

Abstract
The mismatch repair (MMR) complex is composed of MutSα (MSH2-MSH6) and MutLα (MLH1-PMS2) and specifically recognizes mismatched bases during DNA replication. O6-Methylguanine is produced by treatment with alkylating agents, such as N-methyl-N-nitrosourea (MNU), and during DNA replication forms a DNA mismatch (i.e. an O6-methylguanine/thymine pair) and induces a G/C to A/T transition mutation. To prevent this outcome, cells carrying this DNA mismatch are eliminated by MMR-dependent apoptosis, but the underlying molecular mechanism is unclear. In this study, we provide evidence that the chromatin-regulatory and ATP-dependent nucleosome-remodeling protein SMARCAD1 is involved in the induction of MMR-dependent apoptosis in human cells. Unlike control cells, SMARCAD1-knockout cells (ΔSMARCAD1) were MNU-resistant, and the appearance of a sub-G1 population and caspase-9 activation were significantly suppressed in the ΔSMARCAD1 cells. Furthermore, the MNU-induced mutation frequencies were increased in these cells. Immunoprecipitation analyses revealed that the recruitment of MutLα to chromatin-bound MutSα, observed in SMARCAD1-proficient cells, is suppressed in ΔSMARCAD1 cells. Of note, the effect of SMARCAD1 on the recruitment of MutLα exclusively depended on the ATPase activity of the protein. On the basis of these findings, we propose that SMARCAD1 induces apoptosis via its chromatin-remodeling activity, which helps recruit MutLα to MutSα on damaged chromatin.
Keywords: apoptosis, chromatin remodeling, DNA damage response, DNA mismatch repair, mutL homolog 1 (MLH1), CRISPR/Cas9, N-methyl-N-nitrosourea, O6-methylguanine, SMARCAD1, DNA damage signaling, O6-methylguanine, SMARCAD1
Introduction
DNA mismatches are produced constantly at low levels during DNA replication and cause base substitution mutations unless they are repaired. O6-Methylguanine produced by the action of alkylating agents, such as N-methyl-N-nitrosourea (MNU)2 and N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), can pair with thymine as well as cytosine and lead to G/C to A/T transition mutations after the next round of DNA replication (1–3). To prevent such an outcome, organisms—including humans—utilize a specific repair enzyme, O6-methylguanine-DNA methyltransferase (MGMT), which removes a methyl group from the O6-methylguanine moiety, thereby repairing the DNA lesion in a single-step reaction (4, 5). MGMT-defective cells, therefore, accumulate O6-methylguanines on the genome and exhibit hypersensitivity to the killing effect after exposure to alkylating agents (6). An analysis of the response of MGMT-defective cells following the treatment with alkylating agents revealed that O6-methylguanine is cytotoxic and induces apoptosis in a mismatch repair (MMR) protein-dependent manner (7–9).
The MMR system is important for the suppression of tumorigenesis, and MMR defects in humans are frequently associated with inherited cancer predisposition (10–12). In addition to repairing replicational errors, MMR complex, which is composed of MutSα (MSH2-MSH6) and MutLα (MLH1-PMS2) in human cells, recognizes O6-methylguanine/thymine pairs formed during DNA replication and induces apoptosis to eliminate cells carrying mutation-evoking DNA lesions (13–18). It is noteworthy that mice with mutations in both alleles of the Mgmt and Mlh1 genes are as resistant to MNU as are WT mice in terms of the survival but are much more susceptible to MNU-induced tumorigenesis than WT mice (19). Consistent with these results, Mgmt−/− Mlh1−/− cells, derived from the gene-targeted mice, are unable to induce apoptosis and show an elevated mutation frequency after MNU treatment (20).
We recently reported that the high-mobility group A nonhistone chromatin proteins, HMGA1 and HMGA2, are involved in the DNA damage signaling associated with the induction of apoptosis, such as the phosphorylation of ATR and CHK1, although HMGA proteins are dispensable for the formation of MMR complex after the administration of MNU (21). HMGA proteins bound to chromatin are shown to recruit chromatin remodeling factors and histone chaperons, thereby enhancing the alteration of chromatin structure (22). HMGA1- and HMGA2-knockdown cells showed an increased resistance to MNU, and the appearance of a sub-G1 population and caspase-9 activation were suppressed in those cells. In contrast, chromatin assembly factor 1 (CAF-1) is reported to suppress the activity of the MMR system in the cytotoxic response to the alkylating agent, MNNG (23). CAF-1 is the major histone chaperone for the assembly of nucleosomes onto newly replicated DNA (24). It seems that the CAF-1–dependent incorporation of DNA containing the O6-methylguanine/thymine pair into nucleosomes counteracts the activation of the MMR-mediated apoptotic pathway.
SMARCAD1 is an ATP-dependent chromatin remodeling factor that binds and hydrolyzes ATP and regulates histone modification. Indeed, a mutant form of SMARCAD1 that lacks ATPase activity causes aberrant histone modifications and is unable to maintain heterochromatin silencing (25–27). The factor also promotes end resection at the site of DNA double-strand break in yeast and humans and is involved in the repair reaction of the lesion (28–31). SMARCAD1 also interacts with proliferating cell nuclear antigen (PCNA) and MSH2 and is enriched on newly replicative DNA (27, 32, 33). A biochemical analysis using Xenopus egg extract revealed that SMARCAD1 is recruited to mismatch-carrying DNA in a Msh2-dependent manner and assists in the elimination of local nucleosomes to facilitate the repair of mismatch base pair (34). However, the role of SMARCAD1 in MMR-dependent apoptosis in humans remains to be elucidated.
We herein report evidence suggesting that the chromatin remodeler SMARCAD1 is a novel factor associated with the induction of MMR-dependent apoptosis through the enhancement of the interaction of MLH1 with MSH2 on damaged chromatin.
