

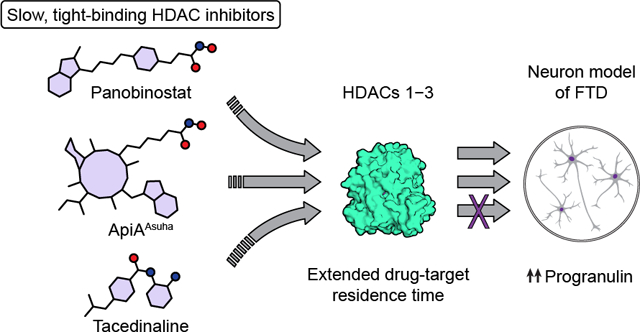
Abstract
Histone deacetylases (HDACs) are enzymes involved in the epigenetic control of gene expression. A handful of HDAC inhibitors have been approved for the treatment of cancer, and HDAC inhibition has also been proposed as a novel therapeutic strategy for neurodegenerative disorders. These disorders include progranulin (PGRN)-deficient forms of frontotemporal dementia caused by mutations in the GRN gene that lead to haploinsufficiency. Hydroxamic acid-based inhibitors of HDACs 1–3, reported to have fast-on/fast-off binding kinetics, induce increased expression of PGRN in human neuronal models, while the benzamide class of slow-binding HDAC inhibitors does not produce this effect. These observations indicate that the kinetics of HDAC inhibitor binding can be tuned for optimal induction of human PGRN expression in neurons. Here, we further expand on these findings using human cortical-like, glutamatergic neurons. We provide evidence that two prototypical, potent hydroxamic acid HDAC inhibitors that induce PGRN (panobinostat and trichostatin A) exhibit an initial fast-binding step followed by a second, slower step, referred to as mechanism B of slow binding, rather than simpler fast-on/fast-off binding kinetics. In addition, we show that trapoxin A, a macrocyclic, epoxyketone-containing class I HDAC inhibitor, exhibits slow binding with high, picomolar potency and also induces PGRN expression in human neurons. Finally, we demonstrate induction of PGRN expression by fast-on/fast-off, highly potent, macrocyclic HDAC inhibitors with ethyl ketone or ethyl ester Zn2+ binding groups. Taken together, these data expand our understanding of HDAC1–3 inhibitor binding kinetics, and further delineate the specific combinations of structural and kinetic features of HDAC inhibitors that are optimal for upregulating PGRN expression in human neurons and thus may have translational relevance in neurodegenerative disease.
Keywords: Epigenetic, Slow-binding inhibitor, Histone deacetylase, Progranulin, Kinetic profiling, Panobinostat
Graphical Abstract
INTRODUCTION
Epigenetic processes are responsible for gene activation and repression1. A complex and highly dynamic epigenetic mechanism is the reversible decoration of the side chains of histone proteins through covalent post-translational modifications (PTMs). These PTMs affect the interaction of histones with DNA and the recruitment of transcriptional machinery, and they can lead to changes between different chromatin condensation states. Residues in the N-terminal histone tail segments are continuously modified by enzymes such as lysine or arginine methyltransferases and lysine acetyltransferases, and reversely, the resulting PTMs are removed by demethylases and deacetylases. In addition, a subset of post-translational histone modifications are recognized by protein domains that recruit transcription factors among other effectors1. Lysine acetylation disrupts electrostatic interactions between histones and DNA, favoring transcription, and it is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs). Moreover, this modification is specifically recognized by bromodomain-containing proteins, which generally serve as transcriptional co-activators1. By altering the acetylation state of histones, as well as non-histone proteins, HDAC inhibitors can thus affect gene expression2. In particular, inhibitors of the Zn2+-dependent HDACs 1–3 (members of class I) have attracted interest since these are the most active deacetylases of histones both in vitro and in cells2, 3. HDACs 1–3 play a critical role in a number of nuclear multiprotein transcriptional repressor complexes with distinct biological roles3. Through affecting the deacetylase activity and the overall epigenetic state, inhibitors of HDACs 1–3 induce growth arrest and differentiation in tumor cells, and they reverse the effects of gene silencing and haploinsufficiency in neurodegenerative disorders1, 2.
Haploinsufficiency of the progranulin gene (GRN) has been identified as a leading cause of familial forms of frontotemporal dementia (FTD), the second most common type of presenile dementia after Alzheimer’s disease4. Even though the molecular pathological mechanisms are still unknown, loss-of-function mutations in one copy of the GRN gene leading to reduced progranulin (PGRN) expression have been shown to lead to ubiquitinated TAR DNA-binding protein 43 (TDP-43) accumulation that is characteristic of ~50% of neuropathologically confirmed cases of FTD4. Furthermore, PGRN restoration in FTD mouse models has proven beneficial for correcting abnormalities associated to the disease5–7. A library screen employing mouse neuronal cells and following studies in induced pluripotent stem cell (iPSC)-derived human neurons identified the broadly acting HDAC inhibitor SAHA (vorinostat, 1, Scheme 1) as an epigenetic promoter of PGRN expression8, 9. More detailed studies employing selective HDAC inhibitors have suggested HDACs 1–3 to be the target of SAHA (1) in relation to this phenotype, after ruling out potential contributions of HDAC6 and HDAC8 that are also inhibited by SAHA (1)10. These studies also gave rise to a proposed mechanism of action for HDAC inhibitor-induced PGRN expression involving enhanced H3K27 acetylation at the GRN promoter region and recruitment of the transcription factor EB (TFEB)10, which has also been identified as an acetylation target11. Most strikingly, not all inhibitors of HDACs 1–3 effectively led to increase in PGRN protein levels. In particular, the ortho-aminoanilides CI-994 (tacedinaline, 3) and Cpd-60 (4) were shown to fail to induce PGRN expression, despite their demonstrated effect on overall histone hyperacetylation in the same human neuronal cell culture system10. Intriguingly, this observation concerning o-aminoanilides was the opposite to that observed for the regulation of the FXN gene encoding frataxin10, 12, which was only enhanced by o-aminoanilides and not by hydroxamic acid-based HDAC inhibitors, thereby providing evidence for locus-selective epigenetic regulation of genes relevant to neurological disorders.
