

ABSTRACT
The fast turnover of membrane components through endocytosis and recycling allows precise control of the composition of the plasma membrane. Endocytic recycling can be rapid, with some molecules returning to the plasma membrane with a half time <5 min. Existing methods to study these trafficking pathways utilize chemical, radioactive or fluorescent labeling of cell surface receptors in pulse-chase experiments, which require tedious washing steps and manual collection of samples. Here, we introduce a live-cell endocytic recycling assay based on a newly designed cell-impermeable fluorogenic ligand for HaloTag, Janelia Fluor 635i (JF635i, where i indicates impermeant), which allows real-time detection of membrane receptor recycling at steady state. We used this method to study the effect of iron depletion on transferrin receptor (TfR) recycling using the chelator desferrioxamine. We found that this perturbation significantly increases the TfR recycling rate. The high temporal resolution and simplicity of this assay provides a clear advantage over extant methods and makes it ideal for large scale cellular imaging studies. This assay can be adapted to examine other cellular kinetic parameters such as protein turnover and biosynthetic trafficking.
KEY WORDS: Endocytic recycling, Fluorophore, Trafficking kinetics
Summary: Introduction to an easy to implement microscopy-based platform enabling accurate quantification of fast endocytic recycling, biosynthetic delivery and protein turnover in real time.
INTRODUCTION
Endocytic recycling pathways comprise fast (t1/2=1–5 min) and slow (t1/2=10–20 min) recycling pathways (Hao and Maxfield, 2000; Mayor et al., 1993) (Fig. 1A) and underlie key plasma membrane-based processes, such as nutrient uptake, controlling ion flux, cell–matrix attachment, cell–cell adhesion, cell migration, signal transduction, cell division and establishment of cell polarity. Endocytic recycling kinetics are typically measured with pulse-chase assays using cell-impermeant probes. These assays involve reversibly blocking endocytosis and recycling using low temperature, which can alter trafficking kinetics. In addition, the temporal resolution of such assays is limited by washing and cooling steps as well as by the manual collection of samples (Fig. 1B; Fig. S1). Ideally, an endocytic recycling assay should selectively detect receptors as they appear at the plasma membrane without aberrantly detecting intracellular receptors. For a fluorescence-based assay, this requires a fluorescent ligand with the following properties: (1) membrane impermeant, (2) fluorogenic upon binding the cell surface receptor, (3) bright and photostable to allow continuous imaging, (4) pH insensitive to prevent quenched states in acidified endosomal compartments, (5) high on-rate (kon), with irreversible chemistry to ensure rapid and complete labeling, (6) usable at nanomolar working concentrations to prevent background signal from bulk endocytosis. We set out to design a ligand that would meet all these requirements, resulting in a single-compound, real-time recycling assay (Fig. 1C). In our proposed assay, the high kon of the ligand ensures rapid saturation of the surface pool of the protein of interest. Over time, unlabeled protein arrives at the surface and is labeled, resulting in an increase in signal, which reflects the kinetics of the fast and slow recycling pathways (Fig. 1D). A recent advance in real-time assays was the development of membrane-impermeable, fluorogenic SNAP-tag ligands based on Förster resonance energy transfer (FRET), which allow continuous measurement of cell-surface SNAP-tagged proteins without washing steps (Komatsu et al., 2011; Leng et al., 2017). However, the relatively low kon (2×103 M−1 s−1) (Komatsu et al., 2011; Leng et al., 2017) and requisite high concentrations of SNAP-tag ligands make them unsuitable for measuring the fast kinetics of endocytic recycling. We sought to remedy this problem by developing a real-time endocytic recycling assay based on HaloTag. HaloTag is a 33 kDa protein tag based on the Rhodococcus dehalogenase (DhaA) protein (Los et al., 2008; Encell et al., 2013), a haloalkane dehalogenase protein that as HaloTag has been modified to irreversibly bind its ligand. The HaloTag ligand, a chloroalkane moiety, can be attached to a plethora of different molecules such as fluorophores, resins or reactive groups. HaloTag ligands have relatively high kon values (Encell et al., 2013) that can be 1000–10,000 times higher than SNAP-tag ligands, which enables labeling at nanomolar concentrations.
Fig. 1.

Assay design. (A) Scheme of the endosomal recycling pathways. After endocytosis and delivery to the sorting endosome (SE), cargo can either be delivered straight back to the plasma membrane (fast recycling) or be delivered to the plasma membrane via the recycling endosome (RE) (slow recycling). (B) Scheme of a commonly used NHS-SS-biotin recycling assay protocol. This pulse-chase assay with endpoint analysis requires cooling steps to stop and restart endocytosis and recycling. (C) Scheme of our proposed recycling assay. (D) Scheme of the proposed recycling assay. Immediately after addition of the ligand, the total surface pool of the protein of interest is labeled (t=0). Over time, ligand-bound protein is endocytosed while unbound protein is deposited to the plasma membrane, immediately becomes labeled and starts emitting. This increases overall fluorescence signal in the sample (tshort). After a longer period, the system becomes saturated, meaning that all protein that was inside the cell is now bound to ligand and the fluorescence increase stops (tlong).
RESULTS
Fluorogenic probe design and synthesis
To allow steady-state recording of endocytic recycling without washing steps, we designed a cell-impermeant, fluorogenic HaloTag ligand that exhibits low background fluorescence in solution but an increase in absorption and quantum yield upon formation of the ligand–protein conjugate. We utilized our recently developed azetidine-containing Janelia Fluor (JF) dyes as a scaffold, which can be fine-tuned by placing different substituents on the four-membered azetidine rings (Grimm et al., 2015). We started with the HaloTag ligand of the Si-rhodamine JF635 (JF635-HaloTag ligand; Fig. 2A), which preferentially adopts a nonfluorescent lactone form in aqueous solution but displays a significant (>100×) fluorescence increase upon binding, allowing no-wash live-cell imaging with low background (Grimm et al., 2017). We reasoned that addition of carboxylic acid moieties would guarantee cell impermeability; we would retain the electronegative fluorine substituents to maintain fluorogenicity. We synthesized JF635i-HaloTag ligand (where i indicates impermeant; Fig. 2C). The compound was modestly chromogenic and fluorogenic in vitro, displaying a sixfold increase in absorbance upon binding to HaloTag (Fig. 2B), similar to first-generation Si-rhodamine dyes (Lukinavičius et al., 2013). The resulting HaloTag conjugate exhibited λmax/λem=640 nm/656 nm, fluorescence quantum yield of 0.75 and high absorptivity (ε=9.6×104 M−1 cm−1). The fluorescence emission of the JF635i-HaloTag conjugate was pH insensitive above pH 3.8 (Fig. 2D), making it suitable for trafficking assays (lysosomal pH is 4–5). Furthermore, the JF635i-HaloTag ligand on-rate was determined using stopped flow analysis. The kon measured was 8.7×105 M−1 s−1 (Fig. S2a), which was about 20-fold lower than the kon of the fastest reported HaloTag ligand (Encell et al., 2013) but high enough to allow >95% labeling in <1 min at a concentration of 200 nM in both a theoretical model and cell-based assays (Fig. S2b).
Fig. 2.

JF635i-HaloTag ligand synthesis. (A) Chemical structure of JF635-HaloTag ligand before and after binding to HaloTag. (B) The absorption and emission spectra of JF635i-HaloTag ligand were measured in the medium as the free ligand and when bound to an excess HaloTag protein to quantify the fluorogenicity of the dye ligand. Normalized absorption (bold lines) and emission (dashed lines) of JF635i-HaloTag ligand in DMEM containing 10% FBS were measured in the presence or the absence of HaloTag protein. (C) JF635i-HaloTag ligand was synthesized in four steps from 6-tert-butoxycarbonylsilafluorescein ditriflate. The methyl ester-protected azetidines were introduced by palladium-catalyzed cross-coupling. Deprotection of the 6-tert-butyl ester S1 with TFA followed by amide coupling afforded intermediate S3, which was deprotected with NaOH to afford the JF635i-HaloTag ligand 4. (D) The pH titration assay showed that the fluorescence emission of JF635i-HaloTag ligand was stable above pH 3.8 (duplicate experiments; error bars represent standard deviation of the mean). (E) Representative images of U2OS cells expressing either H2B-HaloTag (left) or pDisplay-HaloTag (right) labeled with 200 nM JF635i-HaloTag ligand (JF635i-HTL) for 30 min and imaged directly without washing. (F) As positive control, we used U2OS cells expressing the same constructs as C, labeled with 200 nM cell-permeant JF635-HaloTag ligand (JF635-HTL). Images are displayed as a maximum intensity projection of a confocal image stack using Fiji. Scale bars: 50 µm.
