

Abstract
VEK50 is a truncated peptide from a Streptococcal pyogenes surface human plasminogen (hPg) binding M-protein (PAM). VEK50 contains the full A-domain of PAM, which is responsible for its low nanomolar binding to hPg. The interaction of VEK50 with kringle 2, the PAM-binding domain in hPg (K2hPg), has been studied by high-resolution NMR spectroscopy. The data show that each VEK50 monomer in solution contains two tight binding sites for K2hPg, one each in the a1- (RH1; R17H18) and a2- (RH2; R30H31) repeats within the A-domain of VEK50. Two mutant forms of VEK50, viz., VEK50[RH1/AA] (VEK50△RH1) and VEK50[RH2/AA] (VEK50△RH2), were designed by replacing each RH with AA, thus eliminating one of the K2hPg binding sites within VEK50, and allowing separate study of each binding site. Using 13C- and 15N-labeled peptides, NMR-derived solution structures of VEK50 in its complex with K2hPg were solved. We conclude that the A-domain of PAM can accommodate two molecules of K2hPg docked within a short distance of each other, and the strength of the binding is slightly different for each site. The solution structure of the VEK50/K2hPg, complex, which is a reductionist model of the PAM/hPg complex, provides insights for the binding mechanism of PAM to a host protein, a process that is critical to S. pyogenes virulence.
Keywords: Plasminogen binding, 3-D solution structure, bacterial receptor, modular proteins, a-repeats, peptide mutagenesis, NMR structures
Graphical Abstract
1. Introduction
Approximately 250 serotypically distinct strains of Group A Streptococcus pyogenes (GAS) have been identified that present patients with mild to severe symptoms. The different GAS strains are serotyped through the nature of their characteristic surface M- and M-like proteins, with one skin-trophic subgroup (Pattern D) containing a direct human host plasminogen (hPg)/plasmin (hPm) surface M-protein receptor (PAM) (Berge and Sjobring, 1993). Binding of hPg to PAM-type M-proteins greatly facilitates its activation to the protease, hPm, by GAS- secreted streptokinase (SK), a step that is essential for its virulence (Sun et al., 2004). After activation, hPm remains bound to GAS, resulting in exacerbation of GAS pathogenicity through proteolytic disruption of host innate immune barriers to dissemination (Sanderson-Smith et al., 2008), e.g., the extracellular matrix and tight cellular junctions (Lahteenmaki et al., 2000; Sumitomo et al., 2013; Sumitomo et al., 2016). The N-terminal A-domain of various PAM-type M-proteins usually consist of one or two peptide modules, viz., a1- and a2- repeats, having 13–17 residues each (Qiu et al., 2018). Both repeats have been shown to be critical determinants for hPg binding. (Ringdahl et al., 1998; Rios-Steiner et al., 2001; Sanderson-Smith et al., 2007; Schenone et al., 2000; Wang et al., 2010b; Yuan et al., 2017).
Contained within the hPg protein, the lysine binding site (LBS) of its small functionally-independent kringle 2 (K2hPg) domain has been identified as the major receptor site for PAM binding (Wistedt et al., 1998). The LBS of four of the five kringle domains of hPg generally interact with activation effectors (Castellino and Ploplis, 2003), as well as with cellular receptors (Pancholi and Fischetti, 1998), via C-terminal lysine residues of the receptor (Miles et al., 1991). However, the prototypical Pattern D GAS receptor for hPg, e.g., PAM from isolate AP53 (PAMAP53), does not contain a C-terminal lysine residue and interacts with hPg via an internal pseudo-lysine arrangement of amino acid side-chains (Rios-Steiner et al., 2001; Wang et al., 2010b; Yuan et al., 2017). In this regard, adjacent Arg and His residues in both the a1- and a2-repeats of the PAM A-domain have been shown to be essential for hPg binding (Rios-Steiner et al., 2001; Sanderson-Smith et al., 2007; Schenone et al., 2000).
Since the binding domains of PAM and hPg were originally defined (Wistedt et al., 1995; Wistedt et al., 1998), a small peptide, VEK30 from PAMAP53, that contains only its a1-repeat, as well as VKK38 from PAMNS455, that is composed of both the a1- and a2-repeats, along with K2hPg have been employed as minimalistic structural models to investigate the essential aspects of the very specific and tight binding of PAM proteins to hPg. The side-chains of residues R17H18 and E19E20 of VEK30 serve as positive and negative clusters around one turn of an α-helix. The residues are spaced similarly to the α-COOH and ε-NH2 of a free lysine and fit into the LBS of K2hPg. This has been interpreted as an a1-located lysine side-chain isostere (RH1) in PAMAP53 (Rios-Steiner et al., 2001; Wang et al., 2010b). Similarly, the side-chains of the more extended VKK38, containing residues R30H31 and D32H33D34, constitute a second a2-positioned lysine isostere (RH2) in PAMNS455 (Yuan et al., 2017). We showed previously that VKK38 can accommodate two K2hPg peptides on the VKK38 α-helix, likely on opposite faces of this helix (Yuan et al., 2017).
We aimed to elucidate the roles of each of the two hPg/hPm binding sites in PAM in order to assess their equivalency in the structure and function of PAM, especially since other Pattern D GAS strains are highly substituted in this region. Additionally, some Pattern D GAS isolates only contain one hPg/hPm binding site in the PAM A-domain (the a2-repeat), e.g., PAMNS88.2. These PAMs display different hPg acquisition properties than those with two hPg/hPm binding sites (Qiu et al., 2018). Thus, an understanding of the mechanism of binding of hPg to PAM has become an important factor in the study of GAS virulence. In an associated recently submitted paper (Quek et al, submitted for publication), a larger peptide, VEK75, sequentially containing a C-terminal portion of the hypervariable region (HVR)-the full A-domain-and a partial N-terminal portion of the B-domain of PAMAP53 (Bhattacharya et al., 2014), in complex with K2hPg, has been crystallized and the structure solved, showing that the two binding sites in this PAMAP53 peptide can simultaneously bind to K2hPg. In order to resolve potential generic issues surrounding the influence of crystal packing forces in determining the final structure, and/or rearrangement of the conformations for optimal crystal formation, and to obtain a more direct observation of the interchain binding interactions in solution, we have additionally determined the solution structure of a VEK50/K2hPg complex, using a functionally similar, but slightly shorter (for ease in assigning NMR chemical shifts), fragment of PAMAP53 containing its full A-domain. VEK50 is extended in sequence from VEK30 and VKK38 to more rigorously assess possible exosites in binding. The high equivalency of the studies with a variety of peptides from the A domain of PAM clarifies the binding modality of hPg to PAMAP53.
2. Materials and Methods
2.1. Construction of expression plasmids
The construction of plasmids for peptide and protein expression was accomplished as previously described (Wang et al., 2010b). VEK50, cloned from PAMAP53, and VEK50 mutant peptides, generated by site-directed mismatch mutagenesis with synthetic oligonucleotides, were expressed in Escherichia coli BL21 (DE3) cells employing the His6-tagged-GB1 domain fusion expression system (Wang et al., 2010b). The final constructs contained sequentially from the 5’: [ATG initiation codon - purification His6 tag - GB1 domain for enhanced solubility - 9-residue linker - thrombin cleavage site, LVPR/GS, followed by the VEK peptide]. This cassette was inserted into pET-15b (Novagen) (Wang et al., 2010b). Thus, all peptides cleaved with thrombin possessed an exogenous GS dipeptide at the N-terminus. In addition, a codon for a Tyr was intentionally placed at the C-termini of the VEK peptides for 280 nm absorption properties (Bhattacharya et al., 2014). All plasmid inserts were subjected to full nucleotide sequencing and only those identical to the expected sequences were employed for expression.
2.2. Expression and purification of VEK peptides
The expression of VEK50 peptides was carried out in E. coli BL21 using previously published protocols (Wang et al., 2010b). After induction with 0.8 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG), proteins extracted from the cell pellet were loaded onto a Ni2+-Sepharose affinity chromatography column (HisTrap HP; GE Healthcare) at 4° C, washed, and eluted with 60 mM imidazole, pH 8.0. The concentrated eluates were further purified after cleavage with 1000 U of thrombin (ERL). The resulting cleaved fragments were separated using a HisTrap HP affinity column (GE Healthcare). At this step, the flow-through fraction, containing VEK50 peptides, was applied to p-aminobenzamidine agarose (Sigma) to remove thrombin. To prepare 15N- and 15N/13C-VEK50 samples for NMR experiments, expression and purification were similarly accomplished as described previously (Wang et al., 2010a; Wang et al., 2010b). For uniform labeling, 15NH4Cl (99%, Cambridge Isotope Laboratories) and/or 13C-glucose (99%, Isotec) were used as the sole nitrogen and carbon sources, respectively.
