

Abstract
The deafness-associated m.12201T>C mutation affects the A5-U68 base-pairing within the acceptor stem of mitochondrial tRNAHis. The primary defect in this mutation is an alteration in tRNAHis aminoacylation. Here, we further investigate the molecular mechanism of the deafness-associated tRNAHis 12201T>C mutation and test whether the overexpression of the human mitochondrial histidyl-tRNA synthetase gene (HARS2) in cytoplasmic hybrid (cybrid) cells carrying the m.12201T>C mutation reverses mitochondrial dysfunctions. Using molecular dynamics simulations, we demonstrate that the m.12201T>C mutation perturbs the tRNAHis structure and function, supported by decreased melting temperature, conformational changes, and instability of mutated tRNA. We show that the m.12201T>C mutation-induced alteration of aminoacylation tRNAHis causes mitochondrial translational defects and respiratory deficiency. We found that the transfer of HARS2 into the cybrids carrying the m.12201T>C mutation raises the levels of aminoacylated tRNAHis from 56.3 to 75.0% but does not change the aminoacylation of other tRNAs. Strikingly, HARS2 overexpression increased the steady-state levels of tRNAHis and of noncognate tRNAs, including tRNAAla, tRNAGln, tRNAGlu, tRNALeu(UUR), tRNALys, and tRNAMet, in cells bearing the m.12201T>C mutation. This improved tRNA metabolism elevated the efficiency of mitochondrial translation, activities of oxidative phosphorylation complexes, and respiration capacity. Furthermore, HARS2 overexpression markedly increased mitochondrial ATP levels and membrane potential and reduced production of reactive oxygen species in cells carrying the m.12201T>C mutation. These results indicate that HARS2 overexpression corrects the mitochondrial dysfunction caused by the tRNAHis mutation. These findings provide critical insights into the pathophysiology of mitochondrial disease and represent a step toward improved therapeutic interventions for mitochondrial disorders.
Keywords: hearing, transfer RNA (tRNA), mitochondrial respiratory chain complex, translation, mitochondrial membrane potential, mitochondrial DNA (mtDNA), reactive oxygen species (ROS), gene transfer, ATP, aminoacyl tRNA synthetase, aminoacylation, m.12201T>C mutation, mitochondrial histidyl-tRNA synthetase, mitochondrial tRNA mutation, pathophysiology, deafness, rescue, oxidative phosphorylation system (OXPHOS)
Introduction
Mitochondrial DNA (mtDNA)2-dependent defects have been associated with hearing deficit, either the nonsyndromic form (where hearing loss is the only obvious medical problem) or the syndromic form (hearing loss with other medical problems, such as diabetes) (1–5). The majority of these deafness-linked mtDNA mutations are located in the mitochondrial 12S rRNA and tRNA genes (1–6). The m.1555A>G and m.1494C>T mutations in the 12S rRNA gene have been associated with both aminoglycoside-induced and nonsyndromic deafness in many families worldwide (1, 2, 7–9). Mitochondrial tRNAs are the hot spots for mutations associated with hearing loss (3, 10, 11). The syndromic deafness-associated mtDNA mutations are the MELAS-associated tRNALeu(UUR) 3243A>G mutation (12) and MERRF-associated tRNALys 8344A>G mutation (13) and MDD-associated tRNAGlu 14692A>G (14), whereas the nonsyndromic deafness-associated mtDNA mutations included the tRNASer(UCN) 7445A>G, 7505T>C, and 7511T>C; tRNAHis 12201T>C; tRNAAsp 7551A>G; and tRNAIle 4295A>G mutations (15–20). These tRNA mutations have structural and functional consequences, including the processing of the tRNA from the primary transcripts, stability of the folded secondary structure, the charging of the tRNA, or the codon-anticodon interaction in the process of translation (14–19). Of these, the m.12201T>C mutation resided at the uridine at position 68 (U68) and abolished a base-pairing (5A-68U) on the acceptor stem of the tRNAHis that may play an important role in the stability and identity of tRNA (18, 21, 22) (Fig. 1A). The primary defect in this mutation was the aberrant aminoacylation of the tRNAHis (18). The deficient aminoacylation of tRNAHis mainly contributed to a shortage of tRNAHis, thereby causing the reduced rate of mitochondrial protein synthesis and respiration defects (18).
Figure 1.

MD simulations on the acceptor stem of WT and mutated tRNAHis. A, cloverleaf structure of human mitochondrial tRNAHis (18, 22). Arrow, location of the m.12201T>C mutation. Nucleotides in the dashed box in the acceptor stem of tRNASer(UCN) were used for MD simulation analysis. B, time evolution of the root mean square deviation (RMSD) values of all backbone atoms on the acceptor stem for the WT (black lines) and mutant (MT) (red lines) of tRNAHis. C, superimposed models of tertiary structures of acceptor stems of WT (gray) and mutant (steel blue) tRNAHis. The sequences of mutant and WT models were shown in regular mode, respectively. D, hydrogen bonds (black dashes) between A5 and U68 in WT (gray) or A5 and C68 in mutant tRNAHis (steel blue). The occupancies of hydrogen bonds are shown in red.
A specific cognate amino acid is charged or aminoacylated to each tRNA catalyzed by aminoacyl tRNA synthetase (10, 11). Therefore, the deficient aminoacylation of tRNAs may be restored by the transfer of aminoacyl tRNA synthetase in cells carrying the tRNA mutation. In the previous studies, the overexpression of human mitochondrial leucyl-tRNA synthetase in the cybrid cells carrying the tRNALeu(UUR) 3243A>G mutation improved the efficiency of aminoacylation and stability of mitochondrial tRNALeu(UUR) and oxidative phosphorylation (23–26). The aberrant tRNAAla and tRNAVal metabolisms were restored by overexpression of AARS2 and VARS2 in the cells carrying the tRNAAla 5565A>G or tRNAVal 1624C>T mutation, respectively (27, 28). In fact, human mitochondrial histidyl-tRNA synthetase is a highly conserved enzyme composed of 506 amino acids with a mitochondrial signal sequence (29). Thus, it is anticipated that the overexpression of human HARS2 in the cybrid cells carrying the m.12201T>C mutation would improve the aminoacylation capacity of tRNAHis, enhance the stability of tRNA, and then increase the rates of mitochondrial translation and respiration, consequently correcting the mitochondrial dysfunction. To test this hypothesis, stable transfectants were constructed by transferring a human HARS2 cDNA into a cybrid cell line carrying the m.12201T>C mutation and a control cybrid cell line harboring the WT version of tRNAHis. Human HARS2 was further characterized by examining subcellular locations. These stable transfectants were analyzed for the aminoacylation capacity of tRNAs, the stability of the tRNAHis, the rates of mitochondrial translation and respiration, the levels of mitochondrial ATP, and the mitochondrial membrane potential (ΔΨm) as well as the production of reactive oxygen species (ROS).
Results
MD simulation analyses
To assess the impact of m.12201T>C mutation on the tertiary structure of tRNAHis, we carried out the molecular dynamics simulation using the acceptor stems (15 nt) of both WT and mutant tRNAHis by the 100-ns all-atom method. As shown in Fig. 1B, the root mean square deviation (RMSD) curve of the mutated acceptor stem fluctuated more significantly than those of the WT counterpart, suggesting that the mutated acceptor stem exhibited more instability than its WT counterpart. Using the cpptraj program Amber14, the U68 formed a canonical base pair with A5 in the WT tRNAHis through hydrogen bonds with an occupancy of 47 and 64%, respectively (Fig. 1, C and D). In contrast, the newly formed interaction between A5 and C68 in the mutant tRNAHis was decreased to one hydrogen bond with a lower occupancy of 38%. These data implied that the mutant tRNAHis molecule with the mismatch of 5A and C68 may be less stable than those in the WT counterpart.
Aberrant stability and conformation of tRNAHis
To experimentally test whether there was an effect of the m.12201T>C mutation on the stability of tRNAHis, we measured the melting temperatures (Tm) of WT and mutant tRNAHis transcripts by calculating the derivatives of absorbance against a temperature curve. As shown in Fig. 2A, the Tm values of WT (U68) and mutant (C68) transcripts were 47.0 ± 1.0 and 43.0 ± 1.7 °C, respectively. These data were in a good agreement with data of molecular stimulation, indicating that the m.12201T>C mutation led to the instability of tRNAHis.
Figure 2.

Analysis for the stability and conformation of tRNAHis. A, melting profiles of WT and MT tRNAHis transcripts were measured at 260 nm with a heating rate of 1 °C/min from 25 to 95 °C (red curves). First derivatives (dA/dT) against temperature curves are shown to highlight the Tm value transitions (green curves). The calculations were based on three independent determinations. The graph shows the results of a representative experiment. B, assessment of conformational changes by PAGE analysis under native and denaturing conditions. The transcripts of WT and MT tRNAHis were electrophoresed through native or denaturing polyacrylamide gel stained with ethidium bromide. C, Northern blot analysis of tRNAs under native conditions. D, Northern blot analysis of tRNAs from various cell lines under denatured conditions. Five micrograms of total cellular RNAs from mutant and control cell lines were electrophoresed through native polyacrylamide gel, electroblotted, and hybridized with DIG-labeled oligonucleotide probes specific for the tRNAHis, tRNALeu(UUR), and ct-RNAHis, respectively.