Scheme 1. Chemical structure of selected HDAC inhibitors.

(A) Small molecule and (B) peptide-based macrocyclic HDAC inhibitors, with numbers representing different scaffolds and letters indicating ethyl ketone (a), ethyl ester (b), hydroxamic acid (c) or excised (d) Zn2+-binding group.
HDAC inhibitors based on an o-aminoanilide chemotype have attracted significant attention due to potential selectivity towards a subset of HDAC-containing multiprotein complexes3, 13–15 and due to a general exhibition of slow-binding inhibition kinetics14, 16–19. In-depth characterization of the mechanism of action of traditional and recent drugs has brought to light the importance of such time dependency on inhibitory effects20. Most enzyme inhibitors have been thought to compete for the substrate at the time scale of enzymatic catalysis, reaching equilibrium and full inhibition rapidly in a fast-on/fast-off fashion20, 21. However, some enzyme–inhibitor systems, referred to as slow-binding, approach equilibrium gradually during minutes or even hours due to slow release of the inhibitor, which can be attractive for enhancing pharmacological effects through improved residence time22–25. A less commonly studied and often harder to detect mechanism referred to as mechanism B of slow binding is a hybrid between these two scenarios, with inhibitors exhibiting an initial fast-binding step followed by a second, slower binding step and slow release21, 22.
Kinetics of HDAC inhibition appears to play a role in the regulation of GRN, and the o-aminoanilide inhibitors tested so far were unable to induce PGRN production10. Here, we interrogate this observation using a focused collection of macrocyclic HDAC inhibitors with distinct and well-characterized inhibition kinetics, and we describe the ability of macrocycle-based inhibitors with slow-binding properties to promote PGRN expression. Furthermore, we report the surprising finding that the HDAC probes panobinostat (LBH-589, 5) and trichostatin A (TSA, S1, Scheme S1) exhibit slow, tight-binding kinetics, which is unprecedented amongst hydroxamic acid-containing small molecules.
RESULTS
SAHA (1, Scheme 1) is the most widely recognized example of a fast-on/fast-off HDAC inhibitor16, 18, 26, a mechanism tentatively extended to all structurally-related hydroxamic acids. In contrast, the o-aminoanilide Zn2+-binding group has been shown to endow small molecules with longer residence times against class I HDACs14, 16–19. This has also been observed for the trifluoromethyl ketone version of SAHA (SATK, 2)26. We recently reported on the kinetic effect of incorporating various Zn2+-binding groups onto the scaffold of naturally occurring macrocycles such as trapoxin B (7) and apicidin A (9a)27. Surprisingly, hydroxamic acid analogues TpxBAsuha (7c) and ApiAAsuha (9c) exhibited slow-binding kinetics against HDACs 1–3, 6, and 11 with Ki inhibitor constants similar or lower than the enzyme concentration employed (slow, tight-binding)27–29. Intrigued by these observations, we decided to explore the binding kinetics of hydroxamic acids smaller than tetrapeptide macrocycles, but with potencies higher than that of SAHA (1).
In brief, we considered the three kinetic models outlined in Figure 1, namely: 1) fast-on/fast-off inhibition (reaching equilibrium instantly), 2) mechanism A of slow-binding inhibition (with kobs rates of enzyme–inhibitor complex formation proportional to the concentration of inhibitor), and 3) mechanism B of slow-binding inhibition (with kobs fitting a hyperbolic function)22, 27. Slow-binding inhibitors with calculated Ki under the free enzyme concentration range were described as slow, tight-binding inhibitors29. First, we tested tacedinaline (3) as a slow-binding control compound. Unexpectedly, this revealed a mechanism B type of slow-binding inhibition with tight binding against HDACs 1 and 2, and nanomolar potency against HDAC3 (Figure 1C and Table 1). Interestingly, we then found panobinostat (5) and TSA (S1), widely used as HDAC probes30, to also follow slow, tight-binding inhibition profiles (mechanism B) against HDACs 1–3, and effectively irreversible behavior for panobinostat (5) against HDAC2 and for TSA (S1) against HDAC3 (see Tables 1 and S1 for numerical data and Figure S1 for graphical representation). The hydroxamic acid TSA (S1) has been suggested to exhibit slower dissociation rates than SAHA (1) in a reporter displacement assay, but the kinetics were not investigated in detail and mechanism B was not considered in this previous investigation18.
Figure 1. Representative examples of different kinetic mechanisms of HDAC inhibition.

Assay progression curves and secondary plots fitted to the relevant equation (see Supporting Information for more detail). (A) Fast-on/fast-off binding kinetics, exemplified by apicidin (6a), (B) mechanism A of slow-binding kinetics, exemplified by trapoxin A (4), and (C) mechanism B of slow-binding inhibition, exemplified by tacedinaline (3).
Table 1. Summary of the potency and mechanism of HDAC inhibitors, and of their effect on human PGRN and H3K9ac levels at 1 μM concentration.
