

Abstract
Fluoroquinolones (FQ) form the backbone in experimental treatment regimens against drug-susceptible tuberculosis. However, little is known on whether the genetic variation present in natural populations of Mycobacterium tuberculosis (Mtb) affects the evolution of FQ-resistance (FQ-R). To investigate this question, we used nine genetically distinct drug-susceptible clinical isolates of Mtb and measured their frequency of resistance to the FQ ofloxacin (OFX) in vitro. We found that the Mtb genetic background led to differences in the frequency of OFX-resistance (OFX-R) that spanned two orders of magnitude and substantially modulated the observed mutational profiles for OFX-R. Further, in vitro assays showed that the genetic background also influenced the minimum inhibitory concentration and the fitness effect conferred by a given OFX-R mutation. To test the clinical relevance of our in vitro work, we surveyed the mutational profile for FQ-R in publicly available genomic sequences from clinical Mtb isolates, and found substantial Mtb lineage-dependent variability. Comparison of the clinical and the in vitro mutational profiles for FQ-R showed that 51% and 39% of the variability in the clinical frequency of FQ-R gyrA mutation events in Lineage 2 and Lineage 4 strains, respectively, can be attributed to how Mtb evolves FQ-R in vitro. As the Mtb genetic background strongly influenced the evolution of FQ-R in vitro, we conclude that the genetic background of Mtb also impacts the evolution of FQ-R in the clinic.
Keywords: Mycobacterium tuberculosis, antimicrobial resistance, evolution, fluoroquinolones, epistasis, mycobacteria, fitness
Introduction
Antimicrobial resistance (AMR) poses a major threat to our ability to treat infectious diseases (MacGowan 2008; Winston and Mitruka 2012). The rise of AMR is a complex phenomenon with a broad range of contributing socioeconomic and behavioral factors (Dalton et al. 2012; Merker et al. 2015; Alvarez-Uria et al. 2016; Eldholm et al. 2016; Shah et al. 2017). However, the emergence of AMR within any pathogen population is ultimately an evolutionary process (zur Wiesch et al. 2011; Hughes and Andersson 2017). This evolutionary process is influenced by multiple factors, including drug pressure and pathogen genetics. Firstly, the drug type and drug concentration can affect the nature and relative frequencies of AMR mutations observed in a given pathogen population (also known as the mutational profile for AMR) (Zhou et al. 2000; Ford et al. 2013; Lindsey et al. 2013; McGrath et al. 2014; Hughes and Andersson 2017; Huseby et al. 2017). Secondly, pathogen populations comprise genetically distinct strains, and this genetic variation may also influence AMR evolution (Fenner et al. 2012; Vogwill et al. 2014, 2016; Gagneux 2018). Different pathogen genetic backgrounds can have different baseline susceptibilities to a given drug (Ängeby et al. 2010; Coeck et al. 2016), which can consequently affect patient treatment outcomes (Colangeli et al. 2018). The genetic background has also been shown to modulate the acquisition and prevalence of AMR (Borrell and Gagneux 2009; Fenner et al. 2012; Ford et al. 2013; Wollenberg et al. 2017), the mutational profile for AMR (Fenner et al. 2012; Ford et al. 2013; Vogwill et al. 2014; Oppong et al. 2019), and the phenotypic effects of AMR mutations (Gagneux et al. 2006; Decuypere et al. 2012; Angst and Hall 2013; Vogwill et al. 2016). Studying the interplay between pathogen genetics and drug pressure is therefore important in understanding how to restrict the emergence of AMR in pathogen populations.
AMR in Mycobacterium tuberculosis (Mtb), the etiological agent of human tuberculosis (TB), is of particular importance. Mtb infections globally cause the highest rate of mortality due to a single infectious agent both in general, and due to AMR specifically (World Health Organization 2017). Although the genetic variation in Mtb is small compared with other bacterial pathogens (Comas et al. 2010; Gagneux 2018), several studies have shown that this limited genetic variation influences AMR phenotypes and prevalence (Gagneux et al. 2006; Zaczek et al. 2009; Fenner et al. 2012; Gagneux 2018). The global genetic diversity of Mtb comprises seven phylogenetic lineages (Comas et al. 2010; Gagneux 2018), and Mtb strains belonging to the Lineage 2 Beijing/W genetic background have repeatedly been associated with multidrug-resistant TB (MDR-TB; defined as an infection from an Mtb strain that is resistant to at least isoniazid and rifampicin) both in vitro and in clinical settings (Borrell and Gagneux 2009; Fenner et al. 2012; Ford et al. 2013; Merker et al. 2015; Wollenberg et al. 2017).
One strategy to reduce the emergence of AMR in Mtb is the development of new, shorter treatment regimens (Imperial et al. 2018; Vjecha et al. 2018). Many such experimental regimens use third- or fourth-generation fluoroquinolones (FQ) against drug-susceptible Mtb (Gillespie et al. 2014; Jindani et al. 2014; Merle et al. 2014; Imperial et al. 2018; Vjecha et al. 2018). However, FQs have long been integral to treating MDR-TB (Takiff and Guerrero 2011), and the previous use of FQs has led to the emergence of FQ-resistance (FQ-R) in clinical strains of Mtb (Takiff et al. 1994; Maruri et al. 2012; Shah et al. 2017). FQ-R is one of the defining properties of extensively drug-resistant TB (XDR-TB), and XDR-TB accounts for 8.5% of MDR-TB cases (World Health Organization 2017). Understanding how FQ-R is acquired in natural populations of Mtb may allow for the development of tools or strategies to mitigate further increases in FQ-R prevalence.
In Mtb, the sole target of FQ is DNA gyrase (Takiff et al. 1994; Zhou et al. 2000; Piton et al. 2010; Aldred et al. 2016; Blower et al. 2016). Consequently, clinically relevant FQ-R in Mtb is primarily due to a limited set of chromosomal mutations located within the “quinolone-resistance-determining region” (QRDR) of the gyrA and gyrB genes, which encode DNA gyrase (Takiff et al. 1994; Maruri et al. 2012; Wollenberg et al. 2017). No horizontal gene-transfer or plasmid-based resistance to FQ has been documented in Mtb (Boritsch et al. 2016; Gygli et al. 2017). Studying FQ-R evolution in Mtb populations thus provides a promising setting for elucidating how the genetic background may affect the emergence and maintenance of clinically relevant chromosomal AMR mutations in bacterial populations.
While a great deal of literature exists on the biochemical mechanisms leading to the FQ-R phenotype in Mtb (Zhou et al. 2000; Piton et al. 2010; Mustaev et al. 2014; Aldred et al. 2016; Blower et al. 2016), little is known on the evolutionary dynamics of FQ-R in different populations of Mtb. Given that antimicrobial regimens against Mtb infections use standardized, empirical dosing strategies (World Health Organization 2017), it is unclear whether different Mtb genetic backgrounds would acquire FQ-R at the same frequency when exposed to the same antimicrobial concentration. Whether the Mtb genetic background would also modulate the mutational profile for FQ-R, or the phenotypic effects of FQ-R mutations, is unknown. Such knowledge may provide insights on how to maintain or prolong the efficiency of FQs against different genetic variants of Mtb in the clinic.
In this study, we tested whether the Mtb genetic background plays a role in the evolution of FQ-R. We showed that the Mtb genetic background can lead to differences in the frequency of FQ-R emergence that span two orders of magnitude, as well as substantially modulate the mutational profile for FQ-R. We further demonstrated that the phenotypic effects of clinically relevant FQ-R mutations differed depending on the Mtb genetic background they were present in. Analysis of publicly available genomic sequences from clinical Mtb isolates also revealed a positive association between the FQ-R mutational profiles observed in vitro and the mutational profiles observed in the clinic. Taken together, we showed that the Mtb genetic background had a considerable role in evolution of FQ-R in the clinic.
Results
Frequency of Ofloxacin-Resistance in Mtb Is Strain-Dependent
We first tested for whether the Mtb genetic background led to differences in the frequency of FQ-R acquisition. To do so, we performed a Luria–Delbrück fluctuation analysis on nine drug-susceptible and genetically distinct Mtb clinical strains belonging to Lineage 1 (L1), Lineage 2 (L2), and Lineage 4 (L4) (supplementary table S1, Supplementary Material online) (Luria and Delbrück 1943; Comas et al. 2010; Gagneux 2018; Borrell et al. 2019). We measured their frequency of resistance in vitro to the FQ ofloxacin (OFX), as OFX was used extensively to treat MDR-TB patients in the past (Takiff and Guerrero 2011). Given that anti-TB treatment regimens use standardized drug concentrations (World Health Organization 2017), we also measured the frequency of resistance to the same concentration of OFX (4 µg/ml) for all nine strains. We observed significant strain-dependent variation in the frequency of OFX-resistance (OFX-R) at 4 µg/ml, with the difference spanning two orders of magnitude (fig. 1A; P = 2.2 × 10−16, Kruskal–Wallis). Several of the nine drug-susceptible Mtb strains contained missense substitutions in DNA gyrase that are not associated with FQ-R (supplementary table S2, Supplementary Material online) (Borrell et al. 2019). These mutations are phylogenetic markers that reflect the population structure of Mtb and cannot be avoided if strains from different Mtb lineages are used (Comas et al. 2010; Gagneux 2018). We found no evidence for any associations between the presence a given phylogenetic DNA gyrase missense mutation and the frequency of OFX-R acquired.
Fig. 1.

