

Abstract
Purpose: Paclitaxel (PTX) has transpired as a significant agent in the treatment of breast cancer. Meanwhile, polylactic glycolic acid (PLGA) nanoparticles (NPs) are able to increase the anticancer effect of the PTX in the blood.
Methods: Nano-precipitation was used to prepare the PLGA-PTX-VitD3 co-delivery NPs. Drug loading, encapsulation efficiency, in vitro release profile, cell viability, migration, apoptosis, and bcl2 expression of NPs were evaluated.
Results: The average size of co-delivery NPs was 231 ± 46 nm. Observed was a controlled release of the PTX and vitamin D3 from co-delivery NPs between 0.5 and 240 hours. MTT showed the ability of 8 μg.mL-1 of co-delivery NPs to kill 50 % of the MCF-7; likewise, the co-delivery NPs prevented MCF-7 migration. The co-delivery NPs led 46.35 % MCF-7 to enter primary apoptosis. 60.8% of MCF-7 in the control group were able to enter the G (1) phase of the cell cycle. The co-delivery NPs increased expression of bax. In addition to its higher toxicity against MCF-7 than that of PTX, co-delivery NPs were able to release drugs continuously for a long period, which indeed increased the efficiency of the drugs.
Conclusion: The effect of co-delivery NPs on MCF-7 cell viability was different from that in other drugs. In fact, the co-deliver NPs were able to release drugs continuously for a long time, this could induce primary apoptosis in the MCF-7 and decrease the metastasis and toxicity of drugs.
Keywords: Breast cancer, Anzatax, 25-Hydroxyvitamin D2, PLGA compound, Nanoparticle, bax genes, MCF-7 cell
Introduction
Breast cancer is among the most common neoplastic malignancies, and one of the leading causes of women’s death in the world.1 High instances of this malignancy is found in developed countries; the prevalence of breast cancer in Iran is lower than that of developed countries.2 In spite of surgery, chemotherapy and radiotherapy as therapeutic options of this malignancy, mortality rate is still high among patients with breast cancer.2 Currently, the co-delivery of anti-cancer drugs has been developed by assistance of biodegradables nanoparticles (NPs) such as polylactic glycolic acid (PLGA) NPs.3-5
PLGA NPs are one group of the commonly used polymers in manufacturing NPs thanks to their high adaptability to biological environments, ability to degrade into natural metabolites, lower toxicity and higher stability than that of liposomes and some other drug delivery systems. PLGA NPs also have the ability to modify their surface to prevent rapid absorption of this polymer by the reticuloendothelial system of the body. Toxicity and damaging the healthy tissues in the immediate area of the tumor are the result of using high doses of the drug, meanwhile, a biodegradable and biocompatible amphiphilic polymer; in fact, PLGA increases the anti-cancer effect of the drug in the blood by increasing its circulation time. In addition, studies have shown that use of PLGA in active and inactive cerebellar drugs will increase their endocytosis. Moreover, the PLGA-made NPs are of the most promising tools for transferring drugs and genes from the duct-brain.6 The poly vinyl alcohol (PVA) which modifies the size and distribution of small size in NPs is also one of the most widely used polymers for stabilizing NPs.7 Studies show that PVA is tightly connected to PLGA; the justification for this process is the hydrophobic link established between the hydroxyl functional group of PVA. The PLGA-PVA, which amongst others is one of the most successful polymers for making polymer NPs,8,9 could be a suitable candidate for delivery and controlled release of anti-cancer drugs such as paclitaxel (PTX).10
PTX is one of the new anti-cancer drugs used for many human cancers including ovarian and breast cancers. This molecule facilitates accumulation of microtubules from tubulin dimers and stabilizes them by preventing the de-polymerization of microtubules. Preventing the reorganization of the active and natural network of microtubules, this stability is essential for vital interphase and mitotic cell transplantation. In addition, PTX induces an abnormal or cluster arrangement of microtubules during the cell cycle, leading to the formation of multiple lobes of microtubules during mitosis results.11 PTX, in spite of its therapeutic effect, causes side effects. Short metabolized half-life and membrane transducers preventing the concentration of the drug in the cell make repeated use of relatively high doses necessary to achieve the required concentration during the appropriate time periods, which in turn leads to toxicity.11 However, studies have shown that combination of 1, 25 dihydroxy cholecalciferol (VitD3) and PTX can also enhance anti-tumor activity and accelerate apoptosis in breast cancer.12,13 Evidently, many studies have shown that VitD3 has anti-cancer effects on many types of cancer cells, it modulates the immune system and controls the paths, increases apoptosis, decreases cell growth, and regulates biological processes such as angiogenesis and the production of extracellular matrix as well.13,14 These effects result from the inhibition of the G1/S phase of the cell cycle and the effect on many activity regulatory of the cycling.15 VitD3 reduces the expression of anti-apoptotic proteins, and genes involved in the proliferation of uterine fibroids; and the production of extracellular matrix proteins.16
There is no study with respect to the anti-cancer effects of the PLGA-PTX-VitD3 co-delivery NPs on bax expression. We combined the VitD3 and PTX to enhance the anti-cancer function, prolonged release, reduced toxicity, increased expression of bax gene. The cytotoxicity of VitD3 and PTX loaded PLGA NPs against MCF-7 cell lines were examined by MTT method and calculated IC50. The apoptosis, proliferative effects of NPs and the rate of expression of the bax genes were also compared.
