

Abstract
Fibrous dysplasia of bone (FD) is a mosaic disease caused by mutations in GNAS. Constitutive activation of the α-subunit of the Gs stimulatory protein (Gαs) leads to dysregulated proliferation of bone marrow stromal cells (BMSCs), generating expansile lesions of fibrotic tissue and abnormal bone. Local bone remodeling regulation by BMSCs is also altered, and FD tissue is characterized by abundant osteoclast-like cells that may be essential for lesion expansion. Animal models show local expression of RANKL in bone lesions, and treatment with the RANKL neutralizing antibody denosumab decreased lesion expansion rate in a patient with aggressive FD. However, the role of RANKL/osteoprotegerin (OPG) in FD pathophysiology is not yet understood. We measured serum levels of RANKL, OPG, and inactive RANKL-OPG complexes in FD patients of known disease burden and in healthy volunteers (HVs). RANK, RANKL, and Ki67 immunohistochemistry were assessed in FD tissue. Cultured FD and HV BMSCs were stimulated with prostaglandin E2 (PGE2) and 1,25 vitamin D3 to increase RANKL expression, and media levels of RANKL and OPG were measured. Osteoclastogenic induction by FD or HV BMSCs was assessed in co-cultures with HV peripheral monocytes. FD patients showed a 16-fold increase in serum RANKL compared to HVs. OPG was moderately increased (24%), although RANKL/OPG ratio was 12-fold higher in FD patients than in HVs. These measurements were positively correlated with the skeletal burden score (SBS), a validated marker of overall FD burden. No differences in serum inactive RANKL-OPG complexes were observed. In FD tissue, RANKL+ and Ki67+ fibroblastic cells were observed near RANK+ osteoclasts. High levels of RANKL were released by FD BMSCs cultures, but were undetectable in HV cultures. FD BMSC released less OPG than HV BMSCs. FD, but not HV BMSCs, induced osteoclastogenesis in monocyte co-cultures, which was prevented by denosumab addition. These data are consistent with the role of RANKL as a driver in FD-induced osteoclastogenesis.
Keywords: RANKL, RANK, OPG, MCCUNE ALBRIGHT SYNDROME, FIBROUS DYSPLASIA
Introduction
Fibrous dysplasia of bone (FD; OMIM #174800) is a mosaic skeletal disease affecting one or multiple sites in the skeleton, which may be associated with hyperfunctioning endocrinopathies as part of the McCune-Albright Syndrome (MAS), and with skin hyperpigmentation. FD/MAS is caused by gain-of-function post-zygotic point mutations in GNAS, which encodes the α-subunit of the Gs stimulatory protein (Gαs). Gαs is often mutated at the R201, GTPase regulatory residue, causing constitutive signaling through adenylyl cyclase and the cAMP-PKA pathway.(1) These mutations occur during embryogenesis, resulting in altered postnatal skeletal stem cells (SSCs) that generate expansile skeletal lesions during early childhood. In FD lesions, SSC-derived bone marrow stromal cells (BMSCs) acquire a fibroblastic phenotype and proliferate, replacing bone marrow resident cells.(2) Histologically, FD lesions are characterized by the presence of fibrotic tissue that produces undermineralized woven bone and typically includes abundant osteoclasts, a consequence of altered regulation of osteoclastogenesis by FD BMSCs.(3) This dysregulation of local bone resorption may play a key role in FD expansion, and is reflected in marked elevations in serum bone turnover markers.(4) In vitro, human BMSCs transduced with mutant Gαs have shown increased release of RANKL(5) and other osteoclastogenic cytokines,(6) and mouse models of FD exhibit high osteoclastic activity and increased expression of RANKL.(7,8)
Treatment of FD with bisphosphonates seems to decrease bone turnover markers and may relieve FD-associated pain.(9–11) Similar findings were reported by directly targeting RANKL with denosumab,(12) and we reported for the first time that it may also reduce lesion expansion.(13) However, the specific role of these therapies in FD management has not been determined, and a better understanding of the role of RANKL/osteoprotegerin (OPG) regulation in FD is necessary to improve the clinical management of this disorder.
In this study, using the well-characterized NIH cohort of FD patients, we aimed to further understand the role of RANKL/OPG regulation in this disease. We show for the first time in patients with FD that blood levels of RANKL are markedly elevated, RANKL levels are significantly positively correlated with FD burden, and there is overexpression of RANKL by patient-derived BMSCs.