Results
Decreased sensitivity of hSMARCAD1-knockout cells to MNU treatment
Because SMARCAD1 is physically associated with the MMR protein MSH2 (33), this factor may play a role in the MMR-dependent apoptosis. To examine this possibility, an SMARCAD1-knockout cell line (ΔSMARCAD1) derived from HeLa MR was generated using the CRISPR/Cas9 genome-editing system (35). An immunoblotting analysis revealed that the expression of SMARCAD1 was completely abolished in ΔSMARCAD1, but the expression of MMR components, such as MSH2, MSH6, MLH1, and PMS2, were unaffected compared with HeLa MR (Fig. 1A). An MLH1-knockout cell line (ΔMLH1) was also generated using the same genome-editing system, and the expression of related proteins was confirmed by immunoblotting. Using these cell lines, we performed a survival assay with various doses of MNU. As shown in Fig. 1B, ΔSMARCAD1 showed a significantly high level of resistance to MNU compared with the parental cell line HeLa MR. ΔMLH1, on the other hand, completely lost the ability to induce apoptosis. This is also the case for U2OS, another human-derived cell line (Fig. S1). MGMT-proficient U2OS cells were resistant to treatment with MNU. In contrast, an MGMT-knockout cell line (U2OSΔM) readily underwent cell death after exposure to such doses of MNU, indicating that the cell death resulted from O6-methylguanine. SMARCAD1- and MLH1-knockdown cells, into which synthetic siRNAs for each gene had been introduced, also showed increased and complete tolerance, respectively, to treatment with MNU (Fig. S1B). When exposed to other DNA-damaging agents (UV (UV-C), hydrogen peroxide (H2O2), and etoposide, which produce primarily pyrimidine dimers, oxidative bases, and DNA double-strand breaks, respectively), ΔSMARCAD1 cells showed an increased sensitivity to etoposide, as expected from a previous report (28), but a similar degree of sensitivity to UV-C and H2O2 compared with HeLa MR (Fig. 1C). These results imply that ΔSMARCAD1 cells might have a defect in the induction of cell death caused by MNU-induced O6-methylguanine.
Figure 1.

The sensitivities of SMARCAD1-knockout cells to different types of DNA-damaging agents. A, expression of SMARCAD1 and mismatch repair (MMR) proteins in MLH1- and SMARCAD1-knockout cells. The whole-cell extracts were subjected to immunoblotting to detect SMARCAD1 and (MMR) proteins using specific antibodies. β-Actin was a loading control. The molecular weights (× 10−3) are indicated on the left of the panels. B, survival fraction of three types of cell lines after MNU treatment. The cells were treated with MNU for 1 h and then incubated in a drug-free medium for 10 days. The number of colonies was counted, and the survival fractions were determined. Values are the means of at least three independent experiments, and error bars represent S.E. *, significant differences (p < 0.05). C, survival fraction of SMARCAD1-knockout cells treated with other DNA damaging reagents. The cells were treated with etoposide for 12 h or H2O2 for 1 h. For UV irradiation, the cells were exposed to different doses of UV-C. The mean values obtained from at least three independent experiments and the S.E. values are shown. *, significant differences (p < 0.05).
The involvement of SMARCAD1 in MNU-induced apoptosis
To determine whether or not SMARCAD1 is involved in MNU-induced apoptosis, we analyzed the degrees of phosphorylation of ATR, CHK1, ATM, and CHK2, which are known to be activated when the cells are treated with MNU (36, 37). The MNU-induced phosphorylation of these proteins was clearly observed after treatment with MNU in HeLa MR cells, although such phosphorylation was hardly seen in ΔMLH1 cells. In ΔSMARCAD1 cells, the phosphorylation of these proteins was significantly decreased compared with HeLa MR (Fig. 2, A and B). These results indicate that the loss of function of SMARCAD1 impairs MNU-induced DNA damage signaling.
Figure 2.

The effects of SMARCAD1 knockout on the activation of DNA damage signaling. A, activation of DNA damage signaling after exposure to MNU. Three types of cell lines were treated with 0.2 mm MNU for 1 h and then collected at the indicated times. The whole-cell extracts were prepared and subjected to SDS-PAGE, followed by immunoblotting using the specific antibodies indicated. β-Actin was the loading control. The molecular weights (× 10−3) are indicated on the left of the panels. B, relative intensities of signals for the phosphorylation of ATR, CHK1, ATM, and CHK2. The intensities of the bands for pATR, pCHK1, pATM, and pCHK2 at 48 h after MNU treatment were quantified, and the relative intensities compared with that of HeLa MR are shown. The mean values obtained from three independent experiments and the S.E. values (error bars) are shown. *, significant differences (p < 0.01).
We further examined the effect of SMARCAD1-knockout on the appearance of a sub-G1 population, which is known to occur during the process of apoptosis (38). As shown in Fig. 3A, the sub-G1 population increased gradually after treatment with MNU, but the degree of increase in ΔSMARCAD1 cells was significantly lower than that of HeLa MR. In ΔSMARCAD1 cells, the sub-G1 population (24.90%) at day 4 was 70% of that in HeLa MR (35.54%). In ΔMLH1 cells, on the other hand, the sub-G1 population was dramatically diminished even after treatment with MNU. To obtain further evidence that ΔSMARCAD1 cells are defective in the induction of apoptosis, we examined the cleavage of caspase-9 and PARP1 (39–41). An immunoblotting analysis using an antibody that recognizes both pro-caspase-9 and cleaved caspase-9 revealed that the cleavage of caspase-9 started at 72 h and became more evident at 96 h after MNU treatment in HeLa MR (Fig. 3B). In contrast, in ΔSMARCAD1 cells, the ratio of signal for cleaved caspase-9 against pro-caspase-9 at 96 h was significantly lower than that in HeLa MR. Furthermore, cleavage of PARP1 following MNU treatment was also suppressed in ΔSMARCAD1 cells compared with HeLa MR. In contrast, both of these proteins were hardly cleaved in ΔMLH1 cells even after MNU treatment (Fig. 3C). These results support the notion that ΔSMARCAD1 cells have a defect in a certain step of the O6-methylguanine–induced apoptosis pathway.
Figure 3.

The involvement of SMARCAD1 in the MNU-induced apoptosis. A, sub-G1 population in SMARCAD1-knockout cells after MNU treatment. HeLa MR, SMARCAD1-knockout, and MLH1-knockout cells were treated with 0.2 mm MNU for 1 h and incubated for 2, 3, and 4 days. The cells were harvested and subjected to flow cytometry. The mean values of the sub-G1 population obtained from three independent experiments and the S.E. values (error bars) are shown. *, significant differences (p < 0.05). B and C, the cleavage of caspase-9 and PARP1 after MNU treatment in SMARCAD1-knockout and MLH1-knockout cells. HeLa MR and SMARCAD1-knockout cells (B) or HeLa MR and MLH1-knockout cells (C) were treated with 0.2 mm MNU for 1 h and harvested at the indicated time after the treatment. Whole-cell extracts were prepared and subjected to SDS-PAGE followed by immunoblotting using anti-caspase-9 and anti-PARP1 antibodies. β-Actin was the loading control. The molecular weights (× 10−3) are indicated on the left of the panels.
Elevated mutation frequency in ΔSMARCAD1 cells
Because O6-methylguanine is a premutagenic DNA lesion, the mutation frequency would increase if cells carrying O6-methylguanine were not eliminated by apoptosis (20, 42). Therefore, we measured the mutation frequency of ΔSMARCAD1 cells with respect to ouabain resistance, which can arise due to a mutation in the Na+/K+ ATPase locus (Fig. 4). Without MNU treatment, HeLa MR and ΔSMARCAD1 cells exhibited similarly low levels of mutation frequency. After exposure to MNU, the mutation frequencies of both strains increased in a dose-dependent manner, but the extent of the increase in ΔSMARCAD1 cells was significantly greater than that of HeLa MR. This supports the notion that SMARCAD1 may be involved in the induction of apoptosis in cells carrying mutation-evoking DNA lesions.
