

ABSTRACT
Mitophagy is a vital form of autophagy for selective removal of dysfunctional or redundant mitochondria. Accumulating evidence implicates elimination of dysfunctional mitochondria as a powerful means employed by autophagy to keep the immune system in check. The process of mitophagy may restrict inflammatory cytokine secretion and directly regulate mitochondrial antigen presentation and immune cell homeostasis. In this review, we describe distinctive pathways of mammalian mitophagy and highlight recent advances relevant to its function in immunity. In addition, we further discuss the direct and indirect evidence linking mitophagy to inflammation and autoimmunity underlying the pathogenesis of autoimmune diseases including inflammatory bowel diseases (IBD), systemic lupus erythematosus (SLE) and primary biliary cirrhosis (PBC).
Abbreviations: AICD: activation induced cell death; AIM2: absent in melanoma 2; ALPL/HOPS: alkaline phosphatase, biomineralization associated; AMA: anti-mitochondrial antibodies; AMFR: autocrine motility factor receptor; ATG: autophagy-related; BCL2L13: BCL2 like 13; BNIP3: BCL2 interacting protein 3; BNIP3L/NIX: BCL2 interacting protein 3 like; CALCOCO2/NDP52: calcium binding and coiled-coil domain 2; CARD: caspase recruitment domain containing; CASP1: caspase 1; CD: Crohn disease; CGAS: cyclic GMP-AMP synthase; CXCL1: C-X-C motif chemokine ligand 1; DEN: diethylnitrosamine; DLAT/PDC-E2: dihydrolipoamide S-acetyltransferase; DNM1L/Drp1: dynamin 1 like; ESCRT: endosomal sorting complexes required for transport; FKBP8: FKBP prolyl isomerase 8; FUNDC1: Fun14 domain containing 1; GABARAP: GABA type A receptor-associated protein; HMGB1: high mobility group box 1; HPIV3: human parainfluenza virus type 3; IBD: inflammatory bowel diseases; IEC: intestinal epithelial cell; IFN: interferon; IL1B/IL-1β: interleukin 1 beta; iNK: invariant natural killer; IRGM: immunity related GTPase M; LIR: LC3-interacting region; LPS: lipopolysaccharide; LRRK2: leucine rich repeat kinase 2; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MARCH5: membrane associated ring-CH-type finger 5; MAVS: mitochondrial antiviral signaling protein; MDV: mitochondria-derived vesicle; MFN1: mitofusin 1; MHC: major histocompatibility complex; MIF: macrophage migration inhibitory factor; mtAP: mitochondrial antigen presentation; mtDNA: mitochondrial DNA; MTOR: mechanistic target of rapamycin kinase; mtROS: mitochondrial ROS; MUL1: mitochondrial E3 ubiquitin protein ligase 1; NBR1: NBR1 autophagy cargo receptor; NFKB/NF-ĸB: nuclear factor kappa B subunit; NK: natural killer; NLR: NOD-like receptor; NLRC4: NLR family CARD domain containing 4; NLRP3: NLR family pyrin domain containing 3; OGDH: oxoglutarate dehydrogenase; OMM: outer mitochondrial membrane; OPTN: optineurin; ox: oxidized; PARK7: Parkinsonism associated deglycase; PBC: primary biliary cirrhosis; PEX13: peroxisomal biogenesis factor 13; PHB/PHB1: prohibitin; PHB2: prohibitin 2; PIK3C3/VPS34: phosphatidylinositol 3-kinase catalytic subunit type 3; PINK1: PTEN induced kinase 1; PLEKHM1: pleckstrin homology and RUN domain containing M1; PRKN/PARK2: parkin RBR E3 ubiquitin protein ligase; RAB: member RAS oncogene family; RHEB: Ras homolog: mTORC1 binding; RIPK2: receptor interacting serine/threonine kinase 2; RLR: DDX58/RIG-I like receptor; ROS: reactive oxygen species; SBD: small bile ducts; SLC2A1/GLUT1: solute carrier family 2 member 1; SLE: systemic lupus erythematosus; SMURF1: SMAD specific E3 ubiquitin protein ligase 1; SQSTM1/p62: sequestosome 1; TAX1BP1: Tax1 binding protein 1; TCR: T cell receptor; TFAM: transcription factor A: mitochondrial; Th17: T helper 17; TLR9: toll like receptor 9; TMEM173/STING: transmembrane protein 173; TNF/TNF-α: tumor necrosis factor; Ub: ubiquitin; UC: ulcerative colitis; ULK1: unc-51 like autophagy activating kinase 1; WIPI: WD repeat domain: phosphoinositide interacting; ZFYVE1/DFCP1: zinc finger FYVE-type containing 1.
KEYWORDS: Immune cells, inflammatory bowel diseases, inflammatory cytokines, mitophagy, primary biliary cirrhosis, systemic lupus erythematosus
Introduction
Mitochondria are emerging as an essential signaling hub in regulation of both innate and adaptive immunity in eukaryotic cells [1]. Malfunctioning mitochondria are involved in various processes closely related to abnormalities of the immune system. Damaged mitochondria fail to act as a platform for signaling molecules, such as mitochondrial antiviral signaling protein (MAVS), which is involved in DDX58/RIG-I-like receptor (RLR) signaling [1,2]. In addition, intracellular debris, such as mitochondrial DNA (mtDNA), serves as a damage-associated molecular pattern that can elicit abnormal inflammatory responses [3]. Moreover, normal mitochondrial metabolism particularly reactive oxygen species (ROS) generation is disrupted, which further impacts inflammatory cytokine secretion, antimicrobial responses and immune cell activation [4]. Hence, it is crucial to preserve sufficient healthy mitochondria to keep the immune system in check [5].
There are two main methods that are exploited by eukaryotic cells to fix damaged mitochondria. First, when reversible damage occurs, defective mitochondrial subunits are degraded into small peptides by ATPases, and unfolded mitochondrial proteins are targeted for degradation via the proteasome system (UPRmt, the mitochondrial unfolded protein response) [6,7]. Second, unwanted mitochondria can be selectively sequestered and delivered to the lysosome for degradation through mitochondrial-specific autophagy, which is termed mitophagy. Although the precise mechanism of mitophagy is not fully understood, emerging lines of evidence highlight a crucial role for mitophagy in regulation of the immune system and its involvement in inflammation and autoimmunity [5].
Depending on how mitochondria are delivered to the lysosome, mitochondrial elimination occurs via two pathways: macroautophagy and microautophagy (termed macromitophagy and micromitophagy, respectively) [8]. Macromitophagy is a selective form of macroautophagy characterized by the formation of a double membrane vesicle termed an autophagosome that ultimately transfers mitochondria to endosomes/lysosomes via fusion. Micromitophagy, in contrast, represents microautophagy, in which mitochondria are directly sequestered into endosomes/lysosomes for degradation in the absence of autophagosome formation [8,9]. As the latter form of mitophagy is less well understood, here we will focus on the molecular mechanisms of macromitophagy (hereafter referred to as mitophagy) in mammalian cells and its potential role in immune regulation; in addition, we attempt to decipher the association between mitophagy and autoimmune diseases.
Molecular mechanism of mitophagy
Canonical autophagy is the basis of mitophagy, which can be mechanistically divided into multiple steps involving the coordination of various autophagy-related (ATG) proteins. Initially, the ULK1 (unc-51 like autophagy-activating kinase 1) complex and the ATG9A recruits the class III phosphatidylinositol 3-kinase complex, including PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3), BECN1/Beclin 1 and ATG14 for phagophore membrane biogenesis. This in turn promotes the recruitment of the phosphatidylinositol-3-phosphate-binding proteins ZFYVE1/DFCP1 (zinc finger FYVE-type containing 1) and the WIPIs (WD repeat domain, phosphoinositide interacting) to assist in phagophore nucleation. The expansion of the phagophore membrane requires the recruitment of two ubiquitin-like conjugation systems, the ATG12 (ATG12, ATG7, ATG10, ATG5 and ATG16L1) and MAP1LC3 (microtubule-associated protein 1 light chain 3, simply referred to as LC3 below)/GABARAP (GABA type A receptor-associated protein) systems (LC3/GABARAPs, ATG7 and ATG3), respectively, for LC3/GABARAP lipidation and subsequent binding to the phagophore membrane. After the elongation step, the phagophore membrane is sealed to form a complete autophagosome with sequestered contents. Lastly, autophagosomal maturation is mediated by the phagophore GABARAP-preferring, LC3-binding proteins PLEKHM1 (pleckstrin homology and RUN domain containing M1) and ALPL/HOPS (alkaline phosphatase, biomineralization associated), enabling the fusion with RAB7-positive endolysosomal organelles for lysosomal degradation [10–12].
The selectivity of mitophagy is mainly achieved by the ubiquitin (Ub)-dependent or Ub-independent (receptor-dependent) initiating pathways that specifically target the autophagic machinery to the mitochondria. Although the mechanism by which upstream ATG proteins are recruited to selected mitochondria remains elusive, the recruitment of LC3/GABARAPs by mitophagy receptors via LC3-interacting region (LIR) motifs is a characteristic step for mitophagosome (mitochondria-specific autophagosome) formation in the later stages of canonical mitophagy [13] (Figure 1). Apart from the targeting systems, mitochondrial morphology may also affect mitophagy induction and efficiency [14,15]. Elongated mitochondria are spared from mitophagic degradation in response to autophagy induction [15]. In addition, mitochondrial division seems to be a crucial regulator of mitophagy efficiency by segregating damaged mitochondria from the healthy network in order to fit them into autophagosomes [14]. A myraids of studies demonstrated that suppression or ablation of the mitochondrial fission factor DNM1L/Drp1 (dynamin 1 like) attenuates mitophagy, albeit to a heterogenous extent [16–20]. However, Burman et al have recently documented amplified mitophagic flux in the absence of DNM1L [21]. Intriguingly, DNM1L depletion leads to an unexpected increase of basal mitophagy, suggesting that mitochondrial dysfunction, secondary to DNM1L depletion, accumulates to an extent that is sufficient to activate mitophagy [21]. Thus it seemed plausible that even if DNM1L promoted mitophagic efficiency in this study, the potentially inhibitory effect of DNM1L depletion on mitophagy could be masked by concurrently enhanced mitochondrial dysfunction. Further research is needed to determine the impact of DNM1L overexpression on mitophagy rates for better understanding of the inconsistent data. In summary, mitophagy is exquisitely dissected from general autophagy regardless of the shared autophagic machinery.
Figure 1.