Patients and Methods
Subjects
Thirty-six patients with FD were evaluated at the NIH Clinical Center as part of a longstanding natural history protocol (NIH 98-D-0145, in clinicaltrials.gov). The study was approved by the NIDCR Institutional Review Board and all subjects or their parents gave informed consent/assent. Only patients who had not received bisphosphonates or denosumab were studied. FD burden was calculated using a validated method (skeletal burden score [SBS]).(4) Serum samples from 24 healthy volunteers (HVs) were purchased (Valley Biomedical, Winchester, VA, USA). Tissue specimens for histology, immunohistochemistry and BMSC culture were obtained from subjects undergoing biopsy or corrective surgery. BMSCs and monocytes from HVs were obtained according to NIH ethical guidelines (NIH OHSRP exemption #373).
RANKL, OPG, and RANKL-OPG complex determinations
MILLIPLEX MAP Human RANKL kits (EMD Millipore Corporation, Burlington, MA, USA; HBN51K1RANKL) and RayBio® Human Osteoprotegerin ELISA kits (RayBiotech Inc, Norcross, GA, USA; ELH-OPG-001), were used following the manufacturers’ recommendations. RANKL-OPG inactive complexes were measured by a previously described method.(14)
Immunohistochemistry
Tissues from five FD lesions were fixed in 10% formalin, decalcified with 0.25M EDTA (pH 7.3), embedded in paraffin, and sectioned. H&E staining and RANKL (mouse anti-human M366, 1:600, donated by Amgen, Inc., Thousand Oaks, CA, USA), RANK (mouse anti-human N1H8.1, 1:600, donated by Amgen) and Ki67 (M7240, 1:400; Dako, Glostrup, Denmark) immunohistochemistry were performed with heat-induced sodium-citrate antigen retrieval, 2.5% bovine serum albumin (BSA)-PBS blocking buffer, and overnight incubation of primary antibodies at 4°C. The VECTASTAIN ABC kit (Vector Laboratories, Burlingame, CA, USA; PK-4000) and Mayer’s hematoxylin were used.
Cell culture and assays
Primary BMSC cultures were established from four FD biopsy samples and two HV bone marrow aspirates following standard methods(15) using α-MEM (Life Technologies, Grand Island, NY, USA; 12571), 20% lot-selected, non–heat-inactivated fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA, USA; S11150) and 1% penicillin-streptomycin. Cultures were used at passage 3 or lower. Media was replaced twice a week.
For OPG and RANKL measurements, confluent cultures in 60-mm dishes were depleted of FBS (5 hours) and then treated with 1 μM prostaglandin E2 (PGE2; Sigma-Aldrich, St. Louis, MO, USA; P6532) and 10nM 1,25 vitamin D3 (Sigma-Aldrich; D1530) for 48 hours to stimulate RANKL production.(16–18) Conditioned media were concentrated using Amicon Ultra-4 Ultracel-3 centrifugal filter units (EMD Millipore Corporation, Billerica, MA, USA; UFC800324).
For osteoclastogenic co-culture assays, BMSCs were plated on 24-well plates until near confluence in osteogenic media: α-MEM with 100μM ascorbic acid (Sigma-Aldrich; A4544), 10−8M dexamethasone (Sigma), 1% penicillin-streptomycin, and 5% FBS. A total of 500,000 HV monocytes were added per well after isolation by elutriation. Concurrently, 25 ng/mL of recombinant macrophage-colony stimulating factor (rhM-CSF; eBioscience, Santa Clara, CA, USA; 34-8789-82) was added for monocyte stimulation, and PGE2 (1 μM) and 1,25 vitamin D3(10μM) were added to stimulate RANKL expression by BMSCs. In some cultures, denosumab was added at the same time (10 μg/mL). Fourteen days later, tartrate-resistant acid phosphatase (TRAP) staining was performed (Sigma-Aldrich; 387-A).
For proliferation assays, 12-well plates were plated with 8000 BMSCs in 5% FBS α-MEM. Either denosumab (10 μg/mL) or IgG2 (10 μg/mL) was added. Cells were counted with a hemocytometer.
Statistics
Using GraphPad Prism 7.03 (GraphPad Software, Inc., La Jolla, CA, USA), all comparisons were done using the Mann-Whitney test after evaluating normality with the D’Agostino-Pearson test. The Spearman correlation was calculated for serum determinations and SBS. For these analyses, SBS in HVs was considered 0. Linear regression of serum determinations and SBS are plotted in Fig. 1.