Materials and Methods
Cell culture
MCF-7 human breast cancer cell line was cultured in Dulbecco’s modified Eagle medium (Gibco™ DMEM) (Thermo Fisher Scientific, Waltham, MA, USA), containing 10% fetal calf serum (Gibco™ FCS) (Thermo Fisher Scientific, Waltham, MA, USA), and 1% penicillin- streptomycin (Sigma Aldrich, St. Louis, USA) 10000 units/ml at 37°C in a humidified atmosphere of 95% air and 5% CO2.
Preparation of PTX and VitD3-loaded PLGA NPs
Nano-precipitation method was used to prepare the PLGA-PTX-VitD3 co-delivery NPs. The solution of PVA polymer (Thermo Fisher Scientific, Waltham, MA, USA) 1% w/v was prepared in deionized water, as an aqueous phase, under continuous magnetic stirring (FAR TEST, HPMA700) at 70°C for 5 hours to obtain a homogenous solution; likewise, 50 mg of the PLGA (Sigma Aldrich, St. Louis, USA) and 5 mg of the PTX (Sigma Aldrich, St. Louis, USA) were dissolved in 10 mL, and 5 mL of acetone (Merck, Germany), respectively. Next, 1 mL of the VitD3 (Sigma Aldrich, St. Louis, USA) was added to acetone (0.1 % W/V). Subsequently, the PTX was poured in the PLGA solution. Afterward, organic phase was poured in the PVA (100 mL) and stirred overnight at room temperature (around 20 ± 2°C). After the evaporation of the organic phase, the NPs were collected by centrifugation (Eppendorf, MA, USA) at 12 000 rpm for 30 minutes at 4°C and washed twice with deionized water.17
Physiochemical properties of NPs
The hydrodynamic diameter and the median for NPs’ size were obtained using dynamic light scattering (DLS) and zeta potential analyzer (Malvern, UK). The morphology and diameter of NPs were screened by scanning electron microscopy (SEM) as an accelerating voltage of 20.0 kV (Philips XL-30, OH, USA) after sputtering with gold. The NPs suspensions were diluted by sterile distilled water; they were then poured onto the copper plate. After the NPs dried at room temperature, they were coated with a thin layer of gold under vacuum. The diameter of NPs was measured by digitizer software. The Fourier-transform infrared spectroscopy (FTIR)(Nicolet, Magna-IR spectrometer 550) (Thermo Fisher Scientific, Waltham, MA, USA) was used to determine the chemical groups and the interaction between the particles.17
Drug loading, encapsulation efficiency of NPs
Using UV-Visible spectroscopy at the wavelengths of 228 nm and 280 nm for PTX and, VitD3, respectively, we could obtain standard curves (Cecil CE 7250, England). The concentration of PTX and VitD3 encapsulated in the NPs was determined indirectly by measuring the amount of free drug in the supernatant by UV-VIS absorption being used. The supernatant was collected by centrifugation (Eppendorf, MA, USA) at 12 000 rpm for 30 minutes at 4°C and washed twice with deionized water.17
The encapsulation efficiency and drug loading were calculated using the equations below:
Encapsulation Efficiency (%):
Drug loading (%):
In vitro release studies
In vitro release studies of all samples were performed in PBS (PH – 7.4) containing 0.2% Tween 80. In detail, we measured 5 mg of each formulation dispersed in 1 mL of the release media. The suspension was put in a dialysis tubing (cut-off - 12.4 kDa, Cas # - D0405, Sigma Aldrich, USA) and placed in a falcon containing 50 mL of the release media. Afterwards, the falcons were placed in a shacking incubator at 37°C at 100 rpm (Labtech, S. Korea). At predetermined time intervals, falcon tubes were emptied and then replenished by fresh media. The concentration of drugs released from each sample was then determined via UV-VIS spectroscopy at the appropriate wavelengths. The kinetics of release model was Peppa-Korsmeye.18 A cumulative release curve of percent drug release against time was then plotted.
Cell viability assay
MTT assay was applied to evaluate the cytotoxic effect of PLGA-PTX-VitD3 co-delivery NPs, PTX, PLGA-PTX and, PLGA on MCF-7 breast cancer cells.6 To calculate the final concentration of the PLGA-PTX-VitD3 co-delivery NPs, the mole scale of the NPs was converted to milligram scale and so the NPs were measured by weight balance (DORJE, KI-204, and China). Briefly, 1, 12.5, 25, 50, 100 and 200 µM of PLGA-PTX-VitD3 co-delivery NPs were solved into the culture medium. The cells were seeded at a density of 1.2×104 cells/mL and incubated with PLGA-PTX-VitD3 co-delivery NPs, PTX, PLGA-PTX and, PLGA for 24 hours, respectively. Then the DMEM was changed and cells were incubated with MTT solution (Sigma Aldrich, St. Louis, USA) (0.5 mg/mL) for 4 hours. In this study, control group was treated with DMSO 0.1%.