Brief mechanisms of Ub-dependent and Ub-independent mitophagy. (a) Initiation and mitochondrial labeling: Upon mitochondrial depolarization, PINK1 accumulates on the OMM of mitochondria and recruits the E3 ligase PRKN for ubiquitination of OMM proteins which are then recognized by mitophagy receptors. While mitophagy receptors including FUNDC1, BNIP3/BNIP3L, BCL2L13 and FKBP8 are directly increased to tag selected mitochondria in response to mitochondrial depolarization, hypoxia or during reticulocyte development in a Ub-independent pathway. (b) Nucleation and extension: Early ATG proteins such as ULK1 and other core machinery are recruited for nucleation, extension of phagophore membrane and LC3 conjugation. (c) Closure: Mitophagy receptors bind to LC3 bound on the phagophores for further expansion and closure to form mitophagosomes. 4) Maturation: mitophagosomes fuse with lysosomes for mitochondrial degradation.
Ub-dependent pathway
PINK1 (PTEN induced kinase 1)-PRKN/PARK2 (parkin RBR E3 ubiquitin protein ligase) mediated mitophagy is the most extensively studied Ub-dependent pathway employed to clear damaged mitochondria. PINK1 is a serine/threonine kinase that undergoes constitutive degradation by a matrix-processing peptidase [22], PSEN (presenilin)-associated rhomboid-like proteases [23] and the ubiquitin proteasome system [24] in healthy mitochondria. When misfolded proteins accumulate in the mitochondrial matrix [25] or mitochondrial depolarization occurs, PINK1 is stabilized at the outer mitochondrial membrane (OMM) where it phosphorylates ubiquitin for mitochondrial recruitment of PRKN, which is also phosphorylated by PINK1 [26–29]. This process activates the E3 ubiquitin ligase activity of PRKN, leading to ubiquitination of a myriad of OMM proteins [29]. Ubiquitinated mitochondria are then recognized by the ubiquitin-binding receptor proteins OPTN (optineurin) [30,31], CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2) [32], SQSTM1/p62 (sequestosome 1) [31,33], TAX1BP1 (Tax1 binding protein 1) [31] and NBR1 (NBR1 autophagy cargo receptor) [34]. SQSTM1 and NBR1 are likely to support mitophagy by clustering damaged mitochondria together to prepare them for disposal. OPTN and CALCOCO2 recruit the upstream autophagy proteins ULK1, ZFYVE1/DFCP1 and the WIPIs to initiate autophagosome formation on the mitochondrial surface [31]. However, ATG5 is activated downstream of PINK1 for LC3/GABARAP lipidation, but independently of mitophagy receptors [31]. In the later stages, these OMM receptor proteins function synergistically with another indispensable inner mitochondrial membrane protein, PHB2 (prohibitin 2) as mitophagy receptors that associate with lipidated LC3/GABARAPs bound on the nascent mitophagosomes via LC3-interacting region (LIR) motifs [35]. LIR interactions promote the efficiency of mitophagosome (mitochondrial autophagosome) formation and drive mitophagosome-lysosome fusion for mitochondrial degradation [13]. Notably, PINK1 phosphorylated mitofusin 2 has also been implicated in mitochondrial translocation of PRKN but coablation of MFN1 (mitofusin 1) and MFN2 cannot hinder the progression of PINK1-PRKN mediated mitophagy [36]. Thus the role of MFN2 phosphorylation in this process remains elusive. In addition, it is noteworthy that OPTN and CALCOCO2 function redundantly as primary receptors in PINK1-PRKN mediated mitophagy. PINK1 alone can also recruit OPTN and CALCOCO2 to mediate mitophagy in the absence of PRKN, although at a low level [31]. Interestingly, other E3 ubiquitin proteins are also implicated in mitophagy regulation, including SMURF1 (SMAD-specific E3 ubiquitin protein ligase 1) [37,38], AMFR/gp78 (autocrine motility factor receptor) [39] and MUL1 (mitochondrial E3 ubiquitin protein ligase 1) [40,41] although the mechanism is poorly understood [37].
Notably, a newly discovered form of micromitophagy also depends on PRKN to wrap mitochondria into RAB5A-positive endosomes for lysosomal degradation through ESCRT (endosomal sorting complexes required for transport) machinery [42]. Moreover, mitochondria-derived vesicles (MDVs) also require the involvement of PINK1 and PRKN despite independence from ATG5 and LC3 [8,43,44]. This process topologically represents micromitophagy, as it occurs by invagination of the mitochondrial membrane and ends up as late endosomes delivered to lysosomes. These PINK1/PRKN-dependent micromitophagic pathways may complement the macroautophagic degradation of mitochondria.
Ub-independent pathway
In the absence of the Ub-dependent pathway, several LIR-containing mitophagy receptors can directly induce mitophagy, including FUNDC1 (Fun14 domain containing 1) [45], BNIP3 (BCL2 interacting protein 3) [46], BNIP3L/NIX (BCL2 interacting protein 3 like) [47], BCL2L13 (BCL2 like 13) [48] and FKBP8 (FKBP prolyl isomerase 8) [49] in mammals. These receptor proteins are located on the OMM and depend on LIR interaction for mitochondrial clearance. Hence, receptor-mediated mitophagy is regulated at the transcriptional or post-transcriptional level in response to different mitochondrial stresses.
FUNDC1 normally undergoes phosphorylation by casein kinase 2 at serine 13 and SRC kinase at tyrosine 18, inhibiting its LC3 binding activity [50]. When encountering hypoxia or mitochondrial uncouplers, FUNDC1 is phosphorylated by ULK1 at serine 17 [51] and dephosphorylated by phosphoglycerate mutase family member 5 at serine 13, which increases its LC3 binding activity, thereby initiating mitophagy via enhanced LIR interaction [51]. Reciprocally, initial hypoxic stress promotes ubiquitylation and degradation of redundant FUNDC1 through a mitochondrial ubiquitin ligase MARCH5 (membrane associated ring-CH-type finger 5) [52] to prevent exaggerated mitophagy.
BNIP3, BNIP3L and BCL2L13 belong to the BCL2 family with pro-apoptotic activity [53] while FKBP8 is a member of FKBP family having anti-apoptotic function via interaction with BCL2/BCL2L1 (anti-apoptotic BCL2 family members) [54]. Under hypoxic conditions, BNIP3 is upregulated by hypoxia-inducible factor 1, initiating LC3-dependent mitophagy and precluding mtROS overproduction [55–58]. The LIR activity of BNIP3 requires phosphorylation of serine 17 and 24 by an unknown kinase [59]. BNIP3L is initially found in programmed mitophagy during erythroid development [47,60] and later in other cells such as neurons, where it induces mitophagy via LIR interaction in response to hypoxia or starvation [58,61,62]. High levels of oxidative phosphorylation (OXPHOS) also stimulate BNIP3L-dependent mitophagy through mitochondrial translocation of a small GTPase RHEB (Ras homolog, mTORC1 binding), which then interacts with BNIP3L and LC3 to induce mitophagy [63]. Interestingly, both BNIP3 [20] and BNIP3L [64] overexpression can promote mitochondrial translocation of PRKN, facilitating PRKN-mediated mitophagy.
BCL2L13 and FKBP8 are two recently discovered mitophagy receptors by screening for the mammalian homolog of yeast Atg32 [49,65]. Upon mitochondrial depolarization, the expression of BCL2L13 is increased, promoting mitochondrial fission and mitophagy [48]. Similarly, exogenously overexpression of FKBP8 promotes mitochondrial fission and, when combined with LC3, can boost depolarization-induced mitophagy [49]. However, the endogenous role of FKBP8 in mitophagy remains unclear. Each of these LC3 binding receptors can mediate depolarization-induced mitophagy in the absence of PINK1 and PRKN via LIR dependence [53].
Additionally, externalized mitochondrial cardiolipin produced in response to mitochondrial injury can also associate with LC3 for mitophagy induction independent of PRKN in neuronal cells [66]. Similarly, ceramide (waxy lipid molecules) engages mitophagic machinery through interaction with LC3, leading to inhibition of mitochondrial function and oxygen consumption in tumor cells [67].
In addition to the classical pathway described above, alternative macroautophagic pathways are also involved in mitochondrial elimination independent of LC3/GABARAP, as observed during reticulocyte development [68,69] or in HeLa cells in response to starvation or hypoxia [70]. In this scenario, ULK1 and a lysosome membrane-associated protein RAB9A are required, but not ATG5 or ATG7. Interestingly, the mitophagic activity of BNIP3L is controlled by a novel sequence in its cytoplasmic domain for mitophagy in reticulocytes apart from LIR motifs [71]. Whether BNIP3L also participates in alternative mitophagy in reticulocytes remains to be elucidated.
Functions of mitophagy in immunity
Mitophagy and IL1
IL1B/IL-1β (interleukin 1 beta) and IL18 are key members of the IL1 family mediating inflammatory responses through IL1 receptors, leading to fever, migration of neutrophils, promotion of T cell differentiation and function, recruitment of NK cells, and macrophage responses [72–74]. The biological maturation of IL1B and IL18 requires cleavage by CASP1 (caspase 1) following the assembly and activation of inflammasome, an intracellular multiprotein complex containing a core sensor, such as the NOD-like receptors (NLRs) or AIM2 (absent in melanoma 2). Among sensor proteins, NLRP3 (NLR family pyrin domain containing 3) is the best identified NLR that connects autophagy to inflammasome-dependent regulation against IL1B and IL18 hypersecretion [75–78]. Furthermore, there is accumulating evidence attributing the repressive role of autophagy in NLRP3 inflammasome activation to compromised mitophagy [75,76,79,80]. Upon stimulation with various NLRP3 activators, an increase of IL1B secretion is observed in autophagy/mitophagy-deficient macrophages paralleled by accumulation of damaged mitochondria. Reduction of mtROS production or elimination of depolarized mitochondria leads to a cessation of excessive IL1B secretion. The inhibitory effect of mitophagy on IL1B and IL18 secretion can be utilized by viruses to escape pathogen clearance. For example, RIPK2 (receptor interacting serine threonine kinase 2) can upregulate mitophagic activity by facilitating phosphorylation of the mitophagy inducer ULK1 [81]. Abrogation of RIPK2 augments IL18 secretion in influenza A virus infected dendritic cells and mice as a consequence of mitochondrial accumulation, leading to increased morbidity and mortality to viral infection [81].
Alternatively, mitophagy may represent a cell intrinsic anti-inflammatory mechanism by restraining excessive IL1B and IL18 production. Zhong et al have provided elegant evidence supporting this notion and for the first time, the importance of PRKN in regulation of IL1B secretion [79]. In response to NLRP3 agonists, the expression of SQSTM1 is selectively induced by NFKB/NF-κB (nuclear factor kappa B subunit) after expression of NLRP3 and pro-IL1B in lipopolysaccharide (LPS)-treated macrophages. SQSTM1 ablation leads to impaired mitophagy which further increases IL1B secretion via NLRP3 inflammasome activation, indicating that SQSTM1 dependent mitophagy acts as a pivotal brake to restrict NFKB activated IL1B secretion. Similarly, PRKN deficiency results in accumulation of damaged mitochondria, NLRP3 inflammasome activation and IL1B hypersecretion, accompanied by impaired mitochondrial recruitment of SQSTM1. These observations indicate that SQSTM1 dependent regulation of NFKB signaling critically depends on PRKN to ensure mitophagic regulation of IL1B secretion.