Figure 4.

Increased mutation frequency after MNU treatment. HeLa MR and SMARCAD1-knockout cells were treated with 0, 15, or 30 μm MNU. After the treatment, the numbers of viable cells and ouabain-resistant cells were determined, and the mutation frequencies were calculated. The mean values obtained from three experiments and the S.E. values (error bars) are presented. *, significant differences (p < 0.05).
SMARCAD1-mediated recruitment of MutLα to MutSα on damaged chromatin
To assess the interaction of SMARCAD1 with MMR proteins, we prepared chromatin extract from HeLa MR and ΔSMARCAD1 cells following treatment with or without MNU and performed an immunoprecipitation assay using anti-SMARCAD1 antibody. As shown in Fig. 5A, SMARCAD1 bound to MSH2 regardless of MNU treatment, whereas the protein bound to MLH1 preferentially after exposure to MNU. It is noteworthy that the recruitment of SMARCAD1 to damaged chromatin depends on MHS2. As shown in Fig. 5B, SMARCAD1 was loaded onto chromatin with a peak at 12 h after treatment with MNU in HeLa MR cells, whereas such loading was hardly seen in MSH2-knockout derivatives (ΔMSH2) from HeLa MR.
Figure 5.

The effects of SMARCAD1 knockout on the interaction of MSH2 with MLH1 on damaged chromatin. A, interaction of SMARCAD1 with MSH2 and MLH1. HeLa MR and SMARCAD1-knockout cells were treated with or without 0.2 mm MNU for 1 h. The chromatin extracts (Input) prepared from cells collected at 12 and 24 h after treatment were applied to immunoprecipitation with anti-SMARCAD1 antibody conjugated with Protein G–Sepharose beads (Bound). IgG was used as a negative control. The materials were subjected to SDS-PAGE, followed by immunoblotting using specific antibodies. B, MSH2-dependent chromatin loading of SMARCAD1. HeLa MR and MSH2-knockout cells were collected at 0, 12, 24, and 48 h after MNU treatment and subjected to biochemical fractionation. The supernatant and chromatin fractions were subjected to SDS-PAGE followed by immunoblotting using specific antibodies. ORC2 and MEK2 serve as markers of chromatin and supernatant fractions, respectively. C, effects of SMARCAD1 knockout on the formation of MMR complex. The cells were exposed to 0.2 mm MNU and harvested at 0, 6, 12, and 24 h after the treatment. The chromatin extracts (Input) were used for immunoprecipitation with anti-MSH2 antibody beads. The materials (Bound) were subjected to SDS-PAGE and immunoblotted using the indicated antibodies. D, amounts of MLH1 interacting with MSH2 after MNU treatment. Band intensities of MLH1 in the bound fraction in C were divided by that of MSH2, and the relative values after MNU treatment were plotted as means ± S.E. of four independent experiments. *, significant differences (p < 0.05). The molecular weights (× 10−3) are indicated on the left of the panels in A, B, and C.
Next, to analyze the effect of SMARCAD1 deficiency on the formation of MMR complex, chromatin extracts at each time point were employed for an immunoprecipitation assay using anti-MSH2 antibody (Fig. 5C). In SMARCAD1-proficient HeLa MR, chromatin-bound MLH1 gradually increased (Fig. 5C, Input), and MSH2 formed complex with greater amounts of MLH1 on chromatin after MNU treatment (Fig. 5C, Bound). In contrast, the increases in the amounts of MLH1 interacting with MSH2 were significantly suppressed in ΔSMARCAD1 cells. The intensity of bands for MLH1 associated with MSH2 in HeLa MR was increased almost 5-fold at 24 h after MNU treatment, whereas only a 2-fold increase was observed in ΔSMARCAD1 cells (Fig. 5D). When SMARCAD1-knockdown cells derived from U2OS were treated with MNU, a smaller amount of MLH1 was also recruited to MSH2 than to control cells (Fig. S3). These results suggest an important role of SMARCAD1 in recruiting MLH1 on damaged chromatin to form MMR complex that recognizes DNA mismatches. It should be noted that an association between SMARCAD1 and MLH1 was observed in MSH2-proficient cells, but not in ΔMSH2 cells (Fig. S4).
Requirement of ATPase activity of SMARCAD1 for the formation of MMR complex
SMARCAD1 exerts chromatin remodeling activity in an ATP hydrolysis–dependent manner (27, 34). To determine whether or not this activity is required for the induction of apoptosis triggered by O6-methylguanine, we introduced amino acid substitution, altering the conserved lysine residue at position 526 of the Walker A motif to arginine (27), to produce an ATPase-defective form of SMARCAD1, as illustrated in Fig. 6A. Cell lines expressing FLAG-tagged WT SMARCAD1 (WT) or the variant form of SMARCAD1 (K528R) were constructed on a ΔSMARCAD1 cell background, in which the expression of the proteins was comparable with that in HeLa MR cells (Fig. S2). The constructed cells were then used to analyze the effect of these proteins on the sensitivity to the treatment of MNU. As shown in Fig. 6B, two independent SMARCAD1-expressing cell lines (WT #1 and #2) restored the sensitivities to MNU to the same level as in HeLa MR, as expected, whereas cell lines expressing the variant form of SMARCAD1 (K528R #1 and #2) still exhibited an MNU tolerance similar to that of ΔSMARCAD1 cells. An immunoblotting analysis further revealed that SMARCAD1-expressing cells induce the cleavage of both caspase-9 and PARP1 with kinetics similar to those of HeLa MR, whereas such inductions were significantly suppressed in cells expressing the mutant form of the protein (Fig. 6C). Furthermore, the association of MSH2 with MLH1 on the damaged chromatin at 24 h after MNU treatment was significantly suppressed in cells expressing the mutant form of SMARCAD1, to an extent similar to that of ΔSMARCAD1 (Fig. 6D).
Figure 6.

Requirement of ATPase activity of SMARCAD1 for MMR-induced apoptosis. A, structure of human SMARCAD1 protein. The position of amino acid substitution K528R in the ATP-binding motif of the protein is indicated. B, survival fraction of cells expressing WT or mutant SMARCAD1 after MNU treatment. The cells were treated with MNU for 1 h; the number of colonies formed was then counted, and the survival fractions were determined. Values are the means of at least three independent experiments, and error bars indicate S.E. *, significant differences (p < 0.05). C, ATPase activity of SMARCAD1 relevant to apoptotic induction. Whole-cell extracts were prepared from cells treated with MNU and subjected to SDS-PAGE, followed by immunoblotting using anti-caspase-9 and anti-PARP1 antibodies. β-Actin was the loading control. The molecular weights (× 10−3) are indicated on the left of the panels. D, ATPase activity of SMARCAD1 relevant to the formation of MMR complex. The relative amounts of MLH1 co-immunoprecipitated with MSH2 after MNU treatment were calculated as described in the legend to Fig. 5D and are represented as means ± S.E. of six independent experiments. *, significant differences (p < 0.05).
