

Abstract
T cells are critical for protective immune responses to pathogens and tumors. The T-cell receptor (TCR)–CD3 complex is composed of a diverse αβ TCR heterodimer noncovalently associated with the invariant CD3 dimers CD3ϵγ, CD3ϵδ, and CD3ζζ. The TCR mediates recognition of antigenic peptides bound to MHC molecules (pMHC), whereas the CD3 molecules transduce activation signals to the T cell. Whereas much is known about downstream T-cell signaling pathways, the mechanism whereby TCR engagement by pMHC is first communicated to the CD3 signaling apparatus, a process termed early T-cell activation, remains largely a mystery. In this review, we examine the molecular basis for TCR activation in light of the recently determined cryoEM structure of a complete TCR–CD3 complex. This structure provides an unprecedented opportunity to assess various signaling models that have been proposed for the TCR. We review evidence from single-molecule and structural studies for force-induced conformational changes in the TCR–CD3 complex, for dynamically-driven TCR allostery, and for pMHC-induced structural changes in the transmembrane and cytoplasmic regions of CD3 subunits. We identify major knowledge gaps that must be filled in order to arrive at a comprehensive model of TCR activation that explains, at the molecular level, how pMHC-specific information is transmitted across the T-cell membrane to initiate intracellular signaling. An in-depth understanding of this process will accelerate the rational design of immunotherapeutic agents targeting the TCR–CD3 complex.
Keywords: T-cell receptor (TCR), major histocompatibility complex (MHC), signal transduction, cryo-electron microscopy, nuclear magnetic resonance (NMR), molecular dynamics, mechanotransduction, allosteric regulation
Introduction
The ability of cells to respond to their microenvironment is fundamental to most biological processes, including development, differentiation, motility, and immunity. These processes are mediated by multiprotein signaling complexes through which communication may take place over long distances, from the extracellular space through the cytoplasm to the nucleus. A particularly important type of multiprotein signaling complex involves cell-surface receptors that transmit signals upon binding protein ligands on the surface of other cells, thereby enabling direct cell–cell communication. However, the multisubunit composition of these membrane-embedded receptors makes them especially challenging subjects for structural studies.
A premier example of such a multiprotein cell-surface receptor is the T-cell receptor (TCR)3–CD3 complex. This cardinal receptor of the mammalian immune system is expressed on T cells and is essential for protective responses to microbes and cancers. Because of its biomedical relevance, the TCR is one of the most extensively-studied receptors in modern biology. The TCR–CD3 complex is composed of a genetically-diverse αβ (or γδ) TCR heterodimer in noncovalent association with invariant CD3 dimers: CD3ϵγ, CD3ϵδ, and CD3ζζ (1, 2). The TCR mediates recognition of peptide fragments bound to major histocompatibility complex (MHC) molecules on antigen-presenting cells (APCs). These peptides are generated by proteolytic degradation of foreign or self-proteins within cells expressing MHC class I or class II molecules (3). The exquisite sensitivity and specificity of the TCR allow it to recognize a few copies of a single peptide in an ocean of as many as 100,000 other MHC-bound peptides displayed on an APC (4–6).
The α and β chains of the TCR heterodimer each contain an immunoglobulin (Ig)-like extracellular variable (V) and constant (C) domain, a membrane-proximal connecting peptide (CP), a single transmembrane (TM) region, and a short cytoplasmic tail. Similarly, each subunit of the CD3ϵγ and CD3ϵδ heterodimers comprises a single extracellular Ig-like domain, a CP, a TM region, and a long cytoplasmic tail. By contrast, CD3ζ has a short (nine residues) extracellular segment attached to CP, TM, and cytoplasmic regions. The TCR–CD3 complex exists on the T-cell surface in a 1:1:1:1 stoichiometry for the TCRαβ/CD3ϵγ/CD3ϵδ/CD3ζζ dimers (1, 2).
TCRs engage peptide-MHC (pMHC) ligands via their six complementarity-determining region (CDR) loops, three from the Vα domain and three from the Vβ domain (7). The first and second CDRs (CDR1 and CDR2) are encoded within the TCR V segments, whereas CDR3 is formed by DNA recombination involving juxtaposition of Vα and Jα segments for α chain genes and Vβ, D, and Jβ segments for β chain genes. Following TCR binding to pMHC, the CD3 molecules transmit activation signals to the T cell. The TCR α and β chains do not contain intracellular signaling motifs, thus separating pMHC recognition from T-cell activation. By contrast, the CD3 ϵ, γ, δ, and ζ chains each contain one (ϵ, γ, and δ) or three (ζ) immunoreceptor tyrosine-based activation motifs (ITAMs) that undergo phosphorylation by the Src kinase Lck, thereby initiating a downstream T-cell signaling cascade (8).
Despite intensive efforts by many groups over the past 35 years, the molecular mechanism by which signals are transduced from the extracellular domains of the TCR through the T-cell membrane, resulting in phosphorylation of CD3 ITAMs in the cytoplasm, remains poorly understood. A variety of models have been proposed to explain T-cell activation (9–11). However, all these models invoke one or more of four basic mechanisms: aggregation, segregation, mechanosensing, and/or conformational change. Aggregation of TCR–CD3 complexes following TCR ligation could result in enhanced phosphorylation of CD3 ITAMs by increasing the proximity of associated Lck molecules (Fig. 1A) (9). Binding-induced segregation of the inhibitory phosphatase CD45 from the vicinity of TCR–CD3 complexes has also been proposed to explain TCR triggering (Fig. 1B) (12, 13). According to the mechanosensor model, the TCR converts mechanical energy derived from T-cell scanning of APCs into biochemical signals upon engaging pMHC (Fig. 1C) (14, 15). Other models invoke ligand-induced conformational or dynamic changes at TCR sites distant from the pMHC-binding site (allostery) as a mechanism for TCR activation (Fig. 1D) (16, 17).
Figure 1.

Mechanisms of TCR activation. A, in the aggregation model, pMHC binding induces oligomerization of TCR–CD3 complexes. This clustering could increase the proximity of associated Lck molecules, resulting in activation of receptors in the aggregate by trans-autophosphorylation. B, segregation model proposes that TCR binding to pMHC induces zones of close contact at the T-cell–APC interface from which molecules with large ectodomains, such as the inhibitory tyrosine phosphatase CD45, are excluded. Segregation of CD45 favors phosphorylation of CD3 ITAMs by Lck. C, in the mechanosensing model, sliding of the T cell and APC membranes over each other during immune surveillance generates a mechanical force tangential to the T-cell surface that leads to dissociation of CD3 ITAMs from the T-cell membrane, thereby exposing them to phosphorylation by Lck. D, allosteric model postulates that pMHC binding to TCR induces long-range changes in TCR dynamics and/or conformation (represented by color changes in the TCR–CD3 complex) that are transmitted to the cytoplasmic tails of CD3 to expose ITAMs for phosphorylation. This transmission is mediated by allosteric sites in the TCR Cα and Cβ domains. E, in a unified model of TCR activation that combines mechanosensing (C) and allostery (D), mechanical force induces allosteric changes in TCR dynamics and/or conformation that propagate to CD3. Force amplifies allosteric communication between TCR and CD3 following pMHC ligation.
Several excellent reviews of TCR-triggering mechanisms have been published recently (18–22). However, all these reviews were written before the three-dimensional structure of the fully-assembled TCR–CD3 complex became available (23). Accordingly, in this review, we will examine TCR triggering within the context of the new TCR–CD3 structure. Our focus will be on early T-cell activation, which is the process by which TCR ligation by pMHC is first communicated to the CD3 signaling apparatus, thereby enabling phosphorylation of CD3 ITAMs. We first highlight key features of the TCR–CD3 structure of particular relevance to potential TCR-triggering mechanisms. We next review evidence from single-molecule force measurements and NMR spectroscopy that pMHC binding induces allosteric changes in the ectodomains (ECDs) of the TCR–CD3 complex. We then direct our attention inward from the ECDs to the TM and cytoplasmic regions, and to the vexing problem of how pMHC-specific information is transmitted across the T-cell membrane to initiate intracellular signaling. As will become apparent, large gaps remain in our understanding of the structural basis of TCR activation, despite much recent progress.
Structure of the TCR–CD3 complex
The complexity of the octameric membrane-embedded TCRαβ/CD3ϵγ/CD3ϵδ/CD3ζζ receptor has made it an exceptionally difficult target for structural studies. X-ray crystallography and NMR have provided three-dimensional structures of numerous TCR ECDs and TCR–pMHC complexes (7), of CD3ϵγ and CD3ϵδ ECDs (24–27), and of CD3ϵ, CD3ζ, and TCRα TM segments (28–30). However, studies of isolated components of the TCR–CD3 complex have not yielded definitive information on its overall spatial organization or the precise interaction between TCR and CD3 subunits, which is essential for understanding how TCR engagement is coupled to T-cell activation.
