

Abstract
Alisertib, an Aurora kinase A inhibitor, was evaluated in a Phase 1 study in combination with the histone deacetylase inhibitor vorinostat, in patients with relapsed/refractory lymphoid malignancies (N=34; ). Patients received alisertib plus vorinostat in 21-day treatment cycles with escalating doses of alisertib following a continuous or an intermittent schedule. All dose-limiting toxicities (DLTs) were hematologic and there were no study-related deaths. The recommended phase 2 dose (RP2D) of the combination was 20 mg bid of alisertib and 200 mg bid of vorinostat on the intermittent schedule. A 13-patient expansion cohort was treated for a total of 18 patients at the RP2D. There were no DLTs at the RP2D, and toxicities were mainly hematologic. Two patients with DLBCL achieved a durable complete response, and two patients with HL achieved partial response. Alisertib plus vorinostat showed encouraging clinical activity with a manageable safety profile in heavily pretreated patients with advanced disease.
Keywords: alisertib, vorinostat, lymphoma, aurora kinase, histone deacetylase inhibitor
Introduction
The use of aggressive chemoimmunotherapy and stem cell transplantation has led to improvements in outcomes of patients with lymphoid malignancies, yet relapses are frequent and remain a therapeutic challenge. New molecular targeted agents are therefore promising therapeutic options. Aurora kinases (AURK) are a family of serine/threonine kinases including types A, B and C that regulate key functions during mitosis.[1, 2] Aurora kinase A (AURKA) is expressed in most normal cell types and regulates the function of centrosomes, spindles and kinetochores, all required for proper mitotic progression.[3] When overexpressed, AURKA induces centrosome amplification, chromosome instability, and oncogenic transformation in mammalian cells.[4, 5] Amplification or overexpression of AURKA has been reported in multiple malignancies, including breast, ovary, lung, head and neck, and colon cancers and is associated with poorly differentiated tumors, genomic stability, and poor prognosis.[6, 7, 8, 9, 10, 11] Interestingly, AURKA is also overexpressed in non-Hodgkin lymphomas where its expression correlates with disease aggressiveness, with the highest expression reported in high-risk diffuse large B-cell lymphoma (DLBCL) and Burkitt lymphoma.[12]
Alisertib (MLN8237) is a high affinity, selective oral inhibitor of AURKA that induces mitotic spindle defects and mitosis delays, associated with a reduction in cell proliferation in lymphoma and myeloma cell lines and mouse models.[13, 14] It has been evaluated for safety and efficacy in clinical studies of various non-hematologic malignancies[15, 16, 17, 18] and pediatric cancers.[19] In addition, anti-tumor responses were observed with single-agent alisertib in patients with hematologic malignancies, including B-cell non-Hodgkin lymphomas and peripheral T-cell lymphoma (PTCL).[20, 21, 22] Alisertib has also been studied in combination with several chemotherapy agents, where it was also safe and well-tolerated.[23, 24, 25, 26] Vorinostat is an oral histone deacetylase (HDAC) inhibitor that is FDA-approved for the treatment of cutaneous T-cell lymphoma (CTCL)[27], and has been tested in other lymphomas.[28, 29, 30, 31] This pan-HDAC inhibitor inhibits class I and class II enzymes. HDAC inhibitors are a class of anti-cancer agents acting on chromatin structure and function.[32] They exert their anti-tumor effects by promoting the acetylation of histones, the core proteins around which DNA is wound. Histone acetylation renders the DNA more open for transcription, which results in increased expression of several genes that are often silenced in cancer, such as tumor suppressor genes.[33]
In a preclinical study, the addition of the HDAC inhibitor vorinostat to the AURKA inhibitors MK0457 and MK5108 reactivated pro-apoptotic genes and enhanced cell death via epigenetic and protein acetylation mechanisms in various lymphoma cell lines.[34] Synergistic effects of Aurora-B ablation and HDAC inhibition on survival and proliferation of lymphoma cell lines were reported as well.[35] In addition, cytotoxic effects of the combination of alisertib and vorinostat were observed in cancer cell lines in a panel of pediatric cell lines including leukemia, medulloblastoma, and neuroblastoma cell lines.[36] Moreover, romidepsin, another HDAC inhibitor, showed synergy with alisertib through cytokinesis failure in T-cell lymphoma cell lines.[37] Ultimately, HDAC inhibitors have also been shown to decrease expression of AURKA in various cancer cells.[38, 39, 40] Overall, combining an AURKA inhibitor with an HDAC inhibitor such as vorinostat may lead to reactivation of the pro-apoptotic capacity of cells, rendering them more susceptible to apoptosis triggered by cell-cycle inhibition.
Collectively, these data provided a rationale for evaluating alisertib in combination with vorinostat in patients with lymphoid malignancies in a phase 1 multicenter study (). We describe the safety and tolerability, pharmacokinetics, and anti-tumor activity of combined alisertib and vorinostat in relapsed/refractory patients with various lymphoid malignancies.
Materials and methods
Patients
Eligible patients were ≥18 years, with a diagnosis of relapsed or refractory lymphoid malignancy (classical Hodgkin lymphoma, B- and T-cell non-Hodgkin lymphomas) that was histologically or cytologically confirmed, and had measurable disease. Patients were required to have an Eastern Cooperative Oncology Group performance status of ≤2, adequate organ and hematologic function (including an absolute neutrophil count ≥1500/μL and platelets ≥100,000/μL) and to have recovered from toxicity of prior treatments. Prior hematopoietic stem cell transplant patients were allowed if they were past day +100 of transplant, had no active graft-versus-host disease and had been off immunosuppressants for at least 4 weeks. Exclusion criteria included administration of chemotherapy or radiotherapy within 4 weeks (6 weeks for nitrosoureas or mitomycin C) prior to entering the study, known brain metastases, and prior use of valproic acid or any other HDAC inhibitor for lymphoma treatment.
The study (ClinicalTrials.gov: ) was conducted in accordance with the International Conference on Harmonization Guidelines for Good Clinical Practice and the Declaration of Helsinki. Institutional Review Boards and/or independent ethics committees at each participating center reviewed and approved the study protocol. All patients provided written informed consent before the conduct of any study-related procedure.