2.3. Expression and purification of K2hPg
A triple variant of human K2hPg (C4G/E56 D/L72Y) that displays ~5-fold enhanced affinity. without loss of selectivity for PAM, as well as the inability to aggregate through disulfide bonding, compared to WT-K2hPg, was expressed in Pichia pastoris GS115 cells as described in detail previously (Nilsen et al., 1997; Wang et al., 2010a; Wang et al., 2010b). The tighter binding of this variant allowed separation of K2hPg from unbound components during purification. For uniform 15N- and 15N/13C-K2hPg, the medium employed was (15 NH4)2SO4 (99%, Cambridge Isotope Laboratories)/13C-glucose (99%; Isotec)/13C-methanol (99%, Isotec). The K2hPg contained seven exogenous residues at the N-terminus of K2hPg (enumerated as −7 to −1) as a necessary result of the construction of the expression plasmid (Nilsen et al., 1997). The final construct is as follows:
The Y−7VEF is from the polylinker site of the plasmid and SEE represents the linker between K1hPg and K2hPg in hPg numbering begins at C+ and concludes at C 78
The integrity of all labeled and unlabeled proteins and peptides was determined by MALDI-TOF mass spectrometry on an Autoflex III spectrometer (Bruker Daltonics). For all uniformly-labeled 15N- and 13C/15N-peptides, single mass peaks were obtained of the correct molecular weights, indicating complete incorporation of the heavy isotopes.
2.4. Extinction coefficients of peptides and proteins
Precise concentrations of the peptides and proteins are required for accurate titrations of the VEK-peptides and K2hPg. For this, we directly determined extinction coefficients of all samples at 280 nm. Since the VEK peptides are naturally devoid of aromatic residues, a C-terminal Tyr residue was added at the COOH-termini of these peptides to provide an observable A280nm. Next, we determined the A280nm combined with the refractive indices (RI) on the same samples. The procedure made use of a combination of size exclusion chromatography (SEC), to separate any impurities in the sample, with the column flow-through passed in-line through a 2013variable wavelength detector (VWD), set to monitor UV absorbance at 280 nm, a light scattering detector to ensure that the molar mass of the eluted peak matched that of the peptide of interest, followed by a refractive index detector (RID). The equipment used was an Agilent 1260 infinity II HPLC system with a Wyatt WTC-030S5 (7.8 × 300 mm, 5 μm, 300Å) SEC column coupled to Wyatt Treos II multi-angle (three angles) light scattering (MALS) detector, an Agilent VWD, and a Wyatt Optilab T-Rex RID. The flow path, together with the SEC column, was equilibrated with PBS, pH 7.4. The sample (100 μl) in the same buffer was added to the column via an autosampler and passed through the SEC column. Approximately 95% of all samples applied reached the detectors. Since the refractive index increments of the peptides are constant at 0.185 ml/g, the A280nm and RI values allow the extinction coefficient of the sample to be readily calculated (Wen et al., 1997).
The experimental extinction coefficients (ml mg−1 cm−1) determined from this approach (compared to those calculated from ExPASY (Gasteiger et al., 2003) are: K2hPg = 2.38 (2.40); VEK50 = 0.20 (0.24); VEK50△RH1 = 0.21 (0.25); VEK50△RH2 = 0.21 (0.25); hPg (for a positive control) = 1.75 (1.72). In all cases, the experimental extinction coefficients were used for concentration determinations.
2.5. Isothermal titration calorimetry (ITC)
ITC measurements were performed at 25° C on a VP-ITC 200 Microcal calorimeter (Malvern) in 50 mM sodium phosphate, pH 7.4. For VEK50/K2hPg binding, VEK50 peptides (40 μM) was stirred in a cell at ~800 rpm and K2hPg (900 μM) was injected at a rate of 4 μl/s for WT-VEK50 and 2 μl/s for VEK50 mutants with 120 sec equilibration time. Control experiments were performed by titrating K2hPg to buffer instead of VEK peptides. The integrated heats of interaction were normalized to the molar ratio of VEK50 peptide to K2hPg, and the data were fit with Origin-ITC 7.0 software.
2.6. Analytical ultracentrifugation (AUC)
The molecular weights of individual proteins were determined by AUC equilibrium analysis with a Beckman Optima XLI ultracentrifuge. Purified K2hPg (10 μM) and VEK50 peptides (150 μM), at 0.1 – 0.3 μg/ml, were analyzed in 20 mM Tris-Cl/0.1M NaCl, pH 7.4, at 25° C. A280nm data from rotor speeds of 32,000 and 40,000 rpm were recorded every hr until the scans were identical, thus indicating that equilibrium was attained. The partial specific volumes of the VEK50 peptides and K2hPg were calculated from their amino acid sequences using Sednterp (Laue et al., 1992). AUC data were analyzed by the Optima XL-A/XL-I data analysis software (Beckman Coulter), and apparent molecular weights were obtained throughout the concentration gradient generated in the cell (Bhattacharya et al., 2014).
For complexes of K2hPg and each VEK50, the peptides were mixed in a 3:1 molar ratio of K2hPg:VEK50 and loaded onto a S75 gel filtration column (GE Healthcare) equilibrated with 20 mM Tris-Cl/0.1 M NaCl, pH 7.4. The column was operated with an AKTA FPLC system at a flow rate of 0.5 ml/min and elution was monitored by A280nm. The complex was resolved from free K2hPg and the first peak to elute was concentrated by membrane filtration (3K-MWCO), and analyzed on the AUC as described above.
2.7. NMR
2.7.a. Data collection
Protein concentrations used for NMR samples of the apo-VEK50 peptides, apo-K2hPg, and the VEK50/K2hPg complexes ranged from 0.2 –1.0 mM. Purified samples of the VEK50s and K2hPg were dialyzed against NMR sample buffer (20 mM Bis-Tris-d19/2 μM DSS/5% 2H2O/95% H2O, pH 6.7) prior to NMR measurements. In order to solve the bound-form structures of the VEK50 peptides with K2hPg, 15N/13C-VEK50, 15N/13C-VEK50[RH1/AA] (VEK50△RH1), and 15N/13C-VEK50[RH2/AA] (VEK50△RH2) samples were mixed with unlabeled K2hPg at molar ratios of 1:2, 1:1, and 1:1, respectively.
Data were acquired at 298 K on a Bruker AVANCE 800 spectrometer equipped with a 5-mm triple resonance (TCI, 1H, 13C, 15N) cryoprobe. Standard triple resonance NMR experiments were used to assign 15N/13C-labeled VEK50 peptide samples. The following spectra were collected: 15N-HSQC (Kay et al., 1992; Mori et al., 1995), 15N-TOCSY-HSQC (Marion, 1989 #13328} (80 ms mixing time), HNCO/HN(CA)CO (Clubb et al., 1992; Kay et al., 1990), HNCA (Grzesiek and Bax, 1992), HNCACB/CBCA(CO)NH (Grzesiek et al., 1993a; Wittekind and Mueller, 1993), C(CO)NH (Grzesiek et al., 1993b) for the backbone and aliphatic side chain resonance assignments, as well as 15N-NOESY-HSQC (Talluri and Wagner, 1996) and 13C-NOESY-HSQC (Ikura et al., 1990), to collect intramolecular NOE distance constraints for the structure calculations. NMR data were processed using Bruker TopSpin 3.5 software and analyzed using NMRFAM-Sparky (Lee et al., 2009). 1H chemical shifts were referenced to internal DSS, while 13C and 15N chemical shifts were referenced indirectly to DSS (Markley et al., 1998).
For docking studies, intermolecular distance constraints were determined by 15N/13C-half-filtered/edited 3D NOESY-1H-15N/13C-HSQC experiments (120 ms mixing time), performed on samples of 15N/13C-VEK50 peptides complexed to unlabeled K2hPg , and 15N/13 hPg C- K2hPg complexed with unlabeled VEK50 peptides. 2D NOESY experiments with 15N- and 13C-filtered in both dimension (120 ms mixing time) were acquired on the same samples under identical conditions as controls.
2.7.b. Structure calculations of VEK50 peptides
The structures of VEK50 peptides in their apo- and bound-forms were first predicted by the CS-ROSETTA program (Shen et al., 2008) based on their residue-specific chemical shifts obtained from the NMR experiments listed above. Next, the structure from the converged results was used as the template, and further refined by Xplor-NIH 2.36 program (Schweiters et al., 2018). Backbone torsion angle (ϕ and ψ) restraints predicted from TALOS-N distance restraints from NOESY spectra, and RDCs were applied in a simulated annealing protocol in the Xplor-NIH program. For each VEK50 peptide, 200 structures were calculated, from which 20 structures with the lowest energy restraint values were further refined with implicit water (Chen et al., 2004). The quality of the structures was analyzed with PROCHECK 3.5.4 (Laskowski et al., 1993) and MolProbity (Chen et al., 2010). Visualization of the structures was performed using the PyMOL program.
2,7,c. Chemical shift perturbations (CSP)
Titrations for binding studies were carried out by recording 1H/15N-HSQC experiments on 15N-VEK50 peptides (0.2 mM) with increasing molar ratios of unlabeled K2hPg. The amide nitrogen and hydrogen CSPs were mapped for each amino acid according to , where ΔδHN and ΔδN represent the chemical-shift changes of 1H and 15N atoms between the apo- and bound-forms, respectively (Williamson, 2013). CSP values for K2hPg were collected from reversed-labeled samples of the complexes, i.e., 15N- K2hPg titrated with increasing molar ratios of unlabeled VEK50 peptides.