Variation in the frequency of ofloxacin-resistance between genetically distinct, wild-type Mycobacterium tuberculosis strains. (A) Frequency of ofloxacin-resistance at 4 µg/ml ofloxacin (OFX), as measured by fluctuation analysis. Top panel: colored points represent the frequency of ofloxacin-resistance per cell per parallel culture, with darker points representing multiple cultures with the same frequency. Colors denote the lineage that the Mtb strain belongs to (L1, pink; L2, blue; L4, red). Gray points represent the estimated number of drug-resistance mutations per cell per strain as calculated by MSS-MLE, while black bars denote the respective 95% confidence intervals. Bottom panel: the percentage of parallel cultures lacking OFX-resistant mutants. (B) Frequency of ofloxacin-resistance at 2 or 8 µg/ml OFX. ††Due to restrictions on the range of values that the MSS-MLE method is valid for (see Materials and Methods), the estimated number of drug-resistance mutations per cell for N0157 at 2 µg/ml OFX, as presented here, is an underestimate.
The concentration of the antimicrobial can affect the observed frequencies of AMR in Mtb (Zhou et al. 2000; Ford et al. 2013; McGrath et al. 2014). Therefore, we tested whether changing the selective concentration of OFX would affect the relative differences in strain-specific OFX-R frequencies. For the sake of simplicity, we tested only two strains, with each strain at the opposite extremes of the frequency of resistance to 4 µg/ml OFX, as shown in figure 1A: N0157 (high OFX-R frequency) and N0145 (low OFX-R frequency). We found that the frequency of OFX-R remained one to two orders of magnitude higher in N0157 than in N0145 across all the concentrations we tested (fig. 1B, P = 2.46×10−5 for 2 µg/ml OFX, and P = 4.03×10−6 for 8 µg/ml OFX, Wilcoxon rank-sum test). The N0157 strain had nearly confluent growth at 2 µg/ml OFX, which is the OFX concentration that has been shown to inhibit 95% of Mtb strains that have not been previously exposed to OFX, but does not inhibit Mtb strains that are considered resistant to OFX in the clinic (Ängeby et al. 2010; Coeck et al. 2016). This suggested that N0157 has low-level resistance to OFX, despite having no mutation in the QRDR. Meanwhile, at 8 µg/ml OFX, we observed only four resistant colonies for N0145 across all samples, with all colonies arising within the same culture.
The variation in OFX-R frequencies when selecting on the same concentration of OFX may be driven by several, nonexclusive biological factors. Firstly, the Mtb strains we tested may have different baseline DNA mutation rates. Secondly, the number and relative frequency of potential mutations that confer OFX-R may vary depending on the Mtb genetic background. Thirdly, the relative cost of OFX-R mutations may differ between Mtb genetic backgrounds. As the observed frequency of OFX-R in Mtb is likely the result from a combination of multiple factors, we took advantage of the fact that we had identified strains with a range of OFX-R frequencies. We selected three representative strains with significantly different OFX-R frequencies: N0157, N1283, and N0145. These strains had a high-, mid-, and low frequency of OFX-R, respectively (fig. 1A). We then explored the relative contributions of each biological factor listed above in driving the variation in OFX-R across genetically distinct Mtb strains.
Mutation Rate Differences Do Not Drive the In Vitro Variation in OFX-R Frequency in Mtb
We first tested for the presence of differential mutation rates between our panel of Mtb strains in figure 1A. Mutations in dnaE, which encodes the replicative DNA polymerase and serves as the major replicative exonuclease in Mtb, have been shown to confer a hypermutator phenotype in Mtb in the absence of environmental stress (Rock et al. 2015; Baños-Mateos et al. 2017). While dnaE mutations were present in the genomic data of our panel of drug-susceptible Mtb strains (supplementary table S2, Supplementary Material online) (Borrell et al. 2019), none was in the polymerase and histidinol phosphatase domain of DnaE, the region where mutations would impart a hypermutator phenotype (Rock et al. 2015; Baños-Mateos et al. 2017). We did not test for the presence of dnaE mutations in the resistant colonies following the fluctuation analysis, as we reasoned that the likelihood of gaining both a dnaE and a gyrA double mutation within this relatively short period is extremely low as to be considered negligible. To test for mutation rate variation in vitro, we again conducted a fluctuation analysis on N0157, N1283, and N0145 (the high-, mid-, and low-frequency OFX-R strains, respectively), but used streptomycin (STR; 100 µg/ml) instead of OFX. We hypothesized that if the frequency of OFX-R is driven by differential mutation rates, then we should expect similar differences in the frequency of STR-resistance (STR-R). However, we observed no evidence for differences in the frequency of STR-R between the strains tested (fig. 2, P = 0.135, Kruskal–Wallis; supplementary table S3, Supplementary Material online). This suggested that the observed differences in frequency of resistance are specific to OFX, and that there are limited, if any, inherent differences in mutation rates between the Mtb strains tested.
Fig. 2.
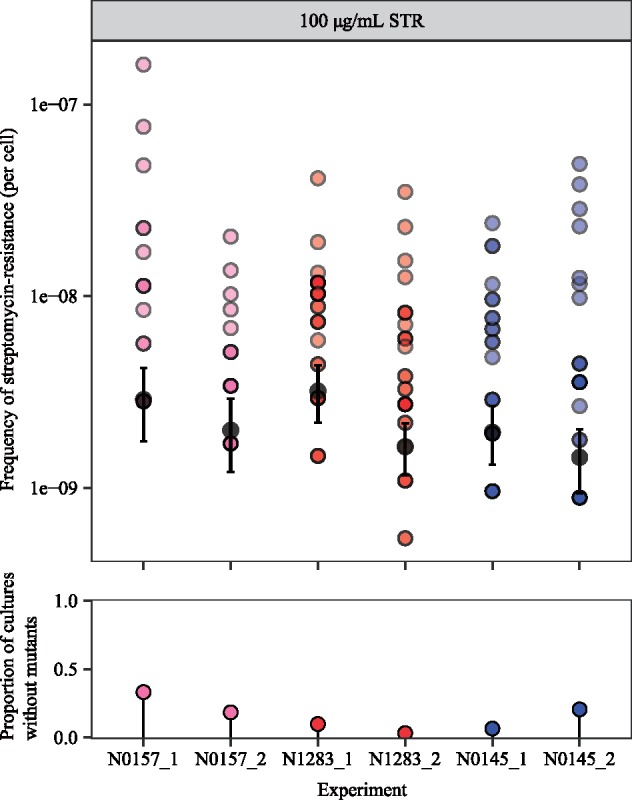
The frequency of streptomycin-resistance at 100 µg/ml streptomycin (STR) for wild-type N0157, N1283, and N0145 Mycobacterium tuberculosis strains, as measured by fluctuation analysis. Top panel: colored points represent the frequency of streptomycin-resistance per cell per parallel culture, with darker points representing multiple cultures with the same frequency. Colors denote the lineage that the Mtb strain belongs to (L1, pink; L2, blue; L4, red). Gray points represent the estimated number of drug-resistance mutations per cell per strain as calculated by MSS-MLE, while black bars denote the respective 95% confidence intervals. Bottom panel: the percentage of parallel cultures lacking STR-resistant mutants. Two biological replicates are presented for each Mtb strain, with each replicate identifier suffixed after the strain name.
Mutational Profile for OFX-R Is Highly Strain-Dependent
We next determined the mutational profile for OFX-R for each strain used in the fluctuation analysis at 4 µg/ml OFX (fig. 1A). The QRDR mutations in 680 gyrA and 590 gyrB sequences were identified in the resistant colonies. We observed that gyrA mutations made up 99.7% of the QRDR mutations observed (645 gyrA mutations, 2 gyrB mutations), and no QRDR double mutants were present (supplementary fig. S1 and tables S4 and S5, Supplementary Material online). The mutational profiles for OFX-R were also highly strain-specific (fig. 3A, P = 5.00 × 10−4, Fisher’s exact test). Specifically, the GyrA A90V mutation was most prevalent in the high-frequency OFX-R strains, while GyrA D94G was most prevalent in all other strains. There was also a slight trend showing that strains with a greater number of unique gyrA mutations present also had higher rates of OFX-R (figs. 1A and 3B).
Fig. 3.
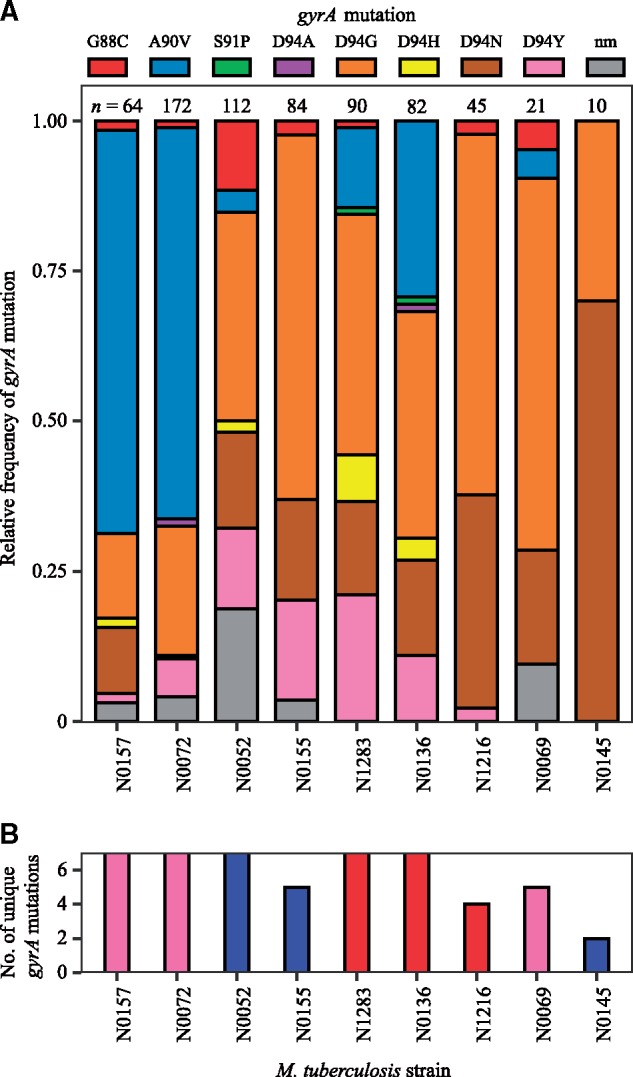
Variation in the mutational profile for ofloxacin-resistance after fluctuation analyses using nine genetically distinct Mycobacterium tuberculosis strains. (A) Mutations in the quinolone-resistance-determining region (QRDR) of gyrA were analyzed in 680 ofloxacin (OFX)-resistant colonies from the fluctuation analysis performed in figure 2A (nm, no identified QRDR gyrA mutations). Strains are ordered left to right based on their frequency of OFX-resistance at 4 µg/ml OFX. Numbers of colonies analyzed per strain are reported directly above each column. (B) The number of unique QRDR gyrA mutations per Mtb strain for OFX-resistance. Bar colors denote the Mtb lineage the strain belongs to (L1, pink; L2, blue; L4, red).
