

Abstract
Infections caused by multi-drug resistant (MDR) bacteria, particularly Gram-negative bacteria, are an escalating global health threat. Often clinicians are forced to administer the last resort antibiotic colistin, however colistin resistance is becoming increasingly prevalent, giving rise to the potential for a situation in which there are no treatment options for MDR Gram-negative infections. The development of adjuvants that circumvent bacterial resistance mechanisms is a promising orthogonal approach to the development of new antibiotics. We recently disclosed that the known IKK-β inhibitor IMD-0354 potently suppresses colistin resistance in several Gram-negative strains. In this report, we explore the structure-activity relationship (SAR) between the IMD-0354 scaffold and colistin resistance suppression, and identify several compounds with more potent activity than the parent against highly colistin resistant strains of Acinetobacter baumannii and Klebsiella pneumoniae.
Keywords: antibiotic resistance, adjuvant, colistin, Gram-negative
Graphical Abstract
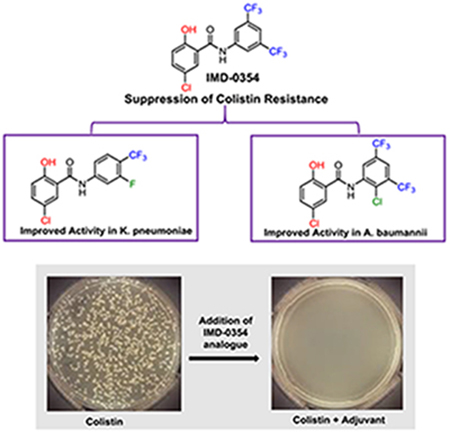
We report a structure-activity relationship (SAR) study on the recently identified colistin adjuvant IMD-0354, and describe several compounds with more potent activity than the parent including one analogue that suppresses the colistin minimum inhibitory concentration (MIC) below the breakpoint level against a highly resistant strain of Klebsiella pneumoniae at 250 nM.
Introduction
The threat to modern medicine caused by rising antibiotic resistance cannot be understated.[1] Of all bacteria, the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) are considered to pose the highest threats to human health.[2] Despite escalating incidence of infections that stem from multi-drug resistant (MDR) strains of these pathogens, the antibiotic discovery pipeline to counter these threats still remains significantly underpopulated.[3] In the case of the Gram-positive members of this group (E. faecium and S. aureus), limited success has been achieved with the introduction of linezolid and daptomycin over the last 20 years.[4] For Gram-negative bacteria, however, the last class of novel antibiotics to be introduced into the clinic was the fluoroquinolone class in the 1960s.[5]
As a result of this current antibiotic landscape, clinicians have become more reliant upon polymyxin antibiotics for the treatment of MDR Gram-negative bacterial infections.[6] Polymyxins are cationic cyclic peptides whose bactericidal activity is driven by physical disruption of the Gram-negative bacterial membrane.[7] As an antibiotic for human use, colistin was introduced into the clinic in 1955 but rapidly fell out of favor due to nephrotoxicity,[8] and was replaced by antibiotics with better safety profiles. However, with limited therapeutic options currently available, coupled with a historically low incidence of resistance due to limited use, colistin is now typically viewed as the antibiotic of last resort for the treatment of MDR Gram-negative bacterial infections.[9]
As colistin treatment has become more common, there has been a corresponding upsurge in colistin resistance observed in the clinic over the past decade. Clinical resistance to colistin, and other polymyxins, is driven by cationic modification of the lipid A component of lipopolysaccharide (LPS) that decorates the outer membrane of Gram-negative bacteria.[10] In A. baumannii, lipid A is typically modified with phosphoethanolamine, while in K. pneumoniae it is typically modified with aminoarabinose.[11] In either case, modification of lipid A serves to decrease the overall net negative charge of LPS, which, in turn, impacts the affinity of colistin for the outer membrane. Historically, colistin resistance has typically been mediated by chromosomal mutations in genes encoding two-component systems (such as PmrAB in A. baumannii) that, combined with colistin’s limited use, meant dissemination of colistin resistant isolates was slow. The situation changed dramatically with the emergence of the plasmid-borne colistin resistance genes mcr-1–9[12] that have raised the possibility of rapid dissemination of colistin resistance into the general pathogen pool via horizontal gene transfer, which would ultimately deprive clinicians of this last resort antibiotic.
With the rising incidence of MDR bacterial infections coupled with a depleted antibiotic pipeline, research into alternatives to traditional antibiotics is warranted. One such approach is the development of small molecule antibiotic adjuvants.[13] Such molecules typically possess little stand-alone microbicidal activity, but instead target antibiotic resistance mechanisms. When paired with the appropriate antibiotic, these antibiotic/adjuvant combinations provide a powerful treatment for MDR bacteria. In the context of colistin resistance, we have identified a number of adjuvants capable of reversing colistin resistance in bacterial strains that contain chromosomally-encoded resistance mechanisms as well as those that harbor the plasmid-borne mcr-1 gene (Compounds A-E Figure 2).[14, 15] Recently, we reported that the non-toxic salicylamide IMD-0354 E, a well-known inhibitor of IKK-β, reversed lipid A modification and potently suppressed colistin resistance in A. baumannii, K. pneumoniae, and Escherichia coli, reducing the minimum inhibitory concentration (MIC) of colistin upwards of 4096-fold at 5 μM (1.9 μg/mL).[15] Given this potency, we became interested in delineating the structural parameters of this scaffold that drive colistin potentiation. Herein, we report our initial exploration into the structure activity relationship (SAR) of this scaffold and the identification of 35 compounds with equivalent or superior activity to IMD-0354 against both the representative strains used in the initial screen.
Figure 2.

Structures of analogues with derivatized benzoic acid moieties.
Results and Discussion
All IMD-0354 analogues, with the following exceptions were synthesized through a one-step PCl3 mediated condensation reaction between a derivatized aniline and corresponding benzoic (salicylic) acid.[16] Compound 2 (Figure 4) was synthesized by methylating the phenolic hydroxyl group of IMD-0354 with methyl iodide in the presence of potassium carbonate.[17] The bis-phenyl derivative 13 (Figure 4) was synthesized from the iodo derivative 10 via a Suzuki coupling with phenylboronic acid in the presence of bis(triphenylphosphine)palladium chloride. Finally, aniline 36 was synthesized via a tin (II) chloride mediated reduction of nitro derivative 35 (Scheme 1).
Figure 4.

Structures of hybrid analogues
Scheme 1: General synthetic route and routes to compounds 2, 13 and 36.

Reagents and conditions: (a) PCl3, toluene, reflux, 16h; (b) CH3I, K2CO3, acetone, rt, 16h; (c) phenyl boronic acid, PdCl2(PPh3), K2CO3, THF, reflux, 48h; (d) SnCl2, HCl/EtOH, 70 °C, 12h.