These transcripts were then assessed for conformational change by PAGE analysis under denaturing and native conditions. As shown in Fig. 2B, electrophoretic patterns showed that the mutant (C68) tRNAHis transcript migrated faster than the WT (U68) tRNAHis transcript under native conditions. However, there were no differences in the migration pattern between WT (U68) and mutant (C68) tRNAHis transcripts under denaturing conditions. To further test whether the m.12201T>C mutation affected the conformation of tRNAHis in vivo, total RNAs isolated from mutant and control cybrids were electrophoresed through 10% native polyacrylamide gel in Tris-glycine buffer and then electroblotted onto a positively charged nylon membrane for hybridization analysis with digoxigenin (DIG)-labeled oligodeoxynucleotide probes for tRNAHis and tRNALeu(UUR), respectively. As shown in Fig. 2C, electrophoretic patterns showed that the tRNAHis in mutant cybrids carrying the m.12201T>C mutation migrated much faster than control cybrids lacking this mutation.
Furthermore, the probes specific for tRNAHis, tRNALeu(UUR), and cytosol-tRNAHis were validated using total RNAs isolated from mutant (E1) and control (C9) cybrids as well as 143B cells and derived mtDNA-less ρo206 cell lines. As shown in Fig. 2D, mitochondrial tRNAHis and tRNALeu(UUR) were only absent in ρ0206 cells, whereas the ct-tRNAHis were present in the ρ0206 as well as 143B, C9, and E1 cells. These data suggested that the structural alterations by the m.12201T>C mutation caused the conformational change of tRNAHis.
Subcellular location of human HARS2
To determine the subcellular localization of human HARS2, pEGFP-N1-HARS2 expressing the HARS2-GFP fusion protein was transfected into the 143B cell line. Fig. 3A shows that the immunofluorescence pattern of transfected 143B cells was double-labeled with an mAb specific for the GFP and Mitotrack probes, which contain a mildly thiol-reactive chloromethyl moiety for labeling mitochondria. A typical mitochondrial staining pattern was observed, and superimposition of two panels showed the complete overlap of two patterns, demonstrating that human HARS2 localizes exclusively at mitochondria.
Figure 3.

Subcellular localization of human HARS2 in 143B cells. A, cells were transiently transfected with a HARS2 cDNA fused with GFP. The fusion protein was visualized by indirect immunofluorescence using antibodies to GFP. Mitotracker Red–stained mitochondria and 4′,6-diamidino-2-phenylindole–stained nuclei were identified by red and blue fluorescence respectively. Scale bars, 50 μm. B, Western blot analysis of mitochondrial aminoacyl-tRNA synthetases in these four transfectants (C9V (vector only), C9H (exogenous HARS2), E1V (vector only), and E1H (exogenous HARS2)) and their parental cell lines E1 and C9. Twenty micrograms of total cellular proteins from various cell lines were electrophoresed through a denaturing polyacrylamide gel, electroblotted, and hybridized with HARS2, LARS2, and SARS2, respectively, and with TOM20 as a loading control.
Construction of stable transfectants expressing the human HARS2
A 1.6-kb human HARS2 cDNA expressed in a pCDH-puro vector or the vector only was transfected into the mutant cybrid cell line (E1) carrying the m.12201T>C mutation and control cell line (C9) lacking this mutation (18). These stable transfectants were isolated by culturing cells in DMEM supplemented with 1 μg/ml puromycin and 10% FBS for 2 weeks.
The expression levels of the HARS2 cDNA in resultant stable transfectants were examined by Western blot analysis, as shown in Fig. 3B. The levels of exogenous HARS2 in transfectants C9 and E1 were more than 8-fold higher than those of our own HARS2. These four transfectants [C9V (vector only), C9H (exogenous HARS2), E1V (vector only), and E1H (exogenous HARS2)] were then used for further characterization.
To test whether HARS2 overexpression affected the expression of other mitochondrial aminoacyl-tRNA synthetases, the levels of LARS2 (leucyl-tRNA synthetase 2) and SARS2 (seryl-tRNA synthetase 2) were determined by Western blot analysis. As illustrated in Fig. 3B, the levels of LARS2 and SARS2 in C9H and E1H cell lines were comparable with those in four other cell lines lacking the exogenous HARS2 expression. This suggested that the overexpression of HARS2 did not affect the expression levels of these mitochondrial aminoacyl-tRNA synthetases.
Enhancing aminoacylation capacity of mitochondrial tRNAHis
We investigated the effects of the transfer of human HARS2 on the aminoacylation of tRNAHis in various cell lines ex vivo. The aminoacylation levels of tRNAHis in these cell lines were determined by using electrophoresis in an acid polyacrylamide/urea gel system to separate uncharged and charged tRNA species, electroblotting and hybridizing with specific probes for tRNAHis as well as tRNALeu(UUR), tRNAMet, tRNAIle, and tRNASer(AGY), respectively. To further distinguish nonaminoacylated tRNA from aminoacylated tRNA, samples of mitochondrial tRNAs were deacylated by being heated for 10 min at 60 °C at pH 8.3 and then run in parallel (30, 31). As shown in Fig. 4, the overexpression of HARS2 increased the aminoacylated levels of tRNAHis mutant cell line E1 but not those of control cell line C9. In particular, the proportions of aminoacylated tRNAs in the E1H cell lines were 75.0, 67.5, 61.6, 43.6, and 47.9% in the tRNAHis, tRNALeu(UUR), tRNAMet, tRNAIle, and tRNASer(AGY), respectively, whereas 56.3% of tRNAHis, 67.6% of tRNALeu(UUR), 59.6% of tRNAMet, 44.3% of tRNAIle, and 47.8% of tRNASer(AGY) were aminoacylated in the parental E1 cell lines. In contrast, there were no significant differences in the aminoacylated levels of these five tRNAs between the C9H cell lines and parental C9 cell lines.
Figure 4.

In vivo aminoacylation assays. A, 10 μg of total cellular RNAs purified from various cell lines under acid conditions were electrophoresed at 4 °C through an acid (pH 5.2) 10% polyacrylamide, 7 m urea gel, electroblotted, and hybridized with a DIG-labeled oligonucleotide probe specific for the tRNAHis. The blots were then stripped and rehybridized with tRNALeu(UUR), tRNAMet, tRNAIle, and tRNASer(AGY), respectively. Samples from control and mutant cell lines were deacylated (DA) by heating for 10 min at 60 °C at pH 8.3, electrophoresed, and hybridized with DIG-labeled oligonucleotide probes specific for the tRNAHis and tRNALeu(UUR). B, in vivo aminoacylated proportions of tRNAHis, tRNALeu(UUR), tRNAMet, tRNAIle, and tRNASer(AGY) in six cell lines. The calculations were based on three independent determinations. Error bars, S.D. values. p indicates the significance, according to the analysis of variance test, of the differences for various cell lines. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
The increasing steady-state levels of tRNAs
To evaluate whether the overexpression of HARS2 enhanced the levels of tRNAHis in transfectants carrying the m.12201T>C mutation, we subjected mitochondrial RNAs from various cell lines to Northern blot analysis and hybridized them with a DIG-labeled oligodeoxynucleotide probe specific for tRNAHis, five other tRNAs and nucleus-encoded 5S rRNA for normalization (32–34). For comparison, the average level of each tRNA in control or mutant cell lines was normalized to the average levels in the same cell line for reference 5S rRNA. As shown in Fig. 5, the steady-state levels of tRNAHis in the C9V, C9H, E1, E1V, and E1H cell lines were 105.4, 119.7, 41.8, 42.3, and 55.9% of the control cell line C9, respectively. Surprisingly, the overexpression of HARS2 also elevated the levels of other tRNAs. As shown in Fig. 5 (A and B), the levels of tRNAAla, tRNAGln, tRNAGlu, tRNALeu(UUR), tRNALys, and tRNAMet in the transfectant cell line E1H were 117.1, 105.6, 103.7, 103.0, 125.0, and 118.3% of the average values of the parental cell line E1, respectively. Similarly, the levels of tRNAAla, tRNAGln, tRNAGlu, tRNALeu(UUR), tRNALys, and tRNAMet in the cell line C9H were 108.4, 95.5, 105.5, 105.9, 127.2, and 122.0% of the average values of the parental cell line C9. This suggested that overexpression of HARS2 increased the levels of tRNAHis and other tRNAs.
Figure 5.

Northern blot analysis of mitochondrial tRNA under denaturing conditions. A, 5 μg of total cellular RNA from various cell lines were electrophoresed through a denaturing polyacrylamide gel, electroblotted, and hybridized with DIG-labeled oligonucleotide probes for the tRNAHis, tRNAAla, tRNAGln, tRNAGlu, tRNALeu(UUR), tRNALys, tRNAMet, and 5S rRNA, respectively. B, quantification of mitochondrial tRNA levels. Average relative tRNA content per cell was normalized to the average content per cell of 5S rRNA. The values for the latter are expressed as percentages of the average values for the control cell line C9. The calculations were based on three independent determinations. C, decay kinetics of tRNAHis in two transfectants (C9H and E1H) and their parental cell lines (C9 and E1). tRNA levels were measured using ImageJ analysis of Northern blots probed for tRNAHis and 5S rRNA using RNA isolated from cells at various time courses during EtBr treatment. RNA levels expressed as a fraction of the signal obtained from a panel of replicates taken at zero time (the time at which EtBr was added) were converted to logarithms on the assumption of first-order decay kinetics. All hybridization signals were normalized to 5S rRNA as the loading control. The data plotted represent the mean ± S.D. (error bars) of three independent experiments. Lines of best fit (least squares method) are shown, r2 for the four panels (clockwise, from top left) being 0.991, 0.987, 0.989, and 0.945, respectively. Graph details and symbols are explained in the legend to Fig. 4.
