

ABSTRACT
Autophagy selectively targets invading bacteria to defend cells, whereas bacterial pathogens counteract autophagy to survive in cells. The initiation of canonical autophagy involves the PIK3C3 complex, but autophagy targeting Group A Streptococcus (GAS) is PIK3C3-independent. We report that GAS infection elicits both PIK3C3-dependent and -independent autophagy, and that the GAS effector NAD-glycohydrolase (Nga) selectively modulates PIK3C3-dependent autophagy. GAS regulates starvation-induced (canonical) PIK3C3-dependent autophagy by secreting streptolysin O and Nga, and Nga also suppresses PIK3C3-dependent GAS-targeting-autophagosome formation during early infection and facilitates intracellular proliferation. This Nga-sensitive autophagosome formation involves the ATG14-containing PIK3C3 complex and RAB1 GTPase, which are both dispensable for Nga-insensitive RAB9A/RAB17-positive autophagosome formation. Furthermore, although MTOR inhibition and subsequent activation of ULK1, BECN1, and ATG14 occur during GAS infection, ATG14 recruitment to GAS is impaired, suggesting that Nga inhibits the recruitment of ATG14-containing PIK3C3 complexes to autophagosome-formation sites. Our findings reveal not only a previously unrecognized GAS-host interaction that modulates canonical autophagy, but also the existence of multiple autophagy pathways, using distinct regulators, targeting bacterial infection.
Abbreviations: ATG5: autophagy related 5; ATG14: autophagy related 14; ATG16L1: autophagy related 16 like 1; BECN1: beclin 1; CALCOCO2: calcium binding and coiled-coil domain 2; GAS: group A streptococcus; GcAV: GAS-containing autophagosome-like vacuole; LAMP1: lysosomal associated membrane protein 1; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MTORC1: mechanistic target of rapamycin kinase complex 1; Nga: NAD-glycohydrolase; PIK3C3: phosphatidylinositol 3-kinase catalytic subunit type 3; PtdIns3P: phosphatidylinositol-3-phosphate; PtdIns4P: phosphatidylinositol-4-phosphate; RAB: RAB, member RAS oncogene GTPases; RAB1A: RAB1A, member RAS oncogene family; RAB11A: RAB11A, member RAS oncogene family; RAB17: RAB17, member RAS oncogene family; RAB24: RAB24, member RAS oncogene family; RPS6KB1: ribosomal protein S6 kinase B1; SLO: streptolysin O; SQSTM1: sequestosome 1; ULK1: unc-51 like autophagy activating kinase 1; WIPI2: WD repeat domain, phosphoinositide interacting 2
KEYWORDS: Autophagy, group A Streptococcus, NAD-glycohydrolase, PIK3C3, RAB GTPase
Introduction
Macroautophagy/autophagy is an essential cellular pathway that degrades various harmful intracellular materials, including protein aggregates, damaged organelles, and invading bacteria [1,2]. The autophagy process is initiated by the formation at ER-associated sites of phagophore membranes, which fuse at their edges to generate the double-membrane vesicles known as autophagosomes; the autophagosomes then fuse with lysosomes, and this eventually leads to the degradation of the sequestered contents by lysosomal enzymes [3].
Autophagy initiation is regulated by ULK1/Atg1 complex, which is recruited to the ER followed by PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3) complex I (PIK3C3-PIK3R4/VPS15-BECN1-ATG14) [4]. ULK1 is phosphorylated by MTORC1 (mechanistic target of rapamycin kinase complex 1), and the phosphorylation prevents the activation of ULK1 [5,6]. The ULK1 complex physically and functionally interacts with PIK3C3 complex I: The ULK1 complex activates the PIK3C3 complex by phosphorylating ATG14 and BECN1 [6–8], and the PIK3C3 complex produces a pool of phosphatidylinositol-3-phosphate (PtdIns3P) and recruits critical autophagy regulators, such as WIPI2, to form the phagophore membrane [9,10]. Therefore, PtdIns3P is essential for various types of autophagy, and this PIK3C3/PtdIns3P-dependent pathway is part of most forms of canonical autophagy, such as starvation-induced autophagy, basal autophagy, and autophagy targeting Salmonella enterica serovar Typhimurium [11,12]. Conversely, the existence of PtdIns3P-independent autophagy has also been suggested by recent studies. For example, in response to glucose depletion, PIK3C3-independent autophagy is activated, whereby PtdIns5P recruits WIPI2 as well as PtdIns3P and regulates autophagosome biogenesis through a PtdIns3P-independent mechanism [13,14]. However, to our knowledge, no study to date has reported the concurrent induction of both PtdIns3P-dependent and -independent autophagy in response to a particular stimulus.
Autophagy specifically targets invading bacteria in host cells and restricts their growth (also called xenophagy). Bacteria internalized through endocytosis/phagocytosis damage the bacterium-surrounding endosomes/phagosomes and escape into the cytosol. The bacteria in the cytosol are ubiquitinated and captured by LC3-positive double membranes through autophagy receptors such as SQSTM1/p62 and CALCOCO2/NDP52, and then delivered to lysosomes for degradation [15]. Thus, autophagy functions as an antibacterial mechanism in cells. However, several bacteria have evolved to evade autophagy. For example, Mycobacterium tuberculosis str. H37Rv inhibits autophagy activation by using Eis, which impedes MAPK/c-JUN N-terminal kinase signaling and subsequent ROS production (which are required for autophagy induction) [16]; Bacillus anthracis and Vibrio cholera inhibit autophagy through cAMP-elevating toxins [17]; and Legionella pneumophila RavZ targets LC3 and thus inhibits autophagosome formation [18].
Group A Streptococcus (GAS), a major human pathogen, enters epithelial cells through endocytosis and then escapes into the cytoplasm by secreting streptolysin O (SLO), a pore-forming toxin produced by GAS [19]. This escaped GAS in the cytoplasm is recognized by the ubiquitin-SQSTM1-CALCOCO2 axis and entrapped by an LC3-positive double-membrane structure, the GAS-containing autophagosome-like vacuole (GcAV) [20,21]. Although serotype M1T1 GAS can evade autophagy by using the cysteine protease SpeB, which degrades SQSTM1 and CALCOCO2, GAS of several serotypes can be targeted by autophagy and eliminated [22]. However, it remains unclear whether the GAS strains targeted by autophagy lack anti-autophagic systems or whether the host cells can defend against and overcome such systems.
GAS-targeting autophagy is ATG5-dependent and involves the ubiquitin-autophagy receptor pathway as well as canonical selective autophagy. However, we have reported that GcAV formation is regulated by distinct sets of RAB GTPases that are dispensable in canonical starvation-induced autophagy [23–25]. Furthermore, we recently showed that GcAV formation occurs through a PtdIns3P-independent mechanism and that PI4KB-mediated PtdIns4P production is critical for GcAV formation, and further that BECN1 and ATG14, two PIK3C3 complex I components, are also dispensable for GcAV formation [26]. Because PIK3C3-dependent autophagy is induced by bacterial pathogens such as Salmonella [11], we suspected that GAS inhibits the canonical PIK3C3-dependent autophagy pathway. Here, we examined the possibility that GAS inhibits PIK3C3-dependent autophagy, and we identified a GAS-secreted protein, NAD-glycohydrolase (Nga), responsible for the inhibition of PIK3C3-dependent autophagy.
Results
GAS inhibits starvation-induced autophagy in a SLO-dependent manner
Starvation-induced formation of LC3 puncta is a widely recognized step in the PIK3C3 complex-dependent autophagy pathway. To investigate whether GAS can inhibit PIK3C3-dependent autophagy, HeLa cells stably expressing GFP-LC3 were infected with GAS JRS4 (a strain that can be targeted by autophagy) for 2 h, and then the cells were incubated in starvation medium for 1 h. We started the incubation in starvation medium at 2 h post-infection because GAS escapes from endosomes into the cytosol at 2 h after infection [19,27]. We detected LC3-positive puncta in response to starvation in non-infected cells, but in the GAS-infected HeLa cells, the LC3 signal was only visible around bacteria and we rarely detected LC3 puncta (Figure 1a,b). Notably, LC3 puncta were not observed even in GAS-infected cells that contained no GcAVs, suggesting that the formation of LC3 puncta is inhibited in GAS-infected cells irrespective of whether GcAVs are formed.