Migration assay
The MCF-7 cell lines were first grown in DMEM supplemented with 10% FBS to test migration assay, then seeded in 6-well plates.19 Next, the width of each well is scratched by sterile pipette tip (1 mm). The wells are washed by the culture medium and the picture of wounds is taken at 0 hour (NIKON, Inverted research microscope). Thereafter, medium was replaced with different concentrations of PLGA-PTX-VitD3 co-delivery NPs, PTX and PLGA-PTX (IC50 concentration) and the cells are allowed to migrate for 24 hours. After 24 hours, the picture of wound is taken from the same regions. The rate of migration (RM) is analyzed by measuring the distance between the scratch edges with digitizer software. The RM is calculated by following equation: RM=(Wi-Wf)/t.20
Cell death with DAPI staining
To evaluate cell death, the MCF-7 cell lines were grown in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin exposed to PLGA-PTX-VitD3 co-delivery NPs (IC50 concentration). The MCF-7 cells were harvested by trypsin-EDTA solution, and then were centrifuged at 2000 rpm for 5 minutes. The cells were stabilized with 4% paraformaldehyde. The cells were washed by adding 500 µL of 4, 6-diamidino-2-phenylindole (DAPI) reagent and were incubated. Next, the MCF-7 cells were centrifuged and after that the buffers (Buffer RLT Plus contains guanidine thiocyanate, buffer RW1 contains a small amount of guanidine thiocyanate, and buffer RW1 Contains ethanol) were added to cell suspension. Excitation wavelength were observed at 340/380 nm by fluorescent microscopy.
Flow cytometer and cell cycle analysis
To analyze the quantitative DNA content in MCF-7 culture cells exposed to PLGA-PTX-VitD3 co-delivery NPs, PTX, and PLGA-PTX, the nucleic acid stain propidium iodide (PI) was used by flow cytometry analysis. In this way, the MCF-7 cells were harvested in the DMEM and washed in PBS. The cells were fixed in cold 70% ethanol for 30 minutes at 4°C. After that, MCF-7 cells were washed with 2X in PBS and spun at 2700 rpm in a centrifuge. The MCF-7 cells were treated with ribonuclease to ensure that only the DNA, and not RNA, is stained. Next, 200 μL PI was added to stock solution. Then, to identify single cells, the forward scatter (FS) and side scatter (SS) are measured by FlowJo softwere.21 The PBS containing MCF-7 cell lines without any NPs was used as a control group.
RNA extraction, cDNA preparation and real-time PCR
Total RNAs were isolated from MCF-7 cells by means of RNeasy plus mini kit )QIAGEN- Cat. No: 74134). Extracted RNA was treated with DNase I enzyme. One microgram of RNA from each sample was used for cDNA synthesis by reverse transcriptase. RNA samples were incubated at 65°C for 5 minutes with 1 μL Oligo dT (0.5 μg/μL) at a final volume of 10 μL, and then, mixed with 4 μL of 5X buffer, 0.5 μL RT Enzyme Mix Ӏ, 0.5 μL of random hexamer primer (6-mer) (100 µM) and, 4.5 μL of RNase Free dH2O. Finally, the 0.2 mL of prepared buffer was used to PCR reaction. After incubation of the buffer at 42°C for 15 min, and at 85°C for 5 minutes, the mixture was transcribed into cDNA. The quality of cDNA is evaluated by electrophoresis assay. 2 μL cDNA of each sample was used for real-time PCR in 16 µL reaction mixtures with 6 µL of SYBR premix ex taq and 0.4 μL of specific forward and reverse primers. The forward β-actin (as a housekeeping gene) primer (5′-GCTCAGGAGGAGCAAT-3’) and reverse β-actin primer (5′-GGCATCCACGAAACTAC-3’) also were used to normalize the target gene expression. The reverse primer sequence of bax primers set was 5’-CAT CTT CTT CCA GAT GGT GA-3’ and forward primer sequence was 5’-GTT TCA TCC AGG ATC GAG CAG-3’. Eventually, 0.25 μL of ROX (50X), 0.8 μL of cDNA and, 8 μL of dH2O are added to buffer. Initial denaturation step in real-time PCR was run at 95°C for 10 minutes. Second step was done with 40 cycles at 95°C for 15 seconds and 60°C for 1 second. The Melt Curve achieved at 95°C for 15 seconds, 60°C for 1 minutes and, 95°C for 15 seconds. The real-time PCR was performed in duplicate, and relative mRNA of each target gene was determined by using the formula 2-ΔΔCT (CT, cycle threshold) where ΔCT = CT (target gene) − CT (β-actin). The comparative expression level of each target gene between different samples was calculated by 2-ΔΔCT. In addition, melting curves used to determine non-specific amplification.
Statistical analysis
Statistical analysis was performed using GraphPad Prism, version 5.0. The significant difference between the experimental groups and controls was assessed by one-way ANOVA and post hoc Tukey test. The results were presented as mean ± standard deviation (SD). P value< 0.05 represented significant differences.
Results and Discussion
Physiochemical properties of NPs were investigated in this study are summarized in Table 1. The morphology and diameter of PLGA-PTX-VitD3 co-delivery NPs were done using SEM at an accelerating voltage of 20.0 KV (Philips XL-30). The diameter of PLGA-PTX-VitD3 co-delivery NPs measured 231 ± 46nm (Figure 1a). The hydrodynamic diameter of PLGA-PTX-VitD3 co-delivery NPs was measured by DLS (Stereoscope-IN A-ONE Enc., Korea) <250 nm as shown in Figure 1b. The NPs were homogeneously dispersed. The zeta potential of NPs was -20 to -10 mV, which related to the presence of polyvinyl alcohol on the surface of the PLGA NPs. The zeta potential prevents the agglomeration of particles by using electrical repulsion (Table 1).
Table 1. Physiochemical properties of PLGA, PLGA-PTX NPs, and PLGA-PTX- VitD3 co-delivery NPs .
| Sample | Size (nm) | PDI | Zeta (mV) | Relative EE % | Relative DL (mg/g) |
| PLGA NPs | 60±09 | 0.02 | -18.4 | - | - |
| PLGA-PTX NPs | 200±27 | 0.04 | -20.3 | 85% | 7 |
| PLGA-PTX-VitD3 NPs | 231±46 | 0.06 | -15.7 | PTX 82% | PTX 6 |
| VitD3 95% | VitD3 1.5 |
PGLA, poly lactic glycolic acid; NPs, nanoparticles; PTX, paclitaxel.
