

Abstract
Context:
Accidental exposure to life-threatening radiation in a nuclear event is a major concern; there is an enormous need for identifying biomarkers for radiation biodosimetry to triage populations and treat critically exposed individuals.
Objective:
To identify dose differentiating miRNA signatures from whole blood samples of whole body irradiated mice.
Methods:
Mice were whole body irradiated with X-rays (2 Gy −15 Gy); blood was collected at various time-points post exposure; total RNA was isolated; miRNA microarrays were performed; miRNAs differentially expressed in irradiated vs. unirradiated controls were identified; feature extraction and classification models were applied to predict dose-differentiating miRNA signature.
Results:
We observed a time and dose responsive alteration in the expression levels of miRNAs. Maximum number of miRNAs were altered at 24 h and 48 h time-points post-irradiation. A 23-miRNA signature was identified using feature selection algorithms and classifier models. An inverse correlation in the expression level changes of miR-17 members and their targets was observed in whole body irradiated mice and non-human primates.
Conclusion:
Whole blood-based miRNA expression signatures might be used for predicting radiation exposures in a mass casualty nuclear incident.
Keywords: Radiation, Bio-dosimetry, biomarker, microRNAs
Introduction
Rapid and reliable methods for conducting biological dosimetry (referred to as biodosimetry) are necessary in the event of a large-scale nuclear or radiological disaster. Biodose refers to a measurable biological outcome (chromosomal, metabolomic, proteomic, transcriptomic or any other physiological change) in response to a known amount of radiation dose when living systems (cultured cells, tissues, or animals) are exposed to it. It is important to differentiate that while the unit gray (Gy) is used for biodose assessment in a person, it is not the same as the physical dose1. In a mass casualty scenario it will be extremely difficult to determine the physical radiation dose because of several confounding factors such as source and type of radiation, total/ partial body exposures, multiple exposures, variations in the radiation field and complications arising because of ingestion of air-borne radiological particles2. Hence, identifying an accurate and precise biological response (biodose) is essential for proper clinical management, triaging and for mitigating to allocate limited resources in the most optimal manner possible. The clinically observed physiological symptoms such as emesis, drop in lymphocyte counts and increased body temperature cannot be extrapolated to calculate biodose because these do not always correlate with the severity of Acute Radiation Syndrome (ARS) in a heterogeneous population. The current “gold standard” assays (dicentric chromosomes and presence of micronuclei) for scoring the absorbed dose rely on chromosomal aberrations observed in cultured lymphocyte cells. Their use in a mass casualty incident is somewhat limited as these assays are expertise intensive and require long preparation times of at least 3 days3. At higher radiation exposures, early manifestation of ARS could occur within hours involving the hematopoietic and gastro-intestinal systems, so that earlier assays are needed in addition to potential use of chromosome-based assays to refine initial dose estimates. Hence, it is extremely important to develop simple-to-use, fast, and cost-effective precision biomarker assays to correctly evaluate and identify an individual’s biodose for proper medical care, triage and resource utilization.
It has been known for decades that only 3 % of the human genome encodes for proteins. It has now become clear that more than 75 % of the genome is encoded into RNA4. Of the huge repertoire of non-coding RNA classes, siRNA, miRNA and piRNA are small RNA molecules (20–30 nt) which function in gene silencing and mRNA decay5. miRNAs are transcribed as long transcripts (>1 kb) called pri-miRNA, which contain a local stem-loop structure6. The pri-miRNA is processed in the nucleus by RNAseIII enzyme, DROSHA into 60 bp stem loop called pre-miRNA. The pre-miRNA is exported to the cytoplasm where it is further processed into the ~22 nt long RNA duplex by the cytoplasmic RNAseIII enzyme, DICER. The miRNA duplex is loaded onto an Ago protein where the duplex is unwound. One of the strands is retained (the guide strand) to form the functional miRISC (miRNA induced silencing complex) while the second strand (the passenger strand) dissociates from the complex. The miRISC complex, mostly targets the 3’ UTRs of protein coding transcripts that have partial complementarity to the seed sequence (2–8 nt) of the miRNA and induces translational repression, de-adenylation, or mRNA degradation. More than 60 % of protein coding genes contain at least one conserved miRNA binding site. These facts suggest that miRNAs play crucial roles in genome regulation affecting developmental, homeostatic, and pathological processes. In fact, alterations in the miRNA biogenesis and expression have been observed in several disease conditions7,8. miRNAs are also shed out into the circulation either inside exosomes or in the naked form bound to proteins and thus play a role in inter-cellular communication9. Owing to their remarkable stability in plasma and over long storage periods, circulating miRNA signatures are being identified for diagnostic purposes in cancers10 including gliomas11, cardiovascular pathologies12, metabolic disorders,13 and infectious diseases14.
miRNA expression profiles are modulated in response to DNA damage15,16. P53 has been shown to transcriptionally activate several miRNAs such as miR-34, miR-15a/16, miR-29, miR-107, miR-145, miR-192, miR-194 and transcriptionally repress the miR-17–92 family. Other DNA damage-responsive transcription factors, c-Myc, c-Jun, NF-κB1, and E2F1 have also been shown to modulate miRNA expression profiles. Several miRNAs in turn can modulate the expression levels of DNA damage response genes. For instance, miR-138 targets H2AX. ATM is a target of several miRNAs such as miR-223, miR-181a, miR-26a, miR-27a, miR-214 and miR-18a. BRCA1, Puma, Bcl2, and Bax etc. are also targets of several miRNAs. It is no surprise that miRNA expression changes are observed as a response to radiation. Several studies in both cancer (PC3, HeLa, HCT116, TK6) and normal cell-lines (keratinocytes, human embryonic stem cells, peripheral blood mononuclear cells) identified miRNA profiles changing in response to radiation17–23.
The above listed characteristics make miRNAs ideal potential candidates for radiation bio-dosimetry. Several groups have already identified serum or plasma-based miRNA signatures in whole body irradiated animals. One of the very first studies identified unique plasma miRNA signatures in total body irradiated (TBI) mice exposed to either 0.5, 2, or 10 Gy at 6 and 24 h post irradiation24. Jacob et al looked at the serum miRNA profiles in TBI mice over a wide range of radiation doses (1 Gy to 12 Gy) at 24 h and 48 h post irradiation25. They identified miR-150 (abundant in lymphocytes) levels to decrease in a dose- and time- dependent fashion indicative of lymphocyte depletion. Another study identified miRNA expression signatures at 24 h post irradiation which successfully differentiated lethal (8 Gy) from sub-lethal (6.5 Gy) dose in TBI mice26. There is no consensus on the miRNA signatures modulated in response to radiation in these studies except a few, notably miR-150–5p. One of the reasons for the disparity could be different doses and time-points studied and the use of different model organisms.
We performed a broad study encompassing a wide range of doses and time-points to address these issues with the goal of utilizing miRNAs as biomarkers for identifying radiation responsive dose-dependent signatures. We performed a whole genome miRnome analysis on TBI mice at 6 h, 24 h, 48 h, and 7 days post irradiation with 2 Gy, 4 Gy, 8 Gy, 12 Gy and 15 Gy of gamma-rays. We identified miRNA signatures for low vs. high doses and for early vs. late time points. We also succeeded in validating a few conserved miRNA expression changes in non-human primate (Macaca mulatta) samples. No single miRNAs are useful across all doses and time points, but we identified miRNAs that would be useful at specific times and/or at specific doses that could be ultimately incorporated into an integrated biomarker panel.
Materials and Methods
Whole body irradiation of mice:
Six to eight weeks old C57BL/6 female mice were whole body irradiated with X-rays using the Small Animal Radiation Research Platform (SARRP Xstrahl Ltd.). Mice were placed in plastic containers and exposed to a single surface dose of 2–15 Gy with a dose rate of 1.05 Gy/min. Control mice (0 Gy) were placed in the same plastic container as the irradiated mice and sham irradiated. 3 animals/dose/time-point have been included in the study. All the mice experiments were performed at Department of Pathology, New York University (NYU) Langone Medical Center as part of a collaborative study.
Terminal blood collection using cardiac puncture from mice:
Blood samples were collected at 6 h, 24 h, 48 h and 7 days post irradiation (2 Gy, 4 Gy, 8 Gy, and 12 Gy). Post 15 Gy irradiation blood samples were collected at 6 h, 24 h, and 48 h time-points. Blood samples were collected using cardiac exsanguination. 100 µL of blood was then transferred into five RNAprotect Animal blood tubes (#76544 or #76554) and gently inverted 8 to 10 times. After an incubation of 2 h at room temperature, the samples were stored at −20°C.