Taken together, these findings indicate that ATPase-related activity of SMARCAD1 is indispensable for facilitating the formation of MMR complex, which is a crucial event at the first step of the induction of apoptosis triggered by O6-methylguanine.
Discussion
MMR protein complex, composed of MutSα and MutLα, plays important roles in tumor suppression by not only repairing mismatched bases formed during DNA replication (43) but also inducing apoptosis to eliminate cells carrying modified bases in DNA, such as O6-methylguanine. However, the molecular mechanism underlying the induction of apoptosis still remains unclear, especially concerning its regulation in the context of chromatin architecture.
In the present study, we showed that the chromatin remodeler SMARCAD1 is a novel factor involved in the induction of MMR-dependent apoptosis triggered by O6-methylguanine. Both SMARCAD1-knockdown and SMARCAD1-knockout cells exhibited higher resistance to MNU than SMARCAD1-proficient cells, concomitant with decreased levels of cleavage of caspase-9 and PARP1, hallmarks of the induction of apoptosis (Fig. 3). After exposure to the drug, SMARCAD1-knockout cells showed increased mutation frequencies in a dose-dependent manner, implying that SMARCAD1 plays a role in preventing genomic instability from O6-methylguanine produced in DNA (Fig. 4). In contrast, SMARCAD1-knockout cells were more sensitive to the treatment with etoposide than the parental control, consistent with the previous report showing that the factor promotes end resection at an early step of DNA double-strand break repair (Fig. 1C) (28).
When O6-methylguanine forms a mispair with thymine during DNA replication, MutSα is believed to act as a genome surveillance factor and recognizes the mismatched base, subsequently facilitating further binding of MutLα to the complex at the initial step of apoptotic induction (44). An immunoprecipitation analysis revealed that SMARCAD1 interacts with MSH2 even under MNU-untreated conditions, as expected (33, 45), and binds to MLH1 after MNU treatment on damaged chromatin (Fig. 5A). Because chromatin loading of SMACAD1 is substantially dependent on MSH2 (Fig. 5B), SMARCAD1 loaded onto chromatin by MSH2 might assist MutSα in recruiting MutLα to O6-methylguanine-carrying DNA in the context of chromatin. Indeed, in SMARCAD1-knockout cells, the levels of MLH1 interacting with MSH2 on chromatin after MNU treatment were severely reduced, and the downstream activation of DNA damage signaling, such as phosphorylation of ATR/CHK1 and ATM/CHK2, was also suppressed, compared with the control HeLa MR (Figs. 2 and 5). These findings clearly indicate that SMARCAD1 plays a role in facilitating the formation of MMR complex that recognizes and processes the O6-methylguanine/thymine pair for the induction of apoptosis.
How does SMARCAD1 assist in recruiting MutLα in the context of chromatin structure? SMARCAD1 is an ATP-dependent chromatin remodeler that is conserved from yeast to humans (46, 47) and interacts with MSH2 even under normal growth conditions. It was recently reported that, in Xenopus egg extracts, Smarcad1 is loaded on mismatch-carrying DNA in an Msh2-dependent manner and facilitates nucleosome exclusion from a region surrounding a mismatched base pair (34). MSH2-dependent chromatin loading of SMARCAD1 in human cells was also demonstrated in this study (Fig. 5B). It is highly possible that the resulting relaxed chromatin structure enhances the efficient binding of MutLα to MutSα. In human cells, we found that an ATP-dependent chromatin remodeling activity is indispensable for SMARCAD1 to assist in recruiting MutLα on damaged chromatin (Fig. 6D). Furthermore, SMARCAD1 is able to interact with MLH1 in an MSH2-dependent manner (Fig. S4). It is therefore probable that the O6-methylguanine/thymine pair produced during DNA replication is recognized by MutSα, and nucleosomes surrounding the mismatched DNA are excluded by MSH2-bound SMARCAD1, thereby enabling the efficient recruitment of MutLα to chromatin-bound MutSα. The resulting MMR complex is able to respond to damaged DNA to induce apoptosis (Fig. 7, left). In contrast, in SMARCAD1-defective cells, a smaller amount of MLH1 interacts with MSH2, and the induction of apoptosis and the activation of DNA damage signaling are both severely suppressed. Nucleosomes positioned at the region surrounding DNA mismatch may very well interfere with the recruitment of MutLα to MutSα on damaged chromatin (Fig. 7, right). This proposed scenario is supported by the recent finding that CAF-1–mediated chromatin assembly coupled with DNA replication conversely counteracts the ability of MMR complex to induce apoptosis upon treatment with an alkylating agent (23).
Figure 7.
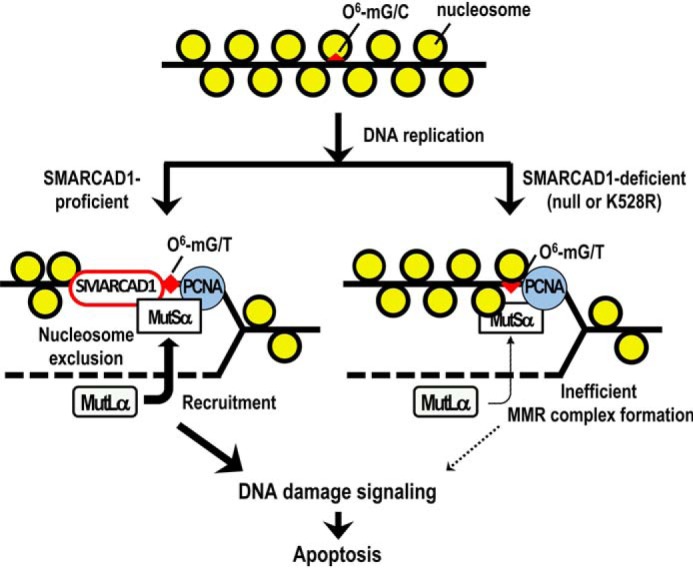
A model of the regulation of chromatin structure for the induction of MMR-dependent apoptosis. A possible role of SMARCAD1 in the induction of MMR-dependent apoptosis is presented. O6-mG, O6-methylguanosine.
Taken together, these findings strongly suggest that temporal and regional exclusion of nucleosomes on damaged chromatin is important for the efficient formation of MMR complex, which is coupled with DNA replication. Further studies are needed to clarify the precise roles played by SMARCAD1 in the induction of MMR-dependent apoptosis.