In a major breakthrough in molecular immunology, Dong et al. (23) used single-particle cryoEM to determine the structure of a complete human TCR–CD3 complex to 3.7 Å resolution (Fig. 2A). Remarkably, this structure, whose overall shape was likened to an ice cream cone, includes full-length TCR α and β chains, and complete ECDs and TM helices of the CD3ϵγ, CD3ϵδ, and CD3ζζ dimers. Of note, the cytoplasmic tails of CD3ϵγ, CD3ϵδ, and CD3ζζ, which contain ITAMs that undergo phosphorylation following pMHC engagement, are poorly visible in the electron density, implying that these intracellular regions, unlike the rest of the TCR–CD3 complex, are highly mobile (23). The TCR Cα/Cβ domains and the ECDs of CD3ϵγ and CD3ϵδ constitute the middle section of the TCR–CD3 complex, with the Vα/Vβ domains projecting from the middle section and positioned furthest from the T-cell membrane (Fig. 2A).
Figure 2.

Structure of the TCR–CD3 complex. A, side views of the overall structure of the TCR–CD3 complex shown in ribbon representation (PDB code 6JXR) (23). B, top view of the TM segments of the TCR–CD3 complex. Side chains of acidic and basic residues forming ionic interactions within the membrane are shown. C, top view of the ECDs of the TCR–CD3 complex.
The eight TM helices of the TCR–CD3 complex are arranged in a parallel orientation (Fig. 2B). They form a compact bundle-like structure with the two TM helices of the TCRαβ heterodimer at its core. Whereas the TM helices of TCRαβ establish extensive, mainly hydrophobic, interactions with each other and with the surrounding six TM helices of CD3ϵγ, CD3ϵδ, and CD3ζζ, there are no contacts between the TM helices of CD3ϵγ and CD3ϵδ. Instead, the TM helices of CD3ζζ form a coiled coil that interacts simultaneously with the TM helices of CD3ϵγ and CD3ϵδ (23). In addition to hydrophobic residues, basic residues in the TM segments of TCRαβ form ionic interactions with acidic residues in the TM segments of CD3ϵγ, CD3ϵδ, and CD3ζζ that further stabilize the 8-helix bundle structure (Fig. 2B).
Assembly of the ECDs of the TCR–CD3 complex is mediated by the Cα/Cβ domains and CPs of TCRαβ that pack against CD3ϵγ and CD3ϵδ to form an arrangement with an approximate 3-fold symmetry proximal to the T-cell membrane (Fig. 2C) (23). CD3δ contacts both Cα and Cβ in their ECDs. Additional interactions occur between the CP of TCRα and CD3δ, whereas the short extracellular segment of one of the two CD3ζ chains packs against the CPs of TCRα and CD3δ. TCR Cβ makes contacts with both subunits of CD3ϵγ that serve to reinforce the extracellular TCR–CD3 assembly. However, the buried surface area between TCRαβ ECDs and CD3 ECDs is considerably smaller than that contributed by other domains, indicating that TM and CP interactions are mainly responsible for maintaining complex integrity.
X-ray crystallographic studies of multiple TCRαβ ECDs in free form and bound to pMHC have identified a broad spectrum of conformational changes in Vα and Vβ CDR loops associated with antigen binding, ranging from small side-chain rearrangements to large backbone displacements (7). By contrast, X-ray crystallography has so far failed to identify clear and consistent conformational changes in the TCR Cα or Cβ domains that can be unambiguously attributed to pMHC ligation. Similarly, superposition of the unbound TCRαβ ECDs from the TCR–CD3 cryoEM structure onto crystal structures of various unbound and pMHC-bound TCRαβ ECDs did not reveal any significant conformational differences (23), although it should be noted the resolution of the TCR–CD3 structure is only moderate (3.7 Å). Whether pMHC binding induces detectable conformational changes anywhere in the TCR–CD3 complex that could be associated with TCR activation must await determination of a TCR–CD3–pMHC structure. An important caveat in this regard is that cryoEM analysis required chemical cross-linking of TCR–CD3 subunits using glutaraldehyde to prevent dissociation. Such cross-linking may prevent the TCR–CD3 complex from assuming alternative conformations upon pMHC engagement. It is also possible, as proposed in the mechanosensor model of TCR triggering (discussed below) (14, 15), that the application of force, which was absent from the cryoEM structure determination, may be necessary to cause rearrangements of TCR–CD3 subunits associated with signaling. Yet another possibility is that the relevant changes may be in protein dynamics (discussed below) (16, 17), a parameter than cannot be accessed through the static snapshots provided by cryoEM or X-ray crystallography. Importantly, these two possibilities are not mutually exclusive but could instead represent interdependent triggering mechanisms, as we hypothesize in this review.
TCR as a mechanosensor
During immune surveillance, T cells bind and crawl over APCs to scan their surface for cognate pMHC ligands. Sliding of the T cell and APC membranes relative to each other generates tensile forces in the piconewton range (15). According to the mechanosensor model, the TCR leverages mechanical energy produced by cell motility to drive biochemical signaling following pMHC engagement. The first experimental evidence that the TCR functions as a mechanosensor was obtained from single-molecule assays using optical tweezers that presented pMHC-coated beads to cell-surface TCRs (14). These measurements showed that pMHC binding alone without force was insufficient for TCR triggering, whereas piconewton force applied tangentially to the T-cell surface with cognate but not irrelevant pMHC resulted in TCR activation, as measured by an increase in calcium flux. Because only tangential force activated the TCR, the receptor behaves as an anisotropic (i.e. directional) mechanosensor (14). Further evidence for TCR mechanosensing came from studies using a micropipette to reveal shear force accompanying activation (31) and a biomembrane force probe (BFP) to demonstrate pulling and pushing associated with T-cell triggering (32).
A major attraction of the mechanosensor model is that it can explain the now well-documented ability of a single pMHC molecule to trigger a T cell (4, 5). It is also consistent with the recent demonstration by single-molecule brightness and coincidence analysis and fluorescence resonance energy transfer (FRET)-based measurements that monomeric, rather than multimeric, TCR–CD3 complexes drive pMHC recognition and intracellular signaling (6). This finding argues against ligand-induced TCR dimerization or oligomerization as a mechanism for physiological T cell stimulation.
Evidence for force-induced conformational changes in TCR–pMHC complexes
A remarkable feature of TCR–pMHC interactions revealed by single-molecule studies using BFP technology is the formation of catch bonds, whose lifetime increases with tensile force applied to the bond (33–36). Normally, bond lifetimes diminish with increasing force (slip bonds). However, in the case of catch bonds, the lifetime of the bond actually increases under load up to a maximum before it decreases at higher forces like in a slip bond. In BFP experiments, the lifetime of the TCR–pMHC bond was measured under a range of forces applied via a pMHC engaged to a TCR on a native T cell (33–36). Force prolonged the lifetimes of TCR–pMHC bonds for agonist pMHC (catch bonds), but shortened them for antagonist pMHC (slip bonds). Moreover, the force that produced catch bonds with the longest lifetimes (∼10 piconewtons) is comparable with the estimated adhesion strength between activated T cells and APCs (15). In general, the functional potency of TCR–pMHC interactions has been found to correlate with catch bond formation.
Steered molecular dynamics (SMD) simulations have provided insights into possible structural changes in TCR CDR loops associated with the acquisition of catch bonds (35, 36). For example, application of a force normal to the interface between TCR 2C and the pMHC agonist R4-H-2Kb increased the frequency of hydrogen bond formation between the MHC-bound R4 peptide and 2C (36). In the absence of force, P4 Arg of R4 formed one hydrogen bond with CDR2β Tyr-50 of 2C, whereas force induced formation of two additional hydrogen bonds between P4 Arg and CDR1β Gly-96 and CDR3α Ser-102, thereby strengthening the TCR–pMHC interaction.
Single-molecule studies have also provided evidence that the TCR–pMHC complex undergoes conformational transitions under force that are not restricted to the TCR CDR loops (34). In these experiments, the TCR–pMHC interaction was isolated to a coverslip surface in a tethered bead configuration, and force was applied to the complex via an optical trap. Bond lifetimes were measured by translating the sample relative to the fixed trap and holding it at a fixed position (and thus force) until bond rupture, identified as an abrupt snap back of the bead position within the trap (34). Displacements of the bead upon applying a pulling force were interpreted as conformational extensions of the TCR–pMHC complex spanning 8–15 nm in the direction of the pull that correlated with ligand potency. However, the exact molecular nature of these surprisingly large extensions cannot be deduced from such studies, which is an intrinsic limitation of single-molecule biophysical methods. In a broad sense, these conformational changes could involve rotations, rearrangements, or elongations of TCR and/or pMHC domains, as well as possible unfolding of the interacting proteins.