Whole body irradiation of non-human primates (NHP) and blood collection:
To examine if similar miRNA changes occur in NHPs, we examined samples available to us from SRI (Study no. R019–16; CiToxLAB Study no. 2017–2243) as part of an agreement with NIAID. All the animal protocols were approved by the respective IACUC committees. NHP (Macaca mulatta) were whole body irradiated using a total dose of 4 Gy using a Co60 source (Theratron 1000) at a dose rate of approximately 60 cGy/min. 500 µl of total blood was collected directly into RNAprotect Animal blood tubes (#76554) and gently inverted 8 to 10 times. Samples were stored at −20°C after an incubation of 2 h at the room temperature.
Ethics:
Experiments using mice were performed at NYU while experiments with NHP were performed at CiToxLAB. Both NYU and CiToxLAB are fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and had obtained IACUC approvals.
RNA isolation:
Total RNA from both mouse and NHP whole blood samples was isolated using the RNAeasy Protect Animal Blood Kit (Cat # 73224, Qiagen) according to the manufacturer’s protocol. Quality and quantity of the RNA samples were assessed using an Agilent Bioanalyzer with the RNA6000 Nano Lab Chip (Agilent Technologies; Santa Clara, CA).
miRNA arrays:
Total RNA was isolated from whole blood preserved in RNAprotect (Qiagen) using Ribopure (Ambion) RNA isolation protocol. Quality and quantity of small and total RNA was assessed using an Agilent Bioanalyzer. Total RNA samples were dephosphorylated, denatured, and end-labeled via the Agilent miRNA labeling kit. Labeled target was applied to Agilent Mouse miRNA 8×60 v19.0 arrays (Design ID 046065) using standard Agilent protocols. Slides were washed and scanned on an Agilent G2566C Microarray Scanner. Initial data analysis was done with Agilent Feature Extraction and GeneSpring Gx v7.3.1 software packages. To compare individual expression values across arrays, raw intensity data from each sample was normalized to the 75th percentile intensity of the array. Probes with intensity values above background in all samples within each group were used for further analysis. Differentially expressed probes were identified by >1.5-fold change and Welch T-test p-values < 0.05 between each treatment group and its control. Whole genome expression profiling was performed as previously reported27. The raw miRNA and mRNA gene expression data was submitted in the Gene Expression Omnibus, an NCI repository (GSE107057 and GSE104121).
Real time RT-PCR analysis of mature miRNAs in NHP blood samples:
200 ng of total RNA was used to convert into cDNA with miScript II RT kit (Qiagen, 218160) to preferentially convert mature miRNAs into cDNA according to the manufacturer’s protocol. miScript Primer Assays (Qiagen, 218300) were subsequently used to detect differential expression in irradiated vs. control samples. Real time PCR reactions were performed in the Applied Biosystems’ thermal cycler (7500). PCR steps included -the holding stage at 95°C for 10 min followed by 40 cycles of alternate denaturation at 95°C for 15 secs, annealing at 55°C for 30 secs and extension at 70°C for 35 secs. A melt curve analysis was performed to ensure the specificity of the corresponding RT-PCR reactions. SnoRNA U6 was used as the normalizing control. Fold change values were derived as follows: Fold change = 2-ddCt where ddCt = dCt (irradiated) - dCt (control); dCt = Ct (miRNA) – Ct (U6); and Ct is the threshold cycle number. All assays were performed in triplicates. Statistical significance was calculated using student’s unpaired t-test. For gene expression analysis, 200 ng of RNA was reverse transcribed using RT2 First Strand synthesis kit (Qiagen, 330401). qPCR assays were performed using RT2 SYBRGreen ROX qPCR Mastermix (Qiagen, 330520) for CDKN1A, THBS1, STAT5, PTEN, and JAK2. GAPDH, 18S and ACTB were used as normalizing genes. Real time PCR reactions were performed in the Applied Biosystems’ thermal cycler (7500). PCR steps included -the holding stage at 95°C for 15 min followed by 40 cycles of alternate denaturation at 95°C for 15 secs, annealing/extension at 60°C for 1 minute. A melt curve analysis was performed to ensure the specificity of the corresponding RT-PCR reactions. Fold change = 2-ddCt where ddCt = dCt (irradiated) - dCt (control); dCt = Ct (gene) – Ct (mean of GAPDH, 18S and ACTB); and Ct is the threshold cycle number. All assays were performed in triplicates. Statistical significance was calculated using student’s unpaired t-test.
Ingenuity Pathway Analysis:
Canonical Pathway Analysis under Expression/Core Analysis was performed in the Ingenuity Pathway Analysis (IPA) software. Significantly altered pathways were selected based on p-values.
Statistical Analysis:
Data were normalized to the 75th percentile intensity of each individual array and 557 probes (FC≥1.5; p≤0.05) were filtered for further comparison. The data passed the Shapiro Wilk normality test. The data were further analyzed using R package (https://www.R-project.org). Two-way ANOVA was performed with dose and time as independent variables, and 187 probes were selected with a dose FDR of 0.05. For identifying dose specific radiation induced targets, ANOVA followed by Dunnett’s test was performed to compare dose-points to controls at each time-point. Summary of the number of probes passing the selection criteria at each step is given in Table 1. For all the results, significant level was set to be 5 % (p value= 0.05).
Table 1:
Details of the selection criteria followed to identify differentially regulated miRNAs in blood in response to whole body irradiation.
| Test performed | miRNA probes passed | |||
|---|---|---|---|---|
| QC passed (Data normalized to 75th percentile; Ratio>1.5; Welch p-value<0.05 at any dose and time-point) | 557 | |||
| Two-Way ANOVA; dose FDR<0.05 | 187 | |||
| One-way ANOVA and Dunnett’s Test (Each dose vs. control at different time-points, p-value<0.05) | 6 h | 24 h | 48 h | 7 d |
| 55 | 243 | 390 | 317 | |
Developing a classifier based on miRNA expression values
Several feature selection algorithms (Boruta, Recursive Feature Elimination, nearest shrunken classifier and the elastic net regression model) were applied to the normalized miRNA expression data (normalized to 75th percentile intensity of each array). Using the number of times each signal was selected by an algorithm as its rank of importance in a variety of predictive models of radiation exposure, a list of 23 miRNAs was selected as a signature for classifying dose and time-points. Using the expression values of these 23 miRNAs, Random Decision Forest28 (RDF) and Support Vector Machine29 (SVM) models were developed by randomly splitting the data-set into a training (75 % data-points) and a test (25 % data-points) set. The RDF model constructs decision trees on training data and outputs the level of radiation exposure that is the mode across all the individual decision trees. SVM is a classification model that maximizes the margin between different classes as each observation is mapped to an n-dimensional space using a function. The RDF and SVM models were constructed on the training data using R open source software packages ‘randomForest’ and ‘e1071’. The SVM model was tuned using four kernel functions (linear, polynomial, radial, and sigmoidal) and with a constant cost of the regularization term ranging from 10−3 to 103. Using an approximation of the leave-p-out cross-validation technique called random sub-sampling validation, a misclassification rate and confusion matrix were computed across 20,000 random splits of the dataset.
Results
miRNA expression profiling in whole blood samples of TBI mice
Dose distribution of the probes induced in response to radiation at each time-point is shown in Figure 1. 79 miRNA probes were differentially expressed in response to an exposure of 15 Gy at 6 h. In contrast, at doses 2–12 Gy, only a handful of miRNA expression changes were observed at the 6 h time-point (Figure 1A). At the 24 h time point, 39 probes were differentially expressed in response to 15 Gy; 63 probes were differentially expressed in response to 12 Gy; 39 probes were differentially expressed in response to 8 Gy; 55 and 33 probes were differentially expressed in response to 4 Gy and 2 Gy respectively; of these 20 miRNA probes were differentially expressed in response to all doses of radiation (Figure 1B). At the 48 h time point, 67 probes were differentially expressed in response to 15 Gy; 66 probes were differentially expressed in response to 12 Gy; 86 probes were differentially expressed in response to 8 Gy; 83 and 66 probes were differentially expressed in response to 4 Gy and 2 Gy respectively; of these 51 miRNA probes were differentially expressed in response to all doses of radiation (Figure 1C). At the 7-d time-point, 66 probes were differentially expressed in response to 12 Gy; 66 probes were differentially expressed in response to 8 Gy; 5 probes were differentially expressed in response to 4Gy; 21 probes were differentially expressed in response to 2 Gy; only 1 miRNA probe was found to be differentially expressed in response to all radiation doses; 54 miRNA probes were commonly differentially regulated in response to both 8 Gy and 12 Gy. (Figure 1D). No mice survived upto 7 days after 15 Gy dose.
