were cultured at 37°C with a humidified atmosphere of 5% CO2 in DMEM (Thermo Fisher) with 10% FBS (Sigma Aldrich) 0.5% HEPES (Thermo Fisher). For RAW 264.7 macrophages stably expressing scramble knockdown and hnRNP M knockdown, cells were transfected with scramble non-targeting shRNA constructs and hnRNP M shRNA constructs targeted toward the 3′ UTR of hnRNP M. After 48 hours, media was supplemented with hygromycin (Invitrogen) to select for cells containing the shRNA plasmid. RAW 264.7 macrophages stably expressing GFP-FL and hnRNP M-FL were transfected for 48 hours and then selected through addition of puromycin (Invivogen).
BMDMs
Eight-week-old male C57BL/6J mice were used to generate bone marrow derived macrophages (BMDMs). All animals were housed, bred at Texas A&M Health Science Center under approved IACUC guidelines. BMDMs were differentiated from BM cells isolated by washing mouse femurs with 10 mL DMEM. Cells were then centrifuged for 5 min at 1000 rpm and resuspended in BMDM media (DMEM, 20% FBS (Millipore), 1mM sodium pyruvate (Lonza), 10% MCSF conditioned media (Waston lab)). BM cells were counted and plated at 3×106 in 15 cm non-TC treated dishes in 30 mL complete BMDM media. Cells were fed with an additional 15 mL of BMDM media on day 3 of culture. Cells were harvested on day 7 with 1 × PBS EDTA (Lonza).
Bacterial strains
Salmonella enterica serovar Typhimurium (SL1344) was obtained from Helene Andrews-Polymenis, TAMHSC. Infections with S. Typhimurium were conducted by plating RAW 264.7 macrophages on tissue-cultured treated 12-well dishes at 7.5 ×105 and incubated overnight. Overnight cultures of S. Typhimurium were diluted 1:20 in LB broth containing 0.3M NaCl and grown until they reached an OD600 of 0.9.
Viral strain
Recombinant Vesicular stomatitis virus (VSV; Indiana serotype) containing a GFP reporter cloned downstream of the VSV G-glyco-protein (VSV-G/GFP) was obtained collaboratively from Dr. John Rose at Yale School of Medicine with Dr. A. Phillip West.
METHOD DETAILS
RNA-SEQ
The RNA-Seq experiment was made up of 12 samples: biological triplicate of SCR uninfected, SCR Salmonella-infected, hnRNP M1 uninfected, and Salmonella-infected hnRNP M1. RNA-Seq and library prep was performed by Texas A&M AgriLife Genomics and Bioinformatics Service. Samples were sequenced on Illumina 4000 using 2 × 75-bp paired-end reads. Raw reads were filtered and trimmed and Fastq data was mapped to the Mus musculus Reference genome (RefSeq) using CLC Genomics Workbench 8.0.1. Differential expression analyses were performed using CLC Genomics Workbench. Relative transcript expression was calculated by counting Reads Per Kilobase of exon model per Million mapped reads (RPKM). The differentially expressed genes were selected as those with p value threshold < 0.05 and a fold change value > 1.5 to include in the heatmaps represented. Follow up RT-qPCR analysis was done with both knockdowns, while the best knockdown (M1) was selected forsequencing, asthe phenotype often tracked with the knockdown efficiency (M1 = ~80%, M2 = ~60%). Genes with p values < 0.05 were displayed in volcano plots and heatmaps using GraphPad Prism software (GraphPad, San Diego, CA).
S. Typhimurium Infection
Unless specified, cell lines at a confluency of 80% were infected with the S. Typhimurium strains at an MOI of 10 for 30 minutes in Hank’s buffered salt solution (HBSS), and subsequently cells were spun for 10 minutes at 1,000rpm, washed twice in HBSS containing 100μg/ml of gentamycin, and refilled with media plus gentamicin (10 μg/ml). Supernatants were collected at 2 hours and 4 hours and analyzed using IL6 ELISA (Biolegend). After removal of supernatant, cells were lysed in Trizol (Thermo Fisher) for RNA collection and analyzed using RT-qPCR.
LPS Treatment
RAW 264.7 macrophages were plated on 12-well tissue-culture treated plates at a density of 7.5×105 and allowed to acclimate over-night. Cells were then treated with E. Coli Lipopolysaccharide (Sigma-Aldrich) at 100ng/mL for the respective time points where supernatants and RNA were collected for analysis.
