

Abstract
Background
Wastewater treatment plants (WWTPs) are considered hotspots for the environmental dissemination of antimicrobial resistance (AMR) determinants. Vancomycin-Resistant Enterococcus (VRE) are candidates for gauging the degree of AMR bacteria in wastewater. Enterococcus faecalis and Enterococcus faecium are recognized indicators of fecal contamination in water. Comparative genomics of enterococci isolated from conventional activated sludge (CAS) and biological aerated filter (BAF) WWTPs was conducted.
Results
VRE isolates, including E. faecalis (n = 24), E. faecium (n = 11), E. casseliflavus (n = 2) and E. gallinarum (n = 2) were selected for sequencing based on WWTP source, species and AMR phenotype. The pangenomes of E. faecium and E. faecalis were both open. The genomic fraction related to the mobilome was positively correlated with genome size in E. faecium (p < 0.001) and E. faecalis (p < 0.001) and with the number of AMR genes in E. faecium (p = 0.005). Genes conferring vancomycin resistance, including vanA and vanM (E. faecium), vanG (E. faecalis), and vanC (E. casseliflavus/E. gallinarum), were detected in 20 genomes. The most prominent functional AMR genes were efflux pumps and transporters. A minimum of 16, 6, 5 and 3 virulence genes were detected in E. faecium, E. faecalis, E. casseliflavus and E. gallinarum, respectively. Virulence genes were more common in E. faecalis and E. faecium, than E. casseliflavus and E. gallinarum. A number of mobile genetic elements were shared among species. Functional CRISPR/Cas arrays were detected in 13 E. faecalis genomes, with all but one also containing a prophage. The lack of a functional CRISPR/Cas arrays was associated with multi-drug resistance in E. faecium. Phylogenetic analysis demonstrated differential clustering of isolates based on original source but not WWTP. Genes related to phage and CRISPR/Cas arrays could potentially serve as environmental biomarkers.
Conclusions
There was no discernible difference between enterococcal genomes from the CAS and BAF WWTPs. E. faecalis and E. faecium have smaller genomes and harbor more virulence, AMR, and mobile genetic elements than other Enterococcus spp.
Keywords: Vancomycin resistant Enterococcus (VRE), Enterococci, Genomics, Antimicrobial resistance, Mobilome, Genome size, Wastewater, Pangenome, Biomarker
Background
Enterococci are ubiquitous in nature and can be found in a variety of environments, including soil, plants, surface water, wastewater, food, and the gastrointestinal tract of animals and humans [43, 60]. Enterococcus faecalis and Enterococcus faecium, are associated with a variety of clinical infections of the urinary tract, heart, surgical wounds, bloodstream and neonates [67] as well as indicators of fecal contamination [10]. The ability to treat infections caused by Enterococcus spp. is hindered by the development and spread of antimicrobial resistance (AMR) [1]. Resistance to antimicrobials of last resort, such as vancomycin, impairs the control of enterococcal infections and is usually accompanied by resistance to other antimicrobials [24, 32].
Enterococci and antimicrobials are excreted in urine and feces, and in urbanized developed nations, most of this waste is transported to and treated in wastewater treatment plants (WWTPs) prior to discharge into surface waters. WWTPs could be considered points of control for the environmental dissemination of AMR and ideal environments to investigate the epidemiology of AMR from a “One Health” perspective [2, 44, 57]. Within this environment, enterococci can not only exchange genes coding for AMR, but also for heavy metal resistance as well as other genes that increase persistence and survival in other environments [3]. This outcome can facilitate the broader dissemination of AMR genes [2]. Comparative genomics has been applied to identify genes responsible for virulence, AMR, metabolism, secondary metabolite production and gene mobility. Comparative genomics can also be used to compare genes from other functional categories, to predict the ecological fitness of strains, and to discern evolutionary relationships among species.
We previously isolated a number of species of enterococci from two WWTPs with different treatment processes, a conventional activated sludge (CAS) and a biological aerated filter (BAF) system, with E. faecalis being the dominant species identified [61]. This work demonstrated changes in AMR phenotypes between wastewater enterococci before and after treatment and between WWTPs. In the current study, we selected 39 wastewater enterococci for sequencing out of 1111 enterococci isolated, including 308 that exhibited vancomycin resistance in broth culture. Isolates were selected so as to be representative of before and after treatment in both WWTPs [61]. We hypothesized that the genomes would not cluster by treatment process but genomes from the BAF system may contain more biofilm-related genes than those from the CAS system. We also proposed that there would be more virulence, AMR, and genetic mobility genes in E. faecalis and E. faecium than other Enterococcus spp. and that the larger genomes in these clinically relevant species would correlate with the number of mobile genetic elements and genes conferring fitness for survival in a broader range of environments.
Results
Sequence statistics and Pan-genomic analysis
A summary of sequencing statistics for the 39 Enterococcus spp. genomes can be found in Table 1. The genomes ranged from 2.48–3.54 Mbp. The genomes of E. casseliflavus and E. gallinarum (3.37–3.54 Mbp, 3161–3344 genes) were larger than those of E. faecalis (2.69–3.09 Mbp, 2528–3051 genes) and E. faecium (2.48–3.02 Mbp, 2374–2992 genes). The GC content of the genomes ranged from 37.3–37.7%, 37.5–38.1%, and 40.4–42.9% for E. faecalis, E. faecium, and E. casseliflavus/E. gallinarum, respectively.
Table 1.
Genome Characteristics of Enterococcus spp. Isolated from Municipal Wastewater Treatment Plants
| Strain | Location | Species | # of Contigs | Size (bp) | %GC | Genes | CDSs | ST* |
|---|---|---|---|---|---|---|---|---|
| B72 | BAF FE | E. casseliflavus | 32 | 3,538,396 | 42.8 | 3344 | 3283 | NA |
| B79 | BAF FE | E. casseliflavus | 49 | 3,527,325 | 42.9 | 3327 | 3268 | NA |
| W41 | CAS PE | E. faecalis | 21 | 2,693,209 | 37.7 | 2528 | 2471 | 116 |
| B139 | BAF FE | E. faecalis | 30 | 2,720,730 | 37.7 | 2553 | 2496 | 138/501 |
| W314 | CAS PE | E. faecalis | 11 | 2,721,427 | 37.6 | 2583 | 2524 | 277 |
| C34 | CAS PE | E. faecalis | 13 | 2,731,087 | 37.6 | 2615 | 2556 | 715 |
| R95 | BAF PE | E. faecalis | 38 | 2,761,310 | 37.6 | 2596 | 2538 | 674 |
| W350 | CAS FE | E. faecalis | 28 | 2,789,796 | 37.6 | 2731 | 2673 | 84 |
| R76 | BAF FE | E. faecalis | 51 | 2,800,339 | 37.6 | 2690 | 2627 | 16 |
| C106 | CAS PE | E. faecalis | 24 | 2,817,683 | 37.4 | 2670 | 2610 | 16 |
| B48 | BAF PE | E. faecalis | 29 | 2,822,491 | 37.5 | 2701 | 2641 | 16 |
| B168 | BAF PE | E. faecalis | 39 | 2,834,215 | 37.5 | 2725 | 2667 | 21 |
| W191 | CAS FE | E. faecalis | 22 | 2,839,739 | 37.6 | 2745 | 2684 | 207 |
| W460 | CAS FE | E. faecalis | 27 | 2,848,194 | 37.4 | 2733 | 2674 | 672 |
| H120S2 | BAF PE | E. faecalis | 21 | 2,853,021 | 37.4 | 2731 | 2670 | 16 |
| C33 | CAS PE | E. faecalis | 19 | 2,860,595 | 37.3 | 2722 | 2662 | 16 |
| R378 | BAF FE | E. faecalis | 50 | 2,892,126 | 37.5 | 2858 | 2796 | 326 |
| W75 | CAS FE | E. faecalis | 59 | 2,901,424 | 37.5 | 2838 | 2776 | 209 |
| B6 | BAF FE | E. faecalis | 48 | 2,906,126 | 37.5 | 2796 | 2738 | 26 |
| R395 | BAF PE | E. faecalis | 35 | 2,951,239 | 37.5 | 2844 | 2785 | 40 |
| W195 | CAS FE | E. faecalis | 30 | 2,970,793 | 37.5 | 2865 | 2806 | 40 |
| H114S2 | BAF PE | E. faecalis | 35 | 2,979,979 | 37.4 | 2881 | 2822 | 40 |
| C379 | CAS FE | E. faecalis | 42 | 2,988,783 | 37.5 | 2902 | 2844 | 40 |
| R61 | BAF FE | E. faecalis | 37 | 3,004,659 | 37.3 | 2969 | 2907 | 16 |
| B150 | BAF FE | E. faecalis | 41 | 3,012,117 | 37.3 | 2955 | 2893 | 16 |
| W37 | CAS PE | E. faecalis | 68 | 3,088,982 | 37.3 | 3051 | 2990 | 768 |
| C329 | CAS FE | E. faecium | 63 | 2,480,628 | 38.1 | 2374 | 2305 | 40 |
| B466 | BAF FE | E. faecium | 71 | 2,553,406 | 38.1 | 2512 | 2443 | 672 |
| C567 | CAS FE | E. faecium | 21 | 2,778,016 | 37.9 | 2714 | 2644 | 1216 |
| C12d | CAS PE | E. faecium | 169 | 2,879,332 | 37.7 | 2823 | 2755 | 18 |
| H101S2 | BAF PE | E. faecium | 177 | 2,884,826 | 37.8 | 2836 | 2770 | 18 |
| H123S2 | BAF PE | E. faecium | 170 | 2,912,775 | 37.7 | 2871 | 2803 | 18 |
| H53S1 | CAS PE | E. faecium | 165 | 2,959,005 | 37.6 | 2915 | 2847 | 18 |
| F11j | CAS PE | E. faecium | 181 | 2,964,368 | 37.6 | 2916 | 2848 | 18 |
| R407 | BAF PE | E. faecium | 176 | 3,005,175 | 37.5 | 2955 | 2887 | 18 |
| B492 | BAF FE | E. faecium | 189 | 3,008,305 | 37.5 | 2961 | 2893 | 18 |
| R337 | BAF PE | E. faecium | 195 | 3,023,784 | 37.5 | 2992 | 2924 | 18 |
| W17 | CAS FE | E. gallinarum | 14 | 3,367,806 | 40.5 | 3161 | 3102 | NA |
| G12 s | CAS PE | E. gallinarum | 30 | 3,442,529 | 40.4 | 3303 | 3244 | NA |
BAF: biological aerated filter, CAS: conventional activated sludge, PE: primary effluent, FE: final effluent, % GC: guanine-cytosine content, CDS: protein coding sequence, ST: sequence type, NA: not available
The range in contigs generated during sequencing was greater in E. faecium (21–195 contigs) than in other species (11–68 contigs), likely due to the presence of repetitive and insertion genetic elements complicating assembly [54]. Genome sizes were greater for vancomycin and multi-drug resistant strains of E. faecium (3.04 Mbp) than for susceptible strains (2.60 Mbp). The genome size of vancomycin-resistant and multi-drug resistant E. faecalis was similar to their susceptible counterparts.