Single-molecule stretching experiments using a BFP were used to confirm pMHC-induced conformational changes (36). The extension increase (16 nm) was similar to that reported on pulling TCR–pMHC complexes using an optical trap (34). SMD simulations were carried out in an attempt to gain insights into the structural basis for this large extension. Application of a force normal to the TCR–pMHC binding interface resulted in an unexpected elongation of the linker between the MHC α1α2 and α3 domains, followed by rupture of the intramolecular association between the MHC α1α2 and β2-microglobulin domains (36). (MHC class I molecules are composed of two subunits: a transmembrane α chain that includes three extracellular domains (α1, α2, and α3) and a noncovalently-associated chain, β2-microglubulin, that consists of a single extracellular domain.) By contrast, the TCR was rigid and stable during mechanical pulling in all SMD simulations.
Assuming these in silico SMD simulations reflect physical reality, the question arises how force-induced conformational changes in the MHC (but apparently not in the TCR) facilitate initial TCR triggering. One possibility is that 10–20-nm elongation of the TCR–pMHC complex reduces its size difference relative to the phosphatase CD45, which is considerably longer than an unstretched TCR–pMHC complex (Fig. 1B), thereby preventing exclusion of CD45 from the region of T cell–APC contacts (36). This may allow CD45 to dephosphorylate the C-terminal tail of Lck, which is required to convert this critical kinase to its active conformation (37). In contrast, CD45 exclusion favors the phosphorylated state of CD3 ITAMs, which promotes T-cell activation (13). Another possibility is that force-induced formation of TCR–pMHC catch bonds enables long-range transmission of changes in TCR dynamics from the CDR loops to associated CD3 molecules in the TCR–CD3 complex. Recent NMR studies have provided support for this allosteric hypothesis, as described next.
NMR and MD analysis of pMHC binding to TCR
As noted above, X-ray crystallography and cryoEM have failed to identify ligand-induced conformational changes in the TCR Cα or Cβ domains that could be communicated to the intimately-associated CD3 subunits (7, 23). Several explanations are possible. One explanation is that such changes simply do not occur and that allosteric mechanisms are not involved in TCR triggering. However, another explanation is that the relevant changes may be in protein dynamics, a parameter that cannot be addressed via the static snapshots provided by X-ray crystallography or cryoEM. Indeed, studies of several protein systems have revealed that ligand binding can alter protein flexibility at distant sites, resulting in long-distance transmission of biological signals, even in the absence of obvious structural changes (38–43). This process is known as dynamic allostery. For example, NMR analysis of catabolite activator protein (CAP), whose role is to bind cyclic AMP (cAMP) to activate transcription, showed that the allosteric response of the system is due to quenching of backbone and side-chain dynamics of CAP upon binding cAMP, without any discernible change in protein conformation (42). Dynamic allostery provides a unifying mechanism for the general phenomenon of allostery and is of special interest in cases where signaling occurs in the absence of apparent structural change (39, 40, 43).
Initial evidence for dynamically-driven TCR signaling came from hydrogen/deuterium (H/D) exchange experiments, which showed that pMHC ligation globally rigidified the TCR (44). More recently, NMR spectroscopy and MD simulations have been used to address the possibility that pMHC binding induces allosteric changes in TCR conformation and/or dynamics that are relayed to CD3. Together, these techniques provide information on protein flexibility over a range of timescales from hundreds of nanoseconds to seconds or longer. In addition, NMR can characterize sparsely-populated conformational states that may be important for biological function (45).
Two TCRs have been studied to date: 1) a mouse TCR (B4.2.3) specific for an HIV-1 gp120-derived peptide (Pro-18–Ile-10) bound to a mouse MHC class I molecule (H2-Dd) (16), and 2) a human antiviral TCR (A6) specific for the Tax peptide of human T-cell lymphotropic virus-1 (HTLV-1) bound to a human MHC class I molecule (HLA-A2) (17). NMR signal perturbations upon ligation of pMHC were examined for the β chain of TCR B4.2.3 and for both the α and β chains of TCR A6. In addition, all atom MD simulations were carried out for TCR A6 in unbound and pMHC-bound states for comparison with NMR results (17).
As expected, many of the chemical shift perturbations (CSPs) and differential peak intensity decreases in TCRs B4.2.3 and A6 induced by pMHC are for residues in the CDR loops at the binding interfaces with Pro-18–Ile-10–H2-Dd and Tax-HLA-A2, respectively (16, 17). In addition, numerous signal perturbations were observed for residues in Vβ and Cβ distant from the pMHC-binding site, mainly in the Vα/Vβ, Vβ/Cβ, and Cα/Cβ interfaces. Strikingly, these spectroscopic changes included long-range effects at the far end of TCR close to the membrane (Fig. 3). Moreover, these structural and/or dynamic changes are conserved in the β chains of B4.2.3 and A6. Most perturbations in the TCR A6 α chain outside the interface with Tax–HLA-A2 are clustered in the Vα/Vβ and Cα/Cβ interfaces (17). However, in sharp contrast to the A6 and B4.2.3 β chains, few perturbations were observed in the Vα/Cα interface, which, as discussed below, is much smaller than the Vβ/Cβ interface.
Figure 3.

Allosteric sites in the TCR constant domains. Three allosteric sites identified by NMR (16, 17) in the Cα and Cβ domains of TCR A6 (PDB code 1QRN) (49) are boxed. Inset 1 shows the Cα AB loop and its surroundings in the TCR–CD3 structure (PDB code 6JXR) (23). The side chains of contacting residues are drawn in stick representation. The Cα AB loop contacts the CD3ϵ subunit of CD3ϵδ. Inset 2 shows the Cβ FG loop and its surroundings in the TCR–CD3 complex. The Cβ FG loop is situated immediately above the CD3ϵγ dimer but makes no direct contacts. Inset 3 shows the Cβ αA helix and its surroundings in the TCR–CD3 structure. The Cβ αA helix contacts the CP of the TCR α chain.
Taken together, independent NMR analyses of two unrelated TCRs, B4.2.3 and A6 (16, 17), provide strong evidence for ligand-induced allosteric signal transmission from the TCR V domains to three distinct sites in the C domains (Fig. 3): 1) the Cα AB loop; 2) the Cβ αA (H3) helix; and 3) the Cβ FG loop. The Cα AB loop and Cβ αA helix are located at the base of the TCR proximal to the T-cell membrane, whereas the Cβ FG loop protrudes from the Vβ/Cβ interface. In the TCR–CD3 cryoEM structure (23), the Cα AB loop contacts the CD3ϵ subunit of the CD3ϵδ dimer (Fig. 3). The Cβ αA helix contacts the CP of TCRα, which links the ECD of TCRα to its TM segment. The Cβ FG loop is positioned directly over the CD3ϵγ dimer (Fig. 2A) but does not make contacts, at least in the unbound TCR–CD3 structure (Fig. 3). Of note, the long (16 residues) Cβ FG loop is a unique structural feature of the Cβ domain that is not found in Cα or in antibody CL or CH1 domains.
A biological role for each of these allosteric sites is supported by mutational and functional studies of TCR signaling. Mutagenesis of residues near the Cβ αA helix dramatically impaired antigen-dependent activation of TCR B.4.2.3 without affecting pMHC affinity, thermal stability, or cell-surface expression (16). Deletion of the Cβ FG loop in TCR transgenic mice, while preserving TCR–CD3 expression on the surface of T cells, attenuated their sensitivity to activation by pMHC (46, 47). In addition, single-molecule measurements have shown the Cβ FG loop allosterically regulates TCR–pMHC bond lifetime (34). Site-directed fluorescence labeling of a human TCR (LC13) specific for a peptide from Epstein-Barr virus bound to HLA-B8 revealed a discrete conformational change in the Cα AB loop associated with pMHC ligation (48). Mutagenesis of residues in the Cα AB loop reduced antigen-specific triggering of LC13, indicating a functional role for this loop in T-cell activation.
Molecular dynamics simulations
All atom MD simulations have been carried out for TCR A6 in unbound and Tax–HLA-A2-bound states (17). As measured by the root mean square fluctuation (RMSF) for the Cα atom of each residue from the start to the end of the simulations, pMHC binding decreased the flexibility of most CDR residues, especially residues in CDR3α and CDR3β (Fig. 4, A and B), which are at the center of the interface with pMHC in the TCR A6–Tax–HLA-A2 complex (49). In addition, the overall decrease in flexibility extended well into the A6 β chain structure, from the Vβ domain, through the Vβ–Cβ linker, and into the Cβ domain, including the membrane-proximal αA and αB helices (Fig. 4C) (17, 23). Strikingly, the Cβ FG loop displayed considerably lower RMSFs in the bound versus unbound state, indicating ligand-induced rigidification (Fig. 4B). Moreover, the decrease in RMSF values for the Cβ FG loop exceeded that for any CDR loop. In contrast to the β chain, pMHC binding induced a combination of decreased and increased flexibility in the α chain (Fig. 3A).