Figure 1: Venn diagrams depicting dose distribution of the radiation regulated miRNAs at the A) 6 h, B) 24 h, C) 48 h and D) 7 days time-points.
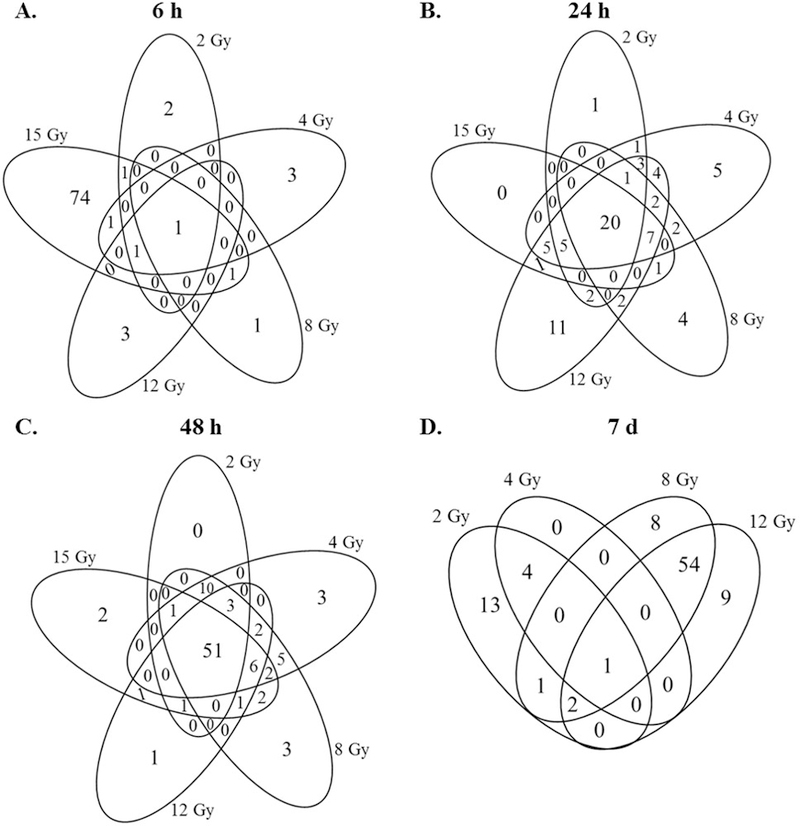
miRNome expression analysis was performed on the total RNA isolated from blood of the irradiated and sham irradiated animals using Agilent arrays. One-way ANOVA analysis for the main effects followed by Dunnett test for treatment vs. control comparisons were made for each time point (6 h, 24 h, 48 h and 7 d) across the respective dose treatments. Overlap of miRNAs were evaluated by Venn diagram using the criteria of adjusted p value ≤0.05 (Dunnett’s test) and fold change of +/− 1.5.
Since the ultimate objective of identifying miRNA biomarkers from mice studies is for radiation biodosimetry applications in humans, we decided to limit our focus to only those miRNAs with a known homolog in humans. Figure 2 highlights the miRNAs conserved between humans and mice selected at FDR threshold of <0.05 (in any of the doses tested). The heatmap illustrates the dose- dependent expression changes of the selected miRNAs for all time-points. For instance, miR-320–3p, miR-574–5p, miR-18a-3p, miR-193b-3p, miR-505–5p, miR-532–3p, miR-150–3p, miR-93–3p, miR-132–3p and miR-676–3p showed low base level expression in all the control samples and were dose responsively upregulated at the 48 h time-point. The induced expression was also observed at 7 d time-point but only in response to higher doses (8 Gy and 12 Gy). In contrast, miR-17–5p, miR-340–5p, miR-150–5p, miR-140–5p, miR-98–5p, miR-22–5p, miR-486–3p, miR-29c-5p and miR-20b-5p showed higher base level expression for all the control samples and were dose-responsively down-regulated at the 24 and 48 h time-points. The down-regulation in the expression levels for these miRNAs was also observed only in response to higher doses (8 Gy and 12 Gy) at the 7 d time-point. For a complete list see supplementary tables 1–4.
Figure 2: Heatmap showing expression values of the most significantly regulated miRNAs with lowest dose FDR across all the samples at all the time-points.

Results from 2-way ANOVA analysis after controlling the effect of time were selected at a FDR cutoff of ≤ 0.05. Hierarchical cluster analysis using correlation distance measure was used to generate the heatmap. The data was converted to z-score (with mean =1; and SD = 0) across the treatments to show relative expression.
Figure 3 highlights the miRNAs conserved between humans and mice selected at FDR threshold of <0.05 (in any of the time-points tested). The heatmap shows the time-dependent expression changes for all doses. The base level expression of all miRNAs shown in figure 3 was low in all the control samples and no induction was observed in response to radiation at the 6 h time-point. In contrast, these miRNAs were induced in a time-responsive manner at the 24 h and 48 h time-points with the magnitude of induction being higher at the 48 h time-point compared to the 24 h time-point for all the doses. At the 7 d time-point, the induction in the expression of miR-423–5p, miR-18a-3p, miR-93–3p, miR-150–3p and miR-92a-3p was observed only in response to 8 Gy and 12 Gy. Supplementary tables 5–9 list all these miRNAs.
Figure 3: Heatmap showing expression values of the most significantly regulated miRNAs (FDR ≤ 0.05) in any of the condition across all the samples at all the dose-points.

The two-way-ANOVA results controlled for dose effect were used after filtering at FDR cutoff of ≤ 0.05. Data is normalized to z-score and subjected to hierarchical cluster analysis using correlation distance measure.
Amongst the significantly down-regulated miRNAs, mmu-miR-140–5p was down-regulated in response to all the radiation doses at 24 h and 48 h and in response to higher doses at the 7-d time-point (Figure 4A). mmu-let-7f-5p was significantly down-regulated in response to all radiation doses at the 48 h time-point and at higher doses at the 7-d time-point (Figure 4B). mmu-miR-223–3p was found to be significantly induced in response to all doses of radiation at 24 h and 48 h while it was induced in response to only higher doses (8 Gy and 12 Gy) at the 7-d time-point (Figure 4C). mmu-let-7b-5p was induced in response to all doses of radiation at the 24 h and 48 h time-points while at the 7-d time-point, induction was observed in response to higher radiation doses (8 Gy and 12 Gy) (Figure 4D). mmu-miR-574–5p was induced in response to all radiation doses at the 48 h time-point while at the 7-d time-point, induction was observed only in response to higher radiation doses of 8 Gy and 12 Gy (Figure 4E).
Figure 4: Short-listed miRNAs from the microarray data.

A) mmu-miR-140–5p was significantly down regulated in response to all the radiation doses at 24 h, 48 h and in response to 8 Gy and 12 Gy of radiation at the 7 d time point. B) mmu-let-7f-5p was significantly repressed in response to all radiation doses at the 48 h time-point and in response to 8 Gy and 12 Gy at the 7 d time-point. C) mmu-miR-223–3p was induced in response to different radiation doses at 6 h, 24 h and 48 h. D) mmu-let7b-5p were significantly induced in response to all radiation doses at 24 and 48 h time-points and I response to 8 Gy and 12 Gy at the 7 d time-point. E) mmu-miR-574–5p was induced in response to all radiation doses at 48 h time-point and in response to 8 Gy and 12 Gy at 7 d time-point.
Inverse regulation of guide and passenger miRNA pairs in response to radiation:
An interesting pattern was observed with the guide and passenger of the same miRNA being regulated in an inverse manner in response to radiation. mmu-miR-150–5p was repressed across all the doses at all the time points except 6 h while mmu-miR-150–3p was induced at 24 h and 48 h time-point for all doses and in response to higher doses at the 7 day time-point (Figure 5A and B). In a similar fashion, mmu-miR-3107–5p, mmu-miR-18a-3p and mmu-miR-93–3p were induced in response all doses of radiation at the 24 h and 48 h time-points and in response to higher doses at the 7 d time point (Figure 5C, E and G). In contrast, mmu-miR-3107–3p, mmu-miR-18a-5p and mmu-miR-93–5p were down-regulated in response to all doses of radiation at the 48 h time-point (Figure 5D, F and H).