Experimental procedures
Cell lines and cell culture
Human-derived HeLa MR, which is defective in the MGMT function, and U2OS cell lines were obtained from our laboratory stocks (48). All of the cell lines were cultivated at 37 °C in 5% CO2 in Dulbecco's modified Eagle's medium (Wako) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Thermo Fisher Scientific).
Plasmid construction and transfection
pX330-U6-Chimeric_BB-CBh-hSpCas9 was a gift from Feng Zhang (Addgene plasmid #42230). TAL effector nuclease expression vectors (Golden Gate TALEN and TAL Effector Kit 2.0 and Yamamoto Lab TALEN Accessory Pack) were a gift from Daniel Voytas and Adam Bogdanove (Addgene kit #1000000024) as well as Takashi Yamamoto (Addgene kit #1000000030). p3xFLAG-CMV-10 was purchased from Sigma-Aldrich. To obtain the SMARCAD1-, MLH1-, and MGMT-knockout cell lines (ΔSMARCAD1, ΔMLH1, and U2OSΔM), oligonucleotides coding gRNA for the SMARCAD1 gene (5′-CACCGTCTCAAAGCGAAAACGGTCC-3′ and 5′-AAATGGACCGTTTTCGCTTTGAGAC-3′), for the MLH1 gene (5′-CACCGCCCTGCCACGAACGACATTT-3′ and 5′-AAACAAATGTCGTTCGTGGCAGGGC-3′), and for the MGMT gene (5′-CACCGTGAAATGAAACGCACCACAC-3′ and 5′-AAACGTGTGGTGCGTTTCATTTCAC-3′) were annealed and inserted into pX330 predigested with BbsI. The resulting plasmids pX330-SMARCAD1 and pX330-MLH1 were transfected into HeLa MR, and pX330-MGMT was transfected into U2OS cells. The cells were cultivated for 24 h, and 500 cells were spread onto a 100-mm dish. The colonies formed were isolated, and the disruption of the SMARCAD1, MLH1, or MGMT gene was analyzed by immunoblotting using specific antibodies. The construction of the MSH2-knockout cell line (ΔMSH2) was described previously (49). In brief, TALE-binding sequences were designed using the web-based software program TAL Effector Nucleotide Targeter 2.0 (https://tale-nt.cac.cornell.edu)3 (50) with the exon 1 sequence of the human MSH2 gene as a query. The RVD array assembly targeting the sequences (TALEN Left, 5′-GAGGAGGTTTCGACATG-3′; TALEN Right, 5′-CTCCAACTGCAGCGTCT-3′) was performed according to the instruction manual of Golden Gate TALEN and the TAL Effector Kit 2.0, and the RVD modules were cloned into the pcDNA-TAL-NC+F vector. The resulting vectors TALEN-MSH2-L and TALEN-MSH2-R were transfected into HeLa MR cells, which were cultivated for 24 h. Five hundred cells were then spread into a 100-mm dish. The next day, the cells were treated with 0.4 mm MNU in serum-free Dulbecco's modified Eagle's medium for 1 h at 37 °C and then incubated in complete medium for 10 days. Colonies were isolated, the expression of MSH2 was analyzed by immunoblotting, and the target locus was sequenced. For siRNA transfection, Silencer siRNA for the SMARCAD1 gene (5′-GAAGGUCCUCUGUUACUAUTT-3′) and Stealth RNAi for the MLH1 gene (5′-UGCACAUUAACAUCCACAUUCUGGG-3′) were purchased from Thermo Fisher Scientific. The cells were transfected with 40 nm siRNA using the Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific) in accordance with the manufacturer's protocol. For the control transfection, Silencer select negative control No. 1 siRNA (Thermo Fisher Scientific) was used. To obtain cell lines stably expressing WT or mutant SMARCAD1, a PCR-amplified DNA fragment with the primers (5′-GTACAAGCTTGGAGGTAATCTTTTCAACCTGGACCG-3′ and 5′-GTACGGATCCTCACAGGCCCATTGATGTTTTTAG-3′) using cDNA for the SMARCAD1 gene, purchased from DNAFORM (Yokohama, Japan) as a template, was inserted into HindIII/BamHI-digested p3xFLAG-CMV-10, and the resulting plasmid was designated p3xFLAG-SMARCAD1-WT. The p3xFLAG-SMARCAD1-K528R plasmid was prepared by swapping the AvrII-EcoNI fragment, corresponding to the Walker A motif, with annealed oligonucleotides, 5′-GGGCCTAGGAAAAACTATTCAAGCCATTGCATTTCTGGCATACCTCTATCAGGAGG-3′ and 5′-CCTCCTGATAGAGGTATGCCAGAAATGCAATGGCTTGAATAGTTTTTCCTAGGCCC-3′. These two plasmids were transfected into ΔSMARCAD1 cells, and the cells were then cultivated for 24 h, followed by further incubation in a medium containing 1 mg/ml G418 for 10 days. Colonies were isolated, and the expression of SMARCAD1 was analyzed by immunoblotting. Transfection with plasmids was performed according to the manufacturer's instructions using Lipofectamine 2000 (Thermo Fisher Scientific).
The colony formation assay
Approximately 500 cells were treated with various concentrations of MNU or H2O2 in serum-free medium for 1 h or with etoposide in a medium containing 10% FBS for 12 h or irradiated with different doses of UV-C. After cultivation with a medium containing 10% FBS for 10 days, the number of colonies was counted, and the survival rate was calculated.
Preparation of whole-cell extract and chromatin fraction
Approximately 1.0 × 106 cells were washed twice with PBS and directly lysed with 2× SDS-PAGE sample buffer (120 mm Tris-HCl (pH 6.8), 4% SDS, 20% glycerol, 200 mm DTT, and 0.002% bromphenol blue). The samples were sonicated, boiled, and then subjected to SDS-PAGE followed by immunoblotting. To prepare the chromatin fraction, cells were washed in hypotonic/sucrose buffer (20 mm Hepes-KOH (pH 7.9), 5 mm KCl, 1.5 mm MgCl2, 0.25 m sucrose) and then suspended in the same buffer containing 0.1% Triton X-100. The cells were incubated for 10 min on ice, and nuclei were collected by low-speed centrifugation. The supernatant was further clarified by high-speed centrifugation and used as the cytoplasmic fraction. Nuclei were washed once in hypotonic/sucrose buffer containing 0.1% Triton X-100 and then lysed in SDS-PAGE sample buffer.
A flow cytometric analysis
Cells (2.5 × 105 to 1.0 × 106 cells/well) cultured in 6-well plates were harvested by treatment with trypsin-EDTA, washed with PBS, and suspended in 500 μl of PBS containing 0.1% Triton X-100, 25 μg/ml propidium iodide, and 0.1 mg/ml of RNase A. The samples were analyzed using a FACSCalibur flow cytometer (BD Biosciences) with 10,000 events per determination.