Figure 4.

Molecular dynamics simulations of TCR in unbound and pMHC-bound states. A, ΔRMSF values for the TCR A6 α chain with positive values indicating regions that become more rigid upon binding Tax–HLA-A2 and negative values indicating increased TCR α chain flexibility (17). B, ΔRMSF values for the A6 β chain. C, ΔRMSF values mapped onto the X-ray structure of the TCR A6–Tax–HLA-A2 complex in surface representation (PDB code 1QRN) (49). Color coding is as follows: HLA-A2 (gray); Tax peptide (magenta); TCR A6 α chain (green); TCR A6 β chain (wheat); ΔRMSF >0.2 Å (blue); 0.1 < ΔRMSF <0.2 Å (light blue); −0.2 < ΔRMSF < −0.1 Å (orange), and ΔRMSF < −0.2 Å (red).
The pattern of allosteric effects in TCR A6 predicted computationally by MD simulations is broadly similar to that seen by NMR (17). Collectively, NMR (16, 17), MD (17), and H/D exchange (44) results indicate that pMHC ligation induces long-range changes in TCR dynamics that may enable allosteric communication with CD3.
In another study, MD simulations were carried out for four TCRs in their free and pMHC-bound states (50). Although the dynamic features of the TCRs differed considerably between the free and bound simulations, these features were not conserved among the TCRs, which would argue against models of TCR triggering involving conserved allosteric changes. However, this conclusion from computational analysis is not in agreement with experimental results from NMR for TCRs B4.2.3 and A6, which revealed conserved long-range dynamic changes upon pMHC ligation (16, 17). We have also observed conserved allosteric changes in an NMR study of a human autoimmune TCR (MS2–3C8) that recognizes a self-peptide from myelin basic protein presented by the MHC class II molecule HLA-DR4.4
Possible pathway for allosteric signal transmission
The unique structure of the TCR β chain may enable allosteric communication between Vβ and Cβ domains. In marked contrast to Vα and Cα domains, and to antibody VL and CL or VH and CH1 domains, Vβ and Cβ are in close contact in all TCR structures reported so far (7). Indeed, the average total buried surface between Vβ and Cβ domains is nearly twice that between Vα and Cα domains (17). Extensive and highly-conserved interdomain contacts between Vβ and Cβ impose on the αβ TCR a rigid conformation that lacks the flexibility in the region homologous to the elbow of antibody Fab fragments (51). A rigid β chain structure could facilitate transmission of allosteric changes that occur in the TCR upon binding pMHC to closely-associated CD3 molecules in the TCR–CD3 complex.
In support of this hypothesis, NMR analysis of both TCRs B.4.2.3 and A6 (16, 17) revealed significant CSPs and/or losses of peak intensity for numerous residues in the large and tightly packed Vβ/Cβ interface, which would provide a clear path for allosteric signal propagation from Vβ to Cβ. In contrast, considerably fewer spectroscopic changes were seen in the much smaller and loosely packed Vα/Cα interface. Therefore, NMR signal perturbations in Vα following pMHC ligation are most likely transmitted to Cα via the β chain rather than across the Vα/Cα interface. This allosteric pathway is further supported by our combined NMR and MD analyses of the human autoimmune TCR MS2–3C8.4
The close juxtaposition between Vβ and Cβ domains seen in the αβ TCR heterodimer is maintained in the pre-TCR, which signals developing T cells to terminate TCR β gene rearrangements (52). Unlike the αβ TCR, the pre-TCR consists of a variable TCR β chain paired with an invariant pre-TCR α chain that lacks a Vα domain. Despite the absence of Vα, the conformation of the β chain in the pre-TCR, including specific interactions across the Vβ/Cβ interface, is virtually indistinguishable from that in the αβ TCR (52), suggesting that allosteric signals may transit similar pathways in pre-TCR and TCR β chains. Notably, the pre-TCR, like the αβ TCR, behaves as an anisotropic mechanosensor (53).
Arguments for and against TCR allostery
Allosteric transmission of biological signals over long distances has now been documented for numerous proteins (54), including cell-surface receptors such as the EphA2 receptor (55). Like mechanosensing, TCR allostery could explain the exceptional ability of a single pMHC molecule binding to an exclusively monomeric TCR to initiate T-cell signaling without any requirement for ligand-induced receptor oligomerization (6) and before formation of the immunological synapse (56). However, the enormous sequence diversity of TCRs presents a major challenge for allosteric models of early T-cell activation. Theoretical estimates of TCR clonal diversity reach as high as 1015 (57), although the actual size of the TCR repertoire in an individual human adult is in the range of 105–108 unique structures (58). How can pMHC binding generate conserved conformational or dynamic changes in the Cα and Cβ domains, given the diversity of Vα and Vβ sequences and of TCR–pMHC interfaces? At least part of the answer is probably that perturbations in the highly variable CDR loops resulting from pMHC ligation are transmitted to the C domains via the Vα/Vβ and Vβ/Cβ interfaces, whose structures are highly conserved. For example, perturbed Vα residues in the interface with Vβ in TCRs A6 and B4.2.3 include Tyr-35 (94% identical in human Vα sequences; 80% identical in mouse Vα sequences), Ala-89 (80%; 92%), and Gly-103 (100%; 100%) (16, 17).
We hypothesize that, following pMHC ligation, mechanical force induces allosteric changes in TCR dynamics and/or conformation that propagate to CD3. According to this unified model (Fig. 1E), mechanical force arising from movement of the T cell relative to the APC (14, 15) is transferred to CD3 through changes in the dynamics of key allosteric sites in the TCR C domains that include the Cα AB loop, the Cβ αA (H3) helix, and the Cβ FG loop (16, 17).
Evidence for pMHC-induced structural changes in the TCR–CD3 TM regions
The mechanism by which pMHC binding to the TCR–CD3 complex transmits specific information across the T-cell membrane to initiate intracellular signaling remains unresolved. Structural changes in the TM regions are expected to occur, but these have only been partially defined.
NMR analysis of an isolated CD3ζ TM peptide in phospholipid micelles revealed a coiled coil formed by two TM α-helices linked by a native disulfide bond and stabilized by both hydrophobic polar interactions across the CD3ζζ dimer interface (28). Importantly, this same dimeric arrangement of CD3ζ TM helices is also observed in the TCR–CD3 cryoEM structure (Figs. 2A and 1B) (23). However, neither study provided information on the long cytoplasmic tail of CD3ζ, which was absent from the peptide used for NMR and is disordered in the TCR–CD3 complex.
Lee et al. (59) have proposed that TCR engagement triggers a change in the spatial relationship between the associated subunits of the CD3ζζ dimer at the junction where they emerge from the membrane into the cytoplasm. CD3ζ chains were engineered with proximity probes at the membrane–cytoplasm junction to measure the distance between CD3ζζ juxtamembrane (JM) regions by FRET via live cell imaging. In unengaged TCR–CD3 complexes, the CD3ζζ JM regions were spatially separated from each other, which is proposed to represent an inactive conformation. Engagement of TCR–CD3 complexes by pMHC brought the CD3ζζ JM regions into close apposition into what is proposed to be the active conformation (59). However, this model does not agree with the cryoEM structure of an unliganded TCR–CD3 complex (23), in which the C termini of the CD3ζζ TM helices are closely juxtaposed in a coiled coil (Fig. 2B), as described above, even in the absence of pMHC. The reason for this apparent discrepancy is unclear.
Brazin et al. (30) determined the NMR structure of an isolated TCRα TM peptide comprising the extracellular CP region, TM segment, and short (five residues) cytoplasmic tail in phospholipid micelles. The CP region is highly mobile and could not be visualized. By contrast, the TM segment adopts an L-shaped α-helical structure composed of two helices connected by a hinge (Fig. 5A). The L-shaped helix exists in conformational exchange with an extended helix (Fig. 5B). Both the hinge region and two basic residues in the TCRα TM segment, Arg-253 and Lys-258, are highly conserved in vertebrates. Brazin et al. (30) propose that mechanical forces operating through the T-cell membrane during pMHC ligation by the TCR (15) induce a conformational transition in the TCRα TM helix from the L-shape to extended state and that the straightened configuration of TCRα TM displaces the CD3ζζ dimer from the TCR–CD3 complex, thereby initiating T-cell activation. However, contrary to what this hypothesis might predict, mutations in the TCRα TM hinge designed to generate a straightened helix did not increase the sensitivity of T cells to pMHC stimulation in a bioassay measuring IL-2 production by mutant versus WT TCRα cell lines as a function of peptide concentration (30).
Figure 5.
