

ABSTRACT
Parkinson disease (PD) is a disabling, incurable disorder with increasing prevalence in the western world. In rare cases PD is caused by mutations in the genes for PINK1 (PTEN induced kinase 1) or PRKN (parkin RBR E3 ubiquitin protein ligase), which impair the selective autophagic elimination of damaged mitochondria (mitophagy). Mutations in the gene encoding LRRK2 (leucine rich repeat kinase 2) are the most common monogenic cause of PD. Here, we report that the LRRK2 kinase substrate RAB10 accumulates on depolarized mitochondria in a PINK1- and PRKN-dependent manner. RAB10 binds the autophagy receptor OPTN (optineurin), promotes OPTN accumulation on depolarized mitochondria and facilitates mitophagy. In PD patients with the two most common LRRK2 mutations (G2019S and R1441C), RAB10 phosphorylation at threonine 73 is enhanced, while RAB10 interaction with OPTN, accumulation of RAB10 and OPTN on depolarized mitochondria, depolarization-induced mitophagy and mitochondrial function are all impaired. These defects in LRRK2 mutant patient cells are rescued by LRRK2 knockdown and LRRK2 kinase inhibition. A phosphomimetic RAB10 mutant showed less OPTN interaction and less translocation to depolarized mitochondria than wild-type RAB10, and failed to rescue mitophagy in LRRK2 mutant cells. These data connect LRRK2 with PINK1- and PRKN-mediated mitophagy via its substrate RAB10, and indicate that the pathogenic effects of mutations in LRRK2, PINK1 and PRKN may converge on a common pathway.
Abbreviations : ACTB: actin beta; ATP5F1B: ATP synthase F1 subunit beta; CALCOCO2: calcium binding and coiled-coil domain 2; CCCP: carbonyl cyanide m-chlorophenylhydrazone; Co-IP: co-immunoprecipitation; EBSS: Earle’s balanced salt solution; GFP: green fluorescent protein; HSPD1: heat shock protein family D (Hsp60) member 1; LAMP1: lysosomal associated membrane protein 1; LRRK2: leucine rich repeat kinase 2; IF: immunofluorescence; MAP1LC3B: microtubule associated protein 1 light chain 3 beta; MFN2: mitofusin 2; OMM: outer mitochondrial membrane; OPTN: optineurin; PD: Parkinson disease; PINK1: PTEN induced kinase 1; PRKN: parkin RBR E3 ubiquitin protein ligase; RHOT1: ras homolog family member T1; ROS: reactive oxygen species; TBK1: TANK binding kinase 1; WB: western blot.
KEYWORDS: Autophagy receptor, mitochondria, optineurin, OPTN, PARK2, parkin, Parkinson disease, PINK1, PRKN, selective autophagy
Introduction
Parkinson disease (PD) is a highly prevalent, disabling neurodegenerative disorder for which no cure exists yet [1]. The number of PD patients in the western world is expected to double over the next two decades, presenting a huge medical, social and economic challenge [2].
Approximately 5% of PD cases have a mendelian cause. Mutations in the gene encoding LRRK2 (leucine rich repeat kinase 2) are by far the most common monogenic cause of PD, accounting for ~10% of autosomal dominant familial cases [3]. LRRK2 mutations are also found in 3.6% of apparently sporadic PD cases and have an incomplete, age-dependent penetrance [3]. In addition, genome-wide association studies identified polymorphisms in the LRRK2 locus as a risk factor for sporadic PD, indicating that pathogenic pathways may be shared between familial and sporadic forms [4]. LRRK2 encodes an enzyme with a kinase domain and a GTPase domain [5]. A recent phosphoproteomics study identified RAB10 and several related members of the RAB family as LRRK2 kinase substrates [6]. The RAB family comprises ~70 small GTPases that cycle between an inactive GDP-bound and an active GTP-bound state as well as between the cytosol and membranes [7]. RABs regulate vesicle formation, trafficking and fusion [7]. PD-causing LRRK2 mutations increase phosphorylation of RAB10, which may disturb its membrane-cytosol equilibrium [6]. How this leads to neurodegeneration, is unknown.
Accumulating evidence implicates impairment of mitophagy as a pathogenic mechanism in PD [8]. Mitophagy is a form of selective autophagy in which damaged or superfluous mitochondria are specifically labeled with ubiquitin and taken up by autophagosomes for degradation in lysosomes [8]. The E3 ubiquitin ligase PRKN (parkin RBR E3 ubiquitin protein ligase) and the mitochondrial kinase PINK1 (PTEN induced kinase 1), both encoded by genes linked to autosomal recessive PD, are critically involved in mitophagy of damaged mitochondria [9–12]. PINK1 accumulates on damaged mitochondria and phosphorylates both PRKN and ubiquitin, hereby activating PRKN [13–15]. PRKN-mediated ubiquitination of outer mitochondrial membrane (OMM) proteins in combination with PINK1-mediated ubiquitin phosphorylation triggers recruitment of autophagy receptors, such as OPTN (optineurin), that tether ubiquitinated mitochondria to LC3 on nascent autophagosomes [16–19]. Loss-of-function mutations in PRKN or PINK1 disrupt mitophagy in vivo and in vitro, leading to deficient mitochondrial quality control, accumulation of dysfunctional mitochondria, oxidative stress and apoptosis [8,20]. Other forms of mitophagy are independent of PINK1 and PRKN and are mediated by factors such as FUNDC1 and cardiolipin [21]. Some mitophagy pathways may mediate steady-state mitochondrial clearance, while others are engaged in programmed mitophagy during differentiation or induced by injurious stimuli [21].
Here we show that mitophagy of depolarized mitochondria is impaired in cells of PD patients with LRRK2 mutations. Our findings implicate the LRRK2 substrate RAB10 in mitophagy via an interaction with OPTN and indicate that the pathogenic effects of LRRK2, PRKN and PINK1 mutations converge on a common pathway.
Results
Depolarization-induced mitophagy and mitochondrial function are impaired in patients with LRRK2 mutations
We have previously established experimental conditions to induce mitophagy in cultured primary human skin fibroblasts expressing endogenous PRKN and PINK1 [22]. In these cells mitophagy is abrogated by PD-causing PRKN and PINK1 mutations [22]. Human skin fibroblasts also express endogenous LRRK2 (Figure S1) [23]. We collected fibroblasts from 2 nonrelated PD patients with the G2019S LRRK2 mutation, 1 PD patient with the R1441C LRRK2 mutation and 5 healthy controls (Table 1). G2019S is the most common LRRK2 mutation and maps to the kinase domain [3]. R1441C is the second most frequent mutation and is located in the GTPase domain [3]. We compared fibroblasts from each LRRK2 mutant patient with control fibroblasts from an age-matched subject (Table 1), because donor age can affect autophagic flux in cultured skin fibroblasts [24]. There was no significant difference in LRRK2 protein levels between LRRK2 mutant and control fibroblasts (Figure S1).