The first compound set we prepared varied the identity of the benzoic acid while retaining the 3,5-bis-trifluoromethylaniline component of IMD-0354 (Figure 4). All derivatives were screened against two representative, highly colistin resistant Gram-negative bacterial strains: A. baumannii 4106 (AB4106) and K. pneumoniae B9 (KPB9). In the absence of adjuvant, AB4106 returned a colistin MIC of 2048 μg/mL while KPB9 returned a colistin MIC of 512 μg/mL. The clinical breakpoint for colistin susceptibility against these bacterial species is 2 μg/mL. We have previously established that at 5 μM, IMD-0354 reduces the colistin MIC against AB4106 to 2 μg/mL (1024-fold) while it reduces the colistin MIC against KPB9 to 0.5 μg/mL (1024-fold). To compare directly to IMD-0354, all analogues were thus screened for activity at 5 μM, with any compound displaying an equal or two-fold variation in MIC labeled as equipotent.
The activity of the lead compounds is summarized in Table 1. Full data can be found in Table S1 in the supporting information. All new analogues exhibited standalone MICs of ≥100 μM, with the exception of compound 60, which exhibited MICs of 50 μM against both strains, compound 25 which exhibited an MIC of 50 μM against AB 4106, compounds 18 and 62, which exhibited an MIC of 50 μM against KP B9, and compounds 9 and 42, which exhibited MICs of 25 μM against KP B9.
Table 1.
Colistin MICs against AB 4106 and KP B9 in the presence of lead IMD-0354 analogues
| Compounda | AB 4106 MIC (μg/mL) (fold reduction) | KP B9 MIC (μg/mL) (fold reduction) |
|---|---|---|
| IMD-0354 | 2 (1024)b | 0.5 (1024)c |
| 9 | 2 (1024) | 0.25 (2048) |
| 10 | 0.5 (4096) | 0.5 (1024) |
| 15 | 1 (2048) | 0.25 (2048) |
| 22 | 2 (1024) | 0.25 (2048) |
| 23 | 2 (1024) | 0.25 (2048) |
| 24 | 0.5 (4096) | 0.5 (1024) |
| 25 | 1 (2048) | 0.25 (2048) |
| 26 | 1 (2048) | 0.25 (2048) |
| 29 | 1 (2048) | 0.5 (1024) |
| 37 | 1 (2048) | 0.25 (2048) |
| 43 | 2 (1024) | 0.125 (4096) |
| 49 | 1 (2048) | 0.25 (2048) |
| 50 | 4 (512) | 0.25 (2048) |
| 51 | 1 (2048) | 0.125 (4096) |
| 52 | 1 (2048) | 0.5 (1024) |
| 55 | 1 (2048) | 0.5 (1024) |
| 56 | 1 (2048) | 0.5 (1024) |
| 60 | 1 (2048) | 0.5 (1024) |
All compounds screened at 5 μM.
MIC of colistin alone is 2048 μg/mL.
MIC of colistin alone is 512 μg/mL.
We first tested the necessity for the phenolic hydroxyl moiety by either complete removal (1) or methylation (2). Both modifications abrogated activity. Next, activity as a function of position of the phenolic hydroxyl group was probed through compounds 3 and 4. Shifting this hydroxyl group from the ortho to meta (in relationship to the amide, 3) resulted in a compound with essentially equipotent activity to IMD-0354, lowering the colistin MIC to 4 μg/mL against AB4106 and 0.5 μg/mL against KPB9. Further rotation of the hydroxyl group to the para position (4), abolished activity. The impact that the chloro substituent had upon activity was probed by changing its relative position, deletion, or substitution with other halogens (5-10). Moving the position of the chlorine (5), deletion of the chlorine (6), or replacement of the chlorine with either fluorine (8), bromine (9), or iodine (10) was tolerated and delivered compounds with essentially equipotent activity to IMD-0354. Interestingly, the iodo derivative, 10, displayed superior activity to IMD-0354 and reduced the AB4106 colistin MIC to 0.5 μg/mL while it retained equipotent activity against KPB9 (reduced colistin MIC to 0.5 μg/mL). Removal of the chlorine in tandem with moving the phenolic hydroxyl (7) was not tolerated and resulted in a derivative devoid of activity. We further probed the impact substitution of the chloro group had upon activity by testing the activity of the methoxy analogue 11. This substitution resulted in a significant impact on activity and returned colistin MICs of 8 μg/mL (AB4106) and 16 μg/mL (KPB9). Steric isosteres at the halogen position were constructed for both IMD-0354 (12) and the iodo derivative 10 (analogue 13). The methyl derivative returned similar colistin MICs to IMD-0354 (4 and 0.5 μg/mL against AB4106 and KPB9 respectively), while the phenyl derivative maintained activity against AB4106 (4 μg/mL) but lost some activity against KPB9 (4 μg/mL). The last three derivatives of this subset prepared were the dichloro analogue 14, the trifluoromethyl analogue 15 and the dihydroxylated derivative 16. We found that 15 exhibited equipotent activity to IMD-0354 (1 and 0.25 μg/mL against AB4106 and KPB9 respectively), while compound 14 was four-fold less active than 15 (yet still considered equipotent to IMD-0354). Finally, we noted that 16 was less active against KPB9 (2 μg/mL) and essentially inactive against AB4106. Of the 16 analogues prepared in this subset, we found nine had equipotent or improved activity against both strains in comparison to IMD-0354, while the phenyl derivative had equivalent activity against AB4106.
Next we probed the structural constraints of the aniline segment of IMD-0354 by fixing the identity of the 5-chlorosalicylic acid and varying the substituents on the aniline ring (Figure 3). We moved the position of the trifluoromethyl substituents from a 3,5 orientation to a 2,5- (17) and a 2,4- (18) substitution pattern. Both compounds retained activity against KPB9 and AB4106 equivalent to IMD-0354. The requirement for the bis-trifluromethyl groups was probed by preparing the corresponding 3,5-dimethoxy and 3,5-dimethyl analogues 19 and 20. Although the dimethoxy derivative was bereft of activity, the dimethyl analogue returned colistin MICs of 2 and 1 μg/mL against AB4106 and KPB9 respectively.
Figure 3.

Structures of analogues with derivatized aniline moieties.