Figure 5: Inverse regulation of guide and passenger miRNA pairs in response to radiation.

A and B) mmu-miR-150–5p was repressed in response to all doses of radiation at 24, 48 h and 7 d time-point; mmu-miR-150–3p was induced in response to all doses of radiation at the 48 h time-point and in response to 8 Gy and 12 Gy at the 7 day time-point; C and D) mmu-miR-3107–5p was induced in response to all doses of radiation at the 24 h and 48 h time-point and in response to 8 Gy and 12 Gy at the 7 d time-point; mmu-miR-3107–3p was repressed in response to all doses of radiation at the 48 h time-point; E and F) mmu-miR-18a0–3p was induced in response to all doses of radiation at the 24 h and 48 h time points and in response to 8 Gy and 12 Gy radiation exposure at the 7 d time-point; mmu-miR-18a-5p was repressed in response to all radiation doses at the 48 h time-point and in response to 8 Gy and 12 Gy at the 7 day time-point; G and H) mmu-miR-93–3p was induced marginally in response to different doses; mmu-miR-93–5p was repressed in response to all doses of radiation at the 48 h time-point and in response to 8 Gy and 12 Gy at the 7 day time-point
Identification of 23-miRNA signature for classification:
Several feature selection algorithms were explored to select the miRNA signals that contained the most information on the level of radiation exposure that the subject received. Algorithms included the Boruta algorithm, which builds a predictive model and ranks subsets of signals by their importance in the model, as well as the Recursive Feature Elimination (RFE) algorithm30, a method that ranks signals by their importance in a model and then iteratively eliminates the lowest ranking signals. Other algorithms used included the nearest shrunken centroid classifier31 and the elastic net regression model, embedded techniques that build a classification model of radiation exposure while eliminating signals that are not supplying predictive information. A classifier signature comprising 23 miRNAs was predicted using such a rigorous analysis (Table 2). As the feature selection model may select features which are statistical anomalies in the data, rather than reflecting an underlying biological change, the roles of the most frequently selected genes were examined for their relevance to radiation injury. Several miRNAs in the classifier have previously been shown to possess hematopoietic roles. Several other miRNAs from the classifier have been shown to play roles in immune related pathways, apoptosis, cell proliferation and growth factor and morphogenesis pathways (Table 2). RDF and SVM classification models were used to generate confusion matrices to compute misclassification rates from the expression data of 23-miRNA signature (Table 3). Both the RDF and SVM models could correctly predict 0 Gy as 0 Gy in 95 % and 86 % of the iterations respectively. In contrast, the dose-points 2 Gy, 4 Gy, 8 Gy, 12 Gy and 15 Gy were correctly predicted in only 38 %, 49 %, 17 %, 35 % and 35 % cases respectively by the RDF model. The SVM model was only slightly better at predicting the doses correctly with 2 Gy, 4 Gy, 8 Gy, 12 Gy and 15 Gy accurately predicted in 55 %, 41 %, 30 %, 37 % and 57 % of the cases respectively. In most of the cases, the wrongly predicted dose is either immediately above it or below it (for instance for a true 2 Gy exposure, the most misclassified classes are 0 Gy and 4 Gy) in both the RDF and SVM models. Some predictions are grossly incorrect. Using RDF model, in 9 % of cases 4 Gy is predicted as 0 Gy; in 21 % of cases 8 Gy is predicted as 0 Gy and so on. Similar observation is seen when using SVM model. The 23-miRNA classifier can be used to differentiate exposed from the unexposed but is a poor tool to classify dose classes correctly. To improve model accuracy, we decided to group the data into three classes (0 Gy/ 2–8 Gy/ 12–15 Gy). Feature selection algorithms picked 49 miRNAs as the optimal classes-differentiating signature. Both the RDF and SVM models performed considerably better to classify these classes (Table 4) with an overall misclassification rate of 18 % and 17 % respectively. Using the SVM model the classes, 0 Gy, 2–8 Gy and 12–15 Gy were correctly predicted 88 %, 84 % and 75 % of the times respectively. The list of the miRNAs comprising the 49-signature is given in supplementary table 10.
Table 2: 23-miRNA expression signature predicted using 18 different feature extraction algorithms.
Column 1 lists the names of miRNAs and column 2 shows the total number of feature selection algorithms in which the corresponding miRNA was predicted as part of the classifier. Column 3 lists the physiological functions and disease correlation of the individual miRNAs identified from miRbase 52–56 and MGI57 (and references therein). PubMed search was also used for identifying functional characterization of some of the miRNAs.
| 23-miRNA classifier | # F-S methods | Physiological Function |
|---|---|---|
| mmu-miR-223–3p | 15 | Hematopoietic |
| mmu-miR-21a-5p | 12 | Role in cell apoptosis, growth, and proliferation |
| mmu-miR-3096b-3p | 11 | Limited data on function, all NGS predicted |
| mmu-miR-5115 | 11 | Fragment of LSU rRNA. Limited data on function, all NGS predicted |
| mmu-miR-150–5p | 9 | Hematopoietic |
| mmu-miR-20b-5p | 9 | Hematopoietic |
| mmu-miR-20a-5p | 8 | Hematopoietic |
| mmu-miR-24–3p | 8 | Hematopoietic |
| mmu-miR-574–5p | 8 | Hematopoietic |
| mmu-miR-222–3p | 8 | Development in heart, skin. Melanoma marker |
| mmu-miR-140–5p | 7 | Tissue homeostasis |
| mmu-miR-1839–3p | 7 | Noncanonical miRNA |
| mmu-miR-29b-3p | 7 | Hematopoietic |
| mmu-miR-466i-5p | 7 | Immune response and inflammation |
| mmu-let-7f-5p | 6 | Hematopoietic response to Cytokines |
| mmu-miR-3082–5p | 6 | Role not clear, downregulation associated with melanoma |
| mmu-miR-30e-5p | 6 | Adipocyte and osteoblast differentiation |
| mmu-miR-98–5p | 6 | Tumor suppression and apoptosis. |
| mmu-miR-22–5p | 6 | Probably Hematopoietic |
| mmu-let-7b-5p | 6 | Hematopoietic response to Cytokines |
| mmu-miR-30d-5p | 6 | Invasion and immunosuppression, autophagy |
| mmu-miR-320–3p | 6 | Hematopoietic |
| mmu-miR-30c-5p | 6 | Hematopoietic |
Table 3: Confusion Matrices using RDF and SVM algorithms for calculating misclassification rates.
Random Decision Forest and Support Vector Machine algorithms were used to compute misclassification rates using the predicted 23-miRNA signature.
| Random Decision Forest | Support Vector Machine | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| True Radiation Dose | True Radiation Dose | ||||||||||||
| 0 Gy | 2 Gy | 4 Gy | 8 Gy | 12 Gy | 15 Gy | 0 Gy | 2 Gy | 4 Gy | 8 Gy | 12 Gy | 15 Gy | ||
| Predicted Radiation Dose | 0 Gy | 95% | 13% | 9% | 21% | 8% | 5% | 86 % | 17 % | 2 % | 14 % | 3 % | 0 % |
| 2 Gy | 0% | 38% | 19% | 9% | 7% | 1% | 4 % | 55 % | 31 % | 13 % | 2 % | 0 % | |
| 4 Gy | 4% | 38% | 49% | 18% | 13% | 6% | 3 % | 21 % | 41 % | 22 % | 3 % | 2 % | |
| 8 Gy | 0% | 10% | 15% | 17% | 14% | 10% | 6 % | 6 % | 16 % | 30 % | 24 % | 12 % | |
| 12 Gy | 0% | 2% | 3% | 32% | 35% | 44% | 1 % | 1 % | 1 % | 21 % | 37 % | 29 % | |
| 15 Gy | 0% | 0% | 5% | 4% | 22% | 35% | 0 % | 0 % | 8 % | 1 % | 30 % | 57 % | |
Table 4:
Confusion Matrix for the 0/2–8 Gy/12–15 Gy classification generated by the SVM model using the predicted 49-miRNA signature from feature selection algorithms.