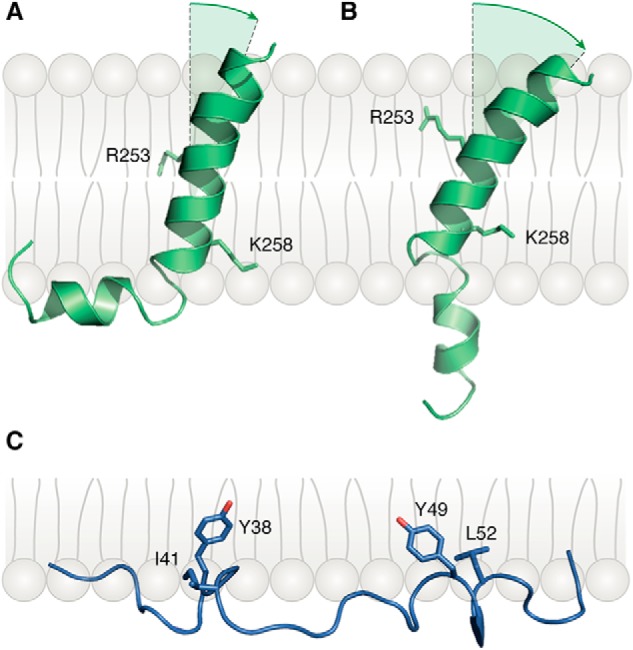
Bent and extended conformations of the TCRα TM helix. A, NMR structure of the TCRα TM helix in the bent conformation (PDB code 6MF8) (30). In the TCR–CD3 structure (23), TCRα Arg-253 and Lys-258 form salt bridges with acidic residues in CD3 TM helices (Fig. 2B). B, extended-state structure of the TCRα TM helix. C, conformation of the CD3ϵ cytoplasmic domain relative to the membrane (PDB code 2K4F) (29). The aromatic side chains of residues Tyr-38 and Tyr-49 of the CD3ϵ ITAM are embedded in the lipid bilayer.
In the TCR–CD3 cryoEM structure (Fig. 2B), TCRα Arg-253 establishes salt bonds with CD3ζ Asp-36, and TCRα Lys-258 forms a bifurcated salt bridge with CD3δ Asp-111 and CD3ϵ Asp-137 (23). These charged–pair interactions, in addition to extensive hydrophobic contacts with the TCRβ TM helix, make it difficult to see how the TCRα TM helix can undergo the large conformational change proposed from NMR analysis of the isolated TCRα TM peptide (Fig. 5, A and B) (30) without disrupting the integrity of the TCR–CD3 complex. Moreover, the TCRα TM segment in the TCR–CD3 structure is entirely α-helical with no indication of a hinge. Therefore, NMR results for the isolated TCRα TM peptide may not apply to the fully-assembled TCR–CD3 complex, with the caveat that the complex may adopt alternative conformations under mechanical force.
Swamy et al. (60) have investigated the role of membrane lipids in regulating signal transduction through the TCR. Using a radioactive cholesterol analog with a UV-inducible cross-linking group, cholesterol was shown to bind specifically to the TCRβ TM region. Cholesterol binding kept the TCR–CD3 complex in an inactive state that could not be phosphorylated by Lck or other tyrosine kinases following stimulation with an anti-CD3ϵ antibody, which was used as a surrogate for pMHC (physiological stimulation by APCs bearing cognate pMHC ligands was not reported). Remarkably, enzymatic removal of cholesterol by treatment with cholesterol oxidase caused the TCR–CD3 complex to switch to an active state that permitted antibody-induced phosphorylation of CD3 ITAMs (60). A similar effect resulted from genetically replacing the cholesterol-binding TM region of TCRβ with the TM region of TCRγ, which does not bind cholesterol. These functional experiments suggested an allosteric model for the control of TCR activity whereby cholesterol binding to the TCR regulates CD3 phosphorylability (60). However, the nature of the conformational changes associated with switching between active and inactive states is not evident from the TCR–CD3 structure (23). It should be noted in this regard that solubilizing the TCR–CD3 complex for cryoEM analysis required replacing its natural membrane lipids with a detergent (digitonin), which could impact the disposition of the TM helices or their ability to undergo possible rearrangements in response to changes in lipid environment or pMHC binding.
Evidence for pMHC-induced structural changes in the TCR–CD3 cytoplasmic tails
Conformational or dynamic changes in the ECDs and TM helices of the TCR–CD3 complex upon pMHC binding must be transmitted to the cytoplasmic tails of CD3 subunits to expose ITAMs for phosphorylation. As noted earlier, the intracellular regions of CDϵγ, CD3ϵδ, and CD3ζζ are not visible in the TCR–CD3 cryoEM structure, most likely due to high flexibility (23). Nevertheless, considerable evidence has been obtained for ligand-induced structural changes in the cytoplasmic tails of CD3ϵ and CD3ζ, although the precise nature of these changes remains to be elucidated.
The CD3ζ chain consists of a small extracellular segment (nine residues), a TM region, and a large cytoplasmic domain (110 residues) containing three ITAMs and three basic rich sequence (BRS) motifs. The first evidence for structural changes at the cytoplasmic face of the TCR–CD3 complex came from a study showing that the cytoplasmic domain of CD3ζ could bind to synthetic lipid vesicles containing acidic phospholipids, despite the absence of the TM region (61). Acidic phospholipids were used in these experiments because the lipid distribution of plasma membranes is asymmetric, with the most abundant negatively charged phospholipids, phosphatidylserine and phosphatidylinositol, localized mainly to the cytoplasmic face (62). CD measurements revealed that binding to synthetic acidic lipid vesicles substantially increased the α-helical content of the CD3ζ cytoplasmic domain (61). By contrast, no change in CD3ζ secondary structure was observed in the presence of vesicles containing zwitterionic (i.e. neutral) lipids. The folding transition was reversible and dependent on acidic phospholipids. In the folded, lipid-bound conformation, CD3ζ was refractory to phosphorylation by Src tyrosine kinase, whereas unstructured CD3ζ was readily phosphorylated. Once phosphorylated, the CD3ζ cytoplasmic domain exhibited neither membrane association nor α-helical structure induction (61).
This study using CD3ζ fragments in artificial membranes was later extended to an intact TCR–CD3 complex in a T-cell membrane (63). A close association between the CD3ζ cytoplasmic domain and the plasma membrane of live resting T cells was demonstrated by FRET between a fluorescent membrane dye and a fluorescent protein attached to the CD3ζ cytoplasmic domain (65). This association was mediated by BRS residues that presumably bind acidic phospholipids in the membrane via electrostatic interactions. Furthermore, T-cell stimulation with anti-CD3 antibody caused dissociation of the CD3ζ cytoplasmic domain from the plasma membrane, which was required for phosphorylation of ITAMs by Lck.
Taken together, these results suggest a mechanism for TCR activation whereby pMHC binding causes a repositioning of tyrosine residues within the ITAMs of the CD3ζ cytoplasmic domain from a relatively inaccessible, membrane-associated conformation that is at least partially structured to an intracellularly oriented, unstructured form that is kinase-accessible. In seeming contradiction of this model, no electron density was observed for the CD3ζ cytoplasmic domains in the unliganded TCR–CD3 cryoEM structure (23), implying that they are unstructured despite not being phosphorylated. However, the nonionic detergent digitonin used to solubilize the TCR–CD3 complex cannot be expected to fully replace the acidic phospholipids that are apparently required for the CD3ζ cytoplasmic domain to adopt a folded conformation (61). In addition, the CD3ζ cytoplasmic domain may display considerable conformational heterogeneity even in a natural lipid environment, as described below for CD3ϵ (64), which would hinder visualization by cryoEM.
Evidence has also been obtained for a reversible conformational change in the 54-residue cytoplasmic domain of CD3ϵ induced by pMHC binding (65, 66). One feature of this structural change is the regulation of accessibility of a proline-rich sequence (PRS) in the CD3ϵ cytoplasmic domain. In the unliganded TCR, the PRS was unable to bind the SH3 domain of the adaptor protein Nck. By contrast, the PRS could bind SH3 in the liganded TCR. As in the case of the CD3ζ cytoplasmic domain (63), an intimate association between the CD3ϵ cytoplasmic domain and the inner leaflet of the plasma membrane was demonstrated in live T cells by FRET (29). TCR triggering by specific pMHC complexes was found to induce dissociation of CD3ϵ ITAMs from the membrane to expose the tyrosines to phosphorylation (68), as also reported for CD3ζ ITAMs (63). Dissociation of CD3ϵ and CD3ζ ITAMs is caused by a local elevation of intracellular Ca2+ concentration that neutralizes the negative charges of acidic lipids (69).