We then tested whether the two carboxylic acid groups were sufficient to prevent HaloTag-independent cellular entry (primarily to determine whether the JF635i ligand was impermeable) and assessed whether the modest increase in fluorescence upon JF635i ligand binding allowed no-wash live-cell imaging. We incubated JF635-HaloTag and JF635i-HaloTag ligand (200 nM, 30 min) with U2OS cells expressing HaloTag either in the nucleus (histone H2B-HaloTag) or on the extracellular side of the plasma membrane (modified pDisplay-HaloTag; see Materials and Methods) and imaged directly without exchanging the media. Incubation with JF635i-HaloTag ligand showed labeling on only the membrane HaloTag protein (Fig. 2E) whereas the cell-permeable JF635-HaloTag labeled both the membrane and nuclear HaloTag (Fig. 2F). A longer (20 h) incubation yielded the same results (Fig. S2c), showing that JF635i-HaloTag ligand is robustly cell impermeant and suitable for experiments that require long-term incubation.
Measuring the kinetics of endogenous TfR recycling in MDCK cells
We then used the JF635i-HaloTag ligand to measure the well-characterized fast and slow recycling kinetics of transferrin receptor (TfR) in MDCK cells (Hao and Maxfield, 2000; Maxfield and McGraw, 2004). A gene-edited cell line was prepared using the CRISPR-Cas9 system to insert the HaloTag protein sequence on the 3′-end of the TFR gene, corresponding to the extracellular domain of the receptor (Fig. S3). To ensure that the C-terminal HaloTag did not interfere with the function of TfR, we incubated cells with transferrin conjugated to CF594 (Tf-594) and imaged the cells using confocal microscopy. Endocytosed transferrin showed high colocalization with TfR-HaloTag in MDCK cells (Fig. S3d), indicating that the function of the TfR is not inhibited by appending a HaloTag protein domain.
With this cell line in hand, we designed an imaging assay to measure TfR-HaloTag recycling in real time using 10 s sampling intervals. Cells were grown in flow chamber slides to allow rapid addition of JF635i-HaloTag ligand. The first time point was taken without adding ligand (t=0); between t=0 and t=1 (10 s) the medium in the culture chamber was replaced with image medium containing 200 nM JF635i-HaloTag ligand. The high on-rate of the HaloTag system ensured that the surface fraction of the TfR-HaloTag was saturated within the first minute (Fig. S2b). The subsequent increase in signal reflected the arrival of unlabeled TfR-HaloTag to the plasma membrane from the fast and slow recycling pathways. This process continued until the system was saturated and all TfR-HaloTag was labeled with JF635i-HaloTag ligand. We compared the recycling rate of TfR-HaloTag in control cells with cells treated with desferrioxamine (DFO), an iron chelator known to increase expression of the receptor (Mattia et al., 1984; Bridges and Cudkowicz, 1984), but its effect on recycling kinetics is unknown. Under standard conditions, the fluorescence signal saturated with a t1/2 of 5.8±0.4 min (Fig. 3A upper panels, quantified in Fig. 3B; Movie 1); however, in cells pretreated with DFO (50 µM, 16 h), the signal saturated substantially faster, with a t1/2 of 3.7±1.0 min (Fig. 3A lower panels, quantified in Fig. 3B; Movie 1). Comparison of the t1/2 of saturation as well as the log transformed data showed that DFO-treated cells had significantly faster recycling kinetics (P<1×10−3 for saturation kinetics and P<1×10−5 for log transformed data; Fig. S4a). These data showed that our method accurately reflects the recycling of TfR and is capable of detecting these fast trafficking processes. Treatment with cytochalasin D, an actin inhibitor, stopped the fluorescence signal accrual beyond the plasma membrane pool, demonstrating that our fluorescence measurements reflect endocytosis and recycling (Fig. 3C; Fig. S4).
Fig. 3.

High temporal resolution endocytic recycling assay using JF635i-HaloTag ligand. (A) Stills from the recycling assay movies show the increase in fluorescence over time and the different kinetics in control versus DFO conditions. (B) Quantification of the fluorescence intensity over background from the movies (n=6 samples per condition, 4 fields per sample) shows the kinetics of the recycling pathway in control and DFO conditions. Data were normalized to the highest value in the set; error bars represent the standard deviation of the mean. (C) Treatment with cytochalasin D (CytoD) completely abolished the kinetics of the system (representative curve of triplicate experiments). (D) Scheme of the kinetic model. X0 represents plasma membrane (PM), X1 the SE, X2 the RE, k01 the endocytic rate, k10 fast recycling rate, k12 the rate of transport between SE and RE and k20 the rate of transport between RE and plasma membrane. (E) Colocalization analysis of TfR-HaloTag (TfR-HT) with transferrin-594 and dextran-488 to determine the relative distribution of TfR-HaloTag in SE (X1) and RE (X2).
A kinetic model for TfR recycling
The resulting kinetic measurements (e.g. Fig. 3B) represent the sum of both fast and slow recycling pathways. To extract the fast and slow recycling rates, we fitted the imaging data to a kinetic model (Fig. 3D). The model considers membrane receptors within the plasma membrane, which can cycle through endosomes and back to the plasma membrane. We denote the plasma membrane as X0, sorting endosome (SE) as X1 and recycling endosome (RE) as X2. Although the receptors circulate, the amount in each compartment is constant over time. Rate constants (k) provide the rates of receptor transfer between compartments (see Materials and Methods). The model assumes instant labeling of cell surface pool by the HaloTag ligand. This assumption was tested by comparing the model including an on-rate of the receptor ligand binding at 200 nM with instant binding. This resulted in near-identical curves (Fig. S5a), allowing us to use the simpler model assuming instant labeling for data fitting. The kinetic rates in this model cannot be calculated with the one-component assay alone but requires the relative fraction of receptor in X1 and X2 as additional parameters. We determined the relative concentrations of X1 and X2 by a colocalization experiment with endocytosed fluorescent markers dextran-488 and transferrin-594 in fixed samples (Fig. 3E). TfR-HaloTag in X1 (SE) co-localizes with both dextran-488 and transferrin-594, whereas TfR-HaloTag in X2 (RE) only co-localizes with transferrin-594 (Baravalle et al., 2005). Colocalization quantification showed an expected high overlap between transferrin-549 and TfR-HaloTag (Manders’ overlap coefficient of 0.93±0.02). The relative distribution of TfR-HaloTag in Tf-594-positive REs was 69.8% and in double-positive SEs 30.2%, resulting in an estimated ratio of HaloTag-positive SE to RE of 1:2. With this parameter, the resulting model showed an excellent fit with the imaging data (Fig. S5b) and allowed us to calculate the specific fast and slow recycling rates and relative concentrations of all compartments (Fig. S5c). Overall, we showed that this method can accurately detect changes in recycling kinetics using a simple experimental setup. The addition of a straightforward fixed-cell imaging experiment allowed determination of specific fast and slow recycling kinetics.
Measuring exocytic trafficking and biosynthesis rates of TfR
To demonstrate the versatility of this system, we then expanded our assay to measure the kinetics of other cellular processes, such as receptor turnover and biosynthetic exocytosis. We used a combination of permeant and impermeant probes to occupy distinct subsets of TfR-HaloTag in pulse-chase experiments, allowing determination of the kinetics of a specific receptor population. To measure the arrival of newly synthesized TfR-HaloTag at the plasma membrane, we incubated TfR-HaloTag MDCK cells with the cell-permeant JF549-HaloTag ligand (200 nM, 6 h; DMEM) to label all HaloTag in the cell. The cells were then washed (3×, prewarmed Hanks' Balanced Salt Solution; HBSS) and incubated with cell-impermeant JF635i-HaloTag ligand (200 nM) with imaging every 10 min for 16 h. In this experiment, nascent TfR-HaloTag became fluorescent when it reached the plasma membrane and was labeled with JF635i (Fig. 4A upper panels, quantified in Fig. 4B); the signal increased linearly with the rate of protein synthesis and was abolished by treatment with cycloheximide (10 µg ml−1, 1 h; Fig. 4A lower panels, quantified in Fig. 4B). We could also extract the time taken for newly synthesized TfR-HaloTag to reach the plasma membrane by measuring the initial “lag-phase” using a sliding window linear regression method (Fig. 4B, Fig. S6a,b). We estimated the time of transport for TfR-HaloTag from synthesis to plasma membrane as 39±5 min, similar to previous reports (Chen et al., 2017; Futter et al., 1995). These results demonstrate that the JF635i assay can accurately measure exocytic trafficking rates in addition to biosynthesis rates.