Immunofluorescence Microscopy
RAW 264.7 macrophages were plated on glass coverslips in 48-well plates. Cells were treated with LPS as described above. At the designated time points, cells were washed with PBS (Thermo Fisher) and then fixed in 4% paraformaldehyde for 10 minutes. Cells were washed with PBS 3x and then permeabilized with 0.2% Triton-X (Thermo Fisher). Coverslips were placed in primary antibody for 1 hour then washed 3x in PBS and placed in secondary antibody. These were washed twice in PBS and twice in deionized water, followed by mounting onto a glass slide using ProLong Diamond antifade mountant (Invitrogen). Images were acquired on a Nikon A1-Confocal Microscope.
Western Blots
Protein samples were run on Any kD Mini-PROTEAN TGX precast protein gels (BioRad) and transferred to 0.45 um nitrocellulose membranes (GE Healthcare). The membranes were incubated in the primary antibody of interest overnight and washed with TBS-Tween 20. Membranes were then incubated in secondary antibody for 1–2 hours and imaged using LI-COR Odyssey FC Imaging System.
siRNA Transfection
BMDMs were plated at 3×105 in 12-well TC-treated plates and incubated overnight. Viromer Blue (Lipocalyx) was used to transfect siRNAs according to the manufacturer’s instructions. The siRNAs directed against Gapdh and hnRNP M were purchased from Thermo Fisher with a negative control present. After 48 hr of siRNA transfection, LPS was added and RNA was collected at the respective time points.
Antibodies
The following primary antibodies were used: rabbit polyclonal hnRNP M (Abcam, #177957), rabbit polyclonal Histone 3 (Abcam, #1791), mouse monoclonal Beta-Actin (Abcam, #6276), mouse monoclonal hnRNP L (Abcam, #6106–100), rabbit polyclonal Beta-Tubulin (Abcam, #179513), mouse monoclonal hnRNP U (Santa-Cruz, sc-32315), DAPI nuclear staining (Thermo Fisher), and mouse monoclonal ANTI-FLAG M2 antibody (Sigma-Aldrich, F3165). Secondary antibodies used were asfollows: IR Dye CW 680 goat anti-rabbit, IR Dye CW800 goat anti-mouse (LI-COR), Alexfluor-488 anti-rabbit and Alexafluo-647 anti-mouse secondary antibodies for immunofluorescence (LI-COR).
Cellular fractionation
Macrophagecellularfractionation was done as described in Pandya-Jonesetal.,2013. Briefly, RAW264.7 macrophages were plated on in 10 cm tissue-culture treated plates at 1–3×107 per plate. Cells and buffers were kept on ice unless noted otherwise. Cells were rinsed twice in cold PBS-EDTA (Lonza) and scraped into 15 mL conical tubes. Cells were spun at 1,000 g for 5 minutes at 4C and resuspended in NP-40 lysis buffer (10 mM Tris-HCl [pH 7.5], 0.05% NP40 [Sigma], 150 mM NaCl, protease inhibitor tablet (Thermo Fisher)) and incubated for 5 min on ice. Lysate was added to 2.5 volumes of a sucrose cushion (Lysis buffer with 24% sucrose) and centrifuged forat 14,000 rpm for 10min at 4C. The supernatant was collected and saved for cytoplasmic protein sample. The nuclear pellet was resuspended in glycerol buffer (20 mM Tris-HCl [pH 7.9], 75 mM NaCl, 0.5 mM EDTA, 0.85 mM DTT, 50% glycerol, protease inhibitor tablet) and lysed with nuclear lysate buffer in equal volume and vortexed 2Xfor2 s(10 mM HEPES [pH 7.6], 1 mM DTT, 7.5 mM MgCl2,0.2 mM EDTA, 0.3 M NaCl, 1 M UREA, 1% NP-40, protease inhibitor tablet). Lysates were chilled on ice for 2 minutes and then spun at 10,000 rpm for 2 minutes at 4C. Supernatant was collected and used for nucleoplasmic protein samples. The remining chromatin pellet was gently rinsed in PBS-EDTA and treated with DNase in DNase buffer for 1 hr at 37C. After incubation, the supernatant was collected for chromatin protein samples. Sample buffer (BIO-RAD) and 2-Mercapoethanol (BIO-RAD) was added to every protein sample with 5 minutes boiling prior to running on gels for western blots. Approximately 10% of sample was loaded for western blots.
RNase Fractionation
For nuclear lysates treated with RNase, nuclear pellets were responded in glycerol buffer. Nuclear lysis buffer was added, and lysates were incubated on ice for 5 minutes. Samples were then divided into two samples with one receiving 1 ul of RNase A (Thermo Fisher) per 50 ul sample and another with no RNase A. Both were incubated at 37C for 30 mins. Lysates were then spun at 10,000rpm for 2 mins and the rest of the fractionation proceeded as described.
