

Abstract
Introduction
Metastasis is a fundamentally physical process in which cells deform through narrow gaps and generate forces to invade surrounding tissues. While it is commonly thought that increased cell deformability is an advantage for invading cells, we previously found that more invasive pancreatic ductal adenocarcinoma (PDAC) cells are stiffer than less invasive PDAC cells. Here we investigate potential mechanisms of the simultaneous increase in PDAC cell stiffness and invasion, focusing on the contributions of myosin II, Arp2/3, and formins.
Method
We measure cell invasion using a 3D scratch wound invasion assay and cell stiffness using atomic force microscopy (AFM). To determine the effects of actin- and myosin-mediated force generation on cell stiffness and invasion, we treat cells with pharmacologic inhibitors of myosin II (blebbistatin), Arp2/3 (CK-666), and formins (SMIFH2).
Results
We find that the activity of myosin II, Arp2/3, and formins all contribute to the stiffness of PDAC cells. Interestingly, we find that the invasion of PDAC cell lines is differentially affected when the activity of myosin II, Arp2/3, or formins is inhibited, suggesting that despite having similar tissue origins, different PDAC cell lines may rely on different mechanisms for invasion.
Conclusions
These findings deepen our knowledge of the factors that regulate cancer cell mechanotype and invasion, and incite further studies to develop therapeutics that target multiple mechanisms of invasion for improved clinical benefit.
Electronic supplementary material
The online version of this article (doi:10.1007/s12195-019-00603-1) contains supplementary material, which is available to authorized users.
Keywords: Mechanobiology, Cytoskeleton, Pancreatic ductal adenocarcinoma, Cell stiffness, Arp2/3, Formins, Traction forces, Cell motility
Introduction
Cell physical phenotypes, including deformability, adhesion, and contractility, are critical in cancer cell invasion.4,108,112 The physical phenotypes of tumor cells may also be implicated in many steps of metastasis: to spread to distant sites, cells are required to invade into the extracellular matrix, intravasate, and extravasate, which all require movement through micron-scale pores smaller than the diameter of a single cell.42,108 Physical phenotypes of cancer cells, such as deformability and adhesion, are emerging as label-free, complementary biomarkers for cancer diagnosis.101 Since the proteins and pathways that regulate cell physical phenotypes also contribute to cell invasion,4,108,112 mechanotransduction,14,36,40,85,113 and chemoresistance,104 understanding the molecular mediators of physical phenotypes can provide mechanistic insight into cancer progression and new therapeutic strategies. For example, cellular contractile force generation is mediated by Rho-associated protein kinase (ROCK). Treatment with the ROCK inhibitor Fasudil reduces invasion in vitro and metastasis in vivo.115,122 Fasudil also decreases the physical tension within the tumor to increase tumor porosity and improve chemotherapy access.104 A deeper knowledge of the relationship between cell physical phenotypes and clinically relevant cell behaviors, such as invasion, could enable identification of new therapeutic strategies to improve patient outcomes.
The mechanical phenotype, or mechanotype, of cells is a physical property that includes cellular deformability, which is the ability of cells to resist shape changes in response to physical forces, and cellular contractile forces, or the magnitude of physical forces cells generate. Mechanotype is emerging as an important property of cancer cells that can change during cancer progression and shows strong associations with in vitro measurements of cell invasion,15,25,43,56,61,74,112,116 which reflect the ability of cells to metastasize in vivo.39 A frequently observed trend is that more invasive tumor cells are more deformable, which has been shown across prostate, ovarian, and breast cancer cell lines in vitro,15,25,56,112 as well as tumor cells of human breast biopsies in situ.51,68 Increased deformability enables cells to transit more easily through narrow gaps,32,81 and may therefore provide a selective advantage in metastasis. Conversely, cells with decreased cellular and nuclear deformability are more likely to occlude narrow gaps32,81 and tend to exhibit reduced invasion.18,72,95,112 However, there are examples where more invasive cells are stiffer, including lung cancer cells,116 pancreatic ductal adenocarcinoma cells (PDAC),61 and cancer cells with increased beta-adrenergic signaling.43 Despite multiple examples of stiffer cells being more invasive, it remains poorly understood what factors contribute to the simultaneous increase in cell stiffness and invasion that is observed in some cell types. With a greater understanding of why stiffer cells are more invasive, we could advance mechanotype as a clinical biomarker and gain insights into novel therapeutic targets.
Cells are complex, dynamic materials, and cellular mechanotype is determined by both intrinsic and extrinsic factors. Intrinsic determinants of cell mechanotype include levels of mechanoregulating proteins, such as cytoskeletal actin8,9 and nuclear lamins,80,96 as well as the organization of higher order structures formed by these proteins such as actin filaments. Cell mechanotype is also determined by the forces that cells generate in response to extrinsic factors, such as soluble molecules, the mechanical stiffness of the extracellular matrix, and the balance of intracellular tension across cell–matrix structures and focal adhesions.107,121 The intrinsic and extrinsic factors that determine cell physical properties also mediate cell motility and invasion.11,30,55,77 For example, actin is a key structural protein that is also crucial in dynamic cellular behaviors, such as the generation of physical forces through the conversion of ATP into mechanical energy. We previously investigated if stiffer PDAC cells are more invasive because they have higher levels of actin.61 However, we found no significant differences in the levels of beta-actin or filamentous (F-)actin across cell lines with varying stiffness and invasive potential. Another possible origin of the increased stiffness of more invasive cells could be the actin-dependent force generation that can influence both cell stiffness8,9,26,95 and invasion.29,37,69 Reducing actomyosin contractility by pharmacologic inhibition of myosin II results in decreased cell migration and invasion,37,69 as well as decreased stiffness of fibroblasts and ovarian cancer cells.9,95 The protrusive forces generated by actin polymerization and branching, which are mediated by the Arp2/3 complex and formins, such as diaphanous proteins, are also critical for certain modes of cancer cell invasion. Reducing the activity of Arp2/3 and formins diminishes the formation of structures involved in motility and invasion such as lamellipodia and invadopodia,31,93,114 and decreases the invasion of head and neck squamous cancer cells.29 The activity of Arp2/3 and formins also contribute to cancer cell mechanotype.28 Thus, we hypothesize that actin-mediated force generation, which is largely regulated by the activity of myosin II, Arp2/3, and formins, contributes to the simultaneous increased invasion and stiffness of cancer cells.
Here we test the hypothesis that more invasive cancer cells are stiffer than their less invasive counterparts due to the activity of myosin II, Arp2/3, and formins. We use a panel of PDAC cell lines as a model system, as they have well-defined mechanotypes and invasive potential61; this allows us to investigate factors that contribute to stiffer cells being more invasive across cell lines that have similar tissue origin.17 We measure cell stiffness using atomic force microscopy (AFM) and invasion using a 3D scratch wound invasion assay, with and without pharmacological inhibition of myosin II, Arp2/3, and formin activity. To develop a more integrated knowledge of PDAC cell invasion, we also measure cellular traction forces and the activity of matrix metalloproteinases (MMPs). Our findings reveal that myosin II, Arp2/3, and formins differentially contribute to cell stiffness and invasion in a cell-type dependent manner.
Experimental Methods
Cell Culture
Pancreatic ductal adenocarcinoma (PDAC) cell lines (Hs766T, MIA PaCa-2, and PANC-1) are from the American Type Culture Collection (ATCC). Cell lines were authenticated using the Promega powerplex16 System recommended by ATCC within 1 year of these experiments. Cells are cultured at 5% CO2 and 37 °C in high glucose, l-glutamine Dulbecco’s Modified Eagle Medium (DMEM) (Life Technologies) with 10% fetal bovine serum and 1% v/v penicillin–streptomycin (Gemini BioProducts). To inhibit myosin II, Arp2/3, and formins, cells are treated with 20 µM, or 50 µM of blebbistatin, CK-666, and SMIFH2; vehicle control is DMSO-treated (0.5% w/w). For AFM and micropillar experiments, cells are treated with drugs for 30 min prior to measurement. For scratch-wound invasion experiments, cells are treated with drugs for the duration of each experiment starting at t = 0 h. To inhibit matrix metalloproteinases (MMPs), we treat cells for 48 h with 10 or 25 µM GM6001 prior to measurement of MMP activity levels; for 3D invasion assays, cells are treated with GM6001 for the duration of the experiment.
Gene Expression and Bioinformatics Analysis
To analyze gene expression, we use publicly available RNA-seq data for Hs766T, MIA PaCa-2, and PANC-1.3 Using STAR v.2.4.2a,19 we align RNA sequence reads to the human reference genome (hg38) with Ensembl v.82 transcriptome annotations. STAR is run with the following parameters: minimum/maximum intron sizes are set to 30 and 500,000; noncanonical, unannotated junctions are removed; maximum tolerated mismatches is set to 10; and the outSAMstrandField intron motif option is enabled. To quantify per sample read abundances we use the Cuffquant command included with Cufflinks v.2.2.1,100 with fragment bias correction and multiread correction enabled. All other options are set to default. Finally, fragments per kilobase of exon per million fragments mapped (FPKM) are calculated using the Cuffnorm command with default parameters. We use these FPKM values to compare expression levels of genes whose protein products are implicated in regulation of cell mechanical properties.16,23,54,91,96,118 We compare arbitrary gene expression values across the 3 cell lines.
Atomic Force Microscopy (AFM)
To measure cell stiffness, AFM is performed as previously described61 using the MFP-3D-BIO system (Asylum Research, Oxford Instruments). Cells are probed with the “C” tip of an MLCT probe (Bruker), which has a nominal spring constant of 0.01 N/m. The exact spring constant for each probe is calibrated before each experiment by indenting clean glass and the spring constant is calculated by the thermal noise method. Force–distance curves are acquired by indenting the cytoplasmic region of 20 to 30 cells for each cell line and drug treatment. Approach and retract speeds for all experiments are 5 µm/s. The elastic modulus for each cell is determined by fitting force–distance curves to the Hertz–Sneddon model using Asylum Research software. Experiments were carried out at 37 °C.