Fig. 4.

Measuring biosynthetic delivery of TfR-HaloTag. (A) Stills from the biosynthetic delivery assay movies show the increase in fluorescence over time and the different kinetics in control versus cycloheximide (CHX) conditions in the biosynthetic delivery assay. (B) Quantification of the fluorescence intensity over background from the biosynthetic delivery assay (n=6 samples per condition, 5 fields per sample) shows the kinetics of biosynthetic delivery in control and CHX conditions. Error bars represent the standard deviation of the mean.
Measuring the half-life rate for TfR
We used a combination of two permeable fluorophores to measure the total population of TfR-HaloTag in the cell in a classical pulse-chase experiment. We first incubated cells with cell-permeant JF635-HaloTag ligand (10 nM, 6 h), followed by JF549-HaloTag ligand (10 nM), imaging the JF635 channel every 30 min for 60 h (Fig. 5A upper panels, quantified in Fig. 5B). The exponential decrease in fluorescence revealed a half-life of TfR-HaloTag of 18.4±4.6 h, similar to previous reports (Rutledge et al., 1991; Perez Bay et al., 2016). Incubation with chloroquine inhibits endosomal acidification and thereby reduces lysosomal degradation of proteins; we observed a delay in the onset of degradation and a reduction in the degradation kinetics (Fig. 5A lower panels, quantified in Fig. 5B, Fig. S4b). These results show that this assay allows determination of the half-life of proteins of interest.
Fig. 5.

Measuring protein half-life of TfR-HaloTag. (A) Stills from the protein turnover assay movies show the decrease in fluorescence over time and the different kinetics in control versus chloroquine (CHQ) conditions. (B) Quantification of the fluorescence intensity over background from the protein turnover assay (n=6 samples per condition, 5 fields per sample) shows the kinetics of protein turnover in control and dose-dependent response to CHQ treatment. Data was normalized to the highest value in the set; error bars represent the standard deviation of the mean.
In summary, we present a new method to accurately measure endocytic recycling. We developed a new compound, the cell-impermeant JF635i-HaloTag ligand, which exhibits the appropriate labeling kinetics and fluorogenicity to allow real-time measurement of receptor insertion and trafficking in living cells. The high temporal resolution and simplicity of the method have distinct advantages over existing techniques and the same experimental setup and cell lines can be used to determine other cellular kinetics such as biosynthesis, exocytosis and protein turnover. We expect these new assays to reveal novel details on receptor trafficking in live cells and enable large scale, high-throughput experiments on recycling kinetics.
DISCUSSION
Studying endocytic recycling kinetics requires the detection of proteins that are delivered to the plasma membrane from endosomal compartments. To do so, all extant recycling assays have two steps: (1) label a pool of molecules in endosomal compartments and (2) detect the relative amount of recycled molecules of that initial pool at a specific time point. We have detailed the existing classes of endocytic recycling assays and compared them with our proposed method (Fig. S1). Independent of the method used to label the molecule of interest, all existing recycling assays rely on a pulse of labeled molecules to fill the endosomal compartments and a chase to detect the relative amount of molecules that have returned to the plasma membrane. In addition, earlier methods all require cooling to stop vesicle trafficking during labeling followed by a reheating step. This temperature change protocol can cause artefacts in the measurements because recycling is highly temperature sensitive (Punnonen et al., 1998) and low temperatures cause disassembly of microtubules (Correia and Williams, 1983). Our assay eliminates the need for pulse-chase experiments and the problematic cooling step by exploiting the rapid labeling kinetics and fluorogenicity of the HaloTag system. The binding rate (kon) of JF635i-HaloTag ligand to HaloTag (8.7×105 M−1 s−1) is high enough to label the entire pool of cell surface receptors in under 1 min (Fig. S2b, blue line). Fast labeling is necessary because receptors can have very short residence times on the plasma membrane; the half-time at the plasma membrane for TfR is 2–5 min (Bleil and Bretscher, 1982; Ciechanover et al., 1983; Hopkins and Trowbridge, 1983). The fastest cell-impermeant SNAP-tag ligand, BGAN-2C (Leng et al., 2017), has a kon of ∼2×103 M−1 s−1. Labeling the entire pool of surface receptors with this probe would take ∼10 min (Fig. S2b, green line), making it unsuitable for study of these fast processes. In addition to rapid labeling kinetics, the use of a fluorogenic probe allows continuous imaging without washing steps. The first-generation JF635i-HaloTag ligand displays a sixfold increase in absorbance upon binding to HaloTag, but this can be tuned to decrease fluorescence in the unbound “dark” state and increase the fluorogenicity upon binding. These improvements will further increase the sensitivity of the assay and allow detection of low expressed proteins.
Increasing the concentration of the ligand decreases the time required to label the total pool of surface receptors, allowing labeling systems with lower kon values to be used in trafficking assays. Nevertheless, using a high concentration of ligand allows receptor-independent cellular entry of the fluorophore by fluid-phase endocytosis – a continuous cellular process that ensures uptake of solutes from the extracellular space. Endocytosis of unbound ligand and entry into the endosomal pathway allows the ligand to bind intracellular receptors that have not yet presented themselves to the plasma membrane, resulting in an erroneous background signal. Although fluid-phase endocytosis is a relatively slow process at 1.20×10−13 µl−1 min−1 µm−2 (Bomsel et al., 1989), using the 5 µM working concentration of a SNAP-tag ligand would result in endocytosis of 722.6 molecules min−1 cell−1 compared with 28.9 molecules min−1 cell−1 using the 200 nM working concentration of JF635i-HaloTag ligand (Table 1). Thus, assuming 50,000 receptors, a 30 min recycling experiment using 5 µM SNAP-tag ligand could result in 43% of receptors erroneously labeled with fluid-phase endocytosed dye compared with just 1.73% of receptors using 200 nM JF635i-HaloTag ligand. These estimates demonstrate the importance of a low working concentration and a high kon for measuring real-time recycling assays using a non-permeable fluorogenic probe.
Table 1.
Contribution of fluid-phase endocytosis to measurement error

The development of SNAP-tag ligands portends the exciting possibility of measuring the recycling kinetics of multiple proteins simultaneously using both SNAP-tag- and HaloTag-conjugated proteins. The recently developed SNAP-tag ligand BGAN-Amino (Qiao et al., 2017) exhibits a nearly tenfold improvement in kon (∼1.4×104 M−1 s−1). Although this specific ligand is cell permeable and therefore unsuitable for our assay, these rates approach kon >5×105 M−1 s−1, which we estimate will permit >95% labeling in <1 min with 200 nM ligand. Further improvements in this and other systems will yield an orthogonal partner for HaloTag to allow multicolor experiments.
We used the recycling assay to study the effect of DFO on the recycling kinetics of TfR. DFO inhibits the binding of iron to transferrin and treatment of cells with DFO results in increases in TfR expression (Mattia et al., 1984; Bridges and Cudkowicz, 1984) and transferrin uptake (Richardson and Baker, 1994). The effect of DFO on TfR recycling kinetics was unknown; however, we discovered that the kinetics of TfR significantly increase after DFO treatment. Upregulation of TfR expression and increased return of endocytosed receptor back to the plasma membrane is probably part of the same response mechanism in an attempt to retrieve more iron from the extracellular milieu. Our recycling assay will allow identification of the regulatory factors in this pathway that control this increased recycling rate.
The high precision and sensitivity of our recycling assay revealed different experimental factors that can change the recycling kinetics of TfR. We discovered that the confluency of the culture and the time between cell plating and imaging can have substantial effects on kinetic rates. This strong dependence on experimental conditions makes it particularly important to maintain a consistent protocol. Furthermore, the lack of precise details in earlier publications makes it difficult to compare our results with other reports of recycling rates. Published values for the t1/2 of TfR recycling range from 4 to 35 min depending on cell type (Mayle et al., 2012). For MDCK cells, the reported recycling kinetics are identical to ours (Fig. S5b), with the exception of the endocytosis rate k01. In addition to differences in confluency and plating time, this discrepancy probably stems from the temperature change protocol used in other recycling assays, causing a delayed start in endocytosis and therefore a higher endocytosis rate. Indeed, adding a 10 min cooling step in our assay prior to recording reduced the recycling rate (data not shown). This strong dependency on experimental conditions complicates the comparison of recycling kinetics measured using different methods and experimental conditions.