Gene Ontology (GO) Canonical Pathway Analysis
To determine the most affected pathwaysin control versus hnRNP M knockdown RAW 264.7 macrophages, canonical pathway analysis was conducted using Ingenuity Pathway Analysis software from QIAGEN Bioinformatics. Genes that were differentially expressed wth a p value < 0.05 from our RNA-SEQ analysis were used as input from uninfected and Salmonella Typhimurium infected cells. The top hits were represented in bar graphs by z-score.
RNA isolation and qPCR analysis
Fortranscript analysis, cells were harvested in Trizol and RNA was isolated using Direct-zol RNA Miniprep kits (Zymo Research) with 1 hrDNase treatment. cDNA was synthesized with iScriptcDNASynthesis Kit (Bio-Rad). CDNAwasdilutedto 1:20foreach sample. A pool of cDNA from each treated or infected sample was used to make a 1:10 standard curve with each standard sample diluted 1:5 to produce a linear curve. RT-qPCR was performed using Power-UpSYBR Green Master Mix (Thermo Fisher) using a Quant Studio Flex 6 (Applied Biosystems). Samples were run in triplicate wellsin a96-well plate. Averagesof the raw values were normalized to average values for the same sample with the control gene, beta-actin. To analyze fold induction, the average of the treated sample was divided by the untreated control sample, which was set at 1.
Chromatin Immunoprecipitation
Chromatin Immunoprecipitation (ChIP) was adapted from Abcam’s protocol. Briefly, two confluent 15 cm dishesof RAW 264.7 macrophages were crosslinked in formaldehyde to a final concentration of 0.75% and rotated for 10 minutes. Glycine was added to stop the cross linking by shaking for 5 minutes at a concentration of 125 mM. Cells were rinsed with PBStwice and then scraped into 5 mL PBS and centrifuged at 1,000 g for 5 min at 4C. Cellular pellets were resuspended in ChIP lysis buffer (750 mL per 1×107 cells) and incubated for 10 min on ice. Cellular lysates were sonicated for 40 minutes (30sec ON, 30sec OFF) on high in a Bioruptor UCD-200 (Diagenode). After sonication, cellular debris was pelleted by centrifugation for 10 min, 4°C, 8,000 × g. Input samples were taken at this step and stored at −80C until decrosslinking. For RNase treated samples, RNase A was added to cell lysates and incubated for 30 mins at 37C. Approximately 25 μg of DNA diluted to 1:10 with RIPA buffer was used forovernight immunoprecipitation. Each ChIP had one sample for the specific antibody and one sample for Protein G beads only which were pre-blocked for 1 hr with single stranded herring sperm DNA (75 ng/μL) and BSA (0.1 μg/μL). The respective primary antibody was added to all samples except the beads-only sample at a concentration of 5 ug and rotated at 4°C overnight. Beads were washed 3× in with a final wash in high salt (500mM NaCl). DNA waseluted with elution buffer and rotated for 15 min at 30C. Centrifuge for 1 min at 2,000 xg and transfer the supernatant into a fresh tube. Supernatant was incubated in NaCl, RNase A (10 mg/mL) and proteinase K (20 mg/mL) and incubated at 65°Cfor 1 h. The DNA was purified using phenol:chloroform extraction. DNA levels were measure by RT-qPCR. Primers were designed by tiling each respective gene every 500 base pairs that were inputted into NCBI primer design.
FLAG Chromatin Immunoprecipitation
In RAW 264.7 macrophages stably expressing FL-hnRNP M or FL-GFP, ChIP was conducted as described above with minor adjustments. Lysates were incubated overnight at 4C with ANTI-FLAG M2 antibody. After washing, DNA was eluted with FLAG peptide (Sigma-Aldrich F4799) by adding 20 ul of 5X FLAG peptide, vortexed at room temperature for 15 mins and supernatants were collected. This process was repeated a total of 3x followed by decrosslinking as described.
Alternative Splicing Analysis
Alternative splicing events were analyzed using MAJIQ and VOILA with the default parameters (Vaquero-Garcia et al., 2016). Briefly, uniquely mapped, junction-spanning reads were used by MAJIQ to construct splice graphs for transcriptsby using the RefSeq annotation supplemented with de-novo detected junctions. Here, de-novo refers to junctions that were not in the RefSeq transcriptome database but had sufficient evidence in the RNA-Seq data. The resulting gene splice graphs were analyzed for all identified local splice variations (LSVs). For every junction in each LSV, MAJIQ then quantified expected percent spliced in (PSI) value in control and hnRNP M knockdown samples and expected change in PSI (dPSI) between control and hnRNP M KD samples. Results from VOILA were then filtered for high confidence changing LSVs (whereby one or more junctions had at least a 95% probability of expected dPSI of at least an absolute value of 20 PSI units (noted as “20% dPSI”) between control and hnRNP M KD) and candidate changing LSVs (95% probability, 10% dPSI). For the high confidence results (dPSI > = 20%), the events were further categorized as single exon cassette, multi-exon cassette, alternative 5’ and/or 3’ splice site, or intron-retention.