An initial NMR analysis of the CD3ϵ cytoplasmic domain in its lipid-bound state used a construct lacking the TM region (29). This analysis revealed a partially folded structure in which protein backbone is localized at the interface between the lipid hydrophobic acyl chain region and the hydrophilic headgroup region (Fig. 5C). The two signature tyrosines of the CD3ϵ ITAM are deeply embedded in the hydrophobic core of the lipid bilayer where they are inaccessible to Src kinases. A subsequent NMR study used a more physiologically relevant construct containing both the TM region and cytoplasmic domain of CD3ϵ in acidic lipid bicelles (64). This analysis, combined with single-molecule atomic force microscopy, gave a more complete (and complex) picture of CD3ϵ conformational dynamics, whereby the CD3ϵ cytoplasmic domain could adopt multiple conformational states with different degrees of exposure of ITAM, BRS, and PRS functional motifs to biochemical modification or binding to downstream signaling molecules. These conformations were generated because different regions of the CD3ϵ cytoplasmic domain exhibited heterogeneous dynamics arising from heterogeneous lipid-binding properties (64).
The ability to visualize the CD3ϵ cytoplasmic domain by NMR but not cryoEM could be due to differences in the solubilizing agents used in each method (acidic phospholipids and nonionic detergents, respectively). However, it remains unclear how antigen engagement can directly induce conformational changes in the CD3ϵ cytoplasmic domain.
Conclusions and future directions
The cryoEM structure of a fully-assembled TCR–CD3 complex has provided a wealth of new information on its overall molecular architecture and the exact interactions between TCR and CD3 subunits (23). Nevertheless, the structural basis for TCR triggering remains largely an enigma. We now know that pMHC binding to TCR results in exposure of ITAMs in the cytoplasmic tails of CD3 subunits to phosphorylation, probably by inducing dissociation of ITAMs from the inner leaflet of the T-cell membrane (29, 61, 63, 64, 68, 69). In addition, there is growing evidence that mechanical force arising from movement of the T cell relative to the APC during immune surveillance provides the energy source for physiological TCR triggering (14, 15, 31–34). However, the molecular mechanism whereby pMHC binding to TCR is actually relayed to CD3 ITAMs is poorly understood.
Here, we have reviewed evidence for force-induced conformational changes in the TCR–CD3 complex (34, 36), for dynamically-driven TCR allostery (16, 17, 44), and for pMHC-induced structural changes in the TM (30, 59, 60) and cytoplasmic (61, 63–69) regions of CD3. It is apparent that arriving at a comprehensive picture of TCR activation that validates or rejects proposed signaling models will require new structure-guided experiments and, possibly, new techniques. Whether pMHC binding induces conformational changes anywhere in the TCR–CD3 complex that could be involved in triggering must await determination of a TCR–CD3–pMHC structure. The unique ability of cryoEM to resolve ensembles of structures to delineate conformational landscapes and identify allosteric transitions may be particularly valuable here, as demonstrated for the chaperonin TRiC/CCT (70). In that study, the authors determined an ensemble of cryoEM structures of yeast TRiC/CCT at various nucleotide concentrations that included both open and closed states, revealing an unforeseen allosteric network at atomic resolution. Visualizing the cytoplasmic domains of CD3 subunits may require cryoEM analysis of the TCR–CD3 complex in nondetergent systems, such as nanodiscs or amphipols, that may more faithfully reproduce a natural lipid environment (67, 71). Finally, none of the conventional structural methods used to study TCR triggering (X-ray crystallography, NMR, and cryoEM) take into account mechanical force, as posited in the mechanosensor model. It is possible that force is necessary to cause rearrangements of TCR–CD3 subunits relevant to triggering or to amplify allosteric communication between TCR V and C domains. New biophysical approaches, possibly combining some existing methods, will likely be needed to provide a clear picture of how the TCR–CD3 complex responds to force. In particular, measuring the effects of force on conformation and dynamics at the atomic level will be required to definitively address such questions not only in T-cell receptor biology but also more generally in receptor biology.
Acknowledgment
We thank Buyong Ma (Frederick National Laboratory for Cancer Research) for valuable discussions.
This work was supported by National Institutes of Health Grant AI129893 (to R. A. M. and J. O.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
R. A. Mariuzza, P. Agnihotri, and J. Orban, unpublished results.
- TCR
- T-cell receptor
- MHC
- major histocompatibility complex
- APC
- antigen-presenting cell
- pMHC
- peptide–MHC
- V
- variable
- C
- constant
- CP
- connecting peptide
- CDR
- complementarity-determining region
- TM
- transmembrane
- ITAM
- immunoreceptor tyrosine-based activation motif
- ECD
- extracellular domain
- cryoEM
- cryo-electron microscopy
- BFP
- biomembrane force probe
- FRET
- fluorescence resonance energy transfer
- SMD
- steered molecular dynamics
- CAP
- catabolite activator protein
- H/D exchange
- hydrogen/deuterium exchange
- MD
- molecular dynamics
- CSP
- chemical shift perturbation
- RMSF
- root mean square fluctuation
- JM
- juxtamembrane
- BRS
- basic rich sequence
- PRS
- proline-rich sequence
- SH3
- Src homology 3
- PDB
- Protein Data Bank
- JM
- juxtamembrane.
References
- 1. Wucherpfennig K. W., Gagnon E., Call M. J., Huseby E. S., and Call M. E. (2010) Structural biology of the T-cell receptor: insights into receptor assembly, ligand recognition, and initiation of signaling. Cold Spring Harb. Perspect. Biol. 2, a005140 10.1101/cshperspect.a005140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kuhns M. S., and Badgandi H. B. (2012) Piecing together the family portrait of TCR–CD3 complexes. Immunol. Rev. 250, 120–143 10.1111/imr.12000 [DOI] [PubMed] [Google Scholar]
- 3. Blum J. S., Wearsch P. A., and Cresswell P. (2013) Pathways of antigen processing. Annu. Rev. Immunol. 31, 443–473 10.1146/annurev-immunol-032712-095910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang J., Brameshuber M., Zeng X., Xie J., Li Q. J., Chien Y. H., Valitutti S., and Davis M. M. (2013) A single peptide–MHC complex ligand triggers digital cytokine secretion in CD4+ T cells. Immunity 39, 846–857 10.1016/j.immuni.2013.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. O'Donoghue G. P., Pielak R. M., Smoligovets A. A., Lin J. J., and Groves J. T. (2013) Direct single molecule measurement of TCR triggering by agonist pMHC in living primary cells. Elife 2, e00778 10.7554/eLife.00778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brameshuber M., Kellner F., Rossboth B. K., Ta H., Alge K., Sevcsik E., Göhring J., Axmann M., Baumgart F., Gascoigne N. R. J., Davis S. J., Stockinger H., Schütz G. J., and Huppa J. B. (2018) Monomeric TCRs drive T cell antigen recognition. Nat. Immunol. 19, 487–496 10.1038/s41590-018-0092-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rossjohn J., Gras S., Miles J. J., Turner S. J., Godfrey D. I., and McCluskey J. (2015) T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 33, 169–200 10.1146/annurev-immunol-032414-112334 [DOI] [PubMed] [Google Scholar]
- 8. Samelson L. E. (2002) Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu. Rev. Immunol. 20, 371–394 10.1146/annurev.immunol.20.092601.111357 [DOI] [PubMed] [Google Scholar]
- 9. van der Merwe P. A., and Dushek O. (2011) Mechanisms for T-cell receptor triggering. Nat. Rev. Immunol. 11, 47–55 10.1038/nri2887 [DOI] [PubMed] [Google Scholar]
- 10. Chakraborty A. K., and Weiss A. (2014) Insights into the initiation of TCR signaling. Nat. Immunol. 15, 798–807 10.1038/ni.2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Malissen B., and Bongrand P. (2015) Early T-cell activation: integrating biochemical, structural, and biophysical cues. Annu. Rev. Immunol. 33, 539–561 10.1146/annurev-immunol-032414-112158 [DOI] [PubMed] [Google Scholar]
- 12. Davis S. J., and van der Merwe P. A. (2006) The kinetic-segregation model: TCR triggering and beyond. Nat. Immunol. 7, 803–809 10.1038/ni1369 [DOI] [PubMed] [Google Scholar]