3D Scratch Wound Invasion and Proliferation Assays
We perform invasion and proliferation assays using the IncuCyte time-lapse imaging system (EssenBioscience). To measure cell invasion through a 3D matrix, we perform scratch wound invasion assays with Matrigel to simulate the ECM. We plate cells in the wells of a 96-well plate at 95% confluency, create a scratch wound (EssenBioscience WoundMaker), overlay the scratch with 8 mg/mL Matrigel (Corning), and acquire phase contrast images every 4 h for 120 h at 5% CO2 and 37 °C (IncuCyte Zoom). We determine the confluence of cells in the wound area at each time point using quantitative image analysis (Essen Bioscience). Since wound confluence may be influenced by cell proliferation, we also measure percent confluence by sparsely plating cells (20% confluency) and acquiring phase contrast images every 2 h for 120 h. To quantify the number of protrusions at the invasion front, the length of each invasion front is measured using the free hand tool of ImageJ.
Cell Rounding Assay
To determine the timescale of cell rounding, which is an indicator of myosin II activity, we measure the rate of change in projected cell area after trypsinization. We plate cells to 40% confluency in a 60 mm petri dish coated with 100 µg/mL Matrigel (overnight at 37 °C), wash twice with 1X phosphate buffered saline (Corning), and then treat with 1× trypsin–EDTA to induce cell rounding (Gemini BioProducts). To quantify changes in cell shape during rounding, we acquire images every 10 s starting immediately before trypsin–EDTA is added (t = 0 s). We assess cell rounding by measuring cell area at each time point using a custom MatLab (Mathworks) script. To determine the rounding time constant, we fit a bounded exponential growth model to our data since the normalized area grows from the origin and reaches an asymptote. We use the form:
where NAf is the asymptotic normalized area (i.e. the final area), t is the time in seconds, and τ is the time constant.
Micropillar Traction Stress Assay
To quantify cellular traction stresses, we use a micropillar assay.99 We fabricate PDMS micropillars as previously described110 and embed gold micro-disks in the top of each pillar to facilitate darkfield imaging with a 20x objective (NA 0.5). We image 10 regions of the pillar array before cell seeding. After seeding for 20 h, we treat cells with drugs (30 m), and fix the cells with 4% paraformaldehyde for 15 min at 37 °C. To delineate cells, we label the plasma membrane with wheat germ agglutinin (WGA), Alexa Fluor 488 conjugate (Invitrogen). The same 10 regions of the micropillar devices are then imaged using fluorescence microscopy (Zeiss Axiovert A1) equipped with a 20x objective (NA 0.5) to identify pillars occupied by cells, and darkfield microscopy to track the displacement of the gold-tipped pillars. The traction force, F, exerted by a cell on a single pillar is determined by:
where E is the elastic modulus of the pillar (2.0 MPa111), r is the radius of the pillar (measured to be 0.875 µm), L is the height of the pillar (measured to be 6.5 µm), and ∆x is the horizontal displacement of the pillar between t0 and tmeasured.
Matrix Metalloproteinase (MMP) Activity Assay
To measure the activity of MMPs, we use the MMP Activity Assay (Fluorometric—Red, abcam). In brief, we retrieve 90 µL of conditioned media from each well of a 96-well plate, in which cells are at ~30% confluency after 18 h of culture. Media is transferred to the wells of a black-walled, clear-bottom 96-well plate (Greiner BioProducts). Absorbance at 540/590 nm is measured on a Molecular Devices Flexstation at 90 min after the addition of the MMP substrate.
Statistical Analysis
All data are obtained from at least three independent experiments. For data with normal distributions, we determine statistical significance using a Student’s t test (Excel, Microsoft). For data that exhibit a non-normal distribution, use the Mann–Whitney U test to determine statistical significance (OriginLab).
Results
To investigate the relationship between cell stiffness, invasion, and the activity of myosin II, Arp2/3, and formins, we use three immortalized PDAC cell lines: Hs766T, MIA PaCa-2, and PANC-1. The MIA PaCa-2 and PANC-1 cell lines have similar founder mutations (KRAS, TP53, and CDKN2A), while the Hs766T cell line has an additional SMAD4 mutation.17 We previously determined that across this panel of PDAC cell lines, more invasive cells tend to be stiffer.61 The Hs766T cells are the stiffest and most invasive of these three cell lines, while the MIA PaCa-2 are the most deformable and least invasive.61
Myosin II Activity has Differential Effects on the Invasion of PDAC Cells
Myosin II is essential for generating forces involved in motility.59,94 The activity of myosin II also contributes to cell stiffness.88,95 Therefore, we first investigate the role of myosin II in the increased invasion of stiffer PDAC cells. Analysis of existing RNA-seq expression data3 reveals higher expression of genes encoding nonmuscle myosin IIA (MYH9) and myosin IIB (MYH10) in Hs766T compared to PANC-1 and MIA PaCa-2 cell lines (Supp. Fig. 1); these findings suggest that Hs766T cells could have increased myosin II activity and thereby generate a larger magnitude of myosin-II dependent forces, which could contribute to their increased invasion.2,52 To test this hypothesis, we determine the effect of myosin II activity on PDAC cell invasion using a 3D scratch-wound assay overlaid with a Matrigel protein matrix. To reduce the activity of myosin II, we treat cells with blebbistatin, a small molecule that inhibits myosin II activity.46 For cells treated with the DMSO vehicle control, we observe that invasion varies across PDAC cell lines, from most to least invasive, Hs766T > PANC1 > MiAPaCa-2 (Fig. 1), which is consistent with our previous findings.61 Interestingly, we find that blebbistatin treatment has cell line-dependent effects on invasion. Both PANC-1 and MIA PaCa-2 show significant, dose-dependent decreases in invasion with increasing concentrations of blebbistatin. For example, at 72 h, MIA PaCa-2 cells show a modest but significant reduction in wound confluence from 19 ± 2% for the DMSO control cells to 11 ± 1% for cells treated with 50 µM blebbistatin (p = 2.1 × 10−4). PANC-1 cells exhibit a greater reduction in wound confluence at 72 h from 30 ± 1% to 13 ± 2% with the 50 µM blebbistatin treatment (p = 5.9 × 10−10) (Fig. 1c). Our results showing reduced invasion of MIA PaCa-2 and PANC-1 cells with decreased myosin II activity are consistent with previous observations that treatment with blebbistatin20,37 and ROCK inhibitors79,103,105 reduces the invasion of cancer cells.79,103,105 By contrast, Hs766T cells show no significant change in wound confluence with 50 µM blebbistatin treatment when compared to the vehicle control at 72 h (50 ± 2% vs. 52 ± 3%, p = 6.1 × 10−1), suggesting that myosin II activity is not required in the dominant mechanism of invasion of these cells.
Figure 1.

Myosin II activity is required for the invasion of MIA PaCa-2 and PANC-1 cells, but not Hs766T cells. (a) Invasion through Matrigel is measured by wound confluence in a 3D scratch wound invasion assay. Scatter plot shows the quantification of wound confluence over time. Cells are treated with blebbistatin or DMSO (ctrl) from t = 0. The dashed lines indicate the 24 h and 72 h time points, which we use to compare wound confluence values for statistical significance. Data points show average invasion over three independent experiments. Bar plot shows average wound confluence at 24 h (b) and 72 h (c). Error bars represent standard error across three independent experiments. Pairwise p values are determined by a Student’s t test. *p < 0.05.
Since the invasion measured in this 3D wound confluence assay can be affected by differences in cell proliferation with and without drug treatment, we measure cell proliferation by tracking the confluence of cells over time. While we find some significant changes in proliferation with blebbistatin treatment (Supp. Fig. 2), these observations cannot fully explain the observed differences in invasion. For example, the decrease in the confluence of MIA PaCa-2 cells over time may contribute to their reduced invasion, but the proliferation of PANC-1 cells is not altered despite their significantly decreased invasion (Fig. 1, Supp. Fig. 2). Thus, differences in cell proliferation across our PDAC cell lines with and without blebbistatin treatment cannot explain the differences we observe in the invasion of these cells when myosin II activity is inhibited. Therefore, our data suggest that while myosin II activity is required for the invasion of the MIA PaCa-2 and PANC-1 cells, other factors mediate the invasion of Hs766T cells.
Hs766T Cells are Slower to Round Upon Detachment, Indicating Reduced Actomyosin Force Generation
After observing that pharmacologic inhibition of myosin II activity does not affect the invasion of Hs766T cells, we next probed myosin II activity using an independent assay: we measure the rate of cell rounding after cells are detached from their substrate with trypsin (Fig. 2). Cells with higher levels of myosin II activity tend to have a faster rounding time compared to cells with reduced myosin II activity.84 We find that MIA PaCa-2 cells have the slowest cell rounding rate, as indicated by the highest rounding time constant of 151 ± 23 s (Fig. 2b); this is consistent with Mia PaCa-2 cells having the slowest invasion rate, which is further decreased by inhibiting myosin II activity. By contrast, the Hs766T and PANC-1 cells round more quickly, as indicated by their shorter rounding time constants of 118 ± 12 s and 82 ± 12 s (Fig. 2b). With blebbistatin treatment, the cell rounding rate is reduced the most for the MIA PaCa-2 and PANC-1 cells. The MIA PaCa-2 cells show a 3-fold increase in rounding time constants, while the PANC-1 exhibit a larger 107-fold increase. These results are consistent with the marked decrease in invasion of these cell lines with myosin II inhibition (Fig. 1). Interestingly, the Hs766T cells show a much smaller ~ 2-fold increase in the rounding time constant with blebbistatin treatment (Fig. 2b), which corroborates our observations of blebbistatin effects on invasion and further substantiates that MIA PaCa-2 and PANC-1 cells are more dependent on myosin II activity to generate forces compared to Hs766T cells. These data further underscore the differential effects of myosin II inhibition across different cell lines and suggest that force generation by Hs766T cells during invasion may occur through another mechanism.