Figure 1.
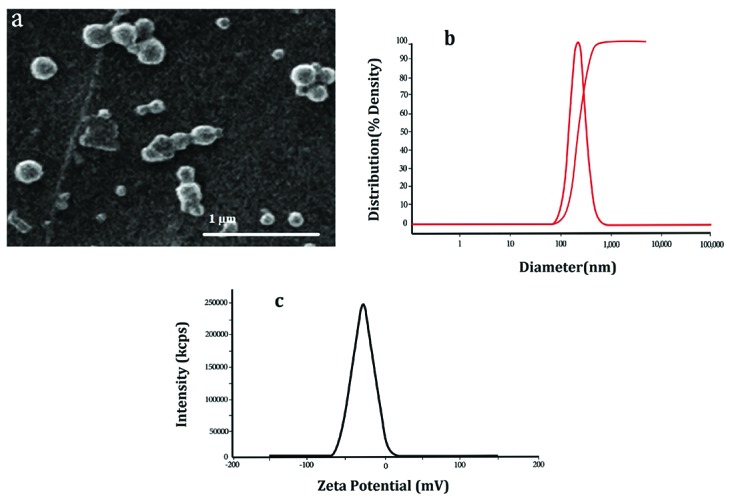
The morphology (a), the hydrodynamic diameter (b) and zeta potential of PLGA-PTX-VitD3 co-delivery NPs (c).
Figure 2 shows FT-IR spectra of PLGA (a), PLGA-PTX NPs (b) and PLGA-PTX- VitD3 co-delivery NPs (c). As shown in Figure 2a, the absorption band between 3600 cm-1 and 3400 cm-1 allocated both the νs (O-H) and νas (O-H) of hydroxyl group in PLGA NPs.22 The extreme absorption band between 2700 cm-1 and 3000 cm-1 allocated tensile vibration at δ (CH, CH2 e CH3). The shoulder between 1750 cm-1 and 1760 cm-1 showed a tensile vibration at δ (C=O).23 The absorption peaks between 1250 cm–1 and 1500 cm–1 indicated change shape ofδ (CH2 e CH3). The absorption peaks between 1150 cm–1 and 1300 cm–1were related to tensile vibration at δ (C=O).24 According to studies, the absorption peaks of PTX were between 3305 cm–1 and 3479 cm–1 of which were related to tensile vibration at δ (N-H). The tensile vibration at δ (C-H) is also shown between 2889 cm–1 and 2970 cm–1. The shoulder between 1703 cm–1 and 1732 cm–1 was in regard to tensile vibration at δ (C=O).25 In current study, the absorption peaks of PTX and vitamin D are not observed in the FTIR spectrum of PLGA-PTX NPs (Figure 2b) and PLGA-PTX-VitD3 co-delivery NPs (Figure 2c), which may indicate the complete encapsulation of PTX and vitamin D into the PLGA NPs.26
Figure 2.
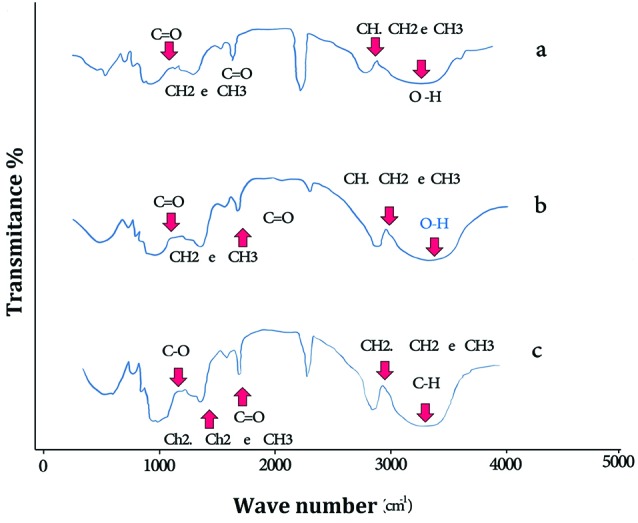
The FT-IR spectra of PLGA (a), PLGA-PTX NPs (b) and PLGA-PTX-VitD3 co-delivery NPs (c).
Cell viability assay revealed that PLGA NPs and PLGA-PTX NPs have no toxic effects on the MCF-7 cell line. As shown in Figure 3, the IC50 of PTX, and PLGA-PTX- VitD3 co-delivery NPs against MCF-7 cell lines calculated 7.5 µg/mL and 8 µg/mL, respectively (Figure 3a). VitD3 did not show toxic effects at concentrations of about 1000 μg/mL (Figure 3b).
Figure 3.

The MCF-7 cell viability in exposure the vitamin D3 (a), PLGA, PLGA-PTX NPs, PTX, DMSO and PLGA-PTX-VitD3 co-delivery NPs (b).