The E. faecalis pangenome consisted of 5708 genes with a core of 2054 genes (36%), a soft-core of 91 genes (1.6%), a shell genome of 1193 (20.9%) and a cloud genome of 2370 genes (41.5%; Fig. 1 a). The E. faecium pangenome consisted of 3950 genes with a core of 1959 genes (49.6%), a shell of 1186 genes (30%) and a cloud genome of 805 genes (20.4%; Fig. 1 b).
Fig. 1.

Enterococcus faecalis (a) and Enterococcus faecium (b) pan-genome illustrated as a matrix with the core SNP tree of the strains on the left and a presence (blue) and absence (white) matrix of core and accessory genes
Multi-locus sequence typing
In the current study, 4 sequence types (STs) for E. faecium and 15 STs for E. faecalis were identified (Table 1). Eight E. faecium genomes belonged to ST18, part of the clonal complex 17 (CC-17). Out of the E. faecalis STs identified in this study, ST16 (n = 7) and ST40 (n = 4) were the most common.
Phenotypic antimicrobial resistance profiles
Sequenced enterococci exhibited a number of phenotypic antimicrobial resistant profiles, with some isolates being resistant to as many as seven antimicrobials (Table 2). VANR, TECR, AMPR, ERYR were among the most common resistant phenotypes found in enterococci.
Table 2.
Phenotypic Antimicrobial Resistance Profiles of sequenced Enterococcus spp isolates
| Isolate | AMR Phenotype* |
|---|---|
| E. casseliflavus B72 | ERYI, Q-DR |
| E. casseliflavus B79 | VANI, ERYI, Q-DI |
| E. faecalis W460 | ERYI, Q-DR |
| E. faecalis W350 | DOXR, ERYI, LZDI |
| E. faecalis W191 | DOXR, Q-DR |
| E. faecalis H120S2 | ERYR, STRI |
| E. faecalis H114S2 | VANR, TECR, AMPR, LVXR |
| E. faecalis C34 | VANR, TECR, ERYI, LZDI, Q-DR |
| E. faecalis C33 | TECR, DOXR, ERYR, LZDI,LVXR, Q-DR |
| E. faecalis B168 | DOXI, ERYI, LZDI, Q-DR |
| E. faecalis B150 | TECR, DOXI, ERYR, LZDR, Q-DR |
| E. faecalis B6 | DOXR, ERYI, Q-DR |
| E. faecalis B48 | DOXR, ERYR, LZDR, Q-DR |
| E. faecalis B139 | VANI, LZDI, Q-DR |
| E. faecalis C106 | TECR, DOXR, ERYR, GENR, Q-DR |
| E. faecalis C379 | DOXR, ERYI, Q-DR |
| E. faecalis R61 | VANI, TECR, DOXR, ERYR, GENI, LZDI, Q-DR |
| E. faecalis R76 | DOXR, ERYR, GENR, Q-DR, STRR |
| E. faecalis R95 | DOXI, ERYR, GENI, LZDI, Q-DR, STRR |
| E. faecalis R378 | ERYI, Q-DI |
| E. faecalis R395 | VANI, TECR, DOXR, ERYI, Q-DR |
| E. faecalis W37 | DOXI, ERYR, LVXR, Q-DI, STRR |
| E. faecalis W41 | VANI, TECR, DOXR, ERYI, Q-DR |
| E. faecalis W75 | VANI, Q-DI |
| E. faecalis W195 | DOXI, ERYI, Q-DR |
| E. faecalis W314 | TECR, ERYI, LZDR, Q-DR |
| E. faecium R407 | VANR, TECR, AMPR, ERYR, GENR, LVXR, STRR |
| E. faecium R337 | VANR, TECR, AMPR, ERYR, NITI, LVXR, STRR |
| E. faecium H53S1 | VANR, TECR, AMPR, ERYR, LVXR, STRI |
| E. faecium F11 J | VANR, TECR, AMPR, ERYR, LVXR, STRR |
| E. faecium C329 | TECR, ERYI, NITR, LZDI, LVXR |
| E. faecium B492 | VANR, TECR, AMPR, ERYR, LVXR, STRR |
| E. faecium B466 | AMPR, DOXR, ERYR, NITI, LVXI |
| E. faecium C12D | VANR, TECR, AMPR, ERYI, LVXR |
| E. faecium C567 | ERYR, NITI, LZDI |
| E. faecium H101S2 | VANR, TECR, AMPR, ERYR, LVXR, STRI |
| E. faecium H123S2 | VANR, TECR, AMPR, ERYR, LVXR, STRI |
| E. gallinarum W17 | VANI |
| E. gallinarum G12S | VANI |
a AMR phenotypic profiles using R for resistant to the antimicrobial and I for intermediately resistant to the antimicrobial. Antimicrobials used for disc susceptibility testing were vancomycin (VAN), teicoplanin (TEC), amipicillin (AMP), doxycycline (DOX), erythromycin (ERY), levofloxacin (LVX), linezolid (LZD), nitrofurantoin (NIT), gentamicin (GEN), streptomycin (STR), quinupristin/dalfopristin (Q-D), and tigecycline (TGC)
Phylogeny
Genomes did not cluster based on WWTP, but all species formed separate monophylogenetic groups (Fig. 2). The majority of wastewater E. faecalis isolates were more closely related to livestock and food-derived E. faecalis genomes, while seven wastewater strains (B139, B168, C34, W37, W75, W191, and W314) clustered with strains isolated from human infections (Fig. 3). None of the E. faecalis wastewater, human, and agriculture (and food-derived) isolates clustered together by source, suggesting that agricultural and human clinical strains are phylogenetically distinct. Vancomycin-resistant E. faecalis isolates also did not cluster as they belonged to different STs, unlike vancomycin-resistant E. faecium, which did cluster as all isolates belonged to CC-17 (Fig. 4). For E. faecium, wastewater strains clustered separately from most clinical strains (Fig. 4). The bovine strain E. faecium F1129F clustered with human clinical strains, whereas the other bovine strain, E. faecium F1213D did not. Three wastewater isolates (E. faecium C567, E. faecium B466, and E. faecium C329) were more closely related to E. faecium F1213D (bovine) and E. faecium NRRL B-2354 (food) than to clinical isolates.
Fig. 2.

Phylogenetic Tree of all Enterococcus spp. isolated from wastewater using Enterococcus faecalis ATCC 29212 as the reference genome
Fig. 3.

Phylogenetic tree of Entercoccus faecalis genome sequences from the present study and complete genome sequences from the NCBI GenBank database based on analysis of single-nucleotide variants (SNVs) of the core genes. Enterococcus faecalis ATCC29212 was used as the reference genome. Origin of Isolates are as indicated in the figures and are grouped by colour into clinical (red), agricultural/food (green) and wastewater/water (blue) groups
Fig. 4.

Phylogenetic tree of Entercoccus faecium genome sequences from the present study and genome sequences from the NCBI GenBank database based on analysis of single-nucleotide variants (SNVs) of the core genes. Entercoccus faecium DO served as the reference genome. Origin of isolates are as indicated in the figures and are grouped by colour into clinical (red), agricultural/food (green) and wastewater/water (blue) groups
Clusters of orthologous groups (COGs): functional categories and genome size
Clusters of Orthologous Groups (COGs) are broad functional categories used to assign proteins to their specific function [69]. Functional categorization of proteins into different COGs revealed variation profiles among Enterococcus spp., but little difference among strains within species, with the exception of the mobilome and genes associated with energy production and conversion (Additional file 1, sheet 6). We assessed which functional categories of genes were disproportionately represented in the isolates collected from the WWTPs with expanded genomes.
Given the variation in genome size between and within species, the relationships between genome size and the number of genes associated with specific functional categories was determined (Fig. 5; Additional file 1, Sheet 6). There were more COGs assigned to carbohydrate transport and metabolism, transcription, cell motility, secondary metabolite biosynthesis, transport, catabolism and signal transduction mechanisms in E. casseliflavus and E. gallinarum compared to enterococci more frequently associated with clinical infections.
Fig. 5.