Figure 2.

Myosin II activity contributions to cell rounding rate vary across cell lines. (a) Representative images show time sequence of MIA PaCa-2 cell shape change following the addition of trypsin. Scale, 10 µm. (b) Quantification of cell area as a function of time for cells treated with DMSO (ctrl) and 50 µM blebbistatin PDAC cells. Insert represents rounding cell over time. Dashed lines indicate fits with a bounded exponential growth model used to extract: (c) rounding time constants, τ. Cells are treated with 50 µM blebbistatin for 30 min prior to trypsinization. Data points show averages of at least 21 cells across three independent experiments. Error bars represent the 95% confidence intervals.
PANC-1 Cells Exert Increased Traction Stresses Compared to Hs766T Cells
The ability of cells to generate traction forces on the surrounding extracellular matrix is critical during many modes of cancer cell invasion.48 More invasive cells tend to exhibit increased traction stresses.47 Thus, since Hs766T cells are the most invasive cell line, we initially predicted that these cells would have increased traction stresses compared to the other PDAC cell lines. However, traction force generation is influenced by myosin II activity, which does not appear to be a dominant mechanism of force generation in Hs766T cells based on our cell invasion and rounding experiments with blebbistatin treatment. To further explore the mechanisms of force generation in PDAC cells, we quantify traction stresses that cells exert on their substrate using a micropillar assay in which cells are plated on polydimethylsiloxane (PDMS) micropillars (Fig. 3a) that have a well-characterized elastic modulus.111 In our micropillar assay, the average force per area (stress) exerted by cells is calculated by measuring the displacements of the micropillars. We find that the average traction stress exerted by PANC-1 cells is 44 ± 7 nN/µm2 while Hs766T cells have a 38% lower average traction stress of 27 ± 7 nN/µm2 (p = 1.0 × 10−5) (Figs. 3b and 3c). These findings corroborate our observations that PANC-1 cells round more quickly than Hs766T cells, as both assays depend on actomyosin activity. We next determine contributions of myosin II to cellular traction forces by treating cells with blebbistatin. For the PANC-1 cells, we find that the average traction stress decreases by 30% following blebbistatin treatment (p = 2.8 × 10−4) (Figs. 3b and 3c). Our results with the blebbistatin-treated PANC-1 cells are consistent with previous studies showing that myosin II activity is a major contributor to traction force generation.7,57 By contrast, the Hs766T cells do not show a significant change in average traction stress with pharmacologic inhibition of myosin II (26 ± 7 nN/µm2 for the vehicle control cells vs. 28 ± 7 nN/µm2 for the blebbistatin-treated cells, p = 3.7 × 10−1) (Figs. 3b and 3c). Thus, our findings substantiate the differential contributions of myosin II activity to the forces generated by PANC-1 and Hs766T cells, and further suggest that Hs766T cells may be using an invasion strategy that does not depend on myosin II.
Figure 3.

PANC-1 cells exert increased traction stresses compared to Hs766T cells. (a) Schematic illustration of the micropillar assay. Device dimensions are indicated for the distance between pillars d, as well as pillar radius r and height h. (b) Representative force map of a cell. Shown here is a PANC-1 cell treated with DMSO (ctrl) for 30 min prior to fixation. Average traction stress per cell is determined by analysis of the displacement of each gold-coated micropillar 18 h after plating of cells. (c) Box plots show the distribution of average traction stress per cell exerted by Hs766T and PANC-1 cells with and without blebbistatin treatment (50 µM). Boxes represent the 25th and 75th percentiles, whiskers represent the 10th and 90th percentiles, and the horizontal line represents the median. p values are determined by a Mann–Whitney U test. *p < 0.05. We measure traction stresses for at least 18 cells across three independent experiments.
Myosin II Activity Contributes to PDAC Cell Stiffness
To further investigate the relationship between cell invasion and cell stiffness, we next determine the contributions of myosin II activity to cellular stiffness. We use atomic force microscopy (AFM) to measure the elastic modulus, E, of PDAC cells with and without the myosin II inhibitor blebbistatin. With the control DMSO treatment, Hs766T cells are nearly twofold stiffer than PANC-1 cells (Fig. 4a), which is consistent with our previous measurements of untreated cells.61 With inhibition of myosin II activity, we observe a significant ~threefold reductions in the stiffness (E) of both Hs766T and PANC-1 cells: Hs766T cell stiffness decreases from a median of 3.5 to 1.1 kPa (p = 3.0 × 10−6), while PANC-1 cell stiffness decreases from a median of 2.0 to 0.7 kPa (p = 1.0 × 10−5) (Fig. 4a). Our results are consistent with other observations that inhibition of myosin II activity makes adherent cells more deformable.95 While we find a significant decrease in Hs766T cell stiffness with blebbistatin treatment, there are no significant effects of myosin II inhibition on the invasion or traction force generation of these cells, suggesting that cell stiffness is not consistently associated with invasive potential.
Figure 4.
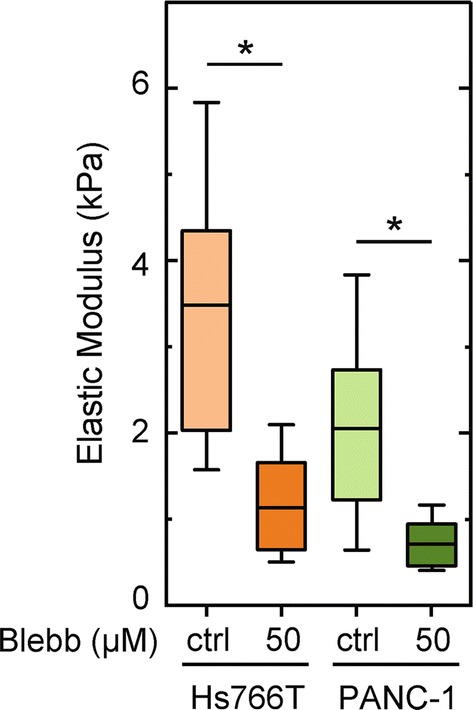
Myosin II activity contributes to PDAC cell stiffness. The elastic modulus, E, of PDAC cells adhered to Matrigel-coated glass is measured using atomic force microscopy (AFM) after cells are treated with DMSO (ctrl) or 50 µM blebbistatin for 30 min. Each cell is measured with one indentation in the cytoplasmic region. Horizontal lines represent the average, boxes represent the 25th and 75th percentiles, and whiskers represent the 10th and 90th percentiles. Statistical significance is determined using a Mann–Whitney U test. *p < 0.05. Data represent 22–37 cells per treatment measured over three independent experiments.
Invasion is not Altered by Matrix Metalloproteinase Inhibition
We next investigate the activity of matrix metalloproteinases (MMPs) across PDAC cells as a possible explanation of why Hs766T cells are stiff and highly invasive but do not rely on myosin II for invasion. MMPs are major contributors to myosin II-independent modes of cancer cell invasion, as these enzymes degrade the surrounding protein matrix and thereby enlarge the size of gaps that cells must deform through during invasion.79,109 The secretion of MMPs is also linked to the formation of invadopodia, which generate forces as they protrude from the plasma membrane.76 If Hs766T cells have increased MMP production, this may explain why these cells are stiffer yet more invasive. To assess levels of MMPs across cell lines, we first analyze existing RNA-seq data.3 We analyze levels of 23 MMP isoforms across Hs766T, MIA PaCa-2, and PANC-1 cell lines (Supp. Fig. 3A). Focusing on levels of the three MMPs that are most strongly implicated in the invasive potential of PDAC cells—MMP2, MMP14, MMP281,73,86,120—we find that Hs766T cells have increased expression of MMP14 compared to MIA PaCa-2 and PANC-1 cells (145-fold and 36-fold higher), and an even higher expression of MMP28 (1716-fold and 1274-fold higher). By contrast, MMP2 expression is 4- and 425-fold higher in PANC-1 cells compared to Hs766T and MiaPaCa-2 cells.
While our gene expression analysis suggests that Hs766T cells have a higher overall level of MMP expression, levels of MMP expression do not always correlate to levels of MMP activity,66 which is a functional measure of matrix degradation. Therefore, we next measure the activity of MMPs secreted from the PDAC cell lines using a fluorescence resonance energy transfer (FRET)-based MMP activity assay. We find that Hs766T cells have a 2.9-fold increase in MMP activity compared to MIA PaCa-2 cells (p = 7.5 × 10−8) and a 1.5-fold increase compared to PANC-1 cells (p = 1.2 × 10−5) (Supp. Fig. 3B). Thus, while Hs766T cells have only a slightly higher MMP activity level than PANC-1 cells, we hypothesized that this elevated MMP activity could contribute to their increased invasion and the insensitivity of their invasion to myosin II inhibition.
To directly test the role of MMP activity in PDAC cell invasion, we performed 3D invasion assays in the presence of the broad-spectrum MMP inhibitor GM6001. While treatment with 10 or 25 µM GM6001 significantly decreases MMP activity across all three PDAC cell lines (Supp. Fig. 3B), we observe no significant effect of MMP inhibition on cell invasion (Supp. Fig. 3C). These results indicate that MMP activity does not contribute significantly to the invasion of these PDAC cell lines as measured by our Matrigel-based invasion assay. Our findings further illustrate that the increased invasion of Hs766T cells compared to other PDAC cell lines cannot be explained alone by their increased MMP activity and further support they are using an alternative force generation mechanism for invasion.