Cell surface receptors regulate important cellular pathways, many of which are defective in a multitude of diseases. It is estimated that about half of modern drugs in clinical use target cell-surface receptors. Current high-throughput screening assays typically measure downstream cell signaling processes and ignore the recycling kinetics of cell-surface receptors. The simplicity of this system – addition of a single ligand with no washing – makes this assay useful for large-scale screening using automated microscopy and opens new opportunities in drug discovery and pharmacodynamics research. More broadly, basic research on intracellular trafficking kinetics has been confined to relatively few laboratories because of the complex and tedious protocols. The ease-of-use of our assay removes a barrier for investigators in measuring this crucial biological process and pushes the frontiers of biological inquiry.
MATERIALS AND METHODS
Chemical synthesis and spectroscopy of JF635i-HaloTag ligand
General
Methyl 3-fluoroazetidine-3-carboxylate hydrochloride was obtained from Synthonix and HaloTag ligand-amine was obtained from Promega. All other commercial reagents were obtained from Fisher Scientific or Sigma-Aldrich and used as received. Solvents used for purification were HPLC grade. Solvents used for organic synthesis were anhydrous and purchased in septum-sealed bottles stored under an inert atmosphere. Reactions were conducted in round-bottomed flasks or septum-capped crimp-top vials containing Teflon-coated magnetic stir bars. Heating of reactions was accomplished with an aluminium reaction block on top of a stirring hotplate equipped with an electronic contact thermometer to maintain the indicated temperatures. Reactions were monitored by liquid chromatography–mass spectrometry (Phenomenex Kinetex 2.1 mm×30 mm 2.6 μm C18 column; 5–10 μl injection; 5–98% MeCN/H2O, linear gradient, with constant 0.1% v/v HCO2H additive; 6 min run; 0.5 ml min−1 flow; ESI; positive ion mode). Reaction products were purified by flash chromatography on an automated purification system using prepacked silica gel columns or by preparative high performance liquid chromatography (HPLC) (Phenomenex Gemini, NX 30×150 mm 5 μm C18 column). Analytical HPLC analysis was performed with a Phenomenex Gemini, NX 4.6×150 mm 5 μm C18 column under the indicated conditions. High-resolution mass spectrometry was obtained from the High-Resolution Mass Spectrometry Facility at the University of Iowa. NMR spectra were recorded on a 400 MHz spectrometer. Deuterated solvents were used as purchased. 1H and 13C chemical shifts (δ) were referenced to tetramethylsilane or residual solvent peaks, and 19F chemical shifts (δ) were referenced to CFCl3. Data for 1H NMR spectra are reported as follows: chemical shift (δ plasma membrane), multiplicity (s, singlet; d, doublet; t, triplet; dd, doublet of doublets; m, multiplet), coupling constant (Hz), integration. Data for 13C NMR spectra are reported by chemical shift (δ ppm) with hydrogen multiplicity (C, CH, CH2, CH3) information obtained from DEPT spectra.
6-tert-butoxycarbonyl-JF635i methyl esters
A vial was charged with 6-tert-butoxycarbonylsilafluorescein ditriflate (see Fig. 2C for structure; 150 mg, 203 μmol), methyl 3-fluoroazetidine-3-carboxylate hydrochloride (104 mg, 609 μmol, 3 eq), Pd2dba3 (18.6 mg, 20.3 μmol, 0.1 eq), XPhos (29 mg, 61 μmol, 0.3 eq), and Cs2CO3 (318 mg, 975 μmol, 4.8 eq). The vial was sealed and degassed with argon. Dioxane (7 ml) was added and the reaction was stirred at 100°C for 5 h. It was subsequently cooled to room temperature, diluted with MeOH, deposited onto Celite, and concentrated to dryness. Purification by silica gel chromatography (0–50% EtOAc/hexanes, linear gradient) afforded S1 (Fig. 2C; 70 mg, 49%) as an off-white solid. 1H NMR (CDCl3, 400 MHz) δ 8.13 (dd, J=8.0, 1.3 Hz, 1H), 7.97 (dd, J=8.0, 0.7 Hz, 1H), 7.83–7.82 (m, 1H), 6.91 (d, J=8.7 Hz, 2H), 6.71 (d, J=2.6 Hz, 2H), 6.37 (dd, J=8.7, 2.7 Hz, 2H), 4.41–4.34 (m, 4H), 4.22–4.13 (m, 4H), 3.88 (s, 6H), 1.55 (s, 9H), 0.67 (s, 3H), 0.60 (s, 3H); 19F NMR (CDCl3, 376 MHz) δ –161.8 (m); 13C NMR (CDCl3, 101 MHz) δ 170.1 (C), 168.9 (C), 168.6 (C), 164.3 (C), 155.1 (C), 149.1 (C), 137.4 (C), 136.3 (C), 134.0 (C), 130.1 (CH), 128.9 (C), 127.8 (CH), 125.9 (CH), 125.0 (CH), 116.3 (CH), 113.3 (CH), 91.2 (C), 88.3 (d, 1JCF=212 Hz, CF), 82.5 (C), 61.3 (d, 2JCF=25 Hz, CH2), 53.3 (CH3), 28.2 (CH3), 0.2 (CH3), −0.7 (CH3). HRMS (ESI) calculated for C37H39N2O8SiF2 [M+H]+ 705.2444, found 705.2446.
JF635i-HaloTag ligand methyl ester
Ester S1 (37 mg, 52 μmol) was taken up in CH2Cl2 (1.5 ml) and then trifluoroacetic acid (TFA; 0.25 ml) added. The reaction was stirred at room temperature for 7 h. Toluene (3 ml) was added, the reaction mixture was concentrated to dryness and then azeotroped with MeOH three times to provide 6-carboxy-JF635i methyl esters as a dark blue solid (TFA salt). The crude acid was combined with HATU (30 mg, 78 μmol, 1.5 eq) and HaloTag ligand-amine (S2 in Fig. 2C; HCl salt, 30 mg, 78 μmol, 1.5 eq). DMF (3 ml) and then DIEA (46 μl, 264 μmol, 5 eq) were added. The reaction was stirred for 1.5 h at room temperature and concentrated to dryness. Purification by silica gel chromatography (30–100% EtOAc/hexanes, linear gradient) afforded ester S3 (Fig. 2C; 40 mg, 90%, two steps) as an off-white solid. 1H NMR (CD3OD, 400 MHz) δ 8.04–8.00 (m, 2H), 7.67–7.66 (m, 1H), 6.84 (d, J=2.7 Hz, 2H), 6.80 (d, J=8.7 Hz, 2H), 6.44 (dd, J=8.8, 2.7 Hz, 2H), 4.40–4.32 (m 4H), 4.17–4.09 (m, 4H), 3.85 (s, 6H), 3.70–3.57 (m, 4H), 3.55–5.52 (m, 4H), 3.48 (t, J=6.6 Hz, 2H), 3.39 (t, J=6.5 Hz, 2H), 1.76–1.62 (m, 2H), 1.50–1.42 (m, 2H), 1.41–1.22 (m, 4H), 0.67 (s, 3H), 0.56 (s, 3H); 19F NMR (CD3OD, 376 MHz) δ –161.0 (m). Analytical HPLC: >98% purity (10–95% CH3CN/H2O, linear gradient, with constant 0.1% v/v TFA additive; 20 min run; 1 ml min−1 flow; ESI; positive ion mode; UV detection at 254 nm). HRMS (ESI) calculated for C43H51N3O9F2SiCl [M+H]+ 854.3051, found 854.3043.