To investigate whether there was an effect of m.12201T>C mutation on the half-life of tRNAHis, we treated the various cell lines with EtBr (250 ng/ml) to block mitochondrial RNA synthesis (35) and extracted total cellular RNA at various time points after the addition of the drug. We then subjected mitochondrial RNAs from various cell lines to Northern blot analysis and hybridized them with a DIG-labeled oligodeoxynucleotide probe specific for tRNAHis and 5S rRNA as a loading control. As shown in Fig. 5C, there were no significant differences in the half-lives of tRNAHis between transfectants and parental mutant and cell lines. This suggested that m.12201T>C mutation and overexpression of HARS2 did not have a significant effect on the half-life of tRNAHis.
Northern blot analysis of mRNA and rRNAs
We then examined whether the m.12201T>C mutation affected the expression/processivity of polycistronic transcripts. RNA transfer hybridization experiments were performed with total cellular RNAs from various mutant and control cell lines, using a set of DIG-labeled RNA probes: ND6 from L-strand transcripts; COX1, COX2, CYTB, 12S rRNA, and 16S rRNA from H-strand transcripts (32, 33); and β-actin as a control, respectively. As shown in Fig. 6, the levels of ND6, COX1, COX2, CYTB, 12S rRNA, and 16S rRNA in mutant cell lines, normalized with respect to those of β-actin mRNA, were comparable with those in the control cell lines. These results indicated that the m.12201T>C mutation did not affect the expression of these mRNAs and rRNAs.
Figure 6.

Northern blot analysis of mitochondrial RNAs. A, 5 μg of total cellular RNA from various mutant and control cell lines were electrophoresed through a 1.5% agarose-formaldehyde gel; transferred onto a positively charged membrane; and hybridized with DIG-labeled RNA probes for ND6 (from L-strand transcript), COX1, COX2, CYTB, 12S rRNA, and 16S rRNA (from H-strand transcript), and β-actin as a control, respectively. B, average relative levels of above mRNAs and rRNAs per cell were normalized to the average level per cell of β-actin in three control cell lines and three mutant cell lines. The values for the latter are expressed as percentages of the average values for the control cell lines. Three independent determinations were used in the calculations. Graph details and symbols are explained in the legend to Fig. 4. Error bars, S.D.
Elevated levels of mtDNA-encoding proteins
To assess whether the overexpression of HARS2 affected the mitochondrial protein synthesis, a Western blot analysis was performed to examine the levels of eight mtDNA-encoding proteins in transfectants as well as their parental cell lines with Tom20 as a loading control. As shown in Fig. 7 (A and B), the overexpression of HARS2 significantly increased the levels of these mitochondrial proteins in the mutant cell line E1. Of these, the levels of ND1, ND4, ND5, and ND6 (subunits 1, 4, 5, and 6 of NADH dehydrogenase (complex I)); CYTB (cytochrome b of ubiquinone cytochrome c oxidoreductase (complex III)); CO2 (subunit 2 of cytochrome c oxidase (complex IV)); and ATP6 and ATP8 (subunits 6 and 8 of the H+-ATPase (complex V)) in E1H cell lines were 66.4, 100.9, 66.0, 97.6, 108, 53.3, 90.0, and 85.1%, with the average 77.4% of the control cell line C9, whereas those in E1 cell lines were 43.7, 55.6, 52.4, 103.7, 70.4, 35.6, 62.0, and 89.8% with the average 43.0% of the control cell line C9. In contrast, the overexpression of HARS2 did not significantly affect the levels of mitochondrial proteins in the control cell line C9.
Figure 7.

Western blot analysis of mitochondrial proteins. A, 20 μg of total cellular proteins from various cell lines were electrophoresed through a denaturing polyacrylamide gel, electroblotted, and hybridized with eight respiratory complex subunits in six cell lines with TOM20 as a loading control. ND1, ND4, ND5, and ND6, subunits 1, 4, 5, and 6 of NADH dehydrogenase (complex I); CYTB, cytochrome b of ubiquinone cytochrome c oxidoreductase (complex III); CO2, subunit 2 of cytochrome c oxidase (complex IV); ATP6 and ATP8, subunit 6 and 8 of the H+-ATPase (complex V). B, quantification of eight respiratory complex subunits. Average relative values of ND1, ND4, ND5, ND6, CYTB, CO2, ATP6, and ATP8 were normalized to the average values of TOM20 in various cell lines. The values for the latter are expressed as percentages of the average values for the control cell line C9. The calculations were based on three independent determinations. C, Western blot analysis of Afg3l2 and Clpp proteins in six cell lines with β-actin as a loading control. Graph details and symbols are explained in the legend to Fig. 4. Error bars, S.D.
To test whether the m.12201T>C-induced deficiency affected the mitochondrial proteostasis, we measured the levels of Clpp involved in mitochondrial ribosome assembly (36) and ATP family gene 3–like 2 (Afg3l2) proteases involved in the turnover of misfolded proteins markers for proteostasis stress (37), in the various cell lines. As shown in Fig. 7C and Fig. S1, there was no significant difference in the levels of Afg3l2 and Clpp among mutant and control cell lines. These data indicated that m.12201T>C mutation may not affect the proteostasis stress.
The restoration of respiration deficiency
To evaluate whether the overexpression of HARS2 rescued the respiratory deficiency caused by the m.12201T>C mutation, we measured the activities of respiratory complexes by the use of isolating mitochondria of various cell lines. Complex I activity was measured by following the oxidation of NADH with ubiquinone as the electron acceptor (38, 39). The activity of complex II (succinate ubiquinone oxidoreductase) exclusively encoded by the nuclear DNA was examined by the artificial electron acceptor DCPIP (40, 41). Complex III (ubiquinone cytochrome c oxidoreductase) activity was measured as the reduction of cytochrome c (III) using d-ubiquinol-2 as the electron donor. The activity of complex IV was monitored by following the oxidation of cytochrome c (II). As shown in Fig. 8, the overexpression of HARS2 enhanced the activities of complex I, III, and IV but not complex II in cells carrying the m.12201T>C mutation. As shown in Table S1, the activities of complexes I, III, and IV in the E1H cell line were 150.2, 145.3, and 209.3% of those in the parental cell line E1, respectively. By contrast, the activities of complexes I, III, and IV in the C9H cell line were 89.4, 107.6, and 139.5% of those in the parental C9 cell line, respectively.
Figure 8.
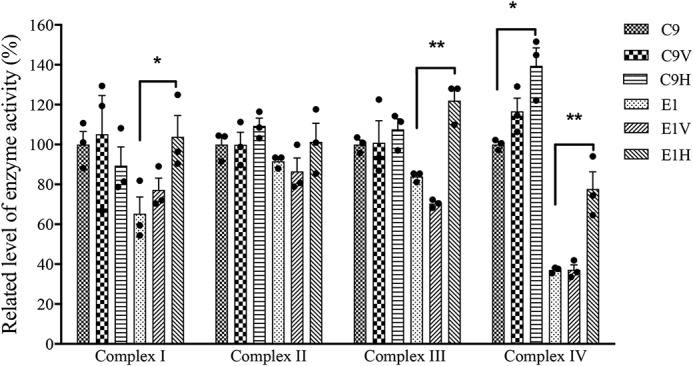
Enzymatic activities of respiratory chain complexes. The activities of respiratory complexes were investigated by enzymatic assay on complexes I–IV in mitochondria isolated from various cell lines. First, specific enzyme activities were normalized to citrate synthase activity. The values for the latter are expressed as percentages of the average values for the control cell line C9. The calculations were based on three independent experiments. Graph details and symbols are explained in the legend to Fig. 4. Error bars, S.D.
Oxygen consumption rate (OCR) is an indicator of mitochondrial respiration. Using a Seahorse Bioscience XF-96 extracellular flux analyzer, we can measure mitochondrial respiratory control, including basal respiration, O2 consumption attributed to ATP production, proton leak, maximum respiratory rate, reserve capacity, and nonmitochondrial respiration (42, 43). As shown in Fig. 9, the basal OCR in the E1H cell line increased 47.9% compared with those in the parental cell line E1, whereas the basal OCR of C9H was comparable with those in the C9 cell line. The drug-insensitive OCRs were then measured after the sequential addition of oligomycin (to inhibit the ATP synthase), carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (to uncouple the mitochondrial inner membrane and allow for maximum electron flux through the ETC), rotenone (to inhibit complex I), and antimycin A (to inhibit complex III). As shown in Fig. 9, the ATP-linked OCR, maximal OCR and reserve capacity OCR in the E1H cell line significantly increased 67.4, 89.5, and 144.7%, as compared with those in the parental cell line E1, respectively. However, basal OCR, ATP-linked OCR, maximal OCR, and reserve capacity OCR in the E1H cell line were significantly lower than those in the C9H cell line. By contrast, the OCRs for C9H cell line were comparable with those of the parental C9 cell line. These data indicated that the respiration of the E1H cell line was significantly improved but not restored to the levels of WT cell line.