Fig. 1.
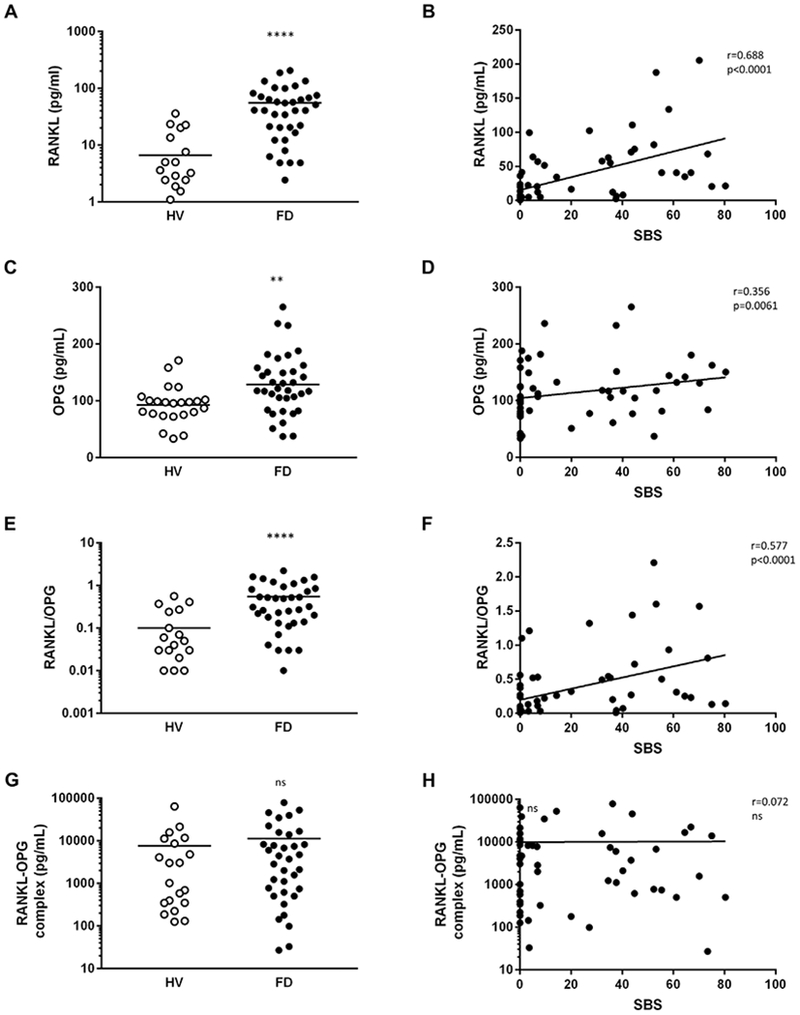
Serum RANKL and OPG, but not inactive RANKL-OPG complex, are altered in FD. Levels of serum RANKL (A), OPG (C), RANKL/OPG ratio (E), and RANKL-OPG complex (G) in FD patients compared to HVs. Spearman correlation and linear regression of RANKL (B), OPG (D), RANKL/OPG ratio (F), and RANKL-OPG complex (H) with FD burden as measured by SBS. **p < 0.01, ****p < 0.0001. FD = fibrous dysplasia; HV = healthy volunteer; SBS = skeletal burden score.
Results
Serum RANKL, OPG, and their ratio are increased in FD patients
FD patients showed a 16-fold increased serum RANKL compared to HVs, which was significantly associated with disease burden (Fig. 1A, B). OPG levels were moderately increased (24%) and also correlated with SBS (Fig. 1C, D). As a result, FD patients exhibited a 12-fold increase in serum RANKL/OPG ratios, which was significantly correlated with SBS (Fig. 1E, F). Serum RANKL-OPG complexes were not different between patients and HVs; indicative of the abundance of inactive RANKL (Fig. 1G, H).
FD BMSCs produce RANKL and promote osteoclastogenesis
In FD lesions, fibroblasts are frequently associated with numerous osteoclasts (Fig. 2A). These regions are often highly proliferative, as evidenced by Ki67 staining (Fig. 2B) of cells that also express RANKL (Fig. 2C) in association with RANK+ osteoclasts (Fig. 2D).
Fig. 2.