2.7.d. RDC measurements
Residual dipolar coupling (RDC) measurements were performed on 15N-VEK50, 15N-VEK50△RH1, and 15N-VEK50△RH2 in their apo-forms and in complexes with unlabeled K2hPg at molar ratios of 1:2, 1:1, and 1:1, respectively. Additionally, 15N-K2hPg, complexed with unlabeled mutant VEK50 peptides, was analyzed at a molar ratio of 1:1. In all cases, the concentration of labeled protein ranged from 0.2 mM to 0.5 mM, and the unlabeled protein was present in sufficient excess to ensure that the labeled protein was entirely in the bound state. 1DNH RDCs were measured using two-dimensional IPAP[15N/1H]HSQC experiments (Ottiger et al., 1998). RDC values for each residue were obtained by taking the difference in the corresponding J-splittings measured in magnetically oriented (~10 mg/ml) Pf1 phage and isotropic in H2O. The magnitudes of the axial component of the tensor, DaNH (−16.5 Hz) and rhombicity, η (0.45), components of the alignment tensor, were determined based on the NMR structure of VEK30/K2hPg (PDB code:2KJ4) using the DC program (https://spin.niddk.nih.gov/bax/). For CSP and RDC data analyses, sequence-specific backbone 15N-1H assignments for K2hPg, published previously (Wang et al., 2010a), were used.
2.7.e. Restraint-based docking of the VEK50/K2hPg complex
Restraint-based docking calculations for the complexes of VEK50 peptides with K2hPg were performed on the HADDOCK web server (van Zundert et al., 2016) and the Xplor-NIH program. The topologies and starting structural coordinate files for input into HADDOCK were generated from the refined NMR structures. For generation of ambiguous interaction restraints, residues R17H18 of VEK50△RH2 and residues R30H31 of VEK50△RH1 that are directly involved in interaction with K2hPg were defined as active-site residues, and residues with significantly different CSP values (△δ > 0.2 ppm) were defined as passive residues. For K2hPg, residues exhibiting CSP values larger than 0.1 ppm were defined as active residues, i.e. the ones at binding site. The passive residues of K2hPg were automatically picked by HADDOCK server program based on its 3D structure. RDC orientation constraints and intermolecular NOEs from VEK50 peptides and K2hPg were used as unambiguous restraints for structure calculation to obtain the complex of VEK50△RH2/K2hPg and VEK50 △RH1/K2hPg at molar ratios of 1:1. Segments of VEK50 peptides and K2hPg that constituted these active and passive residues, plus two sequential residues on either side of the active and passive residues, were kept semiflexible during the docking steps. N- and C-residues of VEK50 peptides (residues 1–8 and 48–50) were kept flexible throughout the docking. The alignment tensor was calculated using the DC program with default parameters for singular value decomposition (SVD) fitting (https://spin.niddk.nih.gov/bax/). Intervector projection angle restraints for VEAN were generated with the python script provided in HADDOCK.
The simulated structure model of the VEK50/K2hPg complex at a molar ratio of 1:2 was obtained from HADDOCK combined with the CSP data from NMR 15N-HSQC titration experiments for VEK50/K2hPg, along with previous results for VEK30/K2hPg (Wang et al., 2010b). The dihedral angle restraints for the bound-form of K2hPg and VEK50 predicted from TALOS-N were also included in HADDOCK calculation. For purposes of docking, intermolecular NOEs and RDCs of K2hPg bound at R17H18 of VEK50△RH2, and K2hPg bound at R30H31 of VEK50△RH1 were used to define the relative positions of the two K2hPg molecules in their complexes with VEK50. The structural models were clustered and the best model with the highest HADDOCK score was selected to present the structure of the complex. The statistical data for the structures derived from HADDOCK are listed in Supporting Information Table S1.
2.8. Data deposition
The coordinates of the calculated structural ensembles for the bound-form of VEK50, as well as the structural ensembles for the complex of K2hPg with VEK50ΔRH1, and K2hPg with VEK50ΔRH2, have been deposited in the Protein Data Bank with accession codes 6OQ9, 6OQJ, and 6OQK respectively. Chemical shift assignments for the VEK50 peptides in complex with K2hPg and the corresponding experimental restraints used in the structure calculation have been deposited in the BioMagResBank with accession numbers 30603, 30605, and 30606, respectively.
3. Results
3.1. Binding stoichiometry of the VEK50 peptides to K2hPg
3.1.a. ITC titrations
VEK50 is a truncated peptide of PAMAP53 that sequentially includes: [the C-terminus of the hypervariable region (HVR) - the entire A-domain, including its component a1a2-repeats and the N-terminus of the flexible B-domain]. This peptide begins at V97 of PAMAP53, which is residue-1 in VEK50 (Table 1). To quantitate binding of VEK50 to K2hPg, ITC measurements were employed. The heats of binding were measured at 25° C as a function of molar ratio of K2hPg to VEK50. As shown in Figure 1A, the binding isotherm is best-fit by a molar stoichiometry of 2:1 for K2hPg:VEK50, with an average Kd value of 9 nM. In order to identify the binding properties of the RH motifs separately, two mutant peptides, VEK50△RH1 and VEK50△RH2, were designed by separately replacing RH1 (R17H18) or RH2 (R30H31) in VEK50 (Table 1) with two Ala residues. This yields peptides, VEK50△RH1 and VEK50△RH2, respectively. Using these peptides in the titrations, a molar binding stoichiometry of 1:1 was observed for K2hPg/VEK50△RH1, with a Kd of 9 ± 2 nM (Figure 1B) and for K2hPg/VEK50△RH2 of 42 ± 4 nM was best-fit to the data (Figure 1C). These results confirmed that the RH motifs are the active-site residues that are directly involved in the binding to K2hPg. The binding affinities of these two mutants also show that K2hPg binds more tightly to RH2 than RH1.
Table 1.
Amino acid sequences of truncated peptides from PAMAP53 and hPg
| Peptides | Amino Acid Sequencesa |
|---|---|
| VEK50 | GS(V1EKLTADAELQRLKNER17H18EEAELERLKSER30H31DHDKKEAERKALEDKLAD)Y |
| VEK50ΔRH1 | GS(V1EKLTADAELQRLKNEA17A18EEAELERLKSER30H31DHDKKEAERKALEDKLAD)Y |
| VEK50ΔRH2 | GS(V1EKLTADAELQRLKNER17H18EEAELERLKSEA30A31DHDKKEAERKALEDKLAD)Y |
| K2hPg | Y-7VEFSEE(C1MHC4SGENYDGKISKTMSGLECQAWDSQSPHAHGYIPSKFPNKNLKKNYCRNPDRE56LRPWCFTTDPNKRWEY72 CDIPRC78)AA |
Residues outside the parentheses in all cases are exogenous to PAM or K2hPg.
Fig. 1. Isothermal titration calorimetric (ITC) analysis of the binding of VEK50 peptides to K2hPg.

Binding isotherms at 25° C for the titration of K2hPg (900 μM stock) into: (A) 40 μM VEK50 in the cell, (B) 40 μM VEK50△RH1 in the cell, and (C) 40 μM VEK50△RH2 in the cell. The heats liberated upon binding were measured as a function of the concentration of K2hPg. The red lines in Panels A-C represent the best-fits of the experimental data (black symbols) to a two-site model (A) and one-site models (B, C).
3.1.b. AUC analyses
The binding stoichiometry between VEK50 peptides and K2hPg was further assessed by determining the molecular masses of the complexes of VEK50/K2hPg using analytical AUC. The complexes of VEK50 peptides with K2hPg were prepared using three different initial concentrations of K2hPg (13 μM, 19 μM, and 25 μM) added to VEK50 (5 μM, 7.5 μM, and 10 μM), each representing a ~2.5-fold molar excess of K2hPg/VEK50 at different total concentrations of the complex. The complexes were separated from the nonbound materials by gel filtration. The theoretical molecular mass of K2hPg is calculated to be 10,151 Da, that of VEK50 is calculated as 6,159, and, for the two mutant VEK50 peptides, the calculated molecular masses are 6,008 Da, all in agreement with the MALDI data listed in Table 2. From the AUC data of Table 2, it is clear that a 2:1 molar complex of K2hPg/VEK50 is found, whereas for the two mutant complexes, viz., VEK50△RH1/K2hPg and VEK50△RH2/K2hPg, the molecular masses of the isolated complexes corresponded to a 1:1 molar stoichiometry (Table 2). In all cases, a single molecular mass species was found throughout the concentration gradient in the centrifugation cell. This single species that exists over a wide concentration range supports the ITC data showing that very tight binding occurs.
Table 2.
Molecular masses of truncated peptides from PAMAP53 and the complexes with K2hPg
| Peptide | Theoretical (Da) | MALDI-TOF (Da)a | AUC (Da)b | |
|---|---|---|---|---|
| Apo-peptides | Peptide + K2hPg | |||
| VEK50 | 6,159 | 6,166 | 6,300 ± 800 | 27,300 ± 850 |
| VEK50ΔRH1 | 6,008 | 6008 | 5,600 ± 500 | 16,150 ± 1,000 |
| VEK50ΔRH2 | 6,008 | 6010 | 5,700 ± 600 | 16,400 ± 1,000 |
| K2hpg | 10,151 | 10,155 | 10,260 ± 800 | above |
Molecular masses of the protein linear sequence from MALDI-TOF.