However, it is interesting to note a more recent study demonstrating that mitophagy deficient pink1−/− and prkn−/− mice display strikingly enhanced type I interferon (IFN) IFNB but unaffected IL1B after exhaustive exercise [82]. By contrast, another study has revealed an increase of NLRP3 expression and IL1B production in rat myocardium following acute strenuous exercise [83]. Hence, one possibility to be tested is that the robust secretion of type I IFN, which has been shown to suppress pro-IL1B expression and NLRP3 inflammasome activation [84,85], may conceal the effect of PINK1 or PRKN deficiency on IL1B release. Another possibility is that IL1B secretion may be hampered by PINK1-PRKN independent mitophagy in this study. Consistently, Li et al have recently provided direct evidence for a role of FUNDC1 mediated mitophagy, independent of Ub, in control of inflammasome-dependent IL1B secretion [80]. However, this hypothesis alone cannot explain why released mtDNA, seen in PINK/PRKN depleted mice, does not induce IL1B hypersecretion via inflammasome activation as revealed by previous reports and the same study by Li et al. Lastly, autophagy can directly target NLRP3 inflammasome components and IL1B for lysosomal degradation [78,86], indicating the existence of other compensatory pathway(s) in regulation of IL1B secretion without mitochondrial involvement. Collectively, the role of specific mitophagy pathways in NLRP3 dependent IL1B secretion is rather complicated and requires further investigation.
Although less understood, mitophagy may also be involved in control of IL1B via other inflammasomes. A previous study of NLRC4 (NLR family CARD domain containing 4) inflammasome activation by the bacterium Pseudomonas aeruginosa infection has revealed that the inhibitory effect of autophagy on IL1B secretion relies on elimination of mitochondria-derived signals, including mtROS and mtDNA [87]. Besides, Li et al have also demonstrated that mitophagy deficient hepatocytes lacking FUNDC1 display enhanced formation of AIM2 inflammasome, in addition to NLRP3 inflammasome, and increased mtDNA after treatment with the chemical carcinogen diethylnitrosamine (DEN) [80]. This corresponds with earlier studies showing that release of mtDNA can activate AIM2 inflammasome by itself [76] and in combination with HMGB1 (high mobility group box 1) to form HMGB1-DNA complex [88,89] which in turn can induce autophagy to limit excessive AIM2 activation [88]. In parallel, the secretion of IL1A, another potent inflammatory member of IL1 family, may also be subject to mitophagic regulation given that ROS/mtROS mediated calpain activation leads to hypersecretion of IL1A following autophagy inhibition [78,90]. Notably, this pathway is independent of inflammasome activation. Thus, mitophagy may be an essential anti-inflammatory pathway through which autophagy restricts the secretion of IL1B/IL18 and IL1A.
Mitophagy and type i IFN
Type I IFNs are a group of pleiotropic cytokines including IFNA and IFNB, promoting antigen presentation, NK cell functions and lymphocyte responses [91]. The ability of mitophagy to suppress type I IFN synthesis was first observed in ATG5-depleted cells, which displayed pools of dysfunctional mitochondria and enhanced type I IFN production in response to vesicular stomatitis virus or synthetic analogs of viral dsRNA [92]. This response is dependent on increased mtROS production and elevated levels of mitochondrial-tethered MAVS, which amplifies RLR signaling [92]. Subsequent studies further demonstrated that distinctive mitophagic pathways can be harnessed by viruses to attenuate type I IFN responses for viral replication. For example, SQSTM1-dependent mitophagy is activated during oncolytic measles virus infection. Ablation or reduction of SQSTM1 enhanced type I IFN production and cell death in infected cells through impaired degradation of mitochondrial-bound MAVS [93]. Alternatively, human parainfluenza virus type 3 (HPIV3) directly induces receptor-mediated mitophagy, as the matrix protein of HPIV3 can translocate to mitochondria and subsequently interact with LC3 to mitigate mitophagy-dependent type I IFN responses [94].
In the absence of viral infection, BNIP3L-dependent mitophagy, one specific mitophagy pathway, is implicated as a crucial mechanism to avoid unprompted production of type I IFN via RLR-MAVS signaling. The polymeric form of MAVS located on the mitochondrial and peroxisomal membranes is required for activation of downstream RLR signaling through its caspase activation and recruitment domain (CARD) via NFKB and interferon regulatory factor [95]. However, full-length MAVS with the CARD is prone to spontaneous aggregation without interaction with shorter isoforms of MAVS lacking CARD, triggering ROS-induced mitophagy [96]. Blocking BNIP3L-mediated mitophagy leads to MAVS accumulation and type I IFN production in the absence of the truncated isoforms [96]. Hence, mitophagy may not only be usurped by viruses for suppression of antiviral immunity but also protects cells from inadvertent type I IFN production under basal conditions via RLR-MAVS signaling.
Besides, a recent study has unraveled an in vivo role of PINK1-PRKN dependent mitophagy in limiting type I IFN production via the TMEM173/STING (transmembrane protein 173) mediated DNA sensing pathway following mitochondrial stress [82]. As mentioned earlier, an unbridled increase in type I IFN but not IL1B was observed in serum of mitophagy compromised pink1−/− and prkn−/− mice after exhaustive exercise. Concurrently elevated was mtDNA, which reportedly binds to the cytosolic DNA sensor, CGAS (cyclic GMP-AMP synthase) that in turn promotes TMEM173 dependent IFN induction by synthesizing cyclic GMP-AMP. This presumed link between mitophagy, mtDNA and the CGAS-TMEM173 pathway was further substantiated by enhanced generation of cyclic GMP-AMP in pink1−/− and prkn−/− mice following exhaustive exercise, and reduced type I IFN production by codeletion of TMEM173 [82]. In contrast, Li et al failed to observe downstream activation of the TMEM173 pathway in mitophagy-defective hepatocytes from fundc1−/− mice after exposure to DEN [80]. Instead, they evidenced enforced activation of NLRP3 and AIM2 inflammasome and increased IL1B secretion. It is conceivable as the CGAS-TMEM173 pathway is reportedly inactive in human and murine hepatocytes owing to loss of TMEM173 expression [97]. Besides, inflammasome activation, induced by classical NLRP3 agonists such as ATP and LPS, reportedly renders the TMEM173 pathway inactive during DNA virus infection through CASP1-dependent cleavage of CGAS even when the TMEM173 pathway is originally intact [98]. Considering the similarity between mtDNA and viral DNA, this discovery may provide clues for why a series of studies using similar NLRP3 agonists, as mentioned in the last section, did not report an elevation of type I IFN production in the presence of augmented mtDNA release and IL1B secretion when autophagy/mitopahgy is blocked. More research detecting both inflammasome components and TMEM173 pathway activity is imperative for better understanding of the regulatory effect of mitophagy on type I IFN production in different contexts.
Mitophagy and other inflammatory cytokines
Control of dysfunctional mitochondria may link mitophagy to other inflammatory cytokines. For example, accumulation of mtROS due to autophagy insufficiency results in excessive production of MIF (macrophage migration inhibitory factor) in human and mouse macrophages upon LPS stimulation [99]. In parallel, in the absence of autophagy, release of mtDNA leads to transcriptionally enhanced production of multiple inflammatory cytokines, including TNF/TNF-α (tumor necrosis factor) and IL6, through activation of the NFKB pathway via TLR9 (toll like receptor 9) [100]. Moreover, stress induced liberation of mtDNA triggers TMEM173 dependent cytokine production such as IL6, other than IFNB, when PINK1-PRKN dependent mitophagy is abolished [82]. At present, it is unknown how mitophagy affects IL23 and IL17 secretion. However, based on the effects of IL1B and IL18 on these cytokines, it is plausible that mitophagy may also restrict the secretion of IL23 and IL17. In support of this idea, augmented release of IL1B can further drive autocrine secretion of IL23 in macrophages and dendritic cells when autophagy is blocked [101]. Similarly, elevated levels of IL1B alone or combined with IL23 have been shown to stimulate the secretion of IL17 by T helper 17 (Th17) and γδT cells in autophagy-compromised macrophages and dendritic cells [101–103]. Concurrently, impaired autophagy in myeloid cells may exert an extra effect on the release of other inflammatory cytokines, including IL12, CXCL1 (C-X-C motif chemokine ligand 1), IL22 and IFNG either by themselves or myeloid T cells [90,101]. The direct relationship between mitophagy and a growing number of inflammatory cytokines warrants more studies using specific mitophagy-modulating methods.
Mitophagy and antigen presentation
Several studies have linked mitophagy to mitochondrial antigen presentation (mtAP) but with seemingly conflicting results. Considering the involvement of autophagy in antigen presentation, it has been speculated that mitophagy may play a similar role in mtAP. In support of this hypothesis, TNF has been shown to activate mitophagy for specific degradation of mitochondrial proteins in macrophages and thus promote the antigen presentation of mitochondrial targeting glycoprotein from herpes simplex virus 1 to CD8+ T cells [104]. The effects of mitophagy on mtAP are further confirmed by the reduction of CD8+ T cell activation seen after treatment with the autophagy inhibitor 3-methyladenine and the corresponding increase observed after treatment with carbonyl cyanide m-chlorophenylhydrazone, a mitochondrial uncoupler commonly used to activate PRKN-mediated mitophagy [104]. However, a recent study has shown that inhibition of mitophagy by 3-methyladenine or genetic silencing of ATG5 or DNM1L does not repress but rather promotes mtAP of glycoproteins induced by heat stress [105]. Alternatively, both in vivo and in vitro data highlight the role of PINK-PRKN regulated MDV in mtAP via the MHC (major histocompatibility complex) class I pathway [105]. Genetic depletion of PINK and other MDV-related elements such as an endosomal GTPase RAB9A promotes mtAP, whereas PRKN overexpression circumvents this process through MDV formation [105]. This raises the possibility that (macro)mitophagy, as an efficient way to eliminate damaged mitochondria, may limit the mtAP on MHC molecules [106]. More studies are required to delineate the differential requirement of distinctive forms of mitophagy in antigen presentation and to characterize the mechanism by which mtAP in MHC class II molecules occurs.