Figure 1.
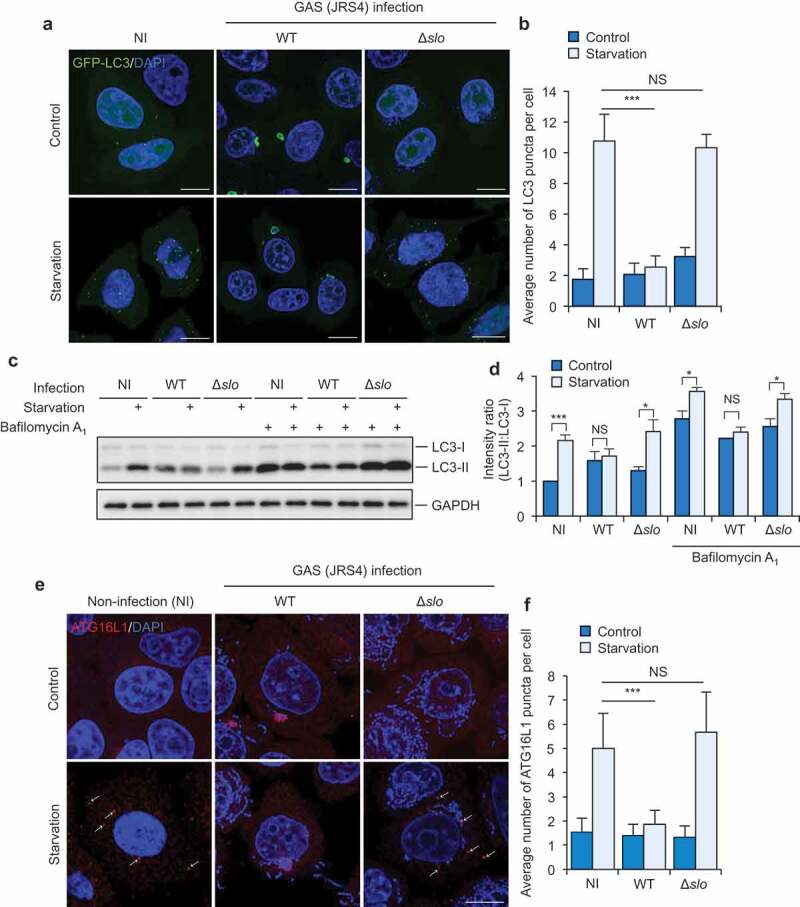
GAS inhibits starvation-induced autophagy in a SLO-dependent mechanism. (a and b) Starvation-induced LC3-puncta formation in GAS-infected or non-infected (NI) cells. Confocal micrographs (a) and quantification (b) of LC3-puncta formation in HeLa cells stably expressing GFP-LC3 infected with GAS JRS4 strain wild-type or isogenic Δslo mutant for 2 h, and subsequently incubated with regular (control) or starvation medium for 1 h. Cells were fixed, and cellular and bacterial DNA was stained with DAPI. Scale bars: 10 μm. (c and d) HeLa infected with indicated GAS strains were incubated with starvation medium with or without bafilomycin A1 and analyzed by immunoblotting with indicated antibodies (c). Intensity ratio of LC3-II:LC3-I were normalized to that in non-infected (NI) cells without bafilomycin A1 (d). (e and f) Starvation-induced phagophore formation in GAS-infected or non-infected cells. Confocal micrographs (e) and quantification (f) of ATG16L1-puncta formation in HeLa cells infected with GAS JRS4 WT or Δslo mutant for 2 h, and subsequently incubated with regular (control) or starvation medium for 1 h. Cells were fixed, and stained with anti-ATG16L1 antibody. Scale bars: 10 μm. Data in (b, d, and f) are mean ± SEM of 3 independent experiments. Data were tested by two-tailed Student’s t-test: ***P < 0.001. NS, not significant.
To determine whether the GAS that escapes into the cytosol inhibits the formation of LC3 puncta, we used an isogenic deletion-mutant of SLO (Δslo). Starvation clearly induced the formation of LC3-positive puncta in Δslo-infected cells (Figure 1a, b), which suggests that the GAS that escapes into the cytosol by using SLO suppresses the formation of LC3 puncta.
To confirm the inhibition of LC3 puncta formation during GAS infection, we examined the conversion of LC3-I to LC3-II during infection. Starvation increased the conversion to LC3-II in non-infected and Δslo-infected cells. However, the conversion to LC3-II was not increased in response to starvation in wild type GAS-infected cells (Figure 1c,d). Furthermore, starvation-induced conversion of LC3-I to LC3-II was not observed under bafilomycin A1-treated condition during GAS infection (Figure 1c, d). These results suggest that GAS inhibits starvation-induced autophagosome formation through SLO.
We further examined whether extracellular GAS can inhibit starvation-induced autophagy through SLO. Under the infection condition tested in the present study, at a multiplicity of infection (MOI) of 100, almost all HeLa cells were infected with GAS and therefore we infected the cells with GAS at MOI 1 and observed starvation-induced LC3-puncta formation in GAS-invaded and -non-invaded cells. Although GAS-invaded cells showed only GcAV, GAS-non-invaded cells had substantial LC3 puncta in response to starvation (Fig. S1A and S1B). Moreover, we infected the cells with fibronectin-binding protein deficient mutants of GAS JRS4 (JRS4 Δspy_0116); this mutant cannot invade HeLa cells [28,29]. As expected, starvation increased LC3-puncta formation during infection of JRS4 Δspy_0116. This demonstrated that invaded GAS into HeLa cells affects starvation-induced autophagosome formation via the SLO-dependent mechanism.
We next examined starvation-induced phagophore formation in GAS-infected cells. Puncta positive for ATG16L1, a phagophore marker, were increased by starvation in non-infected and Δslo-infected cells but not wild-type GAS-infected cells (Figure 1e, f), which indicates that GAS invading cells by using SLO inhibits starvation-induced phagophore formation. Collectively, our results suggest that cytosolic GAS inhibits PIK3C3-dependent starvation-induced autophagy.
GAS effector Nga is involved in inhibiting the formation of LC3 puncta
Bacterial pathogens secrete various effector proteins that affect host physiological activities. Previous studies have shown that Nga, one of the GAS effectors, is translocated into the host cytosol via SLO and promotes the proliferation of intracellular GAS [30]. Nga was detected in the cytosol fraction of GAS-infected HeLa cells but not in Δslo-infected cells (Fig. S2A). We infected the cells with the deletion mutants of Nga (Δnga) and examined starvation-induced autophagy. We found that starvation-induced formation of LC3 puncta occurred in Δnga-infected cells to the same extent as in non-infected cells (Figure 2a, b). Moreover, LC3 puncta were observed in GcAV-harboring cells infected with Δnga (Figure 2a), which suggests that starvation-induced autophagosome formation is inhibited by GAS-secreted Nga. To confirm that Nga is involved in the inhibition of starvation-induced formation of LC3 puncta, we constructed a complemented strain (nga::Δnga, Δnga compl.). In cells infected with the complemented strain, starvation-induced formation of LC3 puncta was again inhibited (Figure 2a, b).
Figure 2.