Previous studies with 2-aminoimidazle-based adjuvants have demonstrated that compounds containing 3,5-dihalogenated benzene rings display various activities; therefore we next studied replacement of the 3,5-bis-trifluoromethyl groups with difluoro, dichloro, and dibromo substituents (21-23). All three analogues were equipotent, suppressing colistin MICs to 2 and 0.25 μg/mL (AB4106 and KPB9). Addition of either a bromine or chlorine at the 2-position of the bis-trifluoroaniline scaffold (24 and 25) again yielded potent compounds returning colistin MICs of 1 and 0.25 μg/mL against AB4106 and KPB9 respectively. Monotrifluoromethyl derivatives 26-30 were then prepared to study the impact retaining one of the trifluoromethyl substituents had upon activity. The para-trifluoromethyl derivative 26, as well as the fluorinated analogues 28 and 29, all returned activity equivalent to IMD-0354. Placement of the trifluoromethyl ortho to the amide (27) suppressed activity such that the colistin AB4106 MIC was only reduced to 8 μg/mL while the KPB9 colistin MIC was 1 μg/mL. Pairing of the trifluoromethyl with a para-methyl was detrimental to activity (30, 64/4 μg/mL against AB4106/KPB9) while the all methyl analogue 31 also showed minimal activity (64/16 μg/mL against AB4106/KPB9). Replacement of one of these methyl groups with a bromo substituent, compound 32, only improved activity against KPB9 (8 μg/mL), while analogues containing an ortho bromo substituent (33 and 34) also showed diminished activity in comparison to IMD-0354 (16/2 and >64/>64 μg/mL against AB4106/KPB9 for 33 and 34 respectively)
We tested further expansion of functional group tolerance on the aniline ring by incorporation of a nitro (35) or an amine (36) substituent in tandem with a para-fluoro substituent. Neither compound displayed significant activity with the exception of 35 against KPB9, which returned an MIC of 1 μg/mL. Next, we focused efforts on monosubstituted aniline building blocks and assembled compounds 37-40. The and meta-chloro derivative (40) was active, returning MICs of 2/0.5 μg/mL against AB4106/KPB9, as was the para-bromo derivative 37 (MICs 1/0.25 against AB4106/KPB9), while the ortho-chloro derivative 39 was less active. Mirroring the results from other analogues in this series, incorporation of the electron donating methoxy group abolished activity (38). Finally, we prepared analogue 41 to test the effects of combining the structural components of two active analogues (22 and 37). This analogue however, showed diminished activity, ca. 4-8 fold over 22 and 37. Overall, we generated 25 derivatives by varying the identity of the aniline substituent, of which 12 showed activity at least equivalent to IMD-0354.
The last compound set we prepared tested the impact of mixing structural elements from both of the previous libraries. The 21 analogues synthesized are depicted in Figure 4. With the exception of the 3,5-dimethyl derivatives (44, 48, and 53) and the trifluoro derivative 42, all analogues prepared showed activity essentially equal to IMD-0354 when the halide identity on the salicylic acid building block was varied. Derivatives that contained a 3,5-dihaloaniline building block and either a hydroxyl trifluoromethyl or dichloro salicylic acid fragment were also equally active to IMD-0354 (54-56 and 62), while again all of the corresponding 3,5-dimethyl analogues were less active (57-59). The 2,5-di-trifluoromethyl aniline coupled with the dichloro salicylic acid fragment (61) also returned equipotent activity. Overall, 13 analogues from this last set were identified to have activity levels similar to IMD-0354 with a clear trend that incorporation of a 3,5-dimethyl substitution pattern was detrimental to activity.
Through screening all compounds for activity against AB4106 and KPB9, we identified 37 salicylamide derivatives that we deemed were worthy of further investigation. To further vet activity, we tested whether compound activity was retained against multiple colistin resistant isolates (Table 2 and Table S2 SI). We chose two additional highly colistin resistant K. pneumoniae strains, KPC3 and KPA5, which have stand-alone colistin MICs of 128 and 2048 μg/mL, respectively, as well as a K. pneumoniae strain harboring the mcr-1 gene (KPF221029mcr−1, colistin MIC = 16 μg/mL).
Table 2.
Dose response activity of lead compounds against KP B9
| Compound | Concentration (μM) | Colistin MIC (μg/mL) (fold reduction) |
|---|---|---|
| IMD-0354 | 5 | 0.5 (1024) |
| 3 | 0.5 (1024) | |
| 1 | 4 (128) | |
| 23 | 5 | 0.25 (2048) |
| 3 | 0.5 (1024) | |
| 1 | 0.5 (1024) | |
| 0.5 | 1 (512) | |
| 0.25 | 4 (128) | |
| 29 | 5 | 0.5 (1024) |
| 3 | 0.5 (1024) | |
| 1 | 0.5 (1024) | |
| 0.5 | 0.5 (1024) | |
| 0.25 | 1 (512) | |
| 51 | 5 | 0.125 (4096) |
| 3 | 0.25 (2048) | |
| 1 | 0.5 (1024) | |
| 0.5 | 1 (512) | |
| 0.25 | 16 (32) | |
| 52 | 5 | 0.5 (1024) |
| 3 | 0.5 (1024) | |
| 1 | 1 (512) | |
| 0.5 | 1 (512) | |
| 0.25 | 16 (32) | |
With the exception of KPA5 and AB4119, IMD-0354 suppresses the colistin MIC of all other isolates to at or below the clinical breakpoint level (2 μg/mL). Against KPA5, IMD-0354 suppresses the colistin MIC to 8 μg/mL and against AB4119 it suppresses it to 4 μg/mL. Of the compounds screened, 15 outperformed IMD-0354 and suppressed the colistin MIC against all strains to at or below breakpoint levels. To further probe activity, we performed a dose response study with these compounds in comparison to IMD-0354 (Table 2 and Tables S3 and S4 in the supporting information) against AB4106 and KPB9. At 3 μM, IMD-0354 suppresses the colistin MIC to 4 μg/mL (AB4106) and 0.5 μg/mL (KPB9), while at 1 μM, it returns colistin MICs of 8 and 4 μg/mL against AB4106 and KPB9. Against AB4106, none of the compounds returned more potent activity than IMD-0354, although 22 compounds performed equally well (MIC within two-fold of that observed with IMD-0354). The most active compounds, 22, 24, 25, 29, and 51 reduce the colistin MIC to 4 μg/mL at 1 μM. Against KPB9, however, 21 analogues outperformed IMD-0354 and returned colistin to at or below breakpoint levels at 1 μM, while 25 derivatives had equipotent activity to IMD-0354. Of the 10 compounds that had superior activity to IMD-0354, four analogues (23, 29, 51 and 52) retained the ability to break colistin resistance at 500 nM, and 29 reduced the colistin MIC below breakpoint levels at 250 nM. All four lead compounds were active against the isolate panel and equally active against AB4106 in comparison to IMD-0354.
To further quantify activity, and to confirm the non-toxic nature of the adjuvants, we constructed time kill curves for colistin and compound 22 as a representative lead compound, against both AB4106 and KPB9 (Figure 5). Mirroring the MIC studies, compound 22 showed significant potentiation of colistin activity.
Figure 5.

Time kill curves for compound 22 and colistin. Top: AB 4106. Bottom: KP B9. Blue: Untreated bacteria. Red: 5 μM 22. Orange 5 μM 22 + 0.5 μg/mL colistin. Green: 5 μM 22 + 4 μg/mL colistin. Purple: 5 μM 22 + 32 μg/mL colistin. Data ± SD from at least four replicates.
Compounds were also tested against four additional A. baumannii strains AB3941, AB3942, AB4112, and AB4119, as well as an A. baumannii strain harboring a plasmid containing the mcr-1 gene (AB17978mcr−1). The colistin MICs of these five A. baumannii strains are 1024, >2048, 2048, 1024, and 64 μg/mL respectively. All compounds were screened for their ability to potentiate colistin at 5 μM and compared to IMD-0354 (Table 3).