Study Design
This open-label study was conducted at 4 centers in the United States (patients were enrolled at City of Hope Comprehensive Cancer Center, UC Davis Comprehensive Cancer Center, and USC Norris Comprehensive Cancer Center; pharmacokinetic studies were performed at UPMC Hillman Cancer Center), and enrolled a total of 35 patients (34 patients were treated). The primary objective was to evaluate the safety and tolerability of the combination of alisertib plus vorinostat, determine the maximum tolerated dose (MTD) of alisertib (MLN8237) when given in combination with vorinostat, and select a recommended Phase 2 dose (RP2D) and schedule in patients with lymphoid malignancies. During the dose escalation stage, patients were enrolled using a novel 3-at-Risk design (see below). Once the MTD was determined, 13 additional subjects were treated at the RP2D to further define the safety and efficacy profiles of the regimen. Secondary objectives involved pharmacokinetics (PK) and clinical response assessment.
Toxicities were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE, Version 4.0). Patients were monitored during the first cycle (21 days) for DLTs defined as: grade ≥3 non-hematologic toxicity, excluding fatigue and inadequately treated nausea, vomiting or diarrhea; grade 4 neutropenia lasting >7 days despite the use of growth factors; grade 4 thrombocytopenia regardless of duration; grade 3 thrombocytopenia lasting >7 days or associated with bleeding and/or requiring transfusion; toxicity within the first cycle that prevents administration of ≥80% of the total planned dose; or any delay in therapy of greater than 14 days. For the purpose of dose escalation, the rule was to replace patients who did not experience a DLT during cycle 1, and yet missed more than 20% of the total planned dose of alisertib or vorinostat for the cycle.
This phase 1 study design, termed the 3-at-Risk design, is a modification of the standard 3+3 phase 1 design.[41] Similar to the 3+3 design, if 1 DLT occurs at a dose level, 6 patients must be treated at that level, however, in the novel design additional patients can be treated as long as at most 3 patients are under observation for first-cycle DLT at any time and the risks are consistent with rules of the 3+3. Similar to the 3+3 design, should 2 DLTs occur at a dose level, the dose would be de-escalated; should no DLT occur in the first 3-5 patients, or if 1 DLT occurs in 6 patients, the dose would be escalated. With this design, the MTD is declared as the highest dose where 6 patients have been treated and at most 1 patient experienced DLT.
Treatment schedules
Dose escalation/de-escalation was performed using 2 different schedules, continuous and intermittent. Table 2 shows the details of individual dose levels for both schedules. Enrollment was initiated on the continuous dosing Schedule I: alisertib (MLN8237, Millennium/Takeda) was administered orally twice a day on days 1-7 and escalated from 30 to 40 mg; 400 mg vorinostat (Merck; FDA-approved daily dose for CTCL) was administered orally on days 1-14 (400 mg at once in dose level 1, then 200 mg twice a day in dose level 2). Due to frequent AEs/dose delays in patients treated on Schedule I, primarily gastrointestinal intolerance and myelosuppression, the dosing was changed to the intermittent dosing Schedule II: alisertib was administered on days 1-3 and 8-10 and de-escalated from 30 to 20 mg; 200 mg vorinostat was administered twice a day on days 1-5 and 8-12. Each cycle was 21-days long on both schedules. Safety assessments included physical examination, vital signs, laboratory test measurements, performance status, and AE reporting.
Table 2.
Treatment table
| Schedule I | Dose Level |
Alisertib D1-D7 |
Vorinostat D1-D14 |
n | DLTs |
|---|---|---|---|---|---|
| 1 | 30 mg po bid | 400 mg po qd | 6 | 1* | |
| 2 | 40 mg po bid | 200 mg po bid | 3 | 2** | |
| Schedule II | Dose Level |
Alisertib D1-D3 and D8-D10 |
Vorinostat D1-D5 and D8-D12 |
n | DLTs |
| 1 | 30 mg po bid | 200 mg po bid | 7 | 2*** | |
| −1 | 20 mg po bid | 200 mg po bid | 5 + 13 | 0 |
Grade 3 thrombocytopenia requiring transfusion
Both grade 4 thrombocytopenia
One grade 4 febrile neutropenia, and one grade 3 thrombocytopenia requiring transfusion
Pharmacokinetic assessments
Pharmacokinetic (PK) sampling was performed before and at 0.5, 1, 2, 3, 4, 6 hours (schedule I) or 0.5, 1, 2, 3, 4, 6, 8, 12 hours (schedule II) following the oral dose of vorinostat and MLN8237 on day 7 for schedule 1, and day 10 for schedule II. At each time point, 8 mL of peripheral blood were collected, with 4 mL placed in red-top vacutainer tubes for serum collection and 4 mL placed in K2EDTA lavender-top tubes for plasma collection. Serum was collected from red-top tubes after clotting for approximately 30 minutes, followed by centrifugation at 2,000 × g for 15 min at 4°C and stored at −70°C until analysis. Plasma was collected from lavender-top tubes by centrifugation at 1,500 × g for 10 min at 4°C and stored at −70°C until analysis. Vorinostat was quantitated in serum using a previously published and validated LC-MS/MS assay.[42] Alisertib was quantitated in plasma using an LC-MS/MS assay developed for the purpose of this study (See Suppl. Materials and Methods).
Efficacy assessments
The efficacy population included all eligible and treated patients. Radiologic evaluations were obtained at screening and every 3 cycles thereafter. Response and progression status were assessed according to the Revised Response Criteria for Malignant Lymphoma.[43] Efficacy endpoints included complete response (CR) and partial response (PR).
Results
Patient characteristics and disposition
Thirty-four patients were enrolled and treated in this study between May 2012 and June 2015. Patient demographics and baseline characteristics are presented in Table 1. All patients had a form of relapsed or refractory lymphoid malignancy including Hodgkin lymphoma, B-, and T-cell non-Hodgkin lymphomas. The most common diagnosis was diffuse large B-cell lymphoma (DLBCL), followed by classical Hodgkin lymphoma (HL) and follicular lymphoma (FL). Median age of patients was 59 years (range 26-82 years) and the majority of patients were male (71%). Most patients had an ECOG performance status of 1 (71%) and were heavily pre-treated, with a median number of prior lines of therapy of 4 (range 1-10). Forty-one percent had undergone prior stem cell transplantation.
Table 1.