The strain-dependent variation in the mutational profile for OFX-R may be due to gyrA mutations conferring different resistance levels depending on the Mtb strain they are present in. To test this hypothesis, we first isolated OFX-R mutants carrying one of four possible GyrA mutations (G88C, A90V, D94G, or D94N) in the three strains used in figure 2: N0157, N1283, and N0145. The OFX MIC was determined for each of the 12 OFX-R mutant strains, along with their respective wild-type ancestors. We found that each parental wild-type strain had different susceptibilities to OFX, with N0157, N1283, and N0145 having OFX MICs of 2, 0.6, and 0.5 µg/ml, respectively (fig. 4A and supplementary table S6, Supplementary Material online). This was consistent with the fluctuation analysis results shown in figure 1B. Furthermore, we observed that the OFX MIC conferred by a given gyrA mutation varied depending on the strain it was present in (fig. 4B and supplementary table S6, Supplementary Material online). For example, mutants in the N0157 strain generally had higher OFX MICs than mutants in either the N0145 or N1283 strains. The only mutation that deviated from this trend was GyrA G88C, which conferred a higher OFX MIC when in the N0145 strain. Notably, the GyrA A90V mutation conferred a resistance level ≥4 µg/ml OFX in the N0157 and N1283 strains, but not in N0145. This was consistent with the presence of GyrA A90V in the OFX-R mutational profile for N0157 and N1283, but not in N0145, in the fluctuation analysis using 4 µg/ml OFX (figs. 1A and 3). In summary, the differences in OFX MIC reflected the strain-dependent mutational profiles for OFX-R in Mtb, as expected.
Fig. 4.
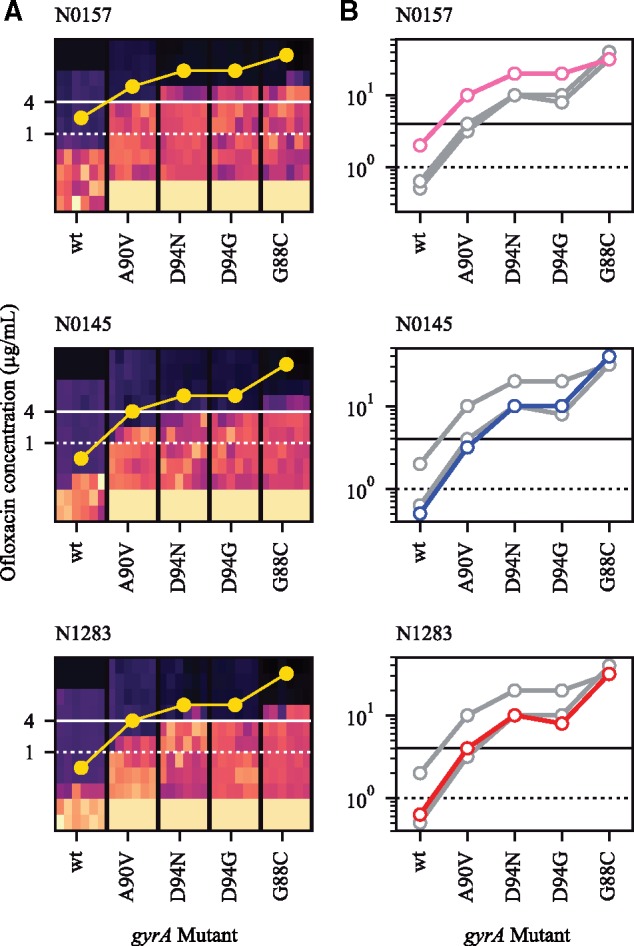
The Mycobacterium tuberculosis genetic background modulates the ofloxacin (OFX) minimum inhibitory concentration (MIC). (A) Heat-map of OFX-susceptibility via Alamar Blue assay for gyrA mutant strains of Mtb, as well as their wild-type ancestor, in three genetic backgrounds (N0157, N0145, or N1283). Light areas represent growing cultures, while dark areas represent nongrowing cultures. Yellow points represent estimates for OFX MIC (≥95% reduction in fluorescence). Areas of solid black colors (at 16+ μg/ml OFX for wild-type) and solid light beige colors (at <0.125 μg/ml OFX for mutants) were not measured and colored in for illustrative purposes. (B) OFX MIC estimates for each strain per genetic background, superimposed. Colored points and lines represent MIC measurements for highlighted genetic background, with the line color denoting the lineage that the strain belongs to (L1, pink, L2, blue, L4, red). Gray points and lines represent the other two genetic backgrounds. Solid horizontal black line denotes 4 μg/ml OFX, while dashed horizontal black line denotes 1 μg/ml OFX.
Fitness of OFX-R Mutations Are Associated with Their Relative Frequency In Vitro
While the OFX MICs may determine which mutations may be observed in a fluctuation analysis, it is not the sole parameter to influence the OFX-R mutational profile for a given strain. We found that while the same gyrA mutation can be observed in two different Mtb strains, their relative frequencies may vary (fig. 3). This variation may be due to the fitness of a given gyrA mutant being different across genetic backgrounds. To test this hypothesis, we used cell growth assays in antibiotic-free conditions to measure the in vitro fitness of our panel of OFX-R mutants relative to their respective parental wild-type ancestors. We observed that the relative fitness of the OFX-R mutants was modulated by both the gyrA mutation and the Mtb strain they were present in (fig. 5A; supplementary figs. S2 and S3 and table S7, Supplementary Material online). Furthermore, there was a positive association between the fitness of a given gyrA mutation with its relative frequency in the fluctuation analysis for the N0157 and N1283 strains (fig. 5B, P = 0.03 for N0157, P = 0.05 for N1283). There was no evidence of an association in the N0145 background due to the lack of GyrA G88C and A90V mutants in its fluctuation analysis.
Fig. 5.

The Mycobacterium tuberculosis genetic background modulates the fitness effect of fluoroquinolone-resistance mutations. (A) Fitness of ofloxacin-resistant Mtb strain with specified gyrA mutation relative to the fitness of their respective wild-type ancestral strain. Fitness was measured by cell growth assay in antibiotic-free conditions. Ancestral strain per gyrA mutant is indicated in the gray bar above each panel. (B) Association between the relative fitness of specified gyrA mutant and their absolute frequency after the fluctuation analysis performed in figure 1A and as reported in figure 3A, in three genetic backgrounds (N0157, N1283, and N0145).
The results from figures 4 and 5, as well as the apparent lack of mutation rate differences between our strains (fig. 2), suggested that differential mutational profiles were an important contributor in the variation in OFX-R frequency in Mtb. These mutational profile differences appear to be driven by the Mtb genetic background’s effect on both the MIC and the relative fitness cost of OFX-R mutations. We next explored whether these in vitro results would be relevant in clinical settings.
Mutational Profile for FQ-R In Vitro Reflects Clinical Observations
To explore the clinical relevance of our in vitro work, we surveyed the FQ-R mutational profile from publicly available Mtb genomes obtained from clinical isolates. FQs are generally used for treatment against MDR-TB (World Health Organization 2017). While it is unclear whether resistance mutations for isoniazid (INH) and/or rifampicin (RIF) predispose a strain to become FQ-R, the prevalence of FQ-R is heavily biased toward MDR-TB strains due to treatment practices. We therefore based our analyses on a collated data set of 3,450 publicly available MDR-TB genomes (supplementary table S8, Supplementary Material online), which we confirmed to be MDR-TB based on the presence of known INH-resistance (INH-R) and RIF-resistance (RIF-R) mutations. This data set provided a reasonable sampling of the overall genetic diversity of Mtb, as six of the seven known phylogenetic Mtb lineages were represented (Lineages 1–6) (Comas et al. 2010; Gagneux 2018). We catalogued their FQ-R mutational profiles, and found 950 FQ-R mutations in 854 genomes (supplementary tables S9 and S10, Supplementary Material online), showing that multiple FQ-R mutations may be present in the genome of a single Mtb clinical isolate. The frequency of FQ-R differed between lineages, with the highest frequencies present in L2 and L4 strains (supplementary table S9, Supplementary Material online, P < 2.20 × 10−16, Chi-square Goodness of Fit Test). Moreover, we noticed a lineage-dependent mutational profile for FQ-R (fig. 6, P = 7.10 × 10−5, Fisher’s exact test; supplementary tables S10 and S11, Supplementary Material online). For example, while the GyrA D94G mutation was most prevalent in strains belonging to L1, L2, and Lineage 3 (L3), the GyrA A90V mutation was most prevalent in L4 and Lineage 6 (L6).
Fig. 6.