Table 1.
Demographic and clinical characteristics of study subjects.
| Subject (ID, gender/age) | Genotype | Ethnic background | Onset age | Comments |
|---|---|---|---|---|
| PD | ||||
| G2019S1, F/39 | G2019S/WT | Moroccan (Berber) | 31 | |
| G2019S2, F/58 | G2019S/WT | Ashkenazi | 50 | Negative for GBA mutations |
| R1441C, M/67 | R1441C/WT | Flemish | 55 | |
| PRKN, F/40 | V3EfsX3/G179LfsX7 | 18 | As described [22] | |
| PINK1, F/68 | W437G/W437G | 38 | As described [22] | |
| Non-manifesting carriers | ||||
| G2019S NM1, F/36 | G2019S/WT | Moroccan (Berber) | NA | |
| G2019S NM2, F/31 | G2019S/WT | Moroccan (Berber) | NA | |
| Controls | ||||
| Control 1, F/40 | WT/WT | Flemish | NA | Control for G2019S1, G2019S NM1 and NM2 |
| Control 2, F/67 | WT/WT | Flemish | NA | Control for R1441C |
| Control 3, M/63 | WT/WT | Ashkenazi | NA | Control for G2019S2 |
| Control 4, F/43 | WT/WT | NA | Control for PRKN | |
| Control 5, F/72 | WT/WT | NA | Control for PINK1 | |
Patient age refers to age at time of biopsy. Onset age refers to age of first motor symptoms of PD. WT, wild-type.
To quantify mitophagy we transfected the fibroblasts with a mitochondrially targeted form of Keima (mito-Keima), a fluorescent protein that is resistant to lysosomal proteases and exhibits pH-dependent excitation [20,25]. The peak of the excitation spectrum of mito-Keima shifts from 440 nm to 586 nm when mitochondria are delivered to acidic lysosomes, which allows live dual-excitation ratiometric quantification of mitophagy [20,25]. Exposure of fibroblasts from healthy controls to the mitochondrial uncoupling agent valinomycin for 48 h resulted in the appearance of punctate structures with high 543 nm:458 nm ratio mito-Keima values that colocalized with the lysosomal dye LysoTracker (Figure 1(a); Figure S2A). Emergence of these high 543:458 ratio puncta was inhibited by the lysosomal inhibitor bafilomycin A1 and by 3-methyladenine, a suppressor of macro-autophagy induction, and was abrogated in fibroblasts from PD patients with PRKN and PINK1 mutations (Figure 1(a-c)). Interestingly, valinomycin-induced mitophagy was consistently impaired in cells from the 2 PD patients with the G2019S mutation and the patient with the R1441C mutation (Figure 1(d,e); Figure S3A,B).
Figure 1.
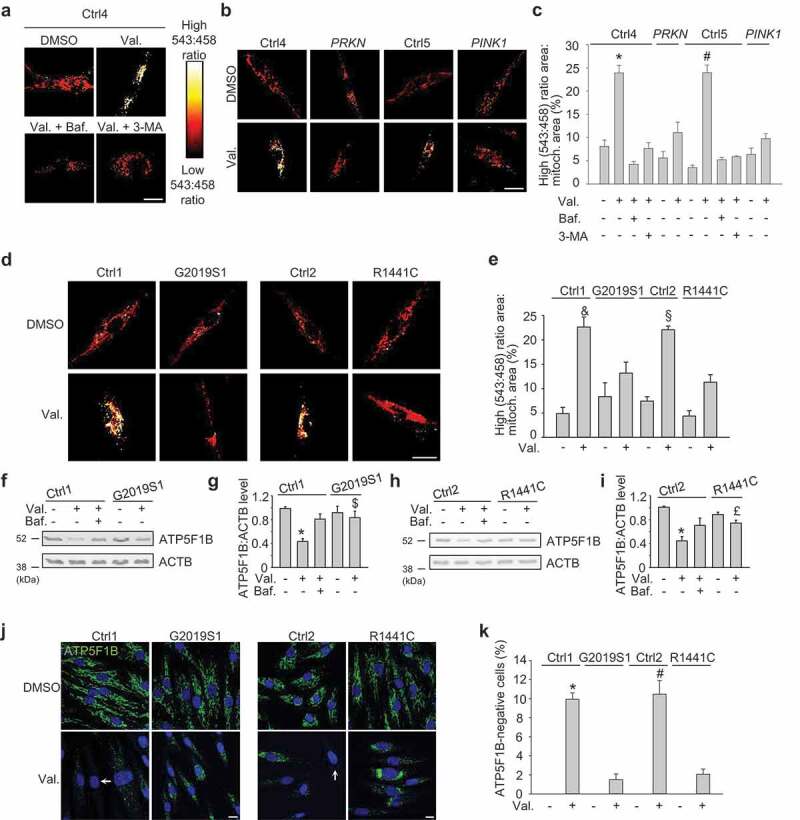
Mitophagy is impaired in fibroblasts from PD patients with LRRK2 mutations. (a-c) Fibroblasts from healthy controls (Ctrl4, Ctrl5), a PD patient with compound heterozygous PRKN mutations and a PD patient with homozygous PINK1 mutations were transfected with mito-Keima and treated for 48 h with DMSO, valinomycin (Val., 1 µM), bafilomycin A1 (Baf., 100 nM) or 3-methyladenine (3-MA, 10 mM) as indicated, followed by live ratiometric imaging. High (543:458) signal corresponds to mito-Keima present in lysosomes. (c) High (543:458) ratio area:total mitochondrial area was quantified as an index of mitophagy (n = 3–6). * P < 0.001 compared with all other conditions in Ctrl4 and PRKN. # P < 0.001 compared with all other conditions in Ctrl5 and PINK1. (d,e) Fibroblasts from a PD patient with the G2019S LRRK2 mutation (G2019S1), a PD patient with the R1441C LRRK2 mutation and age-matched controls (Ctrl1, Ctrl2) were transfected with mito-Keima and treated for 48 h with DMSO or Val. (e) Quantification (n = 4). & P < 0.01 compared with all other Ctrl1 and G2019S1 conditions. § P < 0.01 compared with all other Ctrl2 and R1441C conditions. (f-i) Fibroblasts from controls and PD patients with LRRK2 mutations were treated for 48 h with DMSO, Val. or Baf., as indicated. (f,h) Western blot for the mitochondrial matrix protein ATP5F1B and ACTB. (g,i) Quantification of ATP5F1B:ACTB level (n = 9). * P < 0.001 compared to DMSO in the same subject. $ P < 0.05 compared with the Val. condition in Ctrl1. £ P < 0.05 compared with Ctrl2 Val. (j,k) Immunostaining for ATP5F1B. Nuclei were stained with TOTO-3 (blue). Arrows indicate examples of fibroblasts without detectable ATP5F1B staining. (i) Quantification of the percentage of cells without detectable ATP5F1B (n = 3). * P < 0.001 compared with all other Ctrl1 and G2019S1 conditions. # P < 0.001 compared with all other Ctrl2 and R1441C conditions. Scale bars: 10 μm.