| True Radiation Dose Class | ||||
|---|---|---|---|---|
| 0 Gy | 2–8 Gy | 12–15 Gy | ||
| Predicted Predicted Radiation Dose Exposure | 0 Gy | 88 % | 7 % | 9 % |
| 2–8 Gy | 11 % | 84 % | 16 % | |
| 12–15 Gy | 0 % | 8 % | 75 % | |
miR-17 family repression in response to radiation
We observed several members of the miR-17 family (miR-17–5p, miR-106b-5p, miR-20a-5p and miR-20b-5p) coordinately repressed in response to radiation from 2 Gy onwards at 24 h, 48 h and at higher doses for the 7 d time-point (Figure 6A). Interestingly, experimentally verified targets (identified from miRTarBase) common to miR-17–5p, miR-20a-5p, miR-20b-5p and miR-106b-5p: Cdkn1a, Pten, E2f1, E2f3, and Stat3 were found to be induced in response to radiation in the whole genome expression profiling data (Figure 6B). We selected all the experimentally verified gene targets of miR-17–5p, miR-20a-5p, miR-20b-5p and/or miR-106b-5p identified in either human or mouse studies. We shortlisted those targets which were significantly altered in a radiation responsive manner in our whole genome transcriptomic data. For several of these targets we observed a high negative correlation between the expression levels of miR-17 members and the targets across 2, 4 and 8 Gy dose-points and 24 h and 48 h time-points (Table 5). A preliminary Ingenuity Pathway Analysis (IPA) identified that the negatively correlated genes can positively affect several DNA damage related, immune function related, cancer related and hematopoietic pathways (Table 6).
Figure 6: Expression of miR-17 family members regulated in response to radiation.

A) Members of the miR-17 family sharing a common sequence (mmu-miR-17–5p, mmu-miR-106b-5p, miR-20a/b-5p) were repressed in response to radiation at 24 h and 48 h time-points and in response to higher doses at the 7 d time-point. B) Whole genome expression profiling data identified several miR-17 targets (Cdkn1a, Pten, Stat3, Stat5b, E2f1, E2f3, Thbs1, Jak2) as induced upon different radiation doses at 24 and/or 48 h time-points.
Table 5: Negative Correlation in the expression levels of miR-17 sisters and the gene targets from miRTarBase.
Pairwise pearson’s correlation was performed for the common time-points (24 h and 48 h) and doses (2, 4 and 8 Gy) between miRNA and mRNA expression data. The correlation coefficients and associated correlation p-values are shown for the selected targets
| mmu-miR-17–5p | mmu-miR-20a-5p | mmu-miR-20b-5p | mmu-miR-106b-5p | |||||
|---|---|---|---|---|---|---|---|---|
| CoR_Coeff | P-value | CoR_Coeff | P-value | CoR_Coeff | P-value | CoR_Coeff | P-value | |
| Ulk1 | −0.99 | 0.000 | −0.96 | 0.001 | −0.94 | 0.002 | −0.93 | 0.002 |
| Atg7 | −0.99 | 0.000 | −1.00 | 0.000 | −0.99 | 0.000 | −0.93 | 0.002 |
| Trim8 | −0.99 | 0.000 | −0.97 | 0.000 | −0.96 | 0.001 | −0.96 | 0.001 |
| Mapk14 | −0.98 | 0.000 | −0.98 | 0.000 | −0.96 | 0.000 | −0.95 | 0.001 |
| Egln3 | −0.98 | 0.000 | −0.98 | 0.000 | −0.97 | 0.000 | −0.95 | 0.001 |
| Mfn2 | −0.96 | 0.000 | −0.90 | 0.005 | −0.88 | 0.009 | −0.87 | 0.010 |
| Mdm2 | −0.96 | 0.001 | −0.90 | 0.006 | −0.90 | 0.006 | −0.77 | 0.045 |
| Trp53inp1 | −0.95 | 0.001 | −0.87 | 0.011 | −0.86 | 0.014 | −0.79 | 0.033 |
| Pdlim7 | −0.95 | 0.001 | −0.98 | 0.000 | −0.97 | 0.000 | −0.96 | 0.001 |
| Limk1 | −0.95 | 0.001 | −0.92 | 0.003 | −0.92 | 0.004 | −0.84 | 0.017 |
| Rhoc | −0.94 | 0.002 | −0.95 | 0.001 | −0.96 | 0.001 | −0.85 | 0.016 |
| Epas1 | −0.93 | 0.002 | −0.94 | 0.002 | −0.93 | 0.002 | −0.90 | 0.005 |
| Fyn | −0.93 | 0.003 | −0.91 | 0.005 | −0.88 | 0.008 | −0.93 | 0.002 |
| Fas | −0.93 | 0.003 | −0.89 | 0.008 | −0.88 | 0.009 | −0.83 | 0.022 |
| Akt3 | −0.93 | 0.003 | −0.94 | 0.002 | −0.92 | 0.003 | −0.98 | 0.000 |
| Tnfsf12 | −0.92 | 0.003 | −0.88 | 0.010 | −0.87 | 0.010 | −0.72 | 0.066 |
| Rhoc | −0.92 | 0.003 | −0.93 | 0.002 | −0.94 | 0.001 | −0.82 | 0.025 |
| Gpr137b | −0.92 | 0.003 | −0.85 | 0.015 | −0.84 | 0.017 | −0.76 | 0.049 |
| Dnmt3a | −0.92 | 0.003 | −0.85 | 0.016 | −0.82 | 0.023 | −0.83 | 0.020 |
| Tbc1d2 | −0.91 | 0.004 | −0.94 | 0.002 | −0.92 | 0.003 | −0.99 | 0.000 |
| Mfn2 | −0.90 | 0.006 | −0.86 | 0.014 | −0.83 | 0.022 | −0.90 | 0.006 |
| Map2k3 | −0.90 | 0.006 | −0.90 | 0.006 | −0.88 | 0.009 | −0.92 | 0.003 |
| Timp2 | −0.89 | 0.007 | −0.89 | 0.007 | −0.91 | 0.005 | −0.75 | 0.052 |
| Klf9 | −0.89 | 0.008 | −0.87 | 0.010 | −0.87 | 0.011 | −0.74 | 0.056 |
| Pura | −0.88 | 0.009 | −0.84 | 0.018 | −0.81 | 0.026 | −0.82 | 0.023 |
| Pdlim7 | −0.88 | 0.010 | −0.93 | 0.002 | −0.92 | 0.003 | −0.94 | 0.002 |
| Runx1 | −0.87 | 0.011 | −0.92 | 0.003 | −0.91 | 0.004 | −0.96 | 0.001 |
| Timp2 | −0.86 | 0.013 | −0.82 | 0.023 | −0.83 | 0.022 | −0.71 | 0.074 |
| Jak1 | −0.86 | 0.013 | −0.77 | 0.043 | −0.74 | 0.058 | −0.79 | 0.033 |
| Bmp2 | −0.85 | 0.014 | −0.88 | 0.008 | −0.89 | 0.008 | −0.87 | 0.011 |
| Timp3 | −0.84 | 0.017 | −0.85 | 0.015 | −0.82 | 0.024 | −0.83 | 0.020 |
| Ccnd1 | −0.84 | 0.019 | −0.88 | 0.009 | −0.90 | 0.006 | −0.75 | 0.054 |
| Slc2a3 | −0.83 | 0.021 | −0.90 | 0.005 | −0.90 | 0.006 | −0.93 | 0.002 |
| Rad21 | −0.79 | 0.033 | −0.66 | 0.107 | −0.63 | 0.130 | −0.67 | 0.101 |
| Stat3 | −0.78 | 0.038 | −0.68 | 0.095 | −0.67 | 0.101 | −0.58 | 0.172 |
| Bnip2 | −0.77 | 0.043 | −0.65 | 0.112 | −0.63 | 0.130 | −0.62 | 0.141 |
| Ephb4 | 0.75 | 0.050 | 0.63 | 0.131 | 0.61 | 0.145 | 0.57 | 0.182 |
| Abl2 | −0.72 | 0.069 | −0.56 | 0.187 | −0.54 | 0.210 | −0.50 | 0.253 |
| Cdkn1a | −0.71 | 0.072 | −0.79 | 0.036 | −0.82 | 0.024 | −0.66 | 0.110 |
| Hipk3 | −0.66 | 0.106 | −0.56 | 0.193 | −0.52 | 0.236 | −0.64 | 0.122 |
| Clu | −0.66 | 0.107 | −0.77 | 0.045 | −0.77 | 0.045 | −0.85 | 0.016 |
Table 6: Pathways affected by negatively correlated gene targets identified in table 4.