Mitophagy and immune cells
Apart from influencing immune cells through pro-inflammatory cytokines and mtAP, emerging lines of evidence indicates that mitophagy is directly engaged in the development and differentiation of immune cells including T cells, natural killer (NK) cells and macrophages. This notion derives from earlier discoveries that mitochondrial volume reduction via autophagy is developmentally required for cell viability during the transition of T cells from thymocytes to peripheral naïve T cells and maturation of invariant NK (iNK) T cells in the thymus [107–112]. Mechanistically, ROS accumulation and release of mitochondrial pro-apoptotic components contributes to increased apoptosis in autophagy-deficient naïve/iNK T cells, implying an anti-apoptotic function of mitophagy in T cell maintenance [113]. This pro-survival role of mitophagy has further been substantiated in peripheral T cells undergoing activation-induced cell death (AICD), in which repeated stimulation of T cell receptors (TCRs) drives autonomous apoptosis in order to clear unnecessarily activated T cells [114]. Upon AICD induction, depletion of PRKN, a unique mitophagy effector, promotes AICD through accumulation of damaged and fragmented mitochondria with cristae remodeling and release of CYCS/cytochrome C. Rapamycin, an MTOR (mechanistic target of rapamycin kinase) inhibitor and autophagy inducer, abrogates AICD by reactivation of mitophagy, as indicated by increased colocalization of PRKN and LC3 in mitochondria [114]. Based on these preliminary studies, mitophagy seems to be physiologically regulated in T cell development, activation and death to maintain cell homeostasis while avoiding autoimmunity.
Similarly, mitophagy is utilized by other populations of immune cells, including NK cells [115] and macrophages [116] to resist apoptosis (Table 1). Remarkably, stage-specific regulation of BNIP3- and BNIP3L-mediated mitophagy is essential for differentiation of memory NK cells [115]. In proliferating effector NK cells, mitophagy is temporally suppressed during viral infection, leading to accumulation of damaged mitochondria and apoptosis of most activated NK cells. However, mitophagy is subsequently restored after the expansion phase to rescue a subset of virus-specific NK cells for differentiation into long-lived and quiescent memory cells. The dynamic regulation of mitophagy relies on correspondingly altered expression of BNIP3 and BNIP3L as well as autophagosome-lysosome fusion activity [115]. Intriguingly, similar in vivo kinetics of autophagy has also been observed during effector-memory differentiation of CD8+ virus-specific T cells and B cells in response to viral infection [117–119]. In addition, mitophagy serves as an intrinsic mechanism for maintenance of memory iNK T cells [107] and memory T cells [120]. Hence, the prediction is that induction of mitophagy may be a general mechanism engaged in the differentiation into quiescent memory cells.
Table 1.
Effects of inhibiting autophagy/mitophagy on immune cell survival.
| Immune cells | Experimental model/cells | Effects of autophagy/mitophagy inhibition | Reference |
|---|---|---|---|
| Thymic iNK T cells | Atg7fl/fl – Cd4-Cre mice | A T-cell-intrinsic block of iNK T cell maturation with expanding mitochondria due to increased apoptosis | [107] |
| Resting naïve T (CD4, CD8+ T) in the peripherals |
|
|
[107–112] |
| Peripheral T cells during AICD |
|
Diminished cell death of TCR-restimulated T cells due to restored PRKN-dependent mitophagy | [114] |
| Virus-specific CD8+ T cells | Atg7 fl/fl – Cd4-Cre mice | Increased susceptibility of CD8+ T cells during memory formation, accompanied by increased mitochondrial contents, mtROS, and a higher expression of SLC2A1 | [118] |
| NK cells |
|
Abrogation of NK cell memory formation after viral infection in the absence of BNIP3- and BNIP3L-dependent mitophagy | [115] |
| memory B cells | B/atg7−/− mice | Loss of memory B cells due to accumulation of dysfunctional mitochondria and mtROS | [119] |
| Macrophages |
|
Increased apoptosis of macrophages | [116] |
Interestingly, altered mitochondrial metabolism may also dictate the differentiation of inflammatory immune cells into quiescent cells [1]. Indeed, mature iNK/naïve/memory/regulatory T-cells, NK memory cells and anti-inflammatory (M2) macrophages rely on OXPHOS with limited biosynthetic demands [1,121]. In contrast, immature iNK/effector T cells, activated NK cells and inflammatory (M1) macrophages utilize aerobic glycolysis for energy production [121]. Based on the role of mitophagy in mitochondrial health, it appears likely that mitophagy may impact mitochondrial metabolism during immune cell differentiation. In support of this notion, ATG7-deficient CD8+ T cells display enhanced mitochondrial contents, mtROS and increased expression of the glucose receptor SLC2A1/GLUT1 (solute carrier family 2 member 1; a marker for glycolysis) during memory generation [118]. Furthermore, mitophagy has been shown to guide the differentiation of macrophages through metabolic reprogramming. IL10 mediated inhibition of MTOR activity induces mitophagy but retards glycolysis in LPS stimulated macrophages [122,123]. Mitophagy inhibition by 3-methyladenine promotes the conversion to M1 phenotypes, while mitophagy induction by rapamycin prevents M1 polarization in favor of M2 differentiation in diabetic mouse kidney in response to high glucose treatment [124]. Consistently, mitochondrial dysfunction interferes with normal mitochondrial respiration, leading to blunted repolarization of M2 macrophages [125]. However, alternative evidence demonstrates that BNIP3L deficiency leads to impaired glycolytic switch of macrophages differentiated into M1 phenotypes following LPS stimulation [61]. Importantly, it has also been shown that BNIP3L-dependent mitophagy, a specific mitophagy pathway, is induced by hypoxia in favor of glycolysis during differentiation of retinal ganglion cells. More research is required to clarify the role of mitophagy in macrophage differentiation through integrated mechanisms. In addition, it would be fascinating to investigate how mitophagy affects differentiation of regulatory T cells, which resembles a similar metabolic switch during generation of memory T cells and M2 macrophages.
Mitophagy in autoimmune diseases
IBD and mitophagy
Crohn disease (CD) and ulcerative colitis (UC) are two major clinical entities of inflammatory bowel diseases (IBD), a group of autoimmune diseases characterized by unresolving inflammation in the gastrointestinal tract. The immunopathogenesis of IBD involves the interplay between the immune system and environmental, genetic and microbial factors [126]. Genome-wide searches have identified common autophagy and mitophagy genes for IBD, including ATG16L1, IRGM (immunity-related GTPase M) and LRRK2 (leucine-rich repeat kinase 2) [127]. Compromised mitophagy has further been observed in functional studies of CD-associated risk variants in ATG16L1 and IRGM as manifested by altered mitochondrial morphology, reduction of mitochondrial membrane potential and increased mtROS as well as disruption of LC3 marked autophagic machinery [128,129]. As to LRRK2, the Parkinson disease-linked mutation G2019S could promote mitophagy through induction of mitochondrial fission and interaction with ULK1 [130]. However, whether CD-related variants in LRRK2 disrupt mitophagy remains unclear [130–134]. Interestingly, a recent cross-phenotype study has identified an intronic SNP rs3766606 in PARK7 shared by psoriasis (risk), CD and UC (protective). Similar to LRRK2, PARK7 is a canonical PD risk locus as well as a potential UC candidate gene [135]. PARK7 belongs to the PARK family and has a well-defined role in mitochondrial homeostasis and mitophagy [136]. More specifically, IBD risk genes CALCOCO2, PEX13 and SMURF1 encode proteins acting as mitophagy mediators [137–139]. However, whether IBD-related variants of these genes contribute to disease progression and to what extent this occurs by disrupting mitophagy remains obscure.
Mechanistically, preliminary studies indicate an anti-inflammatory effect of mitophagy on the gut micro-environment in IBD. The CD-associated ATG16L1T300A variant leads to defective mitophagy, mtROS accumulation and correspondingly increased IL1B production [140] and pro-inflammatory macrophage polarization with reduced bacterial killing [129]. Consistently, andrographolide, a small molecule applied for treatment of IBD, was found to protect against colitis progression and colitis-associated carcinoma through mitophagy-mediated inhibition of NLRP3 inflammasome [141]. According to an evaluation of macrophages from murine models of colitis or IBD patients, mitophagy is diminished due to prolonged activation of MTOR by genetic depletion of IL10 or IL10 receptor, leading to amplified inflammasome activation and gut inflammation. In addition, IL10 also favors M2 macrophages through mitochondrial metabolism reprogramming [122,123]. Apart from increased IL1B secretion, Paneth cell abnormalities have been associated with defective mitophagy and autophagy in murine intestinal cells lacking Irgm1 [128,142]. Indeed, human IRGM is a mitochondrial-located protein that induces mitochondrial depolarization and promotes mitochondrial fission, both of which are triggers for mitophagy [143]. The mitochondrial alteration caused by IRGM is crucial to accomplishing autophagic control of mycobacteria, implying the potential involvement of mitophagy in host-microbe interactions.
Alternatively, mitochondrial stress-induced mitophagy may play a role in intestinal epithelial cell (IEC) viability. Diminished expression of PHB/PHB1 (prohibitin), a mitochondrial chaperone for respiration chain proteins, may impair mitochondrial integrity and thus stimulate mitophagy via enhanced intracellular ROS in IECs of IBD patients [144]. Reduction of both PHB and ATG16L1, prevalent in IBD, aggravates mitochondrial depolarization and reduces colonic cell survival [144]. Paradoxically, PHB seems to physically interact with BNIP3L and thus specifically promote mitophagy [145]. Nonetheless, how they might interact is unknown given that PHB is an inner mitochondrial membrane protein while BNIP3L locates on the OMM. In addition, PHB can associate with its homolog PHB2, an essential mitophagy receptor in PRKN-mediated mitophagy [146]. Further studies are required to investigate the complex relationship between PHB, mitochondrial dysfunction and mitophagy.
Taken together, these results indicate mitophagy as a generally protective process in IBD through its regulation of pro-inflammatory cytokine production, macrophage polarization and IEC viability and possibly pathogen clearance.