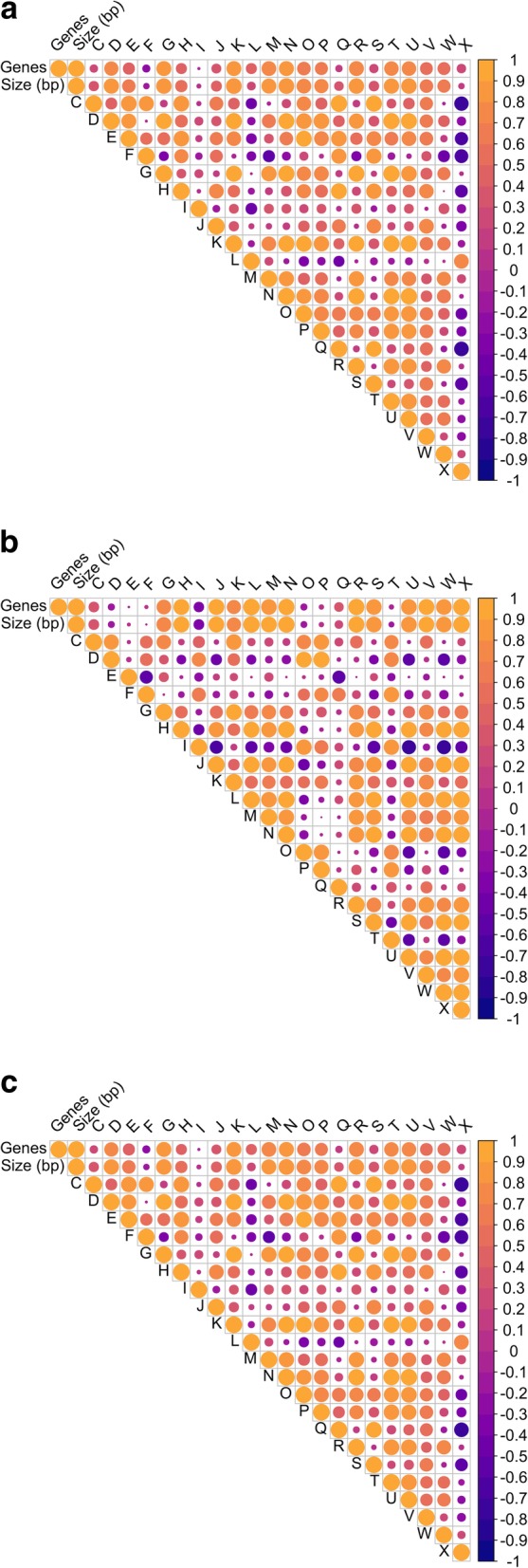
Correlation plots showing the correlations between different COG categories, genome size and number of genes in all of the pooled Enterococcus species (a), E. faecalis, (b) and E. faecium (c)
When all of the wastewater Enterococcus genomes were pooled, there was a strong negative correlation (p < 0.001) between genome size and nucleotide transport and metabolism, lipid metabolism and translation, ribosomal structure and biogenesis and a strong positive correlation (p < 0.001) between genome size and cell motility (Fig. 5 a; Additional file 1, sheet 6). The total number of genes related to cell motility, signal transduction, and carbohydrate transport and metabolism were positively correlated (p < 0.001) with genome size. This is reflective of the greater genome size of environmental species compared to E. faecium and E. faecalis. The total number of genes related to cell division and chromosome partitioning, cell envelope biogenesis, outer membrane and post translational modification, protein turnover, and transcription were negatively correlated (p < 0.001) with genome size.
The species-specific patterns in genomic proportions for each functional category differ from the pooled genomes for the genus. In both E. faecalis and E. faecium, a larger genome was strongly correlated with the mobilome (p < 0.001) (Fig. 5 b and c), a functional category not included in the analysis of Konstantinidis and Tiedje [34]. In contrast, the mobilome was not correlated with genome size in the pooled Enterococcus genomes. There was also a positive correlation (p = 0.005) between the number of unique AMR genes and genome size of E. faecium, suggesting the acquisition of AMR genes occurs through horizontal gene transfer. For example, E. faecium R337 had a genome of 3.02 kbp, 58 genes associated with the mobilome and 23 AMR genes; while E. faecium C329 had a genome of 2.48kbp and 15 genes associated with the mobilome and 3 AMR genes.
The total number of genes related to cell motility (p < 0.001), DNA replication, recombination, and repair (p < 0.001), extracellular structures (p < 0.001), and mobilome (p < 0.001) was positively correlated with genome size in E. faecium. The number of AMR genes also showed a positive correlation (p = 0.002) with the amount of genes related to the mobilome in this species (Fig. 5 c). The eight E. faecium genomes belonged to the same sequence type (CC-17), while E. faecalis genomes were more diverse.
Antimicrobial resistance genes
In this study, we screened 39 multi-antimicrobial resistant enterococci genomes against the CARD database for antimicrobial resistance genes (ARGs) (Additional file 1, Sheet 8) and ten genes (eatAv, emeA, lsaA, efrA, efrB, tetL, efmA, msrC, ermY, and lsaE) associated with multi-drug efflux pumps and other transporters were detected. These efflux proteins may confer intermediate resistance to a variety of antimicrobials.
Genes conferring glycopeptide (vancomycin and teicoplanin) resistance were detected in 20 of the genomes. In E. faecium and E. faecalis, resistance was conferred by vanA and vanM in E. faecium or vanG in E. faecalis. Vancomycin resistance was mediated by vanC, and this was the only ARG detected, in E. casseliflavus and E. gallinarum.
ErmB confers resistance to macrolide-lincosamide-streptogramin B (MLSB) antimicrobials and was found in E. faecium (n = 7) and E. faecalis (n = 4). Other erm genes (ermC, ermG, ermT, ermY) were detected in one E. faecium genome. msrC, which codes for a macrolide efflux pump, was only detected in E. faecium (n = 11). The most common macrolide resistance gene detected in enterococcal genomes was ermB (n = 15).
Thirteen of the enterococci isolates were resistant to high concentrations of gentamicin and streptomycin. In our study, cross-resistance to levofloxacin and the aminoglycosides (gentamicin and streptomycin) occurred in 5 isolates with 3 additional isolates exhibiting intermediate resistance to one or more of these antimicrobials. In our study, additional aminoglycoside genes (ant(9′)-Ia, aad(6′), aph(3′)-IIIa, SAT-4, ant(6′)-Ia, and aac(6′)-Ie-aph(2″)-Ia) were detected in the genomes of up to 5 E. faecalis and 7 E. faecium aminoglycoside resistant isolates. Gentamicin resistance arises as the result of the acquisition of aac(6′)-Ie-aph(2″)-Ia, which was detected in 7 genomes (2 E. faecalis and 5 E. faecium) and confers resistance to all aminoglycosides except streptomycin [42]. The prevalence of streptomycin resistance versus gentamicin resistance differed between species, with streptomycin resistance being more common in E. faecium and gentamicin resistance more common in E. faecalis.
Genes encoding tetracycline resistance were detected in 26 of the genomes, including E. faecium and E. faecalis. In this study, determinants for macrolide and tetracycline were detected together in 16 of the enterococcal genomes. Genes associated with resistance to antimicrobials not included in the disc susceptibility panel were also detected. A gene associated with chloramphenicol resistance, cat, was detected in two E. faecalis genomes. Genes associated with diaminopyrimidine resistance (dfrE, dfrF, and dfrG) were also detected in E. faecium and E. faecalis. Two E. faecalis genomes also had genes associated with lincosamide resistance (InuB and InuG).
Virulence genes
The number of shared virulence genes among genomes of the same species were 16, 6, 5 and 3 for E. faecium, E. casseliflavus, E. faecalis, and E. gallinarum, respectively (Additional file 1, Sheet 9–11). All of the E. faecium isolates contained genes related to adhesion to surfaces (tuf, aga, efaA, and sgrA), cell wall biosynthesis (phosphatase cytidylyltransferase, uppS), cellular defense (lisR), biofilm formation and surface proteins (acm, esp, scm and type A and B pili). Other functions including bile salt degradation (bsh), proteases (tip/ropA), biofilm formation (bopD), enolase (eno), and antiphagocytosis and capsule formation (rfbA-1) were also identified. All of the E. faecalis genomes contained genes for cell adhesion (tuf), carbohydrate metabolism (hyl), endocarditic and biofilm association (ebp) pili (ebpA), Type III secretion proteins (bopD) and fibrinogen-binding proteins (fss1). All of the E. casseliflavus genomes contained the same five virulence genes with functions of: capsule biosynthesis (capE), enolase (eno), leucine aminopeptidase (lap), heat shock protein (hsp60), and protein modification (lplA1). All of the E. gallinarum genomes had an enolase (eno), a flagellar biosynthesis protein (flhA) and a bile salt hydrolase (bsh). One of the E. gallinarum genomes also contained genes related to capsule proteins and another isolated from effluent possessed 2 genes associated with metal transporter (ssaB and psaA) as well as those associated with the CAS system. Hyaluronidase (hyl) genes were detected in all the E. faecalis genomes.
Mobile gene elements
ICE and transposons present in the genomes were identified and described using the ICEberg database (Table 3; Additional file 1, sheet 17). The transposon, Tn917 was identified in 8 of the sequenced E. faecalis genomes. One transposon, Tn6098 was present in all genomes. A multidrug resistance transposon, Tn5385 was also found in all E. faecalis genomes. Other Tn5801 and Tn6013-like ICE elements of unknown function were also present in all E. faecium isolates, in addition to a cadmium and arsenic resistance ICE, ICESde3396. All of the E. gallinarum and E. casseliflavus isolates had Tn916-type transposons (Tn6079, Tn6087 and Tn6084, respectively). Seven out of the unique 27 ICE were present in genomes of more than one Enterococcus species.
Table 3.