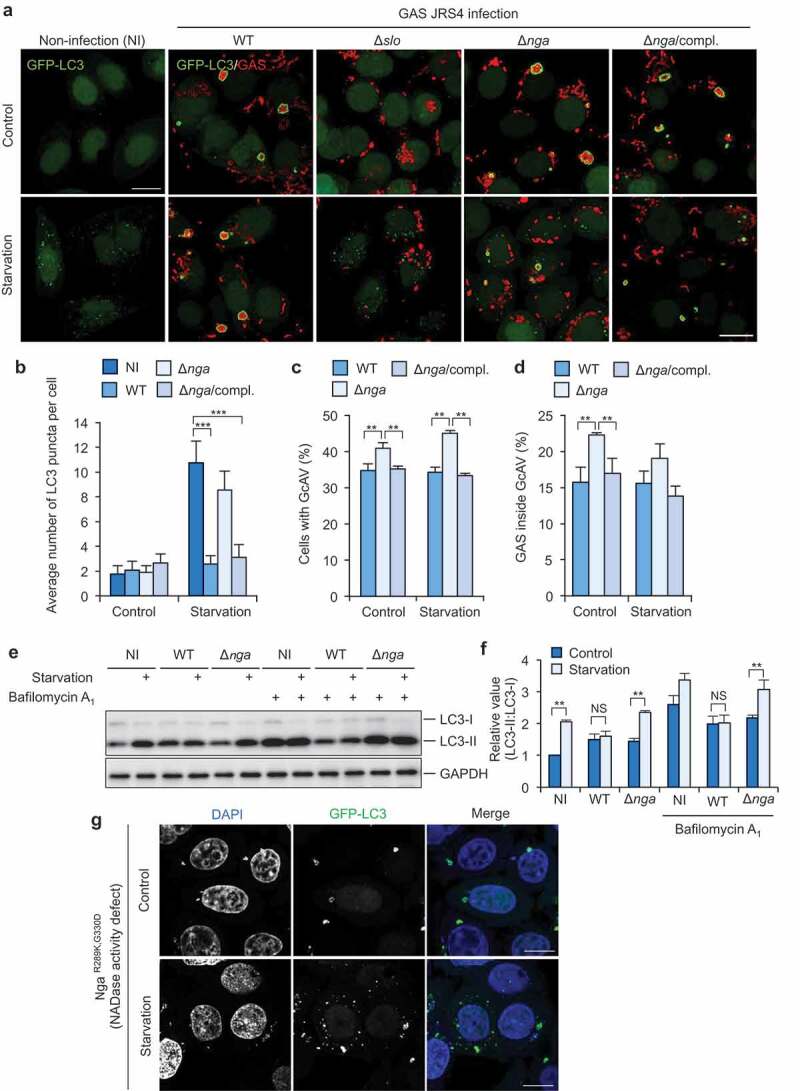
GAS effector Nga inhibits starvation-induced LC3-puncta formation. Confocal micrographs (a) and quantification of starvation-induced LC3-puncta formation (b) and GcAV formation (c and d). HeLa cells stably expressing GFP-LC3 infected with GAS wild-type or its mutants for 2 h, and subsequently incubated with regular (control), starvation medium for 1 h. Cells were fixed and immunostained for GAC (group A carbohydrate) to observe GAS (red). (e and f) Conversion of LC3-I to LC3-II in response to starvation during GAS infection. HeLa cells were infected with indicated GAS strains for 2 h and incubated with regular (control) or starvation medium for 1 h with or without bafilomycin A1. Representative WB images of LC3 and GAPDH (control) (E) and quantification of LC3 conversion (LC3-I to LC3-II) (f). Intensity ratio of LC3-II:LC3-I were normalized to that in non-infected (NI) cells without bafilomycin A1. (g) Confocal micrographs of starvation-induced LC3-puncta formation. HeLa cells stably expressing GFP-LC3 infected with GAS NgaR289K,G330D for 2 h, and subsequently incubated with regular (control), starvation medium for 1 h. Scale bars: 10 μm. Data in (b, c, d, and f) are mean ± SEM of 3 independent experiments. Data were tested by two-tailed Student’s t-test: ***P < 0.001. NS, not significant.
We also examined the recruitment of LC3 to wild type GAS and Δnga during starvation, and found that starvation stimuli did not affect the GcAV formation efficiency during GAS infection (Figure 2c, d). In addition, we found that both the percent of cells with GcAV and that of GAS targeted by GcAV significantly increased during infection with Δnga compared with that during infection with wild type GAS (Figure 2c,d), suggesting that Nga decreases GcAV formation or affects the flux of autophagy against GAS.
To confirm the inhibition of autophagosome formation during starvation by GAS-secreted Nga, we examined the conversion of LC3-I to LC3-II during infection. Although LC3-II was not increased in response to starvation in wild type GAS-infected cells, starvation induced the conversion of LC3-I to LC3-II in Δnga-infected cells (Figure 2e,f). These results suggest that GAS inhibits starvation-induced autophagosome formation through Nga.
To test whether the Nga-mediated inhibition of starvation-induced autophagy is a JRS4 strain-specific event, we repeated the assays by using GAS NIH35 (M28, streptococcal toxic shock syndrome strain), which yielded similar results (Figure S2B, S2C). Therefore, our findings demonstrated that GAS-secreted Nga is the effector responsible for inhibiting starvation-induced formation of LC3 puncta.
Rapamycin treatment is also well known to induce canonical autophagy, and we therefore checked whether GAS Nga inhibits rapamycin-induced autophagy. As shown in Fig. S3A and S3B, rapamycin-induced formation of LC3 puncta occurred in Δnga-infected cells but not in Δnga compl.-infected cells, suggesting that rapamycin-induced autophagy is also inhibited by GAS Nga.
Nga possesses NAD-glycohydrolase activity, which is abolished by the amino acid substitutions R289K and G330D [31]. Thus, we infected cells with NgaR289K,G330D mutant to determine whether the NAD-glycohydrolase activity of Nga is required for inhibiting the formation of LC3 puncta. In NgaR289K,G330D-infected cells, both GcAVs and starvation-induced LC3 puncta were observed (Figure 2g), which suggests that NAD-glycohydrolase activity is critical for the inhibition of starvation-induced LC3-puncta formation.
Protein aggregate clearance is suppressed by GAS effector Nga
PIK3C3-dependent selective autophagy degrades ubiquitinated protein aggregates and is involved in cellular homeostasis [11,32]. Thus, to test whether Nga also inhibits this PIK3C3-dependent autophagic pathway, we examined the clearance of protein aggregates induced by puromycin treatment in GAS-infected cells. Aggregate formation can be triggered by puromycin treatment, which inhibits host transcription and induces premature polypeptide-chain termination and misfolded-protein accumulation [33]. In our aggregate-clearance assay, HeLa cells were incubated with puromycin (5 μg/mL) for 3 h to induce the formation of protein aggregates (which were visualized through ubiquitin staining and confocal microscopy), and the cells were extensively washed to remove the drug and then infected with GAS (Figure 3a). To define the criteria of protein aggregate, we first measured the average size of ubiquitin-positive signals in puromycin-treated or -non-treated cells, and found that < 0.05 μm2 ubiquitin signals were observed even under non-treated condition. We then quantified the percent of cells with > 0.05 μm2 ubiquitin-positive signals (Figure 3b,c). As ubiquitin was also accumulated around GAS (arrowheads in Figure 3b), we manually excluded the ubiquitin signals surrounding GAS from protein aggregates. Among non-infected cells, the percentage of cells containing ubiquitin-positive aggregates gradually decreased, and <5% of the cells were aggregate-positive at 6 h after puromycin removal; by contrast, in autophagy-deficient cells (atg5 knockout cells) or cells infected with wild-type GAS, the decrease was not observed and >80% of the cells remained aggregate-positive at 6 h post-infection (Figure 3b,d). Moreover, aggregate clearance in Δnga-infected and NgaR289K,G330D-infected cells was almost the same as in non-infected cells (Figure 3b,d), which indicates that GAS inhibits the clearance of protein aggregates through Nga activity. We also measured the average size of ubiquitin-signals (excluding GAS-surrounding ubiquitin signals) after puromycin-washout in GAS-infected cells. The reduction in aggregate size was also suppressed in Nga-secreted cells or autophagy-deficient cells (Figure 3e).
Figure 3.