Patient demographics and baseline characteristics
| Characteristics | Population (N = 34) |
|---|---|
| Median age, y (range) | 59 (26-82) |
| Sex, male, n (%) | 24 (71) |
| ECOG performance status, n (%) | |
| 0 | 7 (21) |
| 1 | 24 (71) |
| 2 | 3 (9) |
| Median number of prior therapies, n (range) | 4 (1-10) |
| Prior stem cell transplantation, n (%) | 14 (41) |
| Disease type/subtype, n (%) | |
| Diffuse large B-cell lymphoma (DLBCL) | 12 (41) |
| Classical Hodgkin lymphoma (HL) | 8 (24) |
| Follicular lymphoma (FL) | 5 (12) |
| Mantle cell lymphoma (MCL) | 3 (6) |
| Chronic lymphocytic leukemia (CLL/SLL) | 2 (6) |
| Anaplastic large cell lymphoma (ALCL) | 1 (3) |
| Marginal zone lymphoma (MZL) | 1(3) |
| Natural killer (NK)/T-cell lymphoma (TCL) | 1 (3) |
| Peripheral T-cell lymphoma (PTCL) | 1 (3) |
MTD/RP2D determination
Ten patients were initially enrolled on the continuous dosing Schedule I, with alisertib administered from Day 1 to Day 7 and vorinostat from Day 1 to Day 14 of the cycle. Nine were treated, as one patient was not able to start treatment due to an insufficient neutrophil count at therapy initiation. Three DLTs (one grade 3 thrombocytopenia requiring transfusion at dose level 1 and two grade 4 thrombocytopenia events at dose level 2) were observed on Schedule I (Table 2). At dose level 1 on this schedule, in addition to the DLT, of the remaining 5 patients, one came off treatment after 1 cycle due to patient choice (with grade 3 fatigue), and 2 patients had dose delays during cycle 2 due to myelosuppression, with one of those two patients coming off treatment due to esophagitis after cycle 3. On level 2, the one patient without a DLT came off treatment due to myelosuppression on cycle 2, so all three patients had unacceptable toxicity despite switching vorinostat administration to a bi-daily dosing. To improve tolerance, we changed the dosing to the intermittent Schedule II, where the administration of both alisertib and vorinostat was interrupted for 4 and 2 days each treatment week, respectively, and the total number of days of dosing was reduced from 7 to 6 days for alisertib, and from 14 to 10 days for vorinostat. Twelve patients were treated on Schedule II over two cohorts during the dose-finding portion of the study (Table 2). Seven patients were treated on dose level 1 (one patient was replaced due to receiving <80% of drug without experiencing a DLT). After 2 patients experienced a DLT at dose level 1 (one grade 4 febrile neutropenia and one grade 3 thrombocytopenia requiring transfusion), the dose of alisertib was reduced to dose level −1. This dose and schedule was well tolerated and the MTD of the combination was set at 20 mg alisertib BID and 200 mg vorinostat BID on the intermittent dosing Schedule II. We selected this dose/schedule to be the recommended phase 2 dose (RP2D). An additional 13 patients (expansion cohort) were then treated at the MTD/RP2D.
Our study used an original study design named the 3-at-Risk method, a modification of the standard 3+3 design. This method was designed to adapt to asynchronous patient arrival and evaluation, screen failures and patient inevaluability, with the aim of reducing the duration of Phase 1 studies by increasing speed of accrual. With clinically relevant parameters, simulations comparing the 3-at-risk method to the 3+3 method demonstrated approximately a 1-month reduction in the average duration of a Phase I clinical trial for every year a study is accruing, with only a small (0-2 patient) increase in the median total number of patients required to complete the trial.
Safety
The key adverse events experienced during the dose-finding portion are summarized in Table 2 and were discussed above. Table 3 shows the toxicities experienced at the MTD/RP2D, with 18 patients treated. At the RP2D, there were two grade 4 hematologic toxicities (not dose-limiting) in cycle 1 (1 neutropenia and 1 leukopenia) and a single grade 4 neutropenia starting in cycle 9. The grade 3 treatment-related adverse events observed at the RP2D were anemia (n=3), neutropenia (n=2), thrombocytopenia (n=2), leukopenia (n=2), diarrhea (n=2), lung infection (n=2), hypophosphatemia (n=2), a thromboembolic event, a patient with pyloric stenosis/gastric ulcer with vomiting, and one patient with hypokalemia (Table 3). There were no study-related deaths. All 34 patients have stopped treatment, 21 for progressive disease, 5 for toxicity (of which only 1 was treated at the RP2D), 4 for physician/patient preference, 2 withdrew consent, and 2 patients chose to stop treatment after completing at least 2 further cycles after achieving response (CR). These 2 CR patients both remain in CR to date at 52+ (dose level 1, Schedule II) and 42+ months (RP2D, Schedule II). Alisertib given in combination with vorinostat is thus safe and tolerable in an intermittent dosing schedule among heavily pre-treated patients with relapsed/refractory lymphoid malignancies.
Table 3.
Treatment-related adverse events at the MTD (n=18, highest grade per patient)
| Cycle 1 |
All Cycles |
|||
|---|---|---|---|---|
| Adverse event, n (%) | Grade 3 | Grade 4 | Grade 3 | Grade 4 |
| Any AE | ||||
| Blood and lymphatic system disorders | ||||
| Anemia | 2 (11%) | 0 (0%) | 3 (17%) | 0 (0%) |
| Gastrointestinal disorders | ||||
| Diarrhea | 1 (6%) | 0 (0%) | 2 (11%) | 0 (0%) |
| Gastric ulcer/pyloric stenosis/vomiting | 0 (0%) | 0 (0%) | 1 (6%) | 0 (0%) |
| Infections | ||||
| Lung infection | 1 (6%) | 0 (0%) | 2 (11%) | 0 (0%) |
| Investigations | ||||
| Neutrophil count decreased | 0 (0%) | 1 (6%) | 2 (11%) | 2 (11%) |
| Platelet count decreased | 2 (11%) | 0 (0%) | 2 (11%) | 0 (0%) |
| White blood cell decreased | 1 (6%) | 1 (6%) | 2 (11%) | 1 (6%) |
| Metabolism and nutrition disorders | ||||
| Hypokalemia | 1 (6%) | 0 (0%) | 1 (6%) | 0 (0%) |
| Hypophosphatemia | 2 (11%) | 0 (0%) | 2 (11%) | 0 (0%) |
| Vascular disorders | ||||
| Thromboembolic event | 0 (0%) | 0 (0%) | 1 (6%) | 0 (0%) |
Pharmacokinetics
Vorinostat and alisertib PK data were available for 33 patients (Table 4 and Table 5, respectively). Vorinostat exposure was reduced by half when decreasing the dose from 400 to 200 mg, with an apparent clearance of approximately 293 L/h and a half-life of 2.3 h, which did not monotonically vary with alisertib dose. Alisertib exposure increased when increasing the dose from 20 to 30 mg, while the 40 mg dose had no clear interpretation with only two patients receiving this dose having usable PK data. Overall, apparent clearance was approximately 3.58 L/h and the half-life was 10.1 h.