Arp2/3 and Formin Activity Contribute to the Invasion of Hs766T Cells
While increased myosin II activity can explain why PANC-1 cells are stiffer and more invasive than MIA PaCa-2 cells, it is still unclear what contributes to the concurrent increase in stiffness and invasion of Hs766T cells. We hypothesized that Hs766T cells may utilize alternative mechanisms for invasion that do not strongly rely on myosin II.60 Another key mechanism of force generation is the formation of invadopodia, protrusive structures that contribute to cell invasion.29,31 Two of the main components required for the formation of protrusions are Arp2/3 and formins, which mediate actin nucleation and branching. Thus, we hypothesized that the activity of Arp2/3 and formins contributes to Hs766T cell invasion while myosin II-dependent forces are more critical for the invasion of MIA PaCa-2 and PANC-1 cells.
A characteristic hallmark of Arp2/3-dependent invasion is the formation of protrusions at the leading edge of an invading cell.94 To determine if Hs766T cells exhibit any differences in protrusion formation compared to PANC-1 and MIA PaCa-2 cells, we quantify the length of the invasion front in our 3D scratch wound invasion assays (Supp. Fig. 4); a longer invasion front length indicates a greater number and/or length of protrusions, while a shorter front length reflects that the invading cell front exhibits fewer and/or shorter protrusions. Our analysis reveals that Hs766T cells have an average invasion front length of 6283 ± 1461 µm compared to 3680 ± 454 µm for MIA PaCa-2 cells (p = 9.2 × 10−5) and 3119 ± 574 µm for PANC-1 cells (p = 4.1 × 10−5) (Supp. Fig. 4). The increased length of the Hs766T invasion front supports the hypothesis that the invasion of these cells depends primarily on Arp2/3 activity, while MIA PaCa-2 and PANC-1 largely rely on myosin II activity to invade.
To determine the effects of Arp2/3 activity on PDAC cell invasion, we measure the invasion of cells treated with the Arp2/3 inhibitor, CK-666, which is a small molecule that stabilizes the inactive state of the Arp2/3 complex.35 Comparing invasion across cell lines, we find that inhibition of Arp2/3 activity results in a slight but significant decrease in invasion for the MIA PaCa-2 and PANC-1 cells treated with 50 µM CK-666 (MIA PaCa-2: 11 ± 2% for DMSO-treated cells vs. 9 ± 2% for 50 µM CK-666, p = 3.9 × 10−2; PANC-1: 12 ± 3% for DMSO-treated cells vs. 9 ± 2% for 50 µM CK-666, p = 5.7 × 10−3) (Figs. 5a and 5b). By contrast, we observe a greater, dose-dependent decrease in the invasion of Hs766T cells with increasing CK-666 concentration (23 ± 7% for DMSO-treated cells vs. 19 ± 5% for 20 µM CK-666 vs. 15 ± 5% for 50 µM CK-666; for DMSO vs. 50 µM CK-666 p = 2.1 × 10−4) (Figs. 5a and 5b). We also measure effects of CK-666 on cell proliferation, which could impact the invasion assay findings, but observe there are no significant effects on proliferation over the invasion timescale of 24 h (Supp. Fig. 5A, 5B). Taken together, these results suggest that Arp2/3 activity contributes to the invasion of Hs766T cells but not significantly for PANC-1 or MIA PaCa-2 cells. By contrast, myosin II is a major contributor to the invasion of MIA PaCa-2 and PANC-1 cells, but has no significant effect on Hs766T cell invasion. These findings are consistent with the notion that different cell types rely on different mechanisms for invasion.67
Figure 5.

Activity of Arp2/3 contributes to the invasion of Hs766T cells. Invasion through Matrigel is measured by wound confluence in a 3D scratch wound invasion assay. Scatter plot shows the quantification of wound confluence over time with treatment of (a) the Arp2/3 inhibitor, CK-666 or (c) the formin inhibitor, SMIFH2. The dashed line indicates the 24 h time point, which we use to compare wound confluence values for statistical significance. Cells are treated with CK-666, SMIFH2, or DMSO (ctrl) from t = 0. Data points show average invasion over three independent experiments. Bar plot shows average wound confluence at the 24 h time point for (b) CK-666 or (d) SMIFH2 treated cells. Error bars represent standard error from three independent experiments. Pairwise p values are determined by a Student’s t test. *p < 0.05.
While Arp2/3 is critical for actin nucleation and branching as well as cell motility, formins are also integral in actin nucleation and polymerization.71 We next investigate the role of formin activity in PDAC cell invasion by treating cells with SMIFH2, which broadly inhibits the family of formin proteins that are functionally important for cancer cell motility.41,78 We find there is no significant difference in the invasion of MIA PaCa-2 and PANC-1 cells with SMIFH2 treatment compared to vehicle control (for 50 µM at 24 h; MIA PaCa-2: p = 0.23, PANC-1: p = 0.91) (Figs. 5c and 5d). We observe a small but significant difference in Hs766T cells treated with SMIFH2 compared to vehicle control (p = 2.6 × 10−2). Moreover, we only find a difference in invasion of Hs766T cells with 50 µM SMIFH2, unlike the dose-dependent effects of Arp2/3 inhibition. We further determine the effects of SMIFH2 on cell proliferation, which could impact the invasion assay results, and find there are no significant effects on proliferation over 24 h (Supp. Fig. 5C, 5D)
Taken together our results indicate that the activity of Arp2/3 is a major contributor to the invasion of Hs766T cells, while myosin II activity more heavily influences the invasion of MIA PaCa-2 and PANC-1 cells.
Activity of Arp2/3 and Formins Contributes to Hs766T Cell Stiffness
To determine the extent to which Arp2/3 and formin activity mediate cell stiffness, we next treat cells with either CK-666 or SMIFH2 and measure elastic modulus using AFM. Since Hs766T and PANC-1 cells are the most invasive and stiffest PDAC cell lines of our panel, we focus our measurements on these cell lines. Inhibiting Arp2/3 activity with CK-666 results in a significant decrease in the stiffness of Hs766T cells (p = 3.5 × 10−3), but only a slight, insignificant decrease in PANC-1 stiffness (p = 5.5 × 10−1) (Fig. 6a). Consistent with the effects of CK-666 treatment, inhibition of formin activity results in a greater reduction in stiffness of Hs766T than PANC-1 cells (Hs766T: p = 1.7 × 10−2, PANC-1: p = 2.2 × 10−2) (Fig. 6c). Our AFM results confirm that the activity of both Arp2/3 and formins contribute to PDAC cell stiffness, indicating that actin nucleation, branching, and polymerization play a role in cell mechanotype.
Figure 6.
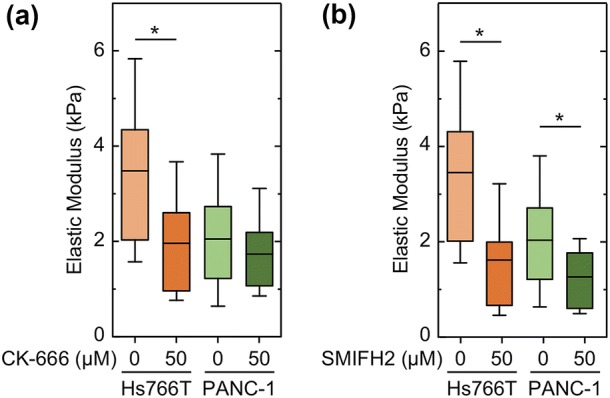
Arp2/3 and formin activity contribute to PDAC cell stiffness. The elastic modulus, E, of PDAC cells adhered to Matrigel-coated glass is measured using atomic force microscopy (AFM) when cells are treated with (a) 50 µM CK-666 or (b) 50 µM SMIFH2 for 30 minutes. Boxes represent the 25th and 75th percentiles, whiskers represent the 10th and 90th percentiles, and the horizontal line represents the median. Statistical significance calculated with Mann Whitney U test. *p < 0.05.
To summarize our findings on the relationship between cell stiffness and invasion, we compile data for cell stiffness and invasion in a single plot (Fig. 7) and determine the strength of correlation across our panel of untreated PDAC cell lines and all drug perturbations at 24 h. We find that median values of invasion and cell stiffness across cell lines and treatment conditions are weakly correlated for Hs766T (R2 = 0.34) and for PANC-1 (R2 = 0.30) cells (Figs. 7a and 7b); compiling data for both cell types in a single plot yields a slightly higher but still weak correlation (R2 = 0.45) (Fig. 7c). While this calculation neglects the sample variance, the lack of strong correlation demonstrates that these drug perturbations affect cell stiffness and invasion differently in different cell lines. Overall, these findings highlight how cell mechanotype and invasion are phenotypes that are influenced by multiple molecular mediators, and the factors that determine the phenotype in one cell type may not be the same for another cell type. For example, the activity of myosin II contributes to both contractile force generation24,65 and actin crosslinking.49 Therefore, the lack of strong correlation between stiffness and invasion could be explained by considering the multiple roles of myosin II: while myosin II inhibition does not affect Hs766T invasion or traction stresses, it may decrease Hs766T cell stiffness by reducing the crosslinking density of their actomyosin network.102 By contrast, inhibition of myosin II activity in PANC-1 cells could reduce both crosslinking of actin and contractile force generation.
Figure 7.

Linear regression of cellular elastic modulus vs. invasion, as measured as wound confluence by 3D scratch-wound invasion for untreated (a) Hs766T and (b) PANC-1 cells, as well as with treatments of blebbistatin, CK-666, or SMIFH2. (c) Compiled data for both Hs766T and PANC-1 cells. Median values and standard deviations are plotted here. R = Pearson’s correlation coefficient determined by linear regression of median elastic modulus and wound confluence values across samples. Note that the sample variances were not included in calculating the correlation.