Figure 9.

Respiration assays. A, analysis of O2 consumption in the various cell lines using different inhibitors. OCRs were first measured on 2 × 104 cells of each cell line under basal conditions, and then sequentially oligomycin (1.5 μm), FCCP (0.5 μm), rotenone (1 μm), and antimycin A (1 μm) were added at the indicated times to determine different parameters of mitochondrial functions. B, graphs present the ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and nonmitochondrial OCR in six cell lines. Nonmitochondrial OCR was determined as the OCR after rotenone/antimycin A treatment. Basal OCR was determined as OCR before oligomycin minus OCR after rotenone/antimycin A. ATP-lined OCR was determined as OCR before oligomycin minus OCR after oligomycin. Proton leak was determined as basal OCR minus ATP-linked OCR. Maximal was determined as the OCR after FCCP minus nonmitochondrial OCR. Reserve capacity was defined as the difference of maximal OCR after FCCP minus basal OCR. The data were based on three determinations for each cell line. Graph details and symbols are explained in the legend to Fig. 4. Error bars, S.D.
Increasing levels of mitochondrial ATP
To further examine the effect of overexpression of HARS2 on the oxidative phosphorylation, we used the luciferin/luciferase assay to measure the levels of cellular and mitochondrial ATP. Populations of cells were incubated in the media in the presence of glucose and of 2-deoxy-d-glucose with pyruvate (18). As shown in Fig. 10, the levels of mitochondrial ATP (the presence of pyruvate and 2-deoxy-d-glucose to inhibit the glycolysis) in the E1H cell line were ∼133% of those in the parental cell line E1, whereas the levels of mitochondrial ATP in the C9H cell line were 103% of those in the parental C9 cell line, respectively. On the contrary, the overexpression of HARS2 did not significantly change the levels of total cellular ATP (the presence of glucose) in the mutant and control cell lines.
Figure 10.
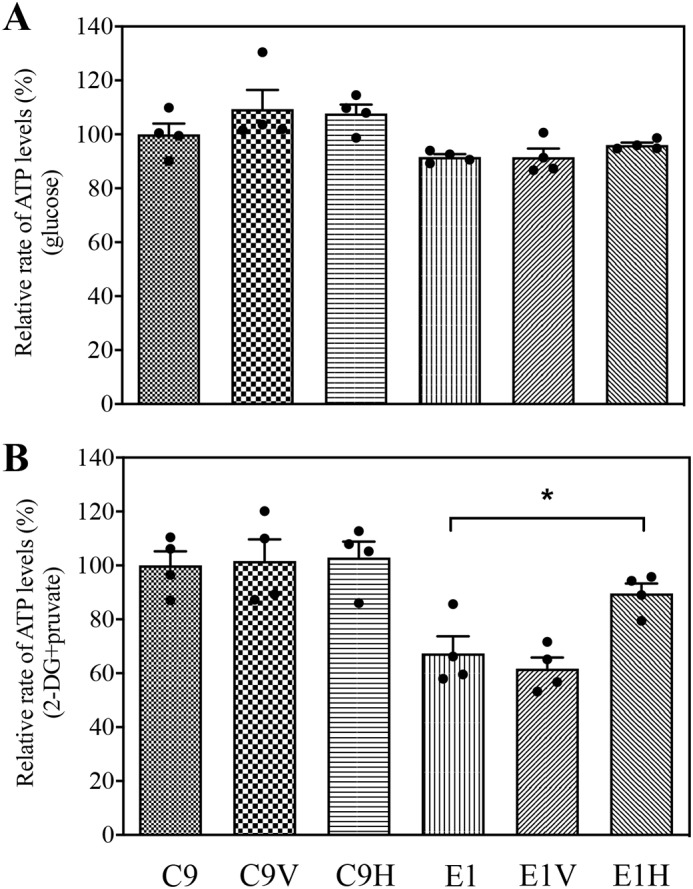
Measurement of cellular and mitochondrial ATP levels using a bioluminescence assay. Cells were incubated with 10 mm glucose or 5 mm 2-deoxy-d-glucose plus 5 mm pyruvate to determine ATP generation under mitochondrial ATP synthesis. Average rates of ATP level per cell line are shown: ATP levels in total cells (A) and ATP levels in mitochondria (B). Four determinations were made for each cell line. Graph details and symbols are explained in the legend to Fig. 4. Error bars, S.D.
Enhancement of mitochondrial membrane potential
The mitochondrial membrane potential (ΔΨm) generated by complexes I, III, and IV is an essential component in the process of energy storage during oxidative phosphorylation (44). The ΔΨm levels in various cell lines were measured using a fluorescence probe JC-10 assay via flow cytometry. As shown in Fig. 11, the ΔΨm of the C9V, C9H, E1, E1V, and E1H cell lines was 101.0, 93.5, 53.6, 60.4, and 86.5% relative to the mean values in the control cell line C9, respectively. In contrast, the populations with a normal level of ΔΨm in these six cell lines were comparable with those in the presence of FCCP.
Figure 11.

Mitochondrial membrane potential analysis. A, ΔΨm was measured in six cell lines by a BD-LSR II flow cytometer system using a fluorescence probe JC-10 assay system. The ratios of fluorescence intensity excitation/emission = 490/590 and 490/530 nm (FL590/FL530) were recorded to delineate the ΔΨm level of each sample. Represented are flow cytometry images of cell lines E1V and E1H with and without 10 μm FCCP. B, the ratios of the population with a normal level of ΔΨm relative to the control cell line C9 were calculated to reflect the level of ΔΨm. The average of three determinations for each cell line is shown. Graph details and symbols are explained in the legend to Fig. 4. Error bars, S.D.
Overexpression of HARS2 reduced the production of mitochondrial ROS
The levels of mitochondrial ROS among these cells were determined using a MitoSOX assay via flow cytometry (45). Geometric mean intensity was recorded to measure and delineate the rate of ROS of each sample. As shown in Fig. 12 (A and B), the levels of mitochondrial ROS production in the cell lines C9V, C9H, E1, E1V, and E1H were 90.5, 87.0, 163.3, 147.7, and 109.2% relative to the mean values in the control cell line C9, respectively.
Figure 12.

Assays for mitochondrial ROS production. A, ratio of geometric mean intensity between levels of the ROS generation in the vital cells. The rates of production in ROS from six cell lines were analyzed by a BD-LSR II flow cytometer system using MitoSox (5 μm). B, the relative ratio of intensity was calculated. The average of three independent determinations for each cell line is shown. C, Western blot analysis of antioxidative enzymes Sod2, Sod1, and catalase in six cell lines with β-actin as a loading control. D, quantification of Sod2, Sod1, and catalase. Average relative values of Sod2, Sod1, and catalase were normalized to the average values of β-actin in various cell lines. The values for the latter are expressed as percentages of the average values for the control cell line C9. The average of three independent determinations for each cell lines is shown. Graph details and symbols are explained in the legend to Fig. 4. Error bars, S.D.
To test whether the m.12201T>C mutation–induced mitochondrial ROS production affected the antioxidant systems, we examined the levels of three antioxidant enzymes, SOD2 in the mitochondrion and SOD1 and catalase in the cytosol (46), in the various cell lines. As shown in Fig. 12C, the mutant cell lines E1 and E1V revealed marked increases in the levels of SOD2 and mild increases in the levels of SOD1 and catalase, as compared with those in the WT cell line C9. Notably, overexpression of HARS2 in the mutant cell line led to pronounced reductions in the level of SOD2 but relatively mild decreases in the levels of SOD1 and catalase.
Discussion
The objective of this study was to further elucidate the molecular pathogenesis of the deafness-associated tRNAHis 12201T>C mutation and to test whether human HARS2 overexpression in the cybrid cells carrying the m.12201T>C mutation reverses the mitochondrial dysfunctions. The m.12201T>C mutation destabilized the canonical A5-U68 base-pairing within the aminoacyl acceptor stem of this mitochondrial tRNAHis (18, 21, 47, 48). Thus, we hypothesized that the m.12201T>C mutation led to the structural and functional effects on this tRNA, including the alteration of conformation, thermal stability, and aminoacylation. MD studies indicated that the U68 formed a canonical base pair with A5 of WT tRNAHis through hydrogen bonds with an occupancy of 47 and 64%, respectively, whereas the newly formed interaction between A5 and C68 in the mutant tRNAHis was decreased to one hydrogen bond with a lower occupancy of 38% (49). These findings indicated that the mutant tRNAHis molecule with the mismatch of 5A and C68 may be less stable than those in the WT counterpart with the canonical A5-U68 base-pairing. In fact, the Tm in mutant tRNAHis molecule was 4 °C lower than those in the WT counterpart. The instability of the mutant tRNA molecule was further evidenced by the drastically reduced level of tRNAHis in the mutant cell lines carrying the m.12201T>C mutation (18). Furthermore, the m.12201T>C mutation caused the conformational change of tRNAHis, as suggested by faster electrophoretic mobility of mutated tRNA with respect to the WT molecule in vitro or ex vivo, consistent with the conformational changes of tRNAs carrying the m.4435A>G and m.3253T>C mutations (50, 51). However, the m.12201T>C mutation did not affect the expression/processivity of polycistronic transcripts, in contrast with the aberrant processing of the polycistronic transcripts observed in the cell lines carrying the tRNALeu(UUR) 3243A>G and tRNASer(UCN) 7445A>G mutations and the m.4401A>G mutation in the precursor of tRNAMet/tRNAGln mutations (15, 24, 52). Moreover, the abolishment of A5:U68 base-pairing substitution may affect the tRNAHis interaction with mitochondrial histidyl-tRNA synthetase, thereby altering the aminoacylation properties of tRNAHis by either charging inefficiently or mischarging with mitochondrial histidyl-tRNA synthetase (53, 54). In this study, the cell line bearing the m.12201T>C mutation displayed increasing levels of aminoacylated tRNAHis and faster electrophoretic mobility of mutated tRNA with respect to the WT cell line (18). The mutant tRNAHis may be metabolically less stable and subject to turnover, thereby lowering the steady-state level of tRNAHis. As a result, a failure in tRNA metabolism is responsible for defective mitochondrial protein synthesis and an impaired oxidative phosphorylation and increasing production of oxidative reactive species (18, 48). The resultant mitochondrial dysfunction would lead to the dysfunction or death of cochlear cells, thereby producing a phenotype of hearing loss.