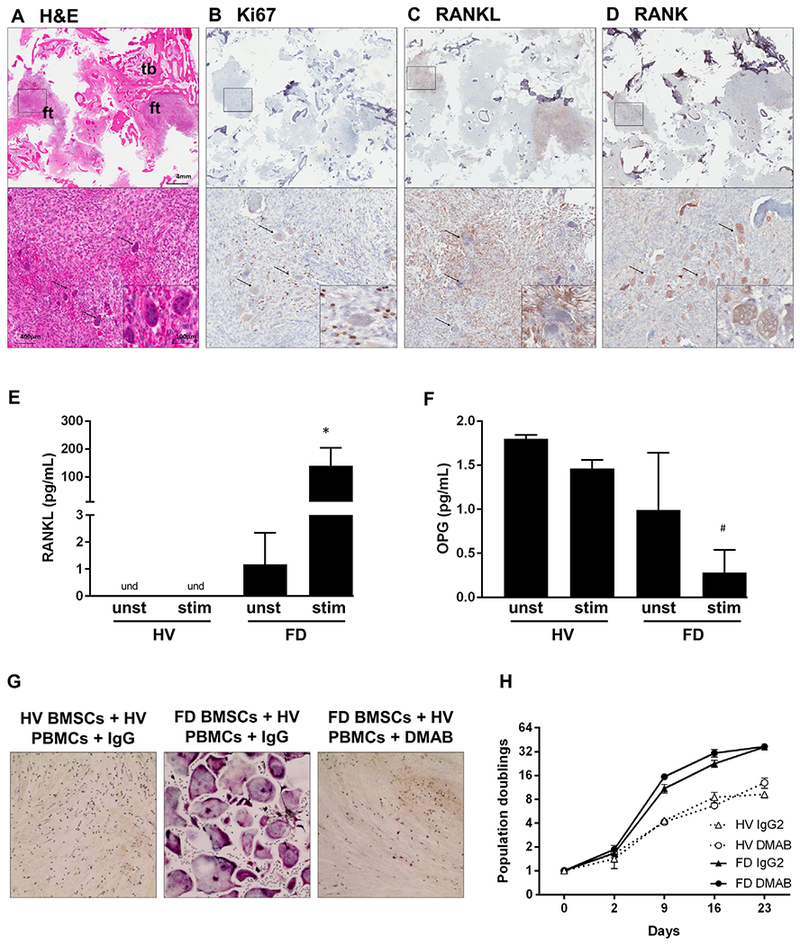
FD BMSCs promote osteoclastogenesis through RANKL overexpression. (A–D) Serial sections of a representative FD specimen showing closely localized Ki67+ (B) and RANKL+ (C) cells with RANK+ (D) osteoclasts; black arrows = osteoclasts. (E, F) RANKL (E) and OPG (F) release by unstimulated (unst) and 1,25 vitamin D3-stimulated + PGE2-stimulated (stim) BMSCs derived from HVs or FD patients. (G) TRAP staining of HV PBMCs co-cultured with HV or FD BMSCs in the presence of DMAB or isotype control antibody (IgG) for 2 weeks. (H) HV and FD BMSCs population doublings with DMAB or IgG2. *p < 0.05 versus unst FD BMSCs. BMSC = bone marrow stromal cell; DMAB = denosumab; FD = fibrous dysplasia; ft = fibrous tissue; HV = healthy volunteer; PBMC = peripheral blood mononuclear cell; stim = stimulated; tb = trabecular bone; unst = unstimulated.
Cultured FD BMSCs released RANKL into the media (on average 1.17 pg/mL, Fig. 2E), which was dramatically increased 48 hours after stimulation with PGE2 and 1,25 vitamin D3 (139.9 pg/mL, Fig. 2E).(16–18) Although HV BMSCs released low levels of RANKL after 7 days of stimulation (not shown), they failed to release detectable levels of RANKL at this time point, and produced 1.8 pg/mL of OPG and 1.46 pg/mL when stimulated (Fig. 2F). FD BMSCs also expressed OPG (1 pg/mL, Fig. 2F), but only residual levels were detected in stimulated cultures (0.28 pg/mL, Fig 2F). OPG and RANKL levels in cell lysates showed similar trends (not shown). Consistent with the above findings, only FD BMSC supported the formation of TRAP+ osteoclasts when co-cultured with HV monocytes (Fig. 2G). Denosumab co-treatment neutralized these effects but had no effect on FD BMSC proliferation compared to the IgG isotype control (Fig. 2G, H).