Integrative conjugative elements (ICE) and transposons identified in the wastewater Enterococcus spp. genomes (n = 39)
| Species | Common ICE | Function | Other Notable ICE | Function |
|---|---|---|---|---|
| E. casseliflavus | Tn6098 Tn6084 Tn6000(EfcTn1) | α-galactoside metabolism Tetracycline resistance Tetracycline resistance | No other ICE detected | |
| E. faecalis | Tn6098 Tn5385 | α-galactoside metabolism Erythromycin, gentamicin, streptomycin, tetracycline, penicillin/β-lactam, mercury resistance |
Tn917 Tn2008 Tn1545 ICESp23FST81 Tn6009 ICESde3396 Tn5301 Tn5276 ICESt1 |
Tetracycline resistance Chloramphenicol, erythromycin, streptomycin, kanamycin resistance Sulfamethoxazole, trimethoprim, chloramphenicol, erythromycin, streptomycin resistance Tetracycline, chloramphenicol resistance, toxin-antitoxin system Mercury resistance Kanamycin, arsenic and cadmium resistance Nisin biosynthesis Nisin biosynthesis, sucrose fermentation Type II restriction modification system |
| E. faecium | Tn6098 Tn5801 Tn6084 Tn6000(EfcTn1) ICESauTW20–2 ICESauT0131–2 ICESauJKD6008–2 ICESpsED99–1 ICESauMu3–1 |
α-galactoside metabolism Tetracycline resistance Tetracycline resistance Tetracycline resistance Unknown Unknown Unknown Unknown Unknown |
Tn2008 Tn1545 ICESde3396 ICESt1 ICEAusCo10a-1 10,750-RD.2 |
See above See above See above See above Toxin-antitoxin system Type II restriction modification, spectinomycin, erythromycin resistance |
| E. gallinarum | Tn6098 Tn6079 Tn6087 ICEsde3396 |
α-galactoside metabolism Tetracycline, erythromycin resistance Tetracycline, antiseptic and antimicrobial resistance (unspecified) Kanamycin, arsenic and cadmium resistance |
Tn2008 Tn1545 |
See above See above |
CRISPR-Cas arrays and bacteriophage
Type II CRISPR-Cas systems were detected in 13 E. faecalis genomes (Fig. 6). Orphan CRISPR arrays (without Cas genes) were identified in 27 of the genomes (Fig. 6). Comparison of CRISPR arrays flanked by Cas genes revealed unique arrays among Enterococcus species, but some arrays were shared among strains of the same species. Arrays identified in the sequenced Enterococcus genomes contained 4 to 20 direct repeat sequences associated with functional CRISPR arrays. An additional 72 unique spacers associated with orphan CRISPR arrays were identified in this study. Eleven E. faecalis and 10 E. faecium genomes lacked CRISPR-Cas systems. Any genomes lacking functional arrays exhibited resistance to 4 or more antimicrobial agents.
Fig. 6.

Pictorial of CRISPR-Cas arrays identified in the analysis of wastewater Enterococcus spp (n = 39) genomes. a Functional CRISPR arrays with the organization of direct repeats (diamonds) and spacers (numbers). b Orphan CRISPR arrays direct repeat and spacer organization. c Legend for numbered direct repeats and the genomes with no CRISPR arrays. The outlined genomes are those that contained both functional CRISPR arrays and prophage. The (*) represents genomes that contained no prophage
Functional CRISPR arrays and intact prophage were identified in 10 E. faecalis genomes, but the combination was not seen in the other 29 genome sequenced in this study. Some of the spacer regions identified in CRISPR arrays were 100% identical to incomplete prophage sequences, but these genomes still contained at least one prophage.
Bacteriophage-mediated transduction of AMR has been demonstrated in enterococci and potential virulence determinants have been identified in phage associated with E. faecalis. Phages found in the genomes were members of the Siphoviridae and Myoviridae (Additional file 1, Sheet 12). Thirty-four of the 39 genomes contained at least one putative phage ranging in size from 19.2 kb to 70.6 kb. A total of 55 unique intact prophages were identified across 34 sequenced genomes. E. faecium and E. faecalis contained up to 3 intact prophages, whereas E. casseliflavus and E. gallinarum contained 1 or 2 intact prophages.
Secondary metabolites
Bacteriocins were identified in 8 E. faecalis and 9 E. faecium genomes in addition to 1 E. gallinarum genome (Additional file 1, Sheet 18). Enterocin A was identified in nine E. faecium genomes. Lantipeptides were identified in 3 E. faecalis genomes as cytolysins, which have both haemolytic and bacteriolytic activities [12]. Lassopeptides were identified in 6 E. faecalis genomes. Terpenes were detected in all E. casseliflavus and E. gallinarum, but not in E. faecalis or E. faecium genomes. Aryl polyene was detected in one E. faecalis (C34) genome.
Biomarker search
The small number of genomes limited the identification of biomarkers, particularly for searches within the same species isolated from different sources (Additional file 1, Sheet 19). These biomarkers are genes or gene fragments only present in one group of genomes and not others making them possible identifiers of the origin of collected isolates. The majority of searches have identified biomarkers with scores below a correlation cut-off of 0.95. However, in our study, E. faecalis from wastewater that clustered with agricultural and animal sources revealed a biomarker associated with CRISPR-associated genes that differentiated (score = 0.8043) these isolates from E. faecalis from wastewater that clustered with human sources. A comparison of E. faecium from clinical (inclusion) and wastewater (exclusion) sources yielded 7 biomarkers with scores greater than 0.80. These were associated with phage (n = 6) and hypothetical proteins (n = 1). A search for potential biomarkers that distinguished among species in this study resulted in 98 signatures for E. faecalis, 130 signatures for E. faecium, and 3 signatures E. casseliflavus and E. gallinarum. These signatures include genes related to various types of nucleotide and carbohydrate metabolism, as well as other functions.
Discussion
Sequence statistics and Pan-genomic analysis
There was considerable variation in the size of the genomes and the number of contigs generated by sequencing each genome. The variation in the size of the genomes within a species could be a result of differences in the size of the chromosome and the presence/absence of plasmids. The variation in the number of contigs is likely due to the presence of repetitive and insertion genetic elements complicating assembly [54]. While the number of genomes used to generate the pan-genome in our study was small, the pan-genome of Enterococcus spp. is considered open as it is continually expanding and acquiring new accessory genome elements from other enterococci and bacterial species [80].
Multi-locus sequence typing
In E. faecium, CC-17 is associated with clinical infections and has been detected in treated and untreated wastewater, [13] suggesting that the majority of E. faecium sequenced from wastewater originated from humans. In E. faecalis, ST16 and ST40 have previously been associated with high level gentamicin resistance in clinical isolates and in isolates from pigs [24, 59]. However, high level gentamicin resistance was not found in any E. faecalis with these sequence types. However, only 5 of the isolates in this study (4 E. faecalis and 1 E. faecium) exhibited high level gentamicin resistance. The association of these sequence types and gentamicin resistance may differ between studies because of geographical location, as gentamicin resistance is transferable, and because it may not be present in all ST16 and ST40 E. faecalis isolates.
Phylogeny
The genomes forming monophylogenetic groups support our previous results of speciation of enterococci based on the groESL locus [61, 79]. The diversity of wastewater strains maybe a reflection of their origin from clinical, companion animal or agricultural sources. There was more genetic diversity in vancomycin-resistant E. faecalis than E. faecium. The distinct clustering between clinical and wastewater strains of E. faecium may be due to the large accessory genome and characterization of these genes may provide insight into the mechanisms whereby enterococci adapt to specific environments.
A disproportionate increase in genes associated with energy conversion, regulatory function, transport and secondary metabolism has been noted with expansion in genome size in previous comparative bacterial genomic studies [6, 34, 66]. So, an analysis of the COGs that are over represented in the expanded genomes of E. faecalis and E. faecium was completed to determine if some of these COGs could be increasing the fitness of multi-drug resistant enterococci. This could ultimately increase the risk of infection with these strains and the transfer of virulence and AMR determinants to other bacteria.
In E. casseliflavus and E. gallinarum some COGs were over represented (i.e., carbohydrate transport and metabolism, transcription, cell motility, secondary metabolite biosynthesis, transport, catabolism and signal transduction mechanisms). These functional categories could allow for higher fitness in aquatic environments where more diverse substrates are typically available at much lower concentrations than in the digestive tract. The increase in cell motility related genes may also enable these species to undertake chemotaxis in aquatic environments where nutrients may be scarce [58]. Compared to E. faecalis and E. faecium, these genomes also contained more genes encoding for secondary metabolites including antimicrobial agents. Although these genes are not required for growth, they can confer competitiveness in diverse environments [31]. E. casseliflavus and E. gallinarum are known to be more environmentally fit than E. faecalis and E. faecium as a result of a variety of mechanisms. For instance, the yellow pigment of E. casseliflavus can protect this species from photo-inactivation in aquatic environments [36]. However, E. faecium and E. faecalis are still the predominant species in wastewater, likely due to the continuous input of fecal waste into these systems.
The number of genes related to the mobilome increased with genome size in E. faecium and E. faecalis and this would suggest that the mobilome is a significant factor in the evolution of these bacteria within wastewater, contributing to genomic expansion and diversity. However, there was a lack of diversity in E. faecium isolates compared to E. faecalis, suggesting that E. faecium isolates may be more specifically adapted to clinical environments.
Antimicrobial resistance genes
Vancomycin-resistant enterococci have been known to exhibit resistance to a number of antimicrobials [32, 74]. Enterococci are also intrinsically resistant to beta-lactams, aminoglycosides and streptogramins and can acquire antimicrobial resistance through horizontal gene transfer [32, 42, 74]. There are a variety of ARGs that confer vancomycin resistance, with vanA, vanB and vanC being the most common in wastewater enterococci. The most common determinant for teicoplanin resistance is vanZ, which can be integrated into the van operon, although it is absent in the vanB operon, and confers resistance to both vancomycin and teicoplanin [19]. As a result, teicoplanin resistance is commonly associated with vancomycin resistance. Although rarely, teicoplanin resistance without vancomycin resistance is likely due to changes in the promoter of the van operon or due to the presence of a different resistance mechanism [14, 21, 35].
Resistance to erythromycin and other macrolides can arise as a result of mutations in the 23S rRNA gene or by efflux pumps [42]. Macrolides are used extensively in both humans and animals. Blanch et al. [9] observed that most wastewater isolates with high-level vancomycin resistance were also resistant to erythromycin, suggesting that erythromycin resistance may favour the persistence of VRE in the environment. The modification of the 23S rRNA target by methylase genes, like ermB, can also confer resistance to streptogramins [42].
Enterococci exhibit intrinsic resistance to low concentrations of aminoglycosides as a result of the presence of aac(6′)-Ii. Gentamicin and streptomycin are clinically-important as they are not inactivated by aac(6′)-Ii; and E. faecium are typically sensitive to these antimicrobials [42]. Aside from cross-resistance to other antimicrobial classes, like fluoroquinolones, resistance to these aminoglycosides is likely acquired. Others have shown that aminoglycoside resistance genes are frequently encoded on plasmids and transposons [42]. Streptomycin resistance either involves the inhibition of the drug at the ribosomal level or enzyme inactivation by an acquired streptomycin adenyltransferase [42].