Madani et al at 2017 prepared PLGA-PTX NPs via single emulsion and precipitation methods with variable parameters including drug concentration, aqueous to organic phase volume ratio, polymer concentration, sonication time and PVA concentration.17 Cerqueira et al at 2017 prepared the PLGA NPs embedded with PTX and coated with hyaluronic acid (HA-PTX-PLGA) to target the drug to breast cancer cells. New formulation of HA-PTX-PLGA NPs showed high potential to decrease IC50 of PTX.27 Turino et al at 2017 prepared the PLGA-NPs, coated with L-ferritin, for the simultaneous delivery of PTX into MCF-7 cells. The results confirmed that NPs decolorated with L-feritin increased PTX cytotoxicity. Moreover, the coating increased NPs stability, thus reducing the fast release of a specific drug before reaching the target. 28 Tran prepared dual anticancer agents such as PTX and artesunate (ART), which were loaded into PLGA NPs by solvent evaporation technique from oil-in-water emulsion, stabilized with Tween 80. The PTX and ART released from NPs in a controlled release pattern. Moreover, compared to free drugs, PTX and ART preparation increased cytotoxicity on breast cancer cell-lines.29
Based on the results, there was no significant difference between the PTX release from PLGA-PTX NPs and the PTX release from PLGA-PTX-VitD3 co-delivery NPs between 0.5 and 240 hours (P > 0.05) (Table 2, Figure 4). No significant difference release of the PTX from PLGA-PTX NPs was also observed between 0.5 and 3 hours (P > 0.05). The release of VitD3 from PLGA-PTX-VitD3 co-delivery NPs between 0.5 and 12 hours did not also show meaningful differences (P > 0.05). Hence, the release of VitD3 from PLGA-PTX-VitD3 co-delivery NPs increased at 240 hours, continuously (P<0.05). The release of PTX and VitD3 from PLGA-PTX-VitD3 co-delivery NPs showed a noticeable difference between 3 and 96 hours (P< 0.05) (Table 2, Figure 4).
Table 2. In vitro drug release from the optimized sample. The percentage of cumulative release of PTX from PLGA-PTX, PTX from Co-delivery NPs and vitamin D from Co delivery NPs against time .
| Time treatment | PTX from PLGA-PTX | PTX from Co-delivery | Vitamin D from Co-delivery |
| 0.5 | 2.04 ± 0.32Ai | 3.00 ± 0.90Ah | 0Ah |
| 1 | 5.67 ± 0.88Ai | 6.00 ± 1.90Ah | 0Ah |
| 2 | 7.67 ± 1.17Ai | 10.73 ± 1.22Agh | 0Ah |
| 3 | 12.23 ± 1.74ABhi | 16.00 ± 5.51Afg | 0Bh |
| 6 | 18.03 ± 2.65Agh | 20.00 ± 8.50Afg | 1.00 ± 0.00Bh |
| 12 | 23.00 ± 4.48Afg | 24.00 ± 3.51Af | 3.00 ± 0.58Bh |
| 24 | 30.00 ± 3.06ABf | 35.00 ± 7.02Ae | 19.30 ± 1.56Bg |
| 48 | 38.00 ± 3.50ABe | 48.00 ± 7.44Ad | 34.00 ± 4.58Bf |
| 72 | 49.03 ± 6.20ABd | 54.00 ± 3.51Ad | 40.97 ± 6.72Bef |
| 96 | 55.97 ± 3.87ABcd | 62.00 ± 4.04Ac | 46.00 ± 4.51Be |
| 120 | 60.00 ± 2.73Ac | 66.00 ± 7.05Ac | 57.00 ± 7.51Ad |
| 144 | 69.00 ± 10.17Ab | 70.00 ± 7.51Ac | 64.97 ± 3.05Ac |
| 168 | 74.00 ± 3.51Ab | 80.17 ± 5.67Ab | 75.00 ± 7.21Ab |
| 240 | 92.00 ± 4.04Aa | 98.00 ± 2.00Aa | 96.00 ± 5.03Aa |
* Values are the means ± standard error (n=3). Different capital letters (A, B, C, ...) in the same row shows significant difference among treatments for each time and different lowercase letters (a, b, c, ...) in the same column shows significant difference among different times (Tukey’s test, P < 0.05).
Figure 4.

PTX release from PLGA NPs (a), PTX release from co-delivery NPs (b) and VitD3 release from co-delivery NPs (c).
Figure 5 shows the migration of MCF-7 cell line at the beginning (as a control) and after 24 hours of exposure, when exposed to PLGA-PTX-VitD3 NPs, PLGA-PTX NPs, and PTX. The results showed that PLGA-PTX-VitD3 co-delivery NPs is able to prevent metastasis of the MCF-7 cell lines.
Figure 5.
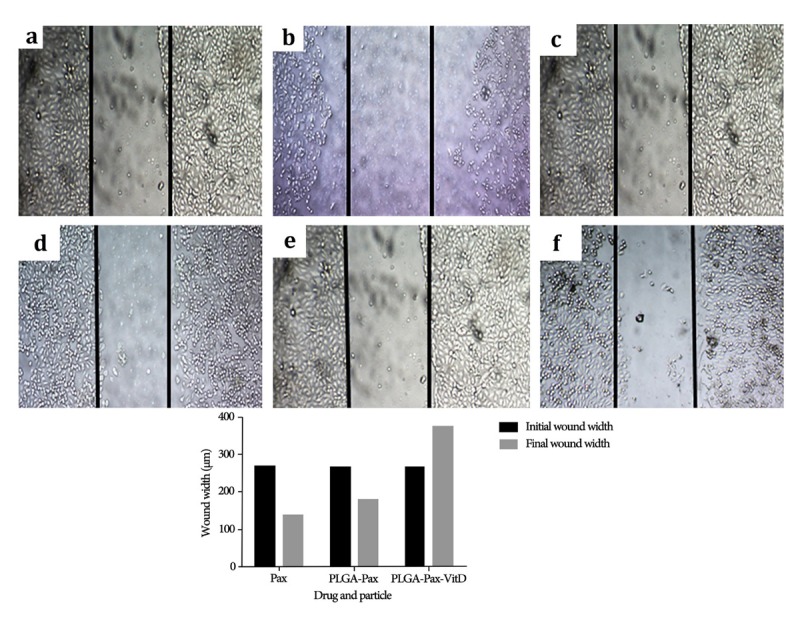
The mean of the distance scratch edges (n = 15) for PLGA-PTX- VitD3 co-delivery NPs (b), PLGA-PTX NPs (d), and PTX (f) at the beginning (a, c and e), and after 24 hours of experience.