Table 3.
Activity of lead compounds against colistin resistant isolate panel
| Compound | Colistin MIC (μg/mL) (fold reduction) |
|||||||
|---|---|---|---|---|---|---|---|---|
| KP C3 | KP A5 | KP F2210291mcr−1 | AB 3941 | AB 3942 | AB 4112 | AB 4119 | AB 17978mcr−1 | |
| - | 128 | 2048 | 16 | 1024 | >2048 | 2048 | 1024 | 64 |
| IMD-0354 | 0.5a (256) | 8 (256) | 0.5 (32) | 2 (512) | 2 (≥2048) | 2 (1024) | 4 (256) | 0.5 (128) |
| 23 | 0.5 (256) | 0.5 (4096) | 0.125 (128) | 2 (512) | 2 (≥2048) | 2 (1024) | 1 (1024) | 0.5 (128) |
| 29 | 1 (128) | 1 (2048) | 0.25 (64) | 1 (1024) | 1 (≥4096) | 1 (2048) | 2 (512) | 0.125 (512) |
| 51 | 0.25 (512) | 0.25 (8192) | 0.25 (64) | 1 (1024) | 1 (≥4096) | 1 (2048) | 0.5 (2048) | 0.125 (512) |
| 52 | 0.5 (256) | 1 (2048) | 0.25 (64) | 1 (1024) | 1 (≥4096) | 1 (2048) | 2 (512) | 0.25 (256) |
All compounds tested at 5 μM
Finally, we tested the cytotoxicity of six compounds (21, 22, 23, 25, 26, and 37) in the absence and presence of 1 μg/mL colistin of the six compounds, 22, 23, 26, and 37 showed no measurable toxicity up to 200 μM. One compound, 21, showed comparable toxicity to IMD-0354 (CT50 = 210 μM) while another (25) was ca. two-fold more toxic than IMD-0354 (CT50 = 110 μM). None of the compounds showed additional toxicity in the presence of 1 μg/mL colistin.
Conclusions
In conclusion, we have presented an initial SAR study on the salicylamide lead IMD-0354. A number of structural trends were elucidated, with the most prominent being that the salicylic OH is required for activity (although the position may vary), while the halogen identity is flexible. On the aniline fragment, the trifluoromethyl groups can be exchanged with halogens, while electron donating groups are generally not tolerated. Through these studies, we have managed to identify a number of analogues with activity similar to or exceeding IMD-0354. Four of these leads (23, 29, 51, and 52) show activity in the nanomolar range, which marks them as some of the most potent colistin adjuvants identified to date. As a potential treatment for colistin resistant infections, IMD-0354 is hampered by solubility and its inhibition of inflammation through targeting IKK-β. To this end, we are currently conducting additional SAR studies on this scaffold in efforts to develop analogues that retain their colistin adjuvant activity, yet are more soluble in aqueous media with muted effect on IKK-β.
Experimental Section
General Chemistry Experimental
All reactions were carried out under an atmosphere of nitrogen using anhydrous solvents unless otherwise specified. All chemical reagents for synthesis were used without further purification. Analytical thin layer chromatography (TLC) was performed using 250 μm Silica Gel 60 F254 pre-coated plates (EMD Chemicals Inc.). Flash column chromatography was performed using 230–400 Mesh 60Å Silica Gel from Sorbent Technologies. NMR spectra were recorded using broadband probes on a Bruker AVANCE III HD 400 Nanobay (400 MHz for 1H and 100 MHz for 13C). All spectra are presented using MestReNova (Mnova) software and 1H NMR are typically displayed from 12 to −0.7 ppm without the use of the signal suppression function. Spectra were obtained in the following solvents (reference peaks also included for 1H and 13C NMRs): CDCl3 (1H NMR: 7.26 ppm; 13C NMR: 77.23 ppm), d6-DMSO (1H NMR: 2.50 ppm; 13C NMR: 39.52 ppm) and d4-MeOD (1H NMR: 3.31 ppm; 13C NMR: 49.00 ppm). All NMR experiments were performed at room temperature. Chemical shift values (δ) are reported in parts per million (ppm) for all 1H NMR and 13C NMR spectra. 1H NMR multiplicities are reported as: s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, br = broad. High-resolution mass spectra were obtained for all new compounds from the mass spectrometry and proteomics facility at university of Notre Dame performed on a Bruker-TOF-ESI spectrometer in positive module using direct infusion in 9:1 acetonitrile: water. IR spectra were recorded on Bruker Alpha II FTIR spectrometer. UV data was taken using a Thermo Scientific, Genesys 10 UV scanning spectrometer.
General procedure for synthesis of N-aryl-2-hydroxybenzamides
To a stirring solution of salicylic acid (1 equiv.) in toluene under nitrogen atmosphere, was added phosphorus trichloride (0.5 equiv.) dropwise and heated to reflux. Substituted aniline (0.9 equiv.) was added in portions over 10 minutes and the reaction mixture was refluxed overnight. The reaction was checked for completion by TLC, then cooled and ethyl acetate (100 mL) added. The suspension was washed with 2N HCl (3x30 mL) followed by brine (30 mL) and then aqueous sodium bicarbonate (3x30 mL). The organic layer was washed with brine (30 mL) then dried using anhydrous sodium sulfate, evaporated and purified via flash chromatography using 1:2 to 1:1 DCM/hexanes to obtain a white solid. Compounds were tested without further purification from stock solutions in biological grade DMSO. Compounds 1, 2, 5, 6, 8-15, 17-28, 30-34, 37-40, 42, 43, 45-51, 53, 56, and 62. have been previously reported and characterization for these compounds can be found in the supporting information.