There are multiple tetracycline resistance genes. Tet(L) encodes an efflux protein and tet(M) and tet(S) encode for ribosomal protection proteins. Disk susceptibility testing revealed that these isolates were resistant to doxycycline, whilst those containing tet(L) were susceptible, suggesting specificity for the tet(L) efflux protein. In general, bacteria that are resistant to doxycycline are also resistant to tetracycline and oxytetracycline [26, 56]. Tetracycline resistance can be due to efflux pumps or ribosomal protection mechanisms, which can be chromosomal and/or plasmid-borne. Co-selection of tetracycline and macrolide resistance in environmental enterococci may occur [39, 40].
Virulence genes
The virulence genes detected have additional functions for improved environmental fitness. For instance, the majority of the virulence genes detected in the genomes from this study were also associated with biofilm formation or adherence to surfaces (i.e., ace, acm, agg, bop, ccf, cob, cpd, ebpABC, ecbA, efaA, esp, fsrABC, gelE, pil, scm, sgrA, sprE, and srt). These genes are ubiquitous as they likely play a role in the fitness of enterococci in both the human digestive tract and WWTPs. A number of capsule protein genes were also common among the genomes and not only confer resistance to phagocytosis in humans and animals [48, 50], but also to predation by amoeba and bacteriophage in aquatic environments [51, 73]. Hyaluronidase (hyl) genes have been associated with increased vancomycin resistance and virulence in mouse peritonitis models [50].
Mobile genetic elements
Mobile genetic elements (MGEs) play an important role in horizontal gene transfer and the spread of AMR among isolates in the environment, humans and animal hosts. MGEs include plasmids, transposable elements, prophages and various genomic islands such as integrative conjugative elements (ICE) [71]. The transposon Tn917 is widely distributed in enterococci [64]. All of these strains exhibited erythromycin resistance and erm(B) was found to be associated with Tn1545 and Tn917 [15]. Transposon Tn6098 was in all of the genomes and possessed genes associated with α-galactoside metabolism. Transposon Tn5385 was found in all of the E. faecalis with these isolates exhibiting erythromycin and doxycycline resistance as this transposon commonly carries these resistance genes [53]. Tn916-type transposons found in E. casseliflavus and E. gallinarum can carry genes coding for tetracycline, minocycline and erythromycin resistance [52, 55]. While these transposons were detected in E. casseliflavus and E. gallinarum, they did not exhibit erythromycin resistance and no associated AMR genes were detected in their genomes.
CRISPR-Cas arrays and bacteriophage
Type II CRISPR-Cas systems are typically described in enterococci. Multiple CRISPR arrays can often be detected in bacterial genomes, but not all arrays are accompanied by Cas genes. The absence of CRISPR/Cas systems may compromise genome defence, increasing the likelihood of acquisition of AMR determinants from bacteriophage and plasmids [47]. When a phage infects a bacterium, it incorporates spacers into the array within the bacterial chromosome and occasionally plasmids. The spacers are expressed as CRISPR RNAs (crRNAs) and provide a surveillance mechanism for descendant cells and guide the CRISPR/Cas system to enable cleavage of the protospacer sequence in the phage genome. The cleaved phage genomes are then cannibalized and can no longer support productive phage infection [5, 68]. CRISPR-Cas systems impact the evolution of both bacteria and phage populations. Transduction dependent horizontal gene transfer is a key driver of bacterial evolution and rapid viral evolution to evade CRISPR-Cas systems [68]. CRISPR/Cas arrays can also provide a record of previous and continued interaction between particular bacteria and phage [5, 65]. Spacers may limit the type of phage that can integrate into the genome, but bacteriophage can develop anti-CRISPR systems to promote their integration into the bacterial genome [11].
Phages found in the genomes were members of the Siphoviridae and Myoviridae. Other prophages in Enterococcus spp. belonging to Podoviridae, Inoviridae, Leviridae, Guttaviridae and Fuselloviridae have also been described [18, 41]. Prophages from the Siphoviridae family were the most prevalent across all species and are also commonly identified in lactic acid bacteria [72].
Secondary metabolites
Bacteriocins are ribosomally synthesized antimicrobial peptides produced by Gram-positive and Gram-negative bacteria that have antimicrobial activity against closely related bacteria. They could provide a competitive advantage to the survival of bacteria in ecological niches that exhibit poor nutrient concentrations, heat and extreme pH [78]. Lantipeptides are also a growing class of bacteriocins with a large diversity of activity, structure, and biosynthetic machinery. Lantipeptides have multiple uses including as a limited class of antimicrobials [33]. Terpenes are most often associated with plants and fungi, and have been described in prokaryotes in only a few instances, including Enterococcus spp [7]. Terpenes can have a variety of functions including as antimicrobials, hormones, pigments, and flavor or odour constituents [45], but their role in Enterococcus spp. is unclear. Aryl polyene biosynthetic clusters produce a pigment that protects the organism from reactive oxygen species [62].
Biomarker search
Biomarkers are genes or gene fragments only present in one group of genomes and not others making them possible identifiers of the origin of collected isolates. For instance, Weigand et al. [77] conducted a search within watershed and enteric enterococcal genomes and found shared phenotype and phylogeny between the two groups, but also identified several biomarkers for both sources. These biomarkers encoded accessory nutrient utilization pathways, including a nickel uptake operon and sugar utilization pathways including xylose were overrepresented in enteric genomes [77]. Genes that serve as biomarker for E. casseliflavus and E. gallinarum include genes related to various types of nucleotide and carbohydrate metabolism, and genes with other functions which can improve environmental fitness, including a variety of transporters and housekeeping genes related to DNA replication, transcription and translation.
Conclusions
In this study, enterococci did not cluster phylogenetically based on point of isolation during wastewater treatment or on the type of WWTPs. Despite being the dominant species in wastewater, E. faecalis and E. faecium have smaller genomes and may be less equipped to survive outside of their target niche than E. casseliflavus and E. gallinarum. However, they do harbor more virulence, AMR, and mobile genetic elements than other Enterococcus spp. A larger genome size in E. faecalis and E. faecium was positively correlated with an expansion in the mobilome. In E. faecium, there was a positive correlation between the number of AMR genes and the mobilome. So, while the larger genome size of E. casseliflavus and E. gallinarum is accompanied by more genes related to metabolism and secondary functions, possibly increasing their fitness in the environment, this was not the case for E. faecium and E. faecalis. This study suggests that the key to understanding the impact of WWTPs on AMR dissemination is likely understanding the mobilome and discerning linkages between enterococci in wastewater and other environmental and clinical sources.
Methods
Isolate selection
Thirty-nine Enterococcus spp., including E. faecalis (n = 24), E. faecium (n = 11), E. casseliflavus (n = 2) and E. gallinarum (n = 2), isolated from wastewater were selected for whole genome sequencing. These were selected from a collection of 308 isolates from the primary and final effluents of two WWTPs in Kingston, Ontario, Canada, a BAF and a CAS system between 2014 and 2016. Isolates were speciated and subsequently underwent disc susceptibility testing for a panel of 12 antimicrobial agents. Nine to ten Enterococcus isolates were chosen from each of the primary and final effluent of the two WWTPs to represent the most prominent species isolated from the samples and the most prominent unique antimicrobial resistance phenotypic profiles. While all of these isolates grew in Todd-Hewitt broth supplemented with vancomycin (≥ 4 mg/L), not all met the requirements for vancomycin resistance using disc susceptibility testing following CLSI and EUCAST guidelines. This procedure used reference strains E. faecium ATCC 700221 (MIC ≥32 mg/L), E. faecalis ATCC 51299 (MIC ≥4 mg/L) and E. faecalis ATCC 29212 (susceptible) and Staphylococcus aureus ATCC 25923. The final isolates selected included 21 vancomycin-susceptible, multi-drug resistant enterococci and 18 enterococci with either intermediate resistance or resistance to vancomycin based on disc susceptibility testing. The AMR phenotypic profiles of the selected isolates are available in Table 2.
DNA extraction and sequencing
Enterococcus spp. were grown on Brain Heart Infusion (BHI) agar (Dalynn Biologicals, Calgary, AB) overnight at 37 °C. Colonies from a freshly grown culture plate were suspended in TE buffer to achieve an OD600 of 2 in order to harvest 2 × 109 cells, and 1 mL was transferred to a microcentrifuge tube and centrifuged for 2 min at 14000 x g. Genomic DNA was extracted using a modified DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) with the addition of an enzymatic lysis step. Bacterial cells were incubated at 37 °C with shaking (150 rpm) in lysis buffer consisting of 20 mM Tris-Cl (pH 8.0), 2 mM sodium EDTA, 1.2% Triton X-100 and 40 mg/mL lysozyme (Sigma Aldrich Canada, Oakville, ON). Proteinase K and 5 μL of 100 mg/mL RNase A were added (Qiagen, Hilden, Germany), and the mixture was incubated at room temperature for 10 min before proceeding to the next step. The quality of the genomic DNA was determined using a Nanodrop One UV-Vis Spectrophotometer (Thermo Scientific, Burlington, ON) and a Qubit fluorometer (Thermo Scientific). Genomic library construction was performed using the Illumina Nextera XT DNA sample preparation kit (Illumina Inc., San Diego, CA) following the manufacturer’s instructions. The library was sequenced on an Illumina MiSeq platform (Illumina, Inc.). FASTA data was filtered for quality and high-quality reads were de novo assembled using SPAdes genome assembler 3.6.0 [4] and annotated using Prokka 1.12 ([63].