Color shadings relate to kinetic mechanisms (cyan: fast- on/fast-off, orange: mechanism A, red: mechanism B of slow-binding kinetics).
| Compound | Mechanism of binding and potency (Ki(,1): nM, dis. t½: min) |
Effect on PGRN (1 μM, NPCs) |
Effect on H3K9ac (1 μM, NPCs) |
||
|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3* | |||
| 1 (SAHA) | Ki = 1.3 (± 0.05)30 | Ki = 1.6 (± 0.05)30 | Ki = 5.0 (± 0.2)30 | >2 fold incr.‡, 10 | >20 fold incr.‡, 10 |
| 2 (SATK) |
Ki = 9800 (± 6700)26
Dis. t½ = 41 (± 17) |
Ki = 11000 (± 4000)26
Dis. t½ = 87 (± 22) |
Ki, 1 = 570 (± 260)26
Ki ~1126 Dis. t½ ≥ 139 |
No change | No change |
| 3 (tacedinaline) |
Ki, 1 = 1450 (± 320) Ki ~1 Dis. t½ ≥ 1386 |
Ki, 1 = 2190 (± 510) Ki ~14 Dis. t½ ≥ 139 |
Ki, 1 = 3560 (± 400) Ki = 105 (± 86) Dis. t½ ≥ 50 |
Decrease‡ | ~18 fold incr‡ |
| 5 (panobinostat) |
Ki, 1 = 33 (± 26) Ki ~0.1 Dis. t½ ≥ 41 |
Ki, 1 = 4.7 (± 2.5) Dis. t½ > 105 |
Ki, 1 = 15 (± 5) Ki ~0.1 Dis. t½ ≥ 63 |
~2.3 fold incr. | ~36 fold incr. |
| 6 (trapoxin A) |
Ki ~0.003 Dis. t½ ~6931 |
Dis. t½ > 105 |
Ki, 1 = 0.59 (± 0.29)27
Dis. t½ > 105 |
~2.4 fold incr. | ~5.0 fold incr. |
| 7a (TpxBAoda) | Ki = 6.6 (± 0.5)27 | Ki = 3.9 (± 0.4)27 | Ki = 0.78 (± 0.05)27 | No change | ~2.8 fold incr. |
| 7b (TpxBAsu(Et))† | Ki = 500 (± 90)27 | Ki = 400 (± 20)27 | Ki = 500 (± 80)27 | No change | ~2.1 fold incr. |
| 7c (TpxBAsuha) |
Ki, 1 = 0.60 (±0.12)27
Dis. t½ > 105 |
Ki, 1 = 0.13 (±0.11 )27
Dis. t½ > 105 |
Ki, 1 = 1.4 (± 0.4)27
Ki ~0.0227 Dis. t½ ≥ 173 |
~2.2 fold incr. | ~6.3 fold incr. |
| 8a (apicidin) | Ki = 0.824 (± 0.003) | Ki = 0.619 (± 0.004) | Ki = 0.051 (± 0.002)27 | ~1.7 fold incr. | ~3.3 fold incr. |
| 9b (ApiAAsu(Et))† | Ki = 10 (± 1)27 | Ki = 11 (± 2)27 | Ki = 15 (± 3)27 | ~1.4 fold incr. | ~2.1 fold incr. |
| 9c (ApiAAsuha) |
Ki, 1 = 0.32 (± 0.06)27
Dis. t½ > 105 |
Ki, 1 = 0.35 (±0.10)27
Dis. t½ > 105 |
Ki, 1 = 1.8 (± 0.6)27 Ki ~0.0427 Dis. t½ ≥ 77 |
~2.5 fold incr. | ~6.4 fold incr. |
| 10 (romidepsin) |
Ki, 1 = 0.88 (± 0.18) Ki ~0.02 Dis. t½ ≥ 26 |
Ki, 1 = 0.47 (± 0.09) Ki ~0.009 Dis. t½ ≥ 41 |
Ki, 1 = 1.3 (± 0.4)27
Dis. t½ > 105 |
~4.1 fold incr. | ~2.3 fold incr. |
| 10d (RomiAla, Dha) | Ki = 690 (± 3)27 | Ki = 370 (± 50)27 | Ki = 800 (± 100)27 | No change | Decrease |
HDAC3 in combination with the DAD domain of NCoR2.
Inhibition data corresponding to the carboxylic acid hydrolyzed species (TpxBAsu and ApiAAsu, respectively)27.
Treatment performed at 10 μM concentration.
HDAC inhibition assay progression curves were recorded as in previous studies by addition of a small amount of trypsin developer (10 μg/mL) to the reaction mixture16, 26, 27, which does not affect the steady-state of the reaction (Figure S1). Here, it came to our attention that trypsin addition resulted in the cleavage of small C- and/or N-terminal fragments of the enzyme without affecting the catalytic domain (see Figure S2 and Methods)31. Gratifyingly, end-point experiments performed without trypsin in the reaction mixture afforded estimated progression curves similar to those recorded in continuous assays (Figure S3), which indicated that these terminal fragments are not required for catalysis or inhibitor binding.
In our previous findings on hydroxamic acid-containing macrocycles, irreversible behavior had only been observed for the natural product trapoxin A (6)32. This compound bears an epoxyketone Zn2+-binding group speculated to form a covalent bond with nucleophilic residues in the HDAC active site32, 33. However, recent co-crystal structures of trapoxin A (6) and of the related natural product HC-toxin (S2, Scheme S1) show that the epoxyketone moiety is intact upon binding to HDAC834 and to the C-terminal domain (CD2) of HDAC635, respectively. Moreover, washout experiments to look for reversibility have not generally been performed for extended time periods. We have previously observed that inhibition of HDAC3 by trapoxin A (6), as well as by HC-toxin (S2), follow a slow, tight-binding profile (mechanism B)27. We further reported mechanism B type of inhibition for HC-toxin (S2) against HDACs 1 and 227, and fast-on/fast-off kinetics against HDAC6 in agreement with the structural findings reported by Christianson and co-workers35. Now, we also report here observing mechanism A kinetics for trapoxin A (6) against HDACs 1 and 2. Interestingly, this distinct kinetic behavior of compounds comparing HDACs 1 and 2 vs HDAC3 has also been seen with SATK (3), and with the o-aminoanilide-containing inhibitor 106 (S3, Scheme S1) developed by Gottesfeld and co-workers16, 26. Lastly, apicidin (8a) and romidepsin (10) were tested against HDACs 1 and 2, confirming fast-on/fast-off binding kinetics for the former27 and slow, tight-binding (mechanism B) for the latter27, 36.