Discussion
The development of effective therapies for FD has been limited by large gaps in knowledge of its pathogenesis. High level of osteoclastogenesis in FD tissue suggest that bone resorption may play a key role in FD progression. Antiresorptive treatment with bisphosphonates has been associated with improvements in FD-associated pain(10) and temporary reduction in bone turnover markers.(11) However, no effects in lesion expansion have been demonstrated with this approach.(19) Direct inhibition of osteoclastogenesis with denosumab has shown promising results in the treatment of FD bone pain, turnover markers,(12,13) and even lesion expansion,(13) in line to reports on the histologically similar giant-cell tumors of bone.(20)
However, RANKL/OPG regulation in FD remains poorly understood. Mouse models of FD describe RANKL overexpression in FD-like tissue, and increased serum RANKL.(7,8) Stable transfection of Ga R201C in human BMSCs leads to overexpression of RANKL.(5) Interestingly, the expression of OPG in these FD models was not different from wild-type,(5,7) or even repressed.(8)
As expected in our study, stimulated FD BMSCs released large amounts of RANKL, which was undetectable in HV BMSCs cultured under similar conditions. These cells also released OPG, but to a lesser degree than HVs, particularly upon stimulation. Stimulated FD BMSCs in osteogenic media induced the differentiation of co-cultured monocytes into TRAP+ osteoclasts, an action prevented by the addition of denosumab, supporting RANKL as the key factor by which FD BMSCs promote local bone resorption that may allow lesion expansion. Stimulated FD BMSCs in non-osteogenic media failed to induce osteoclastogenesis and maintain RANKL expression after 5 days of culture (data not shown), suggesting that although FD BMSCs differentiate abnormally(5,7) they may require some differentiation commitment to induce osteoclastogenesis. Further research about the specific differentiation stage in which BMSCs support osteoclast formation in FD is needed.
In our study, serum levels of RANKL and OPG were increased in FD, but RANKL upregulation was greater by an order of magnitude than that of OPG (16-fold versus 1.2-fold). Consequently, serum RANKL/OPG ratio was 12 times higher in FD patients than in HVs. Serum levels of RANKL and the RANKL/OPG ratio were strongly associated with disease burden, supporting the notion that RANKL expression plays a key role in the FD tissue microenvironment. OPG showed a weaker but significant positive association with disease burden, although there was a lower expression of OPG by FD BMSCs in vitro. This may be due to the mosaic nature of FD. Local unaffected skeletal cells may respond to the FD pro-resorptive microenvironment by increasing expression of RANKL-neutralizing OPG. We previously showed a fourfold increase in the serum RANKL of FD mice,(7) but serum OPG measurement has not been reported in FD mouse models. Future determinations in these models is indicated in light of these new data. FD did not increase the levels of inactive RANKL-OPG complexes, suggesting that the increased serum OPG found in FD patients is insufficient to significantly alter the action of RANKL.
In summary, RANKL is robustly expressed by human FD lesions and cultured FD BMSCs. Circulating levels of active RANKL are markedly increased in FD patients and highly correlated with disease burden. Although OPG serum levels are also slightly increased in FD and correlate with disease burden, the serum RANKL/OPG ratio is increased to a much greater degree in FD, supporting the concept that RANKL expression by FD BMSCs plays a central role in the pathogenesis of FD.
Acknowledgments
This work was supported by the Intramural Research Program of the NIH, DHHS (NIDCR).
Footnotes
Public clinical trial registration: http://clinicaltrials.gov/show/NCT00001727. Screening and Natural History of Patients With Polyostotic Fibrous Dysplasia and the McCune-Albright Syndrome.
Disclosures
All authors state that they have no conflicts of interest.