GAS inhibits the clearance of puromycin-induced protein aggregates through Nga. (a) Schematic of experiments to measure clearance of puromycin-induced protein aggregates during GAS infection. (b) Representative confocal micrographs of puromycin-induced protein aggregates in non-infection or GAS infection condition at indicated time points after puromycin removal. Cells were fixed and aggregates were with antibodies against ubiquitin (red). Nuclei were stained with DAPI (blue). Scale bars: 10 μm. (c) Average size of ubiquitin-positive signals in puromycin-treated or un-treated cells. > 50 signals were measured in each condition. (d) Average size of ubiquitin-positive signals at indicated time points after puromycin was removed. > 50 signals were measured in each condition. (e) Percentage of cells containing protein aggregates (>0.05 μm2 Ub signals) at indicated time points after puromycin was removed. (f) Differential detergent fractionation. HeLa cells after puromycin-treatment for 3 h were washed out and infected with GAS for indicated times. Cells were harvested in Triton X-100 lysis buffer, centrifuged and the supernatants (TX-soluble fraction) were collected. The pelleted material was washed with PBS before extraction with SDS lysis buffer and sonication (TX-insoluble fraction). Data in (c, d, and e) are mean ± SEM of 3 independent experiments.
Misfolded proteins that form large protein aggregates are often insoluble in lysis buffers containing mild detergents [33]. Further, to confirm the inhibition of aggregate clearance via Nga, we examined protein aggregate clearance using a differential detergent extraction approach. In the control cells (non-infected wild-type HeLa cells), SQSTM1 in the Triton X-100 (TX-100) insoluble fraction decreased with time after puromycin washout, whereas it was not decreased in autophagy-deficient cells (Figure 3f), indicating SQSTM1-positive protein aggregates are eliminated through autophagy. During infection with wild type GAS, SQSTM1 in the TX-100 insoluble fraction did not decrease. In contrast, SQSTM1 in the TX-100 insoluble fraction decreased in Δnga- or NgaR289K,G330D-infected cells (Figure 3f). These results suggest that Nga suppresses the clearance of protein aggregates.
GAS inhibits PIK3C3 complex-mediated autophagosome formation to survive in cells
Because GAS inhibited starvation-induced autophagy and selective autophagy against protein aggregates through Nga, we next investigated how Nga affects antibacterial autophagy and intracellular survival of GAS in detail. During wild-type GAS infection, <30% of the cells contained GcAVs at 2 h post-infection and this fraction gradually increased with time, whereas >60% of Δnga-infected cells contained GcAVs at 2 h post-infection and this decreased over time (Figure 4a,b). The nga-complemented strain showed similar kinetics of GcAV formation as wild-type GAS (Figure 4a,b). These results suggest that Nga suppresses GcAV formation at an early stage of infection (2 h post-infection). Furthermore, we examined the intracellular survival of these GAS strains, which revealed that their survival rate at 6 h post-infection was markedly decreased following nga deletion (Figure 4c). Collectively, these findings suggest that GAS delays autophagosome formation and facilitates intracellular bacterial proliferation by secreting Nga.
Figure 4.
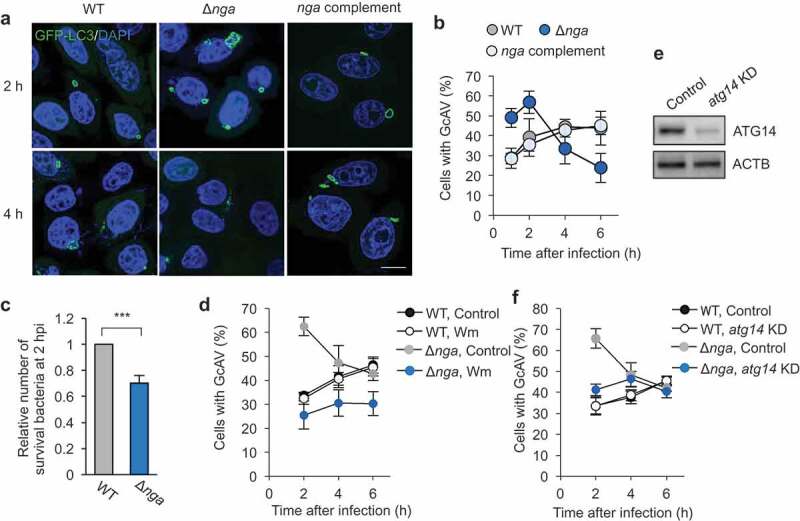
GAS Nga inhibits PIK3C3 complex-dependent autophagosome formation and promotes intracellular proliferation. (a and b) HeLa cells stably expressing GFP-LC3 were infected with GAS JRS4 wild-type or indicated mutants for 2 h or 4 h. Cells were fixed and stained with DAPI. Representative confocal micrographs (a) and quantification (b) of GcAV formation. Scale bars: 10 μm. (c) Intracellular survival rate of GAS. The survival rates of GAS mutants at 2 h post infection were normalized to wild-type. (d) Autophagosome formation against GAS WT and Δnga in wortmannin-treated or non-treated (control) cells. (e) Western blot analysis of atg14 knockdown in HeLa cells. (f) Autophagosome formation against GAS WT and Δnga in control or atg14 knockdown cells. Data in (b, c, and d) were tested by two-tailed Student’s t-test: ***P < 0.001. NS, not significant.
Intriguingly, Nga almost completely inhibited LC3-puncta formation induced by starvation (Figure 2) but only delayed GcAV formation. Moreover, we have reported that GcAV formation targeting wild-type GAS is a PIK3C3 complex-independent mechanism [26]. Thus, we hypothesized that the GcAV formation that occurs at an early stage of Δnga-mutant infection is a step in PIK3C3-dependent autophagy, and we examined how wortmannin, a PIK3C3 inhibitor, affects GcAV formation in Δnga-infected cells: As expected, cells harboring GcAVs targeting Δnga at 2 h decreased to ~30% and then gradually increased over time to the same extent as in cells infected with wild-type GAS (Figure 4d). Furthermore, knockdown of atg14, a component of the PIK3C3 complex, also impaired the formation of GcAVs against Δnga at 2 h (Figure 4e,f). These results suggest that PIK3C3 complex-dependent autophagy is inhibited by Nga, but PIK3C3-independent autophagy targets GAS at later stages of infection.
Nga is reported to inhibit lysosomal fusion and autophagosome acidification in keratinocytes [30]. We therefore investigated lysosomal fusion and acidification of GcAV in HeLa cells. We found a significant difference in the rate of lysosomal fusion; at 2 h post infection, 65% of GcAV were colocalized with LAMP1 during wild-type GAS infection, whereas 85% was colocalized during Δnga infection (Fig. S4A and S4B). In addition, we found that LysoTracker Red-positive GcAVs were markedly increased during Δnga infection in comparison to that during infection with wild-type GAS (Fig. S4C and S4D). These results suggest that Nga not only inhibits canonical PIK3C3-dependent autophagosome formation, but also suppresses autophagosome maturation.
PIK3C3 complex is activated but not recruited to GcAvs during GAS infection
To identify the step in autophagy-initiation signaling that is inhibited by Nga, we first examined MTOR signaling during GAS infection. GAS infection led to decreased both phosphorylation of RPS6KB1, major target of MTOR, and phosphorylated ULK1 (Figure 5a); this suggested that MTOR activity is inhibited by GAS infection. We also analyzed the phosphorylation of ATG14 and BECN1 and found that wild type and Δnga GAS infection induced ATG14 Ser29 and BECN1 Ser15 phosphorylation (Figure 5b). Therefore, although PIK3C3 complex-mediated autophagosome formation was not observed, ULK1-mediated PIK3C3 complex activation might occur during GAS infection.
Figure 5.