Discussion
Here we investigate the relationship between cell stiffness and invasion. We specifically study the role of myosin II, Arp2/3, and formin activity, as we hypothesized that these molecular components could contribute to the simultaneous increase in cell stiffness and invasion since they are involved in force generation. Our findings reveal that the activity of myosin II, Arp2/3, and formins contribute to the increased stiffness of PDAC cell lines; this may result from increased actomyosin contractility, actin polymerization and branching, and/or remodeling of the actin cytoskeleton. Indeed, the activity of Arp2/35 and myosin II44,45 both contribute to the mechanical properties of reconstituted actin networks. Our observation that myosin II, Arp2/3, and formin activity contribute to cell stiffness are also consistent with previous in vitro studies of cancer cell lines.9,28,95 For example, blebbistatin treatment of IGROV and Ovca429TβRIII ovarian cancer cell lines reduce cell stiffness by ~ 50%.95 Since myosin II, Arp2/3, and formins are also essential crosslinkers and mediators of actin nucleation and branching, the decrease in cell stiffness that we observe when these components are inhibited could also result from a restructured actin cytoskeleton.
While inhibition of myosin II, Arp2/3, and formins have consistent effects on decreasing cell stiffness across Hs766T and PANC-1 cells, we discovered that inhibiting these proteins have varying effects on the invasion of different cell lines. We find that myosin II activity is a major contributor to PANC-1 cell invasion, while Hs766T cells are more dependent on the activity of Arp2/3 and formins. Our findings suggest that different pancreatic cancer cell lines utilize different strategies for invasion. It is plausible that these disparities stem from genetic differences, which may influence the response of cancer cells to pharmacologic inhibitors.6,98 The three cancer cell lines that we investigate here—Hs766T, MIA PaCa-2, and PANC-1—are all pancreatic ductal epithelial cells that have undergone oncogenic transformation and have similar tissue origins but different genetic backgrounds. While all three cell lines carry common founder mutations in KRAS, TP53, and CDKN2A, the Hs766T cells have an additional SMAD4 mutation, which is characteristic of higher grade lesions.17SMAD4 encodes for a transcription factor that mediates signaling of the transforming growth factor (TGF)-β superfamily of proteins.98 Loss of Smad4 function in PDAC cells prevents TGF-β-mediated suppression of EGFR promoter activity, triggering a signaling cascade that leads to increased invasion.119 Indeed, EGF-induced ERK activation regulates protrusion formation through the WAVE2 regulatory complex, which then activates Arp2/3.13 It is intriguing to speculate that the Arp2/3-mediated invasion of Hs766T cells results from the loss of Smad4 activity. Indeed, the PANC-1 and MIA PaCa-2 cell lines have more similar genetic background and their invasion depends more strongly on myosin II activity. Our observations are aligned with previous findings that show cell line-specific differences in the influence of the Arp2/3 complex on the migration of pancreatic cancer cells.75 Future studies investigating the relationship between genotype and phenotype across larger numbers of cell lines could enhance our ability to predict treatment response based on a cell’s genotype. For example, our findings that Hs766T invasion is not affected by inhibition of myosin II could have consequences for treatments with inhibitors of Rho and ROCK, which have downstream effects on myosin II activity through a Rho-ROCK-NMII axis. Clinical trials with Fasudil show promise for patient benefit106 and preclinical studies reveal that Rho inhibitors reduce cancer cell invasion in vitro.50,53 Thus, our findings that inhibition of myosin II does not reduce invasion of all cancer cell types could have clinical relevance. A deeper knowledge of genome–phenome relationships, including which specific mutations are associated with specific mechanisms of force generation across cancer subtypes, could guide personalized treatments, in which drug combinations could be targeted to patients based on their genetic profile.
The differential effects of drug treatments on the invasion and stiffness across PDAC cell lines may also be explained by differences in the expression of genes and proteins that contribute to invasion and stiffness.11,117 To determine if Hs766T cells have differential expression of genes encoding key proteins that are essential in cell invasion and stiffness, we analyzed existing RNA-seq data.3 While Hs766T cells exhibited increased MMP expression and activity, we found no differences in invasion with MMP inhibition suggesting that Hs766T cells do not invade more quickly due to their increased MMP levels. Future work will provide more detailed insight into the relationship between MMP expression and the invasion of PDAC cells using knockdown and overexpression of specific MMP-encoding genes. The observed insensitivity of PDAC invasion to MMP inhibitors may also be due to the role of other proteases such as cathepsins, which are highly and specifically expressed in PDAC,33 and could also contribute to the observed invasion. Since the forces that cells generate are critical for cells to invade and deform through a protein matrix, we also investigated expression levels of myosin IIA and IIB, which contribute to cell invasion. Hs766T cells had the highest expression of MYH9 and MYH10, but the PANC-1 cells exhibit faster cell rounding and exert a greater magnitude of traction forces, which are both indicators of increased myosin-II dependent force generation. Our previous analysis of gene expression and protein levels of beta- and F-actin also did not reveal any distinct trends that explain the enhanced invasion of Hs766T cells.61 These discrepancies between expression levels and cellular physical phenotypes highlight how such complex physical properties are challenging to fully explain by analysis of genetics or transcriptomics data alone. Deformability and contractility are emergent phenotypes that are determined by proteins, their higher-order structures, and dynamic remodeling processes. A deeper understanding of the molecules and pathways that regulate cell physical phenotypes, together with more sophisticated expression analysis, should make progress towards predicting physical phenotypes and ultimately cancer cell invasion and tumor progression.
Measurement Effects
In addition to differences in the molecular machinery that influences stiffness and invasion, it is important to acknowledge how measurements of cell mechanotype and invasion may be affected by the experimental method. Measurements of cell stiffness can vary depending on the time and length scale of deformation, as well as the adhesion state of cells. In this study, we use AFM to measure the stiffness of cells that are adhered to a Matrigel-coated, glass surface, revealing that the stiffest to most compliant cells are Hs766T > PANC-1 > MIA PaCa-2. This relationship may differ if cell stiffness is measured using a different method. For example, we previously measured these same cells using the microfluidic-based method, quantitative deformability cytometry (q-DC),61,63 and found that the ranking of cell stiffness from stiffest to most compliant was PANC-1 > Hs766T > MIA PaCa-2. There are differences in the time and length scales of deformations between AFM and q-DC—nanometer-scale deformations over seconds by AFM versus. micron-scale deformations over milliseconds in q-DC—which could impact cell stiffness measurements.63 Furthermore, cells are in a suspended state for q-DC and adhered for AFM, and there are clear differences in the intrinsic and extrinsic factors that determine cell mechanotype when cells are in an adhered versus suspended state. For example, when cells are adhered to their substrate via integrins and focal adhesions, the contractile forces they generate are transduced to the matrix, and bidirectional feedback with matrix stiffness can drive contractile force generation.58,90 Increased myosin II activity subsequently increases cell stiffness and invasion of cancer cells.69 Indeed, cells with increased metastatic potential tend to exert higher levels of traction stresses.47 The degree of cell spreading, which is influenced by Arp2/3 activity,34 could also affect the stiffness of adhered cells. Fluidic methods, such as q-DC, measure cells in suspension, a state in which cortical actin82 and the nucleus32,81 are primary contributors to the deformation of cells through micron-scale pores. The mechanotype of suspended cells is also sensitive to myosin II activity,9,10 suggesting that dynamic remodeling of the cytoskeleton may contribute to the stiffness of cells in a suspended state. Since metastasis requires cancer cells to both invade on solid substrates, such as through the extracellular matrix, and circulate in a suspended state, comparisons of the same cell types using multiple, complementary methods can provide a more integrated understanding of cancer cell physical properties and their impact on disease progression.
Cell invasion measurements may also depend on factors specific to the invasion assay. Here we measured invasion through a Matrigel matrix in a 3D scratch wound assay. Matrigel is rich in laminin and collagen IV, and therefore, has been widely used as a model to study invasion through basement membranes, such as those of the endothelium that cancer cells are required to penetrate in order to seed a metastatic site.38 We cannot exclude that Matrigel might trigger a different invasion mode for Hs766T compared to PANC-1 cells. For example, both Rho/ROCK-dependent and -independent modes of invasion are determined by the spatial organization of surrounding collagen fibers.70 Therefore, the invasion of these cells in a different matrix, such as collagen, which mimics interstitial stromal extracellular matrix,27 could trigger cancer cells to use a different invasion strategy. Considering that PDAC progression also requires cells to invade through the confines of stromal interstitital matrices, future studies of the invasion of PDAC cells through collagen I, which is a major component of the tumor stroma, may capture other behaviors of PDAC cells. Indeed different invasion behaviors have been observed for ovarian cancer cells in a matrix of collagen I versus Matrigel.89 The matrix composition can also impact the role of MMPs in invasion, as cell–matrix adhesions regulate MMP expression21,22,97 and MMP activity may differentially contribute to cell invasion depending on the matrix material.89 The role of MMPs in PDAC invasion also appears to depend on cell type: while we did not observe any effects of MMP inhibition on the invasion of PANC-1, MiaPaCa-2, or Hs766T cells, GM6001 treatment blocked the invasion of the related PDAC cell line, AsPC1.83,92 Cells also adapt distinct invasion modes that utilize different molecular machineries depending on whether the invasion is collective or individual.79 Here we use a 3D scratch wound invasion assay, where cell invasion is measured by the collective movement of cells. While we have observed similar invasion results with 3D scratch wound and transwell migration assays,43 previous reports cite differences in the invasion of cancer cells using transwell, scratch wound, 3D spheroid, and in vivo assays.62,89 In the complex tumor microenvironment, fibroblasts, mast cells, and other cell types produce a number of molecules including cytokines, MMPs, and growth factors—such as vascular endothelial growth factor VEGF and basic fibroblast growth factor bFGF12,87,92—which additionally contribute to PDAC progression.38 Future studies of PDAC invasion across different matrix materials and conditions will provide a more detailed understanding of the context-dependence of PDAC invasion, which could ultimately inform therapeutic interventions.