JF635i-HaloTag ligand
Ester S3 (39 mg, 45 μmol) was dissolved in MeOH (3 ml) and then 1 M NaOH (182 μl, 182 μmol, 4 eq) added. After stirring at room temperature for 15 min, the reaction was acidified with 1 M HCl (210 μl) and directly purified by reverse phase HPLC (40–80% MeCN/H2O, linear gradient, with constant 0.1% v/v TFA additive) to afford JF635i-HaloTag ligand (4 in Fig. 2C; 23 mg, 54%, TFA salt) as a blue solid. 1H NMR (CD3OD, 400 MHz) δ 8.06–8.04 (m, 2H), 7.69 (d, J=1.1 Hz, 1H), 6.87 (d, J=2.7 Hz, 2H), 6.80 (d, J=8.7 Hz, 2H), 6.41 (dd, J=8.8, 2.6 Hz, 2H), 4.45–4.34 (m, 4H), 4.21–4.14 (m, 4H), 3.62–3.50 (m, 8H), 3.47 (t, J=6.6 Hz, 2H), 3.36 (t, J=6.5 Hz, 2H), 1.78–1.61 (m, 2H), 1.47–1.40 (m, 2H), 1.39–1.31 (m, 2H), 1.28–1.18 (m, 2H), 0.67 (s, 3H), 0.57 (s, 3H); 19F NMR (CD3OD, 376 MHz) δ –76.0 (s, 3F), –160.5 (m, 1F). Analytical HPLC: >98% purity (10–95% CH3CN/H2O, linear gradient, with constant 0.1% v/v TFA additive; 20 min run; 1 ml min−1 flow; ESI; positive ion mode; UV detection at 254 nm). HRMS (ESI) calculated for C41H47N3O9F2SiCl [M+H]+ 826.2738, found 826.2733.
UV–visible and fluorescence spectroscopy
All measurements were taken at ambient temperature (23±2°C). Fluorescent molecules were prepared as stock solutions in DMSO and diluted such that the final DMSO concentration did not exceed 1% v/v. Spectroscopy was performed using quartz cuvettes of 1 cm path length. Absorption measurements were recorded on a Cary Model 100 spectrometer (Varian). Fluorescence measurements spectra were recorded on a Cary Eclipse fluorometer (Varian).
The increase in absorption and fluorescence of JF635i-HaloTag ligand upon reaction with HaloTag protein was measured in vitro. HaloTag protein was used as a 100 μM solution in 75 mM NaCl, 50 mM Tris-HCl, pH 7.4. HaloTag ligands were dissolved in DMEM with 10% FBS to a final concentration of 5 μM. An aliquot of HaloTag protein (1.5 equiv) or an equivalent volume of TBS blank was added and the resulting mixture incubated at room temperature until consistent absorbance signal was observed (∼30 min). The solution was diluted by five in DMEM plus 10% FBS for fluorescence spectra and quantum yield measurements.
Determination of kon for JF635i-HaloTag ligand
The binding rate constant kon was determined at 37°C in PBS containing 0.01% CHAPS by stopped-flow analysis on a SX20 spectrometer from Applied Photophysics. JF635i-HaloTag ligand was diluted to 0.2 µM in PBS/CHAPS. Equal volumes of the JF635i-HaloTag ligand solution and HaloTag protein solution (0.2–5 µM in PBS+CHAPS) were mixed. Ligand binding was monitored by recording the fluorescence emission signal by using a 625 nm LED and a 660 nm emission filter. The resulting fluorescence traces were fitted by a single exponential curve to obtain the observed rate constant (kobs) for each different ligand concentration. kon was determined as the slope of the line fitting kobs as a function of the concentration of HaloTag protein. Each measurement was performed in triplicate.
Evaluation of the membrane impermeance of JF635i-HaloTag ligand
General cell culture
The U2OS (ATCC, HTB-96) stable cell line was established by integrating a histone H2B–HaloTag-expressing plasmid via the piggyback transposase, under the selection of 500 μg ml−1 Geneticin (Life Technologies). This “H2B–Halo” cell line undergoes regular mycoplasma testing by the Janelia Cell Culture Facility. U2OS cells (ATCC) for transient transfection and U2OS cells stably expressing a histone H2B-HaloTag protein fusion were cultured in Dulbecco's modified Eagle medium (DMEM, Phenol Red-free; Corning) supplemented with 10% (v/v) fetal bovine serum (Gibco), 2 mM GlutaMAX (Gibco), 1× Pen/Strep solution (10,000 μg ml−1 penicillin, 10,000 µg ml−1 streptomycin; Gibco) and maintained at 37°C in a humidified 5% (v/v) CO2 environment. Cells were authenticated and regularly tested for contamination.
Transient transfection
For extracellular expression of HaloTag, U2OS cells were nucelofected (Lonza) with a variant of the pDisplay vector (Invitrogen) lacking the hemagglutinin tag, termed pCMV(MinDis), containing an IgG kappa secretion signal, HaloTag, the C-terminal transmembrane anchoring domain from platelet-derived growth factor receptor (PDGFR), the self-cleaving P2A peptide and superfolder GFP (sfGFP).
Fluorescence microscopy
At 18–24 h post transfection or plating, U2OS cells expressing nuclear H2B-Halo or extracellular Halo-PDGFR were incubated with either JF635 or JF635i-HaloTag ligand at 37°C for different incubation times (30 min or 3 h) and at different concentrations (200 nM or 1 μM). Cells were imaged live with no wash step on a confocal microscope in the Janelia Imaging Facility (Zeiss LSM800, Plan-Apochromat 20×/0.8 M27) using 640 nm excitation/650–700 nm emission. The images are displayed as a maximum intensity projection of a confocal image stack using Fiji.
Receptor recycling assay
Stable MDCK cell line generation
To generate the MDCK-TfR-HaloTag gene-edited cell line we used the ChopChop tool (Labun et al., 2016) to select a guide RNA close to the STOP codon of the dog TfR gene (gRNA underlined: tgccctctctggtgacatatgg). The homology directed repair (HDR) template was cloned into pUC19 with the insert being flanked on both ends by the same TfR selected gRNA (Nakade et al., 2014). The insert consisted of HaloTag (HT7; Encell et al., 2013) fused to the last codon of TfR via an eight amino acid linker, GPDLVPRG, flanked by homology arms (about 375 bp each) positioned such that there are no extended overhangs on the chromosomal insertion site (Paquet et al., 2016). Furthermore, the gRNA site in the HDR template was minimally mutated with conservative mutations to prevent cleavage of the template by the Cas9/gRNA genome cleaving mechanism (TGCCCTCTCTGGTGACATATGG mutated to TGCCCTCTCTGGTGATATCTGG, where the codons are underlined, and the protospacer-adjacent motif, PAM, is italic, silent mutations to remove the PAM sequence after insertion are indicated in bold).
The gRNA cloned into the pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene 62988) (Ran et al., 2013) and the pUC-based HDR template were co-transfected (5 µg and 10 µg, respectively) into 4×106 wild-type MDCK-II cells using the AMAXA 2B Nucleofector electroporation system (Lonza) and selected for Cas9/gRNA vector expression using 1 µg ml−1 puromycin (Invitrogen). Surviving cells were selected for HaloTag expression using flow cytometry using JF635-HaloTag ligand detection following a 45 min labeling followed by a brief-as-possible trypsinization to reduce recycling HaloTag/JF635 ligand cleavage. To obtain a homozygous population of TfR-HaloTag chromosomal incorporation after expansion of the sorted cells, they were again labeled with the cell-permeable JF635-HaloTag and sorted based on the intensity of their emission into three fluorescence intensity groups: low, mid and high (Fig. S4a). Western blot analysis using a TfR antibody showed a band corresponding to TfR-HaloTag in the mid and high fluorescence groups, whereas the low fluorescence group only had a band corresponding to wild-type TfR (Fig. S2b,c). This showed that incorporation of the tag was successful and that the cells could readily be labeled using HaloTag-compatible JF dyes. The high fluorescence group was further expanded, cryopreserved and used in all consecutive experiments.
Cell culture
Wild-type MDCK-II cells (a gift from Joseph Leighton, Department of Pathology, Medical College of Pennsylvania, Philadelphia, USA) and TfR-HaloTag MDCK cells were grown at 37°C and 5% CO2 in DMEM (Corning) supplemented with 10% (v/v) fetal bovine serum (Invitrogen), 100 U ml−1 penicillin and 100 U ml−1 streptomycin (Invitrogen) and maintained at 37°C in a humidified 5% (v/v) CO2 environment. Cells were authenticated and regularly tested for contamination.
Antibodies and reagents
For detection of TfR-HaloTag using western blotting, mouse anti-TfR antibody (Fitzgerald, 10R-CD71aHU), mouse anti-tubulin antibody (Sigma, T9026), secondary goat anti-rabbit Alexa Fluor 680 (Invitrogen) and secondary goat anti-mouse IRdye-800 (Li-Cor) were used. For colocalization experiments of TfR-HaloTag, cells were incubated with transferrin-CF594 (Biotium) and dextran–Alexa Fluor 488 (MW 10,000; Invitrogen).