Weight-average molecular mass of proteins in solution from AUC experiments at 25° C.
3.1.c. HSQC titrations of VEK50 peptides to K2hPg
1H/15N-HSQC experiments were recorded on 15N-VEK50 peptides with increasing unlabeled K2hPg at 1:0.5, 1:1, 1:1.5, 1:2.0, and 1:2.5 molar ratios of VEK50:K2hPg respectively (Figure 2A–D; Supporting information Figure S1). During the titrations the chemical shifts of residues at the N- and C-terminii, viz., V1-K3 and D49-Y50, respectively, change only slightly and only one peak for each residue is observed in NMR spectra. This suggests that these residues experience fast exchange (Figure 2A). However, the amide resonances of most residues in VEK50 peptides shift significantly and start to appear at two locations. As one example of the data, when K2hPg was titrated to 1:0.5 ratio, residue A48 in VEK50 (Figure 2B), -VEK50△RH1 (Figure 2C), and -VEK50△RH2 (Figure 2D) showed two peaks, corresponding to the amide resonances in the apo- and bound-forms. respectively. After a 1:1 molar ratio was reached, all of the amide resonances in VEK50△RH1 and VEK50△RH2 are shifted to the positions of the bound forms, as exemplified by A48 (yellow) in Figure 2C, D. These residues remained at the same locations when additional unlabeled K2hPg was added to the system. Meanwhile, amide resonances of some residues in VEK50 show three peaks indicating the existence of a mixture containing apo-, 1:1, and 1:2 molar complexes of VEK50 to K2hPg. These residues are located in both the a1- and a2-repeats, suggesting that K2hPg similarly binds to each of these two motifs. The amide resonances were continually shifted until a 1:2 molar ratio of VEK50/K2hPg was reached, as exemplified by A48 (colored blue) in Figure 2B. These results further confirm that the VEK50 peptide within PAMAP53 contains two binding sites for K2hPg and that the binding is mediated by RH motifs in each repeat of the a1a2 domain. For each of the two VEK50 mutants, a 1:1 binding stoichiometry is also confirmed.
Fig. 2. NMR spectroscopic analysis of the interactions between VEK50 peptides and K2hPg.

The stoichiometry between VEK50 and K2hPg was confirmed from chemical shift changes of some residues in overlaid 15N-HSQC spectra of: (A, B) 15N-VEK50; (C) 15N-VEK50△RH1; and (D) 15N-VEK50△RH2 by titrating 15N-labeled VEK50 peptides with unlabeled K2hPg at molar ratios of (VEK50 peptide:K2hPg) of 1:0.5 (orange), 1:1 (yellow), 1.1.5 (green), 1:2 (cyan), and 1:2.5 (blue). The sample buffer was 20 mM Bis-Tris-d19, pH 6.8 at 25° C. (D-F) Chemical-shift differences (△δ ppm) between apo- and bound-forms of VEK50 peptides. The △δ ppm values of: (E) WT-VEK50, (F) VEK50△RH1, and (G) VEK50△RH2 are plotted against the residue numbers. (H–J) Plots of 1DNH RDCs as a function of the residue number for 15N-VEK50 (H), 15N-VEK50△RH1 (I), and 15N-VEK50△RH2 (J), each in complex with K2hPg at molar ratios of 1:2, 1:1, and 1:1. The measurements were performed on samples in filamentous phages Pf1 (10 mg/ml) using two-dimensional IPAP[15N/1H]HSQC experiments (Ottiger et al., 1998). Residues with severe peak overlap or poor signal-to-noise ratios were excluded.
On the basis of these titration experiments, the combined CSPs for the three VEK50 peptides are summarized in Figure 2 (E–G). These data show that residues displaying large chemical shift perturbations upon binding to K2hPg are not limited to R17H18 in the a1-repeat and R 30H31 in the a2-repeat, but also occur in other residues in that region, demonstrating the existence of probable exosites identified using this longer peptide from PAMAP53. The results also demonstrate that conformational transitions occur in VEK50 peptides upon binding to K2hPg. For example, the residues in VEK50△RH1 showing most significant CSPs are in the RH2 binding region, i.e., residues L26-K36 (Figure 2E). In the mutant VEK50△RH2 the chemical shift changes were found between residue A8 to E20, which consists of the RH1 binding motif (Figure 2F). Since WT-VEK50 contains both the RH1 and RH2 binding motifs, most of the residues in VEK50, except those in the N-, and C-terminal regions, show significant chemical shift changes at most locations upon binding to K2hPg (Figure 2D).
In addition, 1H-2H exchange experiments were performed for three VEK50 peptides in the apo-forms and in the complex with K2hpg by dissolving lyophilized sample in 2H2O, followed by temporal acquisition of HSQC spectra. For the apo-form peptides and the complex of two mutant VEK50 peptides with K2hPg, amide resonances disappear during the first 20 min period. Meanwhile, amide resonances of some residues in the WT-VEK50/K2hPg complex remain, and those from residues L26, K27, and E29 are still detectable after 20 hr (Figure 3), indicating that these residues become less exposed to solvent upon VEK50 binding with two K2hPg molecules.
Fig. 3. 1H/2H exchange of VEK50.

Overlays of the 1H-15N HSQC spectra of the complex of 15N-VEK50/unlabeled K2hPg obtained in BisTris-d19, pH 6.8 (black), at 20 min (red) and 24 hr (blue) after the lyophilized complex powder was re-dissolved in 2H2O at 25° C. The assignments of the backbone amide signals are indicated by the respective single-letter codes and residue numbers.
3.2. Binding sites in K2hPg for a1- and a2-repeats
3.2.a. Chemical shift differences show that RH1 and RH2 bind to the same LBS of K2hPg
Chemical shift changes in K2hPg are also ligand-induced, and primarily found in the LBS (Figure 4). The binding of K2hPg to either wild-type VEK50 or its mutant peptides, VEK50△RH1 and VEK50△RH2, is detected in HSQC spectra by monitoring changes in 1H/15N chemical shifts from 15N-labeled K2hPg as a function of the concentration of VEK50 peptides (Supporting Information Figure S2). Three segments in K2hPg, viz., G34-K46, D54-W60, and K68-E71, exhibit the most dramatic CSPs upon binding to VEK50 peptides, suggesting these are binding residues in K2hPg (Figure 4). Several residues in these regions, such as D54 and R55, show weak or very broad signals in the apo-form of K2hPg, but shift to a new position in the bound-form (Supporting Information Figure S2). These results are consistent with previous studies for the K2hPg/VEK30 complex (Wang et al., 2010b), but, additional residues in K2hPg are affected when K2hPg binds to the larger VEK50 peptides, further indicating available exosite interactions. The CSP data also suggest that K2hPg uses the same domain for binding to either VEK50△RH1 or VEK50△RH2 (Figure 4A,B). When three HSQC spectra of K2hPg bound to VEK50 and two mutant peptides were overlaid (Supporting Information Figure S2C), most of peaks appeared at the same chemical shifts. However, in the HSQC spectrum of K2hPg bound to VEK50, the residues with larger CSP always show at two locations with different peak intensities. For examples, D54, L57, and W60 of K2hPg have two fresonance signals corresponding to two K2hPg molecules bound to RH1 or RH2 of VEK50 respectively (Figure 4C). Notably, the signals from K2hPg bound to RH2 show a 3–5-fold stronger peak intensities, suggesting that in the presence of excess of VEK50, RH2 is more favorable for K2hPg binding. This is also consistent with the ITC results for two mutant VEK50 peptides. Thus, the difference of Kd values observed from the mutants truly represents the stronger binding ability of the RH2 site in the WT-VEK50, which is not affected by the steric crowding of two K2hPg molecules in the complex. Slight differences are also observed in the segment comprising residues G6 - N8, which is close to the flexible N-terminus, therefore, the differences are likely not directly related to ligand binding.
Fig. 4. Differences in the binding sites of K2hPg upon its interaction with VEK50△RH1 and VEK50△RH2.

Combined chemical shift changes (△δ ppm) along the sequence of K2hPg between apo-K2hPg and K2hPg bound to: (A) VEK50△RH1 and (B) VEK50△RH2. (C) Residues in the VEK50-bound K2hPg exhibit two resonance signals as shown in the expanded 15N-HSQC spectra. (D) The refined solution structural backbones of K2hPg bound to VEK50△RH1 (red) and VEK50△RH2 (green) are overlaid and shown in as ribbons.