Context-Dependent Relationship Between Cell Stiffness and Invasion
Cellular mechanotype is emerging as a potential complementary biomarker for cancer diagnosis and prognosis. While many studies reveal that more invasive cells tend to be more compliant,15,25,56,112 the positive correlation that we observe between PDAC cell stiffness and invasion is aligned with complementary findings in other cell types, including in lung cancer cells with varying metastatic potential,116 as well as in breast cancer cells treated with agonists to activate beta-adrenergic signaling.43,61,74,116 We previously found a strong correlation between the stiffness and invasion of PANC1, Hs766T, and MIA PaCa-2 cells.61 In this study, we find only a weak correlation between the stiffness and invasion of PDAC cells treated with inhibitors of myosin II (blebbistatin), Arp2/3 (CK-666), or formins (SMIFH2); this suggests that the relationship between stiffness and invasion of cells induced by pharmacologic perturbations may be difficult to predict. A combination of physical phenotypes, such as deformability, contractility, and adhesion, rather than single parameters, could further enhance the use of physical phenotypes as indicators of invasion or metastatic potential. For example, multivariate analysis of physical phenotypes may enhance the ability to predict functional behaviors, such as invasion.64
Conclusion
Our analysis of the relationship between cell mechanotype and invasion across PDAC cells with pharmacologic perturbations of actin and myosin provides deeper insights into mechanisms of cancer cell invasion. The cell-type specific effects of drugs on invasion that we observe reinforces the notion that different cells—even those derived from similar tissue origin—use different strategies for invasion. Given the genetic heterogeneity of cells within a single tumor and across different patients, as well as the phenotypic variability of isogenic cells, a better understanding of clinically relevant phenotypes such as invasion, as well as genotype–phenotype relationships should strengthen clinical strategies to develop therapies that target distinct mechanisms of cell invasion and metastasis.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Electronic supplementary material 1 (PDF 342 kb)
Acknowledgments
We thank our funding sources: the National Science Foundation (CAREER DBI-1254185 and BMMB-1906165 to ACR), the Farber Family Foundation, and UCLA Integrative Biology & Physiology Eureka Scholarship (to AVN), and the National Institutes of Health (R01 GM110482 to MJB). We would also like to thank Timothy Donahue and his laboratory for their insights into PDAC, as well as their generous contributions of the PDAC cell lines used in our studies. We are also grateful to Gordon Robertson and Ewan Gibb for their bioinformatics expertise. The MMP activity assay was performed in the UCLA Molecular Shared Screening Resource in the California NanoSystems Institute with technical support from Robert Damoiseaux and Bobby Tofig.
Conflict of interest
Angelyn V. Nguyen, Brittany Trompetto, Xing Haw Marvin Tan, Michael B. Scott, Kenneth Hsueh-heng Hu, Eric Deeds, Manish J. Butte, Pei Yu Chiou, and Amy C. Rowat have no conflicts of interest.
Ethical standards
No human or animals studies were carried out by the authors for this article.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ali MH, Pearlstein DP, Mathieu CE, Schumacker PT, Mir H, Paul T. Mitochondrial requirement for endothelial responses to cyclic strain: implications for mechanotransduction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;60637:486–496. doi: 10.1152/ajplung.00389.2003. [DOI] [PubMed] [Google Scholar]
- 2.Arjonen A, et al. Mutant p53–associated myosin-X upregulation promotes breast cancer invasion and metastasis. J. Clin. Invest. 2014;124:1069–1082. doi: 10.1172/JCI67280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beadle C, Assanah MC, Monzo P, Vallee R, Rosenfeld SS, Canoll P. The role of myosin II in glioma invasion of the brain. Mol. Biol. Cell. 2008;19:3357–3368. doi: 10.1091/mbc.E08-03-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bieling P, et al. Force feedback controls motor activity and mechanical properties of self-assembling branched actin networks. Cell. 2016;164:115–127. doi: 10.1016/j.cell.2015.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bronte G, et al. Driver mutations and differential sensitivity to targeted therapies: a new approach to the treatment of lung adenocarcinoma. Cancer Treat. Rev. 2010;36(Suppl 3):S21–S29. doi: 10.1016/S0305-7372(10)70016-5. [DOI] [PubMed] [Google Scholar]
- 7.Burridge K, Guilluy C. Focal adhesions, stress fibers and mechanical tension. Exp. Cell Res. 2016;343:14–20. doi: 10.1016/j.yexcr.2015.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calzado-Martín A, Encinar M, Tamayo J, Calleja M, San Paulo A. Effect of actin organization on the stiffness of living breast cancer cells revealed by peak-force modulation atomic force microscopy. ACS Nano. 2016;10:3365–3374. doi: 10.1021/acsnano.5b07162. [DOI] [PubMed] [Google Scholar]
- 9.Cartagena-Rivera AX, Logue JS, Waterman CM, Chadwick RS. Actomyosin cortical mechanical properties in nonadherent cells determined by atomic force microscopy. Biophys. J. 2016;110:2528–2539. doi: 10.1016/j.bpj.2016.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan CJ, et al. Myosin II activity softens cells in suspension. Biophys. J. 2015;108:1856–1869. doi: 10.1016/j.bpj.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan CK, et al. Tumour-suppressor microRNAs regulate ovarian cancer cell physical properties and invasive behaviour. Open Biol. 2016;6:160275. doi: 10.1098/rsob.160275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang DZ. Mast cells in pancreatic ductal adenocarcinoma. OncoImmunology. 2012;1:754–755. doi: 10.4161/onci.19612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y-W, et al. SMAD4 Loss triggers the phenotypic changes of pancreatic ductal adenocarcinoma cells. BMC Cancer. 2014;14:181. doi: 10.1186/1471-2407-14-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chin L, Xia Y, Discher DE, Janmey PA. Mechanotransduction in cancer. Curr. Opin. Chem. Eng. 2016;11:77–84. doi: 10.1016/j.coche.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cross SE, Jin Y-S, Rao J, Gimzewski JK. Nanomechanical analysis of cells from cancer patients. Nat. Nanotechnol. 2007;2:780–783. doi: 10.1038/nnano.2007.388. [DOI] [PubMed] [Google Scholar]
- 16.Cuadrado A, Martin-Moldes Z, Ye J, Lastres-Becker I. Transcription factors NRF2 and NF-κB are coordinated effectors of the rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014;289:15244–15258. doi: 10.1074/jbc.M113.540633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deer EL, et al. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425–435. doi: 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denais CM, et al. Nuclear envelope rupture and repair during cancer cell migration. Science. 2016;352:353–358. doi: 10.1126/science.aad7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duxbury MS, Ashley SW, Whang EE. Inhibition of pancreatic adenocarcinoma cellular invasiveness by blebbistatin: a novel myosin II inhibitor. Biochem. Biophys. Res. Commun. 2004;313:992–997. doi: 10.1016/j.bbrc.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 21.Ellerbroek SM, Wu YI, Overall CM, Stack MS. Functional interplay between type I collagen and cell surface matrix metalloproteinase activity. J. Biol. Chem. 2001;276:24833–24842. doi: 10.1074/jbc.M005631200. [DOI] [PubMed] [Google Scholar]
- 22.Ellerbroek SM, et al. Ovarian carcinoma regulation of matrix metalloproteinase-2 and membrane type 1 matrix metalloproteinase through beta1 integrin. Cancer Res. 1999;59:1635–1641. [PubMed] [Google Scholar]
- 23.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 24.Even-Ram S, Doyle AD, Conti MA, Matsumoto K, Adelstein RS, Yamada KM. Myosin IIA regulates cell motility and actomyosin–microtubule crosstalk. Nat. Cell Biol. 2007;9:299–309. doi: 10.1038/ncb1540. [DOI] [PubMed] [Google Scholar]
- 25.Faria EC, et al. Measurement of elastic properties of prostate cancer cells using AFM. Analyst. 2008;133:1498. doi: 10.1039/b803355b. [DOI] [PubMed] [Google Scholar]
- 26.Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature. 2010;463:485–492. doi: 10.1038/nature08908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J. Cell Sci. 2010;123:4195–4200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fritzsche M, Erlenkämper C, Moeendarbary E, Charras G, Kruse K. Actin kinetics shapes cortical network structure and mechanics. Sci. Adv. 2016;2:e1501337. doi: 10.1126/sciadv.1501337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gardberg M, et al. FHOD1, a formin upregulated in epithelial-mesenchymal transition, participates in cancer cell migration and invasion. PLoS ONE. 2013;8:e74923. doi: 10.1371/journal.pone.0074923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gardel ML, Schneider IC, Aratyn-Schaus Y, Waterman CM. Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 2010;26:315–333. doi: 10.1146/annurev.cellbio.011209.122036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goley ED, Welch MD. The ARP2/3 complex: an actin nucleator comes of age. Nat. Rev. Mol. Cell Biol. 2006;7:713–726. doi: 10.1038/nrm2026. [DOI] [PubMed] [Google Scholar]