Table 4.
Vorinostat pharmacokinetic parameters by schedule and dose level
| Vorinostat (mg) |
Alisertib (mg) |
Cmax (ng/mL) |
Tmax (h) |
AUC0-tau (μg/mL•h) |
Cl/F (L/h) |
t½ (h) |
N |
|---|---|---|---|---|---|---|---|
| Schedule I | |||||||
| 200 | 30 | 504 | 1.00 | 1.40 | 143 | 1.73 | 1 |
| 200 | 40 | 335 (148) | 0.98 (0.04) | 0.956 (0.503) | 243 (128) | 2.31 (1.19) | 2 |
| 200 | All | 391 (143) | 0.98 (0.03) | 1.10 (0.44) | 210 (107) | 2.12 (0.91) | 3 |
| 400 | 30 | 806 (582) | 2.58 (1.86) | 2.94 (1.43) | 163 (70) | 2.61 (0.99) | 6 |
| All | All | 2.05 (1.67) | 180 (82) | 2.43 (0.93) | 9 | ||
| Schedule II | |||||||
| 200 | 20 | 318 (216) | 2.25 (1.58) | 1.03 (0.54) | 369 (504) | 2.16 (1.29) | 18 |
| 200 | 30 | 324 (132) | 2.49 (1.76) | 1.01 (0.35) | 219 (79) | 2.29 (1.06) | 6 |
| 200 | All | 319 (196) | 2.31 (1.59) | 1.03 (0.49) | 331 (440) | 2.19 (1.21) | 24 |
| Both Schedules | |||||||
| All | 20 | 318 (216) | 2.25 (1.58) | 1.03 (0.54) | 369 (504) | 2.16 (1.29) | 18 |
| All | 30 | 560 (455) | 2.42 (1.71) | 1.85 (1.32) | 189 (75) | 2.38 (0.97) | 13 |
| All | 40 | 335 (148) | 0.98 (0.04) | 0.956 (0.503) | 243 (128) | 2.31 (1.19) | 2 |
| All | All | 2.24 (1.59) | 293 (387) | 2.25 (1.14) | 33 | ||
Mean (standard deviation).
Median extrapolated portion of AUC0-tau was 0.51 % (range 0-45%).
Table 5.
Alisertib pharmacokinetic parameters by schedule and dose level
| Vorinostat (mg) |
Alisertib (mg) |
Cmax (μg/mL) |
Tmax (h) |
AUC0-tau (μg/mL•h) |
Cl/F (L/h) |
t½ (h) |
N |
|---|---|---|---|---|---|---|---|
| Schedule I | |||||||
| 200 | 30 | 1.73 | 2.00 | 17.9 | 1.68 | 21.0 | 1 |
| 200 | 40 | 2.36 (0.84) | 3.48 (3.57) | 22.8a | 1.75a | 4.06a | 2 |
| 400 | 30 | 1.29 (0.55) | 2.67 (0.82) | 10.1 (4.3) | 3.40 (1.34) | 11.6 (3.70) | 6 |
| All | 30 | 1.35 (0.53) | 2.57 (0.79) | 13.2 (5.9) | 2.78 (1.41) | 13.2 (5.08) | 7 |
| All | All | 2.77 (1.49) | 2.63 (1.34) | 11.9 (5.77) | 9 | ||
| Schedule II | |||||||
| 200 | 20 | 0.776 (0.317) | 4.39 (2.62) | 7.00 (3.77) | 3.95 (2.78) | 9.50 (3.86) | 18 |
| 200 | 30 | 1.33 (0.71) | 3.81 (1.83) | 11.0 (6.9) | 3.55 (1.82) | 9.72 (3.82) | 6 |
| 200 | All | 4.24 (2.42) | 3.85 (2.55) | 9.55 (3.77) | 24 | ||
| Both Schedules | |||||||
| All | 20 | 0.776 (0.317) | 4.39 (2.62) | 7.00 (3.77) | 3.95 (2.78) | 9.50 (3.86) | 18 |
| All | 30 | 1.34 (0.59) | 3.14 (1.45) | 12.1 (6.2) | 3.16 (1.61) | 11.4 (4.6) | 13 |
| All | 40 | 2.36 (0.84) | 3.48 (3.57) | 22.8a | 1.75a | 4.06a | 2 |
| All | All | 3.84 (2.28) | 3.58 (2.37) | 10.1 (4.30) | 33 | ||
Mean (standard deviation).
Only 1 patient with available parameter.
Median extrapolated portion of AUC0-tau was 6.33% (range 0-52.3%).
Efficacy
All patients were followed until treatment failure or withdrawal of consent (Figure 1). On Schedule I, out of the 9 treated patients, there were no responders, with a median time on treatment of 2 months (range 1-4 months). Among the 25 patients treated on Schedule II, there was 1 CR in a DLBCL patient on dose level 1 (progression-free at 52+ months), 1 CR in another DLBCL patient on the RP2D (dose level −1, progression-free at 42+ months), and 2 PRs in HL patients on the RP2D (dose level −1, PD at 4 months and 9 months after initial response). At the RP2D, the overall response rate was thus 3/18 (16.7%). Of the remaining 15 patients, the median time on treatment was 2 months (range 1-15 months), with 6 patients having at least one assessment of stable disease. The two patients with a durable CR had transformed DLBCL and both halted therapy after completing 2 further cycles of treatment post-CR. They remain in CR to date, without the need to reinstate therapy. Both patients had received prior HSCT, as did the patient with PR who progressed at 9 months.
Figure 1.