3.2.b. The binding interface in K2hPg for RH1 and RH2
Although only one binding domain is detected in K2hPg by NMR titration, the binding interface formed between K2hPg and the a1- or a2-repeat in two mutant VEK50 peptides is somewhat different. The differences are observed from the NMR solution structures of K2hPg in the complex with the two mutant peptides, which are refined by residual dipolar coupling (RDC) data. A total of 63 amide RDCs (1DNH) from 15N-K2hPg/unlabeled VEK50△RH1 and 60 amide RDCs (1DNH) from 15N-K2hPg/unlabeled VEK50△RH2 were obtained from RDC measurements using 2D IPAP- HSQC (Supporting information Figures S3 A, B). The RDC data obtained from these two complexes were fitted to the solution structure of the bound-form of K2hPg in complex with VEK30 (PDB code: 2kj4) (Wang et al., 2010b), which contains only the RH1 binding motif. It was found that residues on N- and C-termini exhibit larger differences (>5 Hz) between the calculated and observed RDCs (1DNH). The residues in the ligand binding region, such as D54-L57 and W60, which show differences in the sign and intensity of the absolute RDC values in Figures S3 A,B, also exhibit larger differences (>3 Hz) when the experimental RDCs are fitted to the bound-form of K2hPg (Figures S3 C,D). Therefore, we used these experimental RDCs to refine the bound-form of K2hPg with VEK30 in order to provide more precise structures for K2hPg with VEK50 peptides. As depicted in Figure 4D, the binding region (colored red and green) in the two complexes can be overlaid with a RMSD of 1.30 Å, while the rest of K2hPg, except the residues at the flexible N- and C-termini, which are exogenous to K2hPg, have a RMSD of 0.6 Å. Additionally, the binding region, D54-P59, has a more open conformation in the complex with VEK50△RH1.
3.3. Changes in solution structure of VEK50 peptides upon binding to K2hPg
3.3.a. Apo-structures of three VEK50 peptides
The solution structures of VEK50 peptides in their apo-forms were calculated based om NMR data (Figure 5). The residues in the N-terminal region, viz., V1 - D7, and in the C-terminal region, viz., D49 - Y50, have only sequential connectivities, suggesting that these regions exist as coils. In apo-VEK50, three short α-helical segments, viz., A8 - K14, E22 - R30, and K37 - K46, are connected through two flexible loops, which contain the RH1 and RH2 motifs (Figure 5A). The overall structures of two mutant peptides are very similar to that of WT-VEK50, although one of the RH groups has been replaced by Ala-Ala residues. The RH2 group of VEK50△RH1 (Figure 5B) and the RH1 group in VEK50△RH2 are also in the loop regions in the apo forms (Figure 5C).
Fig. 5. Comparison of the solution structures of apo- and K2hPg-bound VEK peptides.

(A, D) VEK50; (B, E) VEK50△RH1; and (C, F) VEK50 △RH2. A, B, and C represent the apo-forms of the indicated peptides; D, E, and F represent the peptides bound to K2hPg. The RH1 and RH2 backbones are shown as red sticks and Ala-Ala backbones are colored green in the ribbons.
3.3.b. Bound structures of three VEK50 peptides
The binding regions, RH1 and RH2, in VEK50 undergo significant structural changes and become more rigid upon its binding to K2hPg. Thus, as shown in Figure 5D, the helical segment from residue A6 - D32 in VEK50 is formed and two RH groups (R17H18 and R30H31) are positioned on opposite faces of the helix. The mutant peptides also exhibit conformational changes but those changes mainly occur at the binding regions (Figure 5E,F). The alanine replacements in VEK50△RH1 enhance the rigidity of the region consisting of A6 - E19 in the apo-form. However, the conformation of this region does not significantly change and remains bent in the complex. The conformation of VEK50△RH2 exhibits high similarity to WT-VEK50 in its bound form, especially the RH1 binding regions.
Residual dipolar coupling (RDC) data provide additional structural and dynamic information in a μsec timescale for the bound-form VEK50 peptides. The plot of 1H-15N RDCs as a function of the residue number (Figure 2H–J) confirm that most of the residues in the a1a2- repeat of VEK50 have RDC absolute values above 5 Hz, while the residues at N- and C-terminal are flexibly disordered, presenting significantly smaller RDCs (< 3Hz) (Figure 2H). The plot of the RDCs for VEK50△RH1 show that the residues presenting large RDCs are not limited to those in the binding region, i.e., the a2-repeats (Figure 2I), whereas the residues in a1-repeat of VEK50△RH2 exhibit significantly larger RDCs (Figure 2J), consistent with the more rigid structure formed upon binding.
All three peptides in the bound form contain a helical segment (L23 - E29) between the two RH motifs. The helical feature gained in the bound form of these peptides fades at the end of the a2-repeat. Residues 1–7 at the N-terminus and residues 49–50 at the C-terminus are flexible and show sharp resonance signals in HSQC spectra. But residues 44–48 are always present at two locations in HSQC spectra, indicating that this fragment is in slow exchange between locally different structures in the bound form.
3.4. HADDOCK calculated structural models of the K2hPg/VEK50 complexes
In this study, we present a structural basis of the binding of VEK50, an active fragment of PAMAP53, to K2hPg and provide evidence for the high affinity and specificity of this peptide. From a previous study of the VEK30/K2hPg complex (Wang et al., 2010b), the hydrophobic groove in K2hPg formed by Y35, F40, W60, W70, and Y72 in combination with the anionic center of D 54 and D56, comprised the binding site of K2hPg/RH1. These same residues in K2hPg also exhibit significant chemical shift perturbations upon its binding to either the a1-repeat in VEK50△RH2 or a2-repeat in VEK50△RH1 (Figure 4), suggesting that there is only one potential interaction site for K2hPg in the complex with VEK50 peptides.
For the VEK50 peptides, the active-site residues, which directly participate in binding, were assigned based on the mutations studied previously and the CSPs observed from NMR titrations in this study (Figure 2). It is noted that both VEK50△RH1 and VEK50△RH2 contain the complete a1- and a2-repeats, except that one of the RH groups has been replaced by Ala-Ala, thus inactivating the K2hPg binding site in each repeat. Thus, only the fragment having the intact RH motif exhibits large CSPs upon the binding to K2hPg (Figure 2F, G). This further confirms that the RH motifs are critical residues for tight K2hPg binding. Other residues, i.e., 10–22 in VEK50△RH1 and 24–34 in VEK50△RH2, with dramatic chemical shift changes, were also directly related to binding, and were defined as passive residues for docking calculations.
The interface of VEK50 peptides and K2hPg was also identified by the intermolecular NOEs between 13C/15N-K2hPg and unlabeled VEK50 peptides, or 13C/15N-labeled VEK50s and unlabeled K2hPg. For example, methyl resonances from L26 of VEK50ΔRH1 exhibited NOEs to residues H33, G34, Y35, and I36 of K2hPg. Similar NOEs were also observed form L13 of VEK50ΔRH2 with corresponding residues in K2hPg, suggesting that these residues are in close contact with each other. Using 13C/15N-filtered, 13C/15N-edited NOESY-HSQC experiments, 12 and 14 intermolecular NOEs between K2hPg/VEK50△RH1 and K2hPg/VEK50△RH2 were observed, respectively, and used as unambiguous distance restraints.
In addition to these distance restraints, RDC data collected from 15N-labeled K2hPg (Figure 4C) and 15N-labeled VEK50 peptides (Figure 2 H–J) in the corresponding complex were included to provide the orientation restraints for docking. When the complex samples were oriented in Pf1 phage medium, no noticeable structural perturbations were observed. A total of 90 and 93 RDCs (1DNH) obtained from the complexes of VEK50ΔRH1/K2hPg, and that of VEK50ΔRH2/K2hPg, respectively, were applied as orientation restraints in docking. The resulting structures calculated in HADDOCK showed no distance violations and only residues in the mobile N-terminal and the C-terminal regions in K2hPg and VEK50 peptides show RDC outliners (>3 Hz).
Previous work has shown that the LBS of K2hPg contains an anionic center that interacts with positive side-chains of the pseudo-lysine ligand. In the modeled structures of the pseudo-lysine binding sites, the anionic center of K2hPg (D54, D56) is involved in electrostatic interactions with side-chains of residues, K14, R17, and H18 in the a1-repeat (Figure 6A) and K27, R30 and H31 in the a2-repeat (Figure 6B). In these structural clusters, hydrogen bonds can also form between the side-chains of these same residues in the a1- or a2-repeat with carbonyl groups of D54 and D56 in K2hPg . Meanwhile, in VEK50ΔRH2, acidic residues D9 of the a1-repeat and E20 of the a2- repeat are within close contact distances with basic residues R55 and R69 of K2hPg, respectively (Figure 6A). Although VEK50ΔRH1 only contains one binding motif, viz., RH2, the acidic residue, D9, and basic residues, R12 and K14 in the a1-repeat are all present in this mutant peptide. Since this region is structured as a bent helical fragment, these residues could act as exosites through electrostatic interactions with K2hPg as illustrated between R12 of VEK50ΔRH1 and D54 of K2hPg (Figure 6B). Additionally, D34 in the a2-repeat of VEK50ΔRH1 forms an H-bond with the side-chain of R69 of K2hPg. Overall, more electrostatic interactions were found between VEK50ΔRH1 and K2hPg.
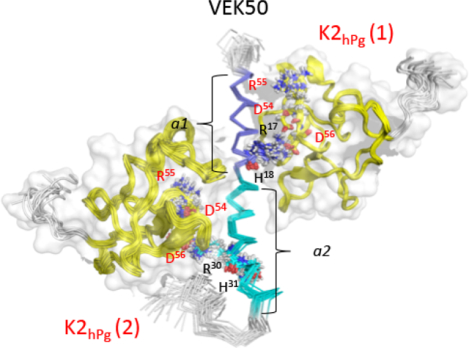
Fig. 6. Solution binding models of the K2hPg/VEK50 peptides derived from Xplor-NIH and HADDOCK.