These kinetic data add to the understanding of HDAC inhibitor binding dynamics. Panobinostat (5), TpxBAsuha (7c), ApiAAsuha (9c), romidepsin (10), and TSA (S1) display dissociative half-life (dis. t½)22 over 20 min, even though they lack the steric characteristics of the o-aminoanilide Zn2+-binding group (see Tables 1 and S1). Thus, the structural basis for this extended residence time should be different from conformational changes in the ‘foot pocket’ of the active site, as observed for other substituted o-aminoanilides31, 37, 38. On the other hand, there is a marked difference between tacedinaline (3), as well as 106 (S3)16, versus the remaining mechanism B type of inhibitors in terms of the order of magnitude of the first and faster step equilibrium constant (Ki,1, Table 1). Both o-aminoanilides exhibit constants in the micromolar range16, whereas all hydroxamic acids and the thiol-containing depsipeptide romidepsin (10) reach nano- and sub-nanomolar Ki,1 values. This still discriminates o-aminoanilides from other Zn2+-binding groups, and it leaves open the possibility for kinetic constants k1 and k-1 to be orders of magnitude different for this compound class. In this regard, more complex and time resolved in vitro systems would be necessary for measuring k1 and k-1 rates for mechanism B type of inhibitors.
We then decided to investigate further our previous observations regarding the effect of binding kinetics of class I HDAC inhibitors on the epigenetic modulation of PGRN production. For this purpose, we adapted culture and compound treatment of human iPSC-derived neural progenitor cells (NPCs) and neurons in 96-well plates. This allowed the performance of simultaneous experiments in high-throughput and optimization of the use of commercially available PGRN ELISA to complement our Western blotting and qRT-PCR assays. Since HDAC inhibition was shown to furnish similar effects on both intracellular and excreted PGRN, only the former was considered for the current study10. While our previous studies on the effect of HDAC inhibitors on human iPSC-derived neurons used a method of neural differentiation involving growth factor withdrawal10, this is only one of many ways to differentiate human neurons and is known to produce a diversity of neuronal subtypes and glia. Here, we used an entirely different method of neuronal differentiation with directed differentiation to an enriched population of cortical-like, excitatory, glutamatergic neurons using induced expression of the bHLH proneural transcription factor Ngn2 (Figure S4)39. Since cortical neurons are the predominant cell type that degenerates in subjects with PGRN-deficient FTD, it renders our current studies, which demonstrate the ability of class I selective HDAC inhibitors to enhance PGRN expression, highly relevant as an ex vivo surrogate of human disease biology.
Previous data on tacedinaline (3), panobinostat (5) and apicidin (8a) were recapitulated (Figure 2A and B, and Table 1) although with a slight decrease in signal which we attribute to a shorter compound treatment (18 h vs 24 h) and the high density, 96-well plate culture format which may have impacted growth rates10. Macrocyclic ethyl esters TpxBAsu(Et) (7b) and ApiAAsu(Et) (9b), which follow fast-on/fast-off kinetic profiles, were able to induce PGRN in both NPCs and neurons as predicted based on their HDAC binding kinetics and potency27. Surprisingly, based upon the inability of the o-aminoanilides to enhance PGRN, we found that all mechanism A (trapoxin A, 6) and mechanism B (TpxBAsuha, 7c; ApiAAsuha, 9c; and romidepsin, 10) slow-binding macrocycles generated a substantial increase in PGRN, even at concentrations below 100 nM (compounds 6 and 9c, Figure 2C). Together with our finding that panobinostat (5) is able to bind HDACs 1–3 in a slow-binding manner, this encourages future development of PGRN enhancers with long resident times that do not rely on an o-aminoanilide moiety as the basis for the prolonged kinetics. A selection of structurally diverse compounds representative of each kinetic binding mode was also tested on HEK293 cells, as this human cell type is known to have HDAC inhibitor-inducible PGRN expression10. This afforded similar results for each of the six compounds as in NPCs, which further corroborates our observations (Figure S5).
Figure 2. Intracellular PGRN levels in response to HDAC inhibitor treatment.

Fold-change in PGRN protein concentration after 18 h compound treatment in (A) human iPSC-derived NPCs and (B) Ngn2 neurons (representation scale was adapted to maximum signal, data represent mean ± SD, see Figure S6 for alternative refined analysis). Dotted lines represent PGRN concentration upon DMSO treatment, and colors represent fast-on/fast-off (cyan), mechanism A (orange) and mechanism B (red) of slow-binding against HDAC1 and HDAC2. †Romidepsin (10) impaired cellular viability at 10 μM concentration. (C) Dose-response curves of tacedinaline (3, EC50 >20 μM), panobinostat (5, EC50 = 0.20±0.01 μM), trapoxin A (6, EC50 = 0.022±0.001 μM), TpxBAsu(Et) (7b, EC50 ≈ 6.0±0.8 μM), TpxBAsuha (7c, EC50 = 0.45±0.01 μM), apicidin (8a, EC50 = 0.79±0.02 μM), ApiAAsu(Et) (9b, EC50 ≈ 9±2 μM), and ApiAAsuha (9c, EC50 = 0.038±0.001 μM).