Novel compound characterization
N-(3,5-bis(trifluoromethyl)phenyl)-3-chloro-5-hydroxybenzamide (3)
The title compound was synthesized following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 3 as a white solid (52 mg, 39%). 1H NMR (400 MHz, d3-Acetonitrile): 9.11 (s, 1H), 8.41 – 8.30 (m, 2H), 7.75 (s, 1H), 7.46 (t, J = 1.4, 0.7 Hz, 1H), 7.30 (t, J = 1.6 Hz, 1H), 7.08 (t, J = 2.0 Hz, 1H). 13C NMR (100 MHz, d3-Acetonitrile): 164.5, 157.8, 140.1, 136.6, 134.3, 131.1 (q, J = 33.3 Hz), 123.1 (q, J = 271.8 Hz), 119.8, 119.8, 118.7, 118.5, 113.1. UV(λmax nm): 265; IRvmax (cm−1): 3324, 1654, 1573, 914; HRMS (ESI): calcd for C15H9ClF6NO2 [M+H]+: 384.0221, found: 384.0213
N-(3,5-bis(trifluoromethyl)phenyl)-3-chloro-4-hydroxybenzamide (4)
The title compound was synthesized following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 4 as a white solid (76 mg, 14%). 1H NMR (400 MHz, d6-DMSO): 11.11 (s, 1H), 10.66 (s, 1H), 8.50 (s, 2H), 8.09 (d, J = 2.3 Hz, 1H), 7.84 (dd, J = 8.6, 2.3 Hz, 1H), 7.80 (s, 1H), 7.11 (d, J = 8.5 Hz, 1H). 13C NMR (100 MHz, d6-DMSO): 164.6, 156.8, 141.2, 130.7 (q, J = 32.7 Hz), 129.7, 128.6, 125.3, 123.4 (q, J = 272.8 Hz), 119.8 (d, J = 4.2 Hz), 119.7, 116.4, 116.2 (m) . UV(λmax nm): 272; IRvmax (cm−1): 3174, 1643, 1118, 681; HRMS (ESI): calcd for C15H9ClF6NO2[M+H]+: 384.0221, found: 384.0194
N-(3,5-bis(trifluoromethyl)phenyl)-3-hydroxybenzamide (7)
The title compound was synthesized following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 7 as a white solid (34 mg, 23%). 1H NMR (400 MHz, d6-DMSO): 10.76 (s, 1H), 9.86 (s, 1H), 8.53 (d, J = 1.8 Hz, 2H), 7.81 (s, 1H), 7.43 (dt, J = 7.6, 1.3 Hz, 1H), 7.40 – 7.33 (m, 2H), 7.03 (ddd, J = 7.9, 2.4, 1.1 Hz, 1H). 13C NMR (100 MHz, d6-DMSO): 13C NMR (100 MHz, d6-DMSO): 166.3, 157.5, 141.2, 135.3, 130.6 (q, J = 32.8 Hz), 129.7, 123.3 (d, J = 272.6 Hz), 119.8 (q, J = 3.4 Hz), 119.3, 118.3, 116.4 (q, J = 7.3 Hz), 114.6. UV (λmax nm): 274; IR vmax (cm−1): 3080, 1564, 1509, 1220; HRMS (ESI): calcd for C14H13ClNO3 [M+H]+: 278.0578, found: 278.0596
N-(3,5-bis(trifluoromethyl)phenyl)-2,5-dihydroxybenzamide (16)
The title compound was synthesized following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 16 as a white solid (70 mg, 26%). 1H NMR (400 MHz, d6-DMSO): 11.04 (s, 1H), 8.41 (s, 1H), 7.82 (s, 1H), 7.17 (t, J = 8.2 Hz, 1H), 6.43 (d, J = 8.2 Hz, 2H). 13C NMR (100 MHz, d6-DMSO): 167.8, 158.3, 140.3, 132.9, 130.8 (q, J = 32.8 Hz), 123.3 (q, J = 272.8 Hz), 120.4, 120.4, 116.8, (q, J = 4.2 Hz), 116.7, 107.5, 107.1. UV(λmax nm): 275; IRvmax (cm−1): 3325, 1644, 1445, 974. HRMS (ESI): calcd for C15H10F6NO3 [M+H]+: 366.0559, found: 366.0541
5-chloro-N-(3-fluoro-4-(trifluoromethyl)phenyl)-2-hydroxybenzamide (29)
The title compound was synthesized following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 29 as a white solid (21 mg, 9%). 1H NMR (400 MHz, d6-DMSO): 11.44 (s, 1H), 10.86 – 10.81 (m, 1H), 7.96 (d, J = 13.4 Hz, 1H), 7.79 (dd, J = 15.3, 5.8 Hz, 2H), 7.67 (d, J = 8.8 Hz, 1H), 7.48 (d, J = 8.8 Hz, 1H), 7.04 (d, J = 8.8 Hz, 1H) . 13C NMR (100 MHz, d6-DMSO): 165.1, 159.1 (d, J = 249.5 Hz), 155.9, 144.2 (d, J = 11.3 Hz), 133.1, 128.8, 127.9 (qd, J = 6.8, 4.5 Hz),122.9, 122.8 (q, J = 271.0 Hz), 120.8, 119.0, 115.9 (d, J = 3.1 Hz), 111.3 (dd, J = 32.7, 12.5 Hz), 107.8 (d, J = 25.4 Hz). UV(λmax nm): 268; IRvmax (cm−1): 2924, 1606, 1122, 695; HRMS (ESI): calcd for C14H9ClF4NO2 [M+H]+:334.0252, found: 334.0232
5-chloro-N-(4-fluoro-3-nitrophenyl)-2-hydroxybenzamide (35)
The title compound was synthesized following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 35 as a white solid (67 mg, 45%). 1H NMR (400 MHz, d6-DMSO): 11.51 (s, 1H), 10.76 (s, 1H), 8.66 (dd, J = 6.9, 2.7 Hz, 1H), 8.05 (dt, J = 9.2, 3.3 Hz, 1H), 7.87 (d, J = 2.7 Hz, 1H), 7.62 (dd, J = 11.2, 9.1 Hz, 1H), 7.48 (dd, J = 8.8, 2.7 Hz, 1H), 7.03 (d, J = 8.8 Hz, 1H). 13C NMR (100 MHz, d6-DMSO): 165.2, 156.4, 150.9 (d, J = 259.3 Hz), 136.4 (d, J = 7.6 Hz), 135.0 (d, J = 3.4 Hz), 133.1, 128.5, 128.0 (d, J = 8.3 Hz), 122.7, 120.2, 119.1, 118.9 (d, J = 21.8 Hz), 117.1 (d, J = 3.0 Hz). UV(λmax nm): 254 ; IRvmax (cm−1): 3291, 1620, 1334, 932; HRMS (ESI): calcd for C13H9ClFN2O4 [M+H]+: 311.0229, found: 311.0241
N-(3-amino-4-fluorophenyl)-5-chloro-2-hydroxybenzamide (36)
To a solution of 5-chloro-N-(4-fluoro-3-nitrophenyl)-2-hydroxybenzamide 35 (1 equiv.) in anhydrous ethanol (0.1 M) was added tin (II) chloride (2 equiv.). The reaction mixture was heated to 70 °C for 12 h, cooled, and concentrated under reduced pressure. The residue was taken in a mixture of 100 mL ethyl acetate and 100 mL saturated aqueous sodium bicarbonate solution and the organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure, and then purified via flash chromatography using 10:1 DCM/methanol to obtain compound 36 as a white solid (112 mg, 71%). 1H NMR (400 MHz, d6-DMSO): 11.94 (s, 1H), 10.22 (s, 1H), 7.95 (d, J = 2.7 Hz, 1H), 7.45 (dd, J = 8.8, 2.7 Hz, 1H), 7.20 (dd, J = 8.4, 2.6 Hz, 1H), 7.00 (d, J = 9.0 Hz, 1H), 6.97 – 6.91 (m, 1H), 6.77 (ddd, J = 8.7, 4.1, 2.6 Hz, 1H), 5.24 (s, 2H). 13C NMR (100 MHz, d6-DMSO): 165.2, 157.5, 148.0 (d, J = 234.7 Hz), 136.9 (d, J = 13.7 Hz), 134.8 (d, J = 2.3 Hz), 133.4, 128.7, 123.1, 119.9, 119.6, 115.1 (d, J = 19.3 Hz), 109.3 (d, J = 4.4 Hz), 108.9 (d, J = 6.5 Hz). UV(λmax nm): 274; IRvmax (cm−1): 3351, 1651, 1128, 933; HRMS (ESI): calcd for C21H14F6NO2 [M+H]+: 281.04876, found: 281.0488
