

ABSTRACT
UBQLNs (ubiquilins) are highly conserved proteins across species, characterized by interactions with proteasomes and ubiquitinated proteins via UBA and UBL domains, respectively. The role of UBQLNs as chaperone proteins of the ubiquitin-proteasome system (UPS) is well-defined; however, the connections between UBQLNs and autophagy remain unclear. A recent study published in Nature Cell Biology from Dr. Hugo J. Bellen’s lab showed a novel role of UBQLNs in macroautophagy/autophagy regulation through v-ATPase-MTOR signaling using Drosophila and mammalian neuronal cells. Notably, the highlighted article also investigated the autophagy phenotype of a common amyotrophic lateral sclerosis (ALS)-associated mutation in the gene encoding UBQLN2, demonstrating the contribution of abnormal v-ATPase-MTOR-mediated autophagy in ALS pathogenesis.
KEYWORDS: Lysosome, macroautophagy, MTOR, stress, v-ATPase
UBQLNs are known as chaperone proteins of the UPS that function independent of autophagy [1]. UBQLNs physically interact with proteasomes and ubiquitinated protein aggregates via an N-terminal UBL domain and C-terminal UBA domain, respectively, shuttling misfolded proteins to the proteasome [2]. Although UBQLNs are shown to interact with LC3 in human cells, it remains unclear how they regulate autophagy [3]. Şentürk et al. [4] recently showed that loss of Ubqn/UBQLN leads to upregulated autophagy induction and impaired autophagy flux. They also determined that Ubqn/UBQLN is required to maintain Torc1/MTORC1 activity and lysosomal acidification. Ubqn/UBQLN interacts with the Vha100-1/V0a/V100 subunit of v-ATPase and promotes stable v-ATPase formation.
UBQLN proteins have been studied mostly in the field of neurodegenerative disorders. Mutations in the genes encoding UBQLNs, particularly UBQLN2, have been implicated in the pathogenic mechanism of ALS and frontotemporal lobar degeneration [5]. Consequently, the authors first explored the neuronal phenotype of a Drosophila ubqn mutant. The authors determined that loss of Ubqn in Drosophila results in severe neuronal and glial degeneration by monitoring neuropil and ultrastructure of the fly eye via hematoxylin and eosin staining and transmission electron microscopy, respectively. They also showed age-dependent phototransduction defects via electroretinogram assays.
In both Drosophila and human neuronal cells, Şentürk et al. determined that Ubqn/UBQLNs loss leads to decreased Tor/MTOR signaling by detecting diminished phosphorylation of Akt1/AKT and S6k/RPS6KB kinase. MTOR signaling negatively regulates autophagy. Thus, the authors further assessed autophagy induction and flux separately. Not surprisingly, an increased number of Atg1-GFP and GFP-Atg8 puncta were observed in Drosophila suggesting enhanced autophagy induction. Fly transmission electron microscopy data showing increased numbers of autophagosomes, amphisomes, autolysosomes, and lysosomes, as well as an LC3 lipidation assay, further confirmed the highly induced autophagy in Ubqn/UBQLNs-deficient Drosophila and mammalian cells.
Şentürk et al. next explored whether autophagy flux is enhanced or abolished through measuring ref(2)P/SQSTM1, a selective autophagy receptor closely correlated with ALS and frontotemporal lobar degeneration [6]. The authors noted an accumulation of ref(2)P/SQSTM1 protein associated with the loss of Ubqn/UBQLNs in both the fly model and human cells. Furthermore, using an mCherry-GFP-Atg8 assay in fly ubqn mutants, the authors showed that the number of acidic puncta is greatly reduced, whereas the number of nonacidic puncta is prominently increased, illustrating impaired autophagic vesicle acidification and defective autophagy flux.
Consistently, abnormal lysosomal morphology (e.g. accumulated and enlarged LAMP1-GFP cellular compartments and aberrant lysosome expansion) as well as compromised lysosomal acidification were observed in the ubqn mutant. Further study in fly demonstrated that Ubqn localizes to the lysosomal surface and physically interacts with Vha100-1, a v-ATPase subunit required for proton translocation, and that loss of Ubqn leads to an accumulation of fragmented Vha100-1. The authors showed that ubqn also genetically interacts with Vha100-1 and ATP6AP2/VhaM8.9, an assembly protein for the v-ATPase. Reduction of the 2 subunits is able to rescue the lysosomal phenotype of the ubqn mutant, including pre-pupal lethality, abnormal lysosomal acidification and impaired autophagy flux [7]. Similarly, the authors demonstrated that UBQLNs in human cells harbor a weak and/or transient binding to ATP6V0A1, the conserved homolog of Vha100-1. Taken together, the results highlighted that Ubqn/UBQLNs modulate v-ATPase activity and lysosome function by regulating the Vha100-1/ATP6V0A1 subunit.