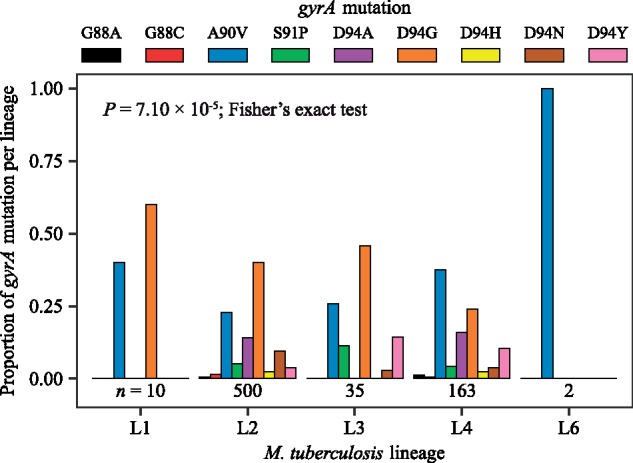
Mutational profile for fluoroquinolone-resistance gyrA mutations is lineage-specific in clinical isolates of Mycobacterium tuberculosis. An initial data set consisting of 3,450 genomes with confirmed MDR-TB mutations were surveyed. About 854 genomes were identified as fluoroquinolone-resistant, with 848 of these genomes containing at least one gyrA mutation. Only fixed fluoroquinolone-resistance mutations in the gyrA gene are enumerated here (n = 710). No fixed mutations were observed in Lineage 5 strains. Numbers of genomes analyzed per lineage are presented directly below their respective bar graph.
We observed that the mutational profile for FQ-R in the fluctuation analysis experiments mimicked published clinical data. Firstly, gyrA mutations made up the large majority of FQ-R mutations in vitro (fig. 3 and supplementary tables S4 and S5, Supplementary Material online) and 944 out of the 950 QRDR mutations in the clinic (99.6%; supplementary table S10, Supplementary Material online). We then tested whether the Mtb genetic background had an impact on FQ-R gyrA mutational profiles in the clinic as it did in vitro. However, transmission events can modulate the frequency of FQ-R gyrA mutations in the clinic, but not in a fluctuation analysis. If each genome from the clinical data is treated as an independent event, then number of FQ-R gyrA mutation events in the clinic would be overestimated compared with the number of mutation events in a fluctuation analysis. Therefore, rather than directly compare the frequency of gyrA mutations from the OFX-R mutational profile in figure 3 to the absolute frequency of gyrA mutations in the genomic data survey, we instead compared the in vitro frequency of gyrA mutations in figure 3 to the frequency of mutation events per gyrA mutation in the genomic data. To do so, we first used differences in the number of single-nucleotide polymorphisms (SNPs) as a measure of genetic distance between two genomes, then defined transmission clusters within the 3,450 MDR-TB genomes via a conservative cutoff of 12 SNPs average distance (Walker et al. 2013). Each unique and fixed FQ-R gyrA mutation present per transmission cluster, as well as each fixed FQ-R gyrA mutations present in nonclustered genomes, were counted as independent mutation events. We limited our analysis to L2 and L4 strains, as these two lineages had the highest clinical frequencies of FQ-R. We observed that the profile for FQ-R gyrA mutation events in L2 strains differed significantly to L4 strains (supplementary fig. S5, Supplementary Material online, P = 0.02, Fisher’s exact test; supplementary table S12, Supplementary Material online). Furthermore, there was a positive association between the frequency of a given FQ-R gyrA mutation in our fluctuation analysis compared with the frequency of its mutation event in the clinic for both L2 and L4 strains (fig. 7, P = 0.02 for L2, P = 0.04 for L4). Based on the adjusted R2 values, 51% of the variability in the clinical frequencies of FQ-R gyrA mutation events in L2 strains and 39% of the variability in L4 strains can be attributed to how FQ-R evolves in Mtb in vitro. As the in vitro evolution of FQ-R is itself modulated by the Mtb genetic background, this provided evidence for the Mtb genetic background’s role in the evolution of FQ-R in the clinic.
Fig. 7.
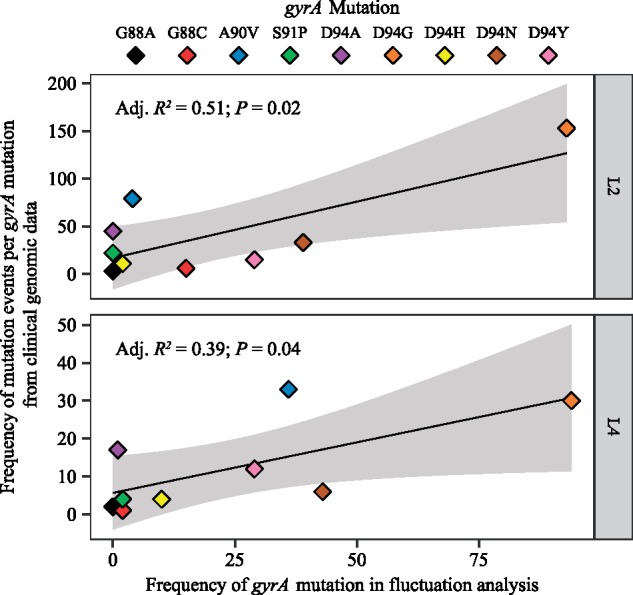
Association between the clinical frequency of mutation events of each fluoroquinolone-resistance (FQ-R) gyrA mutations with their respective in vitro frequencies among Mycobacterium tuberculosis strains belonging to either the L2 or L4 lineages. Mutation events per FQ-R gyrA mutation were enumerated from an initial data set of 3,450 genomes with confirmed MDR-TB mutations. Each unique and fixed FQ-R gyrA mutation present per transmission cluster (cutoff=12 SNPs average distance), as well as each fixed FQ-R gyrA mutation present in nonclustered genomes, were counted as independent mutation events. The in vitro frequencies of FQ-R gyrA mutations presented here are the same as in figure 3A, grouped by lineage.
Discussion
Overall, we illustrate the Mtb genetic background’s considerable role in the evolution of resistance to FQs, a clinically important antimicrobial. We first explored whether the genetic variation among natural populations of Mtb can influence FQ-R evolution in vitro. Specifically, considering that Mtb treatment regimens are based on standardized antimicrobial concentrations (World Health Organization 2017), we tested whether different genetic variants of Mtb would acquire FQ-R at the same frequency when exposed to the same concentration of FQ. Fluctuation analysis on nine, genetically distinct, drug-susceptible Mtb strains showed that the genetic background can have a drastic effect on the rate of OFX-R acquisition when using the same concentration of OFX (fig. 1). Our results provide the first evidence showing that the Mtb genetic background can modulate the frequency of FQ-R acquisition.
However, the effect of the Mtb genetic background on AMR frequencies observed here in the context of OFX-R differed from those reported in previous work focusing on other antibiotics. Past literature has focused on the positive association between MDR-TB and L2 Beijing (Borrell and Gagneux 2009; Fenner et al. 2012; Merker et al. 2015; Eldholm et al. 2016). Initial genetic analysis on a global collection of strains showed that mutations in DNA repair genes were associated with being MDR-TB, and that these mutations were specific to L2 Beijing isolates (Rad et al. 2003). The authors thus hypothesized that L2 Beijing strains may have a hypermutator phenotype, which would lead to higher rates of AMR mutations acquisition (Rad et al. 2003). Based on this L2 Beijing hypermutator hypothesis, one would expect that L2 Beijing strains would also show higher frequencies of FQ-R. However, this was not the case in our fluctuation analysis for OFX-R, as one of our L2 Beijing strains (N0145) repeatedly acquired the lowest frequency of OFX-R (fig. 1). Moreover, we saw minimal, if any, DNA base-pair mutation rate differences between three Mtb strains (one of which was L2 Beijing) with different in vitro OFX-R frequencies (fig. 2). Our results therefore contradict the L2 Beijing hypermutator hypothesis. Published experimental work have also provided varying results. Initial fluctuation analyses showed no difference in the frequency of RIF-R in L2 Beijing strains compared with non-L2 Beijing strains (Werngren and Hoffner 2003). In contrast, a fluctuation analysis performed by Ford et al. (2013) showed that L2 Beijing strains had higher frequencies of resistance for INH, RIF, and ethambutol compared with L4 strains, even after correcting for differences in AMR mutational profiles. Lastly, a more recent fluctuation analysis using the same concentrations of INH or RIF as Ford et al. showed that while different Mtb strains had different frequencies of INH-R, a L2 Beijing strain did not have higher frequencies of INH-R nor RIF-R compared with non-L2 Beijing strains (Carey et al. 2018). Although diverging in their results, these in vitro and genetic studies, together with the study conducted here, highlight the importance of the genetic background when testing for the frequency of AMR in Mtb. Furthermore, these results show that differential DNA mutation rates is not the only parameter relevant in determining the frequency of FQ-R in Mtb.
If DNA mutation rates do not contribute to the variation in OFX-R frequency, we hypothesized that differences in the phenotypic effects of OFX-R mutations, and their consequent effect on the mutational profiles for OFX-R, may be important contributors. By sequencing the QRDR from resistant colonies in our OFX fluctuation analysis, we observed strain-specific patterns in the mutational profiles for OFX-R (fig. 3). This suggested that the mutational profile for FQ-R is not only a function of the FQ type and concentration (Zhou et al. 2000; Malik et al. 2010, 2016; Huseby et al. 2017) but that epistatic interactions between a given FQ-R mutation and the genetic background may also play a role. Similar epistasis between the phenotype of a given AMR mutation and the genetic background have been observed in other bacteria. For example, a given RIF-R rpoB mutation can confer differential MIC and fitness costs depending on the genetic background it occurred in, or on the presence of other AMR mutations, in Escherichia coli (Angst and Hall 2013), Pseudomonas spp. (Vogwill et al. 2014, 2016), Mycobacterium smegmatis (Borrell et al. 2013), and Mtb (Gagneux et al. 2006; Zaczek et al. 2009). In line with these previous studies, we found that the OFX MIC and the fitness effect conferred by a given gyrA mutation varied significantly depending on the Mtb genetic background they occur in (figs. 4 and 5A; supplementary table S6, Supplementary Material online). These results suggest that epistasis plays a role in determining the strain-dependent OFX-R frequencies and mutational profiles observed during our fluctuation analyses (figs. 3 and 5B).