SLE and mitophagy
Systemic lupus erythematosus (SLE) is an antoantibody mediated autoimmune disease resulting in systemic inflammation in multiple organs including skin, joints, heart, kidneys and the nervous system [147]. The pathogenesis of SLE involves aberrant T and B cell signaling, autoantibody production, and deregulated cytokine secretion due to genetic and environmental factors [148]. Although mutations in the ATG genes ATG5, ATG7 and IRGM are implicated in conferring susceptibility to SLE, how these genetic polymorphisms affect mitophagy remains unknown [149]. In lupus T cells, mitochondrial dysfunction characterized by increased mitochondrial mass (megamitochondria), mitochondrial hyperpolarization and ATP depletion contributes to abnormal activation and enhanced necrosis of T cells [150,151]. Afterwards, the necrotic debris of T cells triggers autoantibody production, inflammation and organ damage [150–152]. Multiple mechanisms are responsible for mitochondrial abnormalities in lupus T cells, including enhanced mitochondrial biogenesis and insufficient mitophagy [153]. A pioneer study showed that mitophagy is suppressed in T cells from SLE patients and from lupus-prone mice which characteristically overexpressed a small GTPase RAB4A [154]. This occurs through loss of DNM1L as a consequence of enhanced RAB4A-regulated lysosomal degradation. In line with this, rapamycin, a potent autophagy/mitophagy inducer, ameliorates disease severity and mitochondrial dysfunction via MTOR inhibition [154]. This also corroborates earlier studies reporting autophagy resistance in lupus T cells, as demonstrated by decreased autophagic flux on induction [155]. Moreover, DNM1L restoration by RAB4A blockade reverses mitochondrial accumulation in T and B cells as well as mitochondrial mass expansion, ANA production, and nephritis in lupus-prone mice, implicating mitophagy modulation as a potential therapeutic target [154]. Interestingly, RAB4A is upregulated by oxidative stress via MTOR activation in the setting of SLE [153,156]. Hence, impaired mitophagy as a crucial contributor to mtROS may in turn upregulate RAB4A, hinting at a positive feedback mechanism that causes persistent mitochondrial disruptions in lupus T cells, underlying the SLE pathology.
SLE neutrophils feature mitochondrial retention of oxidized (ox)-mtDNA. When expelled, these ox-mtDNAs stimulate autoantibody production, dendritic cell activation and type I IFN production, contributing to SLE pathogenesis [157]. This response has been attributed to constitutively defective mitophagy and exposure to ribonucleoproteins [157,158]. Unlike most cells, healthy neutrophils fail to complete mitophagy upon mitochondrial depolarization, as indicated by increased mitophagosome formation and decreased mitophagosome-lysosome fusion. Alternatively, ox-mtDNA is routed to lysosomes within cytosolic vesicles, resembling MDV. This micromitophagy-like process is abolished in the presence of type I IFN and anti-Sm and ribonucleotide protein autoantibodies through inhibiting TFAM (transcription factor A, mitochondrial) phosphorylation, ultimately leading to expulsion of highly immunogenic ox-mtDNA [157]. Alternatively, extrusion of mitochondrial contents can be conducted through exocytosis of mitophagosome-lysosomes, as evident in LPS-stimulated hepatocytes [159]. It remains to be clarified whether this process also occurs in lupus neutrophils. Accordingly, proficient mitophagy may have therapeutic effects in SLE through prompt elimination of ox-mtDNA before their extrusion.
PBC and mitophagy
Primary biliary cirrhosis (PBC) is a liver-specific autoimmune disease characterized by the serological presence of serum anti-mitochondrial antibodies (AMA) [160]. Pathologically, PBC involves chronic progressive destruction of small bile ducts (SBD), portal inflammation and ultimately cirrhosis [160]. The abnormal SBD damage in PBC is secondary to aberrant immune responses to mitochondrial autoantigens. The DLAT/PDC-E2 (dihydrolipoamide S-acetyltransferase) and OGDH (oxoglutarate dehydrogenase) complex are the main mitochondrial autoantigens (mtAg), resulting in enhanced CD4+ and CD8+ T cell response in PBC [160,161]. The colocalization of DLAT with autophagy/mitophagy markers LC3 and SQSTM1 in damaged SBDs has been described in PBC patients [162]. Subsequent in vitro studies demonstrated a similar result in cultured bile epithelial cells, and that autophagy induction is correlated with enhanced expression of DLAT on the cell surface [162]. These preliminary results suggest a potential involvement of (macro)mitophagy in the autoimmune pathogenesis of SBD injuries [163]. Interestingly, a recent study indicates that the presentation of another key mtAg OGDH on MHC class I molecules is conducted through PINK1 and PRKN mediated MDV which is blunted rather than promoted by (macro)mitophagy [105,106]. In macrophages encountering heat stress, OGDH presentation is increased by ATG5 depletion and decreased by PRKN overexpression. The negative role of PINK1 and PRKN in OGDH presentation has further been validated in dendritic cells and in vivo mouse model cells stimulated with LPS [105]. In all, mitophagy seems to prevent the autoimmunity involved in PBC, while MDV may be deleterious in facilitating mtAP of autoantigens.
Concluding remarks and future directions
Recent studies highlight the pivotal function of mitophagy in the regulation of inflammatory cytokine production and immune cell homeostasis and differentiation, correlating with the pathogenesis of autoimmune diseases at multiple levels (summarized in Figure 2). Critically, mitophagy serves as an essential intersection between autophagy and production of inflammatory cytokines, such as IL1B and type I IFN, two hallmarks of inflammation and autoimmunity [164]. In addition, mitophagy emerges to be a key player in immune cell development, activation and differentiation. Furthermore, currently available evidence indicates a uniquely protective role for mitophagy in preventing disease progression in IBD, PBC and SLE. However, many puzzles concerning the role of mitophagy in the immune system and disease context remains to be solved. How mitophagy affects the differentiation of immune cells into inflammatory or quiescent cells and the precise mechanisms underlying the dynamic regulation of mitophagy remain obscure. As most relevant studies derive from autophagy-mitophagy-inhibition models, it should be cautioned that hyperactivation of mitophagy may be deleterious in immune cell homeostasis, as evidenced by hematopoietic stem cells [165]. In addition, autophagy and mitophagy are rarely distinguished in many cases of these studies. Notably, the mechanism of mitophagy is extremely complex, involving the integration of multiple ATG proteins, auxiliary mitophagy receptors, endocytic pathways and mitochondrial dynamics. Hence, the functional status and effects of mitophagy modulation cannot be fully extrapolated from studies of autophagic regulation of mitochondria and autoimmunity. Besides, research into the crosstalk of distinctive forms of mitophagy in the regulation of immunity is still lacking. A more thorough understanding of mitophagic pathways in autoimmune diseases is required using specific modulators of mitophagy and mitophagy effectors, such as SQSTM1-mediated mitophagy inducers, combined with multiple approaches to assessing mitophagic activity [166].
Figure 2.
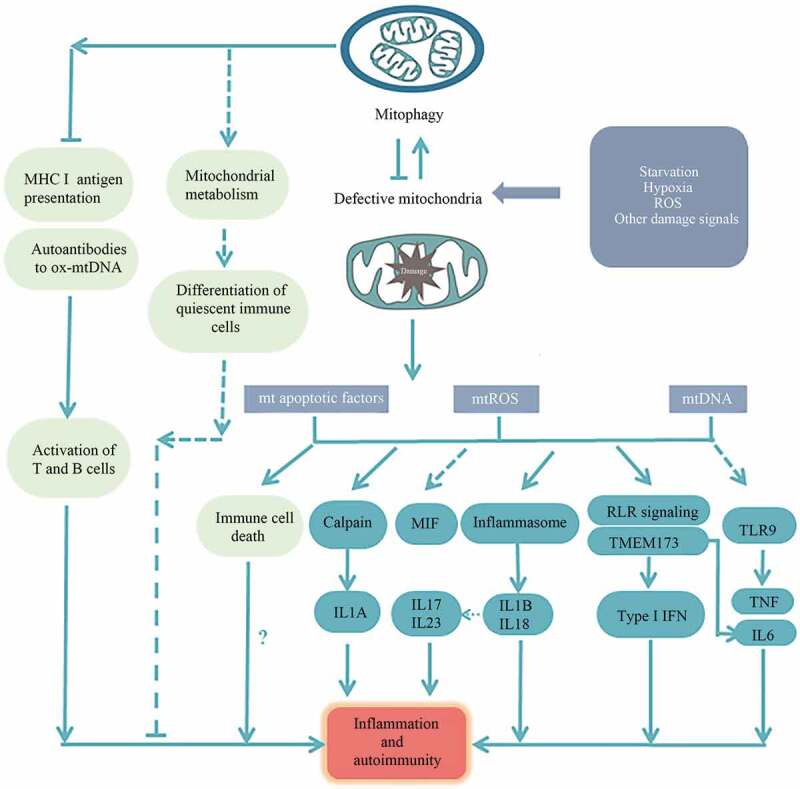
The impact of mitophagy on inflammation and autoimmunity. (a) Mitochondrial dysfunction can cause excessive mtDNA and mtROS production, thus engaged in inflammatory cytokine production including IL1B, IL18 and type I IFN through inflammasome activation, and the RLR signaling/TMEM173 pathway, respectively. Excessive production of IL1A, IL6, TNF, IL17, IL23 and MIF has also been correlated with mitochondrial dysfunction and autophagy/mitophagy deficiency. These inflammatory elements are potent contributors to inflammation and autoimmunity. (b) MtAP seems to be inhibited by mitophagy, directly linking mitophagy to adaptive immunity. Meanwhile, ox-mtDNA can generate autoantibodies which can be removed by mitophagy. (c) Mitophagy is dynamically regulated to ensure the normal development, activation and death of immune cells through blunting mitochondrial apoptosis. Alternatively, mitophagy seems to be involved in regulating mitochondrial metabolism in control of the preferential differentiation of immune cells into quiescent phenotypes that rely on OXPHOS. But the relationship of mitophagy, immune cell differentiation and autoimmunity remains not fully understood.
Funding Statement
This work was supported by the National Natural Science Foundation of China [81670497]; National Natural Science Foundation of China [81470820 & 81770545].