Mutation frequency
HeLa MR and SMARCAD1-knockout cells were treated with 0, 15, or 30 μm MNU and then incubated for 96 h to allow for the expression of ouabain-resistant character. After washing with PBS, cells (2.0 × 106 cells/100 mm-dish) were placed in medium containing 0.2 μm ouabain for 10 days. After staining, the number of resistant colonies was counted. In parallel, a cell suspension containing about 500 cells was plated in several 100-mm dishes, and the number of viable cells was counted. The mutation frequencies were calculated, and the mean values obtained from three experiments and the S.E. values (error bars) are presented. Asterisks indicate significant difference (p < 0.05).
Immunoprecipitation
For ChIP in Fig. 5A and Fig. S3, cells treated with or without 0.2 mm MNU were permeabilized on a dish with hypotonic/sucrose buffer containing 100 μg/ml digitonin and then treated with 1% formaldehyde for 10 min at room temperature. After the addition of 1 m Tris-HCl (pH 8.0), the cells were harvested and collected by centrifugation. The cell pellet was suspended in radioimmune precipitation assay buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS) and then sonicated. The material was centrifuged, and the supernatant fraction was used as the chromatin extract. ChIP in Fig. 5C was performed as described previously (44). The cells were washed with hypotonic/sucrose buffer, permeabilized on a dish with hypotonic/sucrose buffer containing 50 μg/ml digitonin and protease inhibitor mixture (Roche Diagnostics), and then treated with 1 μm 3,3′-dithiobis(sulfosuccinimidyl propionate) for 2 h at 4 °C. After adding 50 mm Tris-HCl (pH 8.0), the cells were harvested and collected by centrifugation. The cell pellet was washed twice with PBS and suspended in NETN buffer (50 mm Tris-HCl (pH 8.0), 0.5% Nonidet P-40, 400 mm NaCl, 1 mm EDTA). Following sonication, the material was centrifuged at 16,000 × g for 15 min at 4 °C, and the supernatant fraction was used as the chromatin extract. For immunoprecipitation, the chromatin extracts were incubated with anti-MSH2 antibody conjugated with protein G-Sepharose beads (GE Healthcare) for 12 h at 4 °C. After extensive washing of the beads with the buffer, the proteins bound to the beads were eluted with 2× SDS-PAGE sample buffer, boiled, and then subjected to immunoblotting. Immunoprecipitation in Fig. S4 was performed as described above using whole-cell extracts prepared from cells directly fixed with 1% formaldehyde, without a permeabilization step by the use of hypotonic/sucrose buffer containing 100 μg/ml digitonin.
Antibodies
Anti-MSH2 (#610918), anti-MLH1 (#554073), anti-PMS2 (#556415), and anti-ORC2 (#559670) antibodies were purchased from BD Biosciences. Anti-SMARCAD1 (#301-593A) was purchased from Bethyl Laboratory, and anti-ATM (#2873), anti-phospho-T68-CHK2 (#2661), anti-caspase-9 (#9502), and anti-HP1α/β (#2623) antibodies were purchased from Cell Signaling Technology. Anti-MSH2 (#337900) antibody was purchased from Life Technologies. Anti-phospho-T1989-ATR (#GTX128145) was purchased from GeneTex. Anti-MEK2 (M24520) was purchased from Transduction Laboratories. Anti-ATR (sc-1887), anti-PARP1 (sc-8007), and anti-actin (A5316) antibodies were purchased from Santa Cruz Biotechnology, Inc., and Sigma-Aldrich, respectively. Horseradish peroxidase–conjugated donkey anti-rabbit and sheep anti-mouse IgG (GE Healthcare) were used as secondary antibodies.
Author contributions
Y. T., R. F., M. R., Y. O., M. S., and M. H. conceptualization; Y. T., R. F., M. R., Y. O., M. S., and M. H. data curation; Y. T., R. F., and M. H. formal analysis; Y. T., R. F., M. S., and M. H. funding acquisition; Y. T., R. F., M. R., Y. O., M. S., and M. H. validation; Y. T., R. F., M. R., Y. O., and M. H. investigation; Y. T., R. F., and M. H. methodology; Y. T., R. F., and M. H. writing-original draft; M. S. and M. H. supervision; M. H. project administration.
Supplementary Material
Acknowledgments
We thank Dr. K. Hayashi (Fukuoka Dental College, Japan) and Dr. Y. Nakatsu for helpful discussion. We also thank Dr. Tatsuro Takahashi (Kyushu University) for sharing unpublished data.
This work was supported by the Private University Research Branding Project (to R. F.), Japan Society for the Promotion of Science (JSPS) KAKENHI Grant-in-Aid for Young Scientist (B) Grant 16K18428 (to Y. T.), JSPS KAKENHI Grant-in-Aid for Young Scientist (B) Grant 26830085 (to R. F.), JSPS KAKENHI Grant-in-Aid for Scientific Research (A) Grant 25241012 (to M.H.), MEXT-Grants-in-Aid and MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2013–2017, Grant S1411042 (to M. S. and M. H.), and the Fukuoka Foundation for Sound Health Cancer Research Fund (to Y. T.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- MNU
- N-methyl-N-nitrosourea
- MMR
- mismatch repair
- MNNG
- N-methyl-N′-nitro-N-nitrosoguanidine
- MGMT
- O6-methylguanine-DNA methyltransferase
- SMARCAD1
- SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A containing DEAD/H box 1
- PCNA
- proliferating cell nuclear antigen
- CAF-1
- chromatin assembly factor 1
- HMGA
- high-mobility group A
- ATR
- ATM and Rad3-related
- ATM
- ataxia telangiectasia-mutated.