As we show in Figure 6, the nucleus of MCF-7 cell lines upon exposure to PLGA-PTX-VitD3 co-delivery NPs become fragment and dense chromatin, which showed MCF-7 cells death.
Figure 6.
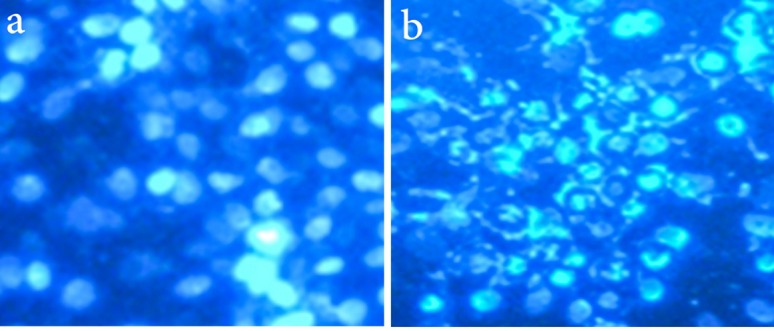
The fragmented nuclei and compact chromatin represent the cell death in the MCF-7 cell lines at present of PLGA-PTX- VitD3 co-delivery NPs (b), and control group (a)
Figure 7A shows the flow cytometer and cell cycle analysis of PLGA (a), PTX (b), PLGA-PTX NPs (c), PLGA-PTX-VitD3 co-delivery NPs (d). We found that there was a significant difference between PTX, PLGA-PTX NPs, PLGA-PTX-VitD3 co-delivery NPs, and the control group in the SUB G (1)/M phase (P = 0.030). The PLGA-PTX-VitD3 co-delivery NPs were able to induce early apoptosis on MCF-7 cells (41.425 ± 6.965) which showed a significant difference from the control group (11.245 ± 0.431) (P = 0.028). The results of this study also showed that PLGA-PTX, PTX and PLGA-PTX-VitD3 co-delivery NPs were able to block the MCF-7 cells at G (1)/M phase, which showed a significant difference in comparison with the control group (P< 0.05).
Figure 7.

(A) The percentage of DNA content of MCF-7 cells in the exposure with control (a), PTX (b), PLGA-PTX (c), and PLGA-PTX- VitD3 co-delivery NPs (d). (B) Expression of bax gene of MCF-7 cells in the exposure with PLGA-PTX, PLGA-PTX- VitD3 co-delivery NPs, PTX, PLGA and control group.
The results of this study showed that the expression of bax gene in the MCF-7 cells exposed to the PLGA-PTX-VitD3co-delivery NPs was significantly higher than the control group. Gene expression level at the PLGA-PTX-VitD3 co-delivery NPs showed an increase in comparison with PTX (Figure 7B). The expression of the bax gene in the PLGA-PTX NPs also showed a significant increase compared to free PTX (Figure 7B).
Kang et al found that anti-tumor activity of PTX from microemulsion containing PLGA was enhanced against MCF-7 cell lines.30 Shiny et al in 2013 prepared the PTX loaded microspheres using the blends of PLGA, poly-caprolactone. Shiny studies showed that blend of PLGA with poly-caprolactone resulted in complete release of the drug in a period of 30 days.31 Jin et al in 2009 also prepared the PTX-loaded PLGA NPs and determined cytotoxicity of released PTX for MCF-7 cell lines. The PTX-loaded PLGA NPs demonstrated that released PTX retained its bioactivity to block cells in G (2)/M phase.32 Vivek et al in 2014 investigated “smart” pH-responsive drug delivery system based on chitosan nano-carrier for its potential intelligent controlled release and enhancing chemotherapeutic efficiency of oxaliplatin.33 Furthermore, it was found that expression of bax gene was significantly up-regulated.
This study does not concentrate on anti-tumor activity of VitD3 (as aconfounder) loaded in the PLGA-PTX-VitD3 co-delivery NPs. Indeed, we did not investigate the synergistic impact of VitD3 and PTX. The cytotoxic effects of vitamin D and PLGA against cancer cells have been studied separately. It would not be cost-efficient to do repetitive tests.
Conclusion
Current study showed that PTX and VitD3 were loaded completely into the PLGA NPs. Despite the fact that the toxicity of the PLGA-PTX-VitD3 co-delivery NPs against MCF-7 cell lines became lower than that of free PTX, the co-deliver NPs were able to release drugs continuously for a long time which could increase the efficiency of drugs. The PLGA-PTX-VitD3 co-delivery NPs not only was able to prevent the formation of metastasis, but also, they induced primary apoptosis in the MCF-7. Furthermore, we found that the PLGA-PTX-VitD3 co-delivery NPs increased expression of bax gene in the MCF-7 cell lines.