- 32.Harada T, et al. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 2014;204:669–682. doi: 10.1083/jcb.201308029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harsha HC, et al. A compendium of potential biomarkers of pancreatic cancer. PLoS Med. 2009;6:e1000046. doi: 10.1371/journal.pmed.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henson JH, et al. Arp2/3 complex inhibition radically alters lamellipodial actin architecture, suspended cell shape, and the cell spreading process. Mol. Biol. Cell. 2015;26:887–900. doi: 10.1091/mbc.E14-07-1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hetrick B, Han MS, Helgeson LA, Nolen BJ. Small molecules CK-666 and CK-869 inhibit actin-related protein 2/3 complex by blocking an activating conformational change. Chem. Biol. 2013;20:701–712. doi: 10.1016/j.chembiol.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaqaman K, Grinstein S. Regulation from within: the cytoskeleton in transmembrane signaling. Trends Cell Biol. 2012;22:515–526. doi: 10.1016/j.tcb.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jimenez Valencia AM, et al. Collective cancer cell invasion induced by coordinated contractile stresses. Oncotarget. 2015;6:43438–43451. doi: 10.18632/oncotarget.5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer. 2003;3:422–433. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- 39.Katt ME, Placone AL, Wong AD, Xu ZS, Searson PC. In vitro tumor models: advantages, disadvantages, variables, and selecting the right platform. Front. Bioeng. Biotechnol. 2016;4:12. doi: 10.3389/fbioe.2016.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim S, Coulombe PA. Emerging role for the cytoskeleton as an organizer and regulator of translation. Nat. Rev. Mol. Cell Biol. 2010;11:75–81. doi: 10.1038/nrm2818. [DOI] [PubMed] [Google Scholar]
- 41.Kim H-C, Jo Y-J, Kim N-H, Namgoong S. Small molecule inhibitor of formin homology 2 domains (SMIFH2) reveals the roles of the formin family of proteins in spindle assembly and asymmetric division in mouse oocytes. PLoS ONE. 2015;10:e0123438. doi: 10.1371/journal.pone.0123438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim T-H, Rowat AC, Sloan EK. Neural regulation of cancer: from mechanobiology to inflammation. Clin. Transl. Immunol. 2016;5:e78. doi: 10.1038/cti.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim T-H, et al. Cancer cells become less deformable and more invasive with activation of β-adrenergic signaling. J. Cell Sci. 2016;129:4563–4575. doi: 10.1242/jcs.194803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koenderink GH, et al. An active biopolymer network controlled by molecular motors. Proc. Natl. Acad. Sci. USA. 2009;106:15192–15197. doi: 10.1073/pnas.0903974106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Köhler S, Bausch AR, Welch M, Peloquin J, Svitkina T. Contraction mechanisms in composite active actin networks. PLoS ONE. 2012;7:e39869. doi: 10.1371/journal.pone.0039869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kovács M, Tóth J, Hetényi C, Málnási-Csizmadia A, Sellers JR. Mechanism of blebbistatin inhibition of myosin II. J. Biol. Chem. 2004;279:35557–35563. doi: 10.1074/jbc.M405319200. [DOI] [PubMed] [Google Scholar]
- 47.Kraning-Rush CM, Califano JP, Reinhart-King CA. Cellular traction stresses increase with increasing metastatic potential. PLoS ONE. 2012;7:e32572. doi: 10.1371/journal.pone.0032572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumar S, Weaver VM. Mechanics, malignancy, and metastasis: the force journey of a tumor cell. Cancer Metastasis Rev. 2009;28:113–127. doi: 10.1007/s10555-008-9173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laevsky G, Knecht DA. Cross-linking of actin filaments by myosin II is a major contributor to cortical integrity and cell motility in restrictive environments. J. Cell Sci. 2003;116:3761–3770. doi: 10.1242/jcs.00684. [DOI] [PubMed] [Google Scholar]
- 50.Liu S, Goldstein RH, Scepansky EM, Rosenblatt M. Inhibition of rho-associated kinase signaling prevents breast cancer metastasis to human bone. Cancer Res. 2009;69:8742–8751. doi: 10.1158/0008-5472.CAN-09-1541. [DOI] [PubMed] [Google Scholar]
- 51.Lopez JI, Kang I, You W-K, McDonald DM, Weaver VM. In situ force mapping of mammary gland transformation. Integr. Biol. 2011;3:910. doi: 10.1039/c1ib00043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maly IV, Domaradzki TM, Gosy VA, Hofmann WA. Myosin isoform expressed in metastatic prostate cancer stimulates cell invasion. Sci. Rep. 2017;7:8476. doi: 10.1038/s41598-017-09158-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matsubara M, Bissell MJ. Inhibitors of Rho kinase (ROCK) signaling revert the malignant phenotype of breast cancer cells in 3D context. Oncotarget. 2016;7:31602–31622. doi: 10.18632/oncotarget.9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell. 2004;6:483–495. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 55.Mendez MG, Kojima S-I, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010;24:1838–1851. doi: 10.1096/fj.09-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mey I, Janshoff A, Rother J, No H. Atomic force microscopy-based microrheology reveals significant differences in the viscoelastic response between malign and benign cell lines. Open Biol. 2014;4:140046. doi: 10.1098/rsob.140046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mierke C, Rosel D, Fabry B, Brabek J. Contractile forces in tumor cell migration. Eur. J. Cell Biol. 2008;87:669–676. doi: 10.1016/j.ejcb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mih JD, Marinkovic A, Liu F, Sharif AS, Tschumperlin DJ. Matrix stiffness reverses the effect of actomyosin tension on cell proliferation. J. Cell Sci. 2012;125:5974–5983. doi: 10.1242/jcs.108886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murrell M, Oakes PW, Lenz M, Gardel ML. Forcing cells into shape: the mechanics of actomyosin contractility. Nat. Rev. Mol. Cell Biol. 2015;16:486–498. doi: 10.1038/nrm4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakayama M, et al. Rho-kinase and myosin II activities are required for cell type and environment specific migration. Genes Cells. 2005;10:107–117. doi: 10.1111/j.1365-2443.2005.00823.x. [DOI] [PubMed] [Google Scholar]
- 61.Nguyen AV, et al. Stiffness of pancreatic cancer cells is associated with increased invasive potential. Integr. Biol. 2016;8:1232–1245. doi: 10.1039/c6ib00135a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Noël AC, et al. Invasion of reconstituted basement membrane matrix is not correlated to the malignant metastatic cell phenotype. Cancer Res. 1991;51:405–414. [PubMed] [Google Scholar]
- 63.Nyberg KD, Hu KH, Kleinman SH, Khismatullin DB, Butte MJ, Rowat AC. Quantitative deformability cytometry (q-DC): rapid measurements of single cell viscoelastic properties. Biophys. J. 2017;113:1574–1584. doi: 10.1016/j.bpj.2017.06.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nyberg KD, et al. Predicting cancer cell invasion by single-cell physical phenotyping. Integr. Biol. 2018;10:218–231. doi: 10.1039/c7ib00222j. [DOI] [PubMed] [Google Scholar]
- 65.Ouderkirk JL, Krendel M. Non-muscle myosins in tumor progression, cancer cell invasion, and metastasis. Cytoskeleton. 2014;71:447–463. doi: 10.1002/cm.21187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krakhmal NV, Zavyalova MV, Denisov EV, Vtorushin SV, Perelmuter VM. Cancer invasion: patterns and mechanisms. Acta Nat. 2015;7:17–28. [PMC free article] [PubMed] [Google Scholar]
- 68.Plodinec M, et al. The nanomechanical signature of breast cancer. Nat. Nanotechnol. 2012;7:757–765. doi: 10.1038/nnano.2012.167. [DOI] [PubMed] [Google Scholar]
- 69.Poincloux R, et al. Contractility of the cell rear drives invasion of breast tumor cells in 3D Matrigel. Proc. Natl. Acad. Sci. USA. 2011;108:1943–1948. doi: 10.1073/pnas.1010396108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Provenzano PP, Inman DR, Eliceiri KW, Trier SM, Keely PJ. Contact guidance mediated three-dimensional cell migration is regulated by Rho/ROCK-dependent matrix reorganization. Biophys. J. 2008;95:5374–5384. doi: 10.1529/biophysj.108.133116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pruyne D, et al. Role of formins in actin assembly: nucleation and barbed-end association. Science. 2002;297:612–615. doi: 10.1126/science.1072309. [DOI] [PubMed] [Google Scholar]
- 72.Raab M, et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science. 2016;352:359–362. doi: 10.1126/science.aad7611. [DOI] [PubMed] [Google Scholar]
- 73.Rasheed ZA, Matsui W, Maitra A. Pathology of pancreatic stroma in PDAC. In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment. Trivandrum: Transworld Research Network; 2012. [PubMed] [Google Scholar]
- 74.Rathje L-SZ, et al. Oncogenes induce a vimentin filament collapse mediated by HDAC6 that is linked to cell stiffness. Proc. Natl. Acad. Sci. USA. 2014;111:1515–1520. doi: 10.1073/pnas.1300238111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rauhala HE, Teppo S, Niemelä S, Kallioniemi A. Silencing of the ARP2/3 complex disturbs pancreatic cancer cell migration. Anticancer Res. 2013;33:45–52. [PubMed] [Google Scholar]
- 76.Revach O-Y, Weiner A, Rechav K, Sabanay I, Livne A, Geiger B. Mechanical interplay between invadopodia and the nucleus in cultured cancer cells. Sci. Rep. 2015;5:9466. doi: 10.1038/srep09466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ridley AJ. RhoA, RhoB and RhoC have different roles in cancer cell migration. J. Microsc. 2013;251:242–249. doi: 10.1111/jmi.12025. [DOI] [PubMed] [Google Scholar]