To further validate the mitophagy defect in the LRRK2 mutant fibroblasts, we measured valinomycin-induced clearance of the mitochondrial matrix proteins ATP5F1B (ATP synthase F1 subunit beta) (Figure 1(f-k)) and HSPD1 (heat shock protein family D [Hsp60] member 1) (Figure S3C-F; Figure S4) by western blot and immunostaining. These assays confirmed that valinomycin-induced mitophagy was defective in the G2019S and R1441C mutant cells (Figure 1(f-k); Figure S3C-F; Figure S4). Basal ATP5F1B or HSPD1 levels did not differ between LRRK2 mutant and control fibroblasts (Figure 1(f-i); Figure S3C,D; Figure S4A-D).
We also induced mitophagy with the protonophore carbonyl cyanide m-chlorophenylhydrazone (CCCP) and with a combination of the ATP synthase inhibitor oligomycin and the complex III inhibitor antimycin A [19]. Both treatments induced mitophagy in wild-type fibroblasts but much less so in the G2019S and R1441C mutant cells (Figure S5). Thus, we observed a similar mitophagy defect in the LRRK2 mutant cells with valinomycin, CCCP and combination of oligomycin and antimycin A.
LRRK2 mutations have an incomplete, age-dependent penetrance [3]. We also assessed mitophagy in fibroblasts collected from 2 young G2019S mutation carriers who did not exhibit clinical signs or symptoms of PD (Table 1). Interestingly, mitophagy was also reduced in these non-manifesting LRRK2 mutation carriers (Figure S3G-N). Taken together, impairment of mitophagy appears to be a consistent feature in carriers of the 2 most common PD-causing LRRK2 mutations (Figure S6A). We found no correlation between mitophagy impairment and age at time of skin biopsy (Figure S6A).
To verify that the mitophagy defect of the LRRK2 mutant fibroblasts could be attributed to mutant LRRK2, we knocked down LRRK2 using four different siRNAs (Figure S7). LRRK2 knockdown with each of these siRNAs corrected the mitophagy defect of the G2019S and R1441C mutant fibroblasts (Figure 2(a-f)). Moreover, the LRRK2 kinase inhibitors LRRK2-IN-1 and PF-06447475 both rescued the mitophagy defect of the G2019S and R1441C mutant fibroblasts (FIGURE 2(g-l)), indicating that this defect was mediated by LRRK2 kinase activity. LRRK2 knockdown or LRRK2 kinase inhibitors had no significant effect on valinomycin-induced mitophagy in wild-type fibroblasts, although there was a trend towards increased mitophagy (Figure S8).
Figure 2.
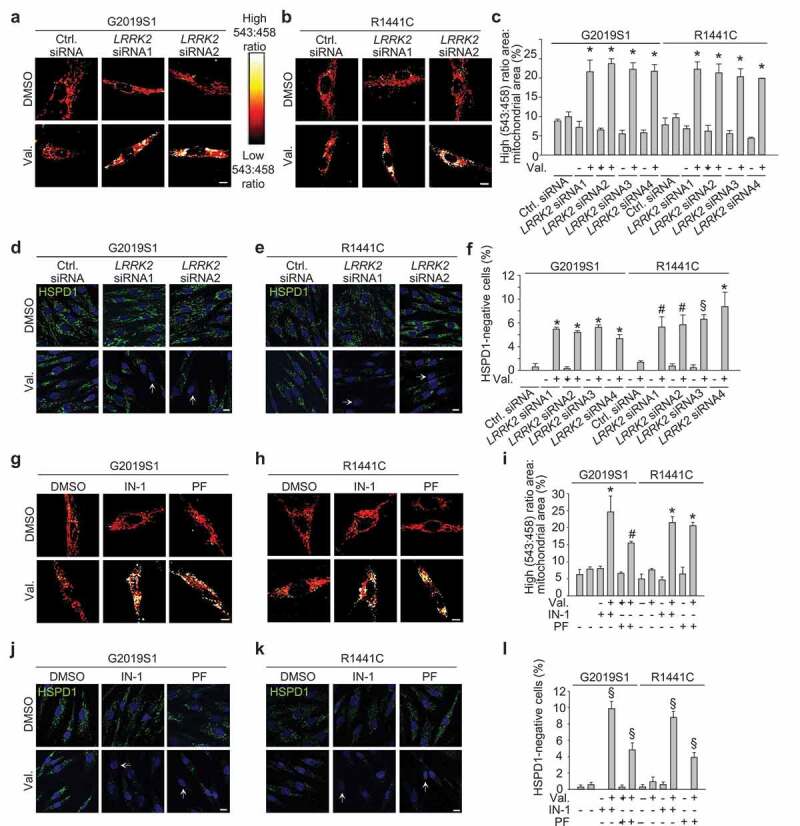
The mitophagy defect of LRRK2 mutant fibroblasts is rescued by LRRK2 knockdown and by LRRK2 kinase inhibitors. (a-c) Fibroblasts from a PD patient with the G2019S LRRK2 mutation (G2019S1) and a PD patient with the R1441C LRRK2 mutation were transfected with mito-Keima and with control siRNA or LRRK2 siRNA1-4. After 72 h cells were treated with DMSO or valinomycin (Val., 1 μM) for 48 h. (c) High (543:458) ratio area:total mitochondrial area was quantified as an index of mitophagy (n = 3). * P < 0.001 compared with control siRNA after Val. in the same subject. (d-f) G2019S and R1441C mutant fibroblasts were transfected with control siRNA or LRRK2 siRNA1-4. After 72 h cells were treated with DMSO or Val. for 48 h and immunostained for HSPD1. Nuclei were stained with TOTO-3 (blue). Arrows in (d) and (e) indicate cells without mitochondria. (f) Quantification of the percentage of cells without HSPD1 staining (n = 3). * P < 0.001, § P < 0.01 and # P < 0.05 compared with Val. in control siRNA in the same subject. (g-i) Mito-Keima-transfected LRRK2 mutant fibroblasts were pre-treated for 24 h with DMSO, LRRK2-IN-1 (1 µM) or PF-06447475 (0.5 µM) and then exposed for 48 h to DMSO or Val. in the continued presence or absence of the LRRK2 kinase inhibitors, followed by live ratiometric imaging. (i) Quantification (n = 4). * P < 0.001 and # P < 0.05 compared with Val. alone in the same subject. (j-l) LRRK2 mutant fibroblasts were pre-treated for 24 h with DMSO, LRRK2-IN-1 (IN-1, 1 µM) or PF-06447475 (PF, 0.5 µM) and then exposed for 48 h to DMSO or Val. in the continued presence or absence of the LRRK2 kinase inhibitors, followed by HSPD1 immunostaining. Arrows in (j) and (k) indicate cells without detectable HSPD1 staining. (l) Quantification (n = 3). § P < 0.01 compared with Val. alone in the same subject. Scale bars: 10 µm.