Comparative analysis
Pangenomic analysis was completed using the contigs extracted from the Genbank file which were re-annotated using Prokka 1.13.3 (Seeman, 2014). This generated GFF files that were used as input to Roary 3.12 [46]. Multi-locus sequence typing (MLST) was performed using online MLST databases. In particular, the Enterococcus faecalis MLST (https://pubmlst.org/ efaecalis/) and Enterococcus faecium MLST (https://pubmlst.org/ efaecium/) based at the University of Oxford [30] and funded by the Wellcome Trust. The phylogenetic trees were constructed based on analysis of single nucleotide variants (SNVs) of the core genes. The phylogenetic analyses were conducted using a single nucleotide variant phylogenomics (SNVPhyl) pipeline [49] using unassembled sequence read data. The paired-end reads for Illumina sequencing of the 39 Enterococcus spp. genomes were aligned to the appropriate reference genome to generate read pileups (SMALT v.0.7.5; http://www.sanger.ac.uk/science/tools/smalt-0). The presence and absence matrices were generated using Phandango [23]. Whole genome sequences of E. faecalis and E. faecium (Additional file 1) were also included in the analysis and were ran through the ART next-generation sequencing read simulator [27] to generate paired-end reads with length and coverage similar to the experimental dataset (2 × 300 base PE and ~50X coverage). The reads were subject to mapping quality filtering (minimum mean mapping quality score of 30) and coverage (15X minimum coverage threshold) estimations. Using a single nucleotide variant (SNV) abundance ratio of 0.75, with no SNV density filtering setting, variant calling, variant consolidation and single nucleotide variant alignment generation of the final phylogeny was run through PhyML [22] using the maximum likelihood method. The resulting tree was visualized using interactive Tree of Life (iTOL) version 4.2.1 (https://itol.embl.de/). Assignment of proteins into clusters of orthologous groups (COGs) was performed using the compare genomes function of DOE Joint Genome Institute Integrated Microbial Genomes & Microbiomes platform [38]. Correlations were calculated using R statistical platform version 3.4.3 (R [16]) and figures were generated using packages Hmisc [25] and corrplot [76].
Draft genome sequences of the 39 Enterococcus spp. were investigated for the presence of putative virulence and AMR genes, mobile gene elements, bacteriophage, and CRISPR/Cas arrays. The contigs of each draft genome were ordered based on alignment against a reference genome using progressive Mauve [17]. Virulence and AMR genes were identified using Virulence Finder version 1.5 [29] and CARD version 2.0.1 [28], respectively. Results for AMR genes were further verified using megaBLAST and hits were manually curated. Genomes were investigated for integrative conjugative elements (ICEs) by homology searches using BLAST against 466 ICEs downloaded from the ICEberg database 1.0 [8]. The genomes were then analyzed for the presence of prophage using PHAST [81]. CRISPR-Cas arrays were identified using the CRISPRdb [20]. Secondary metabolite biosynthetic gene clusters were identified using the Antibiotics and Secondary Metabolite Analysis Shell (antiSMASH) version 3.0 [75].
A biomarker search was carried out with the 39 genomes from this study and an additional 59 genomes retrieved from NCBI using Neptune [37] and a Galaxy instance from the National Microbiology Laboratory in Winnipeg, MB, Canada. The inclusion and exclusion groups are listed in Additional file 1 (Sheet 19). The cut-off score for signatures among species was 95% and the cut-off score for signatures within species from different sources was 80%. The functions related to the genes covered by each signature was identified by mapping the signatures to a reference, then identifying the functions of the genes using UniProt [70]. The reference genomes that were used were E. faecalis V583 (NC_004668), E. faecium DO (NC_017960), and E. casseliflavus B72 (this study).
Supplementary information
Additional file 1: Supplementary Details on Genomic Comparisons. Sheet 1: Complete List of Genomes Used; Sheet 2: Genome Characteristics of Sequenced Genomes; Sheet 3: COG Analysis; Sheet 4: COG Pivot Chart; Sheet 5: COG graphs; Sheet 6: COG correlation and p values; Sheet 7: AMR phenotypic profiles and breakpoints; Sheet 8: AMR genes detected; Sheet 9: Virulence Factors Raw Output; Sheet 10: Virulence Factors by Species; Sheet 11: Virulence Factors Summary; Sheet 12: Phage Detected; Sheet 13: CRISPRfinder Output; Sheet 14: CRISPRfinder Confirmed Systems; Sheet 15: CRISPRdb BLAST Results; Sheet 16: CRISPR Summary Table; Sheet 17: ICEberg Raw Results; Sheet 18: antiSMASH results; Sheet 19: Neptune Biomarker Combinations
Acknowledgements
Thanks to Dr. Chad Liang for his comments on the manuscript.
Abbreviations
- AMR
Antimicrobial resistance
- AntiSMASH
Antibiotics and Secondary Metabolite Analysis Shell
- ARG
Antimicrobial resistance gene
- BAF
Biological aerated filter
- BHI
Blood Heart Infusion
- BLAST
Basic local alignment search tool
- CARD
Comprehensive Antimicrobial Resistance Database
- CAS
Conventional activated sludge
- CLST
Clinical and Laboratory Standards Institute
- COGs
Clusters of orthologous groups
- CRISPR/Cas
Clustered regularly interspaced short palindromic repeats and CRISPR-associated genes
- CRISPRdb
CRISPR database
- CrRNAs
CRISPR RNA
- EUCAST
European Committee on Antimicrobial Resistance Testing
- ICE
Integrated conjugative element
- ITOL
Interactive tree of life
- MGE
Mobile genetic element
- PHAST
Phage search tool
- SNVPhyl
Single nucleotide variants phylogenomics pipeline
- ST
Sequence type
- VRE
Vancomycin-resistant enterococci
- WWTP
Wastewater treatment plant
Authors’ contributions
This work was part of a study that contributed to a PhD thesis by HS. HS, TAM, RSB and SNL designed the experiment. HS extracted the DNA and prepared samples for sequencing. NG sequenced the isolates and KKA selected the platform, sequencing depth / pipeline and contributed funding for sequencing. RZ and ROP performed the assembly, annotation, and phylogenetic analyses of genomes. HS and ROP performed the bioinformatics analysis. HS analyzed the results and drafted the manuscript. TAM RSB, AM and SNL provided funding or equipment necessary to complete the work. AM and RSB selected waste water sampling techniques and contributed to the methodology used to collect isolates. TAM and SNL contributed to the drafting of the manuscript and all authors read and approved the final manuscript for submission.
Funding
Authors are grateful to the Natural Sciences and Engineering Research Council of Canada (NSERC) for financial support, Queen’s University, Public Health Ontario and Agriculture and Agri-food Canada. NSERC, Queen’s University and Public Health Ontario provided funding or equipment vital for the isolation of isolates from wastewater. Agriculture and AgriFood Canada hosted HS at the Lethbridge Research and Development Center (Lethbridge, Alberta) where the isolates were processed, DNA was extracted and the sequencing and analysis took place. HS received Ontario Graduate Student Scholarships and a Queen Elizabeth II Science and Technology Scholarship while completing the work. Support by the Genomics Research and Development Initiative of the Government of Canada made the sequencing of the genomes possible, and BCRC Project FOS 10.13, provided supplies for the handling of isolates and those needed for preparation and extraction of DNA. Funders had no role in the design of the study, interpretation of data or writing of the manuscript.
Availability of data and materials
The genome sequences can be accessed after 2020-02-26 at https://www.ncbi.nlm.nih.gov/bioproject/browse using Bioproject PRJNA524668. Until then, the sequences are available from the corresponding author upon reasonable request.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Haley Sanderson, Email: haley.sanderson@canada.ca.
Rodrigo Ortega-Polo, Email: rodrigo.ortegapolo@canada.ca.
Rahat Zaheer, Email: rahat.zaheer@canada.ca.
Noriko Goji, Email: noriko.goji@canada.ca.
Kingsley K. Amoako, Email: kingsley.amoako@canada.ca
R. Stephen Brown, Email: stephen.brown@chem.queensu.ca.
Anna Majury, Email: anna.majury@oahpp.ca.
Steven N. Liss, Email: steven.liss@ryerson.ca
Tim A. McAllister, Email: tim.mcallister@canada.ca
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12866-019-1683-4.