The lowest-energy conformation was used for the representation of: (A) K2hPg /VEK50ΔRH2 and (B) K2hPg/VEK50ΔRH1. (C) Superposition of backbone traces of the 20 lowest-energy NMR structures of K2hPg /VEK50 is shown. VEK50ΔRH1 and VEK50ΔRH2 bind to K2hPg at a molar ratio of 1:1, whereas VEK50 binds to K2hPg at a molar ratio of 1:2. Residues having H-bond interactions with K2hPg and having close contact (~3 Å) are labeled and shown as sticks. K2hPg is colored yellow whereas the a1- and a2-repeats of VEK50 are colored magenta and cyan, respectively.
To dock two K2hPg molecules onto VEK50, we used the multi-body interface module on the HADDOCK 2.2 web server. As described earlier, a semi-flexible mode of docking was performed, where the side-chains of the active and passive residues are designated as flexible. The bound-form of VEK50 was determined by NMR (Figure 5D), and the bound-form K2hPg was refined using RDCs. At least, 24 intermolecular NOEs collected from VEK50△RH1/K2hPg and VEK50△RH2/K2hPg were used to define each binding interface in VEK50 with K2hPg. The resulting structural model using 20 of the lowest energy structures of VEK50:K2hPg at a molar ratio of 1:2 is shown in Figure 6C. In order to validate the HADDOCK-calculated structures, the observed RDCs of VEK50 were compared with predicted RDCs based on the model and they correlated well with a low Q-factor of 0.5 (Supporting information Figure S4). In these binding models, no further major conformational changes were observed for the NMR-determined bound structures of either VEK50 or K2hPg. The bound-form of VEK50 exists as an α-helix between residue D7-D34, while two K2hPg molecules were bound to opposite faces of the helix. Since both of the a1- and a2-repeats of VEK50 became more structured, these two fragments interact with K2hPg in a very similar manner. For example, the H-bond of R17 and R30 in VEK50 with the LSB of K2hPg can be consistently formed in the simulated structure model. The interactions identified from this structural model are consistent with the observation from NMR experiments, in which the residues in the K2hPg bound to RH2 always show stronger resonance signals. Additionally, L26 and K27 of VEK50 are located in the middle of the helix, are buried in the contact surface of the a2-repeat and K2hPg. This finding is consistent with their slower 1 H/2H exchange rate when present in complexes with K2hPg.
Thus, from these docking calculations, we conclude that two RH motifs have separate clusters to form the required pseudo-lysine ligand that interacts with the LBS of K2hPg. RH1 (R17-H18) in the a1-repeat combines with D9, K14, and E20 with to form the lysine isostere in VEK50△RH2, while RH2 (R30-H31) has another group of residues, including E24, K27, and D34 to form its own lysine isostere in VEK50△RH1.
4. Discussion
The ubiquitous high copy number surface M-protein of GAS, encoded by the emm gene, is a major virulence factor in the pathogenesis of these bacteria, primarily due to its ability to protect against various human host innate responses to infection. Further, the ability of M-proteins to interact with host cells and host proteins confers unique properties to the bacterial cells that allow their invasion and dissemination. There are more than 250 serotypes of M-proteins, primarily identified by the highly variable 5’-nucleotide sequences of the emm gene, and at least five subfamily patterns of M and M-like proteins (A-E), revealed by nucleotide sequences of the more conserved 3’-nucleotide sequences encoding peptidoglycan spanning domains of these proteins (Bessen, 2016). These serotypes and pattern types are of epidemiological value through allowing characterization of infectious breakouts and their possible consequences.
The mature form of M-protein is covalently anchored to the bacterial cell wall at the C-terminus of the protein via a typical sortase A-catalyzed linkage. This leaves the N-terminal region of the elongated M-protein to protrude through the GAS outer capsule to interact with host proteins. The sequence variety at the N-termini of M-proteins allows different types of interactions with the host to occur, and there are further differences depending on the serotype of the M-protein. For example, complement-mediated opsonization inhibitors, e.g., C4BP and Factor H, interact with the N-terminal hypervariable regions (HVR) of some emm-expressed M-proteins of Patterns A-C and E GAS strains (Buffalo et al., 2016; Gustafsson et al., 2013), and in this manner inhibit opsonization of the bacteria. These same complement inhibitors do not bind to the emm gene product (PAM) of Pattern D strains, but do functionally interact with other M-like proteins of these strains, e.g., Enn and Fba, each present along with PAM in the multiple gene activator (mga) regulon, and thereby perform this same function (Agrahari et al., 2013; Liang et al., 2013).
Our major interest in GAS-host interactions is centered in the important hemostasis/inflammation responses to GAS infections. Some Pattern A-C strains of GAS interact with fibrinogen in their centralized B-domain (Glinton et al., 2017) and in this way provide anti-opsonic activity to the microorganism (Courtney et al., 2006; Whitnack and Beachey, 1982). All pattern D strains of M-proteins, e.g., PAM from strain AP53, are unique in their ability to bind hPg tightly and directly to their N-terminal A-domains, with profound consequences to their virulence. Activation of hPg to hPm on the GAS surface by GAS-secreted SK2b generates a surface bound protease, hPm, that can aid dissemination in the host by digesting extracellular matrix proteins and cellular epithelial and endothelial tight junctions through this new proteolytic front.
Pattern D M-proteins exist as elongated fibrous proteins, which, at least in solution, have been identified as irregular weakly associated helical coiled-coils (Ghosh, 2011; McNamara et al., 2008; Qiu et al., 2019; Stewart et al., 2016). The COOH-terminal C- and D-domains of PAM that are present close to the GAS capsule, are primarily responsible for dimerization, with large irregularities in the PAM NH2-terminal HVR-, A-, and B-domains. These irregularities, also seen in coiled-coil proteins such as tropomyosin (Nitanai et al., 2007), appear to allow binding of fibrinogen to B-domains of some M-proteins (McNamara et al., 2008), and thus are of functional significance.
A critical interaction in survival and virulence of Pattern D strains of GAS is the binding of hPg to PAM (Sun et al., 2004) and our work has focused on the nature of this interaction as shown in numerous published studies. The question addressed in this manuscript is the relevance to binding of each of the two adjacent hPg binding sites in the A-domain of PAM. We have employed reductionist approaches in these studies by utilizing peptide secondary structural stretches of PAM, along with the PAM binding K2 domain of hPg. Since PAM is composed solely of secondary structure, with no observed tertiary structure, and K2hPg is an independent domain in hPg (Castellino et al., 1981), this approach should isolate intact binding sites to allow us to examine the properties of each of the regions of interest in these proteins.
In previous studies using X-ray crystallography (PDB code 1I5K) (Rios-Steiner et al., 2001) and NMR (PDB code 2KJ4) (Wang et al., 2010b), very similar structural models of the K2hPg/VEK30 complex have been proposed. The solved structures suggest that there are two interaction clusters formed in the complex. The first is the anionic center of the LBS of K2hPg formed by residues D54 and E/D56, which interact with a cationic cluster of side-chains, viz., K14, R 17, and H18 of VEK30. The second is the cationic locus of the K2hPg binding pocket, R69 which forms a salt-bridge with E20 of VEK30. The role of each residue in the a1-repeat to the binding to K2hPg has been investigated by a variety of mutants, but less has been done for the a2-repeat. From our current study, the interactions between residues K27, R30, and H31 in the a2-repeat act as K14, R17, and H18 in the a1-repeat. The interactions between the LBS of K2hPg and K14, R17, H18, and E20 play major roles in forming a rigid helical structure in the a1-repeat. In the bound form of VEK50, RH1 is in the middle of a rigid helical region from the N-terminal of a1 (D7) to the C-terminal of a2 (D32), while RH2 is close to the flexible C-terminal of a2-repeat. Although both H18 in a1 and H31 in a2 have close contact with the residues of LBS, the H-bond between these two His residues and the LBS are not observed in all models. Meanwhile, the H-bond of R17 and R30 with the LBS of K2hPg can be consistently formed in all of the lowest energy simulated structures.
VEK50 is a truncated peptide from PAMAP53, which contains both the a1- and a2-repeats. The replacement of either RH1 or RH2 by Ala-Ala slightly changes the binding affinity of mutant VEK50 peptides to K2hPg. The differences observed from their binding affinities to K2hPg are related to the secondary structure of these truncated peptides. VEK50 can bind with K2hPg on the a1- and/or a2-repeats, while its mutants can only bind using either a1 or a2. As discussed above, the a2-repeat has more interactions than the a1-repeat with the LBS in K2hPg. Thus, compared to VEK50△RH2, VEK50△RH1 binds to K2hPg tighter via its complete a2-repeat. As shown in the apo-forms, two RH binding motifs are located in flexible regions in apo-VEK50 and become rigid in the bound-form to obtain complete interactions. The interactions between the LBS of K2hPg and K14, R17, H18, and E20 play the major roles in forming a rigid helical structure in the a1-repeat. When the RH1 is inactivated by Ala-Ala substitutions, residues in the a1-repeat remain as a flexible loop. The selective inactivation of RH2 results in a peptide that can only use its a1-repeat to bind to LBS in K2hPg. Similarly, this a1-repeat alone appears to have fewer interactions with K2hPg compared to the a2-repeat. Therefore, VEK50△RH1 has a slightly higher affinity than VEK50△RH2 for K2hPg.