References
- 1.Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med. 1991;325(24): 1688–95. [DOI] [PubMed] [Google Scholar]
- 2.Riminucci M, Saggio I, Robey PG, Bianco P. Fibrous dysplasia as a stem cell disease. J Bone Miner Res. 2006;21 Suppl 2:P125–31. [DOI] [PubMed] [Google Scholar]
- 3.Riminucci M, Liu B, Corsi A, et al. The histopathology of fibrous dysplasia of bone in patients with activating mutations of the Gs alpha gene: site-specific patterns and recurrent histological hall-marks. J Pathol. 1999;187(2):249–58. [DOI] [PubMed] [Google Scholar]
- 4.Collins MT, Kushner H, Reynolds JC, et al. An instrument to measure skeletal burden and predict functional outcome in fibrous dysplasia of bone. J Bone Miner Res. 2005;20(2):219–26. [DOI] [PubMed] [Google Scholar]
- 5.Piersanti S, Remoli C, Saggio I, et al. Transfer, analysis, and reversion of the fibrous dysplasia cellular phenotype in human skeletal progenitors. J Bone Miner Res. 2010;25(5):1103–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamamoto T, Ozono K, Kasayama S, et al. Increased IL-6-production by cells isolated from the fibrous bone dysplasia tissues in patients with McCune-Albright syndrome. J Clin Invest. 1996;98(1):30–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao X, Deng P, Iglesias-Bartolome R, et al. Expression of an active Galphas mutant in skeletal stem cells is sufficient and necessary for fibrous dysplasia initiation and maintenance. Proc Natl Acad Sci U S A. 2018;115(3):E428–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khan SK, Yadav PS, Elliott G, Hu DZ, Xu R, Yang Y. Induced Gnas(R201H) expression from the endogenous Gnas locus causes fibrous dysplasia by up-regulating Wnt/beta-catenin signaling. Proc Natl Acad Sci U S A. 2018;115(3):E418–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyce AM, Kelly MH, Brillante BA, et al. A randomized, double blind, placebo-controlled trial of alendronate treatment for fibrous dysplasia of bone. J Clin Endocrinol Metab. 2014;99(11):4133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelly MH, Brillante B, Collins MT. Pain in fibrous dysplasia of bone: age-related changes and the anatomical distribution of skeletal lesions. Osteoporos Int. 2008;19(1):57–63. [DOI] [PubMed] [Google Scholar]
- 11.Majoor BC, Appelman-Dijkstra NM, Fiocco M, van de Sande MA, Dijkstra PS, Hamdy NA. Outcome of long-term bisphosphonate therapy in McCune-Albright syndrome and polyostotic fibrous dysplasia. J Bone Miner Res. 2017;32(2):264–76. [DOI] [PubMed] [Google Scholar]
- 12.Ganda K, Seibel MJ. Rapid biochemical response to denosumab in fibrous dysplasia of bone: report of two cases. Osteoporos Int. 2014;25(2):777–82. [DOI] [PubMed] [Google Scholar]
- 13.Boyce AM, Chong WH, Yao J, et al. Denosumab treatment for fibrous dysplasia. J Bone Miner Res. 2012;27(7):1462–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Widschwendter M, Burnell M, Fraser L, et al. Osteoprotegerin (OPG), the endogenous inhibitor of receptor activator of NF-kappaB ligand (RANKL), is dysregulated in BRCA mutation carriers. EBioMedicine. 2015;2(10):1331–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robey PG, Kuznetsov SA, Ren J, Klein HG, Sabatino M, Stroncek DF. Generation of clinical grade human bone marrow stromal cells for use in bone regeneration. Bone. 2015;70:87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cong L, Zhang C, Tu G. Osteoblastic NF-kappaB pathway is involved in 1alpha, 25(OH)2D3-induced osteoclast-like cells formation in vitro. Int J Clin Exp Pathol. 2015;8(5):5988–96. [PMC free article] [PubMed] [Google Scholar]
- 17.Park HJ, Baek K, Baek JH, Kim HR. TNFalpha increases RANKL expression via PGE(2)-induced activation of NFATc1. Int J Mol Sci. 2017;18(3):495 DOI: 10.3390/ijms18030495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh PP, van der Kraan AG, Xu J, Gillespie MT, Quinn JM. Membrane-bound receptor activator of NFkappaB ligand (RANKL) activity displayed by osteoblasts is differentially regulated by osteolytic factors. Biochem Biophys Res Commun. 2012;422(1):48–53. [DOI] [PubMed] [Google Scholar]
- 19.Hart ES, Kelly MH, Brillante B, et al. Onset, progression, and plateau of skeletal lesions in fibrous dysplasia and the relationship to functional outcome. J Bone Miner Res. 2007;22(9):1468–74. [DOI] [PubMed] [Google Scholar]
- 20.Thomas D, Henshaw R, Skubitz K, et al. Denosumab in patients with giant-cell tumour of bone: an open-label, phase 2 study. Lancet Oncol. 2010;11(3):275–43. [DOI] [PubMed] [Google Scholar]