References
- 1.Arias CA, Murray BE. Emergence and management of drug-resistant enterococcal infections. Expert Rev Anti-Infect Ther. 2008;6:637–655. doi: 10.1586/14787210.6.5.637. [DOI] [PubMed] [Google Scholar]
- 2.Ashbolt NJ, Amézquita A, Backhaus T, Borriello P, Brandt KK, Collignon P, Coors A, Finley R, Gaze WH, Heberer T, Lawrence JR, Larsson DG, McEwen SA, Ryan JJ, Schonfeld J, Silley P, Snape JR, van den Eede C, Topp E. Human health risk assessment (HHRA) for environmental development and transfer of antibiotic resistance. Environ Health Perspect. 2013;121:993. doi: 10.1289/ehp.1206316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker-Austin C, Wright MS, Stepanauskas R, McArthur JV. Co-selection of antibiotic and metal resistance. Trends Microbiol. 2006;14(4):176–182. doi: 10.1016/j.tim.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrangou R, Horvath P. The CRISPR system protects microbes against phages, plasmids. Microbe. 2009;4:224–230. [Google Scholar]
- 6.Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O’Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3 (2) Nature. 2002;417:141. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 7.Beukers AG, Zaheer R, Goji N, Amoako KK, Chaves AV, Ward MP, McAllister TA. Comparative genomics of Enterococcus spp. isolated from bovine feces. BMC Microbiology. 2017;17:52. doi: 10.1186/s12866-017-0962-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bi D, Xu Z, Harrison EM, Tai C, Wei Y, He X, Jia S, Deng Z, Rajakumar K, Ou HY. ICEberg: a web-based resource for integrative and conjugative elements found in Bacteria. Nucleic Acids Resesearch. 2012;40:D621–D626. doi: 10.1093/nar/gkr846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blanch AR, Caplin JL, Iversen A, Kühn I, Manero A, Taylor HD, Vilanova X. Comparison of enterococcal populations related to urban and hospital wastewater in various climatic and geographic European regions. J Appl Microbiol. 2003;94:994–1002. doi: 10.1046/j.1365-2672.2003.01919.x. [DOI] [PubMed] [Google Scholar]
- 10.Boehm, A. B., & Salubrious, L. M. Enterococci as indicators of environmental fecal contamination. Gilmore MS, Clewell DB, Ike Y, et al., editors. Enterococci: From Commensals to Leading Causes of Drug Resistant Infection [Internet]. Boston: Massachusetts Eye and Ear Infirmary; 2014. Retrieved from < https://www.ncbi.nlm.nih.gov/books/NBK190421/>
- 11.Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013;493:429. doi: 10.1038/nature11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Booth MC, Bogie CP, Sahl HG, Siezen RJ, Hatter KL, Gilmore MS. Structural analysis and proteolytic activation of Enterococcus faecalis cytolysin, a novel lantibiotic. Mol Microbiol. 1996;21:1175–1184. doi: 10.1046/j.1365-2958.1996.831449.x. [DOI] [PubMed] [Google Scholar]
- 13.Caplin JL, Hanlon GW, Taylor HD. Presence of vancomycin and ampicillin-resistant Enterococcus faecium of epidemic clonal complex-17 in wastewaters from the south coast of England. Environ Microbiol. 2008;10:885–892. doi: 10.1111/j.1462-2920.2007.01507.x. [DOI] [PubMed] [Google Scholar]
- 14.Chang YM, Chen CM, Ko TP, Chang-Chien MW, Wang AJ. Structural analysis of the antibiotic-recognition mechanism of MarR proteins. Acta Crystallogr D Biol Crystallogr. 2013;69:1138–1149. doi: 10.1107/S0907444913007117. [DOI] [PubMed] [Google Scholar]
- 15.Chen L, Ruan Z, Wu Q, Zhou T, Li C. Relationship between ermB gene and transposons Tn1545 and Tn917 in Enterococcus faecalis. Chinese Journal of Nosocomiology. 2010;20(6):763–766. [Google Scholar]
- 16.Core Team R. R Foundation for statistical computing. Vienna: Austria.URL; 2013. R: a language and environment for statistical computing. [Google Scholar]
- 17.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PloS one. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duerkop, B. A., Palmer, K. L., & Horsburgh, M. J. Enterococcal bacteriophages and genome defense. 2014. In: Gilmore, M. S., Clewell, D.B., Ike, Y., Shankar, N., Ed. Enterococci: from commensals to leading cause of drug resistant infection. Boston: Massachusetts Eye and Ear Infirmary. 2014; [online]< https://www.ncbi.nlm.nih.gov/books/NBK190419/> [PubMed]
- 19.Faron ML, Ledeboer NA, Buchan BW. Resistance mechanisms, epidemiology, and approaches to screening for vancomycin-resistant Enterococcus in the health care setting. J Clin Microbiol. 2016;54:2436–2447. doi: 10.1128/JCM.00211-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grove A. MarR family transcription factors. Curr Biol. 2013;23:R142–R143. doi: 10.1016/j.cub.2013.01.013. [DOI] [PubMed] [Google Scholar]
- 22.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 23.Hadfield J, Croucher NJ, Goater RJ, Abudahab K, Aanensen DM, Harris SR. Phandango: an interactive viewer for bacterial population genomics. Bioinformatics. 2018;34(2):292–293. doi: 10.1093/bioinformatics/btx610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hammerum AM. Enterococci of animal origin and their significance for public health. Clin Microbiol Infect. 2012;18:619–625. doi: 10.1111/j.1469-0691.2012.03829.x. [DOI] [PubMed] [Google Scholar]
- 25.Harrell Jr, F. E., & Harrell Jr, M. F. E. Package ‘Hmisc’. CRAN2018, 2019; 235–6.
- 26.Hölzel CS, Harms KS, Küchenhoff H, Kunz A, Müller C, Meyer K, Schwaiger K, Bauer J. Phenotypic and genotypic bacterial antimicrobial resistance in liquid pig manure is variously associated with contents of tetracyclines and sulfonamides. J Appl Microbiol. 2010;108:1642–1656. doi: 10.1111/j.1365-2672.2009.04570.x. [DOI] [PubMed] [Google Scholar]
- 27.Huang W, Li L, Myers JR, Marth GT. ART: a next-generation sequencing read simulator. Bioinformatics. 2012;28:593–594. doi: 10.1093/bioinformatics/btr708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, Lago BA, Dave BM, Pereira S, Sharma AN, Doshi S, Courtot M, Lo R, Williams LE, Frye JG, Elsayegh T, Sardar D, Westman EL, Pawlowski AC, Johnson TA, Brinkman FS, Wright GD, McArthur AG. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45:D566–D573. doi: 10.1093/nar/gkw1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joensen KG, Scheutz F, Lund O, Hasman H, Kaas RS, Nielsen EM, Aarestrup FM. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J Clin Microbiol. 2014;52:1501–1510. doi: 10.1128/JCM.03617-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jolley KA, Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kell DB, Kaprelyants AS, Grafen A. Pheromones, social behaviour and the functions of secondary metabolism in bacteria. Proc Natl Acad Sci U S A. 1995;71:2843–2847. doi: 10.1016/s0169-5347(00)89013-8. [DOI] [PubMed] [Google Scholar]
- 32.Klein G. Taxonomy, ecology and antibiotic resistance of enterococci from food and the gastro-intestinal tract. Int J Food Microbiol. 2003;88:123–131. doi: 10.1016/S0168-1605(03)00175-2. [DOI] [PubMed] [Google Scholar]
- 33.Knerr PJ, Van Der Donk WA. Discovery, biosynthesis, and engineering of lantipeptides. Annu Rev Biochem. 2012;81:479–505. doi: 10.1146/annurev-biochem-060110-113521. [DOI] [PubMed] [Google Scholar]
- 34.Konstantinidis KT, Tiedje JM. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc Natl Acad Sci. 2004;101:3160–3165. doi: 10.1073/pnas.0308653100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loong SK, Che Mat Seri NAA, Mahfodz NH, Ahmad Nasrah SN, Akbar SZ, AbuBakar S. Research note a report of Vancomycin-susceptible, Teicoplanin-resistant Enterococcus faecalis ST6 in Malaysia. Trop Biomed. 2016;33:577–582. [PubMed] [Google Scholar]
- 36.Maraccini PA, Ferguson DM, Boehm AB. Diurnal variation in Enterococcus species composition in polluted ocean water and a potential role for the enterococcal carotenoid in protection against photoinactivation. Appl Environ Microbiol. 2012;78(2):305–310. doi: 10.1128/AEM.06821-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marinier E, Zaheer R, Berry C, Weedmark KA, Domaratzki M, Mabon P, Knox NC, Reimer AR, Graham MR, Chui L, Patterson-Fortin L. Neptune: a bioinformatics tool for rapid discovery of genomic variation in bacterial populations. Nucleic acids research. 2017;45(18):e159. doi: 10.1093/nar/gkx702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, Huntemann M, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2011;40:D115–D122. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martins Da Costa P, Vaz-Pires P, Bernardo F. Antimicrobial resistance in Enterococcus spp. isolated in inflow, effluent and sludge from municipal sewage water treatment plants. Water Research. 2006;40:1735–1740. doi: 10.1016/j.watres.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 40.MARTINS DA COSTA PAULO M., VAZ-PIRES PAULO M., BERNARDO FERNANDO M. Antibiotic Resistance of Enterococcus spp. Isolated from Wastewater and Sludge of Poultry Slaughterhouses. Journal of Environmental Science and Health, Part B. 2006;41(8):1393–1403. doi: 10.1080/03601230600964258. [DOI] [PubMed] [Google Scholar]
- 41.Mazaheri Nezhad Fard R, Barton MD, Heuzenroeder MW. Novel bacteriophages in enterococcus spp. Current Microbiology. 2010;60:400–406. doi: 10.1007/s00284-009-9555-z. [DOI] [PubMed] [Google Scholar]
- 42.Miller WR, Munita JM, Arias CA. Mechanisms of antibiotic resistance in enterococci. Expert Rev Anti-Infect Ther. 2014;12:1221–1236. doi: 10.1586/14787210.2014.956092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Müller T, Ulrich A, Ott EM, Müller M. Identification of plant-associated enterococci. J Appl Microbiol. 2001;91:268–278. doi: 10.1046/j.1365-2672.2001.01373.x. [DOI] [PubMed] [Google Scholar]
- 44.Munck C, Albertsen M, Telke A, Ellabaan M, Nielsen PH, Sommer MO. Limited dissemination of the wastewater treatment plant core resistome. Nat Commun. 2015;6:8452. doi: 10.1038/ncomms9452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paduch R, Kandefer-Szerszeń M, Trytek M, Fiedurek J. Terpenes: substances useful in human healthcare. Arch Immunol Ther Exp. 2007;55:315. doi: 10.1007/s00005-007-0039-1. [DOI] [PubMed] [Google Scholar]