Ethical Issues
This study was in accordance with the declaration of Helsinki; however, because we just used leftovers from primary cell line the need for ethical approval was not applicable.
Conflict of Interest
Authors declare no conflict of interest in this study.
Acknowledgments
We thank our colleagues from Guilan University of Medical Siences who provided insight and expertise that greatly assisted the research although they may not agree with all of the interpretations/conclusions of this paper. We would also like to show our gratitude to Dr. Bahram Soltani for sharing their pearls of wisdom with us during the course of this research, and we thank 3 “anonymous” reviewers for their so-called insights. We gratefully acknowledge the dedicated efforts of the investigators and coordinators participated in this study.
References
- 1.Rocha A, Wang L, Penichet M, Martins-Green M. Pomegranate juice and specific components inhibit cell and molecular processes critical for metastasis of breast cancer. Breast Cancer Res Treat. 2012;136(3):647–58. doi: 10.1007/s10549-012-2264-5. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen KT. Targeted nanoparticles for cancer therapy: promises and challenge. J Nanomedic Nanotechnol. 2011;2:103e. doi: 10.4172/2157-7439.1000103e. [DOI] [Google Scholar]
- 3.Zheng N, Lian B, Xu G, Liu X, Li X, Ji J. Development of a Subcellular Semimechanism-Based Pharmacokinetic/Pharmacodynamic Model to Characterize Paclitaxel Effects Delivered by Polymeric Micelles. J Pharm Sci. 2019;108(1):725–31. doi: 10.1016/j.xphs.2018.10.062. [DOI] [PubMed] [Google Scholar]
- 4.Venugopal V, Krishnan S, Palanimuthu VR, Sankarankutty S, Kalaimani JK, Karupiah S. et al. Anti-EGFR anchored paclitaxel loaded PLGA nanoparticles for the treatment of triple negative breast cancer In-vitro and in-vivo anticancer activities. PLoS One. 2018;13(11):e0206109. doi: 10.1371/journal.pone.0206109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vijayan V, Shalini K, Yugesvaran V, Yee TH, Balakrishnan S, Palanimuthu VR. Effect of paclitaxel-loaded PLGA nanoparticles on MDA-MB type cell lines: apoptosis and cytotoxicity studies. Curr Pharm Des. 2018;24(28):3366–75. doi: 10.2174/1381612824666180903110301. [DOI] [PubMed] [Google Scholar]
- 6.Sadat Tabatabaei Mirakabad F, Nejati-Koshki K, Akbarzadeh A, Yamchi MR, Milani M, Zarghami N. et al. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac J Cancer Prev. 2014;15(2):517–35. doi: 10.7314/apjcp.2014.15.2.517. [DOI] [PubMed] [Google Scholar]
- 7.Feng SS, Mu L, Win KY, Huang G. Nanoparticles of biodegradable polymers for clinical administration of paclitaxel. Curr Med Chem. 2004;11(4):413–24. doi: 10.2174/0929867043455909. [DOI] [PubMed] [Google Scholar]
- 8.Grabowski N, Hillaireau H, Vergnaud J, Santiago LA, Kerdine-Romer S, Pallardy M. et al. Toxicity of surface-modified PLGA nanoparticles toward lung alveolar epithelial cells. Int J Pharm. 2013;454(2):686–94. doi: 10.1016/j.ijpharm.2013.05.025. [DOI] [PubMed] [Google Scholar]
- 9.Menon JU, Kona S, Wadajkar AS, Desai F, Vadla A, Nguyen KT. Effects of surfactants on the properties of PLGA nanoparticles. J Biomed Mater Res A. 2012;100(8):1998–2005. doi: 10.1002/jbm.a.34040. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Z, Feng SS. The drug encapsulation efficiency, in vitro drug release, cellular uptake and cytotoxicity of paclitaxel-loaded poly(lactide)-tocopheryl polyethylene glycol succinate nanoparticles. Biomaterials. 2006;27(21):4025–33. doi: 10.1016/j.biomaterials.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 11.Horwitz SB. Taxol (paclitaxel): mechanisms of action. Ann Oncol. 1994;5 Suppl 6:S3–6. [PubMed] [Google Scholar]
- 12.Hershberger PA, Yu WD, Modzelewski RA, Rueger RM, Johnson CS, Trump DL. Calcitriol (1,25-dihydroxycholecalciferol) enhances paclitaxel antitumor activity in vitro and in vivo and accelerates paclitaxel-induced apoptosis. Clin Cancer Res. 2001;7(4):1043–51. [PubMed] [Google Scholar]
- 13.Koshizuka K, Koike M, Asou H, Cho SK, Stephen T, Rude RK. et al. Combined effect of vitamin D3 analogs and paclitaxel on the growth of MCF-7 breast cancer cells in vivo. Breast Cancer Res Treat. 1999;53(2):113–20. doi: 10.1023/a:1006123819675. [DOI] [PubMed] [Google Scholar]
- 14.Krishnan AV, Swami S, Feldman D. Vitamin D and breast cancer: inhibition of estrogen synthesis and signaling. J Steroid Biochem Mol Biol. 2010;121(1-2):343–8. doi: 10.1016/j.jsbmb.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 15.Chakraborti CK. Vitamin D as a promising anticancer agent. Indian J Pharmacol. 2011;43(2):113–20. doi: 10.4103/0253-7613.77335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mathiasen IS, Lademann U, Jäättelä M. Apoptosis induced by vitamin D compounds in breast cancer cells is inhibited by Bcl-2 but does not involve known caspases or p53. Cancer Res. 1999;59(19):4848–56. [PubMed] [Google Scholar]
- 17.Madani F, Esnaashari SS, Mujokoro B, Dorkoosh F, Khosravani M, Adabi M. Investigation of effective parameters on size of paclitaxel loaded PLGA nanoparticles. Adv Pharm Bull. 2018;8(1):77–84. doi: 10.15171/apb.2018.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dash S, Murthy PN, Nath L, Chowdhury P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol Pharm. 2010;67(3):217–23. [PubMed] [Google Scholar]
- 19.Ahmadiankia N, Moghaddam HK, Mishan MA, Bahrami AR, Naderi-Meshkin H, Bidkhori HR. et al. Berberine suppresses migration of MCF-7 breast cancer cells through down-regulation of chemokine receptors. Iran J Basic Med Sci. 2016;19(2):125–31. [PMC free article] [PubMed] [Google Scholar]
- 20.Grada A, Otero-Vinas M, Prieto-Castrillo F, Obagi Z, Falanga V. Research techniques made simple: analysis of collective cell migration using the wound healing assay. J Invest Dermatol. 2017;137(2):e11–e6. doi: 10.1016/j.jid.2016.11.020. [DOI] [PubMed] [Google Scholar]
- 21. Giaretti W. Model of Chromosomal Instability in Oral Carcinogenesis and Progression. Oral Cancer. InTech; 2012. 10.5772/31883. [DOI]
- 22. Wiberley SE, Colthup NB, Daly L. Introduction to Infrared and Raman Spectroscopy. New York: Academic Press; 1975.