The epistasis between the Mtb genetic background and FQ-R mutations may have clinical consequences. A recent study has shown that drug-susceptible Mtb strains with higher MICs to INH and RIF were associated with increased risk of relapse following first-line treatment (Colangeli et al. 2018). FQ-R gyrA mutations that confer higher MICs, such as any gyrA mutation in codon D94 except for D94A, have also been associated with poorer treatment outcomes in MDR-TB patients (Rigouts et al. 2016; Farhat et al. 2017). Considering our observation that the Mtb genetic background affected both the OFX MICs and OFX-R mutational profiles (figs. 3 and 4; supplementary tables S4–S6, Supplementary Material online), the genetic background may therefore contribute to differences in patient treatment outcomes when using FQs as first-line drugs.
Using publicly available genomic data from Mtb clinical isolates, we observed significant lineage-dependent variation in the frequency of and mutational profiles for FQ-R (fig. 6). As expected, the vast majority of FQ-R mutations were observed in gyrA (supplementary tables S10 and S11, Supplementary Material online) (Takiff et al. 1994; Zhou et al. 2000; Piton et al. 2010; Maruri et al. 2012; Aldred et al. 2016; Blower et al. 2016; Wollenberg et al. 2017). FQ-R was also most frequent in L2 and L4. This was also as expected, as strains from the L2 Beijing sublineage are known to associate with MDR-TB (Borrell and Gagneux 2009; Fenner et al. 2012; Merker et al. 2015; Wollenberg et al. 2017), while L4 strains are the most prevalent globally, including in regions classified as high burden for TB (Stucki et al. 2016; World Health Organization 2017; Brynildsrud et al. 2018; Gagneux 2018). Consequently, strains from L2 and L4 would be more exposed to FQs, leading to the higher FQ-R frequencies observed in these two lineages. Furthermore, we observed that more than half of the variability in the clinical frequency of FQ-R gyrA mutation events in L2 strains can be explained by how Mtb evolves in vitro (fig. 7). However, the in vitro FQ-R evolution could only account for 39% of the variability for the frequency of FQ-R gyrA mutation events in clinical L4 strains. This suggested that while the Mtb genetic background can influence the evolution of FQ-R in the clinic, other factors (which may be independent of the Mtb genetic background) likely played strong roles as well. Epidemiological factors including socioeconomic disruptions, health system inefficiencies, and human behavior are well known risk factors for the emergence and transmission of AMR in Mtb (Dalton et al. 2012; Merker et al. 2015; Alvarez-Uria et al. 2016; Eldholm et al. 2016; Shah et al. 2017). Meanwhile, biological factors not explored in this study, such as antibiotic type and concentration (Zhou et al. 2000; Ford et al. 2013; Lindsey et al. 2013; McGrath et al. 2014; Mustaev et al. 2014; Malik et al. 2016), pharmacodynamic and pharmacokinetic features (Pienaar et al. 2017; Sarathy et al. 2018), and the selective pressure of the host immune system (Handel et al. 2009), may also influence the evolution of FQ-R.
How the genetic background modulates FQ-R evolution in the clinic may also differ between different bacterial pathogens. In contrast to Mtb where DNA gyrase is the sole target for FQs, Gram-negative bacteria such as E. coli and Salmonella have two targets for FQs: DNA gyrase and topoisomerase IV (Hooper 1999). In E. coli, the evolutionary trajectory toward high-level FQ-R generally involves the stepwise acquisition of FQ-R mutations in either DNA gyrase or topoisomerase IV (Huseby et al. 2017). Therefore, the genetic background of Gram-negative bacteria may modulate the phenotypes of FQ-R mutations not only in DNA gyrase but in topoisomerase IV as well. Nevertheless, a recent study has shown that a common FQ-R mutation in Gram-negative bacteria, GyrA S83L, confers different phenotypes depending on whether it is present in E. coli or in Salmonella (Apjok et al. 2019). This suggests that the genetic background of Gram-negative bacteria may affect the evolution of FQ-R in the clinic. This type of epistasis is also not restricted to bacteria. The genetic background modulated the phenotypes of AMR mutations in the protozoan parasite Leishmania donovani (Decuypere et al. 2012), and the phenotypes of mutations in yeast when exposed to different environments, including antimicrobial exposure (Mullis et al. 2018). Thus, while the mode of epistasis between the genetic background and the phenotypes of mutations may differ in different organisms and environments, published work and the results of our study provide compelling evidence that this epistasis is a major factor in the evolution of AMR in both prokaryotic and eukaryotic organisms.
Our study is limited by the fact that our survey of clinical FQ-R frequencies involved a genomic data set that was sampled by convenience. This data set was used due to its public availability, and may not be fully representative of FQ-R frequencies in Mtb populations. We noted that lineage-specific frequencies of FQ-R were likely biased due to the overrepresentation L2 and L4 strains. Thus, to acquire a better understanding on which FQ-R mutations appeared and at what frequency they occurred at in different Mtb lineages, either more genomes from clinical isolates from other Mtb lineages must be made available, or a population-based study must be undertaken, preferably in a high-burden MDR-TB region.
Exposure to quinolones have been shown to lead to SOS response-mediated mutagenesis, which can increase the rate of AMR acquisition, including resistance to quinolones themselves (Cirz et al. 2005; Malik et al. 2010; Frenoy and Bonhoeffer 2018). Therefore, the strain-dependent OFX-R acquisition rates (fig. 1) may be due to strain-dependent differences in the magnitude of quinolone-induced mutagenesis. We did not explicitly test for this possibility. However, phylogenetic SNPs present in SOS response-related genes may lead to strain-dependent differences in quinolone-induced mutagenesis, and we observed no such SNPs present across our panel of drug-susceptible Mtb strains (supplementary table S2, Supplementary Material online) (Borrell et al. 2019). Thus, we observed no genetic evidence for strain-specific SOS response-mediated mutagenesis. Furthermore, in E. coli, quinolone-induced quinolone-resistant mutations may only be observed after 5 days of incubation with quinolones, which is equivalent to >220 generations for wild-type E. coli (Cirz et al. 2005; Fujikawa and Morozumi 2005). Meanwhile, our wild-type Mtb strains were incubated for 40 generations at most in the presence of OFX (see Materials and Methods and supplementary table S7, Supplementary Material online), making the likelihood of observing OFX-induced OFX-R mutants in our in vitro system extremely low.
Another limitation of our study is that fluctuation analyses only model AMR emergence. Long-term population dynamics also play an important role in AMR evolution (zur Wiesch et al. 2011; Lindsey et al. 2013; Huseby et al. 2017). For example, population bottleneck events modulate AMR evolution during serial transfer experiments (Comas et al. 2012; Barrick and Lenski 2013; Vogwill et al. 2016; Huseby et al. 2017), and have also been hypothesized to strongly influence Mtb evolution in the clinic (Hershberg et al. 2008). Thus, modeling FQ-R evolution in Mtb in epidemiological settings would benefit from the use of some measure of long-term population dynamics and between-host transmission. Nevertheless, the fitness of AMR mutants is an important factor in determining its evolutionary fate (zur Wiesch et al. 2011; Angst and Hall 2013; Barrick and Lenski 2013; Lindsey et al. 2013; Hughes and Andersson 2017; Huseby et al. 2017) and its potential for between-host transmission (Comas et al. 2012; de Vos et al. 2013). Considering that the Mtb genetic background modulated the fitness effect of FQ-R mutations (fig. 5 and supplementary table S7, Supplementary Material online), the genetic background may modulate how likely FQ-R mutants transmit between patients.
In conclusion, we illustrate how the genetic variation present in natural populations of Mtb modulates FQ-R evolution. Considering the nonrandom geographic distribution of different Mtb genetic variants (Comas et al. 2010; Gagneux 2018), our work suggests that there may be regional differences in the rate of FQ-R emergence and FQ-R prevalence when using FQs as a first-line drug. We therefore highlight the importance of standing genetic variation in determining how FQ-R evolves in Mtb and, in general, how AMR evolves in pathogens.
Materials and Methods
Collection of Drug-Susceptible Clinical Isolates of Mtb Strains for In Vitro Studies
We used nine genetically distinct Mtb strains, with three strains from each of the following Mtb lineages: Lineage 1 (L1; also known as the East-Africa and India Lineage), Lineage 2 (L2; the East Asian Lineage), and Lineage 4 (L4; the Euro-American Lineage) (Comas et al. 2010; Gagneux 2018). All strains were previously isolated from patients, fully drug-susceptible, and previously characterized by Borrell et al. (2019) (supplementary table S1, Supplementary Material online).
Prior to all experimentation, starter cultures for each Mtb strain were prepared by recovering a 20 μl aliquot from frozen stocks into a 10 ml volume of Middlebrook 7H9 broth (BD), supplemented with an albumin (Fraction V, Roche), dextrose (Sigma–Aldrich), catalase (Sigma–Aldrich), and 0.05% Tween 80 (AppliChem) (hereafter designated as 7H9 ADC). These starter cultures were incubated until their optical density at wavelength of 600 nm (OD600) was ∼0.50, and were then used for in vitro assays.
