

Abstract
Juvenile polyposis syndrome (JPS) patients can have a significantly high burden of polyposis. Patient who undergoes prophylactic colectomy for polyps not amenable to endoscopic treatments requires close clinical surveillance to negate risk of malignancy from interval polyposis.
Keywords: adenomatous polyps, colorectal cancer syndromes, hereditary polyposis, juvenile polyps
Juvenile polyposis syndrome (JPS) patients can have a significantly high burden of polyposis. Patient who undergoes prophylactic colectomy for polyps not amenable to endoscopic treatments requires close clinical surveillance to negate risk of malignancy from interval polyposis.
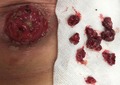
Q: Can you identify the pathology from the picture(s)?
A: Familial adenomatous polyposis.
B: Peutz‐Jeghers polyposis syndrome.
C: Juvenile polyposis syndrome.
D: Benign ileostomy granulomas.
1. CASE REPORT
Juvenile polyposis syndrome (JPS) is characterized by multiple hamartomatous polyps of the GI tract. The lifetime risk of developing malignancy with this condition is approximately 40%. The condition is inherited autosomal dominantly due to SMAD4 or BMPR1A gene mutations.
A 43‐year‐old male who had a history of JPS and underwent a risk‐reducing surgery in the form of restorative proctocolectomy and ileal pouch anal anastomosis at the outside hospital presented to us for further management of his condition. Post–pouch surgery, patient had regular endoscopic surveillance for new polyps. The polyp burden was managed with repeated endoscopic resections. He was also medically treated with sulindac for chemoprevention during the same period. However, despite our best efforts, patient developed a significantly high‐volume disease necessitating close and frequent interval polypectomies. He underwent excision of anal pouch and end ileostomy formation as a last attempt to reduce the risk of future malignancy. After the second procedure, new polyps developed over the ileostomy stump (Figure 1) prompting a close clinical, endoscopic, and radiological (MR small bowel) surveillance. Most of the new polyps on the ileostomy stump were treated with electro‐cautery excision in an office‐based setting (Figure 2). Our case highlights the natural history of disease progression in JPS patients which appears to be different to other bowel polyposis syndromes such as familial adenomatous polyposis (FAP). In this case, clinical surveillance besides regular imaging and endoscopic surveillance after GI surgery helped diagnose and treat new polyps before they turn malignant. This due to an increased risk (50%‐60%) malignancy associated with JPS.
Figure 1.
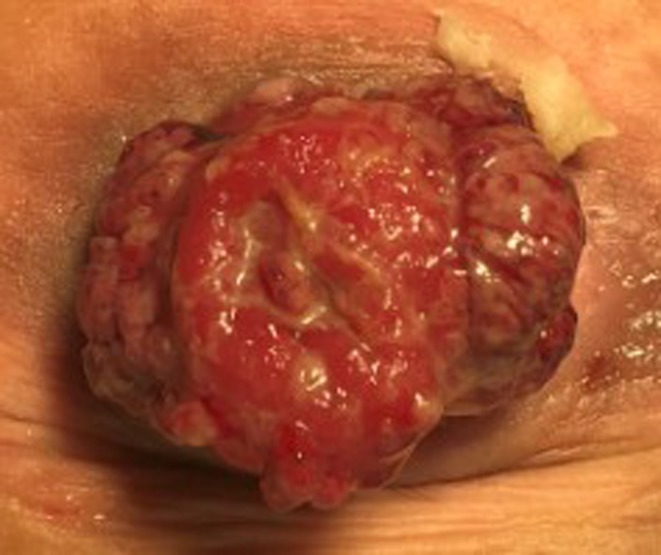
Patient postexcision of pouch and end ileostomy. New juvenile polyps detected on interval clinical surveillance
Figure 2.