References
- 1. Beranek D. T. (1990) Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat. Res. 231, 11–30 10.1016/0027-5107(90)90173-2 [DOI] [PubMed] [Google Scholar]
- 2. Aquilina G., Hess P., Branch P., MacGeoch C., Casciano I., Karran P., and Bignami M. (1994) A mismatch recognition defect in colon carcinoma confers DNA microsatellite instability and a mutator phenotype. Proc. Natl. Acad. Sci. U.S.A. 91, 8905–8909 10.1073/pnas.91.19.8905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Loechler E. L., Green C. L., and Essigmann J. M. (1984) In vivo mutagenesis by O6-methylguanine built into a unique site in a viral genome. Proc. Natl. Acad. Sci. U.S.A. 81, 6271–6275 10.1073/pnas.81.20.6271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maze R., Carney J. P., Kelley M. R., Glassner B. J., Williams D. A., and Samson L. (1996) Increasing DNA repair methyltransferase levels via bone marrow stem cell transduction rescues mice from the toxic effects of 1,3-bis(2-chloroethyl)-1-nitrosourea, a chemotherapeutic alkylating agent. Proc. Natl. Acad. Sci. U.S.A. 93, 206–210 10.1073/pnas.93.1.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davis B. M., Reese J. S., Koç O. N., Lee K., Schupp J. E., and Gerson S. L. (1997) Selection for G156A O6-methylguanine DNA methyltransferase gene-transduced hematopoietic progenitors and protection from lethality in mice treated with O6-benzylguanine and 1,3-bis(2-chloroethyl)-1-nitrosourea. Cancer Res. 57, 5093–5099 [PubMed] [Google Scholar]
- 6. Sakumi K., Shiraishi A., Shimizu S., Tsuzuki T., Ishikawa T., and Sekiguchi M. (1997) Methylnitrosourea-induced tumorigenesis in MGMT gene knockout mice. Cancer Res. 57, 2415–2418 [PubMed] [Google Scholar]
- 7. Qin X., Liu L., and Gerson S. L. (1999) Mice defective in the DNA mismatch gene PMS2 are hypersensitive to MNU induced thymic lymphoma and are partially protected by transgenic expression of human MGMT. Oncogene 18, 4394–4400 10.1038/sj.onc.1202798 [DOI] [PubMed] [Google Scholar]
- 8. Tominaga Y., Tsuzuki T., Shiraishi A., Kawate H., and Sekiguchi M. (1997) Alkylation-induced apoptosis of embryonic stem cells in which the gene for DNA-repair, methyltransferase, had been disrupted by gene targeting. Carcinogenesis 18, 889–896 10.1093/carcin/18.5.889 [DOI] [PubMed] [Google Scholar]
- 9. Kaina B., Ziouta A., Ochs K., and Coquerelle T. (1997) Chromosomal instability, reproductive cell death and apoptosis induced by O6-methylguanine in Mex−, Mex+ and methylation-tolerant mismatch repair compromised cells: facts and models. Mutat. Res. 381, 227–241 10.1016/S0027-5107(97)00187-5 [DOI] [PubMed] [Google Scholar]
- 10. Fishel R., Lescoe M. K., Rao M. R., Copeland N. G., Jenkins N. A., Garber J., Kane M., and Kolodner R. (1993) The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 75, 1027–1038 10.1016/0092-8674(93)90546-3 [DOI] [PubMed] [Google Scholar]
- 11. Leach F. S., Nicolaides N. C., Papadopoulos N., Liu B., Jen J., Parsons R., Peltomäki P., Sistonen P., Aaltonen L. A., and Nyström-Lahti M. (1993) Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 75, 1215–1225 10.1016/0092-8674(93)90330-S [DOI] [PubMed] [Google Scholar]
- 12. Parsons R., Li G. M., Longley M. J., Fang W. H., Papadopoulos N., Jen J., de la Chapelle A., Kinzler K. W., Vogelstein B., and Modrich P. (1993) Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell 75, 1227–1236 10.1016/0092-8674(93)90331-J [DOI] [PubMed] [Google Scholar]
- 13. Modrich P. (1997) Strand-specific mismatch repair in mammalian cells. J. Biol. Chem. 272, 24727–24730 10.1074/jbc.272.40.24727 [DOI] [PubMed] [Google Scholar]
- 14. Buermeyer A. B., Deschênes S. M., Baker S. M., and Liskay R. M. (1999) Mammalian DNA mismatch repair. Annu. Rev. Genet. 33, 533–564 10.1146/annurev.genet.33.1.533 [DOI] [PubMed] [Google Scholar]
- 15. Kolodner R. D., and Marsischky G. T. (1999) Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 9, 89–96 10.1016/S0959-437X(99)80013-6 [DOI] [PubMed] [Google Scholar]
- 16. Branch P., Aquilina G., Bignami M., and Karran P. (1993) Defective mismatch binding and a mutator phenotype in cells tolerant to DNA damage. Nature 362, 652–654 10.1038/362652a0 [DOI] [PubMed] [Google Scholar]
- 17. Kat A., Thilly W. G., Fang W. H., Longley M. J., Li G. M., and Modrich P. (1993) An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair. Proc. Natl. Acad. Sci. U.S.A. 90, 6424–6428 10.1073/pnas.90.14.6424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu J., Gu L., Wang H., Geacintov N. E., and Li G. M. (1999) Mismatch repair processing of carcinogen-DNA adducts triggers apoptosis. Mol. Cell Biol. 19, 8292–8301 10.1128/MCB.19.12.8292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kawate H., Sakumi K., Tsuzuki T., Nakatsuru Y., Ishikawa T., Takahashi S., Takano H., Noda T., and Sekiguchi M. (1998) Separation of killing and tumorigenic effects of an alkylating agent in mice defective in two of the DNA repair genes. Proc. Natl. Acad. Sci. U.S.A. 95, 5116–5120 10.1073/pnas.95.9.5116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takagi Y., Takahashi M., Sanada M., Ito R., Yamaizumi M., and Sekiguchi M. (2003) Roles of MGMT and MLH1 proteins in alkylation-induced apoptosis and mutagenesis. DNA Repair (Amst.) 2, 1135–1146 10.1016/S1568-7864(03)00134-4 [DOI] [PubMed] [Google Scholar]
- 21. Fujikane R., Komori K., Sekiguchi M., and Hidaka M. (2016) Function of high-mobility group A proteins in the DNA damage signaling for the induction of apoptosis. Sci. Rep. 6, 31714 10.1038/srep31714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cleynen I., and Van de Ven W. J. (2008) The HMGA proteins: a myriad of functions (Review). Int. J. Oncol. 32, 289–305 [PubMed] [Google Scholar]
- 23. Kadyrova L. Y., Dahal B. K., and Kadyrov F. A. (2016) The major replicative histone chaperone CAF-1 suppresses the activity of the DNA mismatch repair system in the cytotoxic response to a DNA-methylating agent. J. Biol. Chem. 291, 27298–27312 10.1074/jbc.M116.760561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taddei A., Roche D., Sibarita J. B., Turner B. M., and Almouzni G. (1999) Duplication and maintenance of heterochromatin domains. J. Cell Biol. 147, 1153–1166 10.1083/jcb.147.6.1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Strålfors A., Walfridsson J., Bhuiyan H., and Ekwall K. (2011) The FUN30 chromatin remodeler, Fft3, protects centromeric and subtelomeric domains from euchromatin formation. PLoS Genet. 7, e1001334 10.1371/journal.pgen.1001334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taneja N., Zofall M., Balachandran V., Thillainadesan G., Sugiyama T., Wheeler D., Zhou M., and Grewal S. I. (2017) SNF2 family protein Fft3 suppresses nucleosome turnover to promote epigenetic inheritance and proper replication. Mol. Cell 66, 50–62.e6 10.1016/j.molcel.2017.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rowbotham S. P., Barki L., Neves-Costa A., Santos F., Dean W., Hawkes N., Choudhary P., Will W. R., Webster J., Oxley D., Green C. M., Varga-Weisz P., and Mermoud J. E. (2011) Maintenance of silent chromatin through replication requires SWI/SNF-like chromatin remodeler SMARCAD1. Mol. Cell 42, 285–296 10.1016/j.molcel.2011.02.036 [DOI] [PubMed] [Google Scholar]