Since SATK (2, mechanism A) and tacedinaline (3, mechanism B) were the only slow-binding inhibitors that were not able to affect PGRN production (an effect also reported for Cpd-60, 4)10, we next evaluated the influence of the tested compounds on overall histone acetylation in cells to ensure cellular target engagement. In particular, we measured histone 3 (H3) acetylation at lysines (K) 9 and 27, which have been shown to be enriched upon HDAC inhibition10. The intensity of histone marks were increased to different extents by all inhibitors (Figure 3 and S7), with the exception of SATK (2) and RomiAla,Dha (10d), which may suffer from poor potency, stability and/or ability to penetrate the cell membrane26, 27. Overall, mechanism A and mechanism B slow-binding inhibitors outperformed fast-on/fast-off inhibitors in terms of the magnitude of induction in cellular assays, highlighting the functional advantage of an extended enzyme residence time22, 23. Tacedinaline (3) induced H3 acetylation to levels comparable to panobinostat (5) at concentrations where PGRN levels remained unchanged, in agreement with previous observations10. We thus found o-aminoanilides to be the only case in which effects on PGRN expression and HDAC inhibition do not correlate, independent of the mechanism of binding.
Figure 3. Relationship of PGRN expression to histone acetylation after HDAC inhibitor treatment.

Two-dimensional plots of changes in intracellular PGRN vs. changes in (A) H3K9 acetylation or (B) H3K27 acetylation upon 18 h compound treatment at 1 μM concentration. Colors relate to kinetic mechanisms against HDACs 1 and 2 (cyan: fast-on/fast-off, orange: mechanism A, red: mechanism B of slow-binding kinetics). Histone acetylation Western Blot data and images are included in Figure S7 and S8. *Tacedinaline (3) was dosed at 10 μM concentration.
Panobinostat (5), and not tacedinaline (3), has been reported to increase histone acetylation at the promoter of the GRN gene10. This could indicate that the SIN3 family of HDAC1/2-containing multiprotein complexes is responsible for the regulation of GRN transcription, as tacedinaline (3) and other o-aminoanilides are not able to inhibit them13, 15. This possibility might be testable by genetic or chemical-biological perturbation of SIN3 complex components. On the other hand, and although GRN mRNA levels are upregulated by SAHA (1), panobinostat (5), apicidin (8a) and other HDAC inhibitors to quantitatively similar levels as compared to PGRN protein levels8, 10, enhanced protein stability could also contribute to PGRN accumulation. PGRN contains two lysine residues that can be targeted for ubiquitination40, and future studies will be needed to assess whether HDAC inhibition could lead to acetylation of these lysines and stabilization of PGRN protein.
In conclusion, we provide data to support the hypothesis that slow-binding HDAC inhibitors that are not based upon an o-aminoanilide scaffold can provide valid candidates for restoring PGRN levels in GRN-haploinsufficient FTD cases. This feature may be beneficial for maximizing biological effects and can be found repeatedly amongst clinically effective, approved drugs20, 25. In addition, we report previously unknown mechanism B slow, tight-binding properties of certain hydroxamic acid-containing HDAC inhibitors, which we envision might influence future analysis of their behavior. The PGRN enhancement was achieved not only with hydroxamic acids, but also with compounds bearing ethyl ketone (7a and 8a) and ethyl ester (7c and 9c) Zn2+-binding groups. These are attractive due to better theoretical membrane penetrating properties and as potential prodrugs, respectively27. Tacedinaline (3) with its mechanism B slow, tight-binding HDAC inhibitory properties, but inability to enhance PGRN expression, remains an intriguing exception in the epigenetic regulation of PGRN10. Further investigations into inhibitor kinetics and selectivity towards different physiological HDAC-containing complexes13–15 will be necessary to fully understand the modes of action in HDAC inhibitor upregulation of PGRN.
Methods
More detailed experimental methods are included in the Supporting Information.
HDAC Inhibition Continuous Assays.
Kinetic in vitro assays were performed in Tris buffer pH 8.0 composed of 50 mM Tris/Cl, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2 and 0.5 mg/mL bovine serum albumin (BSA), with additional 0.2 mM tris(2-carboxyethyl)phosphine (TCEP) for assays including romidepsin (10). The assay was performed in 96-well plate format in a final volume of 50 μL per well, where dilution series of the inhibitor (2-fold dilutions) were incubated with peptide substrate (Ac-LGKac-AMC, 20 μM), trypsin (10 μg/mL) and HDAC enzyme (3.5 nM HDAC1, 1.8 nM HDAC2, or 1.2 nM HDAC3/NCoR2). HDAC integrity was assessed via steady-state substrate conversion (Figure S1) and gel electrophoresis (Figure S2). HDACs 1–3 were found to be ~7 kDa smaller than the full length protein (HDAC1assay: 49 kDa, HDAC2assay: 48 kDa, HDAC3assay: 43 kDa, Figure S2) under the experimental conditions employed here and in previous studies16, 26, 27. This would correspond to cleavage of the C-terminal His tag (and C-terminal HDAC1 FLAG tag) as well as of small C- and/or N-terminal fragments by trypsin without affecting the catalytic domain (36 kDa), as supported by the linear conversion rates observed in control experiments (Figure S1). Moreover, inhibitor kinetic profiling resulted similar in end-point assays without trypsin in the reaction mixture (Figure S3). In situ fluorophore release was monitored immediately by fluorescence readings recorded continuously every 30 s for 30–60 min at 25 °C. Data were fitted to the relevant equations as reported in the Supporting Information.
Human Neural Progenitor Cell Culture.