N-(4-bromo-3,5-dichlorophenyl)-5-chloro-2-hydroxybenzamide (41)
The title compound was following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 41 as a white solid (53 mg, 19%). 1H NMR (400 MHz, d6-DMSO): 11.43 (s, 1H), 10.60 (s, 1H), 8.03 (s, 2H), 7.81 (d, J = 2.7 Hz, 1H), 7.48 (dd, J = 8.8, 2.7 Hz, 1H), 7.03 (d, J = 8.8 Hz, 1H) . 13C NMR (100 MHz, d6-DMSO): 165.1, 156.0, 139.1, 135.1, 133.2, 128.7, 122.8, 120.5, 120.2, 119.0, 116.1 . UV(λmax nm): 248; IRvmax (cm−1): 3149, 1626, 1208, 655; HRMS (ESI): calcd for C13H8BrCl3NO2 [M+H]+: 393.8799, found: 393.8808
N-(3,5-dimethylphenyl)-5-fluoro-2-hydroxybenzamide (44)
The title compound was synthesized following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 44 as a white solid (58 mg, 43%). 1H NMR (400 MHz, d6-DMSO): 11.7 (s, 1H), 10.3 (s, 1H), 7.8 (dd, J = 9.8, 3.2 Hz, 1H), 7.3 (d, J = 1.4 Hz, 2H), 7.3 – 7.3 (m, 1H), 7.0 (dd, J = 9.0, 4.6 Hz, 1H), 6.8 (s, 1H), 2.3 (s, 6H). 13C NMR (100 MHz, d6-DMSO): 165.3 (d, J = 2.5 Hz), 155.3 (d, J = 235.0 Hz), 154.9, 138.3 (d, J = 2.8 Hz), 126.3, 121.0, 120.8, 119.1 (d, J = 7.6 Hz), 119.0, 118.9 (d, J = 6.9 Hz), 115.2 (d, J = 24.4 Hz), 21.5. UV(λmax nm): 271; IRvmax (cm−1): 3086, 1634, 1578, 912; HRMS (ESI): calcd for C15H15FNO2 [M+H]+: 260.1081, found: 260.1064
N-(3,5-dibromophenyl)-2-hydroxy-5-iodobenzamide (52)
The title compound was synthesized following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 52 as a white solid (53 mg, 13%). 1H NMR (400 MHz, d6-DMSO): 11.46 (s, 1H), 10.59 (s, 1H), 8.08 (d, J = 2.3 Hz, 1H), 8.00 (d, J = 1.7 Hz, 2H), 7.71 (dd, J = 8.7, 2.3 Hz, 1H), 7.59 (t, J = 1.7 Hz, 1H), 6.83 (d, J = 8.6 Hz, 1H) . 13C NMR (100 MHz, d6-DMSO): 165.1, 158.6, 141.5, 141.2, 137.2, 128.4, 122.3, 121.7, 120.3, 79.9. UV(λmax nm): 236; IRvmax (cm−1): 3079, 1636, 1109, 664; HRMS (ESI): calcd for C13H9Br2INO2[M+H]+: 495.8039, found: 495.8059
N-(3,5-difluorophenyl)-2-hydroxy-4-(trifluoromethyl)benzamide (54)
The title compound was synthesized following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 54 as a white solid (191 mg, 50%).1H NMR (400 MHz, d6-DMSO): 11.54 (s, 1H), 10.70 (s, 1H), 7.88 (d, J = 7.9 Hz, 1H), 7.53 – 7.45 (m, 2H), 7.29 (d, J = 8.5 Hz, 2H), 7.00 (tt, J = 9.4, 2.4 Hz, 1H) . 13C NMR (100 MHz, d6-DMSO): 164.9, 162.5 (dd, J = 243.2, 15.3 Hz), 156.5, 141.0 (t, J = 13.7 Hz), 132.5 (q, J = 31.8 Hz), 130.8, 124.5, 123.6 (q, J = 272.7 Hz), 115.5 (q, J = 3.8 Hz), 113.3 (q, J = 3.9 Hz), 103.0 (dd, J = 20.8, 8.9 Hz), 99.2 (t, J = 26.2 Hz) . UV(λmax nm): 268; IRvmax (cm−1): 3173, 1620, 1113, 841; HRMS (ESI): calcd for C14H9F5NO2 [M+H]+: 318.0548, found: 318.0536.
N-(3,5-dichlorophenyl)-2-hydroxy-4-(trifluoromethyl)benzamide (55)
The title compound was synthesized following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 55 as a white solid (239 mg, 56%). 1H NMR (400 MHz, d6-DMSO): 11.54 (s, 1H), 10.65 (s, 1H), 7.89 (d, J = 7.9 Hz, 1H), 7.84 (d, J = 1.9 Hz, 2H), 7.37 (t, J = 2.0 Hz, 1H), 7.29 (d, J = 8.7 Hz, 2H) . 13C NMR (100 MHz, d6-DMSO): 165.0, 156.6, 140.8, 134.1, 132.6 (q, J = 31.9 Hz), 130.8, 124.3, 123.6 (q, J = 272.8 Hz), 123.3, 118.3, 115.5 (q, J = 3.7 Hz), 113.3 (q, J = 4.0 Hz). UV(λmax nm): 270; IRvmax (cm−1): 3081, 1640, 1110, 664; HRMS (ESI): calcd for C14H9Cl2F3NO2[M+H]+: 349.9957, found: 349.9935
N-(3,5-dimethylphenyl)-2-hydroxy-4-(trifluoromethyl)benzamide (57)
The title compound was synthesized following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 57 as a white solid (25 mg, 14%). 1H NMR (400 MHz, d6-DMSO): 11.91 (s, 1H), 10.32 (s, 1H), 8.02 (d, J = 8.0 Hz, 1H), 7.34 (s, 2H), 7.29 (d, J = 9.3 Hz, 2H), 6.79 (s, 1H), 2.27 (s, 6H) . 13C NMR (100 MHz, d6-DMSO): 164.6, 157.4, 138.1, 137.9, 132.6 (q, J = 32.0 Hz), 130.7, 125.9, 123.6 (q, J = 272.8 Hz), 123.4, 118.3, 115.4 (q, J = 3.6 Hz), 113.6 (q, J = 3.8 Hz), 21.1 . UV(λmax nm): 302; IRvmax (cm−1): 2588, 1620, 1119, 681; HRMS (ESI): calcd for C16H15F3NO2 [M+H]+: 310.1049, found: 310.1046
3-chloro-N-(3,5-dimethylphenyl)-2-hydroxybenzamide (58)
The title compound was synthesized following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 58 as a white solid (45 mg, 27%). 1H NMR (400 MHz, d6-DMSO): 12.9 (s, 1H), 10.5 (s, 1H), 8.0 (dd, J = 8.1, 1.5 Hz, 1H), 7.7 (dd, J = 7.9, 1.4 Hz, 1H), 7.3 (s, 2H), 7.0 (t, J = 8.0 Hz, 1H), 6.8 (s, 1H), 2.3 (s, 6H). 13C NMR (100 MHz, d6-DMSO): 167.8, 156.2, 137.8, 137.2, 134.0, 126.8, 126.4, 121.3, 119.6, 119.1, 117.2, 21.0. UV(λmax nm): 271 ; IRvmax (cm−1): 3380, 1614, 1549, 1117; HRMS (ESI): calcd for C15H15ClNO2 [M+H]+: 276.0786, found: 276.0767.