VSV infection
7×105 RAW cells were seeded in 12-well plates 16h before infection. Cells were infected with VSV-GFP virus(Dalton and Rose, 2001) at multiplicityof infection (MOI) of 1 and 0.1 in serum-free DMEM (HyCloneSH30022.01). After 1h ofincubation with media containing virus, supernatant was removed, and fresh DMEM plus 10% FBS was added to each well. At indicated times post infection, cells were harvested with Trizol and prepared for RNA isolation.
QUANTITATION AND STATISTICAL ANALYSIS
Statistical analysis of data was performed using GraphPad Prism software. Two-tailed unpaired Student’s t tests were used for statistica! analyses, and unlessotherwise noted, all results are representative of at leastthree biological experiments (mean ± SEM (n = 3 per group)).
DATA AND CODE AVAILABILITY
This article contains all datasets generated during this study. All coding for Majiq and Voila was standard and is available for academic download. Raw read files for RNA-seq can be found on GEO (Accession #GSE137603).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal hnRNP M | Abcam | #177957 |
| Mouse monoclonal β-Actin | Abcam | Cat#6276; RRID:AB_2223210 |
| Rabbit polyclonal Histone 3 | Abcam | Cat#1791 |
| Mouse monoclonal hnRNP L | Abcam | Cat#6106–100; RRID:AB_305294 |
| Rabbit polyclonal β-Tubulin | Abcam | Cat#179513 |
| Mouse Monoclonal ANTI-FLAG M2 | Sigma-Aldrich | Cat#F3165; RRID:AB_259529 |
| Mouse monoclonal hnRNP U | Santa-Cruz | Cat#sc-32315 |
| Alexa Fluor-488 goat anti-rabbit | Invitrogen | Cat#A-11034 |
| IR Dye CW800 goat anti-mouse | LI-COR | Cat#925–32210 |
| Alexa Fluor-647 goat anti-mouse | Invitrogen | Cat#A21235 |
| IR Dye CW 680 goat anti-rabbit | LI-COR | Cat#926–68071 |
| Bacterial and Virus Strains | ||
| Salmonella enterica (SL1344) | Dr. Helene Andrews-Polymenis, TAMHSC | N/A |
| Recombinant Vesicular stomatitis virus (VSV; Indiana serotype) containing a GFP reporter cloned downstream of the VSV G-glycoprotein (VSV-G/GFP) | Dr. John Rose, Yale School of Medicine | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Flag Peptide | Sigma-Aldrich | Cat#F4799 |
| Critical Commercial Assays | ||
| ELISA MAX Mouse Standard Set IL-6 | BioLegend | Cat#431301 |
| Viromer Blue | Lipocalyx | Cat#VB-01LB-0 |
| Experimental Models: Cell Lines | ||
| RAW 264.7 | ATCC | N/A |
| 3xFLAG_hnRNPM RAW264.7 macrophages | This paper | N/A |
| hnRNPM shRNA SCR RAW 264.7 macrophages | This paper | N/A |
| hnRNPM shRNA M1 KD RAW 264.7 macrophages | This paper | N/A |
| hnRNPM shRNA M2 KD RAW 264.7 macrophages | This paper | N/A |
| 3xFLAG_hnRNPM S85A RAW264.7 macrophages | This paper | N/A |
| 3xFLAG_hnRNPM S85D RAW264.7 macrophages | This paper | N/A |
| 3xFLAG_hnRNPM S431A RAW264.7 macrophages | This paper | N/A |
| 3xFLAG_hnRNPM S431D RAW264.7 macrophages | This paper | N/A |
| 3xFLAG_hnRNPM S480A RAW264.7 macrophages | This paper | N/A |
| 3xFLAG_hnRNPM S480D RAW264.7 macrophages | This paper | N/A |
| 3xFLAG_hnRNPM S574A RAW264.7 macrophages | This paper | N/A |
| 3xFLAG_hnRNPM S574D RAW264.7 macrophages | This paper | N/A |
| 3xFLAG_hnRNPM S636A RAW264.7 macrophages | This paper | N/A |
| 3xFLAG_hnRNPM S636D RAW264.7 macrophages | This paper | N/A |
| Oligonucleotides | ||
| See Table S4 | N/A | |
| Recombinant DNA/RNA | ||
| Silencer Select GAPDH Positive Control siRNA | ThermoFisher | Cat#4390849 |
| Silencer Select Negative Control No. 1 siRNA | ThermoFisher | Cat#4390843 |
| heterogeneous nuclear ribonucleoprotein M siRNA | ThermoFisher | Cat#4390771 |
| Software and Algorithms | ||
| CLC Genomics Workbench 8.0.1 | QIAGEN Bioinformatics | https://www.qiagenbioinformatics.com/products/clc-genomics-workbench/ |
| Ingenuity Pathway Analysis | QIAGEN Bioinformatics | N/A |
| MAJIQ | Vaquero-Garcia et al., 2016 | https://majiq.biociphers.org/ |
| VOILA | Vaquero-Garcia et al., 2016 | https://majiq.biociphers.org/ |
| Other | ||
| RNA-seq rawand filtered datafrom hnRNP M/SCR resting and Salmonella infected macrophages | This paper | GEO #GSE137603 |
Highlights.