In the present investigation, we have shown that the overexpression of human HARS2 in the cybrid cells carrying the m.12201T>C mutation corrected the mitochondrial dysfunctions. In particular, the overexpression of HARS2 in the mutant cybrids bearing the m.12201T>C mutation raised the level of aminoacylated tRNAHis from 56.3 to 75.0% but did not change the aminoacylation levels of other tRNAs. Furthermore, overexpression of HARS2 did not increase significantly the level of aminoacylated tRNAHis in the WT cybrids, suggesting that 50% of aminoacylated tRNAHis in cells appeared to be the maximum threshold level to maintain the normal function, as in the case of overexpression of LARS2 in the WT cybrid cell line HSI (24). The increasing proportions of aminoacylated tRNA led to the elevating steady-state level of tRNAHis in the cybrids harboring the m.12201T>C mutation from 41.8 to 55.9% of those in the control cybrids lacking the mtDNA mutation. The increasing levels of mutant tRNAHis were likely due to the improvement of stability and function of mutant tRNAHis. It is worthwhile to note that overexpression of HARS2 also enhanced the steady-state levels of noncognate tRNAs, such as tRNAAla, tRNAGln, tRNAGlu, tRNALeu(UUR), tRNALys, and tRNAMet, but did not affect the levels of mRNAs and rRNAs in cells bearing the m.12201T>C mutation. In fact, overexpression of LARS2 increased the levels of mutated tRNAVal in the cells in the cybrid cell line carrying the m.1624C>T mutation (26). These findings suggested that the overexpression of HARS2 may mediate the noncognate tRNA metabolisms in cells carrying the pathogenic tRNA mutation. The improved efficiency of aminoacylation and stability of mitochondrial tRNAs by overexpression of HARS2 resulted in ∼80% increasing levels of mitochondrial translation in the cybrid cell line harboring the m.12201T>C mutation. The restoration of deficient mitochondrial translation by overexpression of HARS2 appeared to be more efficient than those in myoblasts carrying the m.3243A>G mutation by overexpression of EFTu and EFG2 (55). The facilitated synthesis of these mtDNA-encoding polypeptides improved the respiration capacity. In particular, the enhancing levels of OCR and mitochondrial ATP in the cybrid bearing the m.12201T>C mutation by overexpression of HARS2 were correlated well with the increasing levels of mitochondrial protein, suggesting that the improvement of mitochondrial translation was responsible for the ameliorated oxidative phosphorylation deficiency. Furthermore, the elevating activities of respiratory chain complexes caused by overexpression of HARS2 yielded ∼61.4% increase of ΔΨm in the cell line bearing m.12201T>C mutation. Indeed, ΔΨm reflects the pumping of hydrogen ions across the inner membrane during the process of electron transport and oxidative phosphorylation (44). The improvement of both OXPHOS and ΔΨm would reduce the production of ROS and expression of SOD2 in mutant cells carrying the m.12201T>C mutation. The lower production of ROS can reduce a vicious cycle of oxidative stress in the mitochondria, thereby decreasing the damage of mitochondrial and cellular proteins, lipids, and nuclear acids (56).
In summary, our results demonstrated that overexpression of HARS2 corrected the mitochondrial dysfunction caused by the deafness-associated tRNAHis 12201T>C mutation. The biochemical phenotypes manifested by interplay between tRNAHis 12201T>C mutation and HARS2 gene may provide new insights into the pathophysiology of maternally inherited deafness. The restoration of m.12201T>C mutation–induced mitochondrial dysfunctions by overexpression of HARS2 may be a step toward therapeutic interventions for these disorders.
Experimental procedures
Cell lines and culture conditions
The 143B.TK− cell line, the mutant cybrid cell lines (E1) carrying the m.12201T>C mutation, and control cybrid cell lines (C9) belonging to the same mtDNA haplogroup Z3 but lacking the mutation (H7) were grown in DMEM (containing 4.5 mg of glucose and 0.11 mg of pyruvate per ml), supplemented with 5% FBS (18). The mtDNA-less ρ°206 cell line, derived from 143B.TK− (57) was grown under the same conditions as the parental line, except for the addition of 50 μg of uridine/ml. The stable transfectants were grown in DMEM supplemented with 10% FBS and 1 μg/ml puromycin.
Molecular dynamics simulation procedure
The acceptor stem of tRNAHis containing 15 nucleotide bases spanning the A5-68U/C base-pairing (Fig. 1A) was constructed as the model for simulations. The initial coordinates for WT were extracted from the crystal structure of human mitochondrial tRNAPhe-PheRS complex (Protein Data Bank entry 3TUP). The coordinates of the backbones were maintained, and nucleotide bases were substituted with the mitochondrial tRNAHis sequence by Chimera (58). In the mutated structure, the coordinates of U to C substitution at position 68 were generated by Chimera using the established WT model.
The acceptor stems of WT and mutant tRNAHis were simulated using the same methods. The ff14SB force field parameters were employed in MD simulations with Amber14 (59). The initial models were surrounded by TIP3P water molecules and ions. The simulation systems were maintained in a solution containing 50 mm NaCl, made by the addition of 3Na+ and 3Cl−, and then 13 Na+ for neuralization. The SHAKE algorithm was used to constrain hydrogen atoms (60). Alternately, we minimized and equilibrated the whole system to relieve all unfavorable interactions of the initial model. The procedure of equilibration in NVT ensemble was heated to 300 K and was not subjected to pressure. NPT ensemble was applied to equilibrate the solvent at the final equilibration step, in which the temperature was kept at 300 K and pressure at 1 bar in periodic boundary conditions. Subsequently, 100-ns production simulations were performed for both systems with a time step of 2 fs. The MD trajectory of each system was observed with VMD (61). Extraction and analysis of trajectories were performed using the cpptraj program in Amber14. Distance cutoff for hydrogen bond and angle cutoff were set to 3.0 Å and 135°, respectively.
Measurement of melting temperature
UV melting assays were carried out as described previously (50, 51). The WT and mutant tRNAHis transcripts were produced using in vitro transcription by T7 RNA polymerase according to previous protocols (62). The tRNAHis transcripts were dissolved in 50 mm sodium phosphate buffer (pH 7.0), containing 50 mm NaCl, 5 mm MgCl2, and 0.1 mm EDTA. Absorbance against melting temperature curves was measured at 260 nm with a heating rate of 1 °C/min from 25 to 95 °C via an Agilent Cary 100 UV spectrophotometer.
Isolation of human HARS2 cDNA
To construct the plasmid pHARS2 containing the entire coding region of HARS2 cDNA, RT-PCR was performed by using TaqDNA polymerase (Promega) and total RNA isolated from 143B cells as template, with the primers 5′-ATAATGTCTGACCCGCCTCCTT-3′ (nt 159–176) and 5′-CCTTGGTGGCTAGTTGTTGTGA (nt 1705–1720) (GenBankTM accession no. NM_001278732.1). The predominant PCR product was purified by agarose gel electrophoresis and subsequently cloned into a pGEM-T vector (Promega). Nucleotide sequence was determined by Sanger sequencing.
Subcellular localization of human HARS2
The coding region of HARS2 cDNA lacking its natural stop codon was obtained by PCR using pHARS2 cDNA as the template. Primers 5′-CCGCTCGAGCCGGCGTCCTGCCGC (nt 204–220) and 5′-CCGACCGGTGATCCAGACTCAGACAG (nt 2870–2891) were used for the PCR amplification. PCR products were digested with AgeI and XhoI and then cloned into pEGFP-N1. After sequence determination, Resultant constructs were transfected into 143B cells using the jetPRIMETM transfection reagent (Polyplus Transfection) according to the manufacturer's protocol. Immunofluorescence analysis was performed as detailed elsewhere (63, 64).