Fluctuation Analyses
Fluctuation analyses were performed as described by Luria and Delbrück (1943). Briefly, an aliquot from the starter cultures for each strain was used to inoculate 350 ml of 7H9 ADC to have an initial bacterial density of 5,000 colony forming units (CFU) per milliliter. This was immediately divided into 33 parallel cultures, each with 10 ml of culture volume aliquoted into individual 50 ml Falcon Conical Centrifuge Tubes (Corning Inc.). The parallel cultures were incubated at 37 °C on standing racks, with resuspension by vortexing (Bio Vortex V1, Biosan) every 24 h. Cultures were grown until an OD600 of between 0.40 and 0.65. Once at this density, final cell counts (Nt) from three randomly chosen parallel cultures were calculated by serial dilution and plating on Middlebrook 7H11 (BD), supplemented with oleic acid (AppliChem), albumin, and catalase (hereafter referred to as 7H11 OADC). To calculate the number of resistant colonies (r), the remaining 30 parallel cultures not used for Nt determination were pelleted at 800×g for 10 min at 4 °C using the Allegra X-15R Benchtop Centrifuge (Beckmann Coulter). The supernatants were discarded, and the bacterial pellets resuspended in 300 μl of 7H9 ADC. The resuspensions were spread on 7H11 OADC plates supplemented with the relevant drug concentration (2, 4, or 8 µg/ml of ofloxacin, or 100 µg/ml STR; Sigma). Resistant colonies were observed and enumerated after 21–35 days of incubation, depending on the Mtb strain. The frequency of drug-resistant mutants per culture (r) was enumerated, and the estimated number of drug-resistance mutations per culture (m) was estimated from the distribution of the r values (rdist) using the Ma, Sarkar, Sandri-Maximum Likelihood Estimator method (MSS-MLE) (Rosche and Foster 2000). Values of r that were >300 were simply given a value of 300, as this would not change the precision of the calculated m value using the MSS-MLE method (Rosche and Foster 2000). The MSS-MLE method is also only valid for a range of m values between 0.3 and 20 (Rosche and Foster 2000). The frequency of drug-resistance mutations acquired per cell (F) per strain was then calculated by dividing the calculated m values by their respective Nt values. The 95% confidence intervals for each F were calculated as previously described by Rosche and Foster (2000). Hypotheses testing for significant differences between the rdist between strains for the fluctuation analyses at 4 µg/ml of OFX (fig. 1A) and at 100 µg/ml of STR (fig. 2) were performed using the Kruskal–Wallis test; significant differences in the rdist between strains in the fluctuation analyses at 2 and 8 µg/ml (fig. 1B) were tested for using the Wilcoxon rank-sum test. Statistical analyses were performed using the R statistical software (v.3.5.1) (R Core Team 2018).
Determining the Mutational Profile for OFX-R In Vitro
From the parallel cultures plated on 4 µg/ml of OFX (fig. 1A), up to 120 resistant colonies per strain (at least 1 colony per plated parallel culture if colonies were present, to a maximum of 6) were transferred into 100 μl of sterile deionized H2O placed in Falcon 96-well Clear Microplate (Corning Inc.). The bacterial suspensions were then heat-inactivated at 95 °C for 1 h, and used as PCR templates to amplify the QRDR in gyrA and gyrB using primers designed by Feuerriegel et al. (2009). PCR products were sent to Macrogen, Inc. or Microsynth AG for Sanger sequencing, and QRDR mutations were determined by aligning the PCR product sequences against the H37Rv reference sequence (Cole et al. 1998). Sequence alignments were performed using the Staden Package (Staden 1996), while the amino acid substitutions identification were performed using the Molecular Evolutionary Genetics Analysis Version 6.0 package (Tamura et al. 2013). Fisher’s exact test was used to test for significant differences between the strains’ mutational profiles for OFX-R. Data analyses were performed using the R statistical software (v.3.5.1) (R Core Team 2018), and figures were produced using the ggplot2 package (Wickham, 2016).
Isolation of Spontaneous Ofloxacin-Resistant Mutants
Spontaneous OFX-resistant mutants were isolated from strains belonging one of three genetic backgrounds: N0157 (L1, Manila sublineage), N1283 (L4, Ural sublineage), and N0145 (L2, Beijing sublineage) (Borrell et al. 2019). To begin, we transferred 50 μl of starter cultures for each strain into separate culture tubes containing 10 ml of fresh 7H9 ADC. Cultures were incubated at 37 °C until OD600 of ∼0.80, and pelleted at 800×g for 5 min at 4 °C. The supernatant was discarded, and the pellet resuspended in 300 μl of 7H9 ADC. The resuspension was plated on 7H11 OADC (BD) supplemented with 2 μg/ml of OFX, and incubated until resistant colonies appeared (∼14–21 days). Resistant colonies were picked and resuspended in fresh 10 ml 7H9 ADC, and incubated at 37 °C. Once the culture reached early stationary phase, two aliquots were prepared. The first aliquot was heat-inactivated at 95 °C for 1 h, and the gyrA mutation identified by PCR and Sanger sequencing, as described in the mutational profile for OFX-R assay. If the first aliquot harbored one of four OFX-R gyrA mutations (GyrAD94G, GyrAD94N, GyrAA90V, or GyrAG88C), the second aliquot was stored in −80 °C for future use.
Prior to further experimentation with the spontaneously OFX-resistant mutant strains, starter cultures were prepared in the same manner as for the drug-susceptible strains.
Drug Susceptibility Assay
We determined the OFX-susceptibility levels of our spontaneous OFX-resistant mutants and their respective drug-susceptible ancestors by performing the colorimetric, microtiter plate-based Alamar Blue assay (Franzblau et al. 1998). Briefly, we used a Falcon 96-well Clear Microplate, featuring a serial 2-fold dilution of OFX. For drug-susceptible strains, a range of OFX concentration from 15 to 0.058 μg/ml was used. Meanwhile, for OFX-resistant strains, a range of 60–0.234 μg/ml was used. Each well was inoculated with a 10 μl volume of starter culture to have a final inoculum of ∼5 × 106 CFU/ml. The plates were incubated at 37 °C for 10 days. Following incubation, 10 μl of Resazurin (Sigma) were added to each well, and the plates were incubated for another 24 h at 37 °C. After this incubation period, plates were inactivated by adding 100 μl of 4% formaldehyde to every well. Measurement of fluorescence produced by viable cells was performed on SpectraMAX GeminiXPS Microplate Reader (Molecular Devices). The excitation wavelength was set at 536 nm, and the emission wavelength at 588 nm was measured. Minimum inhibitory concentration (MIC) for OFX was determined by first fitting a Hill curve to the distribution of fluorescence, and then defining the MIC as the lowest OFX concentration where the fitted Hill curve showed a ≥ 95% reduction in fluorescence. Two sets of experiments were performed for every strain, with three technical replicates per experiment. Analyses of MIC data were performed and figures created using the numpy, scipy, pandas, and matplotlib modules for the Python programming language.
Cell Growth Assay
We set up three or four 1,000 ml roller bottles with 90 ml of 7H9 ADC and 10 ml borosilicate beads. Each bottle was inoculated with a volume of starter cultures so that the initial bacterial density was at an OD600 of 5 × 10−7. The inoculated bottles were then placed in a roller incubator set to 37 °C, and incubated for 12–18 days with continuous rolling. OD600 measurements were taken once or twice every 24 h. Two independent experiments in either triplicates or quadruplicates were performed per strain.
We defined the exponential phase as the bacterial growth phase where we observed a log2-linear relationship between OD600 and time; specifically, we used a Pearson’s R2 value ≥ 0.98 as the threshold. The growth rate of a particular strain was then defined as the slope of the linear regression model. The relative fitness of a given spontaneous OFX-R mutant was defined by taking the growth rate of the OFX-resistant mutant strain and dividing it by the growth rate of its respective drug-susceptible ancestor. Linear regression models for the cell growth assays data were performed using the numpy, scipy, pandas, and matplotlib modules for the Python programming language, as well as the R statistical software (v.3.5.1) and the ggplot2 package (Wickham, 2016; R Core Team 2018).
Surveying the FQ-R Profile from Publicly Available Mtb Genomes
We screened public databases to download global representatives of Mtb genomes, as described by Menardo et al. (2018). We selected genomes that were classified as MDR-TB based on the presence of both INH-R and RIF-R mutations. This provided a data set of 3,450 genomes with confirmed MDR-TB; their accession numbers are reported in supplementary table S8, Supplementary Material online. These MDR-TB genomes were then screened for the presence of FQ-R mutations, and we identified 854 genomes that were classified as FQ-R.
The INH-R, RIF-R, and FQ-R mutations used for screening are the same mutations used by Payne et al. (2019), and are listed in supplementary table S13, Supplementary Material online. A drug-resistance mutation was defined as “fixed” in the population when it reached a frequency of ≥90%. Meanwhile, a drug-resistance mutation was considered “variable” in the population when its frequency was between 10% and 90%; thus, multiple drug-resistance mutations may be present in the genomic data from a single Mtb clinical isolate.
Defining Transmission Clusters and Determining the Frequency of FQ-R gyrA Mutation Events
To define transmission clusters, the differences in the number of SNPs were used as a measure of genetic distance between two Mtb genomes. Using the haplotypes package (v.1.0) for the R statistical software (v.3.5.1) (R Core Team 2018), a genetic distance matrix was then inferred for the 3,450 MDR-TB genomes. Insertions/deletions were considered missing data. Agglomerative clustering was performed using the agnes function from the cluster package (v.2.0.6) for the R statistical software (R Core Team 2018). A conservative threshold of 12 SNPs average distance was used to define likely patient-to-patient transmission (Walker et al. 2013), and the tree was cut at a height of 12 SNPs using the hclust function. All resulting transmission clusters, with a minimum size of two clustered genomes, were used for further analysis. For every transmission cluster, each unique and fixed FQ-R gyrA mutation was treated as an independent mutation event. Fixed FQ-R gyrA mutations in nonclustered Mtb genomes were also treated as an independent mutation event. Figures were produced using the ggplot2 package (Wickham, 2016).
Supplementary Material
Acknowledgments
The authors would like to thank Sebastian Bonhoeffer, Daniel Angst, and Diarmaid Hughes for providing critical comments on the article. Calculations were performed at sciCORE (http://scicore.unibas.ch/) scientific computing core facility at University of Basel. Library preparation and sequencing was carried out in the Genomics Facility Basel. This work was supported by the Swiss National Science Foundation (grants 310030_166687, IZRJZ3_164171, IZLSZ3_170834, and CRSII5_177163), the European Research Council (309540-EVODRTB), and SystemsX.ch.