- 46.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, Parkhill J. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31(22):3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palmer, K. L., & Gilmore, M. S. 2010. Multidrug-resistant enterococci lack CRISPR-cas. MBio. 2010; 1, e00227-10. [DOI] [PMC free article] [PubMed]
- 48.Palmer KL, Godfrey P, Griggs A, Kos VN, Zucker J, Desjardins C, Cerqueira G, Gevers D, Walker S, Wortman J, Feldgarden M, Haas B, Birren B, Gilmore MS. Comparative genomics of enterococci: variation in Enterococcus faecalis, clade structure in E. faecium, and defining characteristics of E. gallinarum and E. casseliflavus. MBio. 2012;3:e00318–e00311. doi: 10.1128/mBio.00318-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petkau A, Mabon P, Sieffert C, Knox NC, Cabral J, Iskander M, Iskander M, Weedmark K, Zaheer R, Katz LS, Nadon C, Reimer A, Taboada E, Beiko RG, Hsiao W, Brinkman F, Graaham M, van Domselaar G. SNVPhyl: a single nucleotide variant phylogenomics pipeline for microbial genomic epidemiology. Microbial Genomics. 2017;3. [DOI] [PMC free article] [PubMed]
- 50.Qin X, Galloway-Peña JR, Sillanpaa J, Roh JH, Nallapareddy SR, Chowdhury S, Bourgogne A, Choudhury T, Muzny DM, Buhay CJ, Ding Y, Dugan-Rocha S, Liu W, Kovar C, Sodergren E, Highlander S, Petrosino JF, Worley KC, Gibbs RA, Weinstock GM, Murray BE. Complete genome sequence of Enterococcus faecium strain TX16 and comparative genomic analysis of Enterococcus faecium genomes. BMC Microbiol. 2012;12:135. doi: 10.1186/1471-2180-12-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rendueles O, Garcia-Garcerà M, Néron B, Touchon M, Rocha EP. Abundance and co-occurrence of extracellular capsules increase environmental breadth: implications for the emergence of pathogens. PLoS Pathog. 2017;13:e1006525. doi: 10.1371/journal.ppat.1006525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rice LB. Tn916 family conjugative transposons and dissemination of antimicrobial resistance determinants. Antimicrob Agents Chemother. 1998;42:1871–1877. doi: 10.1128/AAC.42.8.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rice LB, Carias LL. Transfer of Tn5385, a composite, multiresistance chromosomal element from Enterococcus faecalis. J Bacteriol. 1998;180:714–721. doi: 10.1128/JB.180.3.714-721.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ricker N, Qian H, Fulthorpe RR. The limitations of draft assemblies for understanding prokaryotic adaptation and evolution. Genomics. 2012;100:167–175. doi: 10.1016/j.ygeno.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 55.Roberts AP, Mullany P. Tn 916-like genetic elements: a diverse group of modular mobile elements conferring antibiotic resistance. FEMS Microbiol Rev. 2011;35:856–871. doi: 10.1111/j.1574-6976.2011.00283.x. [DOI] [PubMed] [Google Scholar]
- 56.Roberts MC. Resistance to tetracycline, macrolide-lincosamide-streptogramin, trimethoprim, and sulfonamide drug classes. Mol Biotechnol. 2002;20:261–283. doi: 10.1385/MB:20:3:261. [DOI] [PubMed] [Google Scholar]
- 57.Roca I, Akova M, Baquero F, Carlet J, Cavaleri M, Coenen S, Cohen J, Findlay D, Gyssens I, Heure OE, Kahlmeter G, Kruse H, Laxminarayan R, Liebana E, Lopez-Cerero L, MacGowen A, Martins M, Rodriguez-Bano J, Rolain JM, Segavia C, Sigauque B, Tacconelli E, Wellingtim E, Vila J. The global threat of antimicrobial resistance: science for intervention. New Microbes and New Infections. 2015;6:22–29. doi: 10.1016/j.nmni.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roszak DB, Colwell RR. Survival strategies of bacteria in the natural environment. Microbiol Rev. 1987;51:365. doi: 10.1128/mr.51.3.365-379.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ruiz-Garbajosa P, Bonten MJ, Robinson DA, Top J, Nallapareddy SR, Torres C, Coque TM, Canton R, Baquero F, Murray BE, del Campo R, Willems RJL. Multilocus sequence typing scheme for Enterococcus faecalis reveals hospital-adapted genetic complexes in a background of high rates of recombination. J Clin Microbiol. 2006;44:2220–2228. doi: 10.1128/JCM.02596-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sanderson H, Fricker C, Brown RS, Majury A, Liss SN. Antibiotic resistance genes as an emerging environmental contaminant. Environ Rev. 2016;24:205–218. doi: 10.1139/er-2015-0069. [DOI] [Google Scholar]
- 61.Sanderson H, Ortega-Polo R, McDermott K, Zaheer R, Brown RS, Majury A, McAllister TA, Liss SN. Comparison of biochemical and genotypic speciation methods for vancomycin-resistant enterococci isolated from urban wastewater treatment plants. J Microbiol Methods. 2019;161:102–110. doi: 10.1016/j.mimet.2019.04.019. [DOI] [PubMed] [Google Scholar]
- 62.Schöner TA, Gassel S, Osawa A, Tobias NJ, Okuno Y, Sakakibara Y, Shindo K, Sandmann G, Bode HB. Aryl polyenes, a highly abundant class of bacterial natural products, are functionally related to antioxidative carotenoids. ChemBioChem. 2016;17:247–253. doi: 10.1002/cbic.201500474. [DOI] [PubMed] [Google Scholar]
- 63.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 64.Shaw JH, Clewell DB. Complete nucleotide sequence of macrolide-lincosamide-streptogramin B-resistance transposon Tn917 in Streptococcus faecalis. J Bacteriol. 1985;164:782–796. doi: 10.1128/JB.164.2.782-796.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shmakov SA, Sitnik V, Makarova KS, Wolf YI, Severinov KV, Koonin EV. The CRISPR spacer space is dominated by sequences from species-specific mobilomes. MBio. 2017;8(5):e01397–e01317. doi: 10.1128/mBio.01397-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stover C. K., Pham X. Q., Erwin A. L., Mizoguchi S. D., Warrener P., Hickey M. J., Brinkman F.S. L., Hufnagle W. O., Kowalik D. J., Lagrou M., Garber R. L., Goltry L., Tolentino E., Westbrock-Wadman S., Yuan Y., Brody L. L., Coulter S. N., Folger K. R., Kas A., Larbig K., Lim R., Smith K., Spencer D., Wong G. K.-S., Wu Z., Paulsen I. T., Reizer J., Saier M. H., Hancock R. E. W., Lory S., Olson M. V. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406(6799):959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 67.Strateva T, Atanasova D, Savov E, Petrova G, Mitov I. Incidence of virulence determinants in clinical Enterococcus faecalis and Enterococcus faecium isolates collected in Bulgaria. Braz J Infect Dis. 2016;20:127–133. doi: 10.1016/j.bjid.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tao P, Wu X, Rao V. Unexpected evolutionary benefit to phages imparted by bacterial CRISPR-Cas9. Science Advances. 2018;4:eaar4134. doi: 10.1126/sciadv.aar4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res. 2014;43(D1):D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Elsas J, Bailey MJ. The ecology of transfer of mobile genetic elements. FEMS Microbiol Ecol. 2002;42:187–197. doi: 10.1111/j.1574-6941.2002.tb01008.x. [DOI] [PubMed] [Google Scholar]
- 72.van Schaik W, Top J, Riley DR, Boekhorst J, Vrijenhoek JE, Schapendonk CM, Hendrickx APA, Nijman IJ, Bonten MJM, Tettelin H, Willems RJL. Pyrosequencing-based comparative genome analysis of the nosocomial pathogen Enterococcus faecium and identification of a large transferable pathogenicity island. BMC Genomics. 2010;11:239. doi: 10.1186/1471-2164-11-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van Tyne D, Gilmore MS. Friend turned foe: evolution of enterococcal virulence and antibiotic resistance. Annu Rev Microbiol. 2014;68:337–356. doi: 10.1146/annurev-micro-091213-113003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Varela AR, Ferro G, Vredenburg J, Yanık M, Vieira L, Rizzo L, Lameiras C, Manaia CM. Vancomycin resistant enterococci: from the hospital effluent to the urban wastewater treatment plant. Sci Total Environ. 2013;450:155–161. doi: 10.1016/j.scitotenv.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 75.Weber T, Blin K, Duddela S, Krug D, Kim H. U., Bruccoleri, R., lee, S. Y., Fischbach, M. a., Muller, R., Wohlleben, W., Breitling, R., Takano, E., & Medema, M. H. antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237–W243. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wei T, Simko V, Levy M, Xie Y, Jin Y, Zemla J. (2017). Package ‘corrplot’. Statistician. 2017;56:316–324. [Google Scholar]
- 77.Weigand MR, Ashbolt NJ, Konstantinidis KT, Santo Domingo JW. Genome sequencing reveals the environmental origin of enterococci and potential biomarkers for water quality monitoring. Environmental science & technology. 2014;48(7):3707–3714. doi: 10.1021/es4054835. [DOI] [PubMed] [Google Scholar]
- 78.Yang SC, Lin CH, Sung CT, Fang JY. Antibacterial activities of bacteriocins: application in foods and pharmaceuticals. Front Microbiol. 2014;5:241. doi: 10.3389/fmicb.2014.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zaheer R, Yanke LJ, Church D, Topp E, Read RR, McAllister TA. High-throughput species identification of enterococci using pyrosequencing. J Microbiol Methods. 2012;89:174–178. doi: 10.1016/j.mimet.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 80.Zhong Z, Zhang W, Song Y, Liu W, Xu H, Xi X, Menghe B, Zhang H, Sun Z. Comparative genomic analysis of the genus Enterococcus. Microbiol Res. 2017;196:95–105. doi: 10.1016/j.micres.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 81.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary Details on Genomic Comparisons. Sheet 1: Complete List of Genomes Used; Sheet 2: Genome Characteristics of Sequenced Genomes; Sheet 3: COG Analysis; Sheet 4: COG Pivot Chart; Sheet 5: COG graphs; Sheet 6: COG correlation and p values; Sheet 7: AMR phenotypic profiles and breakpoints; Sheet 8: AMR genes detected; Sheet 9: Virulence Factors Raw Output; Sheet 10: Virulence Factors by Species; Sheet 11: Virulence Factors Summary; Sheet 12: Phage Detected; Sheet 13: CRISPRfinder Output; Sheet 14: CRISPRfinder Confirmed Systems; Sheet 15: CRISPRdb BLAST Results; Sheet 16: CRISPR Summary Table; Sheet 17: ICEberg Raw Results; Sheet 18: antiSMASH results; Sheet 19: Neptune Biomarker Combinations
Data Availability Statement
The genome sequences can be accessed after 2020-02-26 at https://www.ncbi.nlm.nih.gov/bioproject/browse using Bioproject PRJNA524668. Until then, the sequences are available from the corresponding author upon reasonable request.