Acknowledgments
We would like to acknowledge support from National Science Foundation of China (No. 81470820, 81670497 & 81770545).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Mills EL, Kelly B, O‘Neill LAJ.. Mitochondria are the powerhouses of immunity. Nat Immunol. 2017. April 18;18(5):488–498. PubMed PMID: 28418387. [DOI] [PubMed] [Google Scholar]
- [2].Tait SW, Green DR. Mitochondria and cell signalling. J Cell Sci. 2012. February 15;125(Pt 4):807–815. PubMed PMID: 22448037; PubMed Central PMCID: PMC3311926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. 2015. March 17;42(3):406–417. PubMed PMID: 25786173; PubMed Central PMCID: PMC4365295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012. October 26;48(2):158–167. PubMed PMID: 23102266; PubMed Central PMCID: PMC3484374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lazarou M. Keeping the immune system in check: a role for mitophagy. Immunol Cell Biol. 2015. January;93(1):3–10. PubMed PMID: 25267485 [DOI] [PubMed] [Google Scholar]
- [6].Leonhard K, Guiard B, Pellecchia G, et al. Membrane protein degradation by AAA proteases in mitochondria: extraction of substrates from either membrane surface. Mol Cell. 2000. April;5(4):629–638. PubMed PMID: 10882099. [DOI] [PubMed] [Google Scholar]
- [7].Haynes CM, Ron D. The mitochondrial UPR-protecting organelle protein homeostasis. J Cell Sci. 2010. November 15;123(Pt 22):3849–3855. PubMed PMID: 21048161. [DOI] [PubMed] [Google Scholar]
- [8].Lemasters JJ. Variants of mitochondrial autophagy: types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. 2014;2:749–754. PubMed PMID: 25009776; PubMed Central PMCID: PMC4085350. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Disatnik MH, Hwang S, Ferreira JC, et al. New therapeutics to modulate mitochondrial dynamics and mitophagy in cardiac diseases. J Mol Med (Berl). 2015. March;93(3):279–287. PubMed PMID: 25652199; PubMed Central PMCID: PMC4333238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yoshii SR, Mizushima N. Autophagy machinery in the context of mammalian mitophagy. Biochim Biophys Acta. 2015. October;1853(10 Pt B):2797–2801. .PubMed PMID: 25634658 [DOI] [PubMed] [Google Scholar]
- [11].Rogov VV, Stolz A, Ravichandran AC, et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep. 2017. August;18(8):1382–1396. PubMed PMID: 28655748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nguyen TN, Padman BS. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol. 2016. December 19;215(6):857–874. PubMed PMID: 27864321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Padman BS, Nguyen TN, Lazarou M. Autophagosome formation and cargo sequestration in the absence of LC3/GABARAPs. Autophagy. 2017. April 3;13(4):772–774. PubMed PMID: 28165849; PubMed Central PMCID: PMC5388231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ni HM, Williams JA, Ding WX. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015;4(C):6–13. PubMed PMID: 25479550; PubMed Central PMCID: PMC4309858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011. May;13(5):589–598. PubMed PMID: 21478857; PubMed Central PMCID: PMC3088644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Twig G, Elorza A, Molina AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J. 2008. January 23;27(2):433–446. PubMed PMID: 18200046; PubMed Central PMCID: PMC2234339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tanaka A, Cleland MM, Xu S, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010. December 27;191(7):1367–1380. PubMed PMID: 21173115; PubMed Central PMCID: PMC3010068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].MacVicar TD, Lane JD. Impaired OMA1-dependent cleavage of OPA1 and reduced DRP1 fission activity combine to prevent mitophagy in cells that are dependent on oxidative phosphorylation. J Cell Sci. 2014. May 15;127(Pt 10):2313–2325. PubMed PMID: 24634514; PubMed Central PMCID: PMCPmc4021475. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wu W, Lin C, Wu K, et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 2016. July 1;35(13):1368–1384. PubMed PMID: 27145933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lee Y, Lee HY, Hanna RA, et al. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2011. November;301(5):H1924–31. PubMed PMID: 21890690; PubMed Central PMCID: PMCPmc3213962. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Burman JL, Pickles S, Wang C. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol. 2017. October 2;216(10) :3231–3247. PubMed PMID: 28893839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Greene AW, Grenier K, Aguileta MA, et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012. April;13(4):378–385. PubMed PMID: 22354088; PubMed Central PMCID: PMC3321149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jin SM, Lazarou M, Wang C, et al. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010. November 29;191(5):933–942. PubMed PMID: 21115803; PubMed Central PMCID: PMC2995166. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy. 2013. November 1;9(11):1758–1769. PubMed PMID: 24121706; PubMed Central PMCID: PMC4028335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy. 2013. November 1;9(11):1750–1757. PubMed PMID: 24149988; PubMed Central PMCID: PMC4028334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen Y, Dorn GW 2nd.. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013. April 26;340(6131):471–475. PubMed PMID: 23620051; PubMed Central PMCID: PMC3774525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Koyano F, Okatsu K, Kosako H, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014. June 5;510(7503):162–166. PubMed PMID: 24784582. [DOI] [PubMed] [Google Scholar]
- [28].Nguyen TN, Padman BS, Lazarou M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016. October;26(10):733–744. PubMed PMID: 27291334 [DOI] [PubMed] [Google Scholar]
- [29].Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010. January 26;8(1):e1000298. PubMed PMID: 20126261; PubMed Central PMCID: PMC2811155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014. October 21;111(42):E4439–48. PubMed PMID: 25294927; PubMed Central PMCID: PMC4210283. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015. August 20;524(7565):309–314. PubMed PMID: 26266977; PubMed Central PMCID: PMC5018156. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Heo JM, Ordureau A, Paulo JA, et al. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. 2015. October 1;60(1):7–20. PubMed PMID: 26365381; PubMed Central PMCID: PMC4592482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Geisler S, Holmstrom KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010. February;12(2):119–131. PubMed PMID: 20098416; eng. [DOI] [PubMed] [Google Scholar]
- [34].Sarraf SA, Raman M, Guarani-Pereira V, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013. April 18;496(7445):372–376. PubMed PMID: 23503661; PubMed Central PMCID: PMC3641819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wei Y, Chiang WC, Jr SR, et al. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168(1–2):224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Narendra D, Tanaka A, Suen DF, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008. December 1;183(5):795–803. PubMed PMID: 19029340; PubMed Central PMCID: PMCPmc2592826. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yamaguchi O, Murakawa T, Nishida K, et al. Receptor-mediated mitophagy. J Mol Cell Cardiol. 2016. June;95:50–56. PubMed PMID: 27021519. [DOI] [PubMed] [Google Scholar]
- [38].Orvedahl A, Sumpter R Jr., Xiao G, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011. December 1;480(7375):113–117. PubMed PMID: 22020285; PubMed Central PMCID: PMC3229641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Fu M, St-Pierre P, Shankar J, et al. Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol Biol Cell. 2013. April;24(8):1153–1162. PubMed PMID: 23427266; PubMed Central PMCID: PMC3623636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Attaix D, Taillandier D. The missing link: mul1 signals mitophagy and muscle wasting. Cell Metab. 2012. November 7;16(5):551–552. PubMed PMID: 23140636. [DOI] [PubMed] [Google Scholar]
- [41].Li J, Qi W, Chen G, et al. Mitochondrial outer-membrane E3 ligase MUL1 ubiquitinates ULK1 and regulates selenite-induced mitophagy. Autophagy. 2015;11(8):1216–1229. PubMed PMID: 26018823; PubMed Central PMCID: PMC4590677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hammerling BC, Najor RH, Cortez MQ, et al. A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat Commun. 2017. January 30;8:14050. PubMed PMID: 28134239; PubMed Central PMCID: PMC5290275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Soubannier V, McLelland GL, Zunino R, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012. January 24;22(2):135–141. PubMed PMID: 22226745. [DOI] [PubMed] [Google Scholar]
- [44].McLelland GL, Soubannier V, Chen CX, et al. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. Embo J. 2014. February 18;33(4):282–295. PubMed PMID: 24446486; PubMed Central PMCID: PMC3989637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012. January 22;14(2):177–185. PubMed PMID: 22267086; eng. [DOI] [PubMed] [Google Scholar]
- [46].Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008. April 18;283(16):10892–10903. PubMed PMID: 18281291; PubMed Central PMCID: PMC2447655. eng. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [47].Sandoval H, Thiagarajan P, Dasgupta SK, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008. July 10;454(7201):232–235. PubMed PMID: 18454133; PubMed Central PMCID: PMCPmc2570948. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Murakawa T, Yamaguchi O, Hashimoto A, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. 2015. July 6;6:7527. PubMed PMID: 26146385; PubMed Central PMCID: PMC4501433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bhujabal Z, Birgisdottir AB, Sjottem E, et al. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017. June;18(6):947–961. PubMed PMID: 28381481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chen G, Han Z, Feng D, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014. May 8;54(3):362–377. PubMed PMID: 24746696. [DOI] [PubMed] [Google Scholar]
- [51].Wu W, Tian W, Hu Z, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014. May;15(5):566–575. PubMed PMID: 24671035; PubMed Central PMCID: PMC4210082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chen Z, Liu L, Cheng Q, et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017. March;18(3):495–509. PubMed PMID: 28104734; PubMed Central PMCID: PMC5331199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hamacher-Brady A, Brady NR. Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci. 2016. February;73(4):775–795. PubMed PMID: 26611876; PubMed Central PMCID: PMC4735260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Shirane-Kitsuji M, Nakayama KI. Mitochondria: FKBP38 and mitochondrial degradation. Int J Biochem Cell Biol. 2014. June;51:19–22. PubMed PMID: 24657651; eng. [DOI] [PubMed] [Google Scholar]
- [55].Quinsay MN, Thomas RL, Lee Y, et al. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy. 2014;6(7):855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tracy K, Dibling BC, Spike BT, et al. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol. 2007. September;27(17):6229–6242. PubMed PMID: 17576813; PubMed Central PMCID: PMC1952167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009. July;16(7):939–946. PubMed PMID: 19229244; PubMed Central PMCID: PMC2768230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Shi RY, Zhu SH, Li V, et al. BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci Ther. 2014. December;20(12):1045–1055. PubMed PMID: 25230377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhu Y, Massen S, Terenzio M, et al. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem. 2013. January 11;288(2):1099–1113. PubMed PMID: 23209295; PubMed Central PMCID: PMC3542995. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Schweers RL, Zhang J, Randall MS, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA. 2007. December 4;104(49):19500–19505. PubMed PMID: 18048346; PubMed Central PMCID: PMC2148318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Esteban-Martinez L, Sierra-Filardi E, McGreal RS, et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. Embo J. 2017. June 14;36(12):1688–1706. PubMed PMID: 28465321; PubMed Central PMCID: PMC5470043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Li W, Zhang X, Zhuang H, et al. MicroRNA-137 is a novel hypoxia-responsive microRNA that inhibits mitophagy via regulation of two mitophagy receptors FUNDC1 and NIX. J Biol Chem. 2014. April 11;289(15):10691–10701. PubMed PMID: 24573672; PubMed Central PMCID: PMC4036186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Melser S, Chatelain EH, Lavie J, et al. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab. 2013. May 7;17(5):719–730. PubMed PMID: 23602449. [DOI] [PubMed] [Google Scholar]