Construction of stable transfectants
The insert of pHARS2 was subcloned into pCDH-puro (Invitrogen). The resultant constructs or vector only were transfected into E1 and C9 cell lines using the jetPRIMETM transfection reagent (Polyplus Transfection) according to the manufacturer's protocol. The stable transfectants were isolated by culturing cells in DMEM supplemented with 1 μg/ml puromycin and 10% FBS for 2 weeks. The resultant clones were examined for the expression of HARS2 by Western blot analysis.
Mitochondrial tRNA analysis
For the tRNA Northern blot analysis, the isolation of total cellular RNAs, gel electrophoresis, electroblotting, and hybridization were as detailed elsewhere (18, 24, 45, 65–67). DIG-labeled probes of tRNAHis, tRNAAla, tRNAGln, tRNAGlu, tRNALeu(UUR), tRNALys, tRNAMet, and 5S rRNA for hybridization were as described elsewhere (18, 24, 45, 52). Quantification of density in each band was made as detailed previously (18, 24, 45).
For the tRNA mobility shift assay, 2 or 5 μg of RNAs were electrophoresed through a 10% polyacrylamide native gel at 4 °C with 50 mm Tris-glycine buffer. After electrophoresis, the gels were treated according to the Northern blot analysis as described above (50, 51).
The aminoacylation assays including the isolation of total cellular and gel electrophoresis were as detailed elsewhere (30, 45). The gels were then electroblotted onto a positively charged nylon membrane (Roche Applied Science) for the hybridization analysis with oligodeoxynucleotide probes as described above. Quantification of the density in each band was performed as detailed previously (30, 45).
tRNA half-life measurements were performed as detailed elsewhere (35). Briefly, various cell lines were incubated in fresh medium containing 250 ng/ml EtBr for the times indicated in Fig. 5C. Ten micrograms of total cellular RNAs, extracted as above, were subjected to Northern blot analysis as detailed above.
Mitochondrial RNA Northern blot analysis
Eight micrograms of total cellular RNAs were fractionated by electrophoresis through a 1.5% agarose-formaldehyde gel, transferred onto a positively charged membrane (Roche Applied Science), and hybridized with DIG-labeled RNA probes: ND6, COX1, COX2, CYTB, 12S rRNA, 16S rRNA, and β-actin as a control, respectively. Probes were synthesized on the corresponding restriction enzyme linearized plasmid using a DIG RNA-labeling kit (Roche Applied Science). The plasmids used for RNA probes were constructed by PCR-amplifying fragments of ND6 (positions 14343–14618), COX1 (positions 7146–7425), COX2 (positions 7823–8156), CYTB (positions 14824–15208), 12S rRNA (positions 1201–1235), 16S rRNA (positions 2245–2635), and β-actin (positions 69–618, NM 001101.5) and cloning these fragments into the pCRII-TOPO vector (16, 52, 68).
Western blot analysis
Western blot analysis was carried out as detailed previously (18, 67, 69, 70). Twenty micrograms of total proteins obtained from lysed mitochondria were denatured and loaded onto SDS-polyacrylamide gels. The gels were electroblotted onto a polyvinylidene difluoride membrane for hybridization. The antibodies used for this investigation were from Abcam (TOM20 (ab56783), ND1 (ab74257), ND5 (ab92624), A6 (ab101908), and CO2 (ab110258)), Santa Cruz Biotechnology, Inc. (ND4 (sc-20499-R) and ND6 (sc-20667)), Proteintech (CYTB (55090-1-AP), ATP8 (26723-1-AP), HARS2 (11301-1-AP), LASRS2 (17097-1-AP), SARS2 (17258-1-AP), AFG3L2 (14631-1-AP), and CLPP (15698-1-AP)), and Cell Signaling Technology (SOD2 (13141), SOD1 (4266), and catalase (12980)). Peroxidase Affini Pure goat anti-mouse IgG and goat anti-rabbit IgG (Jackson) were used as secondary antibodies, and protein signals were detected using the ECL system (Millipore). Quantification of density in each band was performed as detailed previously (18, 50).
Enzymatic assays
The enzymatic activities of complexes I–IV were measured as detailed elsewhere (38–40, 67).
Measurements of oxygen consumption
The rates of oxygen consumption in lymphoblastoid cell lines were assayed with a Seahorse Bioscience XF-96 extracellular flux analyzer (Seahorse Bioscience), as detailed previously (42, 43, 67). The protein content of each well was then measured to normalize OCR values.
Measurements of ATP levels
The Cell Titer-Glo® luminescent cell viability assay kit (Promega) was used for the measurement of cellular and mitochondrial ATP levels, following the modified manufacturer's instructions (18, 51).
Assessment of mitochondrial membrane potential
Mitochondrial membrane potential was assessed with the JC-10 assay kit-microplate (Abcam) according to the manufacturer's general recommendations with some modifications, as detailed elsewhere (44, 51).
ROS measurements
ROS measurements were conducted as detailed previously (45, 51).
Statistical analysis
Statistical analysis was performed by the analysis of variance test contained in the StatView program SAS (version 9.4) (SAS Institute) and entering individual replicate values. Unless indicated otherwise, a p value < 0.05 was considered statistically significant.
Author contributions
S. G., X. W., F. M., L. C., Q. Y., Q. Z., X. C., Z. C., and M.-X. G. data curation; S. G. and M.-X. G. funding acquisition; S. G. validation; S. G., X. W., and M.-X. G. investigation; S. G., F. M., Q. Y., and X. C. methodology; S. G. writing-original draft; X. W. and J. Q. M. visualization; F. M., L. C., Q. Y., Q. Z., X. C., J. Q. M., and Y. L. formal analysis; Y. L. and M.-X. G. resources; M.-X. G. conceptualization; M.-X. G. supervision; M.-X. G. project administration; M.-X. G. writing-review and editing.
Supplementary Material
Acknowledgments
The MD simulation was carried out at the National Supercomputer Center in Tianjin, and the calculations were performed on TianHe-1A.
This work was supported by Ministry of Science and Technology of Zhejiang Province Grant 2018C03026; National Key Technologies R&D Program Grant 2014CB541704 from the Ministry of Science and Technology of China (to M. X. G.); National Natural Science Foundation of China Grants 81330024 (to M. X. G), 31671305, 81470685, and 81500804 (to S. S. G.); Natural Science Foundation of Zhejiang Province, China, Grants LY18C060003 (to S. S. G.) and LY18C050002 (to X. H. C.); and Science and Technology Bureau of Taizhou City, Zhejiang Province, China, Grant 1901ky78 (to S. S. G.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Table S1 and Fig. S1.
- mtDNA
- mitochondrial DNA
- ROS
- reactive oxygen species
- DIG
- digoxigenin
- FBS
- fetal bovine serum
- OCR
- oxygen consumption rate
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- nt
- nucleotides.