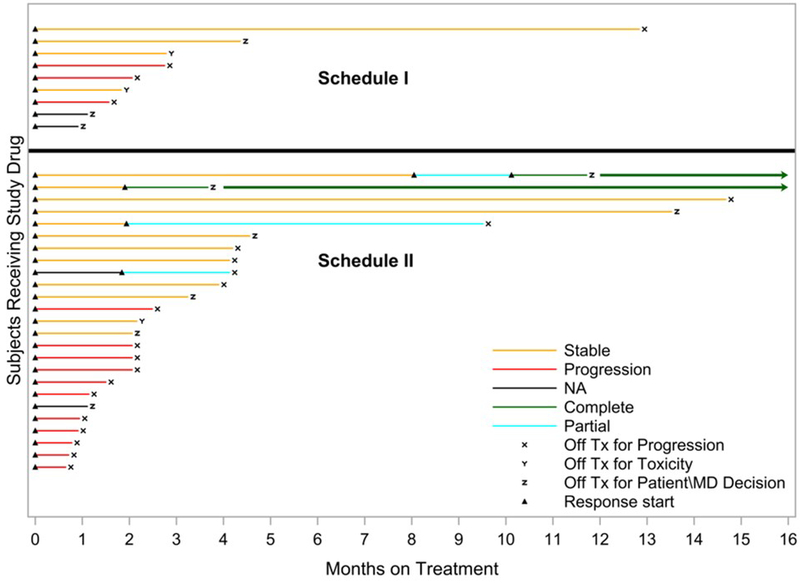
Duration of treatment and response – each bar represents one patient in the study. With the exception of the two patients who continue in CR without additional treatment on or off protocol (see text), the plots do not reflect survival after the patients discontinued protocol-specified treatment.
Discussion
Relapsed/refractory lymphomas pose a challenge as they tend not to respond well to various treatment modalities. Novel therapeutic options such as chimeric antigen receptor (CAR)-T cells are gaining traction in the aggressive B-cell non-Hodgkin lymphoma patient population, primarily DLBCL, but the remaining NHL subtypes and Hodgkin lymphoma still need better therapeutic options. The combination of alisertib and vorinostat in patients with lymphoid malignancies was motivated by evidence of synergistic effects between the two agents in preclinical studies and their clinical activity in lymphomas as single agents. Starting dose and schedule were based on MTD data available from phase 1 studies of alisertib, and on the FDA-approved daily dose of vorinostat for continuous dosing in CTCL patients. Combining these two agents also had the convenience of being an exclusively oral therapy.
In previous studies, the dominant toxicities observed with continuous administration of alisertib included blood and lymphatic system disorders as well as gastro-intestinal events, consistent with its anti-proliferative effects. As toxicities were mainly appearing from Day 8 onward in these studies, a 7-day administration schedule followed by a 2-week therapy-free period to allow recovery prior to starting a new cycle was eventually favored.[44, 45, 46] Reported adverse events for single agent vorinostat therapy included gastrointestinal and hematologic toxicities that were manageable as they resolved quickly after drug administration was interrupted,[47] but could potentially overlap with the anticipated side effects of alisertib. Our initial schedule, which featured a continuous, yet restricted in time, administration of both agents was not well tolerated, and excessive adverse events prompted us to transition to an intermittent schedule. The MTD on the intermittent schedule was declared after one dose reduction of alisertib, and no DLT was observed in the expansion cohort. Overall, adverse events observed when combining alisertib and vorinostat were mainly hematologic, in accordance with toxicities reported for each agent in the literature.
Alisertib and vorinostat share glucuronidation as a metabolic pathway.[42, 48] Vorinostat Cmax and AUC0-tau both increased proportionally with dose and parameter values (Cmax 319 ng/mL at 200 mg; clearance 331 L/h) and were consistent with previously published data (Cmax 295 ng/mL at 200 mg; clearance 271 L/h), indicating that alisertib did not affect vorinostat pharmacokinetics.[47] Alisertib Cmax and AUC0-tau also increased with dose. Our estimate of the half-life at 10.1 h is slightly lower than previous reports (14.9 h),[49] which is likely due to our schedule preventing sampling out longer than a single half-life, rather than due to a drug-drug-interaction with vorinostat. Indeed, our estimate of alisertib apparent clearance (3.58 L/h) was consistent with a previously published population estimate (4.25 L/h), indicating that alisertib did not affect vorinostat pharmacokinetics either.[48]
Although alisertib failed to show a significant benefit over a treatment based on investigator’s choice in PTCL patients, it still demonstrated clinical activity against this disease, with an ORR of 30%.[20, 21, 50] In other lymphoma subtypes, single agent alisertib led to partial responses in follicular lymphoma and DLBCL.[21, 22] Single agent vorinostat has demonstrated limited activity in relapsed/refractory Hodgkin lymphoma,[30, 51] and led to some responses in DLBCL and transformed DLBCL.[51] By contrast, two phase 2 studies in non-Hodgkin lymphomas showed an overall response rate of 47-49% in follicular lymphoma.[29, 31] The recently published phase 1 study combining alisertib and romidepsin in relapsed/refractory aggressive B-cell and T-cell lymphomas reported an ORR of 28%.[52] In this study most responses were observed in patients with HL (5 responders among 7 HL patients), while the longest PFS was 21 months. In our multicenter phase 1 study, the 2 patients who achieved CR both had DLBCL transformed from an indolent NHL (MZL and FL) and were both treated on the intermittent dosing Schedule II. As of today, these two patients remain in CR, one 52+ months later and the other 42+ months later, although they had previously failed multiple lines of therapy for their aggressive disease. The long-term response observed in these two DLBCL patients warrants further study, potentially in selected patients. Interestingly, a recent study reported some clinical activity of alisertib combined with rituximab +/− vincristine in non-germinal center B-cell (non-GCB) DLBCL.[35]
Since alisertib may work better in rapidly dividing tumor cells, this agent is now being studied in acute leukemias. It could still be a good option for patients with aggressive B- and T-cell lymphomas and seems to have some benefit in Hodgkin lymphoma, where it could potentially be assessed as part of maintenance therapy. In conclusion, alisertib in combination with vorinostat demonstrated an acceptable safety profile and showed some clinical activity in lymphoma patients. This study supports further clinical evaluation of the combination of alisertib with HDAC inhibitors especially in a select group of lymphoma patients.