References
- Aldred KJ, Blower TR, Kerns RJ, Berger JM, Osheroff N.. 2016. Fluoroquinolone interactions with Mycobacterium tuberculosis gyrase: enhancing drug activity against wild-type and resistant gyrase. Proc Natl Acad Sci U S A. 113(7):E839–E846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Uria G, Gandra S, Laxminarayan R.. 2016. Poverty and prevalence of antimicrobial resistance in invasive isolates. Int J Infect Dis. 52:59–61. [DOI] [PubMed] [Google Scholar]
- Ängeby KA, Jureen P, Giske CG, Chryssanthou E, Sturegård E, Nordvall M, Johansson AG, Werngren J, Kahlmeter G, Hoffner SE, et al. 2010. Wild-type MIC distributions of four fluoroquinolones active against Mycobacterium tuberculosis in relation to current critical concentrations and available pharmacokinetic and pharmacodynamic data. J Antimicrob Chemother. 65:946–952. [DOI] [PubMed] [Google Scholar]
- Angst DC, Hall AR.. 2013. The cost of antibiotic resistance depends on evolutionary history in Escherichia coli. BMC Evol Biol. 13:163.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apjok G, Boross G, Nyerges Á, Fekete G, Lázár V, Papp B, Pál C, Csörgő B.. 2019. Limited evolutionary conservation of the phenotypic effects of antibiotic resistance mutations. Mol Biol Evol. 36(8):1601–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baños-Mateos S, Roon AMV, Lang UF, Maslen SL, Skehel JM, Lamers MH.. 2017. High-fidelity DNA replication in Mycobacterium tuberculosis relies on a trinuclear zinc center. Nat Commun. 8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, Lenski RE.. 2013. Genome dynamics during experimental evolution. Nat Rev Genet. 14(12):827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower TR, Williamson BH, Kerns RJ, Berger JM.. 2016. Crystal structure and stability of gyrase–fluoroquinolone cleaved complexes from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 113(7):1706–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boritsch EC, Khanna V, Pawlik A, Honoré N, Navas VH, Ma L, Bouchier C, Seemann T, Supply P, Stinear TP, et al. 2016. Key experimental evidence of chromosomal DNA transfer among selected tuberculosis-causing mycobacteria. Proc Natl Acad Sci U S A. 113(35):9876–9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrell S, Gagneux S.. 2009. Infectiousness, reproductive fitness and evolution of drug-resistant Mycobacterium tuberculosis. Int J Tuberc Lung Dis. 13:1456–1466. [PubMed] [Google Scholar]
- Borrell S, Teo Y, Giardina F, Streicher EM, Klopper M, Feldmann J, Müller B, Victor TC, Gagneux S.. 2013. Epistasis between antibiotic resistance mutations drives the evolution of extensively drug-resistant tuberculosis. Evol Med Public Health. 2013(1):65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrell S, Trauner A, Brites D, Rigouts L, Loiseau C, Coscolla M, Niemann S, Jong BD, Yeboah-Manu D, Kato-Maeda M, et al. 2019. Reference set of Mycobacterium tuberculosis clinical strains: a tool for research and product development. PLoS One 14(3):e0214088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brynildsrud OB, Pepperell CS, Suffys P, Grandjean L, Monteserin J, Debech N, Bohlin J, Alfsnes K, Pettersson J-H, Kirkeleite I, et al. 2018. Global expansion of Mycobacterium tuberculosis lineage 4 shaped by colonial migration and local adaptation. Sci Adv. 4(10):eaat5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey AF, Rock JM, Krieger IV, Chase MR, Fernandez-Suarez M, Gagneux S, Sacchettini JC, Ioerger TR, Fortune SM.. 2018. TnSeq of Mycobacterium tuberculosis clinical isolates reveals strain-specific antibiotic liabilities. PLoS Pathog. 14(3):e1006939.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirz RT, Chin JK, Andes DR, Crécy-Lagard VD, Craig WA, Romesberg FE.. 2005. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 3(6):e176.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coeck N, de Jong BC, Diels M, de Rijk P, Ardizzoni E, Van Deun A, Rigouts L.. 2016. Correlation of different phenotypic drug susceptibility testing methods for four fluoroquinolones in Mycobacterium tuberculosis. J Antimicrob Chemother. 71(5):1233–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colangeli R, Jedrey H, Kim S, Connell R, Ma S, Chippada Venkata UD, Chakravorty S, Gupta A, Sizemore EE, Diem L, et al. 2018. Bacterial factors that predict relapse after tuberculosis therapy. N Engl J Med. 379(9):823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393(6685):537–544. [DOI] [PubMed] [Google Scholar]
- Comas I, Borrell S, Roetzer A, Rose G, Malla B, Kato-Maeda M, Galagan J, Niemann S, Gagneux S.. 2012. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet. 44(1):106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, Ernst JD, Gagneux S.. 2010. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet. 42(6):498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton T, Cegielski P, Akksilp S, Asencios L, Caoili JC, Cho S-N, Erokhin VV, Ershova J, Gler MT, Kazennyy BY, et al. 2012. Prevalence of and risk factors for resistance to second-line drugs in people with multidrug-resistant tuberculosis in eight countries: a prospective cohort study. Lancet 380(9851):1406–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vos M, Müller B, Borrell S, Black PA, van Helden PD, Warren RM, Gagneux S, Victor TC.. 2013. Putative compensatory mutations in the rpoC gene of rifampin-resistant Mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob Agents Chemother. 57(2):827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decuypere S, Vanaerschot M, Brunker K, Imamura H, Müller S, Khanal B, Rijal S, Dujardin J-C, Coombs GH.. 2012. Molecular mechanisms of drug resistance in natural Leishmania populations vary with genetic background. PLoS Negl Trop Dis. 6(2):e1514.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldholm V, Pettersson J-O, Brynildsrud OB, Kitchen A, Rasmussen EM, Lillebaek T, Rønning JO, Crudu V, Mengshoel AT, Debech N, et al. 2016. Armed conflict and population displacement as drivers of the evolution and dispersal of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 113(48):13881–13886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhat MR, Jacobson KR, Franke MF, Kaur D, Murray M, Mitnick CD.. 2017. Fluoroquinolone resistance mutation detection is equivalent to culture-based drug sensitivity testing for predicting multidrug-resistant tuberculosis treatment outcome: a Retrospective Cohort Study. Clin Infect Dis. 65(8):1364–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenner L, Egger M, Bodmer T, Altpeter E, Zwahlen M, Jaton K, Pfyffer GE, Borrell S, Dubuis O, Bruderer T, et al. 2012. Effect of mutation and genetic background on drug resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 56(6):3047–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerriegel S, Cox HS, Zarkua N, Karimovich HA, Braker K, Rüsch-Gerdes S, Niemann S.. 2009. Sequence analyses of just four genes to detect extensively drug-resistant Mycobacterium tuberculosis strains in multidrug-resistant tuberculosis patients undergoing treatment. Antimicrob Agents Chemother. 53(8):3353–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CB, Shah RR, Maeda MK, Gagneux S, Murray MB, Cohen T, Johnston JC, Gardy J, Lipsitch M, Fortune SM.. 2013. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat Genet. 45(7):784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzblau SG, Witzig RS, McLaughlin JC, Torres P, Madico G, Hernandez A, Degnan MT, Cook MB, Quenzer VK, Ferguson RM, et al. 1998. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the Microplate Alamar Blue assay. J Clin Microbiol. 36(2):362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenoy A, Bonhoeffer S.. 2018. Death and population dynamics affect mutation rate estimates and evolvability under stress in bacteria. PLoS Biol. 16(5):e2005056.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa H, Morozumi S.. 2005. Modeling surface growth of Escherichia coli on agar plates. Appl Environ Microbiol. 71(12):7920–7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux S. 2018. Ecology and evolution of Mycobacterium tuberculosis. Nat Rev Microbiol. 16(4):202–213. [DOI] [PubMed] [Google Scholar]
- Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan B.. 2006. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 312(5782):1944–1946. [DOI] [PubMed] [Google Scholar]
- Gillespie SH, Crook AM, McHugh TD, Mendel CM, Meredith SK, Murray SR, Pappas F, Phillips PPJ, Nunn AJ.. 2014. Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. N Engl J Med. 371(17):1577–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygli SM, Borrell S, Trauner A, Gagneux S.. 2017. Antimicrobial resistance in Mycobacterium tuberculosis: mechanistic and evolutionary perspectives. FEMS Microbiol Rev. 41(3):354–373. [DOI] [PubMed] [Google Scholar]
- Handel A, Margolis E, Levin BR.. 2009. Exploring the role of the immune response in preventing antibiotic resistance. J Theor Biol. 256(4):655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Lipatov M, Small PM, Sheffer H, Niemann S, Homolka S, Roach JC, Kremer K, Petrov DA, Feldman MW, et al. 2008. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. 6(12):e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper DC. 1999. Mechanisms of fluoroquinolone resistance. Drug Resist Updat. 2(1):38–55. [DOI] [PubMed] [Google Scholar]
- Hughes D, Andersson DI.. 2017. Evolutionary trajectories to antibiotic resistance. Annu Rev Microbiol. 71:579–596. [DOI] [PubMed] [Google Scholar]