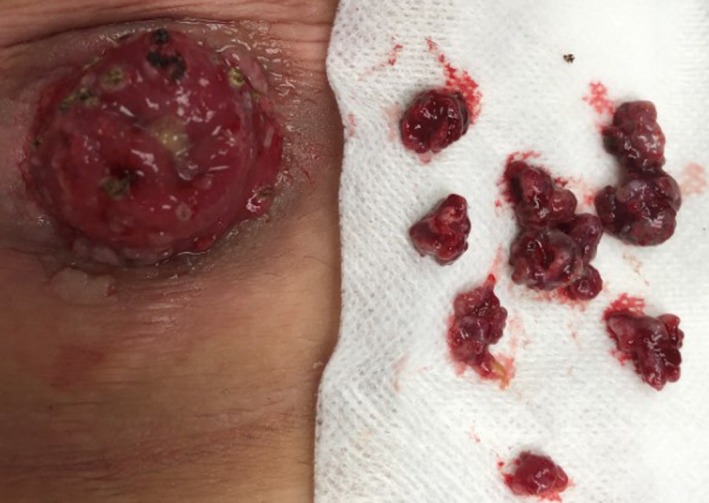
Patient had excision of polyps using electro‐cautery in the clinic for a definitive pathological diagnosis of dysplasia or cancer
2. DISCUSSION
Juvenile polyposis syndrome (JPS) is characterized by hamartomatous polyps in the gastrointestinal (GI) tract. The polyp formation usually involves the stomach, small intestine, colon, and rectum. The term “Generalized Juvenile Polyposis” refers to polyp of upper and lower GI tract. Juvenile polyposis coli refer to polyps in the colon only. The term “Juvenile” is usually a misnomer as it is not related to the patient age of onset of polyp but the histopathology of the polyp itself. Most juvenile polyps are benign and symptomatic ones usually present with bleeding, anemia, and/or bowel obstruction depending on the polyp burden in the GI tract. However, polyps in some patients can turn malignant with a literature incidence reported to range from 9% to 50%.1 The etiology of the JPS is akin to some other known colorectal cancer syndromes such as FAP and Peutz‐Jeghers polyposis with autosomal inheritance pattern. Alongside with JPS, these cancer syndromes form the family of hereditary colorectal cancer syndromes. However, approximately 67% of JP patients have no previous history of polyps in the family and may have the disorder as the result of a de novo pathogenic variant.1 The genes commonly implicated in the etiology and pathogenesis of JPS are SMAD4 and BMPR1A.1, 2 However, some pilot studies have shown ENG germ line mutations to be associated with early‐onset JPS.2 Patient with JPS can have different phenotypic variations with same gene mutation, one such variant being a combined syndrome of JPS and hereditary hemorrhagic telangiectasia (JPS/HHT) with SMAD4 pathogenic variant.3, 4 Furthermore, variations in polyp burden are also known to exist with JPS as some patients develop 4 to 5 polyps over lifetime and others may have more than 100 at a very young age itself. Therefore, it becomes important to identify family‐specific pathogenic variant to offer appropriate molecular genetic testing on at‐risk family members in decades of life. Such an approach can help family members who will benefit from early surveillance and intervention. Genetic testing also helps separate JPS from other conditions in which juvenile polyps form, especially Cowden syndrome (CS) and Bannayan‐Riley‐Ruvalcaba syndrome.2, 5, 6 The generally accepted clinical criteria for JPS include the following5, 6:
at least five juvenile polyps in the colorectum;
juvenile polyps in other parts of the GI tract; or
any number of juvenile polyps in a person with a known family history of juvenile polyps
The clinical diagnosis of JPS can be made in patients presenting with anemia, GI bleeding or prolapse of rectal polyp. However, in a selected proband of patients the diagnosis can be established with clinical features of either more than five juvenile polyps of the colon or rectum; multiple juvenile polyps of the upper and lower GI tract; any number of juvenile polyps and a family history of juvenile polyposis and identification of a heterozygous pathogenic variant in one of the SMAD4 or BMPRA1A genes.1, 3, 4 The clinical presentation of polyposis of infancy can have a varied presentation with symptoms of GI bleeding, obstruction, hyponatremia, anasarca, and failure to thrive. Clinical management of patients with symptomatic polyposis depends primarily on the anatomy of the GI tract involved and polyp burden. Diagnosis can be made with high index of suspicion in patients presenting with above‐mentioned symptoms in early years of life and/or a family history of JPS. Patient can be screened using endoscopy of the upper and lower GI tract to identify polyps and remove them for both diagnostic and therapeutic purposes. Capsule and push endoscopy can be used as screening tools for small bowel polyps.7 MR small bowel series can be used complimentarily with endoscopic techniques and/or for surveillance in patients with an established diagnosis.6, 7 Treatment of JPS involves removing polyps if few in number. To relieve symptoms, or if a large number of polyps are present, part of the stomach and bowel might need to be removed. There are no medicines to treat JPS; however, COX 2 inhibitor therapy with sulindac may be tried for chemoprevention. The most important strategy in the treatment armamentarium involves screening patients with an established diagnosis and/or family history for early management. The screening recommendations to follow are as follows7, 8:
Screening starts with continuous monitoring for symptoms in people with JPS.
Screening including a blood test, colonoscopy, and an upper endoscopy should be done by the time the person with JPS is 15 years old or when symptoms first appear. If results are negative, screening should be repeated in 3 years.
If only a few polyps are found, polyps should be removed and screening should be done every year until no polyps are found. Then screening can be done every 3 years.
If surgery was needed, screening should be done on a yearly basis until no more polyps are found, then screening can be done every 3 years.