Mitophagy contributes to mitochondrial quality control. As expected in cells with impaired mitophagy, mitochondrial quality in G2019S and R1441C mutant fibroblasts was impaired as evidenced by reduced average mitochondrial membrane potential and increased mitochondrial production of reactive oxygen species (ROS) (Figure S9A,B). These mitochondrial defects were also rescued by LRRK2 kinase inhibition (Figure S9C,D).
Non-selective autophagy is preserved in patients with LRRK2 mutations
In addition to selective forms of autophagy such as mitophagy, cells have the capacity for non-selective (bulk) autophagy in which cytosolic components and organelles are degraded indiscriminately via mechanisms that are independent of autophagy receptors [26]. A classical trigger for non-selective autophagy is amino acid starvation. Surprisingly, starvation-induced autophagy, as measured by lipidated MAP1LC3B (LC3-II):ACTB (actin beta) ratios on western blot, was preserved in cells from the 2 G2019S patients and the R1441C patient compared with healthy controls (Figure 3(a,b)). This is consistent with previous work showing intact starvation-induced autophagy in LRRK2 mutant patient fibroblasts as assessed by LC3-II western blot relative to ACTB or GAPDH [27,28].
Figure 3.
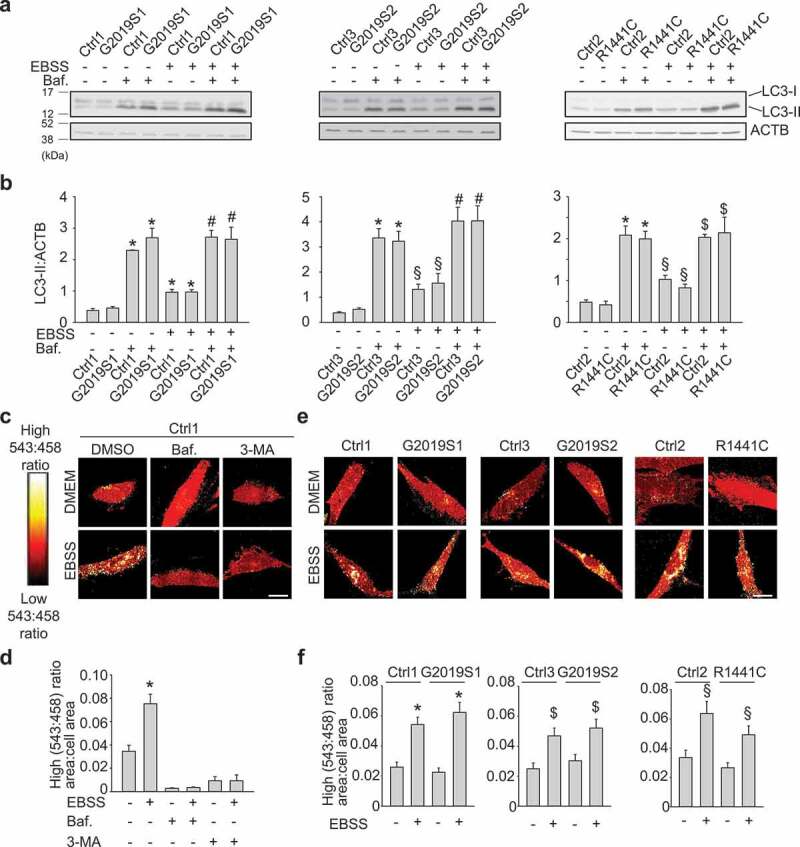
Non-selective autophagy is preserved in PD patients with LRRK2 mutations. (a,b) Fibroblasts from 2 PD patients with the G2019S mutation (G2019S1, G2019S2), a PD patient with the R1441C LRRK2 mutation and 3 controls (Ctrl1, Ctrl2, Ctrl3) were incubated either in Earle’s balanced salt solution (EBSS) for 4 h to trigger starvation-induced autophagy, or in standard culture medium (DMEM), in the presence or absence of bafilomycin A1 (Baf., 100 nM). (a) Western blot showing LC3-I, LC3-II and ACTB levels. (b) Quantification of LC3-II:ACTB (n = 3–8). * P < 0.001 and § P < 0.05 compared with DMEM without Baf. in the same subject. # P < 0.001 and $ P < 0.01 compared with EBSS without Baf. in the same subject. (c,d) Fibroblasts from Ctrl1 were transfected with Keima and incubated for 4 h in DMEM or EBSS in the presence or absence of Baf. (100 nM) and 3-methyladenine (3-MA, 10 mM), as indicated, followed by ratiometric live imaging. High (543:458) signal corresponds to Keima present inside lysosomes. (d) Quantification (n = 3). * P < 0.001 compared with all other conditions. (e-f) Control and LRRK2 mutant fibroblasts were transfected with Keima and incubated in DMEM or EBSS for 4 h, followed by ratiometric live imaging. (f) High (543:458) ratio area:total cell area was quantified as an index of autophagy (n = 4). * P < 0.001, $ P < 0.01 and § P < 0.05 compared with DMEM in the same subject. Scale bars: 10 μm.
As an additional assay for non-selective autophagy, we transfected fibroblasts with Keima, i.e. the same probe as used for mitophagy imaging (Figure 1(a-e)) except that it lacked the mitochondrial targeting sequence [25]. Starvation of control cells induced high 543:458 ratio Keima puncta that colocalized with LysoTracker (Figure S2B) and were suppressed by treatment with bafilomycin A1 and 3-methyladenine (Figure 3(c,d)). Strikingly, Keima imaging showed intact starvation-induced autophagy in G2019S and R1441C LRRK2 mutant patients (Figure 3(e,f)), in contrast to the mitophagy defect observed with mito-Keima imaging (Figure 1(d,e); Figure S3A,B). Taken together, these data show that fibroblasts from patients with G2019S and R1441C mutations have impaired mitophagy, but preserved non-selective autophagy.
OPTN recruitment to depolarized mitochondria is impaired in patients with LRRK2 mutations
Once autophagosomes are completely closed around their cargo, they are probably processed similarly along their path to fusion with lysosomes irrespective of whether they have been formed by selective or non-selective autophagy. Our finding of impaired mitophagy and preserved non-selective autophagy in LRRK2 mutant cells therefore suggested that LRRK2 mutations disrupted mitophagy at a site upstream of closure of autophagosomal membranes around the mitochondria. To determine at which stage mitophagy was affected by LRRK2 mutations, we first assessed accumulation of PINK1 after mitochondrial depolarization. Depolarized mitochondria fail to import and degrade PINK1, resulting in stabilization of PINK1 on the OMM [8]. Endogenous PINK1 accumulated to a similar extent in LRRK2 mutant and wild-type fibroblasts after mitochondrial depolarization (Figure 4(a-d); Figure S10A-D).