References
- 1. Fischel-Ghodsian N. (1999) Mitochondrial deafness mutations reviewed. Hum. Mutat. 13, 261–270 [DOI] [PubMed] [Google Scholar]
- 2. Guan M. X. (2011) Mitochondrial 12S rRNA mutations associated with aminoglycoside ototoxicity. Mitochondrion 11, 237–245 10.1016/j.mito.2010.10.006 [DOI] [PubMed] [Google Scholar]
- 3. Zheng J., Ji Y., and Guan M. X. (2012) Mitochondrial tRNA mutations associated with deafness. Mitochondrion 12, 406–413 10.1016/j.mito.2012.04.001 [DOI] [PubMed] [Google Scholar]
- 4. Area-Gomez E., and Schon E. A. (2014) Mitochondrial genetics and disease. J. Child Neurol. 29, 1208–1215 10.1177/0883073814539561 [DOI] [PubMed] [Google Scholar]
- 5. Ruiz-Pesini E., Lott M. T., Procaccio V., Poole J. C., Brandon M. C., Mishmar D., Yi C., Kreuziger J., Baldi P., and Wallace D. C. (2007) An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 35, D823–D828 10.1093/nar/gkl927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guan M. X. (2004) Molecular pathogenetic mechanism of maternally inherited deafness. Ann. N.Y. Acad. Sci. 1011, 259–271 10.1196/annals.1293.025 [DOI] [PubMed] [Google Scholar]
- 7. Prezant T. R., Agapian J. V., Bohlman M. C., Bu X., Oztas S., Qiu W. Q., Arnos K. S., Cortopassi G. A., Jaber L., Rotter J. I., Shohat M., and Fischel-Ghosian N. (1993) Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat. Genet. 4, 289–294 10.1038/ng0793-289 [DOI] [PubMed] [Google Scholar]
- 8. Zhao H., Li R., Wang Q., Yan Q., Deng J. H., Han D., Bai Y., Young W. Y., and Guan M. X. (2004) Maternally inherited aminoglycoside-induced and nonsyndromic deafness is associated with the novel C1494T mutation in the mitochondrial 12S rRNA gene in a large Chinese family. Am. J. Hum. Genet. 74, 139–152 10.1086/381133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lu J., Li Z., Zhu Y., Yang A., Li R., Zheng J., Cai Q., Peng G., Zheng W., Tang X., Chen B., Chen J., Liao Z., Yang L., Li Y., et al. (2010) Mitochondrial 12S rRNA variants in 1642 Han Chinese pediatric subjects with aminoglycoside-induced and nonsyndromic hearing loss. Mitochondrion 10, 380–390 10.1016/j.mito.2010.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abbott J. A., Francklyn C. S., and Robey-Bond S. M. (2014) Transfer RNA and human disease. Front. Genet. 5, 158 10.3389/fgene.2014.00158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suzuki T., Nagao A., and Suzuki T. (2011) Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet. 45, 299–329 10.1146/annurev-genet-110410-132531 [DOI] [PubMed] [Google Scholar]
- 12. Goto Y., Nonaka I., and Horai S. (1990) A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348, 651–653 10.1038/348651a0 [DOI] [PubMed] [Google Scholar]
- 13. Mancuso M., Orsucci D., Angelini C., Bertini E., Carelli V., Comi G. P., Minetti C., Moggio M., Mongini T., Servidei S., Tonin P., Toscano A., Uziel G., Bruno C., Caldarazzo Ienco E., et al. (2013) Phenotypic heterogeneity of the 8344A>G mtDNA “MERRF” mutation. Neurology 80, 2049–2054 10.1212/WNL.0b013e318294b44c [DOI] [PubMed] [Google Scholar]
- 14. Wang M., Liu H., Zheng J., Chen B., Zhou M., Fan W., Wang H., Liang X., Zhou X., Eriani G., Jiang P., and Guan M. X. (2016) A deafness and diabetes associated tRNA mutation caused the deficient pseudouridinylation at position 55 in tRNAGlu and mitochondrial dysfunction. J. Biol. Chem. 291, 21029–21041 10.1074/jbc.M116.739482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guan M. X., Enriquez J. A., Fischel-Ghodsian N., Puranam R. S., Lin C. P., Maw M. A., and Attardi G. (1998) The deafness-associated mitochondrial DNA mutation at position 7445, which affects tRNASer(UCN) precursor processing, has long-range effects on NADH dehydrogenase subunit ND6 gene expression. Mol. Cell. Biol. 18, 5868–5879 10.1128/MCB.18.10.5868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li X., Fischel-Ghodsian N., Schwartz F., Yan Q., Friedman R. A., and Guan M. X. (2004) Biochemical characterization of the mitochondrial tRNASer(UCN) T7511C mutation associated with nonsyndromic deafness. Nucleic Acids Res. 32, 867–877 10.1093/nar/gkh226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xue L., Chen Y., Tang X., Yao J., Huang H., Wang M., Ye S., Wang M., and Guan M. X. (2019) A deafness-associated mitochondrial DNA mutation altered the tRNASer(UCN) metabolism and mitochondrial function. Mitochondrion 46, 370–379 10.1016/j.mito.2018.10.001 [DOI] [PubMed] [Google Scholar]
- 18. Gong S., Peng Y., Jiang P., Wang M., Fan M., Wang X., Zhou H., Li H., Yan Q., Huang T., and Guan M. X. (2014) A deafness-associated tRNAHis mutation alters the mitochondrial function, ROS production and membrane potential. Nucleic Acids Res. 42, 8039–8048 10.1093/nar/gku466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang M., Peng Y., Zheng J., Zheng B., Jin X., Liu H., Wang Y., Tang X., Huang T., Jiang P., and Guan M. X. (2016) A deafness-associated tRNAAsp mutation alters the m1G37 modification, aminoacylation, and stability of tRNAAsp and mitochondrial function. Nucleic Acids Res. 44, 10974–10985 10.1093/nar/gkw726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gutiérrez Cortés N., Pertuiset C., Dumon E., Börlin M., Hebert-Chatelain E., Pierron D., Feldmann D., Jonard L., Marlin S., Letellier T., and Rocher C. (2012) Novel mitochondrial DNA mutations responsible for maternally inherited nonsyndromic hearing loss. Hum. Mutat. 33, 681–689 10.1002/humu.22023 [DOI] [PubMed] [Google Scholar]
- 21. Lovato M. A., Chihade J. W., and Schimmel P. (2001) Translocation within the acceptor helix of a major tRNA identity determinant. EMBO J. 20, 4846–4853 10.1093/emboj/20.17.4846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Giegé R. Sissler M. and Florentz C. (1998) Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 26, 5017–5035 10.1093/nar/26.22.5017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Park H., Davidson E., and King M. P. (2008) Overexpressed mitochondrial leucyl-tRNA synthetase suppresses the A3243G mutation in the mitochondrial tRNALeu(UUR) gene. RNA 14, 2407–2416 10.1261/rna.1208808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li R., and Guan M. X. (2010) Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes. Mol. Cell. Biol. 30, 2147–2154 10.1128/MCB.01614-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perli E., Giordano C., Pisano A., Montanari A., Campese A. F., Reyes A., Ghezzi D., Nasca A., Tuppen H. A., Orlandi M., Di Micco P., Poser E., Taylor R. W., Colotti G., Francisci S., et al. (2014) The isolated carboxy-terminal domain of human mitochondrial leucyl-tRNA synthetase rescues the pathological phenotype of mitochondrial tRNA mutations in human cells. EMBO Mol. Med. 6, 169–182 10.1002/emmm.201303198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hornig-Do H. T., Montanari A., Rozanska A., Tuppen H. A., Almalki A. A., Abg-Kamaludin D. P., Frontali L., Francisci S., Lightowlers R. N., and Chrzanowska-Lightowlers Z. M. (2014) Human mitochondrial leucyl tRNA synthetase can suppress non cognate pathogenic mt-tRNA mutations. EMBO Mol. Med. 6, 183–193 10.1002/emmm.201303202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rorbach J., Yusoff A. A., Tuppen H., Abg-Kamaludin D. P., Chrzanowska-Lightowlers Z. M., Taylor R. W., Turnbull D. M., McFarland R., and Lightowlers R. N. (2008) Overexpression of human mitochondrial valyl tRNA synthetase can partially restore levels of cognate mt-tRNAVal carrying the pathogenic C25U mutation. Nucleic Acids Res. 36, 3065–3074 10.1093/nar/gkn147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao X., Han J., Zhu L., Xiao Y., Wang C., Hong F., Jiang P., and Guan M. X. (2018) Overexpression of human mitochondrial alanyl-tRNA synthetase suppresses biochemical defects of the mt-tRNAAla mutation in cybrids. Int. J. Biol. Sci. 14, 1437–1444 10.7150/ijbs.27043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pierce S. B., Chisholm K. M., Lynch E. D., Lee M. K., Walsh T., Opitz J. M., Li W., Klevit R. E., and King M. C. (2011) Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. U.S.A. 108, 6543–6548 10.1073/pnas.1103471108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Enríquez J. A., and Attardi G. (1996) Analysis of aminoacylation of human mitochondrial tRNAs. Methods Enzymol. 264, 183–196 10.1016/S0076-6879(96)64019-1 [DOI] [PubMed] [Google Scholar]
- 31. Jia Z., Zhang Y., Li Q., Ye Z., Liu Y., Fu C., Cang X., Wang M., and Guan M. X. (2019) A coronary artery disease-associated tRNAThr mutation altered mitochondrial function, apoptosis and angiogenesis. Nucleic Acids Res. 47, 2056–2074 10.1093/nar/gky1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ojala D., Montoya J., and Attardi G. (1981) tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474 10.1038/290470a0 [DOI] [PubMed] [Google Scholar]
- 33. Mercer T. R., Neph S., Dinger M. E., Crawford J., Smith M. A., Shearwood A. M., Haugen E., Bracken C. P., Rackham O., Stamatoyannopoulos J. A., Filipovska A., and Mattick J. S. (2011) The human mitochondrial transcriptome. Cell 146, 645–658 10.1016/j.cell.2011.06.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Magalhães P. J., Andreu A. L., and Schon E. A. (1998) Evidence for the presence of 5S rRNA in mammalian mitochondria. Mol. Biol. Cell 9, 2375–2382 10.1091/mbc.9.9.2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Toompuu M., Yasukawa T., Suzuki T., Hakkinen T., Spelbrink J. N., Watanabe K., and Jacobs H. T. (2002) The 7472insC mitochondrial DNA mutation impairs the synthesis and extent of aminoacylation of tRNASer(UCN) but not its structure or rate of turnover. J. Biol. Chem. 277, 22240–22250 10.1074/jbc.M200338200 [DOI] [PubMed] [Google Scholar]