Human neural progenitor cells (NPCs) were derived from reprogrammed induced pluripotent stem cells (iPSCs), as described in the literature39, 41. This cell line was transfected with an inducible Neurogenin-2 (Ngn2) construct for differentiation purposes as reported in Cheng et al., 201739. Neural proliferation medium (NPM) was composed of 70% Dulbecco’s Modified Eagle’s Medium, 30% Ham’s F12 with L-glutamine, with 1% Pen/Strep and 2% B-27 Supplement. NPM was then supplemented with Epidermal Growth Factor (EGF, 20 ng/mL), basic Fibroblast Growth Factor (bFGF, 20 ng/mL) and heparin (5 μg/mL) before use (complete NPM). NPCs were maintained in 6-well plates coated with poly-ornithine and laminin, with complete NPM, and passaged every 3 days.
Human Neuron Culture.
Human iPSC-derived neurons were obtained from NPCs by neurogenin-2 (Ngn2)-induced differentiation as described in Cheng et al., 201739. iNgn2 neural medium (N3aM) was composed of 48% Neurobasal medium, 48% DMEM/F12, 1% B-27 Supplement, 0.5% N-2 Supplement, 0.75% GlutaMax, 1% Pen/Strep, 0.5% MEM non-essential amino acids solution and 50 μM 2-mercaptoethanol. N3aM was then supplemented with 0.2% BSA, doxycycline (2 μg/mL), Brain-Derived Neurotrophic Factor (BDNF, 10 ng/mL) and Neurotrophin-3 (NT3, 10 ng/mL) just before use (complete N3aM). NPCs were plated in coated 96-well tissue culture plates in complete N3aM at 30,000 cells/well density (200 μL/well), and fed every 2 days with half media change. On day 2 and day 4, cells were fed with complete N3aM containing puromycin (1 μg/mL); on day 6, with complete N3aM containing cytosine arabinoside (AraC, 5 μM); and on days 8–12, with fresh complete N3aM. On day 13 cells were treated by adding compounds at 2× concentration in N3aM.
Enzyme-Linked ImmunoSorbent Assay (ELISA).
ELISA was performed with a commercial Progranulin (human) ELISA kit (AdipoGen, #AG-45A-0018YPP-KI01) following the guidelines provided. For NPC experiments, cells were transferred to coated 96-well plates in complete NPM at 30,000 or 60,000 cells/well density, allowed to grow to 95–100% confluency, and then treated by half media change with compounds at 2× concentration. For neuron experiments, treatment was performed on day 13 of differentiation in N3aM. Treatment was allowed to proceed for 18 h, followed by media removal and cell lysis at 4 °C for 30 min. Then, 12.5 μL/well (NPCs) or 25 μL/well (neurons) of lysate were transferred to ELISA strips and analyzed. Three internal replicates of each treatment were included, and each experiment was performed at least twice. Data were analyzed using GraphPad Prism software, and reported as mean ± SD of the biological replicates.
Western Blot.
Cell lysates were prepared for Western blot analysis by adding 4× NuPAGE LDS Sample Buffer and 30× dithiothreitol (DTT), heating and sonicating on PCR plates. 15 μL/sample were separated by gel electrophoresis in MOPS SDS Running buffer and transferred onto activated 0.45 μm PVDF membranes. Membranes were then blocked with 5% non-fat dry milk and probed overnight at 4 °C with H3K9ac (rabbit polyclonal, MilliPore, #07–352, Lot 2203126) or H3K27ac (rabbit polyclonal, Cell Signaling, #4353T, Lot 1) antibodies at 1:5000 dilution in TBST with 5% BSA and 0.02% sodium azide. Secondary HRP-conjugated antibody incubation was performed at 1:5000 dilution in TBST containing 5% milk for 1 h at room temperature, and blots were developed with chemiluminiscence reagents. Thereafter, membranes were probed at room temperature for 4 h with GAPDH antibody (mouse monoclonal, abcam, #ab8245, Lot GR3207992–1) at 1:5000 dilution in TBST with 5% milk and 0.02% sodium azide, incubated with secondary HRP-conjugated antibody and developed. Western Blot images were obtained by exposing the membranes to autoradiography films in the dark. The three internal replicates from a single experiment were analyzed in the same membrane, and relative histone acetylation fold increase is reported as mean ± SD of the three.
Supplementary Material
Acknowledgement
We thank Dr. Andreas S. Madsen for fruitful discussion of kinetic inhibitory data, members of the Haggarty Laboratory for experimental assistance, and the Pless Laboratory at the University of Copenhagen for donation of the HEK293 cell line.
Funding Sources
This work was supported by the University of Copenhagen (Ph.D. fellowship for C.M.-Y.), the Lundbeck Foundation (R289-2018-2074, C.A.O.), the Bluefield Project to Cure FTD (S.J.H., J.H.), the National Institute of Neurological Disorders & Stroke (R01NS108115, S.J.H., J.H.), the National Institute of Health (R37 HL63762, R01 NS093382, R01 NS108115 and RF1 AG053391, J.H.), the Brighfocus Foundation (J.H.), the Harrington Scholar Innovator Award (J.H.) and the Stuart & Suzanne Steele MGH Research Scholars Program (S.J.H.). Travel grants were provided by the University of Copenhagen (Drug Research Academy) and Dagmar Marshalls Fond (C.M.-Y.).
Footnotes
The authors declare the following competing financial interest(s): S.J.H. has financial interest in Rodin Therapeutics, is a member of its scientific advisory board, and is an inventor on IP licensed to this entity that is not directly related to this study. S.J.H. is also member of the scientific advisory board of Frequency Therapeutics, Psy Therapeutics, and Souvien Therapeutics, none of who were involved in this study. JH is a cofounder of Reelin Therapeutics, a company that pursues interests entirely unrelated to this study.