Supplementary Material
Acknowledgments
Alisertib was supplied by Millennium Pharmaceuticals, Inc. and distributed by the CTEP, DCTD, NCI. Vorinostat was supplied by Merck and Co., Inc. and distributed by the CTEP, DCTD, NCI.
The authors wish to acknowledge assistance with protocol management by Stella Khoo, M.B.A., and Diana Calcanas-Perez in the California Cancer Consortium Data Coordinating Center at City of Hope.
Funding information
This trial was supported in part by the National Cancer Institute (UM1CA186717, UM1CA186690) and Leidos Biomedical Research Inc. (BOA 14X251-TO1). Research reported in this publication included work performed in the City of Hope Comprehensive Cancer Center Pathology Core supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. This project used the UPMC Hillman Cancer Center Cancer Pharmacokinetics and Pharmacodynamics Facility (CPPF) and was supported in part by award P30CA047904, and R50CA211241. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Disclosure of interest
TS: Speaker for Pharmacyclics/Janssen and Seattle Genetics; consultant for Juno Therapeutics, Pharmacyclics, BeiGene, and Astra Zeneca. RC: Millennium Pharmaceuticals: Consultancy, Research Funding; Merck & Co., Inc.: Consultancy, Research Funding, Speakers Bureau; Genentech Inc.: Consultancy; Affimed: Research Funding; Seattle Genetics: Consultancy, Honoraria, Research Funding, Speakers Bureau; Pharmacyclics: Consultancy, Research Funding; Bristol-Myers Squibb: Consultancy, Research Funding. KK: Speaker for Pharmacyclics/Janssen, Seattle Genetics, Bayer, Gilead, Novartis; consultant for Agios, Amgen, Teva and Jazz, research funding from Takeda (not for the current study). SA: Research funding: Pharmacyclics and Advisory Board/Consultant: Celgene, Takeda, Janssen, Amgen, Novartis. PK: Speaker for Bristol Myers Squibb and Incyte Pharmaceuticals.
The other authors declare no conflict of interest in regards to the current manuscript.
References
- 1.Carvajal RD, Tse A, Schwartz GK. Aurora kinases: new targets for cancer therapy. Clin Cancer Res. 2006. December 01;12(23):6869–75. doi: 10.1158/1078-0432.CCR-06-1405. PubMed PMID: 17145803. [DOI] [PubMed] [Google Scholar]
- 2.Fu J, Bian M, Jiang Q, et al. Roles of Aurora kinases in mitosis and tumorigenesis. Molecular cancer research : MCR. 2007. January;5(1):1–10. doi: 10.1158/1541-7786.MCR-06-0208. PubMed PMID: 17259342. [DOI] [PubMed] [Google Scholar]
- 3.Marumoto T, Zhang D, Saya H. Aurora-A - a guardian of poles. Nature reviews Cancer. 2005. January;5(1):42–50. doi: 10.1038/nrc1526. PubMed PMID: 15630414. [DOI] [PubMed] [Google Scholar]
- 4.Boss DS, Beijnen JH, Schellens JH. Clinical experience with aurora kinase inhibitors: a review. The oncologist. 2009. August;14(8):780–93. doi: 10.1634/theoncologist.2009-0019. PubMed PMID: 19684075. [DOI] [PubMed] [Google Scholar]
- 5.Zhou H, Kuang J, Zhong L, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nature genetics. 1998. October;20(2):189–93. doi: 10.1038/2496. PubMed PMID: 9771714. [DOI] [PubMed] [Google Scholar]
- 6.Gritsko TM, Coppola D, Paciga JE, et al. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin Cancer Res. 2003. April;9(4):1420–6. PubMed PMID: 12684414. [PubMed] [Google Scholar]
- 7.Nadler Y, Camp RL, Schwartz C, et al. Expression of Aurora A (but not Aurora B) is predictive of survival in breast cancer. Clin Cancer Res. 2008. July 15;14(14):4455–62. doi: 10.1158/1078-0432.CCR-07-5268. PubMed PMID: 18628459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishida N, Nagasaka T, Kashiwagi K, et al. High copy amplification of the Aurora-A gene is associated with chromosomal instability phenotype in human colorectal cancers. Cancer biology & therapy. 2007. April;6(4):525–33. PubMed PMID: 17457043. [DOI] [PubMed] [Google Scholar]
- 9.Reiter R, Gais P, Jutting U, et al. Aurora kinase A messenger RNA overexpression is correlated with tumor progression and shortened survival in head and neck squamous cell carcinoma. Clin Cancer Res. 2006. September 01;12(17):5136–41. doi: 10.1158/1078-0432.CCR-05-1650. PubMed PMID: 16951231. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka T, Kimura M, Matsunaga K, et al. Centrosomal kinase AIK1 is overexpressed in invasive ductal carcinoma of the breast. Cancer Res. 1999. May 01;59(9):2041–4. PubMed PMID: 10232583. [PubMed] [Google Scholar]
- 11.Xu HT, Ma L, Qi FJ, et al. Expression of serine threonine kinase 15 is associated with poor differentiation in lung squamous cell carcinoma and adenocarcinoma. Pathology international. 2006. July;56(7):375–80. doi: 10.1111/j.1440-1827.2006.01974.x. PubMed PMID: 16792546. [DOI] [PubMed] [Google Scholar]
- 12.Yakushijin Y, Hamada M, Yasukawa M. The expression of the aurora-A gene and its significance with tumorgenesis in non-Hodgkin’s lymphoma. Leukemia & lymphoma. 2004. September;45(9):1741–6. doi: 10.1080/10428190410001683615. PubMed PMID: 15223631. [DOI] [PubMed] [Google Scholar]
- 13.Gorgun G, Calabrese E, Hideshima T, et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood. 2010. June 24;115(25):5202–13. doi: 10.1182/blood-2009-12-259523. PubMed PMID: 20382844; PubMed Central PMCID: PMC2892955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manfredi MG, Ecsedy JA, Chakravarty A, et al. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res. 2011. December 15;17(24):7614–24. doi: 10.1158/1078-0432.CCR-11-1536. PubMed PMID: 22016509. [DOI] [PubMed] [Google Scholar]
- 15.Cervantes A, Elez E, Roda D, et al. Phase I pharmacokinetic/pharmacodynamic study of MLN8237, an investigational, oral, selective aurora a kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2012. September 01;18(17):4764–74. doi: 10.1158/1078-0432.CCR-12-0571. PubMed PMID: 22753585. [DOI] [PubMed] [Google Scholar]