Further affirmation that the entire hPg binding ability is contained within the A-domain of PAM-type M-proteins is evidenced by studies with another class of M-protein, M1 from a Pattern A GAS strain. M1 does not directly interact with hPg, but instead tightly binds to fibrinogen via its B-domain. We replaced the entire B-domain of M1 with the A domain of PAMAP53. This mutated M1 no longer interacted with fibrinogen but interacted with hPg nearly identically to PAMAP53 (Chandrahas et al., 2015). These results affirm that the a1a2 module in PAM proteins presents the complete epitope with regard to hPg binding, whereas effects from other domains in PAM are of little influence in this regard. In the wider scope, we suggest that the domains of M-proteins have evolved independently and have been incorporated into other M-proteins by recombination to generate new GAS strains with advantages for their survival against the immune responses of the host (Bao et al., 2016a; Bao et al., 2016b). The variability of M-proteins, that are relatively rapidly genetically adapted to survive in various host human niches, lies at the basis of the numerous serotypes of GAS that are singularly directed to humans, who cannot evolve as rapidly to combat these bacteria. In addition, the ability of GAS to undergo rapid genetic adaptation is detrimental to M-protein-directed vaccines to combat these infections.
5. Conclusions
Group A Streptococcus (GAS) is a bacterial pathogen unique in its remarkable ability to exploit a range of cellular and tissue environments to establish colonization and progress to a wide spectrum of disease states in the human host. A major determinant of GAS host tropism and virulence is the M- and M-like protein (PAM), expressed on the surface of all GAS strains. M-protein forms have evolved to serve highly specialized roles in GAS survival, including its specific ability to recruit and activate hPg. In this report, we provide a detailed structural view of the VEK50/K2hPg complex, a reductionist approach to understanding the nature of the PAM/hPg complex, that offers insights at the molecular scale for the binding mechanism of PAM to hPg. Broadly considered, our findings suggest that the domains of M-proteins have likely been exchanged through horizontal recombination events and independently further evolved in order that emerging GAS strains optimize survival advantages against the host immune response (Bao et al., 2016a; Bao et al., 2016b). The ability of GAS to utilize multiple genetic mechanisms to produce highly variable M-protein forms that successfully allow GAS to adapt to survive in various host human niches, lies at the basis of the numerous serotypes of GAS that are singularly directed to humans, who cannot evolve as rapidly or as specifically to combat these bacteria. Studies aimed at elucidating the mechanistic details of how GAS virulence determinants, such as PAM, engage host targets will provide future insights into improved anti-virulence strategies as well as alternative approaches to improve current vaccine strategies against GAS.
Supplementary Material
Highlights.
Solution structural determination by high-resolution NMR of the A-domain of PAM reveals occupancy by two molecules of K2hPg on opposite faces of the A-domain helix.
K2hPg binds to PAM through repeating sequences (a1a2-repeats) in the A domain of PAM and uses the same lysine binding site for its interaction with each of the two a-repeats.
K2hPg binds more tightly to the a2-region than the a1-region due to different sets of exosite interactions.
6. Funding
This work was supported by National Institutes of Health Grant HL013423.
Abbreviations used:
- GAS
Group A Streptococcus pyogenes
- hPm
human plasmin
- hPg
human plasminogen
- K2hPg
kringle 2 domain of human plasminogen
- LBS
lysine binding site
- PAM
plasminogen-binding group A streptococcal M-like protein
- SPR
surface plasmon resonance
- CSP
chemical shift perturbation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare that they do not have conflicts of interest.
REFERENCES
- Agrahari G, Liang Z, Mayfield JA, Balsara RD, Ploplis VA, Castellino FJ, 2013. Complement-mediated opsonization of invasive Group A Streptococcus pyogenes strain AP53 is regulated by the bacterial two-component cluster of virulence responder/sensor (CovRS) system. J. Biol. Chem 288, 27494–27504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao YJ, Shapiro BJ, Lee SW, Ploplis VA, Castellino FJ, 2016a. Phenotypic differentiation of Streptococcus pyogenes populations is induced by recombination-driven gene-specific sweeps. Sci. Rep 6, 36644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao YJ, Liang Z, Mayfield JA, Donahue DL, Carothers KE, Lee SW, Ploplis VA, Castellino FJ, 2016b. Genomic characterization of a Pattern D Streptococcus pyogenes emm53 isolate reveals a genetic rationale for invasive skin tropicity. J. Bacteriol 198, 1712–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge A, Sjobring U, 1993. PAM, a novel plasminogen-binding protein from Streptococcus pyogenes. J. Biol. Chem 268, 25417–25424. [PubMed] [Google Scholar]
- Bessen DE, 2016. Molecular basis of serotyping and the underlying genetic organization of Streptococcus pyogenes in Streptococcus pyogenes: Basic Biology to Clinical Manifestations, Ferretti JJ, Stevens DL, Fischetti VA, editors. February, 2016. [PubMed] [Google Scholar]
- Bhattacharya S, Liang Z, Quek AJ, Ploplis VA, Law R, Castellino FJ, 2014. Dimerization is not a determining factor for functional high affinity human plasminogen binding by the group A streptococcal virulence factor PAM and is mediated by specific residues within the PAM a1a2 domain. J. Biol. Chem 289, 21684–21693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffalo C, Bahn-Suh AJ, Hirakis SP, Biswas T, Amaro RE, Nizet V, Ghosh P, 2016. Conserved patterns hidden within group A streptococcus M protein hypervariability recognize human C4b-binding protein. Nat. Microbiol 1, 16155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellino FJ, Ploplis VA, 2003. Human plasminogen: structure, activation, and function. Plasminogen Structure, activation, and regulation Kluwer Academic/Plenum Publishers, 3–17. [Google Scholar]
- Castellino FJ, Ploplis VA, Powell JR, Strickland DK, 1981. The existence of independent domain structures in human lys77-plasminogen. J. Biol. Chem 256, 4778–4782. [PubMed] [Google Scholar]
- Chandrahas V, Glinton K, Liang Z, Donahue DL, Ploplis VA, Castellino FJ, 2015. Direct host plasminogen binding to bacterial surface M-protein in Pattern D strains of Streptococcus pyogenes Is required for activation by Its natural coinherited SK2b protein. J. Biol. Chem 290, 18833–18842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Im W, Brooks CL, 2004. Refinement of NMR structures using implicit solvent and advanced sampling techniques. J. Am. Chem. Soc 126, 16038–16047. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC, 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clubb RT, Thanabal V, Wagner G, 1992. A new 3D HN(CA)HA experiment for obtaining fingerprint HN-Halpha peaks in 15N- and 13C-labeled proteins. J. Biomol. NMR 2, 203–210. [DOI] [PubMed] [Google Scholar]
- Courtney HS, Hasty DL, Dale JB, 2006. Anti-phagocytic mechanisms of Streptococcus pyogenes: binding of fibrinogen to M-related protein. Mol .Microbiol 59, 936–947. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A, 2003. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucl. Acids Res 31, 3784–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P, 2011. The nonideal coiled coil of M protein and its multifarious functions in pathogenesis. Adv. Exp. Med. Biol 715, 197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinton K, Beck J, Liang Z, Qiu C, Lee SW, Ploplis VA, Castellino FJ, 2017. Variable region in streptococcal M-proteins provides stable binding with host fibrinogen for plasminogen-mediated bacterial invasion. J. Biol. Chem 292, 6775–6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesiek S, Bax A, 1992. Correlatting backbone amide and side-chain resonances in larger proteins by multiple relayed thriple resonance NMR. J. Am. Chem. Soc 114, 6291–6293. [Google Scholar]
- Grzesiek S, Anglister J, Bax A, 1993a. Correlation of backbone amide and aliphatic side-chain resonances in C-13/N-15-enriched proteins by isotropic mixing of C-13 magnetization. J. Magn. Reson 101, 114–119. [Google Scholar]
- Grzesiek S, Anglister J, Ren H, Bax A, 1993b. 13C line narrowing by 2H decoupling in 2H/13C/15N-enriched proteins. Application to triple resonance 4D J connectivity of sequential amides. J. Am. Chem. Soc 115, 4369–4370. [Google Scholar]
- Gustafsson MC, Lannergård J, Nilsson OR, Kristensen BM, Olsen JE, Harris CL, Ufret-Vincenty RL, Stålhammar-Carlemalm M, Lindahl G, 2013. Factor H binds to the hypervariable region of many Streptococcus pyogenes M proteins but does not promote phagocytosis resistance or acute virulence. PLoS Pathog. 9, e1003323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura M, Kay LE, Tschudin R, Bax A, 1990. 3-Dimensional NOESY-HMQC spectroscopy of a C-13-labeled protein. J. Magn. Reson 86, 204–209. [Google Scholar]