Figure 4.
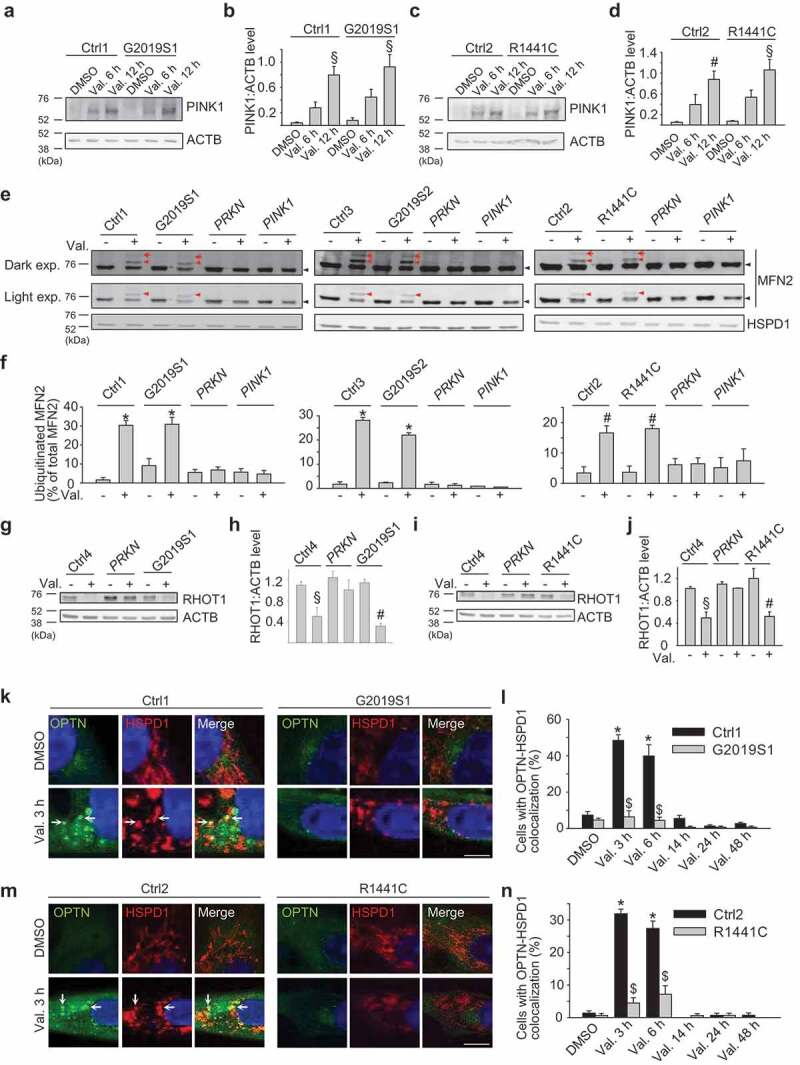
OPTN recruitment to depolarized mitochondria is impaired in PD patients with LRRK2 mutations despite preserved mitochondrial ubiquitination. (a-d) Untransfected control (Ctrl1 and Ctrl2) and LRRK2 mutant (G2019S1 and R1441C) fibroblasts were treated with DMSO or valinomycin (Val., 1 µM) for 6 or 12 h. (a,c) Western blot for endogenous PINK1 and ACTB. (b) Quantification of PINK1:ACTB (n = 3–4). § P < 0.01 and # P < 0.05 compared with DMSO condition in the same subject. (e,f) Untransfected control (Ctrl1, Ctrl2, Ctrl3), LRRK2 (G2019S1, G2019S2, R1441C), PRKN and PINK1 mutant fibroblasts were treated with DMSO or valinomycin (Val., 1 µM) for 3 h. (E) Western blot of mitochondrial fractions for MFN2 and HSPD1. The same MFN2 blots are shown after either light or dark exposure (exp.). Red arrows indicate diubiquitinated MFN2, red arrowheads monoubiquitinated MFN2 and black arrowheads non-ubiquitinated MFN2. (f) Quantification of ubiquitinated MFN2 relative to total MFN2 (n = 3–4). * P < 0.001 and #P < 0.05 compared with DMSO condition in the same subject. (g-j) Control, PRKN and LRRK2 mutant fibroblasts were treated with DMSO or Val. (1 µM) for 6 h. (g,i) Western blot for RHOT1 and ACTB. (h,j) Quantification of RHOT1:ACTB (n = 3). § P < 0.01 and # P < 0.05 compared with DMSO in the same subject. (k-n) Control and LRRK2 mutant fibroblasts were transfected with GFP-tagged OPTN. After 24 h cells were treated with DMSO or Val. (1 µM) for 3, 6, 14, 24 or 48 h. Fibroblasts were immunostained for HSPD1 and nuclei were stained with TOTO-3. Arrows in (k) and (m) indicate mitochondria that colocalize with OPTN. (l,n) Quantification of the percentage of cells with OPTN-HSPD1 colocalization (n = 3). * P < 0.001 compared with DMSO in the same subject. $ P < 0.001 compared with the corresponding Val. condition in the control subject. Scale bars: 10 µm.
As a next step in the mitophagy pathway, we examined PINK1-mediated activation of PRKN at the OMM of depolarized mitochondria using a MFN2 (mitofusin 2) ubiquitination assay. MFN2 is an OMM protein that is ubiquitinated by activated PRKN after mitochondrial depolarization [16,29]. Ubiquitinated MFN2 is then extracted from the OMM and degraded by the proteasome [16]. Valinomycin treatment for 3 h in control fibroblasts expressing endogenous PRKN resulted in ubiquitination of endogenous MFN2 (Figure 4(e,f); Figure S10E,F). This was abrogated in PRKN and PINK1 mutant fibroblasts, as expected, but was fully preserved in G2019S and R1441C LRRK2 mutant cells (Figure 4(e,f); Figure S10E,F), indicating that PINK1 and PRKN activity at the OMM of depolarized mitochondria was not affected by LRRK2 mutations.