Recycling assay setup
TfR-HaloTag MDCK cells were grown in six-chamber µ-slides VI (IBIDI, 80606) to subconfluent density. The assay was performed ∼48 h after plating the cells. Immediately before the assay, the chamber was flushed with image medium and a syringe with 0.5 ml image medium containing 200 nM JF635i-HaloTag ligand was connected to the chamber. Four regions of cells were selected, and the assay run with time points every 10 s for 45 min. After the first time point, the medium in the chamber was replaced with image medium containing JF635i-HaloTag ligand. To correct for fluorophore bleaching of the ligand and the cellular autofluorescence background during the recycling assay, fully labeled cells were imaged with the same settings as used for the recycling assay and the signal decrease over background was calculated and added back to the recycling curves.
Biosynthetic delivery assay
A 40/60 mix of wild-type to TfR-HaloTag MDCK cells were grown in 24-well glass-bottom dishes (Cellvis, P24-1.5H-N) to confluent density. Wild-type cells were used for background correction during image analysis. Cells were incubated for 6 h with permeable JF549-HaloTag ligand to label all TfR-HaloTag in the cells. Next, the cells were washed three times with prewarmed HBSS and incubated with image medium containing 10 nM JF635i-HaloTag ligand. The cells from five regions per well were then imaged for 16 h with a time point every 10 min. The cycloheximide-treated samples were incubated with 10 µg ml−1 cycloheximide (Sigma) for 1 h before the assay and during the assay.
Protein turnover assay
A 40/60 mix of wild-type to TfR-HaloTag MDCK cells were grown in 24-well glass-bottom dishes (Cellvis, P24-1.5H-N) to confluent density. Wild-type cells were used for background correction during image analysis. Cells were incubated for 6 h with permeable JF635i-HaloTag ligand to label all TfR-HaloTag in the cells. Next, the cells were washed three times with pre-warmed HBSS and incubated with image medium containing 10 nM permeable JF549-HaloTag ligand. The cells from five regions per well were then imaged for 60 h with a time point every 30 min. The chloroquine-treated samples were incubated with 25 µM chloroquine (Sigma) 4 h before assay and during assay in image medium.
Fluorescence microscopy
Live cell imaging of MDCK-TfR-HaloTag cells was performed in image medium consisting of Phenol Red-free HBSS with calcium and magnesium (Corning), 10% FBS (Invitrogen) and 25 ng ml−1 Trolox (Sigma). Acquisition of JF635i-HaloTag ligand was performed using a Zeiss AxioObserverZ.1 widefield system using the Colibri2 625 nm LED at 50% LED power and 100 ms excitation time. The system was equipped with a CO2, humidity and temperature incubation system (PeCon). The microscope setup included a Plan-Apochromat 20×/0.8 M27 objective and Hamamatsu Flash4.0 V2 sCMOS camera for acquisition of the fluorescence signal of the assays. A Zeiss Cell Observer SD confocal with a Yokagawa CSU-X1 spinning disk and Plan-Apochromat 63×/1.4 M27 objective paired with a 1.2× adapter to a Photometrics Evolve 512 EMCCD camera were used for image acquisition of fix samples used for colocalization and compartment quantification.
Image analysis
For the recycling assay, images were analyzed with a custom plugin on FIJI v1.52e (Schindelin et al., 2012). The total movie was converted into a single average image using Z-projection (average intensity). In this image, the area occupied by the cells during the course of the movie was selected and slightly increased (dilated). The inverse of this selection was used as the background area. Then, the total intensity and the background area were measured for each frame of the movie. The mean of the background intensity was then subtracted from the mean of the total intensity to get a measurement of the background-corrected signal for each frame.
For the turnover and biosynthetic delivery assay, because of the duration of the movie, the cells move too much for the same approach as the recycling assay. We used a different custom plugin on FIJI ImageJ. For each frame of the movie, the total intensity was measured and then a threshold was selected using the percentile method to determine the signal of the image. The inverse of this selection (the regions in the image not occupied with the signal) was used as the background area and measured. The mean of the background intensity was then subtracted from the total image intensity to obtain a measurement of the background-corrected signal for each frame.
Colocalization analysis was performed using the JACOP plugin (Bolte and Cordelières, 2006) for FIJI and quantified using the Manders' overlap coefficient.
Flow cytometry
To select clones of cells positive for TfR-HaloTag, we incubated the cells with permeable JF635 for 2 h. Then, the cells were trypsinized and washed in HBSS containing 10% fetal bovine serum and 2 mM EDTA to inhibit trypsin and washed again in the same buffer. The signal from JF635 was measured using the 640 nm laser on an Aria II FACS sorter (BD) and sorted based on the gates as shown in Fig. S4a.
Western blotting
Cells were lysed in RIPA lysis buffer for 20 min at 4°C and clarified by centrifugation for 10 min at 16,000 g at 4°C. Protein samples were loaded in 4–12% pre-cast gradient polyacrylamide gels, run for 90 min at 100 mV and transferred to nitrocellulose membranes using iBlot transfer stacks (Invitrogen). Uncropped blots are shown in Fig. S4c.
Statistical analysis
Data collected for the recycling assay (n=6), the protein turnover assay (n=6) and the biosynthetic pathway assay (n=6) were analyzed in R version 3.5.1 (https://www.r-project.org/). The recycling and protein turnover assay data were negative natural log-transformed to achieve a positive linear relationship between time and the normalized intensity for the region of interest. Non-independent, within sample replicates (separate movies from one sample) were averaged prior to data analysis. All independent replicates (different samples) were utilized when fitting the linear models. To identify the linear region of the curve in an unbiased manner, sliding window linear regression was conducted using the rollapply function within the R package ZOO (Zeileis and Grothendieck, 2005). The resulting slope coefficients were categorized into linear and nonlinear groups using k-means clustering. Analysis of covariance (ANCOVA) tests were performed to compare regression slopes and intercepts between experimental groups; Tukey's HSD multiple comparison test was conducted at a significance level of P<0.05.
Kinetic model calculations
We consider a membrane receptor within the plasma membrane, which can cycle through the sorting endosome (SE) to the recycling endosome (RE), and back to the membrane. We denote these three compartments with subscripts, i=0.3, with xi being the absolute quantity of receptor in each. Although the receptors circulate, the amount in each compartment is constant over time. Rate constants kij provide the rates of receptor transfer from compartment i to j so that, with the assumption that receptor is neither created nor destroyed during the experiments, there are two independent mass conservation relations for SE and RE:
| (1.1) |
| (1.2) |
For the experiments under consideration, fluorescing label is added to the external bath so that the plasma membrane resident carrier is fully labeled at all times. Over time, this label is transferred to carriers within the SE and RE, so that compartment label increases. We denote by ηi(t) the ratio of labeled carrier to total carrier (xi) within compartment i. Then, by the assumptions of this experiment:
| (1.3) |
| (1.4) |
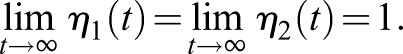 |
(1.5) |
If we identify the fluorescence of each compartment with its component of labeled receptor ηi(t)xi, then the total fluorescence of the cell F(t) can be written as:
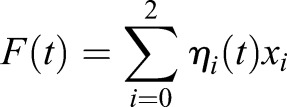 |
(1.6) |
and the initial fluorescence is plasma membrane carrier, and the final fluorescence is total carrier:
| (1.7) |
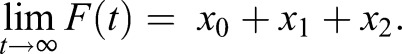 |
(1.8) |
Mass conservation of label provides two equations for fluorescent fraction in x1 and x2:
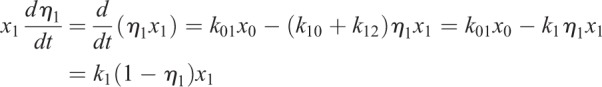 |
(1.9) |
| (1.10) |
The last equalities of each line come from substituting the kinetic relations for total carrier and allow elimination of the absolute carrier content from each equation:
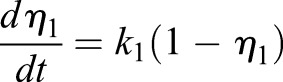 |
(1.11) |
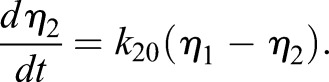 |
(1.12) |
For the specified initial conditions, these equations have the following solutions:
| (1.13) |
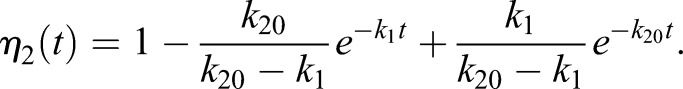 |
(1.14) |
Preliminaries for data analysis:
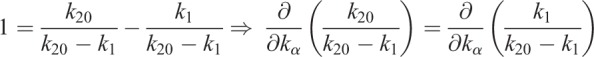 |
(1.15) |
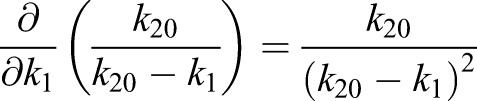 |
(1.16) |
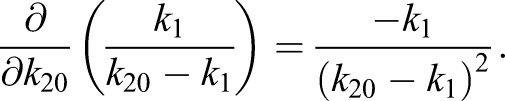 |
(1.17) |
Thus, the derivatives of η1 with respect to the model parameters kα are
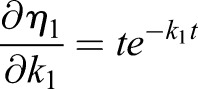 |
(1.18) |
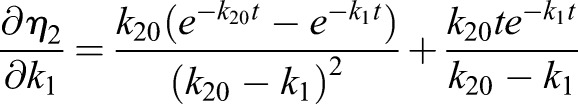 |
(1.19) |
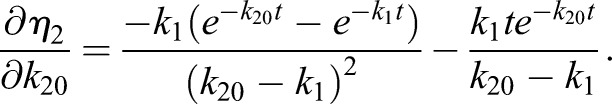 |
(1.20) |
Separate experiments provide values for the ratio x1/x2.