Associated Content
Supporting Information: kinetic parameters and Ki values of HDAC inhibitors, chemical structures of TSA (S1), HC toxin (S2) and 106 (S3), HDAC assay progression and data fitting curves, SDS-PAGE and His-tag Western blot of HDACs 1–3 after trypsin treatment, HDAC assay progression curves estimated from end-point assays without trypsin, immunocytochemical characterization of 10-day differentiated neurons, neuron PGRN data corrected for row effects, HEK293 PGRN data, Western Blot data and images, and Supplemental Methods.
References
- [1].Arrowsmith CH, Bountra C, Fish PV, Lee K, and Schapira M (2012) Epigenetic protein families: a new frontier for drug discovery, Nat. Rev. Drug Discov. 11, 384–400. [DOI] [PubMed] [Google Scholar]
- [2].Falkenberg KJ, and Johnstone RW (2014) Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders, Nat. Rev. Drug Discov. 13, 673–691. [DOI] [PubMed] [Google Scholar]
- [3].Millard CJ, Watson PJ, Fairall L, and Schwabe JW (2017) Targeting class I histone deacetylases in a “complex” environment, Trends Pharmacol. Sci. 38, 363–377. [DOI] [PubMed] [Google Scholar]
- [4].Van Swieten JC, and Heutink P (2008) Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia, Lancet Neurol. 7, 965–974. [DOI] [PubMed] [Google Scholar]
- [5].Gass J, Lee WC, Cook C, Finch N, Stetler C, Jansen-West K, Lewis J, Link CD, Rademakers R, Nykjær A, and Petrucelli L (2012) Progranulin regulates neuronal outgrowth independent of sortilin, Mol. Neurodegener. 7, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Arrant AE, Filiano AJ, Unger DE, Young AH, and Roberson ED (2017) Restoring neuronal progranulin reverses deficits in a mouse model of frontotemporal dementia, Brain 140, 1447–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Arrant AE, Onyilo VC, Unger DE, and Roberson ED (2018) Progranulin gene therapy improves lysosomal dysfunction and microglial pathology associated with frontotemporal dementia and neuronal ceroid lipofuscinosis, J. Neurosci. 38, 2341–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cenik B, Sephton CF, Dewey CM, Xian X, Wei S, Yu K, Niu W, Coppola G, Coughlin SE, Lee SE, Dries DR, Almeida S, Geschwind DH, Gao F-B, Miller BL, Farese RV Jr., Posner BA, Yu G, and Herz J (2011) Suberoylanilide hydroxamic acid (Vorinostat) up-regulates progranulin transcription, J. Biol. Chem. 286, 16101–16108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Almeida S, Gao F, Coppola G, and Gao F-B (2016) Suberoylanilide hydroxamic acid increases progranulin production in iPSC-derived cortical neurons of frontotemporal dementia patients, Neurobiol. Aging 42, 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].She A, Kurtser I, Reis SA, Hennig K, Lai J, Lang A, Zhao W-N, Mazitschek R, Dickerson BC, Herz J, and Haggarty SJ (2017) Selectivity and kinetic requirements of HDAC inhibitors as progranulin enhancers for treating frontotemporal dementia, Cell Chem. Biol. 24, 892–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang J, Wang J, Zhou Z, Park J-E, Wang L, Wu S, Sun X, Lu L, Wang T, Lin Q, Sze SK, Huang D, and Shen H-M (2018) Importance of TFEB acetylation in control of its transcriptional activity and lysosomal function in response to histone deacetylase inhibitors, Autophagy 14, 1043–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Herman D, Jenssen K, Burnett R, Soragni E, Perlman SL, and Gottesfeld JM (2006) Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia, Nat. Chem. Biol. 2, 551–558. [DOI] [PubMed] [Google Scholar]
- [13].Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon A-M, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dümpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, and Drewes G (2011) Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes, Nat. Biotechnol. 29, 255–265. [DOI] [PubMed] [Google Scholar]
- [14].Becher I, Dittmann A, Savitski MM, Hopf C, Drewes G, and Bantscheff M (2014) Chemoproteomics reveals time-dependent binding of histone deacetylase inhibitors to endogenous repressor complexes, ACS Chem. Biol. 9, 1736–1746. [DOI] [PubMed] [Google Scholar]
- [15].Fuller NO, Pirone A, Lynch BA, Hewitt MC, Quinton MS, McKee TD, and Ivarsson M (2019) CoREST complex-selective HDAC inhibitors show pro-synaptic effects and an improved safety profile to enable treatment of synaptopathies, ACS Chem. Neurosci. 10, 1729–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chou CJ, Herman D, and Gottesfeld JM (2008) Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases, J. Biol. Chem. 283, 35402–35409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schroeder FA, Lewis MC, Fass DM, Wagner FF, Zhang Y-L, Hennig KM, Gale J, Zhao W-N, Reis S, Barker DD, Berry-Scott E, Kim SW, Clore EL, Hooker JM, Holson EB, Haggarty SJ, and Petryshen TL (2013) A selective HDAC 1/2 inhibitor modulates chromatin and gene expression in brain and alters mouse behavior in two mood-related tests, PLoS One 8, e71323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lauffer BE, Mintzer R, Fong R, Mukund S, Tam C, Zilberleyb I, Flicke B, Ritscher A, Fedorowicz G, Vallero R, Ortwine DF, Gunzner J, Modrusan Z, Neumann L, Koth CM, Lupardus PJ, Kaminker JS, Heise CE, and Steiner P (2013) Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability, J. Biol. Chem. 288, 26926–26943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wagner FF, Zhang Y-L, Fass DM, Joseph N, Gale JP, Weïwer M, McCarren P, Fisher SL, Kaya T, Zhao W-N, Reis SA, Hennig KM, Thomas M, Lemercier BC, Lewis MC, Guan JS, Moyer MP, Scolnick E, Haggarty SJ, Tsai L-H, and Holson EB (2015) Kinetically selective inhibitors of histone deacetylase 2 (HDAC2) as cognition enhancers, Chemical science 6, 804–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Holdgate GA, Meek TD, and Grimley RL (2018) Mechanistic enzymology in drug discovery: a fresh perspective, Nat. Rev. Drug Discov. 17, 115–132. [DOI] [PubMed] [Google Scholar]