Mitochondrial depolarization also triggers PINK1- and PRKN-dependent proteasomal degradation of the OMM protein RHOT1 (ras homolog family member T1) (also known as MIRO1) [16,30]. Hsieh et al. recently reported defective mitophagy in fibroblasts from patients with the G2019S LRRK2 mutation [31], consistent with our findings, and attributed this defect to G2019S LRRK2-mediated disruption of RHOT1 degradation [31]. We measured degradation of endogenous RHOT1 in the G2019S and R1441C LRRK2 and PRKN mutant fibroblasts in response to valinomycin and also, as in the study by Hsieh et al. [31], to CCCP. RHOT1 degradation after mitochondrial depolarization was abrogated in the PRKN mutant fibroblasts (Figure 4(g-j); Figure S10G-J). However, there was no difference in RHOT1 degradation between LRRK2 mutant and control cells (Figure 4(g-j); Figure S10G-J). Hence, the observed mitophagy defect of LRRK2 mutant fibroblasts could not be explained by disrupted RHOT1 degradation.
PINK1- and PRKN-mediated generation of phospho-ubiquitin on depolarized mitochondria induces recruitment of autophagy receptors that tether the mitochondria to nascent autophagosomal membranes [19]. Recent work has revealed a critical role for the autophagy receptor OPTN in mitophagy [18,19]. OPTN associates with ubiquitinated mitochondria via its ubiquitin-binding domain and recruits LC3 via its LC3 interaction region to promote mitochondria engulfment by autophagosomes. In basal conditions green fluorescent protein (GFP)-tagged OPTN was predominantly cytosolic both in control and LRRK2 mutant fibroblasts (Figure 4(k-n); Figure S10K-N; Figure S11). After mitochondrial depolarization OPTN accumulated on mitochondria in control cells. However, in G2019S and R1441C mutant cells OPTN recruitment to depolarized mitochondria was severely and consistently reduced (Figure 4(k-n); Figure S6B; Figure S10K-N; Figure S11). Endogenous OPTN was also recruited to a subset of mitochondria in wild-type fibroblasts after mitochondrial depolarization, and this recruitment was substantially impaired in G2019S and R1441C mutant fibroblasts (Figure S12). Thus, although PINK1 accumulation and PRKN activity at depolarized mitochondria were preserved in LRRK2 mutant fibroblasts, these mutations interfered with OPTN recruitment.
The time course of colocalization of endogenous OPTN with depolarized mitochondria in wild-type fibroblasts was more prolonged (Figure S13A) than that of overexpressed GFP-OPTN (Figure 4(l,n)). This difference in kinetics can be explained by the fact that overexpression of OPTN strongly accelerated the clearance of mitochondria (Figure S13B,C). As a result, a large percentage of GFP-OPTN-overexpressing cells were already devoid of mitochondria at earlier time points, explaining the earlier and steeper decline in the percentage of cells displaying colocalization of GFP-OPTN with mitochondria.
LC3 recruitment to mitochondria (Figure S14) and mitochondrial colocalization with the lysosomal marker LAMP1 (lysosomal associated membrane protein 1) (Figure S15) after valinomycin treatment were also impaired in LRRK2 mutant cells.
RAB10 mitigates defects in OPTN recruitment, mitophagy and mitochondrial function in LRRK2 mutant cells
RAB10 and several related members of the RAB GTPase family were recently identified as substrates of the LRRK2 kinase [6,32]. Wild-type LRRK2 phosphorylates RAB10 on residue T73, and the G2019S and R1441C mutations further enhance RAB10 phosphorylation in cells [6]. As LRRK2 kinase inhibition rescued mitophagy in the G2019S and R1441C mutant cells (Figure 2(g-l)), we wondered if RAB10 or other RAB substrates of LRRK2 were involved in the mitophagy defect of these cells.
First, we tested whether RAB10 is phosphorylated at T73 in human fibroblasts, as this has not been shown before. Using a novel monoclonal antibody against RAB10 phosphorylated at T73 [33] we indeed detected endogenous T73-phosphorylated RAB10 in control fibroblasts (Figure 5(a-d); Figure S16). The phospho-RAB10 signal was dissipated after treatment with the LRRK2 kinase inhibitors PF-06447475 and MLi-2, confirming the specificity of the antibody (Figure 5(a-d)). Importantly, the ratio of endogenous T73-phosphorylated RAB10 over endogenous total RAB10 was significantly higher in the G2019S and R1441C mutant fibroblasts, consistent with increased kinase activity of the mutant protein (Figure 5(a-d); Figure S16). In PRKN and PINK1 mutant fibroblasts, T73 phosphorylation of RAB10 was not enhanced compared with wild-type cells (Figure S16).
Figure 5.
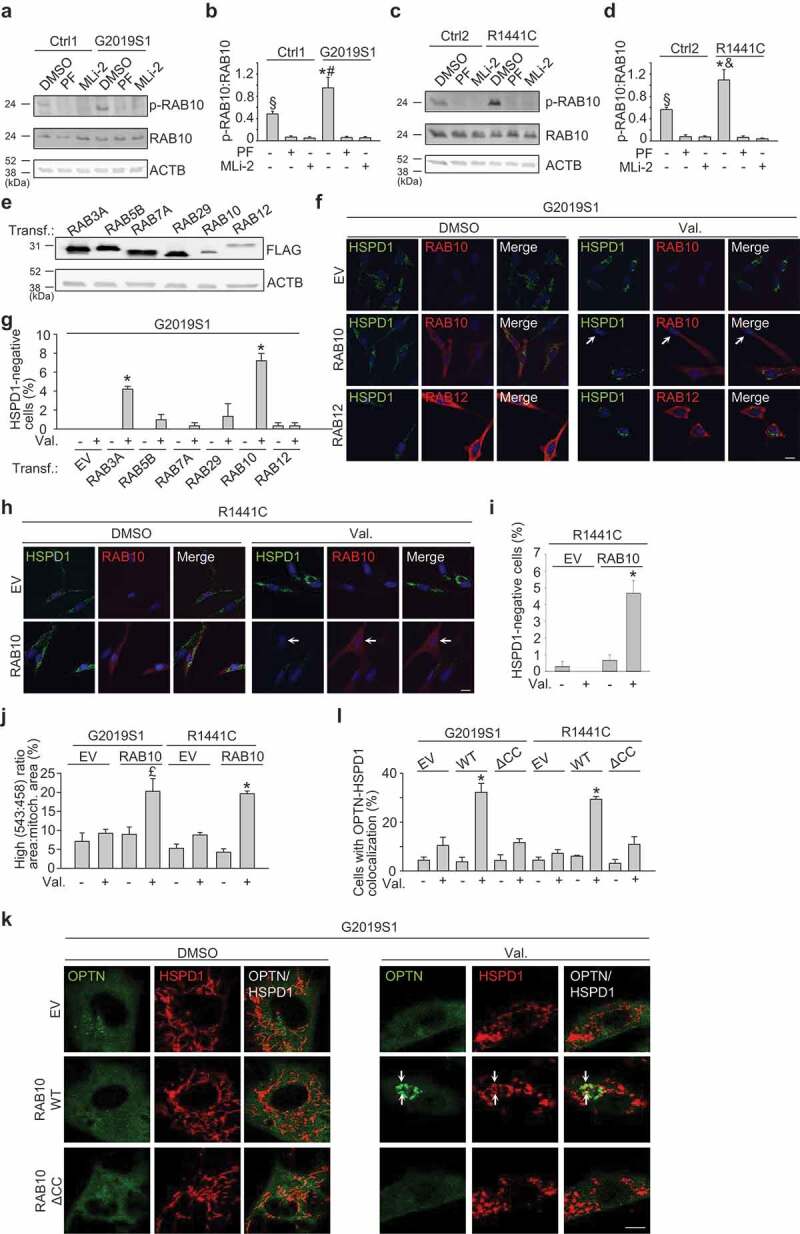