- 16.Dees EC, Cohen RB, von Mehren M, et al. Phase I study of aurora A kinase inhibitor MLN8237 in advanced solid tumors: safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin Cancer Res. 2012. September 01;18(17):4775–84. doi: 10.1158/1078-0432.CCR-12-0589. PubMed PMID: 22767670. [DOI] [PubMed] [Google Scholar]
- 17.Matulonis UA, Sharma S, Ghamande S, et al. Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecologic oncology. 2012. October;127(1):63–9. doi: 10.1016/j.ygyno.2012.06.040. PubMed PMID: 22772063. [DOI] [PubMed] [Google Scholar]
- 18.Melichar B, Adenis A, Lockhart AC, et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. The Lancet Oncology. 2015. April;16(4):395–405. doi: 10.1016/S1470-2045(15)70051-3. PubMed PMID: 25728526. [DOI] [PubMed] [Google Scholar]
- 19.Mosse YP, Lipsitz E, Fox E, et al. Pediatric phase I trial and pharmacokinetic study of MLN8237, an investigational oral selective small-molecule inhibitor of Aurora kinase A: a Children’s Oncology Group Phase I Consortium study. Clin Cancer Res. 2012. November 01;18(21):6058–64. doi: 10.1158/1078-0432.CCR-11-3251. PubMed PMID: 22988055; PubMed Central PMCID: PMC4008248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barr PM, Li H, Spier C, et al. Phase II Intergroup Trial of Alisertib in Relapsed and Refractory Peripheral T-Cell Lymphoma and Transformed Mycosis Fungoides: SWOG 1108. J Clin Oncol. 2015. July 20;33(21):2399–404. doi: 10.1200/JCO.2014.60.6327. PubMed PMID: 26077240; PubMed Central PMCID: PMC4500834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friedberg JW, Mahadevan D, Cebula E, et al. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol. 2014. January 01;32(1):44–50. doi: 10.1200/JCO.2012.46.8793. PubMed PMID: 24043741; PubMed Central PMCID: PMC3867644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelly KR, Shea TC, Goy A, et al. Phase I study of MLN8237--investigational Aurora A kinase inhibitor--in relapsed/refractory multiple myeloma, non-Hodgkin lymphoma and chronic lymphocytic leukemia. Investigational new drugs. 2014. June;32(3):489–99. doi: 10.1007/s10637-013-0050-9. PubMed PMID: 24352795; PubMed Central PMCID: PMC4045308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DuBois SG, Marachelian A, Fox E, et al. Phase I Study of the Aurora A Kinase Inhibitor Alisertib in Combination With Irinotecan and Temozolomide for Patients With Relapsed or Refractory Neuroblastoma: A NANT (New Approaches to Neuroblastoma Therapy) Trial. J Clin Oncol. 2016. April 20;34(12):1368–75. doi: 10.1200/JCO.2015.65.4889. PubMed PMID: 26884555; PubMed Central PMCID: PMC4872349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fathi AT, Wander SA, Blonquist TM, et al. Phase I study of the aurora A kinase inhibitor alisertib with induction chemotherapy in patients with acute myeloid leukemia. Haematologica. 2017. April;102(4):719–727. doi: 10.3324/haematol.2016.158394. PubMed PMID: 28034990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graff JN, Higano CS, Hahn NM, et al. Open-label, multicenter, phase 1 study of alisertib (MLN8237), an aurora A kinase inhibitor, with docetaxel in patients with solid tumors. Cancer. 2016. August 15;122(16):2524–33. doi: 10.1002/cncr.30073. PubMed PMID: 27192055. [DOI] [PubMed] [Google Scholar]
- 26.Persky DO, Mahadevan D, Miller TP, et al. Phase 1 Study Of Investigational Agent MLN8237 (Alisertib) + Rituximab ± Vincristine In Patients (Pts) With Relapsed/Refractory (Rel/Ref) Aggressive B-Cell Lymphomas. Blood. 2013;122(21):3027–3027. [Google Scholar]
- 27.Mann BS, Johnson JR, Cohen MH, et al. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. The oncologist. 2007. October;12(10):1247–52. doi: 10.1634/theoncologist.12-10-1247. PubMed PMID: 17962618. [DOI] [PubMed] [Google Scholar]
- 28.Apuri S, Sokol L. An overview of investigational Histone deacetylase inhibitors (HDACis) for the treatment of non-Hodgkin’s lymphoma. Expert opinion on investigational drugs. 2016. June;25(6):687–96. doi: 10.1517/13543784.2016.1164140. PubMed PMID: 26954526. [DOI] [PubMed] [Google Scholar]
- 29.Kirschbaum M, Frankel P, Popplewell L, et al. Phase II study of vorinostat for treatment of relapsed or refractory indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol. 2011. March 20;29(9):1198–203. doi: 10.1200/JCO.2010.32.1398. PubMed PMID: 21300924; PubMed Central PMCID: PMC3083875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirschbaum MH, Goldman BH, Zain JM, et al. A phase 2 study of vorinostat for treatment of relapsed or refractory Hodgkin lymphoma: Southwest Oncology Group Study S0517. Leukemia & lymphoma. 2012. February;53(2):259–62. doi: 10.3109/10428194.2011.608448. PubMed PMID: 21823829; PubMed Central PMCID: PMC3477846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogura M, Ando K, Suzuki T, et al. A multicentre phase II study of vorinostat in patients with relapsed or refractory indolent B-cell non-Hodgkin lymphoma and mantle cell lymphoma. British journal of haematology. 2014. June;165(6):768–76. doi: 10.1111/bjh.12819. PubMed PMID: 24617454; PubMed Central PMCID: PMC4282031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nature reviews Drug discovery. 2002. April;1(4):287–99. doi: 10.1038/nrd772. PubMed PMID: 12120280. [DOI] [PubMed] [Google Scholar]
- 33.Johnstone RW, Licht JD. Histone deacetylase inhibitors in cancer therapy: is transcription the primary target? Cancer cell. 2003. July;4(1):13–8. PubMed PMID: 12892709. [DOI] [PubMed] [Google Scholar]