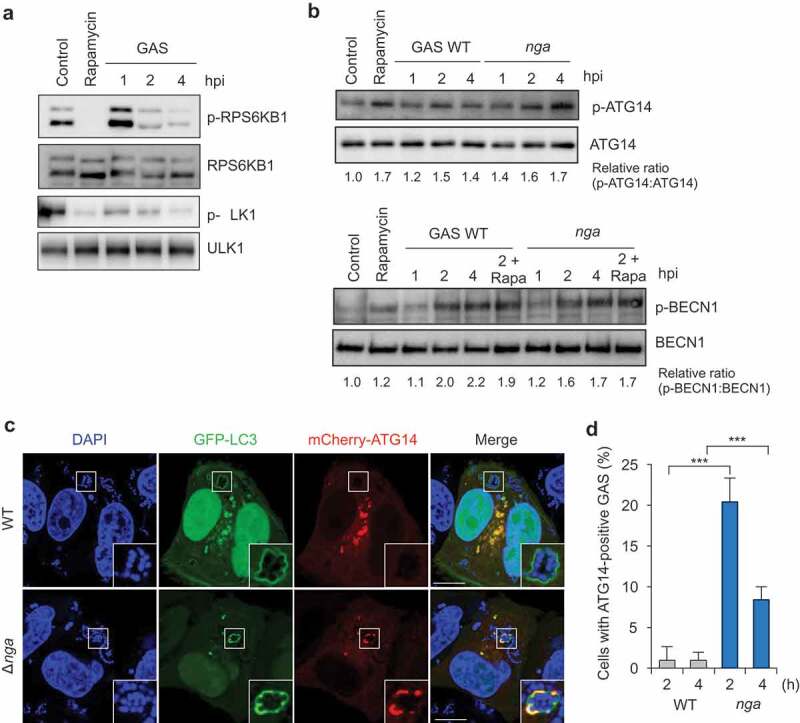
GAS inhibits ATG14 recruitment to GcAVs. (a) MTORC1 activity is decreased by GAS infection. HeLa cells were treated with rapamycin (100 nM) or infected with GAS JRS4 wild-type for the indicated times. The phosphorylation states at Thr389 of endogenous RPS6KB1, and at Ser757 of endogenous ULK1 in cell lysate were analyzed by western blot. (b) BECN1 and ATG14 are activated during GAS infection. HeLa cells were treated with rapamycin (100 nM) or infected with GAS strains for indicated times. The phosphorylation states at Ser29 of endogenous ATG14 and at Ser15 of endogenous BECN1 in cell lysate were analyzed by western blot. (c) HeLa cells expressing GFP-LC3 and mCherry-ATG14 were infected with GAS WT and Δnga for 2 or 4 h. Representative images of ATG14 localization to GAS at 2 h (c) and quantification of ATG14-positive GAS (d). Scale bars: 10 μm. Data in (d) were tested by two-tailed Student’s t-test: ***P < 0.001.
Next, we examined whether the ATG14-containing PIK3C3 complex localizes around GAS: ATG14 was rarely recruited to intracellular wild-type GAS but was efficiently localized to Δnga GAS and colocalized GcAVs (Figure 5c,d). These results imply that GAS Nga inhibits ATG14 recruitment to GcAV-formation sites.
As WIPI2 is a PtdIns3P-binding protein and required for canonical autophagosome formation, we examined the recruitment of WIPI2 to GAS. EmGFP-WIPI2 was not colocalized with GcAV during wild-type GAS infection, but clearly colocalized with GcAV against Δnga GAS (Fig. S5), suggesting that WIPI2 recruitment to GAS is also inhibited by Nga.
GAS Nga inhibits RAB1-dependent RAB24-positive autophagosome formation
We previously showed that largely distinct sets of RAB GTPases function in GcAV formation and in canonical starvation-induced autophagy, and that RAB1A, RAB11A, and RAB24, which are associated with starvation-induced autophagy, are not recruited to GcAVs [23]. Here, we examined RAB GTPase localization during infection with GAS nga mutants. Canonical autophagic RAB proteins (RAB1A, RAB11A, and RAB24) did not colocalize with GcAVs targeting wild-type GAS, in accord with our previous report, but these RABs were efficiently recruited to GcAVs against Δnga or NgaR289K,G330D GAS (Figure S6,A). Although we observed frequent colocalization between RAB1 and RAB24 during Δnga infection, we detected no GAS-containing vacuoles positive for both RAB17 and RAB24 (Figure 6b). RAB17-positive GAS-containing vacuoles were also RAB1A- and RAB11A-negative (Fig. S7). There is one potential cause for the distinct localization, that is RAB17 is recruited to GcAVs after RAB24 dissociates from the membrane; RAB24 and RAB17 were preferentially recruited at 2 and 4 h, respectively. However, we have previously shown that RAB17 is recruited to the phagophore membrane; thus, they localize to both autophagosomes and autolysosomes during GAS infection [24]. Notably, RAB1A knockdown severely impaired the formation of RAB24-positive GcAVs but did not affect RAB17-positive GcAVs (Figure 6c,d). Furthermore, atg14 knockdown caused a disappearance of RAB24-positive but not RAB17-positive GcAVs (Figure 6e,f). These findings suggest that RAB24-positive GcAVs and RAB17-positive GcAVs are different compartments and that their formation is RAB1- and PIK3C3-dependent and -independent, respectively (Figure 6g).
Figure 6.
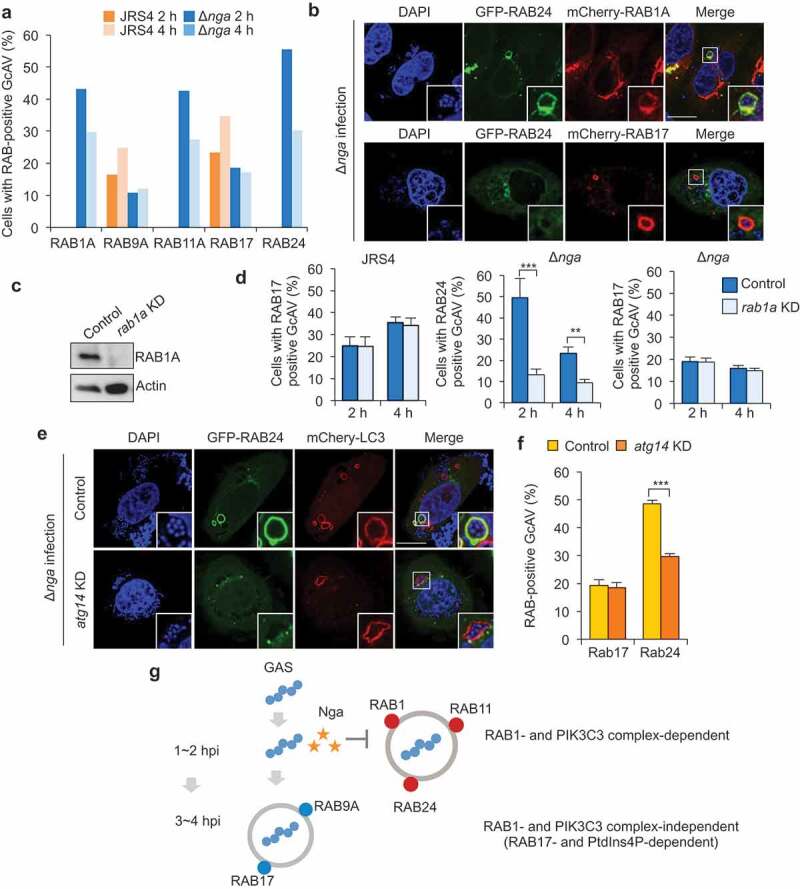
GAS Nga inhibits RAB1-dependent RAB24-positive GcAV formation. (a and b) RAB proteins recruitment to GcAVs in wild-type or Δnga mutant infected cells. Confocal micrographs of GcAVs (A) and quantification of RAB-positive GcAVs (b) at 2 and 4 h after infection. Scale bars: 10 μm. (c) Western blot of rab1a-knockdown HeLa cells. (d) Quantification of RAB24- or RAB17-positive GcAVs in rab1a-knockdown cells against GAS Δnga mutant. (e) Confocal micrographs of RAB24 and GcAVs during Δnga. Scale bars: 10 μm. (f) Quantification of RAB24- or RAB17-positive GcAVs against GAS Δnga mutant. (g) Proposed model of the interaction between invading GAS and autophagy. Data in (d and f) were tested by two-tailed Student’s t-test: **P < 0.01, ***P < 0.001.