- 78.Rizvi SA, et al. Identification and characterization of a small molecule inhibitor of formin-mediated actin assembly. Chem. Biol. 2009;16:1158–1168. doi: 10.1016/j.chembiol.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rodriguez-Hernandez I, Cantelli G, Bruce F, Sanz-Moreno V. Rho, ROCK and actomyosin contractility in metastasis as drug targets. F1000Research. 2016;5:783. doi: 10.12688/f1000research.7909.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rowat AC, Lammerding J, Herrmann H, Aebi U. Towards an integrated understanding of the structure and mechanics of the cell nucleus. BioEssays. 2008;30:226–236. doi: 10.1002/bies.20720. [DOI] [PubMed] [Google Scholar]
- 81.Rowat AC, et al. Nuclear envelope composition determines the ability of neutrophil-type cells to passage through micron-scale constrictions. J. Biol. Chem. 2013;288:8610–8618. doi: 10.1074/jbc.M112.441535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Salbreux G, Charras G, Paluch E. Actin cortex mechanics and cellular morphogenesis. Trends Cell Biol. 2012;22:536–545. doi: 10.1016/j.tcb.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 83.Sato N, Maehara N, Su GH, Goggins M. Effects of 5-Aza-2’-deoxycytidine on matrix metalloproteinase expression and pancreatic cancer cell invasiveness. J. Natl. Cancer Inst. 2003;95:327–330. doi: 10.1093/jnci/95.4.327. [DOI] [PubMed] [Google Scholar]
- 84.Sen S, Kumar S. Cell-matrix de-adhesion dynamics reflect contractile mechanics. Cell. Mol. Bioeng. 2009;2:218–230. doi: 10.1007/s12195-009-0057-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shi Q, et al. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol. Carcinog. 2007;46:488–496. doi: 10.1002/mc.20297. [DOI] [PubMed] [Google Scholar]
- 86.Shields MA, Dangi-Garimella S, Krantz SB, Bentrem DJ, Munshi HG. Pancreatic cancer cells respond to type I collagen by inducing snail expression to promote membrane type 1 matrix metalloproteinase-dependent collagen invasion. J. Biol. Chem. 2011;286:10495–10504. doi: 10.1074/jbc.M110.195628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sipos B, et al. Vascular endothelial growth factor mediated angiogenic potential of pancreatic ductal carcinomas enhanced by hypoxia: an in vitro and in vivo study. Int. J. Cancer. 2002;102:592–600. doi: 10.1002/ijc.10753. [DOI] [PubMed] [Google Scholar]
- 88.Smith BA, Tolloczko B, Martin JG, Grütter P. Probing the viscoelastic behavior of cultured airway smooth muscle cells with atomic force microscopy: stiffening induced by contractile agonist. Biophys. J. 2005;88:2994–3007. doi: 10.1529/biophysj.104.046649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sodek KL, Brown TJ, Ringuette MJ. Collagen I but not Matrigel matrices provide an MMP-dependent barrier to ovarian cancer cell penetration. BMC Cancer. 2008;8:223. doi: 10.1186/1471-2407-8-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Southern BD, et al. Matrix-driven myosin II mediates the pro-fibrotic fibroblast phenotype. J. Biol. Chem. 2016;291:6083–6095. doi: 10.1074/jbc.M115.712380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stossel TP, Hartwig JH. Filling gaps in signaling to actin cytoskeletal remodeling. Dev. Cell. 2003;4:444–445. doi: 10.1016/s1534-5807(03)00098-4. [DOI] [PubMed] [Google Scholar]
- 92.Strouch MJ, et al. Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin. Cancer Res. 2010;16:2257–2265. doi: 10.1158/1078-0432.CCR-09-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Suraneni P, Rubinstein B, Unruh JR, Durnin M, Hanein D, Li R. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J. Cell Biol. 2012;197:239–251. doi: 10.1083/jcb.201112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Suraneni P, et al. A mechanism of leading-edge protrusion in the absence of Arp2/3 complex. Mol. Biol. Cell. 2015;26:901–912. doi: 10.1091/mbc.E14-07-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Swaminathan V, Mythreye K, Tim O’Brien E, Berchuck A, Blobe GC, Superfine R. Mechanical stiffness grades metastatic potential in patient tumor cells and in cancer cell lines. Cancer Res. 2011;71:5075–5080. doi: 10.1158/0008-5472.CAN-11-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Swift J, et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341:1240104. doi: 10.1126/science.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Symowicz J, et al. Engagement of collagen-binding integrins promotes matrix metalloproteinase-9-dependent E-cadherin ectodomain shedding in ovarian carcinoma cells. Cancer Res. 2007;67:2030–2039. doi: 10.1158/0008-5472.CAN-06-2808. [DOI] [PubMed] [Google Scholar]
- 98.Takai E, Yachida S. Genomic alterations in pancreatic cancer and their relevance to therapy. World J. Gastrointest. Oncol. 2015;7:250–258. doi: 10.4251/wjgo.v7.i10.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS. Cells lying on a bed of microneedles: An approach to isolate mechanical force. Proc. Natl. Acad. Sci. USA. 2003;100:1484–1489. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tse HTK, et al. Quantitative diagnosis of malignant pleural effusions by single-cell mechanophenotyping. Sci. Transl. Med. 2013;5:212ra163. doi: 10.1126/scitranslmed.3006559. [DOI] [PubMed] [Google Scholar]
- 102.Tseng Y, et al. How actin crosslinking and bundling proteins cooperate to generate an enhanced cell mechanical response. Biochem. Biophys. Res. Commun. 2005;334:183–192. doi: 10.1016/j.bbrc.2005.05.205. [DOI] [PubMed] [Google Scholar]
- 103.Unbekandt M, et al. A novel small-molecule MRCK inhibitor blocks cancer cell invasion. Cell Commun. Signal. 2014;12:1–15. doi: 10.1186/s12964-014-0054-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vennin C, et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci. Transl. Med. 2017;9:eaai8504. doi: 10.1126/scitranslmed.aai8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang Z-M, Yang D-S, Liu J, Liu H-B, Ye M, Zhang Y-F. ROCK inhibitor Y-27632 inhibits the growth, migration, and invasion of Tca8113 and CAL-27 cells in tongue squamous cell carcinoma. Tumor Biol. 2016;37:3757–3764. doi: 10.1007/s13277-015-4115-6. [DOI] [PubMed] [Google Scholar]
- 106.Wei L, Surma M, Shi S, Lambert-Cheatham N, Shi J. Novel insights into the roles of rho kinase in cancer. Arch. Immunol. Ther. Exp. (Warsz) 2016;64:259–278. doi: 10.1007/s00005-015-0382-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Weng S, Shao Y, Chen W, Fu J. Mechanosensitive subcellular rheostasis drives emergent single-cell mechanical homeostasis. Nat. Mater. 2016;15:961–967. doi: 10.1038/nmat4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wirtz D, Konstantopoulos K, Searson PC. The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer. 2011;11:512–522. doi: 10.1038/nrc3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wolf K, et al. Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 2013;201:1069–1084. doi: 10.1083/jcb.201210152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xiao F, Wen X, Chiou PY. Plasmonic micropillars for massively parallel precision cell force measurement. Micro Electro Mech. Syst. 2017;1:243–246. [Google Scholar]
- 111.Xiao F, Wen X, Tan XHM, Chiou P-Y. Plasmonic micropillars for precision cell force measurement across a large field-of-view. Appl. Phys. Lett. 2018;112:033701. doi: 10.1063/1.5005525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xu W, Mezencev R, Kim B, Wang L, McDonald J, Sulchek T. Cell stiffness is a biomarker of the metastatic potential of ovarian cancer cells. PLoS ONE. 2012;7:e46609. doi: 10.1371/journal.pone.0046609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yachida S, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta. 2007;1773:642–652. doi: 10.1016/j.bbamcr.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ying H, et al. The Rho kinase inhibitor fasudil inhibits tumor progression in human and rat tumor models. Mol. Cancer Ther. 2006;5:2158–2164. doi: 10.1158/1535-7163.MCT-05-0440. [DOI] [PubMed] [Google Scholar]
- 116.Yu HW, et al. PIX controls intracellular viscoelasticity to regulate lung cancer cell migration. J. Cell Mol. Med. 2015;19:934–947. doi: 10.1111/jcmm.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zaidel-Bar R, Zhenhuan G, Luxenburg C. The contractome—a systems view of actomyosin contractility in non-muscle cells. J. Cell Sci. 2015;128:1–9. doi: 10.1242/jcs.170068. [DOI] [PubMed] [Google Scholar]
- 118.Zhang W, et al. Microfluidics separation reveals the stem-cell-like deformability of tumor-initiating cells. Proc. Natl. Acad. Sci. USA. 2012;109:18707–18712. doi: 10.1073/pnas.1209893109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhao S, Wang Y, Cao L, Ouellette MM, Freeman JW. Expression of oncogenic K-ras and loss of Smad4 cooperate to induce the expression of EGFR and to promote invasion of immortalized human pancreas ductal cells. Int. J. Cancer. 2010;127:2076–2087. doi: 10.1002/ijc.25412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhao X, et al. Hypoxia-Inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-Bundling protein fascin. Cancer Res. 2014;74:2455–2464. doi: 10.1158/0008-5472.CAN-13-3009. [DOI] [PubMed] [Google Scholar]
- 121.Zhou L, et al. Theoretical modeling of mechanical homeostasis of a mammalian cell under gravity-directed vector. Biomech. Model. Mechanobiol. 2018;17:191–203. doi: 10.1007/s10237-017-0954-y. [DOI] [PubMed] [Google Scholar]
- 122.Zhu F, et al. Rho kinase inhibitor fasudil suppresses migration and invasion though down-regulating the expression of VEGF in lung cancer cell line A549. Med. Oncol. 2011;28:565–571. doi: 10.1007/s12032-010-9468-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Electronic supplementary material 1 (PDF 342 kb)