hnRNP M represses a cohort of innate immune transcripts in macrophages
hnRNP M associates with the IL6 genomic locus to co-transcriptionally repress splicing
Following infection, phosphorylation of hnRNP M at S574 can relieve this repression
Loss of hnRNP M enhances macrophages’ ability to control viral infection
ACKNOWLEDGMENTS
We would like to thank Drs. Jeffery Cox, Bennett Penn, and Jonathan Budzik for sharing M. tuberculosis phosphoproteomics data and members of the Watson and Patrick labs for critical reading of the manuscript and invaluable feedback. We would also like to thank the Texas A&M AgriLife Genomics & Bioinformatics Services for performing RNA sequencing experiments and providing computing resources. This work was funded by R01AI25512 (R.O.W.) and R21AI4004 (R.O.W. and K.L.P.). A.P.W. and S.T.-O. were supported by the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Programs under award number W81XWH-17-1-0052. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.09.078.
REFERENCES
- Allemand E, Guil S, Myers M, Moscat J, Cáceres JF, and Krainer AR (2005). Regulation of heterogenous nuclear ribonucleoprotein A1 transport by phosphorylation in cellsstressed by osmoticshock. Proc. Natl. Acad. Sci. USA 102, 3605–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atianand MK, Hu W, Satpathy AT, Shen Y, Ricci EP, Alvarez-Dominguez JR, Bhatta A, Schattgen SA, McGowan JD, Blin J, et al. (2016). A Long Noncoding RNA lincRNA-EPS Acts as a Transcriptional Brake to Restrain Inflammation. Cell 165, 1672–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DM, Pandya-Jones A, Tong A-J, Barozzi I, Lissner MM, Natoli G, Black DL, and Smale ST (2012). Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell 150, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieberstein NI, Straube K, and Neugebauer KM (2014). Chromatin immunoprecipitation approaches to determine co-transcriptional nature of splicing. Methods Mol. Biol 1126, 315–323. [DOI] [PubMed] [Google Scholar]
- Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, Byron M, Monks B, Henry-Bezy M, Lawrence JB, et al. (2013). A long non-coding RNA mediates both activation and repression of immune response genes. Science 341, 789–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-L, Jiang Y-W, Su Y-L, Lee S-C, Chang M-S, and Chang C-J (2013). Transcriptional regulation of tristetraprolin by NF-κB signaling in LPS-stimulated macrophages. Mol. Biol. Rep 40, 2867–2877. [DOI] [PubMed] [Google Scholar]
- Chen S, Zhang J, Duan L, Zhang Y, Li C, Liu D, Ouyang C, Lu F, and Liu X (2014). Identification of HnRNP M as a novel biomarker for colorectal carcinoma by quantitative proteomics. Am. J. Physiol. Gastrointest. Liver Physiol 306, G394–G403. [DOI] [PubMed] [Google Scholar]
- Chen W-Y, Lin C-L, Chuang J-H, Chiu F-Y, Sun Y-Y, Liang M-C, and Lin Y (2017). Heterogeneous nuclear ribonucleoprotein M associates with mTORC2 and regulates muscle differentiation. Sci. Rep 7, 41159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Moon H, Loh TJ, Oh HK, Cho S, Choy HE, Song WK, Chun J-S, Zheng X, and Shen H (2014). hnRNP M facilitates exon 7 inclusion of SMN2 pre-mRNA in spinal muscularatrophy bytargeting an enhanceron exon 7. Biochim. Biophys. Acta 1839, 306–315. [DOI] [PubMed] [Google Scholar]
- Cobianchi F, Calvio C, Stoppini M, Buvoli M, and Riva S (1993). Phosphorylation of human hnRNP protein A1 abrogates in vitro strand annealing activity. Nucleic Acids Res. 21, 949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Pereira AP (2014). Regulation of IL-6-type cytokine responses by MAPKs. Biochem. Soc. Trans 42, 59–62. [DOI] [PubMed] [Google Scholar]
- Dalton KP, and Rose JK (2001). Vesicular stomatitisvirus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology 279, 414–421. [DOI] [PubMed] [Google Scholar]
- de Almeida SF, Grosso AR, Koch F, Fenouil R, Carvalho S,Andrade J, Levezinho H, Gut M, Eick D, Gut I, et al. (2011). Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nat. Struct. Mol. Biol 18, 977–983. [DOI] [PubMed] [Google Scholar]