The target gene-list from table 5 was input in the Ingenuity Pathway Analysis with fold change data at each dose-point and time-point (2 Gy, 4 Gy and 8 Gy at 24 h and 48 h time-points compared to 0 Gy controls). IPA computes z-scores and p-values. The significantly affected pathways are listed here along with corresponding -log (p values) and the genes (from target list in table 5) involved in the corresponding pathway.
| Ingenuity Canonical Pathways | -log (p-value) | Molecules Associated |
|---|---|---|
| Molecular Mechanisms of Cancer | 11.5 | MAPK14,AKT3,FYN,CCND1,CDKN1A,RHOC,MAP2K3,JAK1,MDM2,BMP2,FAS |
| p53 Signaling | 9.77 | MAPK14,AKT3,CCND1,CDKN1A,MDM2,TP53INP1,FAS |
| Pancreatic Adenocarcinoma Signaling | 7.8 | STAT3,AKT3,CCND1,CDKN1A,JAK1,MDM2 |
| IL-22 Signaling | 7.46 | MAPK14,STAT3,AKT3,JAK1 |
| Tec Kinase Signaling | 6.9 | STAT3,FYN,TNFSF12,RHOC,JAK1,FAS |
| Oncostatin M Signaling | 6.83 | STAT3,TIMP3,JAK1,EPAS1 |
| IL-7 Signaling Pathway | 6.76 | MAPK14,AKT3,FYN,CCND1,JAK1 |
| Acute Myeloid Leukemia Signaling | 6.71 | STAT3,AKT3,CCND1,RUNX1,MAP2K3 |
| AMPK Signaling | 6.29 | ULK1,MAPK14,AKT3,CCND1,CDKN1A,MAP2K3 |
| HIF1α Signaling | 6.2 | MAPK14,AKT3,SLC2A3,EGLN3,MDM2 |
| PI3K/AKT Signaling | 6.07 | AKT3,CCND1,CDKN1A,JAK1,MDM2 |
| Melanoma Signaling | 5.97 | AKT3,CCND1,CDKN1A,MDM2 |
| Colorectal Cancer Metastasis Signaling | 5.94 | STAT3,AKT3,TLR7,CCND1,RHOC,JAK1 |
| EGF Signaling | 5.6 | MAPK14,STAT3,AKT3,JAK1 |
| IL-10 Signaling | 5.57 | MAPK14,STAT3,MAP2K3,JAK1 |
| Glioblastoma Multiforme Signaling | 5.52 | AKT3,CCND1,CDKN1A,RHOC,MDM2 |
| GM-CSF Signaling | 5.47 | STAT3,AKT3,CCND1,RUNX1 |
| IL-15 Signaling | 5.4 | MAPK14,STAT3,AKT3,JAK1 |
| IL-17A Signaling in Airway Cells | 5.36 | MAPK14,STAT3,AKT3,JAK1 |
| Role of Macrophages, Fibroblasts and Endothelial Cells in Rheumatoid Arthritis | 5.36 | MAPK14,STAT3,AKT3,TLR7,CCND1,MAP2K3 |
| JAK/Stat Signaling | 5.25 | STAT3,AKT3,CDKN1A,JAK1 |
| Role of JAK family kinases in IL-6-type Cytokine Signaling | 5.24 | MAPK14,STAT3,JAK1 |
| IL-17 Signaling | 5.21 | MAPK14,AKT3,MAP2K3,JAK1 |
| HER-2 Signaling in Breast Cancer | 5.15 | AKT3,CCND1,CDKN1A,MDM2 |
| Production of Nitric Oxide and Reactive Oxygen Species in Macrophages | 5.13 | MAPK14,AKT3,CLU,RHOC,JAK1 |
| FGF Signaling | 5.09 | MAPK14,STAT3,AKT3,MAP2K3 |
| Sumoylation Pathway | 5 | DNMT3A,RHOC,MDM2,FAS |
| Prostate Cancer Signaling | 4.98 | AKT3,CCND1,CDKN1A,MDM2 |
| Mouse Embryonic Stem Cell Pluripotency | 4.83 | MAPK14,STAT3,AKT3,JAK1 |
| Chronic Myeloid Leukemia Signaling | 4.83 | AKT3,CCND1,CDKN1A,MDM2 |
| Inhibition of Angiogenesis by TSP1 | 4.83 | MAPK14,AKT3,FYN |
| Type I Diabetes Mellitus Signaling | 4.75 | MAPK14,MAP2K3,JAK1,FAS |
| Glioma Signaling | 4.71 | AKT3,CCND1,CDKN1A,MDM2 |
| HGF Signaling | 4.69 | STAT3,AKT3,CCND1,CDKN1A |
| Fc Epsilon RI Signaling | 4.63 | MAPK14,AKT3,FYN,MAP2K3 |
| Role of NANOG in Mammalian Embryonic Stem Cell Pluripotency | 4.59 | STAT3,AKT3,JAK1,BMP2 |
| Role of Tissue Factor in Cancer | 4.58 | MAPK14,AKT3,FYN,LIMK1 |
| IL-6 Signaling | 4.51 | MAPK14,STAT3,AKT3,MAP2K3 |
| HMGB1 Signaling | 4.44 | MAPK14,AKT3,RHOC,MAP2K3 |
| Aryl Hydrocarbon Receptor Signaling | 4.34 | CCND1,CDKN1A,MDM2,FAS |
| Semaphorin Signaling in Neurons | 4.24 | FYN,LIMK1,RHOC |
| Neuroinflammation Signaling Pathway | 4.15 | MAPK14,AKT3,TLR7,JAK1,FAS |
| Acute Phase Response Signaling | 4.03 | MAPK14,STAT3,AKT3,MAP2K3 |
| Germ Cell-Sertoli Cell Junction Signaling | 4 | MAPK14,LIMK1,RHOC,MAP2K3 |
| Ephrin Receptor Signaling | 3.98 | STAT3,AKT3,FYN,LIMK1 |
| Glucocorticoid Receptor Signaling | 3.98 | MAPK14,STAT3,AKT3,CDKN1A,JAK1 |
| Cell Cycle: G1/S Checkpoint Regulation | 3.96 | CCND1,CDKN1A,MDM2 |
| Glioma Invasiveness Signaling | 3.88 | TIMP3,RHOC,TIMP2 |
| Regulation of the Epithelial-Mesenchymal Transition Pathway | 3.85 | STAT3,AKT3,MAP2K3,JAK1 |
| Role of MAPK Signaling in the Pathogenesis of Influenza | 3.84 | MAPK14,AKT3,MAP2K3 |
| STAT3 Pathway | 3.81 | MAPK14,STAT3,CDKN1A |
| TREM1 Signaling | 3.79 | STAT3,AKT3,TLR7 |
| IL-8 Signaling | 3.78 | AKT3,CCND1,LIMK1,RHOC |
| ILK Signaling | 3.78 | AKT3,CCND1,RHOC,BMP2 |
| Toll-like Receptor Signaling | 3.77 | MAPK14,TLR7,MAP2K3 |
| CD40 Signaling | 3.72 | MAPK14,STAT3,MAP2K3 |
| IL-3 Signaling | 3.66 | STAT3,AKT3,JAK1 |
| FLT3 Signaling in Hematopoietic Progenitor Cells | 3.61 | MAPK14,STAT3,AKT3 |
| PEDF Signaling | 3.6 | MAPK14,AKT3,FAS |
| TGF-β Signaling | 3.6 | MAPK14,MAP2K3,BMP2 |
| Bladder Cancer Signaling | 3.58 | CCND1,CDKN1A,MDM2 |
| Death Receptor Signaling | 3.51 | TNFSF12,LIMK1,FAS |
| Role of Osteoblasts, Osteoclasts and Chondrocytes in Rheumatoid Arthritis | 3.5 | MAPK14,AKT3,MAP2K3,BMP2 |
| GADD45 Signaling | 3.49 | CCND1,CDKN1A |
| ATM Signaling | 3.45 | MAPK14,CDKN1A,MDM2 |
| Cholecystokinin/Gastrin-mediated Signaling | 3.41 | MAPK14,RHOC,MAP2K3 |
| IGF-1 Signaling | 3.35 | STAT3,AKT3,JAK1 |
| Role of JAK1, JAK2 and TYK2 in Interferon Signaling | 3.28 | STAT3,JAK1 |
| Estrogen-mediated S-phase Entry | 3.21 | CCND1,CDKN1A |
| PTEN Signaling | 3.2 | AKT3,CCND1,CDKN1A |
| p38 MAPK Signaling | 3.19 | MAPK14,MAP2K3,FAS |
qRT-PCR analysis of radiation responsive miRNAs in the blood samples of whole body irradiated non-human primates (NHP)
To establish whether the signatures identified as radiation responsive in mouse blood would be regulated in a comparable way in higher primates, we analyzed the expression levels in the blood samples of non-human primates after they were whole body irradiated with a total dose of 4 Gy. We observed down-regulation of miR-17–5p, miR-106b-5p and miR-20a-5p in response to radiation (Figure 7A). As seen in mouse, in NHP samples also an inverse correlation was observed in the expression alteration of miR-17 members and gene targets such as CDKN1A, PTEN, STAT5B, THBS1 and JAK2 (Figure 7B).