Discussion
We have shown that the cell-invading GAS that escapes into the cytosol inhibits canonical RAB1- and PIK3C3-dependent autophagy by secreting Nga, but that the alternative RAB1- and PIK3C3-independent autophagy pathway is Nga-insensitive (Figure 6g). These findings explain why GcAVs targeting wild-type GAS (nga-harboring strain) presents unique characteristics in terms of requirement of RAB proteins and phosphoinositides. To our knowledge, this is the first report showing that both PIK3C3-dependent and -independent autophagy cooperate to defend cells against pathogens.
We demonstrated that the GAS effector Nga modulates starvation-induced autophagosome formation, clearance of protein aggregates, and PIK3C3-dependent GcAV formation by potentially affecting ATG14 trafficking. Nga is translocated across the host cell membrane into the cytosol by SLO [34] and is suggested to be involved in the pathogenesis of invasive GAS infections such as streptococcal toxic shock syndrome [35,36]. Nga cleaves β-NAD at the ribose-nicotinamide bond, and thus depletes intracellular NAD pools, produces the potent vasoactive compound nicotinamide, and causes ATP depletion in cells. Accordingly, Nga has been reported to inhibit the lysosomal fusion of autophagosomes potentially through ATP depletion in keratinocytes and macrophages [37,38]. Although we showed that Nga regulates canonical-autophagy initiation through its NADase activity, how NADase activity affects PIK3C3-mediated autophagy initiation remains unknown.
Because PIK3C3-independent GcAV formation was induced even during infection by Nga-positive wild-type GAS, LC3 lipidation or the autophagic membrane-formation machinery is not likely the target of Nga. Infection with Shigella, Salmonella, and Listeria causes a rapid induction of cytosolic amino acid starvation due to membrane damage, resulting in MTOR inhibition and autophagy induction [39,40]. We also showed that GAS infection caused MTOR inhibition and subsequent ULK1 activation, which indicates that Nga could suppress MTOR signaling at distinct points. Furthermore, because ATG14 and BECN1 phosphorylation was also clearly induced by GAS infection, PIK3C3 complex activation is not likely suppressed by Nga. Notably, although ATG14 phosphorylation was induced, ATG14 recruitment to GAS was inhibited by Nga, which indicates that Nga might affect the mechanism underlying ATG14 recruitment to sites of autophagosome formation. ATG14 recruitment to autophagosome-formation sites at the ER involves Atg13 in yeast [41], but ATG14 Ser29 phosphorylation by ULK1 is regulated by ATG13 in mammalian cells; thus, Nga might inhibit a mechanism distinct from ATG13-mediated ATG14 recruitment. Moreover, the ER-Golgi intermediate compartment is required for ATG14 recruitment and autophagosome biogenesis [42]. Considering our previous data indicating that the Golgi compartment is influenced by GAS infection, we suggest that Nga inhibits PIK3C3 complex-mediated autophagy by affecting Golgi-associated trafficking.
In this study, we noticed that the ATG14 signals in Δnga-targeting GcAV were partially colocalized with LC3 (Figure 5c). As GcAV formation is mediated by the fusion between phagophores [43] and ATG14 is required for phagophore formation [44], the ATG14-containing complex might be specifically localized in the initiation site of autophagosome biogenesis against GAS. In previous studies, the morphology of GcAV against wild-type GAS was investigated by electron microscopy and it was shown to be a double membrane structure [19,27,30]. To further investigate the difference between Nga-sensitive and -insensitive GcAVs, we would need to examine the morphology of GcAVs in depth.
RAB1- and PIK3C3-independent autophagy (RAB17-positive GcAV formation) was observed during infection by not only wild type GAS but also Δnga GAS, which suggests that this alternative pathway is not induced by the inhibition of the canonical pathway. As mentioned earlier in this section, MTOR activity was inhibited during GAS infection, which suggests that canonical PIK3C3-dependent autophagy during GAS infection is induced through MTOR-ULK1 signaling. However, the mechanism by which PIK3C3-independent alternative autophagy is induced is unknown. Because GcAV formation during wild-type GAS infection is also SLO-dependent, membrane damage by SLO or invading bacterial components could serve as the trigger for the autophagy targeting GAS. Notably, PIK3C3-dependent GcAV formation was observed at an early infection stage, whereas PIK3C3-independent (RAB17-positive) GcAVs appeared at a later stage; moreover, fewer RAB17-positive GcAVs were observed during Δnga infection than wild-type GAS infection (Figure 6a,f). These results suggest that PIK3C3-dependent and -independent GcAV formation is induced through different mechanisms. Given that RAB17-regulated PIK3C3-independent autophagy has not been reported under other conditions, the PIK3C3-independent pathway might be associated with specific bacterial components or host response to GAS invasion.
Wortmannin-sensitive (PtdIns3K-dependent) GcAVs colocalized with RAB1, RAB11, and RAB24, which have been reported to function in starvation-induced autophagy, whereas wortmannin-insensitive GcAVs did not harbor these RAB proteins and instead contained RAB9A and RAB17. Although both GcAVs were observed during Δnga infection, we detected no colocalization of RAB1, RAB11, and RAB24 with RAB9A and RAB17, which suggests that the two types of GcAVs exist concurrently and that their membrane characteristics are largely different. The principal reason underlying the selective recruitment of different sets of RAB proteins to the distinct GcAVs remains unclear, and elucidation of this recruitment should lead to enhanced understanding of the autophagosome formation mechanism. Intriguingly, the Salmonella effector SopB alters the phagosome-membrane surface charge by affecting PtdIns(4,5)P2 levels and also affects RAB recruitment [45]. Because the alternative PIK3C3-independent GcAVs are PtdIns4P-rich, GcAV surface charge might represent one of the factors responsible for differential RAB recruitment. Furthermore, because RAB recruitment is regulated through RAB guanine-nucleotide exchange factor proteins [46,47], comprehensive analysis of RAB GEFs would be crucial for elucidating how the GcAV-associated RAB pattern is regulated.
In conclusion, our results demonstrated that the GAS effector Nga selectively inhibits canonical RAB1- and PIK3C3-dependent autophagy. Our discovery of autophagy inhibition by Nga potentially reveals a previously unrecognized autophagy mechanism.
Materials and methods
Cell culture and transfection
HeLa cells were maintained in Dulbecco’s modified Eagle’s medium (Nacalai Tesque, 18,459–64) supplemented with 10% fetal bovine serum (Gibco, 26,140,079) and 50 μg/mL gentamicin (Nacalai Tesque, 11,980–14) in a 5% CO2 incubator at 3700B0C. The transfection reagents used were polyethylenimine (Polysciences, 23,966–2) and Lipofectamine 3000 (Invitrogen Corporation, L3000001).
Bacterial strains
GAS strains JRS4 (M6) and NIH35 (M28) were grown in Todd-Hewitt broth (BD Diagnostic Systems, 249,240) supplemented with 0.2% yeast extract (Nacalai Tesque, 15,838–45), as described previously [19].