The authors hypothesized that failure in autophagic vesicle acidification by v-ATPase is the primary reason for impaired autophagy degradation. They exploited the poly(DL-lactic-co-glycolic acid) (PLGA) nanoparticles, which are able to deliver acidic properties to lysosomes, to investigate the effects of lysosomal acidification in the ubqn mutant [8]. Not surprisingly, restoration of acidification and elevated autophagic flux was identified in flies feeding on medium containing the acidic nanoparticles.
Last, Şentürk et al. tested the pathological role of Ubqn in ALS development. In Drosophila, the overexpression of ALS-associated mutant UBQLN2P497H results in impaired autophagy degradation, abnormal Vha100-1 level and failure in lysosomal acidification, demonstrating the participation of UBQLN2P497H in autophagy deficits through the v-ATPase-MTOR pathway in ALS.
Overall, the work from the Bellen lab provides novel insight into the mechanism of how Ubqn/UBQLNs mediate the clearance of protein aggregates. So far, two Ubqn/UBQLN-mediated pathways, including the UPS and autophagy, have been revealed to contribute to neuronal maintenance (Figure 1). In the autophagy pathway highlighted here, the novel role of Ubqn/UBQLNs is identified and linked with v-ATPase and MTOR signaling. Moreover, the elucidation of how ALS-associated mutations in UBQLNs impair cellular protein clearance sheds light on our understanding of ALS pathogenesis and may ultimately contribute to therapeutic design in the future.
Figure 1.
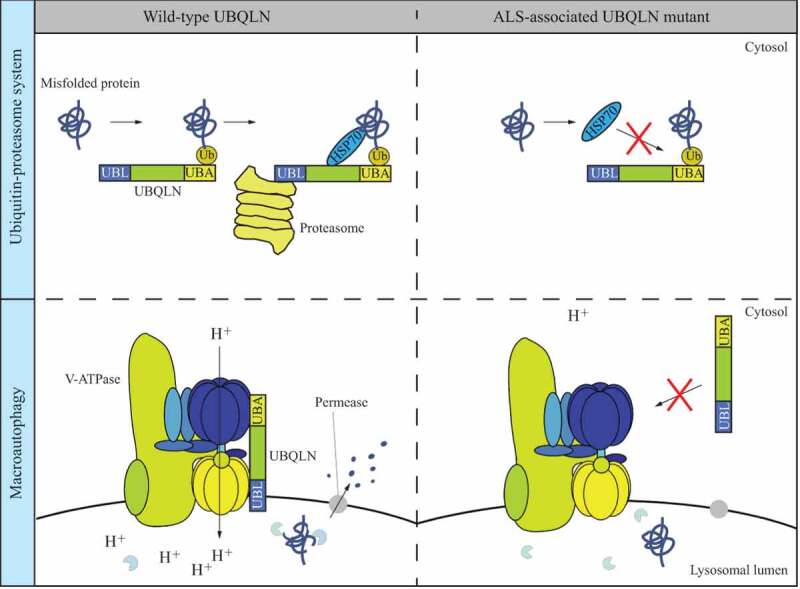
Dual roles of Ubqn/UBQLNs in the UPS and autophagy. In the UPS, UBQLN binds to a polyubiquitinated-misfolded protein, HSP70 and the proteasome via its UBA domain, HSP70-binding domain and UBL domain, respectively, allowing the degradation of the misfolded protein. The common UBQLN mutant found in ALS patients is associated with abnormal HSP70 binding and thus failure in protein degradation through the UPS. Ubqn/UBQLN mediates auto-phagy through the interaction with the v-ATPase. The ALS-associated UBQLN mutant fails to bind to v-ATPase and is less efficient in autophagy degradation in autolysosomes with a feature of elevated lysosomal pH.
Funding Statement
This work was supported by the National Institute of General Medical Sciences [GM131919].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Hjerpe R, Bett JS, Keuss MJ, et al. UBQLN2 mediates autophagy-independent protein aggregate clearance by the proteasome. Cell. 2016;166(4):935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Itakura E, Zavodszky E, Shao S, et al. Ubiquilins chaperone and triage mitochondrial membrane proteins for degradation. Mol Cell. 2016;63(1):21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhang KY, Yang S, Warraich ST, et al. Ubiquilin 2: a component of the ubiquitin–proteasome system with an emerging role in neurodegeneration. Int J Biochem Cell Biol. 2014;50:123–126. [DOI] [PubMed] [Google Scholar]
- [4].Şentürk M, Lin G, Zuo Z, et al. Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat Cell Biol. 2019;21(3):384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Deng H-X, Chen W, Hong S-T, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477(7363):211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Goode A, Butler K, Long J, et al. Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy. 2016;12(7):1094–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hiesinger PR, Fayyazuddin A, Mehta SQ, et al. The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell. 2005;121(4):607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bourdenx M, Daniel J, Genin E, et al. Nanoparticles restore lysosomal acidification defects: implications for Parkinson and other lysosomal-related diseases. Autophagy. 2016;12(3):472–483. [DOI] [PMC free article] [PubMed] [Google Scholar]