Overexpression of RAB10 mitigates the defect in mitophagy and OPTN recruitment in LRRK2 mutant cells. (a-d) Untransfected fibroblasts were treated with DMSO, PF-06447475 (0.5 μM) or MLi-2 (150 nM) for 24 h. (a,c) Western blot for endogenous RAB10 phosphorylated at T73 (p-RAB10), total RAB10 and ACTB. (B) Quantification of p-RAB10:total RAB10 (n = 5–7). * P < 0.001 and § P < 0.01 compared to all other conditions in the same subject. # P < 0.01 and & P < 0.001 compared to DMSO in the corresponding control subject. (e-g) Fibroblasts were transfected (Transf.) with empty vector (EV) or FLAG-RAB3A, -RAB5B, -RAB7A, -RAB29, -RAB10 or -RAB12. (e) Western blot showing expression levels of overexpressed FLAG-RAB proteins. (f,g) G2019S1 fibroblasts transfected with the indicated FLAG-tagged RAB were treated with DMSO or valinomcyin (Val., 1 µM) for 48 h, followed by immunostaining for FLAG and HSPD1. Nuclei were stained with TOTO-3 (blue). Arrows in (f) indicate cell without detectable HSPD1. (g) Quantification (n = 3–8). *P < 0.001 compared with Val. EV. (h,i) R1441C fibroblasts were transfected with EV or FLAG-RAB10, treated with DMSO or Val. (1 µM) for 48 h and immunostained for FLAG and HSPD1. Arrow in (h) indicates cell without detectable HSPD1. (i) Quantification (n = 3). * P < 0.001 compared with all other conditions. (j) G2019S1 and R1441C fibroblasts were transfected with mito-Keima and either EV or GFP-RAB10 and treated with DMSO or Val. (1 µM) for 48 h, followed by live ratiometric imaging. High (543:458) ratio area:total mitochondrial area was quantified (n ≥ 3). £ P < 0.05 compared to all other conditions in G2019S1. * P < 0.001 compared to all other conditions in R1441C. (k,l) G2019S1 and R1441C mutant fibroblasts were transfected with GFP-tagged OPTN and either EV, FLAG-tagged wild-type RAB10 or FLAG-tagged RAB10 lacking the 2 C-terminal cysteine residues (ΔCC RAB10). After 24 cells were treated with DMSO or Val. (1 µM) for 3 h. Fibroblasts were immunostained for HSPD1. Arrows in (k) indicate mitochondria that colocalize with OPTN. (l) Quantification of the percentage of cells with OPTN and HSPD1 colocalization (n = 3). * P < 0.001 compared to all other conditions in the same subject. Scale bars: 10 µm.
To explore whether RAB10 was involved in the mitophagy defect of the LRRK2 mutant fibroblasts, we overexpressed RAB10 and several other RAB substrates of LRRK2: RAB3A, RAB5B, RAB12 and RAB29 (Figure 5(e-g)) [6,32]. In addition to these LRRK2 substrates, we also overexpressed RAB7A because of its reported functional link with LRRK2 (Figure 5(e,g)) [34]. If the mitophagy defect of LRRK2 mutant cells was due to enhanced phosphorylation of RAB10, we reasoned that overexpression of RAB10 might mitigate the mitophagy defect, because the low abundance of endogenous LRRK2 protein would be too limiting to phosphorylate the excess of overexpressed substrate. Indeed, overexpression of FLAG-tagged RAB10 in LRRK2 mutant cells caused a large increase in total RAB10, but only a mild increase in phospho-RAB10, implying that it mostly enhanced the amount of non-phosphorylated RAB10 (Figure S17). Interestingly, overexpression of RAB10 mitigated the mitophagy deficit of G2019S and R1441C mutant fibroblasts (Figure 5(f-j)). Overexpression of RAB3A also had a significant, although smaller, rescuing effect, whereas RAB5B, RAB7A, RAB12 and RAB29 had no effect (Figure 5(g)). RAB10 overexpression in G2019S and R1441C fibroblasts also improved recruitment of OPTN (Figure 5(k,l)) and LC3 (Figure S18), ameliorated mitochondrial membrane potential (Figure S9E) and mitigated mitochondrial ROS production (Figure S9F).
RAB10 facilitates mitophagy in wild-type cells
Next we knocked down RAB10 in wild-type fibroblasts to determine whether endogenous RAB10 regulated mitophagy. Our siRNA approach effectively reduced RAB10 protein levels (Figure S19A-D). RAB10 knockdown in wild-type cells impaired valinomycin-induced mitophagy, as shown by HSPD1 immunocytochemistry (Figure 6(a,b)) and live mito-Keima imaging (Figure 6(c,d)), while knockdown of RAB3A (Figure S19E-H) had no effect (Figure 6(a,b)).
Figure 6.
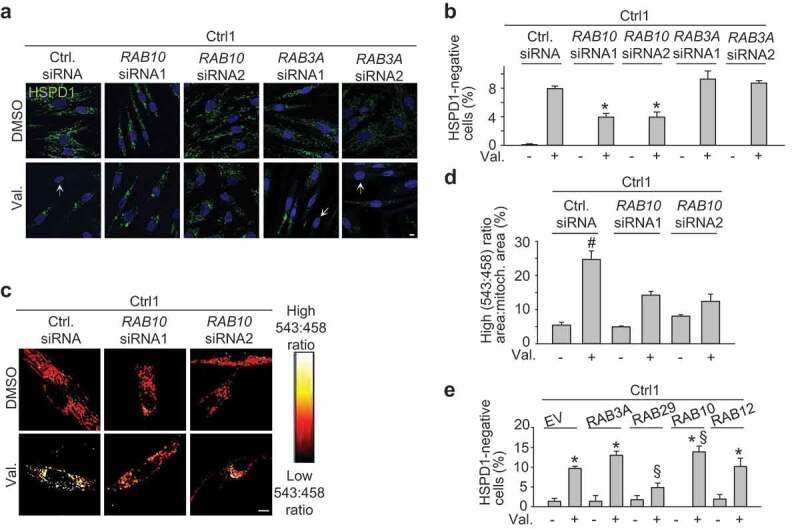
Endogenous RAB10 facilitates mitophagy in wild-type cells. (a-b) Control 1 (Ctrl1) fibroblasts were transfected with control siRNA, RAB10 siRNA1, RAB10 siRNA2, RAB3A siRNA1 or RAB3A siRNA2. After 72 h cells were treated with DMSO or valinomycin (Val., 1 µM) for 48 h and immunostained for HSPD1. Nuclei were stained with TOTO-3 (blue). Arrows in (a) indicate cells without mitochondria. (b) Quantification (n = 3–9). * P < 0.005 compared with Val. in control siRNA. (c,d) Ctrl1 fibroblasts were transfected with mito-Keima and either control siRNA, RAB10 siRNA1 or RAB10 siRNA2. After 72 h, cells were treated with DMSO or Val. (1 µM) for 48 h, followed by ratiometric imaging. (d) Quantification (n = 3–6). # P < 0.01 compared with all other conditions. (e) Ctrl1 fibroblasts were transfected with empty vector (EV) or FLAG-tagged RAB3A, RAB29, RAB10 or RAB12, treated with DMSO or Val. (1 µM) for 48 h and immunostained for HSPD1 and FLAG. The percentage of cells without HSPD1 staining was quantified (n = 3–8). * P < 0.005 compared with the corresponding DMSO condition. § P < 0.05 compared with Val. in EV. Scale bars: 10 µm.