- 23.Silva ATCR, Cardoso BCO, e Silva MESR, Freitas RFS, Sousa RG. Synthesis, characterization, and study of PLGA copolymer in vitro degradation. J Biomater Nanobiotechnol. 2015;6(1):8–19. doi: 10.4236/jbnb.2015.61002. [DOI] [Google Scholar]
- 24.Hummel DO, Scholl F, Solti A. Atlas of polymer and plastics analysis: plastics, fibres, rubbers, resins; starting and auxiliary materials, degradation products. Carl Hanser. 1988 [Google Scholar]
- 25.Yerlikaya F, Ozgen A, Vural I, Guven O, Karaagaoglu E, Khan MA. et al. Development and evaluation of paclitaxel nanoparticles using a quality-by-design approach. J Pharm Sci. 2013;102(10):3748–61. doi: 10.1002/jps.23686. [DOI] [PubMed] [Google Scholar]
- 26.Alipour S, Montaseri H, Tafaghodi M. Preparation and characterization of biodegradable paclitaxel loaded alginate microparticles for pulmonary delivery. Colloids Surf B Biointerfaces. 2010;81(2):521–9. doi: 10.1016/j.colsurfb.2010.07.050. [DOI] [PubMed] [Google Scholar]
- 27.Cerqueira BBS, Lasham A, Shelling AN, Al-Kassas R. Development of biodegradable PLGA nanoparticles surface engineered with hyaluronic acid for targeted delivery of paclitaxel to triple negative breast cancer cells. Mater Sci Eng C Mater Biol Appl. 2017;76:593–600. doi: 10.1016/j.msec.2017.03.121. [DOI] [PubMed] [Google Scholar]
- 28.Turino LN, Ruggiero MR, Stefania R, Cutrin JC, Aime S, Geninatti Crich S. Ferritin decorated PLGA/paclitaxel loaded nanoparticles endowed with an enhanced toxicity toward MCF-7 breast tumor cells. Bioconjug Chem. 2017;28(4):1283–90. doi: 10.1021/acs.bioconjchem.7b00096. [DOI] [PubMed] [Google Scholar]
- 29.Tran BN, Nguyen HT, Kim JO, Yong CS, Nguyen CN. Developing combination of artesunate with paclitaxel loaded into poly-d,l-lactic-co-glycolic acid nanoparticle for systemic delivery to exhibit synergic chemotherapeutic response. Drug Dev Ind Pharm. 2017;43(12):1952–62. doi: 10.1080/03639045.2017.1357729. [DOI] [PubMed] [Google Scholar]
- 30.Kang BK, Chon SK, Kim SH, Jeong SY, Kim MS, Cho SH. et al. Controlled release of paclitaxel from microemulsion containing PLGA and evaluation of anti-tumor activity in vitro and in vivo. Int J Pharm. 2004;286(1-2):147–56. doi: 10.1016/j.ijpharm.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 31.Shiny J, Ramchander T, Goverdhan P, Habibuddin M, Aukunuru JV. Development and evaluation of a novel biodegradable sustained release microsphere formulation of paclitaxel intended to treat breast cancer. Int J Pharm Investig. 2013;3(3):119–25. doi: 10.4103/2230-973x.119212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin C, Bai L, Wu H, Song W, Guo G, Dou K. Cytotoxicity of paclitaxel incorporated in PLGA nanoparticles on hypoxic human tumor cells. Pharm Res. 2009;26(7):1776–84. doi: 10.1007/s11095-009-9889-z. [DOI] [PubMed] [Google Scholar]
- 33.Vivek R, Thangam R, Nipunbabu V, Ponraj T, Kannan S. Oxaliplatin-chitosan nanoparticles induced intrinsic apoptotic signaling pathway: a “smart” drug delivery system to breast cancer cell therapy. Int J Biol Macromol. 2014;65:289–97. doi: 10.1016/j.ijbiomac.2014.01.054. [DOI] [PubMed] [Google Scholar]