- 34.Kretzner L, Scuto A, Dino PM, et al. Combining histone deacetylase inhibitor vorinostat with aurora kinase inhibitors enhances lymphoma cell killing with repression of c-Myc, hTERT, and microRNA levels. Cancer Res. 2011. June 01;71(11):3912–20. doi: 10.1158/0008-5472.CAN-10-2259. PubMed PMID: 21502403; PubMed Central PMCID: PMC3107377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang C, Chen J, Cao W, et al. Aurora-B and HDAC synergistically regulate survival and proliferation of lymphoma cell via AKT, mTOR and Notch pathways. European journal of pharmacology. 2016. May 15;779:1–7. doi: 10.1016/j.ejphar.2015.11.049. PubMed PMID: 26638998. [DOI] [PubMed] [Google Scholar]
- 36.Muscal JA, Scorsone KA, Zhang L, et al. Additive effects of vorinostat and MLN8237 in pediatric leukemia, medulloblastoma, and neuroblastoma cell lines. Investigational new drugs. 2013. February;31(1):39–45. doi: 10.1007/s10637-012-9831-9. PubMed PMID: 22669335; PubMed Central PMCID: PMC3655801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zullo KM, Guo Y, Cooke L, et al. Aurora A Kinase Inhibition Selectively Synergizes with Histone Deacetylase Inhibitor through Cytokinesis Failure in T-cell Lymphoma. Clin Cancer Res. 2015. September 15;21(18):4097–109. doi: 10.1158/1078-0432.CCR-15-0033. PubMed PMID: 25878331; PubMed Central PMCID: PMC4581881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cha TL, Chuang MJ, Wu ST, et al. Dual degradation of aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2-M arrest and apoptosis of renal cancer cells. Clin Cancer Res. 2009. February 01;15(3):840–50. doi: 10.1158/1078-0432.CCR-08-1918. PubMed PMID: 19188154. [DOI] [PubMed] [Google Scholar]
- 39.Park JH, Jong HS, Kim SG, et al. Inhibitors of histone deacetylases induce tumor-selective cytotoxicity through modulating Aurora-A kinase. Journal of molecular medicine. 2008. January;86(1):117–28. doi: 10.1007/s00109-007-0260-8. PubMed PMID: 17851643. [DOI] [PubMed] [Google Scholar]
- 40.Zhang XH, Rao M, Loprieato JA, et al. Aurora A, Aurora B and survivin are novel targets of transcriptional regulation by histone deacetylase inhibitors in non-small cell lung cancer. Cancer biology & therapy. 2008. September;7(9):1388–97. PubMed PMID: 18708766. [DOI] [PubMed] [Google Scholar]
- 41.Frankel P, Longmate J, Sposto R, et al. The Three-at-Risk Design: Reducing the Duration of Phase I Trials with a Queue-Based Method Proceedings of the American Statistical Association, Biopharmaceutical Section, Alexandria, VA: American Statistical Association; 2016;Abstract #321198. [Google Scholar]
- 42.Parise RA, Holleran JL, Beumer JH, et al. A liquid chromatography-electrospray ionization tandem mass spectrometric assay for quantitation of the histone deacetylase inhibitor, vorinostat (suberoylanilide hydroxamicacid, SAHA), and its metabolites in human serum. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2006. August 18;840(2):108–15. doi: 10.1016/j.jchromb.2006.04.044. PubMed PMID: 16725386. [DOI] [PubMed] [Google Scholar]
- 43.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007. February 10;25(5):579–86. doi: 10.1200/JCO.2006.09.2403. PubMed PMID: 17242396. [DOI] [PubMed] [Google Scholar]
- 44.Dees EC, Infante JR, Burris HA, et al. Phase I study of the investigational drug MLN8237, an Aurora A kinase (AAK) inhibitor, in patients (pts) with solid tumors. Vol. 28 2010. [Google Scholar]
- 45.Goldberg SL, Fenaux P, Craig MD, et al. Phase 2 Study of MLN8237, An Investigational Aurora A Kinase (AAK) Inhibitor In Patients with Acute Myelogenous Leukemia (AML) or Myelodysplastic Syndromes (MDS). Blood. 2010;116(21):3273–3273. [Google Scholar]
- 46.Sharma S, Kurzrock R, Gouw L, et al. Phase I dose-escalation study of the investigational Aurora A kinase (AAK) inhibitor MLN8237 as an enteric-coated tablet (ECT) formulation in patients with nonhematologic malignancies. Vol. 29 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelly WK, O’Connor OA, Krug LM, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005. June 10;23(17):3923–31. doi: 10.1200/JCO.2005.14.167. PubMed PMID: 15897550; PubMed Central PMCID: PMC1855284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Venkatakrishnan K, Zhou X, Ecsedy J, et al. Dose selection for the investigational anticancer agent alisertib (MLN8237): Pharmacokinetics, pharmacodynamics, and exposure-safety relationships. Journal of clinical pharmacology. 2015. March;55(3):336–47. doi: 10.1002/jcph.410. PubMed PMID: 25302940. [DOI] [PubMed] [Google Scholar]
- 49.Falchook GS, Zhou X, Venkatakrishnan K, et al. Effect of Food on the Pharmacokinetics of the Investigational Aurora A Kinase Inhibitor Alisertib (MLN8237) in Patients with Advanced Solid Tumors. Drugs in R&D. 2016. March;16(1):45–52. doi: 10.1007/s40268-015-0114-8. PubMed PMID: 26689566; PubMed Central PMCID: PMC4767718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Connor OA, Ozcan M, Jacobsen ED, et al. Randomized Phase III Study of Alisertib or Investigator’s Choice (Selected Single Agent) in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma. J Clin Oncol. 2019. March 10;37(8):613–623. doi: 10.1200/JCO.18.00899. PubMed PMID: 30707661; PubMed Central PMCID: PMC6494247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Connor OA, Heaney ML, Schwartz L, et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J Clin Oncol. 2006. January 1;24(1):166–73. doi: 10.1200/JCO.2005.01.9679. PubMed PMID: 16330674. [DOI] [PubMed] [Google Scholar]
- 52.Strati P, Nastoupil LJ, Davis RE, et al. A phase 1 trial of alisertib and romidepsin for relapsed/refractory aggressive B-cell and T-cell lymphomas. Haematologica. 2019. May 9. doi: 10.3324/haematol.2019.220012. PubMed PMID: 31073068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.