Most polyps are treated by removing them with endoscopic polypectomy. But when the polyps are very large or there too many, or if they present a risk for cancer, then surgery may be necessary. The histopathology of the juvenile polyps shows a classical appearance of normal epithelium with a dense stroma, an inflammatory infiltrate, and a smooth surface with dilated, mucus‐filled cystic glands in the lamina propria. Muscle fibers and the proliferative characteristics of adenomas are typically not seen in juvenile polyps as juvenile polyps are hamartomas that develop from an abnormal collection of tissue elements normally present at this site.7
Most juvenile polyps are benign; however, malignant transformation can occur. Lifetime estimated risk of developing GI cancers in families with JPS range from 9% to 50% 5, 8, 9 of individuals treated surgically and followed with surveillance. In a study by Aytac et al, 4/27 individuals with SMAD4 pathogenic variants and 0/8 individuals with BMPR1A pathogenic variants developed cancer.10, 11 Most of the increased risk is attributed to colon cancer followed by cancers of the stomach, upper GI tract, and pancreas. The incidence of colorectal cancer is 17%‐22% by age 35 years and approaches 68% by age 60 years.9, 10, 11 The median age at diagnosis is 42 years. In the study by Brosens et al, the relative risk for colorectal cancer was 34.0 in individuals with JPS. The mean age of diagnosis of colorectal cancer was 43.9 years, with a cumulative lifetime risk of 38.7%.10 Therefore, due to the heightened risk of GI malignancies in JPS patients and families, a vigilant surveillance and robust management approach should be undertaken to mitigate the risks. The American College of Gastroenterology6 (ACS) recommends the following steps for the management and surveillance for JPS patients:
Surveillance of the GI tract in affected or at‐risk JPS patients should include screening for colon, stomach, and small bowel cancers.
Colectomy and IRA or proctocolectomy and IPAA is indicated for polyp‐related symptoms, or when the polyps cannot be managed endoscopically.
Cardiovascular examination for and evaluation for hereditary hemorrhagic telangiectasia should be considered for SMAD4 mutation carriers (conditional recommendation).
3. CONCLUSION
Our case highlights the importance of combined clinical, endoscopic, and radiological follow‐up of JPS patients. The complimentary clinical follow‐up approach helps mitigate some of the shortfalls of the screening modalities and identify new interval polyps’ in‐time before they turn malignant.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
This manuscript has been jointly produced by TH and JC. Both authors declare no conflict of interest. Both authors have reviewed the manuscript and accepted it in the current form for publication. TH has written the manuscript and did the literature searches. The case belongs to JC and he has reviewed the manuscript and approved it.
Hussain T, Church JM. Juvenile polyposis syndrome. Clin Case Rep. 2020;8:92–95. 10.1002/ccr3.2616
REFERENCES
- 1. Howe JR, Roth S, Ringold JC, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998b;280:1086‐1088. [DOI] [PubMed] [Google Scholar]
- 2. Ngeow J, Heald B, Rybicki LA, et al. Prevalence of Germline PTEN, BMPR1A, SMAD4, STK11, and ENG Mutations in Patients With Moderate‐Load Colorectal Polyps. Gastroenterology. 2013;144:1402‐1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wain KE, Ellingson MS, McDonald J, et al. Appreciating the broad clinical features of SMAD4 mutation carriers: a multicenter chart review. Genet Med. 2014;16:588‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. O'Malley M, LaGuardia L, Kalady MF, et al. The prevalence of hereditary hemorrhagic telangiectasia in juvenile polyposis syndrome. Dis Colon Rectum. 2012;55:886‐892. [DOI] [PubMed] [Google Scholar]
- 5. Latchford AR, Neale K, Phillips RK, Clark SK. Juvenile polyposis syndrome: a study of genotype, phenotype, and long‐term outcome. Dis Colon Rectum. 2012;55:1038‐1043. [DOI] [PubMed] [Google Scholar]
- 6. Syngal S, Brand RE, Church JM, et al. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110(2):223‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Larsen Haidle J, Howe JR. Juvenile Polyposis Syndrome. Seattle, WA: University of Washington; 1993. [Google Scholar]
- 8. Dove‐Edwin I, Sasieni P, Adams J, et al. Prevention of colorectal cancer by colonoscopic surveillance in individuals with a family history of colorectal cancer: 16 year, prospective, follow‐up study. BMJ. 2005;331:1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aretz S, Stienen D, Uhlhaas S, et al. High proportion of large genomic deletions and a genotype phenotype update in 80 unrelated families with juvenile polyposis syndrome. J Med Genet. 2007;44:702‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brosens LA, van Hattem A, Hylind LM, et al. Risk of colorectal cancer in juvenile polyposis. Gut. 2007;56:965‐967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aytac E, Sulu B, Heald B, et al. Genotype‐defined cancer risk in juvenile polyposis syndrome. Br J Surg. 2015;102:114‐118. [DOI] [PubMed] [Google Scholar]