Figure 7: qRT-PCR analysis of radiation responsive miRNAs in the blood samples of whole body irradiated non-human primates (NHP).

NHP were whole body irradiated with a total dose of 4 Gy. Blood samples were drawn at D-8 (8 days prior to radiation), D1 (1-day post-radiation) and D2 (2 days post radiation). Total RNA was isolated from whole blood and 200 ng of total RNA was reverse transcribed using miScript RT II kit. A) qPCR was performed using miScript SYBRGreen and miScript primer assays for miR-17–5p, miR-106b-5p, miR-20a-5p. B) 200 ng of total RNA was reverse transcribed using Qiagen First Strand Synthesis Kit. qPCR assays were performed using Qiagen SYBR green master mix and qPCR primer assays against Rhesus monkey CDKN1A, THBS1, STAT5B, PTEN and JAK2.
Discussion
miRNAs form an important distinct layer of post-transcriptional gene regulation where they exert their effect by inducing either mRNA degradation or translational repression. In animals, only partial complementarity is needed suggesting that each miRNA has multiple targets and each mRNA can be targeted by more than one miRNA32. This would mean that loss of a small sub-set of miRNAs could have profound effects on cellular functions. Indeed, loss of miRNA expression can lead to increased cellular plasticity, de-differentiation and cellular transformation33–35. Ample evidence for the stability of miRNAs in body fluids make them important diagnostic biomarkers for several disease conditions including cancers. It is now widely known that tumors secrete miRNAs as part of exosomes and micro-vesicles which stay for extended periods of time in the circulation enabling assay development for diagnostic purposes36,37.
A plethora of studies in the last decade has focused on the role miRNAs play in DNA damage response. Cells with defects in miRNA biogenesis machinery have defective DDR pathways38. A generalized miRNA response signature to three kinds of genotoxic stress - radiation, H2O2 and etoposide - was identified in human dermal fibroblast cells suggesting that these miRNAs played crucial roles in cellular defense mechanisms39. It follows that ionizing radiation would also cause changes in the miRNA expression profiles and indeed, treatment of several normal and cancer cell-lines to ionizing radiation lead to induction of distinct miRNA profiles18,19,40. Different miRNAs have also been implicated in development of either radio-resistance41 or radio-sensitivity42. The recent explosion in the knowledge about miRNAs in radiation response has led to several efforts to identify miRNA biomarker signatures for the purposes of radiation biodosimetry. One of the very first studies by Liu et al in 2010 identified increased levels of miR-34a in the plasma as indicative of radiation injury43. Jacob et al reported in 2013 that plasma levels of miR-150 depleted in a dose-dependent manner25. Plasma and serum miRNA profiles were successfully shown to discriminate low vs. high and lethal vs. sub-lethal doses in two separate studies using the murine model24,26.
We report here identification of a miRNA-based signature for various doses and time points in whole blood samples drawn from whole body irradiated female C57Bl6 mice. We did not find any miRNA which could be used as a sole biomarker for assessing radiation exposure for any dose and any time point in mice. A 23-miRNA signature was identified to differentiate different radiation classes. The applicability of the classifier is limited to distinguish the unexposed (0 Gy) from any dose exposure. The misclassification rates for each of the dose classes were too high to apply this classifier signature as a biodosimeter. Misclassification rates were considerably lower when we grouped the data into higher classes (0 Gy / 2–8 Gy / 12–15 Gy). This apparent discrepancy from the earlier published studies where dose-differentiating signatures were identified with higher accuracies could be because of several reasons. All the previous studies were based on serum and plasma samples while in our study we used whole blood RNA. The lesser number of animals per dose class also limits the ability of the feature extraction algorithms and classification models. Prediction accuracies could be substantially improved by expanding the study to higher number of animals. We also intend to explore the possibility of utilizing the gene and miRNA expression profiling data together to identify integrated signatures.
We observed that the guide and passenger miRNAs of miR-150, miR-3107, miR-93 and miR-18a were altered in an opposing manner in response to radiation This indicates regulation of the miRNA biogenesis machinery or some of its components as a direct consequence of radiation. It might also be a consequence of the changes observed in the cellular composition of the blood in response to radiation such as loss of lymphocytes and relative increase in the myeloid cells.
An important finding of this study is the down-regulation of several miR-17 sisters in response to whole body radiation exposure. We have previously shown that fractionated radiation repressed the expression levels of the miR-17–92 cluster in the p53 positive prostate cancer cell-lines and not in the p53 negative cells44. Indeed miR-17–92 cluster is widely known as the ‘Oncomir1’ and is transcriptionally repressed by p5345,46. The miR-17–92 cluster is often dysregulated in hematopoietic and solid cancers47. Interestingly several of the targets of the miR-17 sisters (miR-17–5p, miR-20a-5p, miR-20b-5p and miR-106b-5p) were found induced in response to radiation in the whole genome expression profiling data. Many of the targets, such as Pten, Cdkn1a, E2f factors play crucial roles in cell-cycle arrest48–50. Indeed, an Ingenuity Pathway Analysis identified DNA damage, immune function, cancer and hematopoiesis related functions for the inversely correlated gene targets. This interesting negative correlation in the expression perturbations in the miR-17 sisters and the target genes in response to radiation was observed in both mice and NHP models. We previously had reported an inverse correlation of radiation responsiveness of miR-17 and its targets in normal human coronary artery endothelial cells51. This inverse correlation serves as an example for our proposed integrated signature approach alluded to earlier.
Conclusion
Although robust transcriptional level changes have been observed in the miRNA levels after whole body irradiation in mouse blood, classifier based on miRNA expression data alone did not perform with the level of accuracy needed for biodosimetry applications. Because of the interdependent regulation of miRNA, mRNA and lncRNA, we hypothesize that an integrated signature based on expression levels of miRNAs, lncRNAs and the target mRNAs might be more elucidative and suitable for biodosimetry purposes.
Supplementary Material
Clinical significance:
Findings from the present work would not only enable accurate biodosimetry but might also have applications for predicting normal tissue injury sustained during cancer radiotherapy.
Acknowledgement:
This study was supported by the NIH Intramural Research Program, National Cancer Institute, Center for Cancer Research and funded by NIAID (IAA #NRC-13028). We thank SRI International, US (Study no. R019–16) and CiToxLAB, Canada (Study no. 2017–2243) for providing the non-human primate samples. The authors thank Patricia Rivera-Solis and Katherine Wilsdon for their excellent technical assistance.
Financial Support: This study was supported by the NIH Intramural Research Program, National Cancer Institute, Center for Cancer Research, grant ZIA BC 010670 and funded by NIAID (IAA #NRC-13028)
Footnotes
Disclosure of potential conflicts of interest
The authors report no conflicting interests.