3,5-dichloro-N-(3,5-dimethylphenyl)-2-hydroxybenzamide (59)
The title compound was synthesized following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 59 as a white solid (65 mg, 44%). 1H NMR (400 MHz, d6-DMSO): 12.9 (s, 1H), 10.7 (s, 1H), 8.1 (t, J = 2.9, 1.5 Hz, 1H), 7.8 (d, J = 2.6 Hz, 1H), 7.3 (s, 2H), 6.8 (s, 1H), 2.3 (s, 6H). 13C NMR (100 MHz, d6-DMSO): 166.4, 155.5, 137.8, 137.2, 133.0, 126.4, 122.7, 121.8, 119.3, 118.3, 118.2, 21.0. UV(λmax nm): 265; IRvmax (cm−1): 3234, 1632, 1572, 1168. HRMS (ESI): calcd for C15H14Cl2NO2 [M+H]+: 310.0396, found: 310.0385.
3,5-dichloro-N-(3,5-dibromophenyl)-2-hydroxybenzamide (60)
The title compound was synthesized following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 60 as a white solid (25 mg, 19%). 1H NMR (400 MHz, d6-DMSO): 14.75 (s, 1H), 7.92 (d, J = 1.7 Hz, 2H), 7.64 (d, J = 3.0 Hz, 1H), 7.44 (t, J = 1.7 Hz, 1H), 7.34 (d, J = 3.0 Hz, 1H). 13C NMR (100 MHz, d6-DMSO): 166.3, 163.9, 143.0, 131.7, 127.5, 127.4, 126.1, 122.9, 121.3, 119.3, 113.3. UV(λmax nm): 278; IRvmax (cm−1): 3293, 1640, 1579, 145;. HRMS (ESI): calcd for C13H8Br2Cl2NO2 [M+H]+: 437.8293, found: 437.8261.
N-(2,5-bis(trifluoromethyl)phenyl)-3,5-dichloro-2-hydroxybenzamide (61)
The title compound was synthesized following the general procedure for synthesis of N-aryl-2-hydroxybenzamides to afford 61 as a white solid (21 mg, 12%). 1H NMR (400 MHz, d3-Acetonitrile): 9.0 (s, 1H), 7.8 (d, J = 8.3 Hz, 1H), 7.7 (d, J = 3.0 Hz, 1H), 7.5 – 7.4 (m, 1H), 7.3 (d, J = 3.0 Hz, 1H). 13C NMR (100 MHz, d3-Acetonitrile): 167.6, 166.4, 140.3, 134.5, 134.2, 132.3, 128.7, 128.0 (q, J = 5.8 Hz), 127.2, 124.8 (q, J = 271.9 Hz), 124.6 (q, J = 271.9 Hz), 121.8 (q, J = 4.1 Hz), 119.9, 119.4 (q, J = 3.8 Hz), 113.3. UV(λmax nm): 273; IRvmax (cm−1): 3291, 1640, 1542, 1120; HRMS (ESI): calcd for C15H7Cl2F6NO2Na [M+Na]+: 439.9650, found: 439.9659.
General Biological Experimental
Bacterial strains, media, and antibiotics
A. baumannii strains 4106, 4112, 4119, 3941 and 3942 were obtained from Walter Reed Army Institute for Research (WRAIR). A. baumannii strain 17978mcr−1 and K. pneumoniae strains B9, A5, C3 and F2210291mcr−1 were obtained from Professor Robert Ernst at The University of Maryland, Baltimore. Stock cultures were stored in 25% glycerol and maintained at −80 °C. Prior to use, colonies were grown on LB (Lennox) agar. CAMHB was purchased from BD Diagnostics. Colistin sulfate salt was purchased from Sigma Aldrich (Cat# C4461). All assays were run in duplicate and repeated at least two separate times.
Broth microdilution method for the determination of minimum inhibitory concentrations
Bacteria were cultured for 4 to 6 hours in CAMHB and subcultured to 1.04 x 106 CFU/mL for AB 4106 and 1.96 x 106 CFU/mL for KP B9 in fresh CAMHB. To aliquots (1 mL) was added compound from stock solutions in DMSO, such that the compound concentration equaled the highest concentration tested. Samples were then dispensed (200 μL) into the first row of a 96-well microtiter plate in which all but the final row of subsequent wells were prefilled with 100 μL of the untreated bacterial subculture. The final row was filled with media to act as a sterility control and blank. Row one wells were mixed 6-7 times, then, 100 μL was withdrawn and transferred to row two. Row two wells were mixed 6-7 times followed by a 100 μL transfer from row two to row three. This procedure was used to serially dilute the rest of the rows of the microtiter plate, excluding the last prefilled row, which was used to measure growth in the absence of compound. Plates were then sealed with GLAD Press’n Seal and incubated under stationary conditions at 37 °C. After 16 hours, the plates were removed, and MIC values were measured by recording the OD600 of each well. MIC values were determined as the minimum concentration required to achieve 90% growth inhibition compared to growth in untreated wells.
Broth microdilution method for measurement of colistin potentiation
Bacteria were cultured for 4 to 6 hours in CAMHB and diluted to 1.04 x 106 CFU/mL for AB 4106 and 1.96 x 106 CFU/mL for KP B9 in fresh CAMHB. To aliquots (4 mL) was added compound from stock solutions in DMSO. One aliquot was not dosed to allow measurement of the colistin MIC in the absence of compound. A 1 mL aliquot of each sample was dosed with colistin, and from this 200 μL was dispensed into the first row of a 96-well microtiter plate in which all but the final row of subsequent wells was prefilled with 100 μL of the corresponding compound dosed bacterial suspension The final row was filled with media to act as a sterility control and blank. Row one wells were mixed 6-7 times, then, 100 μL was withdrawn and transferred to row two. Row two wells were mixed 6-7 times followed by a 100 μL transfer from row two to row three. This procedure was used to serially dilute the rest of the rows of the microtiter plate, excluding the last prefilled row, which was used to measure growth in the presence of compound alone. Plates were then sealed with GLAD Press’n Seal and incubated under stationary conditions at 37 °C. After 16 hours, the plates were removed, and MIC values were measured by recording the OD600 of each well. MIC values were determined as the minimum concentration required to achieve 90% growth inhibition compared to growth in untreated wells.