- De Arras L, and Alper S (2013). Limiting of the innate immune response by SF3A-dependent control of MyD88 alternative mRNA splicing. PLoS Genet. 9,e1003855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Arras L, Seng A, Lackford B, Keikhaee MR, Bowerman B, Freedman JH, Schwartz DA, and Alper S (2013). An evolutionarily conserved innate immunity protein interaction network. J. Biol. Chem 288,1967–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray P, Michelsen KS, Sirois CM, Lowe E, Shimada K, Crother TR, Chen S, Brikos C, Bulut Y, Latz E, et al. (2010). Identification of a novel human MD-2 splice variant that negatively regulates Lipopolysaccharide-induced TLR4 signaling. J. Immunol 184, 6359–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higa M, Oka M, Fujihara Y, Masuda K,Yoneda Y, and Kishimoto T (2018). Regulation of inflammatory responses by dynamic subcellular localization of RNA-binding protein Arid5a. Proc. Natl. Acad. Sci. USA 115, E1214–E1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovhannisyan RH, and Carstens RP (2007). Heterogeneous ribonucleo-protein m is a splicing regulatory protein that can enhance or silence splicing of alternatively spliced exons. J. Biol. Chem 282, 36265–36274. [DOI] [PubMed] [Google Scholar]
- Huang Y, Yario TA, and Steitz JA (2004). A molecularlink between SR protein dephosphorylation and mRNA export. Proc. Natl. Acad. Sci. USA 101, 9666–9670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelga SC, Vu AQ, Arnold JD, Liang TY, Liu PP, Yan BY, Donohue JP, Shiue L, Hoon S, Brenner S, et al. (2012). Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 1, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S, Burns K, Vercammen E, Tschopp J, and Beyaert R (2003). MyD88S, a splice variant of MyD88, differentially modulates NF-kappaB- and AP-1-dependent gene expression. FEBS Lett. 548, 103–107. [DOI] [PubMed] [Google Scholar]
- Kalam H, Fontana MF, and Kumar D (2017). Alternate splicing of transcripts shape macrophage response to Mycobacterium tuberculosis infection. PLoS Pathog. 13, e1006236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandasamy RK, Vladimer GI, Snijder B, Müller AC, Rebsamen M, Bi-genzahn JW, Moskovskich A, Sabler M, Stefanovic A, Scorzoni S, et al. (2016). A time-resolved molecular map of the macrophage response to VSV infection. NPJ Syst. Biol. Appl 2, 16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim H, Fong N, Erickson B, and Bentley DL (2011). Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc. Natl. Acad. Sci. USA 108, 13564–13569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi S, Hasebe M, Tomita M, and Yanagawa H (2009). Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction ofcomposite motifs. Proc. Natl. Acad. Sci. USA 106,10171–10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein M, Guo W, and Tartakoff AM (2001). Control ofnuclearexport of hnRNP A1. Traffic 2, 261–267. [DOI] [PubMed] [Google Scholar]
- Liepelt A, Mossanen JC, Denecke B, Heymann F, De Santis R, Tacke F, Marx G, Ostareck DH, and Ostareck-Lederer A (2014).Translation control of TAK1 mRNA by hnRNP K modulates LPS-induced macrophage activation. RNA 20, 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling JP, Pletnikova O, Troncoso JC, and Wong PC (2015). TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling JP, Chhabra R, Merran JD, Schaughency PM, Wheelan SJ, Corden JL, and Wong PC (2016). PTBP1 and PTBP2 Repress Nonconserved Cryptic Exons. Cell Rep. 17, 104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llères D, Denegri M, Biggiogera M, Ajuh P, and Lamond AI (2010). Direct interaction between hnRNP-M and CDC5L/PLRG1 proteins affects alternative splice site choice. EMBO Rep. 11, 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marko M, Leichter M, Patrinou-Georgoula M, and Guialis A (2010). hnRNP M interacts with PSF and p54(nrb) and co-localizeswithin defined nuclear structures. Exp. Cell Res. 316, 390–400. [DOI] [PubMed] [Google Scholar]
- Martins SB, Rino J, Carvalho T, Carvalho C, Yoshida M, Klose JM, de Almeida SF, and Carmo-Fonseca M (2011). Spliceosome assembly is coupled to RNA polymerase II dynamics at the 3′ end of human genes. Nat. Struct. Mol. Biol 18,1115–1123. [DOI] [PubMed] [Google Scholar]