- 13. Chang V. T., Fernandes R. A., Ganzinger K. A., Lee S. F., Siebold C., McColl J., Jönsson P., Palayret M., Harlos K., Coles C. H., Jones E. Y., Lui Y., Huang E., Gilbert R. J. C., Klenerman D., et al. (2016) Initiation of T cell signaling by CD45 segregation at 'close contacts'. Nat. Immunol. 17, 574–582 10.1038/ni.3392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim S. T., Takeuchi K., Sun Z. Y., Touma M., Castro C. E., Fahmy A., Lang M. J., Wagner G., and Reinherz E. L. (2009) The αβ T-cell receptor is an anisotropic mechanosensor. J. Biol. Chem. 284, 31028–31037 10.1074/jbc.M109.052712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brazin K. N., Mallis R. J., Das D. K., Feng Y., Hwang W., Wang J. H., Wagner G., Lang M. J., and Reinherz E. L. (2015) Structural features of the αβTCR mechanotransduction apparatus that promote pMHC discrimination. Front. Immunol. 6, 441 10.3389/fimmu.2015.00441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Natarajan K., McShan A. C., Jiang J., Kumirov V. K., Wang R., Zhao H., Schuck P., Tilahun M. E., Boyd L. F., Ying J., Bax A., Margulies D. H., and Sgourakis N. G. (2017) An allosteric site in the T-cell receptor Cβ domain plays a critical signalling role. Nat. Commun. 8, 15260 10.1038/ncomms15260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rangarajan S., He Y., Chen Y., Kerzic M. C., Ma B., Gowthaman R., Pierce B. G., Nussinov R., Mariuzza R. A., and Orban J. (2018) Peptide-MHC (pMHC) binding to a human antiviral T-cell receptor induces long-range allosteric communication between pMHC- and CD3-binding sites. J. Biol. Chem. 293, 15991–16005 10.1074/jbc.RA118.003832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Natarajan K., Jiang J., May N. A., Mage M. G., Boyd L. F., McShan A. C., Sgourakis N. G., Bax A., and Margulies D. H. (2018) The role of molecular flexibility in antigen presentation and T-cell receptor-mediated signaling. Front. Immunol. 9, 1657 10.3389/fimmu.2018.01657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buckle A. M., and Borg N. A. (2018) Integrating experiment and theory to understand TCR–pMHC dynamics. Front. Immunol. 9, 2898 10.3389/fimmu.2018.02898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schamel W. W., Alarcon B., and Minguet S. (2019) The TCR is an allosterically regulated macromolecular machinery changing its conformation while working. Immunol. Rev. 291, 8–25 10.1111/imr.12788 [DOI] [PubMed] [Google Scholar]
- 21. Connolly A., and Gagnon E. (2019) Electrostatic interactions: from immune receptor assembly to signaling. Immunol. Rev. 291, 26–43 10.1111/imr.12769 [DOI] [PubMed] [Google Scholar]
- 22. Mallis R. J., Brazin K. N., Duke-Cohan J. S., Hwang W., Wang J. H., Wagner G., Arthanari H., Lang M. J., and Reinherz E. L. (2019) NMR: an essential structural tool for integrative studies of T cell development, pMHC ligand recognition and TCR mechanobiology. J. Biomol. NMR 73, 319–332 10.1007/s10858-019-00234-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dong, Zheng L., Lin J., Zhang B., Zhu Y., Li N., Xie S., Wang Y., Gao N., and Huang Z. (2019) Structural basis of assembly of the human T-cell receptor–CD3 complex. Nature 573, 546–552 10.1038/s41586-019-1537-0 [DOI] [PubMed] [Google Scholar]
- 24. Sun Z. J., Kim K. S., Wagner G., and Reinherz E. L. (2001) Mechanisms contributing to T-cell receptor signaling and assembly revealed by the solution structure of an ectodomain fragment of the CD3ϵγ heterodimer. Cell 105, 913–923 10.1016/S0092-8674(01)00395-6 [DOI] [PubMed] [Google Scholar]
- 25. Kjer-Nielsen L., Dunstone M. A., Kostenko L., Ely L. K., Beddoe T., Mifsud N. A., Purcell A. W., Brooks A. G., McCluskey J., and Rossjohn J. (2004) Crystal structure of the human T-cell receptor CD3ϵγ heterodimer complexed to the therapeutic mAb OKT3. Proc. Natl. Acad. Sci. U.S.A. 101, 7675–7680 10.1073/pnas.0402295101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sun Z. Y., Kim S. T., Kim I. C., Fahmy A., Reinherz E. L., and Wagner G. (2004) Solution structure of the CD3ϵδ ectodomain and comparison with CD3ϵγ as a basis for modeling T-cell receptor topology and signaling. Proc. Natl. Acad. Sci. U.S.A. 101, 16867–16872 10.1073/pnas.0407576101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arnett K. L., Harrison S. C., and Wiley D. C. (2004) Crystal structure of a human CD3-ϵ/δ dimer in complex with a UCHT1 single chain antibody fragment. Proc. Natl. Acad. Sci. U.S.A. 101, 16268–16273 10.1073/pnas.0407359101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Call M. E., Schnell J. R., Xu C., Lutz R. A., Chou J. J., and Wucherpfennig K. W. (2006) The structure of the ζζ transmembrane dimer reveals features essential for its assembly with the T-cell receptor. Cell 127, 355–368 10.1016/j.cell.2006.08.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu C., Gagnon E., Call M. E., Schnell J. R., Schwieters C. D., Carman C. V., Chou J. J., and Wucherpfennig K. W. (2008) Regulation of T-cell receptor activation by dynamic membrane binding of the CD3ϵ cytoplasmic tyrosine-based motif. Cell 135, 702–713 10.1016/j.cell.2008.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brazin K. N., Mallis R. J., Boeszoermenyi A., Feng Y., Yoshizawa A., Reche P. A., Kaur P., Bi K., Hussey R. E., Duke-Cohan J. S., Song L., Wagner G., Arthanari H., Lang M. J., and Reinherz E. L. (2018) The T cell antigen receptor α transmembrane domain coordinates triggering through regulation of bilayer immersion and CD3 subunit associations. Immunity 49, 829–841.e6 10.1016/j.immuni.2018.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Y. C., Chen B. M., Wu P. C., Cheng T. L., Kao L. S., Tao M. H., Lieber A., and Roffler S. R. (2010) Cutting edge: mechanical forces acting on T cells immobilized via the TCR complex can trigger TCR signaling. J. Immunol. 184, 5959–5963 10.4049/jimmunol.0900775 [DOI] [PubMed] [Google Scholar]
- 32. Husson J., Chemin K., Bohineust A., Hivroz C., and Henry N. (2011) Force generation upon T-cell receptor engagement. PLoS ONE 6, e19680 10.1371/journal.pone.0019680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu B., Chen W., Evavold B. D., and Zhu C. (2014) Accumulation of dynamic catch bonds between TCR and agonist peptide–MHC triggers T cell signaling. Cell 157, 357–368 10.1016/j.cell.2014.02.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Das D. K., Feng Y., Mallis R. J., Li X., Keskin D. B., Hussey R. E., Brady S. K., Wang J. H., Wagner G., Reinherz E. L., and Lang M. J. (2015) Force-dependent transition in the T-cell receptor β-subunit allosterically regulates peptide discrimination and pMHC bond lifetime. Proc. Natl. Acad. Sci. U.S.A. 112, 1517–1522 10.1073/pnas.1424829112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sibener L. V., Fernandes R. A., Kolawole E. M., Carbone C. B., Liu F., McAffee D., Birnbaum M. E., Yang X., Su L. F., Yu W., Dong S., Gee M. H., Jude K. M., Davis M. M., Groves J. T., et al. (2018) Isolation of a structural mechanism for uncoupling T-cell receptor signaling from peptide-MHC binding. Cell 174, 672–687.e27 10.1016/j.cell.2018.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu P., Zhang T., Liu B., Fei P., Cui L., Qin R., Zhu H., Yao D., Martinez R. J., Hu W., An C., Zhang Y., Liu J., Shi J., Fan J., et al. (2019) Mechano-regulation of peptide-MHC class I conformations determines TCR antigen recognition. Mol. Cell 73, 1015–1027.e7 10.1016/j.molcel.2018.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Courtney A. H., Amacher J. F., Kadlecek T. A., Mollenauer M. N., Au-Yeung B. B., Kuriyan J., and Weiss A. (2017) A phosphosite within the SH2 domain of Lck regulates its activation by CD45. Mol. Cell 67, 498–511.e6 10.1016/j.molcel.2017.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tzeng S. R., and Kalodimos C. G. (2009) Dynamic activation of an allosteric regulatory protein. Nature 462, 368–372 10.1038/nature08560 [DOI] [PubMed] [Google Scholar]
- 39. Smock R. G., and Gierasch L. M. (2009) Sending signals dynamically. Science 324, 198–203 10.1126/science.1169377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Motlagh H. N., Wrabl J. O., Li J., and Hilser V. J. (2014) The ensemble nature of allostery. Nature 508, 331–339 10.1038/nature13001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McLeish T. C., Cann M. J., and Rodgers T. L. (2015) Dynamic transmission of protein allostery without structural change: spatial pathways or global modes? Biophys. J. 109, 1240–1250 10.1016/j.bpj.2015.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tzeng S. R., and Kalodimos C. G. (2012) Protein activity regulation by conformational entropy. Nature 488, 236–240 10.1038/nature11271 [DOI] [PubMed] [Google Scholar]