Total fluorescence data F(t) are used to determine the kinetic parameters k1 and k20, and the cell membrane transport pool x0. What is reported is
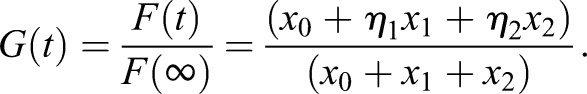 |
(1.21) |
Setting yi=xi/(x0+x1+x2) and α=x1/x2,
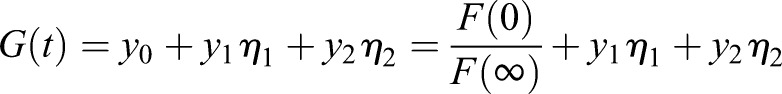 |
(1.22) |
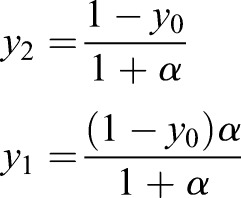 |
(1.23) |
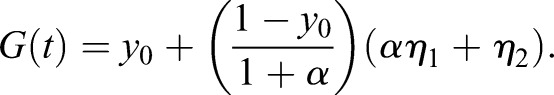 |
(1.24) |
The error E of the model is given by
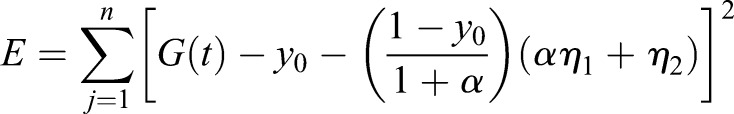 |
(1.25) |
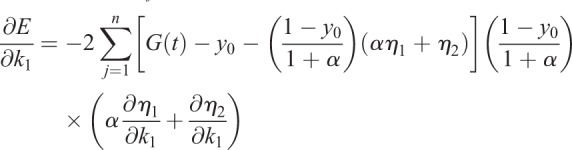 |
(1.26) |
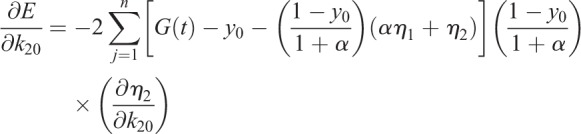 |
(1.27) |
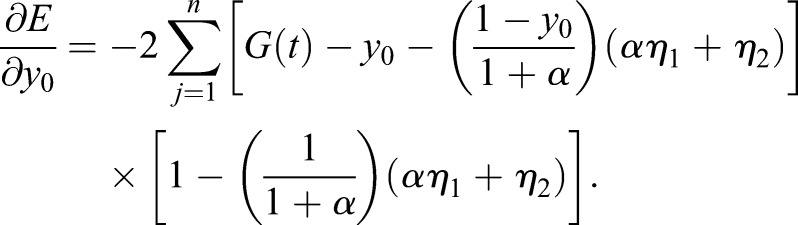 |
(1.28) |
Finally, the optimal k1 and k20 and y0 are determined as solutions to the equations
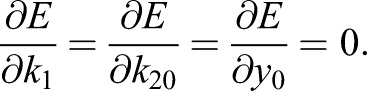 |
(1.29) |
With k1 and k20 from the data fit, and y1 and y2 specified, the remaining kinetic parameters are
| (1.30) |
| (1.31) |
| (1.32) |
Calculating receptor ligand binding rates for SNAP-tag ligand and HaloTag ligand
Reaction of a receptor R and a ligand L to a receptor–ligand complex RL can be described as
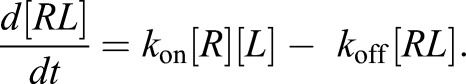 |
(2.1) |
The total number of receptors bound and unbound is constant, neglecting endocytosis. The sum of the bound receptors [RL] and unbound receptors [R] is constant at the total number of receptors [RT]:
| (2.2) |
Considering the large extracellular bath in these experiments, ligand concentration is also constant at
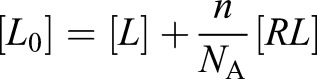 |
(2.3) |
where n is the number of cells per unit volume and NA is Avogadro's number.
Neglecting endocytosis and receptor synthesis, and substituting [R] and [L] from Eqn 2.1 we obtain
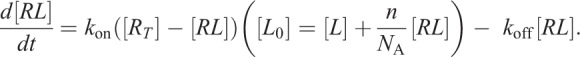 |
(2.4) |
Because the ligand concentration remains constant under the experimental conditions, [L]=[L0] and the equation can be simplified to
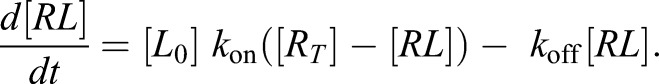 |
(2.5) |
This solves to
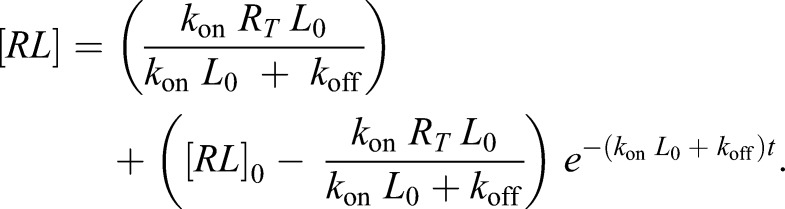 |
(2.6) |
Assuming koff=0 and [RL0]=0, this simplifies to
| (2.7) |
Supplementary Material
Acknowledgements
We thank Diego Gravotta, Paulo Caceres Puzzella and Christin Hanke-Gogokhia for fruitful discussions and their valuable input. We also acknowledge the Visual Function Core at Weill Cornell Medicine and the Light Microscopy and Cell Culture Shared Resources at Janelia for their assistance with cell culture and microscopy.
Footnotes
Competing interests
The authors declare competing interests. L.D.L. has filed patent and patent applications (e.g. US Patent 9,933,417) covering azetidine-containing fluorophores such as JF635 whose value may be affected by this publication.
Author contributions
Conceptualization: C.T.H.J., C.D., A.M.W., L.D.L., R.S.; Methodology: C.T.H.J., C.D., P.J.Z., A.N.T., A.M.W., L.D.L., R.S.; Software: C.T.H.J., P.J.Z., A.M.W.; Validation: C.T.H.J., C.D., P.J.Z., L.D.L., R.S.; Formal analysis: C.T.H.J., C.D., P.J.Z., A.N.T., A.M.W., R.S.; Investigation: C.T.H.J., C.D., P.J.Z., A.N.T., A.M.W., L.D.L., R.S.; Resources: C.T.H.J., C.D., E.R.-B., L.D.L., R.S.; Data curation: C.T.H.J., C.D., P.J.Z., A.N.T., L.D.L., R.S.; Writing - original draft: C.T.H.J., C.D., A.M.W., L.D.L.; Writing - review & editing: C.T.H.J., C.D., A.M.W., E.R., L.D.L., R.S.; Visualization: C.T.H.J., C.D., P.J.Z., R.S.; Supervision: E.R.-B., L.D.L., R.S.; Project administration: E.R.-B., L.D.L., R.S.; Funding acquisition: A.M.W., E.R.-B., L.D.L.