- [64].Ding WX, Ni HM, Li M, et al. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010. September 3;285(36):27879–27890. PubMed PMID: 20573959; PubMed Central PMCID: PMC2934655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Otsu K, Murakawa T, Yamaguchi O. BCL2L13 is a mammalian homolog of the yeast mitophagy receptor Atg32. Autophagy. 2015;11(10):1932–1933. PubMed PMID: 26506896; PubMed Central PMCID: PMC4824574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013. October;15(10):1197–1205. PubMed PMID: 24036476; PubMed Central PMCID: PMC3806088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Sentelle RD, Senkal CE, Jiang W, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012. October;8(10):831–838. PubMed PMID: 22922758; PubMed Central PMCID: PMC3689583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Honda S, Arakawa S, Nishida Y, et al. Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat Commun. 2014. June 4;5:4004. PubMed PMID: 24895007. [DOI] [PubMed] [Google Scholar]
- [69].Nishida Y, Arakawa S, Fujitani K, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009. October 1;461(7264):654–658. PubMed PMID: 19794493. [DOI] [PubMed] [Google Scholar]
- [70].Hirota Y, Yamashita S, Kurihara Y, et al. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy. 2015;11(2):332–343. PubMed PMID: 25831013; PubMed Central PMCID: PMC4502654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zhang J, Loyd MR, Randall MS, et al. A short linear motif in BNIP3L (NIX) mediates mitochondrial clearance in reticulocytes. Autophagy. 2012. September;8(9):1325–1332. PubMed PMID: 22906961; PubMed Central PMCID: PMC3442879. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. PubMed PMID: 19302047. [DOI] [PubMed] [Google Scholar]
- [73].Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. 2010. February;10(2):89–102. PubMed PMID: 20081871 [DOI] [PubMed] [Google Scholar]
- [74].Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013. December 12;39(6):1003–1018. PubMed PMID: 24332029; PubMed Central PMCID: PMC3933951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zhou R, Yazdi AS, Menu P, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011. January 13;469(7329):221–225. PubMed PMID: 21124315. [DOI] [PubMed] [Google Scholar]
- [76].Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011. March;12(3):222–230. PubMed PMID: 21151103; PubMed Central PMCID: PMCPmc3079381. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008. November 13;456(7219):264–268. PubMed PMID: 18849965. [DOI] [PubMed] [Google Scholar]
- [78].Harris J, Hartman M, Roche C, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011. March 18;286(11):9587–9597. PubMed PMID: 21228274; PubMed Central PMCID: PMC3058966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016. February 25;164(5):896–910. PubMed PMID: 26919428; PubMed Central PMCID: PMC4769378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Li W, Li Y, Siraj S, et al. FUN14 domain-containing 1-mediated mitophagy suppresses hepatocarcinogenesis by inhibition of inflammasome activation in mice. Hepatology. 2019. February;69(2):604–621. PubMed PMID: 30053328. [DOI] [PubMed] [Google Scholar]
- [81].Lupfer C, Thomas PG, Anand PK, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol. 2013. May;14(5):480–488. PubMed PMID: 23525089; PubMed Central PMCID: PMC3631456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Sliter DA, Martinez J, Hao L, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018. September;561(7722):258–262. PubMed PMID: 30135585; eng. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [83].Li H, Miao W, Ma J, et al. Acute exercise-induced mitochondrial stress triggers an inflammatory response in the myocardium via NLRP3 inflammasome activation with mitophagy. Oxid Med Cell Longev. 2016;2016:1987149. PubMed PMID: 26770647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Guarda G, Braun M, Staehli F, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011. February 25;34(2):213–223. PubMed PMID: 21349431; eng. [DOI] [PubMed] [Google Scholar]
- [85].Reboldi A, Dang EV, McDonald JG, et al. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science. 2014. August 8;345(6197):679–684. PubMed PMID: 25104388; PubMed Central PMCID: PMCPmc4289637. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Shi CS, Shenderov K, Huang NN, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012. January 29;13(3):255–263. PubMed PMID: 22286270; PubMed Central PMCID: PMCPmc4116819. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Jabir MS, Hopkins L, Ritchie ND, et al. Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy. 2015;11(1):166–182. PubMed PMID: 25700738; PubMed Central PMCID: PMCPmc4502769. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Liu L, Yang M, Kang R, et al. HMGB1-DNA complex-induced autophagy limits AIM2 inflammasome activation through RAGE. Biochem Biophys Res Commun. 2014. July 18;450(1):851–856. PubMed PMID: 24971542; PubMed Central PMCID: PMCPmc4107148. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Liu Y, Yan W, Tohme S, et al. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J Hepatol. 2015. July;63(1):114–121. PubMed PMID: 25681553; PubMed Central PMCID: PMCPmc4475488. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Castillo EF, Dekonenko A, Arko-Mensah J, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A. 2012. November 13;109(46):E3168–76. PubMed PMID: 23093667; PubMed Central PMCID: PMCPmc3503152. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014. January;14(1):36–49. PubMed PMID: 24362405; PubMed Central PMCID: PMC4084561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Tal MC, Sasai M, Lee HK, et al. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci USA. 2009. February 24;106(8):2770–2775. PubMed PMID: 19196953; PubMed Central PMCID: PMC2650341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Xia M, Gonzalez P, Li C, et al. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J Virol. 2014. May;88(9):5152–5164. PubMed PMID: 24574393; PubMed Central PMCID: PMC3993837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Ding B, Zhang L, Li Z, et al. The matrix protein of human parainfluenza virus type 3 induces mitophagy that suppresses interferon responses. Cell Host Microbe. 2017. April 12;21(4):538–547 e4. PubMed PMID: 28407488. [DOI] [PubMed] [Google Scholar]
- [95].Seth RB, Sun L, Ea CK, et al. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005. September 9;122(5):669–682. PubMed PMID: 16125763. [DOI] [PubMed] [Google Scholar]
- [96].Qi N, Shi Y, Zhang R, et al. Multiple truncated isoforms of MAVS prevent its spontaneous aggregation in antiviral innate immune signalling. Nat Commun. 2017. June 13;8:15676. PubMed PMID: 28607490; PubMed Central PMCID: PMC5474743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Thomsen MK, Nandakumar R, Stadler D, et al. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology. 2016. September;64(3):746–759. PubMed PMID: 27312012; eng. [DOI] [PubMed] [Google Scholar]
- [98].Wang Y, Ning X, Gao P, et al. Inflammasome activation triggers caspase-1-mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity. 2017. March 21;46(3):393–404. PubMed PMID: 28314590; eng. [DOI] [PubMed] [Google Scholar]
- [99].Lee JP, Foote A, Fan H, et al. Loss of autophagy enhances MIF/macrophage migration inhibitory factor release by macrophages. Autophagy. 2016. June 2;12(6):907–916. PubMed PMID: 27163877; PubMed Central PMCID: PMC4922441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Oka T, Hikoso S, Yamaguchi O, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012. May 10;485(7397):251–255. PubMed PMID: 22535248; PubMed Central PMCID: PMC3378041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Peral de Castro C, Jones SA, Ni Cheallaigh C, et al. Autophagy regulates IL-23 secretion and innate T cell responses through effects on IL-1 secretion. J Immunol. 2012. October 15;189(8):4144–4153. PubMed PMID: 22972933. [DOI] [PubMed] [Google Scholar]
- [102].Mills KH, Dungan LS, Jones SA, et al. The role of inflammasome-derived IL-1 in driving IL-17 responses. J Leukoc Biol. 2013. April;93(4):489–497. PubMed PMID: 23271701. [DOI] [PubMed] [Google Scholar]
- [103].Reed M, Morris SH, Owczarczyk AB, et al. Deficiency of autophagy protein Map1-LC3b mediates IL-17-dependent lung pathology during respiratory viral infection via ER stress-associated IL-1. Mucosal Immunol. 2015. September;8(5):1118–1130. PubMed PMID: 25669150; PubMed Central PMCID: PMC4532659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Bell C, English L, Boulais J, et al. Quantitative proteomics reveals the induction of mitophagy in tumor necrosis factor-alpha-activated (TNFalpha) macrophages. Mol Cell Proteomics. 2013. September;12(9):2394–2407. PubMed PMID: 23674617; PubMed Central PMCID: PMC3769319. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Matheoud D, Sugiura A, Bellemare-Pelletier A, et al. Parkinson‘s disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell. 2016. July 14;166(2):314–327. PubMed PMID: 27345367. [DOI] [PubMed] [Google Scholar]
- [106].Baden P, Deleidi M. Mitochondrial antigen presentation: a vacuolar path to autoimmunity in Parkinson‘s disease. Trends Immunol. 2016. November;37(11):719–721. PubMed PMID: 27638127 [DOI] [PubMed] [Google Scholar]
- [107].Salio M, Puleston DJ, Mathan TS, et al. Essential role for autophagy during invariant NKT cell development. Proc Natl Acad Sci U S A. 2014. December 30;111(52):E5678–87. PubMed PMID: 25512546; PubMed Central PMCID: PMC4284579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Pua HH, Guo J, Komatsu M, et al. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. 2009. April 1;182(7):4046–4055. PubMed PMID: 19299702. [DOI] [PubMed] [Google Scholar]
- [109].Willinger T, Flavell RA. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc Natl Acad Sci USA. 2012. May 29;109(22):8670–8675. PubMed PMID: 22592798; PubMed Central PMCID: PMC3365213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Parekh VV, Wu L, Boyd KL, et al. Impaired autophagy, defective T cell homeostasis, and a wasting syndrome in mice with a T cell-specific deletion of Vps34. J Immunol. 2013;190(10):5086–5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Stephenson LM, Miller BC, Ng A, et al. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy. 2009. July;5(5):625–635. PubMed PMID: 19276668; PubMed Central PMCID: PMC3737142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Jia W, Pua HH, Li QJ, et al. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol. 2011. February 1;186(3):1564–1574. PubMed PMID: 21191072; PubMed Central PMCID: PMC3285458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011. August 26;333(6046):1109–1112. PubMed PMID: 21868666; PubMed Central PMCID: PMC3405151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Corrado M, Campello S. Autophagy inhibition and mitochondrial remodeling join forces to amplify apoptosis in activation-induced cell death. Autophagy. 2016. December;12(12):2496–2497. PubMed PMID: 27629285; PubMed Central PMCID: PMC5173281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].O‘Sullivan TE, Johnson LR, Kang HH, et al. BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer cell memory. Immunity. 2015. August 18;43(2):331–342. PubMed PMID: 26253785; PubMed Central PMCID: PMC5737626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Larson-Casey JL, Deshane JS, Ryan AJ, et al. Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity. 2016. March 15;44(3):582–596. PubMed PMID: 26921108; PubMed Central PMCID: PMC4794358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Xu X, Araki K, Li S, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol. 2014. December;15(12):1152–1161. PubMed PMID: 25362489; PubMed Central PMCID: PMC4232981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Puleston DJ, Zhang H, Powell TJ, et al. Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife. 2014. November 11;3(3). PubMed PMID: 25385531; PubMed Central PMCID: PMC4225493. DOI: 10.7554/eLife.03706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Chen M, Hong MJ, Sun H, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med. 2014. May;20(5):503–510. PubMed PMID: 24747745; PubMed Central PMCID: PMC4066663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Bronietzki AW, Schuster M, Schmitz I. Autophagy in T-cell development, activation and differentiation. Immunol Cell Biol. 2015. January;93(1):25–34. .PubMed PMID: 25287445 [DOI] [PubMed] [Google Scholar]