- Huseby DL, Pietsch F, Brandis G, Garoff L, Tegehall A, Hughes D.. 2017. Mutation supply and relative fitness shape the genotypes of ciprofloxacin-resistant Escherichia coli. Mol Biol Evol. 34(5):1029–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperial MZ, Nahid P, Phillips PPJ, Davies GR, Fielding K, Hanna D, Hermann D, Wallis RS, Johnson JL, Lienhardt C, et al. 2018. A patient-level pooled analysis of treatment-shortening regimens for drug-susceptible pulmonary tuberculosis. Nat Med. 24(11):1708–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindani A, Harrison TS, Nunn AJ, Phillips PPJ, Churchyard GJ, Charalambous S, Hatherill M, Geldenhuys H, McIlleron HM, Zvada SP, et al. 2014. High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. N Engl J Med. 371(17):1599–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey HA, Gallie J, Taylor S, Kerr B.. 2013. Evolutionary rescue from extinction is contingent on a lower rate of environmental change. Nature 494(7438):463–467. [DOI] [PubMed] [Google Scholar]
- Luria SE, Delbrück M.. 1943. Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28(6):491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGowan AP. 2008. Clinical implications of antimicrobial resistance for therapy. J Antimicrob Chemother. 62(Suppl 2):ii105–ii114. [DOI] [PubMed] [Google Scholar]
- Malik M, Hoatam G, Chavda K, Kerns RJ, Drlica K.. 2010. Novel approach for comparing the abilities of quinolones to restrict the emergence of resistant mutants during quinolone exposure. Antimicrob Agents Chemother. 54(1):149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik M, Mustaev A, Schwanz HA, Luan G, Shah N, Oppegard LM, de Souza EC, Hiasa H, Zhao X, Kerns RJ, et al. 2016. Suppression of gyrase-mediated resistance by C7 aryl fluoroquinolones. Nucleic Acids Res. 44(7):3304–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruri F, Sterling TR, Kaiga AW, Blackman A, van der Heijden YF, Mayer C, Cambau E, Aubry A.. 2012. A systematic review of gyrase mutations associated with fluoroquinolone-resistant Mycobacterium tuberculosis and a proposed gyrase numbering system. J Antimicrob Chemother. 67(4):819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath M, Gey van Pittius NC, van Helden PD, Warren RM, Warner DF.. 2014. Mutation rate and the emergence of drug resistance in Mycobacterium tuberculosis. J Antimicrob Chemother. 69(2):292–302. [DOI] [PubMed] [Google Scholar]
- Menardo F, Loiseau C, Brites D, Coscolla M, Gygli SM, Rutaihwa LK, Trauner A, Beisel C, Borrell S, Gagneux S.. 2018. Treemmer: a tool to reduce large phylogenetic datasets with minimal loss of diversity. BMC Bioinformatics 19(1):164.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merker M, Blin C, Mona S, Duforet-Frebourg N, Lecher S, Willery E, Blum MGB, Rüsch-Gerdes S, Mokrousov I, Aleksic E, et al. 2015. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat Genet. 47(3):242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merle CS, Fielding K, Sow OB, Gninafon M, Lo MB, Mthiyane T, Odhiambo J, Amukoye E, Bah B, Kassa F, et al. 2014. A four-month gatifloxacin-containing regimen for treating tuberculosis. N Engl J Med. 371(17):1588–1598. [DOI] [PubMed] [Google Scholar]
- Mullis MN, Matsui T, Schell R, Foree R, Ehrenreich IM.. 2018. The complex underpinnings of genetic background effects. Nat Commun. 9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustaev A, Malik M, Zhao X, Kurepina N, Luan G, Oppegard LM, Hiasa H, Marks KR, Kerns RJ, Berger JM, et al. 2014. Fluoroquinolone-gyrase-DNA complexes two modes of drug binding. J Biol Chem. 289(18):12300–12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppong YEA, Phelan J, Perdigão J, Machado D, Miranda A, Portugal I, Viveiros M, Clark TG, Hibberd ML.. 2019. Genome-wide analysis of Mycobacterium tuberculosis polymorphisms reveals lineage-specific associations with drug resistance. BMC Genomics 20(1):252.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JL, Menardo F, Trauner A, Borrell S, Gygli SM, Loiseau C, Gagneux S, Hall AR.. 2019. Transition bias influences the evolution of antibiotic resistance in Mycobacterium tuberculosis. PLoS Biol. 17(5):e3000265.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pienaar E, Sarathy J, Prideaux B, Dietzold J, Dartois V, Kirschner DE, Linderman JJ.. 2017. Comparing efficacies of moxifloxacin, levofloxacin and gatifloxacin in tuberculosis granulomas using a multi-scale systems pharmacology approach. PLoS Comput Biol. 13(8):e1005650.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piton J, Petrella S, Delarue M, André-Leroux G, Jarlier V, Aubry A, Mayer C.. 2010. Structural insights into the quinolone resistance mechanism of Mycobacterium tuberculosis DNA gyrase. PLoS One 5(8):e12245.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2018. R: a language and environment for statistical computing. Vienna (Austria: ): R Foundation for Statistical Computing; Available from: https://www.R-project.org/; last accessed September 9, 2019. [Google Scholar]
- Rad ME, Bifani P, Martin C, Kremer K, Samper S, Rauzier J, Kreiswirth BN, Blazquez J, Jouan M, Soolingen DV, et al. 2003. Mutations in putative mutator genes of Mycobacterium tuberculosis strains of the W-Beijing family. Emerg Infect Dis. 9:838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigouts L, Coeck N, Gumusboga M, de Rijk WB, Aung KJM, Hossain MA, Fissette K, Rieder HL, Meehan CJ, de Jong BC, et al. 2016. Specific gyrA gene mutations predict poor treatment outcome in MDR-TB. J Antimicrob Chemother. 71(2):314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JM, Lang UF, Chase MR, Ford CB, Gerrick ER, Gawande R, Coscolla M, Gagneux S, Fortune SM, Lamers MH.. 2015. DNA replication fidelity in Mycobacterium tuberculosis is mediated by an ancestral prokaryotic proofreader. Nat Genet. 47(6):677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosche WA, Foster PL.. 2000. Determining mutation rates in bacterial populations. Methods 20(1):4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarathy JP, Via LE, Weiner D, Blanc L, Boshoff H, Eugenin EA, Barry CE, Dartois VA.. 2018. Extreme drug tolerance of Mycobacterium tuberculosis in caseum. Antimicrob Agents Chemother. 62:e02266–e02317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NS, Auld SC, Brust JCM, Mathema B, Ismail N, Moodley P, Mlisana K, Allana S, Campbell A, Mthiyane T, et al. 2017. Transmission of extensively drug-resistant tuberculosis in South Africa. N Engl J Med. 376(3):243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staden R. 1996. The staden sequence analysis package. Mol Biotechnol. 5(3):233.. [DOI] [PubMed] [Google Scholar]
- Stucki D, Brites D, Jeljeli L, Coscolla M, Liu Q, Trauner A, Fenner L, Rutaihwa L, Borrell S, Luo T, et al. 2016. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat Genet. 48(12):1535–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takiff H, Guerrero E.. 2011. Current prospects for the fluoroquinolones as first-line tuberculosis therapy. Antimicrob Agents Chemother. 55(12):5421–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takiff HE, Salazar L, Guerrero C, Philipp W, Huang WM, Kreiswirth B, Cole ST, Jacobs WR, Telenti A.. 1994. Cloning and nucleotide sequence of Mycobacterium tuberculosis gyrA and gyrB genes and detection of quinolone resistance mutations. Antimicrob Agents Chemother. 38(4):773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S.. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30(12):2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vjecha MJ, Tiberi S, Zumla A.. 2018. Accelerating the development of therapeutic strategies for drug-resistant tuberculosis. Nat Rev Drug Discov. 17(9):607–608. [DOI] [PubMed] [Google Scholar]
- Vogwill T, Kojadinovic M, Furió V, MacLean RC.. 2014. Testing the role of genetic background in parallel evolution using the comparative experimental evolution of antibiotic resistance. Mol Biol Evol. 31(12):3314–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogwill T, Kojadinovic M, MacLean RC.. 2016. Epistasis between antibiotic resistance mutations and genetic background shape the fitness effect of resistance across species of Pseudomonas. Proc R Soc B. 283(1830):20160151.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, Eyre DW, Wilson DJ, Hawkey PM, Crook DW, et al. 2013. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 13(2):137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werngren J, Hoffner SE.. 2003. Drug-susceptible Mycobacterium tuberculosis Beijing genotype does not develop mutation-conferred resistance to rifampin at an elevated rate. J Clin Microbiol. 41(4):1520–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. 2009. ggplot2: elegant graphics for data analysis. New York: Springer. [Google Scholar]
- Winston CA, Mitruka K.. 2012. Treatment duration for patients with drug-resistant tuberculosis, United States. Emerg Infect Dis. 18(7):1201–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollenberg KR, Desjardins CA, Zalutskaya A, Slodovnikova V, Oler AJ, Quiñones M, Abeel T, Chapman SB, Tartakovsky M, Gabrielian A, et al. 2017. Whole-genome sequencing of Mycobacterium tuberculosis provides insight into the evolution and genetic composition of drug-resistant tuberculosis in Belarus. J Clin Microbiol. 55(2):457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. 2017. Global tuberculosis report 2018. Geneva (Switzerland). Available from: http://www.who.int/tb/publications/global_report/en/; last accessed June 3, 2019.
- Zaczek A, Brzostek A, Augustynowicz-Kopec E, Zwolska Z, Dziadek J.. 2009. Genetic evaluation of relationship between mutations in rpoB and resistance of Mycobacterium tuberculosis to rifampin. BMC Microbiol. 9:10.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Dong Y, Zhao X, Lee S, Amin A, Ramaswamy S, Domagala J, Musser JM, Drlica K.. 2000. Selection of antibiotic-resistant bacterial mutants: allelic diversity among fluoroquinolone-resistant mutations. J Infect Dis. 182(2):517–525. [DOI] [PubMed] [Google Scholar]
- zur Wiesch PA, Kouyos R, Engelstädter J, Regoes RR, Bonhoeffer S.. 2011. Population biological principles of drug-resistance evolution in infectious diseases. Lancet Infect Dis. 11(3):236–247. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.