Antibodies and other reagents
The following primary antibodies were used: rabbit polyclonal anti-LC3B (Abcam, ab51520), goat polyclonal anti-Streptococcus pyogenes group A carbohydrate (Abcam, ab9191), rabbit polyclonal anti-ATG16L (MBL Life Science, PM040), rabbit monoclonal anti-phospho-ATG14 (Ser29; Cell Signaling Technology, 13,155), rabbit monoclonal anti-ATG14 (Cell Signaling Technology, 5504), rabbit monoclonal anti-phospho-BECN1 (Ser15;, D4B7R; Cell Signaling Technology, 84,966), anti-BECN1 (D40C5; Cell Signaling Technology, 3495), rabbit monoclonal anti-phospho-RPS6KB1 (T389,; 108D2; Cell Signaling Technology, 9234), rabbit monoclonal anti-RPS6KB1 (Cell Signaling Technology, 9202), rabbit monoclonal anti-phospho-ULK1 (Ser757; Cell Signaling Technology, 6888), rabbit monoclonal anti-ULK1 (D8H5; Cell Signaling Technology, 8054), rabbit monoclonal anti-β-actin (D6A8; Cell Signaling Technology, 8457), rabbit monoclonal anti-RAB1A (D3X9S; Cell Signaling Technology, 13,075), rabbit polyclonal anti-RAB1B (G20; Cell Signaling Technology, sc-599), mouse monoclonal anti-LAMP1 (H4A3; Santa Cruz Biotechnology, sc-20,011), mouse monoclonal anti-GAPDH (6C5; Santa Cruz Biotechnology, sc-32,233), mouse monoclonal anti-SQSTM1 (D-3; Santa Cruz Biotechnology, sc-28,359), mouse monoclonal anti-FLAG (M2; Sigma-Aldrich, A2220), and mouse monoclonal anti-GFP (GF200; Nacalai Tesque, 04363–24). The secondary antibodies used were the following: for immunoblotting, HRP-conjugated anti-rabbit and anti-mouse IgG (Jackson ImmunoResearch Laboratories, 115–035-146 and 111–035-144); for immunostaining, Cy5-goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, 111–175-152), and anti-mouse or anti-rabbit IgG conjugated with Alexa Fluor 488/594 (Molecular Probes/Invitrogen Corporation, A11001, A11008, A11005, and A11012) and anti-mouse IgG conjugated with Alexa Fluor 350/647 (Molecular Probes/Invitrogen Corporation, A21120 and A28181). The following reagents were used: Bafilomycin A1 (InvivoGen, tlrl-baf1), wortmannin (Tocris Biaoscience, 1232), rapamycin (Nacalai Tesque, 30,037–94), LysoTracker Red DND-99 (Molecular Probes/Invitrogen Corporation, L7528), puromycin (InvivoGen, ant-pr-1). Reagents were used at the following concentration: Rapamycin at 100 nM, wortmannin at 100 nM, bafilomycin A1 at 100 nM, LysoTracker Red DND-99 at 150 nM.
Plasmids
Human RAB1A, ATG14 and WIPI2 cDNAs were amplified by PCR from HeLa total mRNA, cloned into a pENTR/D‐TOPO vector using a pENTR Directional TOPO cloning kit (Invitrogen corporation, K2400-20) or pENTR1A (Invitrogen corporation, A10462) and sub-cloned into pcDNA6.2/N‐EmGFP‐DEST (Invitrogen Corporation, V35620). pcDNA-6.2/N-3xFLAG-DEST (N-terminal tagged), pcDNA-6.2/N-mCherry-DEST (N-terminal tagged) vectors were made by replacement from pcDNA-6.2/N-EmGFP to the 3xFLAG corresponding oligonucleotide or mCherry gene fragment (kindly provided from Dr. Tsien, University of California) for compatibility with the Gateway system, respectively. A BLOCK-iT Pol II miR-RNAi expression vector kit (Invitrogen, K493500) was used to knock down RAB1A expression, with this targeting sequence: 5′-TACAAGTACTCGGCAAGTTGT-3′. The miRNA double-stranded sequence was ligated into the plasmid to generate pcDNA6.2-GW/miR, as per supplier instructions; pcDNA6.2-GW/miR-neg (Invitrogen Corporation, K493500) was used as the miRNA control. The plasmids were transfected into HeLa cells using Lipofectamine 3000. For silencing ATG14 expression, we used a small interfering RNA (siRNA; 5′-CCACUGCAUACCCUCAGGAAUCUAA-3′; stealth RNAiTM siRNA, Invitrogen Corporation); a nonspecific scrambled siRNA (Invitrogen Corporation, 12,935,200) was used as a control. Knockdowns were confirmed through immunoblotting.
Bacterial infection
Cells were infected with GAS as described previously [23]. Briefly, bacteria were incubated with cell cultures for 1 h at a multiplicity of infection of 100 in the absence of antibiotics. The infected cells were washed with phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.47 mM KH2PO4; pH 7.4) and then the antibiotic gentamicin (100 μg/mL) was added for an appropriate period to kill extracellular bacteria.
Fluorescence microscopy and image analyses
For confocal microscopy analysis, cells were cultured on 12-mm-diameter glass coverslips (Matsunami Glass Co., C012001) in 24-well culture plates (VIOLAMO, 2–8588-03). For immunostaining, cells were washed with PBS, fixed with 4% paraformaldehyde (Nacalai Tesque, 26,126–25) in PBS for 15 min, permeabilized with 0.1% Triton X-100 (Nacalai Tesque, 35,501–15) in PBS for 10 min, washed again with PBS, and then incubated in skim-milk blocking buffer (5% skim milk, 2.5% goat serum [Sigma-Aldrich, G9023], 2.5% donkey serum [Millipore, S30], 0.1% gelatin/PBS) or BSA blocking buffer (2% BSA [Sigma-Aldrich, A4503] in PBS) at room temperature for 1 h. Subsequently, the cells were incubated (room temperature, 1 h) with primary antibodies diluted with blocking solution, washed with PBS, and then probed with secondary antibodies. For LysoTracker Red staining, cells were incubated with 150 nM LysoTracker Red DND-99 for 30 min before fixation, and fixed with 4% paraformaldehyde and 0.5% glutaraldehyde (Nacalai Tesque, 17,003–92) in PBS at room temperature for 30 min. Bacterial and cellular DNAs were stained with 4′,6-diamidino-2-phenylindole (DAPI; Nacalai Tesque, 11,034–56). All fluorescence micrographs shown here are confocal images acquired using an FV1000 laser-scanning microscope (Olympus). For quantitative analysis, cell images were captured at random using the confocal microscope.
Bacterial viability assay
HeLa cells (5 × 104 cells/well) were cultured in 24-well culture plates and infected as described under ‘Bacterial Infection.’ After an appropriate incubation period, cells were lysed in sterile distilled water, and then serial dilutions of the lysates were plated on THY agar plates. Colony counting was used to determine the numbers of invaded and surviving GAS; the invasion data are presented as the ratio of ‘total intracellular GAS at 2 h’ to ‘total adhered and internalized GAS at 1 h,’ and the survival data are presented as the ratio of ‘intracellular live GAS at 6 h’ to ‘total intracellular GAS at 2 h.’
Differential detergent extraction
To examine the levels and solubility of SQSTM1 upon puromycin treatment, HeLa cells were plated in a 6-cm plate and treated with 5 μg/mL puromycin for 3 h. Cells were harvested in 200 μL Triton X-100 lysis buffer (50 mM NaCl, 10 mM Tris pH 7.5, 5 mM EDTA and 1% Triton X-100), centrifuged at 17,000 x g for 10 min at 4°C and the supernatants (TX-soluble fraction) were collected. The pellet was washed with phosphate buffered saline (PBS) before extraction with 100 μL SDS lysis buffer (2% SDS, 50 mM Tris pH 7.5 and 1 mM EDTA) and sonication (TX-insoluble fraction). Samples were analyzed by SDS–PAGE and western blotting.
Statistical analysis
Colocalization and GcAV formation were quantified through direct visualization performed using confocal microscopy. Unless indicated otherwise, at least 50 GcAVs or 200 GAS-infected cells were counted per condition in each experiment, and at least 3 independent experiments were performed for each trial. Values, including those displayed in graphs, are means ± SEM. Statistical analysis was performed using a two-tailed Student’s t test. P < 0.05 was considered statistically significant.
Funding Statement
This work was supported by the Japan Agency for Medical Research and Development [18fk0108073h0001]; Japan Agency for Medical Research and Development [18fm0208030h0002]; Japan Society for the Promotion of Science [16K08775]; Japan Society for the Promotion of Science [26462776]; Japan Society for the Promotion of Science [17K19552]; Japan Society for the Promotion of Science [16H05188]; Japan Society for the Promotion of Science [15K15130]; Japan Society for the Promotion of Science [18K07109]; Yakult Bio-Science Foundation [N/A]; Japan Agency for Medical Research and Development [19fk0108073h0002]; Japan Agency for Medical Research and Development [19fm0208030h0003]; Japan Society for the Promotion of Science [19H03471]; Joint Research Project of the Institute of Medical Science, the University of Tokyo [N/A]; Japan Agency for Medical Research and Development [19fk0108044h0203]; Takeda Science Foundation [N/A].