- Kay L, Keifer P, Saarinen T, 1992. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc 114, 10663–10665. [Google Scholar]
- Kay LE, Ikura M, Tschudin R, Bax A, 1990. Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins. J. Magn. Reson 89, 496–514. [DOI] [PubMed] [Google Scholar]
- Lahteenmaki K, Kuusela P, Korhonen TK, 2000. Plasminogen activation in degradation and penetration of extracellular matrices and basement membranes by invasive bacteria. Methods 21, 125–132. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM, 1993. PROCHEK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst 26, 283–291. [Google Scholar]
- Laue TM, Shah BD, Pelletier SL, 1992. Computer-aided interpretation of analytical sedimentation data for proteins, p. 90–125, in: Harding SE, et al. , Eds.), Analytical Ultracentrifugation in Biochemistry and Polymer Science, Royal Society of Chemistry, Cambridge, UK. [Google Scholar]
- Lee W, Westler WM, Bahrami A, Eghbalnia HR, Markley JL, 2009. PINE-SPARKY: graphical interface for evaluating automated probabilistic peak assignments in protein NMR spectroscopy. Bioinformatics 25, 2085–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Zhang Y, Agrahari G, Chandrahas V, Glinton K, Donahue DL, Balsara RD, Ploplis VA, Castellino FJ, 2013. A natural inactivating mutation in the CovS component of the CovRS regulatory operon in a pattern D Streptococcal pyogenes strain influences virulence-associated genes. J. Biol. Chem 288, 6561–6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markley JL, Bax A, Arata Y, Hilbers CW, Kaptein R, Sykes BD, Wright PE, Wüthrich K, 1998. Recommendations for the presentation of NMR structures of proteins and nucleic acids. IUPAC-IUBMB-IUPAB Inter-Union Task Group on the Standardization of Data Bases of Protein and Nucleic Acid Structures Determined by NMR Spectroscopy. J. Biomol. NMR 12, 1–23. [DOI] [PubMed] [Google Scholar]
- McNamara C, Zinkernagel AS, Macheboeuf P, Cunningham MW, Nizet V, Ghosh P, 2008. Coiled-coil irregularities and instabilities in group A Streptococcus M1 are required for virulence. Science 319, 1405–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles LA, Dahlberg CM, Plescia J, Felez J, Kato K, Plow EF, 1991. Role of cell-surface lysines in plasminogen binding to cells: Identification of a-enolase as a candidate plasminogen receptor. Biochemistry 30, 1682–1691. [DOI] [PubMed] [Google Scholar]
- Mori S, Abeygunawardana C, Johnson MO, van Zijl PC, 1995. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. B 108, 94–98. [DOI] [PubMed] [Google Scholar]
- Nilsen SL, DeFord ME, Prorok M, Chibber BAK, Bretthauer RK, Castellino FJ, 1997. High-level secretion in Pichia pastoris and biochemical characterization of the recombinant kringle 2 domain of tissue-type plasminogen activator. Biotech. Appl. Biochem 25, 63–74. [DOI] [PubMed] [Google Scholar]
- Nitanai Y, Minakata S, Maeda K, Oda N, Maéda Y, 2007. Crystal structures of tropomyosin: flexible coiled-coil. Adv. Exp. Med. Biol 592, 135–151. [DOI] [PubMed] [Google Scholar]
- Ottiger M, Delaglio F, Bax A, 1998. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson 131, 373–378. [DOI] [PubMed] [Google Scholar]
- Pancholi V, Fischetti VA, 1998. alpha-enolase, a novel strong plasmin(ogen) binding protein on the surface of pathogenic streptococci. J. Biol. Chem 273, 14503–14515. [DOI] [PubMed] [Google Scholar]
- Qiu C, Yuan Y, Liang Z, Lee SW, Ploplis VA, Castellino FJ, 2019. Variations in the secondary structures of PAM proteins influence their binding affinities to human plasminogen. J. Struct. Biol 206, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu C, Yuan Y, Zajicek J, Liang Z, Balsara RD, Brito-Robionson T, Lee SW, Ploplis VA, Castellino FJ, 2018. Contributions of different modules of the plasminogen-binding Streptococcus pyogenes M-protein that mediate its functional dimerization. J. Struct. Biol 204, 151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringdahl U, Svensson M, Wistedt AC, Renn T, Kellner R, Muller-Esterl W, Sjobring U, 1998. Molecular co-operation between protein PAM and streptokinase for plasmin acquisition by Streptococcus pyogenes. J. Biol. Chem 272, 6424–6230. [DOI] [PubMed] [Google Scholar]
- Rios-Steiner JL, Schenone M, Mochalkin I, Tulinsky A, Castellino FJ, 2001. Structure and binding determinants of the recombinant kringle-2 domain of human plasminogen to an internal peptide from a group A Streptococcal surface protein. J. Mol. Biol 308, 705–719. [DOI] [PubMed] [Google Scholar]
- Sanderson-Smith ML, Dowton M, Ranson M, Walker MJ, 2007. The plasminogen-binding group A streptococcal M protein-related protein Prp binds plasminogen via arginine and histidine residues. J. Bacteriol 189, 1435–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson-Smith ML, Dinkla K, Cole JN, Cork AJ, Maamary PG, McArthur JD, Chhatwal GS, Walker MJ, 2008. M protein-mediated plasminogen binding is essential for the virulence of an invasive Streptococcus pyogenes isolate. FASEB J. 22, 2715–2722. [DOI] [PubMed] [Google Scholar]
- Schenone MM, Warder SE, Martin JA, Prorok M, Castellino FJ, 2000. An internal histidine residue from the bacterial surface protein, PAM, mediates its binding to the kringle-2 domain of human plasminogen. J. Pept. Res 56, 438–445. [DOI] [PubMed] [Google Scholar]
- Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu G, Eletsky A, Wu Y, Singarapu KK, Lemak A, Ignatchenko A, Arrowsmith CH, Szyperski T, Montelione GT, Baker DM, Bax A, 2008. Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. USA 105, 4685–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CM, Buffalo CZ, Valderrama JA, Henningham A, Cole JN, Nizet V, Ghosh P, 2016. Coiled-coil destabilizing residues in the group A Streptococcus M1 protein are required for functional interaction. Proc. Natl. Acad. Sci. USA 113, 9515–9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumitomo T, Nakata M, Higashino M, Terao Y, Kawabata S, 2013. Group A streptococcal cysteine protease cleaves epithelial junctions and contributes to bacterial translocation. J. Biol. Chem 288, 13317–13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumitomo T, Nakata M, Higashino M, Yamaguchi M, Kawabata S, 2016. Group A Streptococcus exploits human plasminogen for bacterial translocation across epithelial barrier via tricellular tight junctions. Sci. Rep 7, 20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Ringdahl U, Homeister JW, Fay WP, Engelberg NC, Yang AY, Rozek LS, Wang X, Sjobring U, Ginsburg D, 2004. Plasminogen is a critical host pathogenicity factor for Group A streptococcal infection. Science 305, 1283–1286. [DOI] [PubMed] [Google Scholar]
- Talluri S, Wagner G, 1996. An optimized 3D NOESY-HSQC. J. Magn. Reson. Ser. B 112, 200–205. [DOI] [PubMed] [Google Scholar]
- van Zundert GCP, Rodrigues JPGLM, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, van Dijk M, de Vries SJ, Bonvin AMJJ, 2016. The HADDOCK2.2 web server: User-friendly integrative modeling of biomolecularcomplexes. J. Mol. Biol 428, 720–725. [DOI] [PubMed] [Google Scholar]
- Wang M, Prorok M, Castellino FJ, 2010a. NMR backbone dynamics of VEK-30 bound to the human plasminogen kringle 2 domain. Biophys. J 99, 302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Zajicek J, Geiger JH, Prorok M, Castellino FJ, 2010b. Solution structure of the complex of VEK-30 and plasminogen kringle 2. J. Struct. Biol 169, 349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Arakawa T, Wypych J, Langley KE, Schwartz MG, Philo JS, 1997. Chromatographic determination of extinction coefficients of non-glycosylated proteins using refractive index (RI) and UV absorbance (UV) detectors: Applications for studying protein interactions by size exclusion chromatography with light-scattering, UV, and RI detectors. Tech. Prot. Chem 8, 113–119. [Google Scholar]
- Whitnack E, Beachey EH, 1982. Antiopsonic activity of fibrinogen bound to M protein on the surface of group A streptococci. J Clin Invest. 69, 1042–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson MP, 2013. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc 73, 1–16. [DOI] [PubMed] [Google Scholar]
- Wistedt AC, Ringdahl U, Müller-Esterl W, Sjøbring U, 1995. Identification of a plasminogen-binding motif in PAM, a bacterial surface protein. Mol. Microbiol 18, 569–578. [DOI] [PubMed] [Google Scholar]
- Wistedt AC, Kotarsky H, Marti D, Ringdahl U, Castellino FJ, Schaller J, Sjobring U, 1998. Kringle 2 mediates high affinity binding of plasminogen to an internal sequence in streptococcal surface protein PAM. J. Biol. Chem 273, 24420–24424. [DOI] [PubMed] [Google Scholar]
- Wittekind M, Mueller L, 1993. HNCACB, A high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha-carbon and beta-carbon resonances in proteins. J. Magn. Reson 101, 201–205. [Google Scholar]
- Yuan Y, Zajicek J, Qiu C, Chandrahas V, Lee SW, Ploplis VA, Castellino FJ, 2017. Conformationally organized lysine isosteres in Streptococcus pyogenes M protein mediate direct high-affinity binding to human plasminogen. J. Biol. Chem 292, 15016–15027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.