- 43. Wand A. J. (2013) The dark energy of proteins comes to light: conformational entropy and its role in protein function revealed by NMR relaxation. Curr. Opin. Struct. Biol. 23, 75–81 10.1016/j.sbi.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hawse W. F., Champion M. M., Joyce M. V., Hellman L. M., Hossain M., Ryan V., Pierce B. G., Weng Z., and Baker B. M. (2012) Cutting edge: evidence for a dynamically driven T cell signaling mechanism. J. Immunol. 188, 5819–5823 10.4049/jimmunol.1200952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Anthis N. J., and Clore G. M. (2015) Visualizing transient dark states by NMR spectroscopy. Q. Rev. Biophys. 48, 35–116 10.1017/S0033583514000122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sasada T., Touma M., Chang H. C., Clayton L. K., Wang J. H., and Reinherz E. L. (2002) Involvement of the TCR Cβ FG loop in thymic selection and T cell function. J. Exp. Med. 195, 1419–1431 10.1084/jem.20020119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Touma M., Chang H. C., Sasada T., Handley M., Clayton L. K., and Reinherz E. L. (2006) The TCR Cβ FG loop regulates αβ T cell development. J. Immunol. 176, 6812–6823 10.4049/jimmunol.176.11.6812 [DOI] [PubMed] [Google Scholar]
- 48. Beddoe T., Chen Z., Clements C. S., Ely L. K., Bushell S. R., Vivian J. P., Kjer-Nielsen L., Pang S. S., Dunstone M. A., Liu Y. C., Macdonald W. A., Perugini M. A., Wilce M. C., Burrows S. R., Purcell A. W., Tiganis T., et al. (2009) Antigen ligation triggers a conformational change within the constant domain of the αβ T-cell receptor. Immunity 30, 777–788 10.1016/j.immuni.2009.03.018 [DOI] [PubMed] [Google Scholar]
- 49. Ding Y. H., Baker B. M., Garboczi D. N., Biddison W. E., and Wiley D. C. (1999) Four A6-TCR/peptide/HLA-A2 structures that generate very different T cell signals are nearly identical. Immunity 11, 45–56 10.1016/S1074-7613(00)80080-1 [DOI] [PubMed] [Google Scholar]
- 50. Knapp B., van der Merwe P. A., Dushek O., and Deane C. M. (2019) MHC binding affects the dynamics of different T-cell receptors in different ways. PLoS Comp. Biol. 15, e1007338 10.1371/journal.pcbi.1007338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bentley G. A., Boulot G., Karjalainen K., and Mariuzza R. A. (1995) Crystal structure of the β chain of a T cell antigen receptor. Science 267, 1984–1987 10.1126/science.7701320 [DOI] [PubMed] [Google Scholar]
- 52. Pang S. S., Berry R., Chen Z., Kjer-Nielsen L., Perugini M. A., King G. F., Wang C., Chew S. H., La Gruta N. L., Williams N. K., Beddoe T., Tiganis T., Cowieson N. P., Godfrey D. I., Purcell A. W., et al. (2010) The structural basis for autonomous dimerization of the pre-T-cell antigen receptor. Nature 467, 844–848 10.1038/nature09448 [DOI] [PubMed] [Google Scholar]
- 53. Mallis R. J., Bai K., Arthanari H., Hussey R. E., Handley M., Li Z., Chingozha L., Duke-Cohan J. S., Lu H., Wang J. H., Zhu C., Wagner G., and Reinherz E. L. (2015) Pre-TCR ligand binding impacts thymocyte development before αβTCR expression. Proc. Natl. Acad. Sci. U.S.A. 112, 8373–8378 10.1073/pnas.1504971112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nussinov R., Tsai C.-J., and Ma B. (2013) The underappreciated role of allostery in the cellular network. Annu. Rev. Biophys. 42, 169–189 10.1146/annurev-biophys-083012-130257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chavent M., Seiradake E., Jones E. Y., and Sansom M. S. (2016) Structures of the EphA2 receptor at the membrane: role of lipid interactions. Structure 24, 337–347 10.1016/j.str.2015.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fooksman D. R., Vardhana S., Vasiliver-Shamis G., Liese J., Blair D. A., Waite J., Sacristán C., Victora G. D., Zanin-Zhorov A., and Dustin M. L. (2010) Functional anatomy of T-cell activation and synapse formation. Annu. Rev. Immunol. 28, 79–105 10.1146/annurev-immunol-030409-101308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nikolich-Zugich J., Slifka M. K., and Messaoudi I. (2004) The many important facets of T-cell receptor diversity. Nat. Rev. Immunol. 4, 123–132 10.1038/nri1292 [DOI] [PubMed] [Google Scholar]
- 58. Qi Q., Liu Y., Cheng Y., Glanville J., Zhang D., Lee J. Y., Olshen R. A., Weyand C. M., Boyd S. D., and Goronzy J. J. (2014) Diversity and clonal selection in the human T-cell repertoire. Proc. Natl. Acad. Sci. U.S.A. 111, 13139–13144 10.1073/pnas.1409155111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee M. S., Glassman C. R., Deshpande N. R., Badgandi H. B., Parrish H. L., Uttamapinant C., Stawski P. S., Ting A. Y., and Kuhns M. S. (2015) A mechanical switch couples T-cell receptor triggering to the cytoplasmic juxtamembrane regions of CD3ζζ. Immunity 43, 227–239 10.1016/j.immuni.2015.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Swamy M., Beck-Garcia K., Beck-Garcia E., Hartl F. A., Morath A., Yousefi O. S., Dopfer E. P., Molnár E., Schulze A. K., Blanco R., Borroto A., Martín-Blanco N., Alarcon B., Höfer T., Minguet S., and Schamel W. W. (2016) A cholesterol-based allostery model of T-cell receptor phosphorylation. Immunity 44, 1091–1101 10.1016/j.immuni.2016.04.011 [DOI] [PubMed] [Google Scholar]
- 61. Aivazian D., and Stern L. J. (2000) Phosphrylation of T-cell receptor ζ is regulated by a lipid dependent folding transition. Nat. Struct. Biol. 7, 1023–1026 10.1038/80930 [DOI] [PubMed] [Google Scholar]
- 62. Devaux P. F. (1991) Static and dynamic lipid asymmetry in cell membranes. Biochemistry 30, 1163–1173 10.1021/bi00219a001 [DOI] [PubMed] [Google Scholar]
- 63. Zhang H., Cordoba S.-P., Dushek O., and van der Merwe P. A. (2011) Basic residues in the T-cell receptor ζ cytoplasmic domain mediate membrane association and modulate signaling. Proc. Natl. Acad. Sci. U.S.A. 108, 19323–19328 10.1073/pnas.1108052108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guo X., Yan C., Li H., Huang W., Shi X., Huang M., Wang Y., Pan W., Cai M., Li L., Wu W., Bai Y., Zhang C., Liu Z., Wang X., et al. (2017) Lipid-dependent conformational dynamics underlie the functional versatility of T-cell receptor. Cell Res. 27, 505–525 10.1038/cr.2017.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gil D., Schamel W. W., Montoya M., Sánchez-Madrid F., and Alarcón B. (2002) Recruitment of Nck by CD3ϵ reveals a ligand-induced conformational change essential for T-cell receptor signaling and synapse formation. Cell 109, 901–912 10.1016/S0092-8674(02)00799-7 [DOI] [PubMed] [Google Scholar]
- 66. Risueño R. M., Gil D., Fernández E., Sánchez-Madrid F., and Alarcón B. (2005) Ligand-induced conformational change in the T-cell receptor associated with productive immune synapses. Blood 106, 601–608 10.1182/blood-2004-12-4763 [DOI] [PubMed] [Google Scholar]
- 67. Puthenveetil R., and Vinogradova O. (2019) Solution NMR: a powerful tool for structural and functional studies of membrane proteins in reconstituted environments. J. Biol. Chem. 294, 15914–15931 10.1074/jbc.REV119.009178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gagnon E., Schubert D. A., Gordo S., Chu H. H., and Wucherpfennig K. W. (2012) Local changes in lipid environment of TCR microclusters regulate membrane binding by the CD3ϵ cytoplasmic domain. J. Exp. Med. 209, 2423–2439 10.1084/jem.20120790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shi X., Bi Y., Yang W., Guo X., Jiang Y., Wan C., Li L., Bai Y., Guo J., Wang Y., Chen X., Wu B., Sun H., Liu W., Wang J., and Xu C. (2013) Ca2+ regulates T-cell receptor activation by modulating the charge property of lipids. Nature 493, 111–115 10.1038/nature11699 [DOI] [PubMed] [Google Scholar]
- 70. Jin M., Han W., Liu C., Zang Y., Li J., Wang F., Wang Y., and Cong Y. (2019) An ensemble of cryo-EM structures reveal its conformational landscape and subunit specificity. Proc. Natl. Acad. Sci. U.S.A. 116, 19513–19522 10.1073/pnas.1903976116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Thonghin N., Kargas V., Clews J., and Ford R. C. (2018) Cryo-electron microscopy of membrane proteins. Methods 147, 176–186 10.1016/j.ymeth.2018.04.018 [DOI] [PubMed] [Google Scholar]