- [121].Riffelmacher T, Richter FC, Simon AK. Autophagy dictates metabolism and differentiation of inflammatory immune cells. Autophagy. 2018;14(2):199–206. PubMed PMID: 28806133; PubMed Central PMCID: PMC5902226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Ip WKE, Hoshi N, Shouval DS, et al. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017. May 5;356(6337):513–519. PubMed PMID: 28473584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Patel J. IL-10 reprogramming of metabolism in macrophages through mitophagy. Cardiovasc Res. 2017. September 1;113(11):e40–e41. PubMed PMID: 28859306. [DOI] [PubMed] [Google Scholar]
- [124].Zhao Y, Guo Y, Jiang Y, et al. Mitophagy regulates macrophage phenotype in diabetic nephropathy rats. Biochem Biophys Res Commun. 2017. December 9;494(1–2):42–50. PubMed PMID: 29061302. [DOI] [PubMed] [Google Scholar]
- [125].Van Den Bossche J, Baardman J, Otto NA, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. 2016. October 11;17(3):684–696. PubMed PMID: 27732846. [DOI] [PubMed] [Google Scholar]
- [126].de Souza HS, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. 2016. January;13(1):13–27. PubMed PMID: 26627550 [DOI] [PubMed] [Google Scholar]
- [127].Kabi A, Nickerson KP, Homer CR, et al. Digesting the genetics of inflammatory bowel disease: insights from studies of autophagy risk genes. Inflamm Bowel Dis. 2012. April;18(4):782–792. PubMed PMID: 21936032; PubMed Central PMCID: PMC3245781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Liu B, Gulati AS, Cantillana V, et al. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2013. October 15;305(8):G573–84. PubMed PMID: 23989005; PubMed Central PMCID: PMC3798734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Zhang H, Zheng L, McGovern DP, et al. Myeloid ATG16L1 facilitates host-bacteria interactions in maintaining intestinal homeostasis. J Immunol. 2017. March 1;198(5):2133–2146. PubMed PMID: 28130498; PubMed Central PMCID: PMC5322190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Zhu Y, Wang C, Yu M, et al. ULK1 and JNK are involved in mitophagy incurred by LRRK2 G2019S expression. Protein Cell. 2013. September;4(9):711–721. PubMed PMID: 27023913; PubMed Central PMCID: PMC4875534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Hui KY, Fernandez-Hernandez H, Hu J, et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn‘s disease and Parkinson‘s disease. Sci Transl Med. 2018. January 10;10(423):eaai7795. PubMed PMID: 29321258; PubMed Central PMCID: PMC6028002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Liu Z, Lenardo MJ. The role of LRRK2 in inflammatory bowel disease. Cell Res. 2012. July;22(7):1092–1094. PubMed PMID: 22430149; PubMed Central PMCID: PMC3391018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Liu TC, Naito T, Liu Z, et al. LRRK2 but not ATG16L1 is associated with Paneth cell defect in Japanese Crohn‘s disease patients. JCI Insight. 2017. March 23;2(6):e91917. PubMed PMID: 28352666; PubMed Central PMCID: PMC5358495. Corporation during the conduct of the study. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Hsieh CH, Shaltouki A, Gonzalez AE, et al. Functional impairment in Miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson‘s disease. Cell Stem Cell. 2016. December 1;19(6):709–724. PubMed PMID: 27618216; PubMed Central PMCID: PMC5135570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Anderson CA, Boucher G, Lees CW, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011. March;43(3):246–252. PubMed PMID: 21297633; PubMed Central PMCID: PMC3084597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Gan-Or Z, Dion PA, Rouleau GA. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy. 2015;11(9):1443–1457. PubMed PMID: 26207393; PubMed Central PMCID: PMC4590678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Lassen KG, Xavier RJ. Genetic control of autophagy underlies pathogenesis of inflammatory bowel disease. Mucosal Immunol. 2017. May;10(3):589–597. PubMed PMID: 28327616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Franke A, Balschun T, Sina C, et al. Genome-wide association study for ulcerative colitis identifies risk loci at 7q22 and 22q13 (IL17REL). Nat Genet. 2010. April;42(4):292–294. PubMed PMID: 20228798. [DOI] [PubMed] [Google Scholar]
- [139].Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012. November 1;491(7422):119–124. PubMed PMID: 23128233; PubMed Central PMCID: PMC3491803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Lassen KG, Kuballa P, Conway KL, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A. 2014. May 27;111(21):7741–7746. PubMed PMID: 24821797; PubMed Central PMCID: PMC4040621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Guo W, Sun Y, Liu W, et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy. 2014. June;10(6):972–985. PubMed PMID: 24879148; PubMed Central PMCID: PMC4091180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Georges M. The long and winding road from correlation to causation. Nat Genet. 2011. March;43(3):180–181. PubMed PMID: 21350497 [DOI] [PubMed] [Google Scholar]
- [143].Singh SB, Ornatowski W, Vergne I, et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol. 2010. December;12(12):1154–1165. PubMed PMID: 21102437; PubMed Central PMCID: PMC2996476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Kathiria AS, Butcher LD, Feagins LA, et al. Prohibitin 1 modulates mitochondrial stress-related autophagy in human colonic epithelial cells. PLoS One. 2012;7(2):e31231. PubMed PMID: 22363587; PubMed Central PMCID: PMC3281932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Panopoulos M, Han J, Theiss AL. Tu1848 Prohibitin deficiency leads to defective mitochondrial clearance by mitophagy in intestinal epithelial cells. Gastroenterology. 2015;148(4):S-918–S-919. [Google Scholar]
- [146].Artal-Sanz M, Tavernarakis N. Prohibitin and mitochondrial biology. Trends Endocrinol Metab. 2009. October;20(8):394–401. PubMed PMID: 19733482 [DOI] [PubMed] [Google Scholar]
- [147].Murphy G, Lisnevskaia L, Isenberg D. Systemic lupus erythematosus and other autoimmune rheumatic diseases: challenges to treatment. Lancet. 2013;382(9894):809–818. [DOI] [PubMed] [Google Scholar]
- [148].Pierdominici M, Vomero M, Barbati C, et al. Role of autophagy in immunity and autoimmunity, with a special focus on systemic lupus erythematosus. Faseb J. 2012. April;26(4):1400–1412. PubMed PMID: 22247332. [DOI] [PubMed] [Google Scholar]
- [149].Zhou XJ, Lu XL, Lv JC, et al. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis. 2011. July;70(7):1330–1337. PubMed PMID: 21622776. [DOI] [PubMed] [Google Scholar]
- [150].Leishangthem BD, Sharma A, Bhatnagar A. Role of altered mitochondria functions in the pathogenesis of systemic lupus erythematosus. Lupus. 2016. March;25(3):272–281. PubMed PMID: 26385216 [DOI] [PubMed] [Google Scholar]
- [151].Caza TN, Talaber G, Perl A. Metabolic regulation of organelle homeostasis in lupus T cells. Clin Immunol. 2012. September;144(3):200–213. PubMed PMID: 22836085; PubMed Central PMCID: PMC3423541. eng [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Lee HT, Wu TH, Lin CS, et al. The pathogenesis of systemic lupus erythematosus - From the viewpoint of oxidative stress and mitochondrial dysfunction. Mitochondrion. 2016. September;30:1–7. PubMed PMID: 27235747. [DOI] [PubMed] [Google Scholar]
- [153].Perl A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat Rev Rheumatol. 2013. November;9(11):674–686. PubMed PMID: 24100461; PubMed Central PMCID: PMC4046645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Caza TN, Fernandez DR, Talaber G, et al. HRES-1/Rab4-mediated depletion of Drp1 impairs mitochondrial homeostasis and represents a target for treatment in SLE. Ann Rheum Dis. 2014. October;73(10):1888–1897. PubMed PMID: 23897774; PubMed Central PMCID: PMC4047212. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Alessandri C, Barbati C, Vacirca D, et al. T lymphocytes from patients with systemic lupus erythematosus are resistant to induction of autophagy. Faseb J. 2012. November;26(11):4722–4732. PubMed PMID: 22835828; PubMed Central PMCID: PMC3475261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Oaks Z, Winans T, Caza T, et al. Mitochondrial dysfunction in the liver and antiphospholipid antibody production precede disease onset and respond to rapamycin in lupus-prone mice. Arthritis Rheumatol. 2016. November;68(11):2728–2739. PubMed PMID: 27332042; PubMed Central PMCID: PMC5083168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Caielli S, Athale S, Domic B, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. 2016. May 2;213(5):697–713. PubMed PMID: 27091841; PubMed Central PMCID: PMC4854735. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Konig MF, Andrade F. A critical reappraisal of neutrophil extracellular traps and NETosis mimics based on differential requirements for protein citrullination. Front Immunol. 2016;7(7):461. PubMed PMID: 27867381; PubMed Central PMCID: PMC5095114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Unuma K, Aki T, Funakoshi T, et al. Extrusion of mitochondrial contents from lipopolysaccharide-stimulated cells: involvement of autophagy. Autophagy. 2015;11(9):1520–1536. PubMed PMID: 26102061; PubMed Central PMCID: PMC4590602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Hirschfield GM, Gershwin ME. The immunobiology and pathophysiology of primary biliary cirrhosis. Annu Rev Pathol. 2013. January 24;8(8):303–330. PubMed PMID: 23347352. [DOI] [PubMed] [Google Scholar]
- [161].Berg PA. The role of the innate immune recognition system in the pathogenesis of primary biliary cirrhosis: a conceptual view. Liver Int. 2011. August;31(7):920–931. PubMed PMID: 21733082 [DOI] [PubMed] [Google Scholar]
- [162].Sasaki M, Miyakoshi M, Sato Y, et al. Increased expression of mitochondrial proteins associated with autophagy in biliary epithelial lesions in primary biliary cirrhosis. Liver Int. 2013. February;33(2):312–320. PubMed PMID: 23231002. [DOI] [PubMed] [Google Scholar]
- [163].van de Graaf S, Beuers U. Autophagy - another piece of the puzzle towards understanding primary biliary cirrhosis?. Liver Int. 2014. April;34(4):481–483. PubMed PMID: 24612169 [DOI] [PubMed] [Google Scholar]
- [164].van Kempen TS, Wenink MH, Leijten EF, et al. Perception of self: distinguishing autoimmunity from autoinflammation. Nat Rev Rheumatol. 2015. August;11(8):483–492. PubMed PMID: 25963881. [DOI] [PubMed] [Google Scholar]
- [165].Jin G, Xu C, Zhang X, et al. Atad3a suppresses Pink1-dependent mitophagy to maintain homeostasis of hematopoietic progenitor cells. Nat Immunol. 2018. January;19(1):29–40. PubMed PMID: 29242539; PubMed Central PMCID: PMC5905408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].East DA, Fagiani F, Crosby J, et al. PMI: a DeltaPsim independent pharmacological regulator of mitophagy. Chem Biol. 2014. November 20;21(11):1585–1596. PubMed PMID: 25455860; PubMed Central PMCID: PMC4245710. [DOI] [PMC free article] [PubMed] [Google Scholar]