Funding
This work was supported by the National Institutes of Health (NIH) [R01GM025662 to C.T.H.J., P.J.Z., R.S. and E.R.-B.] and the Howard Hughes Medical Institute [C.D. and L.D.L.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.231225.supplemental
References
- Baravalle G., Schober D., Huber M., Bayer N., Murphy R. F. and Fuchs R. (2005). Transferrin recycling and dextran transport to lysosomes is differentially affected by bafilomycin, nocodazole, and low temperature. Cell Tissue Res. 320, 99-113. 10.1007/s00441-004-1060-x [DOI] [PubMed] [Google Scholar]
- Bleil J. D. and Bretscher M. S. (1982). Transferrin receptor and its recycling in HeLa cells. EMBO J. 1, 351-355. 10.1002/j.1460-2075.1982.tb01173.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S. and Cordelières F. P. (2006). A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213-232. 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- Bomsel M., Prydz K., Parton R. G., Gruenberg J. and Simons K. (1989). Endocytosis in filter-grown Madin-Darby canine kidney cells. J. Cell Biol. 109, 3243-3258. 10.1083/jcb.109.6.3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges K. R. and Cudkowicz A. (1984). Effect of iron chelators on the transferrin receptor in K562 cells. J. Biol. Chem. 259, 12970-12977. [PubMed] [Google Scholar]
- Chen Y., Gershlick D. C., Park S. Y. and Bonifacino J. S. (2017). Segregation in the Golgi complex precedes export of endolysosomal proteins in distinct transport carriers. J. Cell Biol. 216, 4141-4151. 10.1083/jcb.201707172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A., Schwartz A. L., Dautry-Varsat A. and Lodish H. F. (1983). Kinetics of internalization and recycling of transferrin and the transferrin receptor in a human hepatoma cell line. Effect of lysosomotropic agents . J. Biol. Chem. 258, 9681-9689. [PubMed] [Google Scholar]
- Correia J. J. and Williams R. C. (1983). Mechanisms of Assembly and Disassembly of Microtubules. Annu. Rev. Biophys. Bioeng. 12, 211-235. 10.1146/annurev.bb.12.060183.001235 [DOI] [PubMed] [Google Scholar]
- Encell L. P., Ohana R. F., Zimmerman K., Otto P., Vidugiris G., Wood M. G., Los G. V., McDougall M. G., Zimprich C. et al. (2013). Development of a dehalogenase-based protein fusion tag capable of rapid, selective and covalent attachment to customizable ligands. Curr. Chem. Genomics 6, 55-71. 10.2174/1875397301206010055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futter C. E., Connolly C. N., Cutler D. F. and Hopkins C. R. (1995). Newly synthesized transferrin receptors can be detected in the endosome before they appear on the cell surface. J. Biol. Chem. 270, 10999-11003. 10.1074/jbc.270.18.10999 [DOI] [PubMed] [Google Scholar]
- Grimm J. B., English B. P., Chen J., Slaughter J. P., Zhang Z., Revyakin A., Patel R., Macklin J. J., Normanno D., Singer R. H. et al. (2015). A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods 12, 244-250. 10.1038/nmeth.3256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B., Muthusamy A. K., Liang Y., Brown T. A., Lemon W. C., Patel R., Lu R., Macklin J. J., Keller P. J., Ji N. et al. (2017). A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 14, 987-994. 10.1038/nmeth.4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao M. and Maxfield F. R. (2000). Characterization of rapid membrane internalization and recycling. J. Biol. Chem. 275, 15279-15286. 10.1074/jbc.275.20.15279 [DOI] [PubMed] [Google Scholar]
- Hopkins C. R. and Trowbridge I. S. (1983). Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J. Cell Biol. 97, 508-521. 10.1083/jcb.97.2.508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu T., Johnsson K., Okuno H., Bito H., Inoue T., Nagano T. and Urano Y. (2011). Real-time measurements of protein dynamics using fluorescence activation-coupled protein labeling method. J. Am. Chem. Soc. 133, 6745-6751. 10.1021/ja200225m [DOI] [PubMed] [Google Scholar]
- Labun K., Montague T. G., Gagnon J. A., Thyme S. B. and Valen E. (2016). CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 44, W272-W276. 10.1093/nar/gkw398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng S., Qiao Q., Miao L., Deng W., Cui J. and Xu Z. (2017). A wash-free SNAP-tag fluorogenic probe based on the additive effects of quencher release and environmental sensitivity. Chem. Commun. 53, 6448-6451. 10.1039/C7CC01483J [DOI] [PubMed] [Google Scholar]
- Los G. V., Encell L. P., Mcdougall M. G., Hartzell D. D., Karassina N., Zimprich C., Wood M. G., Learish R., Ohana R. F., Urh M. et al. (2008). HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3, 373-382. 10.1021/cb800025k [DOI] [PubMed] [Google Scholar]
- Lukinavičius G., Umezawa K., Olivier N., Honigmann A., Yang G., Plass T., Mueller V., Reymond L., Corrêa I. R. Jr, Luo Z.-G. et al. (2013). A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat. Chem. 5, 132-139. 10.1038/nchem.1546 [DOI] [PubMed] [Google Scholar]
- Mattia E., Rao K., Shapiro D. S., Sussman H. H. and Klausner R. D. (1984). Biosynthetic regulation of the human transferrin receptor by desferrioxamine in K562 cells. J. Biol. Chem. 259, 2689-2692. [PubMed] [Google Scholar]
- Maxfield F. R. and Mcgraw T. E. (2004). Endocytic recycling. Nat. Rev. Mol. Cell Biol. 5, 121-132. 10.1038/nrm1315 [DOI] [PubMed] [Google Scholar]
- Mayle K. M., Le A. M. and Kamei D. T. (2012). The intracellular trafficking pathway of transferrin. Biochim. Biophys. Acta Gen. Subjects 1820, 264-281. 10.1016/j.bbagen.2011.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor S., Presley J. F. and Maxfield F. R. (1993). Sorting of membrane components from endosomes and subsequent recycling to the cell surface occurs by a bulk flow process. J. Cell Biol. 121, 1257-1269. 10.1083/jcb.121.6.1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakade S., Tsubota T., Sakane Y., Kume S., Sakamoto N., Obara M., Daimon T., Sezutsu H., Yamamoto T., Sakuma T. et al. (2014). Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat. Commun. 5, 5560 10.1038/ncomms6560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet D., Kwart D., Chen A., Sproul A., Jacob S., Teo S., Olsen K. M., Gregg A., Noggle S. and Tessier-Lavigne M. (2016). Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533, 125-129. 10.1038/nature17664 [DOI] [PubMed] [Google Scholar]
- Perez Bay A. E., Schreiner R., Benedicto I., Paz Marzolo M., Banfelder J., Weinstein A. M. and Rodriguez-Boulan E. J. (2016). The fast-recycling receptor Megalin defines the apical recycling pathway of epithelial cells. Nat. Commun. 7, 11550 10.1038/ncomms11550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punnonen E.-L., Ryhänen K. and Marjomi V. S. (1998). At reduced temperature, endocytic membrane traffic is blocked in multivesicular carrier endosomes in rat cardiac myocytes. Eur. J. Cell Biol. 75, 344-352. 10.1016/S0171-9335(98)80067-8 [DOI] [PubMed] [Google Scholar]
- Qiao Q., Liu W., Chen J., Zhou W., Yin W., Miao L., Cui J. and Xu Z. (2017). A naphthalimide-derived fluorogenic probe for SNAP-tag with a fast record labeling rate. Dye. Pigment. 147, 327-333. 10.1016/j.dyepig.2017.08.032 [DOI] [Google Scholar]
- Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A. and Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281-2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson D. R. and Baker E. (1994). Two saturable mechanisms of iron uptake from transferrin in human melanoma cells: The effect of transferrin concentration, chelators, and metabolic probes on transferrin and iron uptake. J. Cell. Physiol. 161, 160-168. 10.1002/jcp.1041610119 [DOI] [PubMed] [Google Scholar]
- Rutledge E. A., Mikoryak C. A. and Draper R. K. (1991). Turnover of the transferrin receptor is not influenced by removing most of the extracellular domain. J. Biol. Chem. 266, 21125-21130. [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676-682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeileis A. and Grothendieck G. (2005). Zoo: S3 infrastructure for regular and irregular time series. J. Stat. Softw. 14, 1-27. 10.18637/jss.v014.i06 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.