Time-kill Curves
Strains were cultured overnight in CAMHB and subcultured to 1.04 x 106 CFU/mL for AB 4106 and 1.96 x 106 CFU/mL for KP B9 in fresh CAMHB. The subculture was then transferred to culture tubes in 3 mL aliquots, which were dosed with adjuvant, colistin, or adjuvant plus colistin. One aliquot was not dosed to serve as a control. All samples were then incubated at 37 °C with shaking. At 2, 4, 6, 8, and 24-hour time points, 100 μL was taken from each sample and ten-fold diluted in CAMHB up to 7 times. 100 μL of diluted culture was plated on LB (Lennox) agar and incubated at 37 °C overnight. The total number of bacterial colonies on each plate was determined using a SphereFlash colony counter (NEUTEC Group Inc.)
Cell line Toxicity
4T1 cells (ATCC Manassas, VA) were plated at a density of 1 x 104 cells/well in 96-well plates in Roswell Park Memorial Institute Media 1640 (RPMI) (Gibco, Gaithersburg, MD) supplemented with 10% Fetal Bovine Serum (Gibco), 2 mM GlutaMAX (Gibco) and 50 μM 2-mercaptoethanol (Sigma Aldrich, St. Louis, MO) and incubated at 37°C under a 5% CO2 atmosphere in the dark for 18 h. Cell cultures were treated with serial dilutions of compounds in the presence or absence of 1 μg/mL colistin (3 replicates per condition) and incubated for an additional 18 hours. The following control conditions were used: media only (blank), 1% Triton X100 (0% cell viability), 0.5% DMSO (100% cell viability). Each condition was then treated with 10% volume of a 5 mg/mL solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma Aldrich) in sterile filtered 1X phosphate buffered saline (PBS) and incubated for 2 h. at 37° C in 5% CO2, after which the media was aspirated and the resulting formazan crystals were resuspended in 100 μL acidified (4 mM HCl) isopropanol. The 96-well plate was then read at 540 nm on a FLUOstar Optima (BMG Labtech Cary, NC) microplate reader. Cell viability was calculated as a percentage using the two previously mentioned controls.
Supplementary Material
Figure 1.

Previously identified compounds that break colistin resistance
Acknowledgements
The authors thank the NIH (1R01AI136904) for funding.
Footnotes
Supporting information for this article is given via a link at the end of the document
References:
- [1].O’Neill J, 2014.
- [2].Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J, Clin Infect Dis 2009, 48(1), 1–12. [DOI] [PubMed] [Google Scholar]
- [3].Kmietowicz Z, BMJ 2017, 358, j4339. [DOI] [PubMed] [Google Scholar]
- [4].Stevens DL, Herr D, Lampiris H, Hunt JL, Batts DH, Hafkin B, Clin Infect Dis 2002, 34(11), 1481–1490; [DOI] [PubMed] [Google Scholar]; Fowler VG, Boucher HW, Corey GR, Abrutyn E, Karchmer AW, Rupp ME, Levine DP, Chambers HF, Tally FP, Vigliani GA, Cabell CH, Link AS, DeMeyer I, Filler SG, Zervos M, Cook P, Parsonnet J, Bernstein JM, Price CS, Forrest GN, Fätkenheuer G, Gareca M, Rehm SJ, Brodt HR, Tice A, Cosgrove SE, S. a. E. a. B. S. Group, N Engl J Med 2006, 355(7), 653–665. [DOI] [PubMed] [Google Scholar]
- [5].Richter MF, Hergenrother PJ, Reaction: Broad-Spectrum Antibiotics, a Call for Chemists, Vol. 3, Chem 2017, pp. 8–14. [Google Scholar]
- [6].Nation RL, Li J, Cars O, Couet W, Dudley MN, Kaye KS, Mouton JW, Paterson DL, Tam VH, Theuretzbacher U, Tsuji BT, Turnidge JD, Lancet Infect Dis 2015, 15(2), 225–234. [DOI] [PubMed] [Google Scholar]
- [7].Biswas S, Brunel JM, Dubus JC, Reynaud-Gaubert M, Rolain JM, Expert Rev Anti Infect Ther 2012, 10(8), 917–934. [DOI] [PubMed] [Google Scholar]
- [8].Ozkan G, Ulusoy S, Orem A, Alkanat M, Mungan S, Yulug E, Yucesan FB, Antimicrob Agents Chemother 2013, 57(8), 3463–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Petrosillo N, Taglietti F, Granata G, J Clin Med 2019, 8(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pelletier MR, Casella LG, Jones JW, Adams MD, Zurawski DV, Hazlett KR, Doi Y, Ernst RK, Antimicrob Agents Chemother 2013, 57(10), 4831–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Olaitan AO, Morand S, Rolain JM, Front Microbiol 2014, 5, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Caliendo AM, Gilbert DN, Ginocchio CC, Hanson KE, May L, Quinn TC, Tenover FC, Alland D, Blaschke AJ, Bonomo RA, Carroll KC, Ferraro MJ, Hirschhorn LR, Joseph WP, Karchmer T, MacIntyre AT, Reller LB, Jackson AF, A. Infectious Diseases Society of, Clin Infect Dis 2013, 57 Suppl 3, S139–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Melander RJ, Melander C, ACS Infect Dis 2017, 3(8), 559–563; [DOI] [PMC free article] [PubMed] [Google Scholar]; Wright GD, Trends Microbiol 2016, 24(11), 862–871. [DOI] [PubMed] [Google Scholar]
- [14].Huggins WM, Barker WT, Baker JT, Hahn NA, Melander RJ, Melander C, ACS Med Chem Lett 2018, 9(7), 702–707; [DOI] [PMC free article] [PubMed] [Google Scholar]; Barker WT, Chandler CE, Melander RJ, Ernst RK, Melander C, Bioorg Med Chem 2019, 27(9), 1776–1788; [DOI] [PMC free article] [PubMed] [Google Scholar]; Minrovic BM, Hubble VB, Barker WT, Jania LA, Melander RJ, Koller BH, Melander C, ACS Med Chem Lett 2019, 10(5), 828–833; [DOI] [PMC free article] [PubMed] [Google Scholar]; Brackett CM, Furlani RE, Anderson RG, Krishnamurthy A, Melander RJ, Moskowitz SM, Ernst RK, Melander C, Tetrahedron 2016, 72(25), 3549–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Barker WT, Nemeth AM, Brackett SM, Basak AK, Chandler CE, Jania LA, Zuercher WJ, Melander RJ, Koller BH, Ernst RK, Melander C, ACS Infect Dis 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kang S, Min HJ, Kang MS, Jung MG, Kim S, Bioorg Med Chem Lett 2013, 23(6), 1748–1751. [DOI] [PubMed] [Google Scholar]
- [17].Lindsey EA, Worthington RJ, Alcaraz C, Melander C, Org Biomol Chem 2012, 10(13), 2552–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.