- [21].Morrison JF, and Walsh CT (1988) The behavior and significance of slow-binding enzyme inhibitors, Adv. Enzymol. Relat. Areas Mol. Biol. 61, 201–301. [DOI] [PubMed] [Google Scholar]
- [22].Copeland RA, Pompliano DL, and Meek TD (2006) Drug–target residence time and its implications for lead optimization, Nat. Rev. Drug Discov. 5, 730–739. [DOI] [PubMed] [Google Scholar]
- [23].Copeland RA (2016) The drug–target residence time model: a 10-year retrospective, Nat. Rev. Drug Discov. 15, 87–95. [DOI] [PubMed] [Google Scholar]
- [24].Folmer RH (2018) Drug target residence time: a misleading concept, Drug Discovery Today 23, 12–16. [DOI] [PubMed] [Google Scholar]
- [25].Georgi V, Schiele F, Berger B-T, Steffen A, Marin Zapata PA, Briem H, Menz S, Preusse C, Vasta JD, Robers MB, Brands M, Knapp S, and Fernández-Montalván A (2018) Binding kinetics survey of the drugged kinome, J. Am. Chem. Soc. 140, 15774–15782. [DOI] [PubMed] [Google Scholar]
- [26].Madsen AS, and Olsen CA (2016) A potent trifluoromethyl ketone histone deacetylase inhibitor exhibits class-dependent mechanism of action, MedChemComm 7, 464–470. [Google Scholar]
- [27].Kitir B. l., Maolanon AR, Ohm RG, Colaço AR, Fristrup P, Madsen AS, and Olsen CA (2017) Chemical editing of macrocyclic natural products and kinetic profiling reveal slow, tight-binding histone deacetylase inhibitors with picomolar affinities, Biochemistry 56, 5134–5146. [DOI] [PubMed] [Google Scholar]
- [28].Moreno-Yruela C, Galleano I, Madsen AS, and Olsen CA (2018) Histone deacetylase 11 is an ε-N-myristoyllysine hydrolase, Cell Chem. Biol. 25, 849–856. [DOI] [PubMed] [Google Scholar]
- [29].Stein RL (2011) Kinetics of enzyme action: essential principles for drug hunters, John Wiley & Sons. [Google Scholar]
- [30].Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, and Mazitschek R (2010) Chemical phylogenetics of histone deacetylases, Nat. Chem. Biol. 6, 238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Maolanon AR, Madsen AS, and Olsen CA (2016) Innovative strategies for selective inhibition of histone deacetylases, Cell Chem. Biol. 23, 759–768. [DOI] [PubMed] [Google Scholar]
- [32].Kijima M, Yoshida M, Sugita K, Horinouchi S, and Beppu T (1993) Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase, J. Biol. Chem. 268, 22429–22435. [PubMed] [Google Scholar]
- [33].Taunton J, Hassig CA, and Schreiber SL (1996) A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p, Science 272, 408–410. [DOI] [PubMed] [Google Scholar]
- [34].Porter NJ, and Christianson DW (2017) Binding of the microbial cyclic tetrapeptide trapoxin A to the class I histone deacetylase HDAC8, ACS Chem. Biol. 12, 2281–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hai Y, and Christianson DW (2016) Histone deacetylase 6 structure and molecular basis of catalysis and inhibition, Nat. Chem. Biol. 12, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Robers MB, Dart ML, Woodroofe CC, Zimprich CA, Kirkland TA, Machleidt T, Kupcho KR, Levin S, Hartnett JR, Zimmerman K, Niles AL, Ohana RF, Daniels DL, Slater M, Wood MG, Cong M, Cheng Y-Q, and Wood KV (2015) Target engagement and drug residence time can be observed in living cells with BRET, Nature communications 6, 10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bressi JC, Jennings AJ, Skene R, Wu Y, Melkus R, De Jong R, O’Connell S, Grimshaw CE, Navre M, and Gangloff AR (2010) Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl) benzamides, Bioorg. Med. Chem. Lett. 20, 3142–3145. [DOI] [PubMed] [Google Scholar]
- [38].Wagner FF, Weïwer M, Steinbacher S, Schomburg A, Reinemer P, Gale JP, Campbell AJ, Fisher SL, Zhao W-N, Reis SA, Hennig KM, Thomas M, Müller P, Jefson MR, Fass DM, Haggarty SJ, Zhang Y-L, and Holson EB (2016) Kinetic and structural insights into the binding of histone deacetylase 1 and 2 (HDAC1, 2) inhibitors, Biorg. Med. Chem. 24, 4008–4015. [DOI] [PubMed] [Google Scholar]
- [39].Cheng C, Fass DM, Folz-Donahue K, MacDonald ME, and Haggarty SJ (2017) Highly expandable human iPS cell–derived neural progenitor cells (NPC) and neurons for central nervous system disease modeling and high-throughput screening, Curr. Protoc. Hum. Genet. 92, 21.8.1–21.8.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations, Nucleic Acids Res. 43, D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Silva MC, Cheng C, Mair W, Almeida S, Fong H, Biswas MHU, Zhang Z, Huang Y, Temple S, Coppola G, Geschwind DH, Karydas A, Miller BL, Kosik KS, Gao F-B, Steen JA, and Haggarty SJ (2016) Human iPSC-derived neuronal model of tau-A152T frontotemporal dementia reveals tau-mediated mechanisms of neuronal vulnerability, Stem Cell Reports 7, 325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.