- 28. Costelloe T., Louge R., Tomimatsu N., Mukherjee B., Martini E., Khadaroo B., Dubois K., Wiegant W. W., Thierry A., Burma S., van Attikum H., and Llorente B. (2012) The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 489, 581–584 10.1038/nature11353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen X., Cui D., Papusha A., Zhang X., Chu C. D., Tang J., Chen K., Pan X., and Ira G. (2012) The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature 489, 576–580 10.1038/nature11355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eapen V. V., Sugawara N., Tsabar M., Wu W. H., and Haber J. E. (2012) The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol. Cell Biol. 32, 4727–4740 10.1128/MCB.00566-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Densham R. M., Garvin A. J., Stone H. R., Strachan J., Baldock R. A., Daza-Martin M., Fletcher A., Blair-Reid S., Beesley J., Johal B., Pearl L. H., Neely R., Keep N. H., Watts F. Z., and Morris J. R. (2016) Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol. 23, 647–655 10.1038/nsmb.3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sirbu B. M., McDonald W. H., Dungrawala H., Badu-Nkansah A., Kavanaugh G. M., Chen Y., Tabb D. L., and Cortez D. (2013) Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem. 288, 31458–31467 10.1074/jbc.M113.511337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Okazaki N., Ikeda S., Ohara R., Shimada K., Yanagawa T., Nagase T., Ohara O., and Koga H. (2008) The novel protein complex with SMARCAD1/KIAA1122 binds to the vicinity of TSS. J. Mol. Biol. 382, 257–265 10.1016/j.jmb.2008.07.031 [DOI] [PubMed] [Google Scholar]
- 34. Terui R., Nagao K., Kawasoe Y., Taki K., Higashi T. L., Tanaka S., Nakagawa T., Obuse C., Masukata H., and Takahashi T. S. (2018) Nucleosomes around a mismatched base pair are excluded via an Msh2-dependent reaction with the aid of SNF2 family ATPase Smarcad1. Genes Dev. 32, 806–821 10.1101/gad.310995.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., and Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fujikane R., Sanada M., Sekiguchi M., and Hidaka M. (2012) The identification of a novel gene, MAPO2, that is involved in the induction of apoptosis triggered by O6-methylguanine. PLoS One 7, e44817 10.1371/journal.pone.0044817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stojic L., Mojas N., Cejka P., Di Pietro M., Ferrari S., Marra G., and Jiricny J. (2004) Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 18, 1331–1344 10.1101/gad.294404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Darzynkiewicz Z., Bruno S., Del Bino G., Gorczyca W., Hotz M. A., Lassota P., and Traganos F. (1992) Features of apoptotic cells measured by flow cytometry. Cytometry 13, 795–808 10.1002/cyto.990130802 [DOI] [PubMed] [Google Scholar]
- 39. Liu X., Kim C. N., Yang J., Jemmerson R., and Wang X. (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157 10.1016/S0092-8674(00)80085-9 [DOI] [PubMed] [Google Scholar]
- 40. Nicholson D. W., Ali A., Thornberry N. A., Vaillancourt J. P., Ding C. K., Gallant M., Gareau Y., Griffin P. R., Labelle M., and Lazebnik Y. A. (1995) Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376, 37–43 10.1038/376037a0 [DOI] [PubMed] [Google Scholar]
- 41. Tewari M., Quan L. T., O'Rourke K., Desnoyers S., Zeng Z., Beidler D. R., Poirier G. G., Salvesen G. S., and Dixit V. M. (1995) Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 81, 801–809 10.1016/0092-8674(95)90541-3 [DOI] [PubMed] [Google Scholar]
- 42. Sanada M., Hidaka M., Takagi Y., Takano T. Y., Nakatsu Y., Tsuzuki T., and Sekiguchi M. (2007) Modes of actions of two types of anti-neoplastic drugs, dacarbazine and ACNU, to induce apoptosis. Carcinogenesis 28, 2657–2663 10.1093/carcin/bgm188 [DOI] [PubMed] [Google Scholar]
- 43. Gradia S., Acharya S., and Fishel R. (1997) The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell 91, 995–1005 10.1016/S0092-8674(00)80490-0 [DOI] [PubMed] [Google Scholar]
- 44. Hidaka M., Takagi Y., Takano T. Y., and Sekiguchi M. (2005) PCNA-MutSalpha-mediated binding of MutLα to replicative DNA with mismatched bases to induce apoptosis in human cells. Nucleic Acids Res. 33, 5703–5712 10.1093/nar/gki878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen Z., Tran M., Tang M., Wang W., Gong Z., and Chen J. (2016) Proteomic analysis reveals a novel mutator S (MutS) partner involved in mismatch repair pathway. Mol. Cell Proteomics 15, 1299–1308 10.1074/mcp.M115.056093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schoor M., Schuster-Gossler K., and Gossler A. (1993) The Etl-1 gene encodes a nuclear protein differentially expressed during early mouse development. Dev. Dyn. 197, 227–237 10.1002/aja.1001970307 [DOI] [PubMed] [Google Scholar]
- 47. Adra C. N., Donato J. L., Badovinac R., Syed F., Kheraj R., Cai H., Moran C., Kolker M. T., Turner H., Weremowicz S., Shirakawa T., Morton C. C., Schnipper L. E., and Drews R. (2000) SMARCAD1, a novel human helicase family-defining member associated with genetic instability: cloning, expression, and mapping to 4q22-q23, a band rich in breakpoints and deletion mutants involved in several human diseases. Genomics 69, 162–173 10.1006/geno.2000.6281 [DOI] [PubMed] [Google Scholar]
- 48. Hayakawa H., Koike G., and Sekiguchi M. (1990) Expression and cloning of complementary DNA for a human enzyme that repairs O6-methylguanine in DNA. J. Mol. Biol. 213, 739–747 10.1016/S0022-2836(05)80260-8 [DOI] [PubMed] [Google Scholar]
- 49. Hayashida G., Shioi S., Hidaka K., Fujikane R., Hidaka M., Tsurimoto T., Tsuzuki T., Oda S., and Nakatsu Y. (2019) Differential genomic destabilisation in human cells with pathogenic MSH2 mutations introduced by genome editing. Exp. Cell Res. 377, 24–35 10.1016/j.yexcr.2019.02.020 [DOI] [PubMed] [Google Scholar]
- 50. Doyle E. L., Booher N. J., Standage D. S., Voytas D. F., Brendel V. P., Vandyk J. K., and Bogdanove A. J. (2012) TAL effector-nucleotide targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 40, W117–W122 10.1093/nar/gks608 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.