Acknowledgments
This research was supported in part by Grants-in-Aid for Scientific Research (16H05188, 15K15130, 26462776, 16K08775, 17K19552, 18K07109), the Takeda Science Foundation (T.N.), Yakult Bio-Science Foundation, Joint Research Project of the Institute of Medical Science, the University of Tokyo (I.N.), and the Research Program on Emerging and Re-emerging Infectious Diseases (18fk0108073h0001) and J-PRIDE (18fm0208030h0002) from Japan Agency for Medical Research and Development, AMED (I.N.).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental Materials data for this article can be accessed here.
References
- [1].Levine B, Kroemer G.. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Levine B, Mizushima N, Virgin HW.. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shibutani ST, Yoshimori T. A current perspective of autophagosome biogenesis. Cell Res. 2014;24:58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mercer TJ, Gubas A, Tooze SA. A molecular perspective of mammalian autophagosome biogenesis. J Biol Chem. 2018;293:5386–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wold MS, Lim J, Lachance V, et al. ULK1-mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington’s disease models. Mol Neurodegener. 2016;11:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Park JM, Jung CH, Seo M, et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy. 2016;12:547–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dooley HC, Razi M, Polson HE, et al. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell. 2014;55:238–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Polson HE, de Lartigue J, Rigden DJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6:506–522. [DOI] [PubMed] [Google Scholar]
- [11].Huang J, Birmingham CL, Shahnazari S, et al. Antibacterial autophagy occurs at PI(3)P-enriched domains of the endoplasmic reticulum and requires Rab1 GTPase. Autophagy. 2011;7:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Vergne I, Deretic V. The role of PI3P phosphatases in the regulation of autophagy. FEBS Lett. 2010;584:1313–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].McAlpine F, Williamson LE, Tooze SA, et al. Regulation of nutrient-sensitive autophagy by uncoordinated 51-like kinases 1 and 2. Autophagy. 2013;9:361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vicinanza M, Korolchuk VI, Ashkenazi A, et al. PI(5)P regulates autophagosome biogenesis. Mol Cell. 2015;57:219–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol. 2014;12:101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shin DM, Jeon BY, Lee HM, et al. Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog. 2010;6:e1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shahnazari S, Namolovan A, Mogridge J, et al. Bacterial toxins can inhibit host cell autophagy through cAMP generation. Autophagy. 2011;7:957–965. [DOI] [PubMed] [Google Scholar]
- [18].Choy A, Dancourt J, Mugo B, et al. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science. 2012;338:1072–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nakagawa I, Amano A, Mizushima N, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. [DOI] [PubMed] [Google Scholar]
- [20].Ito C, Saito Y, Nozawa T, et al. Endogenous nitrated nucleotide is a key mediator of autophagy and innate defense against bacteria. Mol Cell. 2013;52:794–804. [DOI] [PubMed] [Google Scholar]
- [21].Minowa-Nozawa A, Nozawa T, Okamoto-Furuta K, et al. Rab35 GTPase recruits NDP52 to autophagy targets. Embo J. 2017;36:2790–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Barnett TC, Liebl D, Seymour LM, et al. The globally disseminated M1T1 clone of group A Streptococcus evades autophagy for intracellular replication. Cell Host Microbe. 2013;14:675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nozawa T, Aikawa C, Goda A, et al. The small GTPases Rab9A and Rab23 function at distinct steps in autophagy during group A Streptococcus infection. Cell Microbiol. 2012;14:1149–1165. [DOI] [PubMed] [Google Scholar]
- [24].Haobam B, Nozawa T, Minowa-Nozawa A, et al. Rab17-mediated recycling endosomes contribute to autophagosome formation in response to group A Streptococcus invasion. Cell Microbiol. 2014;16:1806–1821. [DOI] [PubMed] [Google Scholar]
- [25].Oda S, Nozawa T, Nozawa-Minowa A, et al. Golgi-resident GTPase Rab30 promotes the biogenesis of pathogen-containing autophagosomes. PLoS One. 2016;11:e0147061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nakajima S, Aikawa C, Nozawa T, et al. Bcl-xL affects group A Streptococcus-induced autophagy directly, by inhibiting fusion between autophagosomes and lysosomes, and indirectly, by inhibiting bacterial internalization via interaction with beclin 1-UVRAG. PLoS One. 2017;12:e0170138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sakurai A, Maruyama F, Funao J, et al. Specific behavior of intracellular Streptococcus pyogenes that has undergone autophagic degradation is associated with bacterial streptolysin O and host small G proteins Rab5 and Rab7. J Biol Chem. 2010;285:22666–22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jadoun J, Ozeri V, Burstein E, et al. Protein F1 is required for efficient entry of Streptococcus pyogenes into epithelial cells. J Infect Dis. 1998;178:147–158. [DOI] [PubMed] [Google Scholar]
- [29].Ozeri V, Rosenshine I, Mosher DF, et al. Roles of integrins and fibronectin in the entry of Streptococcus pyogenes into cells via protein F1. Mol Microbiol. 1998;30:625–637. [DOI] [PubMed] [Google Scholar]
- [30].O’Seaghdha M, Wessels MR. Streptolysin O and its co-toxin NAD-glycohydrolase protect group A Streptococcus from Xenophagic killing. PLoS Pathog. 2013;9:e1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chandrasekaran S, Ghosh J, Port GC, et al. Analysis of polymorphic residues reveals distinct enzymatic and cytotoxic activities of the Streptococcus pyogenes NAD+ glycohydrolase. J Biol Chem. 2013;288:20064–20075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lystad AH, Simonsen A. Assays to monitor aggrephagy. Methods. 2015;75:112–119. [DOI] [PubMed] [Google Scholar]
- [34].Madden JC, Ruiz N, Caparon M. Cytolysin-mediated translocation (CMT): a functional equivalent of type III secretion in gram-positive bacteria. Cell. 2001;104:143–152. [DOI] [PubMed] [Google Scholar]
- [35].Meehl MA, Pinkner JS, Anderson PJ, et al. A novel endogenous inhibitor of the secreted streptococcal NAD-glycohydrolase. PLoS Pathog. 2005;1:e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cunningham MW. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev. 2000;13:470–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bastiat-Sempe B, Love JF, Lomayesva N, et al. Streptolysin O and NAD-glycohydrolase prevent phagolysosome acidification and promote group A Streptococcus survival in macrophages. mBio. 2014;5:e01690–01614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sharma O, O’Seaghdha M, Velarde JJ, et al. NAD+-glycohydrolase promotes intracellular survival of group A Streptococcus. PLoS Pathog. 2016;12:e1005468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tattoli I, MT Sorbara, Vuckovic D, et al. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe. 2012;11:563–575. [DOI] [PubMed] [Google Scholar]
- [40].Tattoli I, Sorbara MT, Yang C, et al. Listeria phospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures. Embo J. 2013;32:3066–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jao CC, Ragusa MJ, Stanley RE, et al. A HORMA domain in Atg13 mediates PI 3-kinase recruitment in autophagy. Proc Natl Acad Sci U S A. 2013;110:5486–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ge L, Melville D, Zhang M, et al. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife. 2013;2:e00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yamaguchi H, Nakagawa I, Yamamoto A, et al. An initial step of GAS-containing autophagosome-like vacuoles formation requires Rab7. PLoS Pathog. 2009;5:e1000670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Itakura E, Kishi C, Inoue K, et al. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bakowski MA, Braun V, Lam GY, et al. The phosphoinositide phosphatase SopB manipulates membrane surface charge and trafficking of the Salmonella-containing vacuole. Cell Host Microbe. 2010;7:453–462. [DOI] [PubMed] [Google Scholar]
- [46].Gerondopoulos A, Langemeyer L, Liang JR, et al. BLOC-3 mutated in hermansky-pudlak syndrome is a Rab32/38 guanine nucleotide exchange factor. Curr Biol. 2012;22:2135–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Blumer J, Rey J, Dehmelt L, et al. RabGEFs are a major determinant for specific Rab membrane targeting. J Cell Biol. 2013;200:287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.