References
- 1.Coleman CN & Koerner JF Biodosimetry: Medicine, Science, and Systems to Support the Medical Decision-Maker Following a Large Scale Nuclear or Radiation Incident. Radiat. Prot. Dosimetry 172, 38–46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sproull M & Camphausen K State-of-the-Art Advances in Radiation Biodosimetry for Mass Casualty Events Involving Radiation Exposure. Radiat. Res 186, 423–435 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Lemos Pinto MMP, Santos NFG & Amaral A Current status of biodosimetry based on standard cytogenetic methods. Radiat. Environ. Biophys 49, 567–581 (2010). [DOI] [PubMed] [Google Scholar]
- 4.The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang B & Zhang R Regulatory non-coding RNAs : revolutionizing the RNA world 3915–3923 (2014). doi: 10.1007/s11033-014-3259-6 [DOI] [PubMed] [Google Scholar]
- 6.Rnas A, Daugaard I & Hansen TB Biogenesis and Function of. Trends Genet 33, 208–219 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Sand M The Pathways of miRNA Maturation (Humana Press, 2014). [DOI] [PubMed] [Google Scholar]
- 8.Mendell JT & Olson EN Review MicroRNAs in Stress Signaling and Human Disease. Cell 148, 1172–1187 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amin M et al. Cell-free microRNAs as cancer biomarkers : the odyssey of miRNAs through body fluids. Med Oncol 31, (2014). [DOI] [PubMed] [Google Scholar]
- 10.Schwarzenbach H, Nishida N, Calin GA & Pantel K Clinical relevance of circulating cell-free microRNAs in cancer. Nat. Publ. Gr 11, 145–156 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Santangelo A, Tamanini A, Cabrini G & Dechecchi MC Circulating microRNAs as emerging non-invasive biomarkers for gliomas. Ann. Transl. Med 5, 1–8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nouraee N & Mowla SJ miRNA therapeutics in cardiovascular diseases : promises and problems. Front. Genet 6, 1–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rottiers V & Naar A Nature Reviews Mol Cell Bio. Focus Metab 13, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Correia CN, Nalpas NC, Mcloughlin KE, Browne JA & Machugh DE Circulating microRNAs as Potential Biomarkers of infectious Disease. Front. Immunol 8, 1–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma V & Misteli T Non-coding RNAs in DNA damage and repair. FEBS Lett 587, 1832–1839 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang C & Peng G Non-coding RNAs: An emerging player in DNA damage response. Mutat. Res. - Rev. Mutat. Res 763, 202–211 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Leung C, Li S, Chen T, Ho M & Hu L Comprehensive microRNA profiling of prostate cancer cells after ionizing radiation treatment. Oncol. Rep 31, 1067–1078 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beer L et al. High dose ionizing radiation regulates micro RNA and gene expression changes in human peripheral blood mononuclear cells. BMC Genomics 15, 1–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joly-tonetti N, Viñuelas J, Gandrillon O & Lamartine J Differential miRNA expression profiles in proliferating or differentiated keratinocytes in response to gamma irradiation. BMC Genomica 14, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shin S et al. MicroRNAs are significantly influenced by p53 and radiation in HCT116 human colon carcinoma cells. Int. J. Oncol 34, 1645–1652 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Chaudhry MA, Omaruddin RA, Brumbaugh CD, Tariq MA & Pourmand N Identification of radiation-induced microRNA transcriptome by next-generation massively parallel sequencing. J. Radiat. Res 54, 808–822 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding N et al. Detection of Novel Human MiRNAs Responding to X-ray Irradiation. J. Radiat. reaearch 52, 425–432 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Sokolov MV, Panyutin IV & Neumann RD Unraveling the Global microRNAome Responses to Ionizing Radiation in Human Embryonic Stem Cells. PLosOne 7, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cui W, Ma J, Wang Y & Biswal S Plasma miRNA as Biomarkers for Assessment of Total- Body Radiation Exposure Dosimetry. PLoS One 6, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacob NK et al. Identification of Sensitive Serum microRNA Biomarkers for Radiation Biodosimetry. PLoS One 8, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Acharya SS et al. Serum microRNAs are early indicators of survival after radiation-induced hematopoietic injury. Sci. Transl. Med 7, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aryankalayil MJ et al. Radiation-Induced Long Noncoding RNAs in a Mouse Model after Whole-Body Irradiation. Radiat. Res RR14891.1 (2018). doi: 10.1667/RR14891.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hastie T, Tibshirani R & Friedman J Springer Series in Statistics The Elements of Statistical Learning (Springer; ). [Google Scholar]
- 29.Cortes C & Vapnik V Support-Vector Networks. Mach. Learn 20, 273–297 (1995). [Google Scholar]
- 30.Kuhn M, and contributions from Jed Wing SW caret: Classification and Regression Training (R package version 6.0–78). retrevied from https://cran.r-project.org/package=caret.
- 31.Tibshirani R, Hastie T, Narasimhan B, Chu G Class Prediction by Nearest Shrunken Centroids, with Applications to DNA Microarrays. Stat. Sci 18, 104–117 (2003). [Google Scholar]
- 32.Ha M & Kim VN Regulation of microRNA biogenesis. Nat. Publ. Gr 15, 509–524 (2014). [DOI] [PubMed] [Google Scholar]
- 33.D’Amato NC, Howe EN & Richer JK MicroRNA regulation of epithelial plasticity in cancer. Cancer Lett 341, 46–55 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Monticelli S MicroRNAs in T helper cell differentiation and plasticity. Semin. Immunol 25, 291–298 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Zoni E, van der Pluijm G, Gray PC & Kruithof-de Julio M Epithelial Plasticity in Cancer: Unmasking a MicroRNA Network for TGF-β-, Notch-, and Wnt-Mediated EMT. J. Oncol 2015, 198967 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao L, Liu W, Xiao J & Cao B The role of exosomes and ‘ exosomal shuttle microRNA ‘ in tumorigenesis and drug resistance. Cancer Lett 356, 339–346 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Valentino A, Reclusa P, Sirera R & Giallombardo M Exosomal microRNAs in liquid biopsies : future biomarkers for prostate cancer. Clin. Transl. Oncol (2016). doi: 10.1007/s12094-016-1599-5 [DOI] [PubMed] [Google Scholar]
- 38.Hu H & Gatti RA MicroRNAs : new players in the DNA damage response. J. Mol. Cell Biol 3, 151–158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simone NL et al. Ionizing Radiation-Induced Oxidative Stress Alters miRNA Expression. PLoS One 4, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vincenti S et al. HUVEC Respond to Radiation by Inducing the Expression of Pro-angiogenic MicroRNAs HUVEC Respond to Radiation by Inducing the Expression of Pro-angiogenic MicroRNAs. Radiat. Res 175, 535–546 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Lee Y et al. MicroRNA142–3p Promotes Tumor-Initiating and Radioresistant Properties in Malignant Pediatric Brain Tumors. Cell Transplant 23, 669–690 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Arora H, Qureshi R, Jin S, Park A & Park W miR-9 and let-7g enhance the sensitivity to ionizing radiation by suppression of NF κ B1. Exp. Mol. Med 43, 298–304 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu C et al. MiR-34a in Age and Tissue Related Radio-Sensitivity and Serum miR-34a as a Novel Indicator of Radiation Injury. Int J Biol Sci 7, 221–233 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.John-Aryankalayil M et al. Fractionated Radiation Therapy Can Induce a Molecular Profile for Therapeutic Targeting. Radiat. Res 174, 446–458 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Olive Virginie, Qijing Li LH miR-17–92, a polycistronic oncomir with pleiotropic functions. Immunol Rev 253, 158–166 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan H et al. Repression of the miR-17–92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J 28, 2719–2732 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mogilyansky E & Rigoutsos I The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ 20, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karimian A, Ahmadi Y & Yousefi B Multiple functions of p21 in cell cycle , apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst) 42, 63–71 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Stevens C & Thangue NB La. E2F and cell cycle control: a double-edged sword. Arch. Biochem. Biophys 412 412, 157–169 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Worby CA & Dixon JE PTEN. Annu. Rev. Biochem 83, 641–69 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Palayoor ST, John-aryankalayil M, Makinde AY & Falduto MT Differential Expression of Stress and Immune Response Pathway Transcripts and miRNAs in Normal Human Endothelial Cells Subjected to Fractionated or Single-Dose Radiation. Mol. Cancer Res 12, 1002–1016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Griffiths-Jones S MiRBase: MicroRNA sequences and annotation. Curr. Protoc. Bioinforma 34, 1291–12910 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Kozomara A & Griffiths-Jones S MiRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42, 68–73 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A & Enright AJ miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 34, D140–D144 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kozomara A & Griffiths-Jones S miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39, D152–D157 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Griffiths-Jones S, Saini HK, van Dongen S & Enright AJ miRBase: tools for microRNA genomics. Nucleic Acids Res 36, D154–D158 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith CL, Blake JA, Kadin JA, Richardson JE & Bult CJ OUP accepted manuscript. Nucleic Acids Res 46, D836–D842 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.