- 36. Szczepanowska K., Maiti P., Kukat A., Hofsetz E., Nolte H., Senft K., Becker C., Ruzzenente B., Hornig-Do H. T., Wibom R., Wiesner R. J., Krüger M., and Trifunovic A. (2016) CLPP coordinatesmitoribosomal nucleotides is required for cloverleaf folding of a human mitochondrial tRNA assembly through the regulation of ERAL1 levels. EMBO J. 35, 2566–2583 10.15252/embj.201694253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nolden M., Ehses S., Koppen M., Bernacchia A., Rugarli E. I., and Langer T. (2005) The m-AAA maturation proteasedefective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell 123, 277–289 10.1016/j.cell.2005.08.003 [DOI] [PubMed] [Google Scholar]
- 38. Zhang J., Ji Y., Lu Y., Fu R., Xu M., Liu X., and Guan M. X. (2018) Leber's hereditary optic neuropathy (LHON)-associated ND5 12338T>C mutation altered the assembly and function of complex I, apoptosis and mitophagy. Hum. Mol. Genet. 27, 1999–2011 10.1093/hmg/ddy107 [DOI] [PubMed] [Google Scholar]
- 39. Li Y., D'Aurelio M., Deng J. H., Park J. S., Manfredi G., Hu P., Lu J., and Bai Y. (2007) An assembled complex IV maintains the stability and activity of complex I in mammalian mitochondria. J. Biol. Chem. 282, 17557–17562 10.1074/jbc.M701056200 [DOI] [PubMed] [Google Scholar]
- 40. Thorburn D. R., Chow C. W., and Kirby D. M. (2004) Respiratory chain enzyme analysis in muscle and liver. Mitochondrion 4, 363–375 10.1016/j.mito.2004.07.003 [DOI] [PubMed] [Google Scholar]
- 41. Scheffler I. E. (2015) Mitochondrial disease associated with complex I (NADH-CoQ oxidoreductase) deficiency. J. Inherit. Metab. Dis. 38, 405–415 10.1007/s10545-014-9768-6 [DOI] [PubMed] [Google Scholar]
- 42. Dranka B. P., Benavides G. A., Diers A. R., Giordano S., Zelickson B. R., Reily C., Zou L., Chatham J. C., Hill B. G., Zhang J., Landar A., and Darley-Usmar V. M. (2011) Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med. 51, 1621–1635 10.1016/j.freeradbiomed.2011.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brand M. D., and Nicholls D. G. (2011) Assessing mitochondrial dysfunction in cells. Biochem. J. 435, 297–312 10.1042/BJ20110162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reers M., Smiley S. T., Mottola-Hartshorn C., Chen A., Lin M., and Chen L. B. (1995) Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 260, 406–417 10.1016/0076-6879(95)60154-6 [DOI] [PubMed] [Google Scholar]
- 45. Jiang P., Wang M., Xue L., Xiao Y., Yu J., Wang H., Yao J., Liu H., Peng Y., Liu H., Li H., Chen Y., and Guan M. X. (2016) A hypertension-associated tRNAAla mutation alters tRNA metabolism and mitochondrial function. Mol. Cell. Biol. 36, 1920–1930 10.1128/MCB.00199-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schieber M., and Chandel N. S. (2014) ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–R462 10.1016/j.cub.2014.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Florentz C., Sohm B., Tryoen-Tóth P., Pütz J., and Sissler M. (2003) Human mitochondrial tRNAs in health and disease. Cell Mol. Life Sci. 60, 1356–1375 10.1007/s00018-003-2343-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yan X., Wang X., Wang Z., Sun S., Chen G., He Y., Mo J. Q., Li R., Jiang P., Lin Q., Sun M., Li W., Bai Y., Zhang J., Zhu Y., et al. (2011) Maternally transmitted late-onset non-syndromic deafness is associated with the novel heteroplasmic T12201C mutation in the mitochondrial tRNAHis gene. J. Med. Genet. 48, 682–690 10.1136/jmedgenet-2011-100219 [DOI] [PubMed] [Google Scholar]
- 49. Grosjean H., and Westhof E. (2016) An integrated, structure- and energy-based view of the genetic code. Nucleic Acids Res. 44, 8020–8040 10.1093/nar/gkw608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou M., Wang M., Xue L., Lin Z., He Q., Shi W., Chen Y., Jin X., Li H., Jiang P., and Guan M. X. (2017) A hypertension-associated mitochondrial DNA mutation alters the tertiary interaction and function of tRNALeu(UUR). J. Biol. Chem. 292, 13934–13946 10.1074/jbc.M117.787028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou M., Xue L., Chen Y., Li H., He Q., Wang B., Meng F., Wang M., and Guan M. X. (2018) A hypertension-associated mitochondrial DNA mutation introduces an m1G37 modification into tRNAMet, altering its structure and function. J. Biol. Chem. 293, 1425–1438 10.1074/jbc.RA117.000317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhao X., Cui L., Xiao Y., Mao Q., Aishanjiang M., Kong W., Liu Chen Y. H., Hong H., Jia Z., Wang M., Jiang P., and Guan M. X. (2019) Hypertension-associated mitochondrial DNA 4401A>G mutation caused the aberrant processing of tRNAMet, all 8 tRNAs and ND6 mRNA in the light-strand transcript. Nucleic Acids Res. 47, 10340–10356 10.1093/nar/gkz742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kuncha S. K., Mazeed M., Singh R., Kattula B., Routh S. B., and Sankaranarayanan R. (2018) A chiral selectivity relaxed paralog of DTD for proofreading tRNA mischarging in Animalia. Nat. Commun. 9, 511 10.1038/s41467-017-02204-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hao R., Zhao M. W., Hao Z. X., Yao Y. N., and Wang E. D. (2005) A T-stem slip in human mitochondrial tRNALeu(CUN) governs its charging capacity. Nucleic Acids Res. 33, 3606–3613 10.1093/nar/gki677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sasarman F., Antonicka H., and Shoubridge E. A. (2008) The A3243G tRNALeu(UUR) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially suppressed by overexpression of EFTu and EFG2. Hum. Mol. Genet. 17, 3697–3707 10.1093/hmg/ddn265 [DOI] [PubMed] [Google Scholar]
- 56. Raimundo N., Song L., Shutt T. E., McKay S. E., Cotney J., Guan M. X., Gilliland T. C., Hohuan D., Santos-Sacchi J., and Shadel G. S. (2012) Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell 148, 716–726 10.1016/j.cell.2011.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. King M. P., and Attadi G. (1996) Mitochondria-mediated transformation of human rho0 cells. Methods Enzymol. 264, 313–334 10.1016/S0076-6879(96)64030-0 [DOI] [PubMed] [Google Scholar]
- 58. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 59. Case D. A., Cheatham T. E. 3rd, Darden T., Gohlke H., Luo R., Merz K. M. Jr., Onufriev A., Simmerling C., Wang B., and Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 10.1002/jcc.20290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ryckaert J. P., Ciccotti G., and Berendsen H. J. C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 10.1016/0021-9991(77)90098-5 [DOI] [Google Scholar]
- 61. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–27 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 62. Li Y., Chen J., Wang E., and Wang Y. (1999) T7 RNA polymerase transcription of Escherichia coli isoacceptors tRNALeu. Sci. China C Life Sci. 42, 185–190 10.1007/BF02880055 [DOI] [PubMed] [Google Scholar]
- 63. Li X., Li R., Lin X., and Guan M. X. (2002) Isolation and characterization of the putative nuclear modifier gene MTO1 involved in the pathogenesis of deafness-associated mitochondrial 12S rRNA A1555G mutation. J. Biol. Chem. 277, 27256–27264 10.1074/jbc.M203267200 [DOI] [PubMed] [Google Scholar]
- 64. Li X., and Guan M. X. (2002) A human mitochondrial GTP binding protein related to tRNA modification may modulate the phenotypic expression of the deafness-associated mitochondrial 12S rRNA mutation. Mol. Cell. Biol. 22, 7701–7711 10.1128/MCB.22.21.7701-7711.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. King M. P., and Attardi G. (1993) Post-transcriptional regulation of the steady-state levels of mitochondrial tRNAs in HeLa cells. J. Biol. Chem. 268, 10228–10237 [PubMed] [Google Scholar]
- 66. Fan W., Zheng J., Kong W., Cui L., Aishanjiang M., Yi Q., Wang M., Cang X., Tang X., Chen Y., Mo J. Q., Sondheimer N., Ge W., and Guan M. X. (2019) Contribution of a mitochondrial tyrosyl-tRNA synthetase mutation to the phenotypic expression of the deafness-associated tRNASer(UCN) 7511A>G mutation. J. Biol. Chem. 294, 19292–19305 10.1074/jbc.RA119.010598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Meng F., He Z., Tang X., Zheng J., Jin X., Zhu Y., Ren X., Zhou M., Wang M., Gong S., Mo J. Q., Shu Q., and Guan M. X. (2018) Contribution of the tRNAIle 4317A>G mutation to the phenotypic manifestation of the deafness-associated mitochondrial 12S rRNA 1555A>G mutation. J. Biol. Chem. 293, 3321–3334 10.1074/jbc.RA117.000530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Andrews R. M., Kubacka I., Chinnery P. F., Lightowlers R. N., Turnbull D. M., and Howell N. (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 23, 147 10.1038/13779 [DOI] [PubMed] [Google Scholar]
- 69. Jiang P., Jin X., Peng Y., Wang M., Liu H., Liu X., Zhang Z., Ji Y., Zhang J., Liang M., Zhao F., Sun Y. H., Zhang M., Zhou X., Chen Y., et al. (2016) The exome sequencing identified the mutation in YARS2 encoding the mitochondrial tyrosyl-tRNA synthetase as a nuclear modifier for the phenotypic manifestation of Leber's hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum. Mol. Genet. 25, 584–596 10.1093/hmg/ddv498 [DOI] [PubMed] [Google Scholar]
- 70. Meng F., Cang X., Peng Y., Li R., Zhang Z., Li F., Fan Q., Guan A. S., Fischel-Ghosian N., Zhao X., and Guan M. X. (2017) Biochemical evidence for a nuclear modifier allele (A10S) in TRMU (Methylaminomethyl-2-thiouridylate-methyltransferase) related to mitochondrial tRNA modification in the phenotypic manifestation of deafness-associated 12S rRNA mutation. J. Biol. Chem. 292, 2881–2892 10.1074/jbc.M116.749374 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.