- Masuda K, Ripley B, Nishimura R, Mino T, Takeuchi O, Shioi G, Kiyonari H, and Kishimoto T (2013). Arid5a controls IL-6 mRNA stability, which contributes to elevation of IL-6 level in vivo. Proc. Natl. Acad. Sci. USA 110,9409–9414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehle EA, Ryan CJ, Krogan NJ, Kress TL, and Guthrie C (2012). The yeast SR-like protein Npl3 links chromatin modiffcation to mRNA processing. PLoS Genet. 8,e1003101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehle EA, Braberg H, Krogan NJ, and Guthrie C (2014). Adventures in time and space: splicing efficiency and RNA polymerase II elongation rate. RNABiol. 11,313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves LT, Douglass S, Spreafico R, Venkataramanan S, Kress TL, and Johnson TL (2017). The histone variant H2A.Z promotes efficient cotranscriptional splicing in S. cerevisiae. Genes Dev. 31, 702–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen KE, Homer CM, Ryan CJ, Shales M, Krogan NJ, Patrick KL, and Guthrie C (2017). The histone variant H2A.Z promotes splicing of weak introns. Genes Dev. 31, 688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima T, Gomes T, Carmo-Fonseca M, and Proudfoot NJ (2016). Mammalian NET-seq analysis defines nascent RNA proffles and associated RNA processing genome-wide. Nat. Protoc 11, 413–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostareck DH, and Ostareck-Lederer A (2019). RNA-Binding Proteins in the Control of LPS-Induced Macrophage Response. Front. Genet 10, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostareck-Lederer A, Ostareck DH, Cans C, Neubauer G, Bomsztyk K, Superti-Furga G, and Hentze MW (2002). c-Src-mediated phosphorylation of hnRNP K drives translational activation of specifically silenced mRNAs. Mol. Cell.Biol 22, 4535–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai AA, Baharian G, Pagé Sabourin A, Brinkworth JF, Nédélec Y, Foley JW, Grenier J-C, Siddle KJ, Dumaine A, Yotova V, et al. (2016). Wide-spread Shortening of 3′ Untranslated Regions and Increased Exon Inclusion Are Evolutionarily Conserved Features of Innate Immune Responses to Infection. PLoS Genet. 12, e1006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A, Ding SL, Qin Q-M, Gupta R, Gomez G, Lin F, Feng X, Fachini da Costa L, Chaki SP, Katepalli M, et al. (2017). Global Reprogramming of Host Kinase Signaling in Response to Fungal Infection. Cell Host Microbe 21, 637–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya-Jones A, and Black DL (2009). Co-transcriptional splicing of constitutive and alternative exons. RNA 15, 1896–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya-Jones A, Bhatt DM, Lin C-H, Tong A-J, Smale ST, and Black DL (2013). Splicing kineticsand transcript releasefrom the chromatin compartment limit the rate of Lipid A-induced gene expression. RNA 19, 811–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Iaccarino C, Lee J, Kwon I, Baik SM, Kim M, Seong JY, Son GH, Borrelli E, and Kim K (2011). Regulatory roles of heterogeneous nuclear ribonucleoprotein M and Nova-1 protein in alternative splicing of dopamine D2 receptor pre-mRNA. J. Biol. Chem 286, 25301–25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passacantilli I, Frisone P, De Paola E, Fidaleo M, and Paronetto MP (2017). hnRNPM guides an alternative splicing program in response to inhibition of the PI3K/AKT/mTOR pathway in Ewing sarcoma cells. Nucleic Acids Res. 45, 12270–12284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick KL, Ryan CJ, Xu J, Lipp JJ, Nissen KE, Roguev A, Shales M, Krogan NJ, and Guthrie C (2015). Genetic interaction mapping reveals a role for the SWI/SNF nucleosome remodeler in spliceosome activation in fission yeast. PLoS Genet. 11, e1005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn BH, Netter Z, Johnson JR, Von Dollen J, Jang GM, Johnson T, Ohol YM, Maher C, Bell SL, Geiger K, et al. (2018). An Mtb-Human Protein-Protein Interaction Map Identifies a Switch between Host Antiviral and Antibacterial Responses. Mol. Cell 71, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PettitKneller EL,Connor JH, and Lyles DS(2009). hnRNPsRelocalizetothe cytoplasmfollowing infectionwithvesicularstomatitisvirus. J.Virol 83,770–780.19004954 [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. (1998). Defective LPS signaling in C3H/ HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088. [DOI] [PubMed] [Google Scholar]
- Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, Black JC, Hoffmann A, Carey M, and Smale ST (2009). A unifying model fortheselective regulation ofinducibletranscription byCpG islands and nucleosome remodeling. Cell 138, 114–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao N, Nguyen S, Ngo K, and Fung-Leung W-P (2005). A novel splice variant of interleukin-1 receptor (IL-1R)-associated kinase 1 plays a negative regulatory role in Toll/IL-1R-induced inflammatory signaling. Mol. Cell. Biol 25, 6521–6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldi T, Cortazar MA, Sheridan RM, and Bentley DL (2016). Coupling of RNA Polymerase II Transcription Elongation with Pre-mRNA Splicing. J. Mol. Biol 428, 2623–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J-W, Yang E-J, Kim SH, and Choi I-H (2015). An inhibitory alternative splice isoform of Toll-like receptor 3 is induced by type I interferons in human astrocyte cell lines. BMB Rep. 48, 696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, Feng Y, and Manley JL (2004). Dephosphorylated SRp38 acts as a splicing repressor in response to heat shock. Nature 427, 553–558. [DOI] [PubMed] [Google Scholar]
- Shin D-L, Hatesuer B, Bergmann S, Nedelko T, and Schughart K (2015). Protection from Severe Influenza Virus Infections in Mice Carrying the Mx1 Influenza Virus Resistance Gene Strongly Depends on Genetic Background. J.Virol 89, 9998–10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm S (2008). Regulation ofalternative splicing by reversible protein phosphorylation. J. Biol. Chem 283, 1223–1227. [DOI] [PubMed] [Google Scholar]
- Stetson DB, and Medzhitov R (2006). Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103. [DOI] [PubMed] [Google Scholar]
- Sun H, Kamanova J, Lara-Tejero M, and Galan JE (2018). Salmonella stimulates pro-inflammatory signalling through p21-activated kinases bypassing innate immune receptors. Nat. Microbiol 3, 1122–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P, Forse RA, and Bajenova O (2011). Carcinoembryonic antigen (CEA) and its receptor hnRNP M are mediators of metastasis and the inflammatory response in the liver. Clin. Exp. Metastasis 28, 923–932. [DOI] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. (2016). Robust transcriptome-wide discovery of RNA-binding protein binding siteswith enhanced CLIP(eCLIP). Nat. Methods 13, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquero-Garcia J, Barrera A, Gazzara MR, González-Vallinas J, Lahens NF, Hogenesch JB, Lynch KW, and Barash Y (2016). A new view of transcriptome complexity and regulation through the lens of local splicing variations. eLife 5, e11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viktorovskaya OV, Greco TM, Cristea IM, and Thompson SR (2016). Identification of RNA Binding Proteins Associated with Dengue Virus RNA in Infected Cells Reveals Temporally Distinct Host Factor Requirements. PLoS Negl. Trop. Dis 10, e0004921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Gao XD, Lee J-H, Huang H, Tan H, Ahn J, Reinke LM, Peter ME, Feng Y, Gius D, et al. (2014). Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev. 28, 1191–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachdav G, Kloppmann E, Kajan L, Hecht M, Goldberg T, Hamp T, Honigschmid P, Schafferhans A, Roos M, Bernhofer M, et al. (2014). PredictProtein-an open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 42, W337–W343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, and Akira S (2003). Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 307, 640–643. [DOI] [PubMed] [Google Scholar]
- Zhao W, Wang L, Zhang M, Wang P, Qi J, Zhang L, and Gao C (2012). Nuclear to cytoplasmic translocation of heterogeneous nuclear ribonucleoprotein U enhances TLR-induced proinflammatory cytokine production by stabilizing mRNAs in macrophages. J. Immunol 188, 3179–3187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This article contains all datasets generated during this study. All coding for Majiq and Voila was standard and is available for academic download. Raw read files for RNA-seq can be found on GEO (Accession #GSE137603).