We also determined the effect of overexpression of RAB10 and other RAB family members on mitophagy in wild-type cells (Figure 6(e)). RAB10 overexpression enhanced mitophagy in wild-type cells, while RAB12 overexpression had no effect (Figure 6(e)). Overexpression of RAB3A also had no significant effect on mitophagy, although there was a trend towards an increase (Figure 6(e)). Interestingly, overexpression of RAB29 inhibited mitophagy (Figure 6(e)). RAB29 was recently shown to stimulate the kinase activity of LRRK2 [35]. Thus, the inhibitory effect of RAB29 on mitophagy fits with our finding that LRRK2 kinase activity inhibits mitophagy (Figure 2(g-l)).
RAB10 accumulates on depolarized mitochondria in wild-type cells
To search for the mechanism underlying facilitation of mitophagy by RAB10, we analyzed its subcellular localization in wild-type cells. In baseline conditions, FLAG-tagged RAB10 had a predominantly cytosolic localization (Figure 7(a,b)). After mitochondrial depolarization, however, FLAG-RAB10 formed puncta that showed striking colocalization with mitochondria (Figure 7(a,b)). Accumulation of FLAG-RAB10 on depolarized mitochondria was also confirmed by western blotting after mitochondrial fractionation (Figure 7(d,e)). Overexpressed untagged RAB10 (Figure S20) and endogenous RAB10 (Figure 7(f,g)) also accumulated on mitochondria after valinomycin treatment. By contrast, FLAG-tagged RAB12 did not show detectable translocation to depolarized mitochondria (Figure 7(c)). Next, we tested whether PRKN and RAB10 localized to the same subset of depolarized mitochondria. As the available PRKN-specific antibodies do not detect the low level of endogenous PRKN in fibroblasts, we overexpressed HA-tagged PRKN for this experiment. RAB10 indeed accumulated on depolarized mitochondria that also showed strong PRKN accumulation, whereas it was not detectable on mitochondria that were devoid of PRKN signal (Figure 7(h)).
Figure 7.
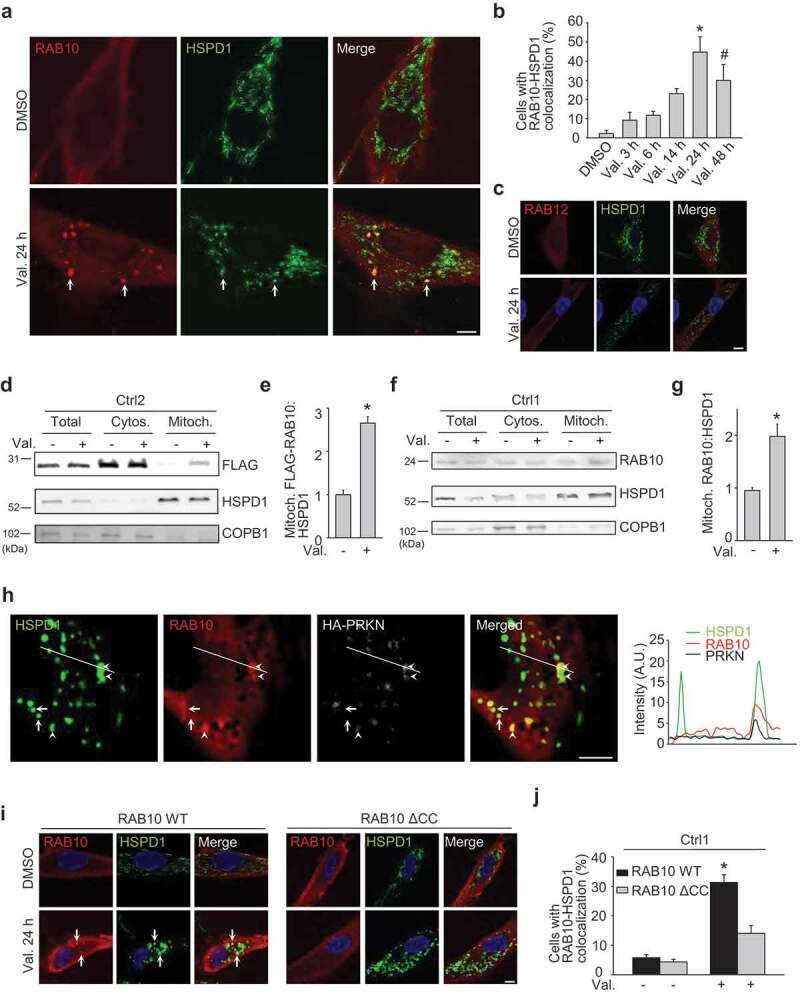
RAB10 accumulates on depolarized mitochondria in wild-type cells. (a-c) Fibroblasts from Ctrl1 were transfected with FLAG-RAB10 (A, B) or FLAG-RAB12 (c). After 24 h cells were treated with DMSO or valinomycin (Val., 1 µM) for the indicated time and immunostained for FLAG and HSPD1. (b) Quantification of the percentage of cells with colocalization of FLAG-RAB10 and HSPD1 (n = 4). * P < 0.001 and # P < 0.05 compared with DMSO. Arrows in (a) indicate examples of RAB10 puncta that colocalize with mitochondria. (d,e) Ctrl2 fibroblasts transfected with FLAG-RAB10 were treated with DMSO or Val. for 14 h, followed by subcellular fractionation and western blotting of total, cytosolic (Cytos.) and mitochondrial (Mitoch.) fractions for FLAG, HSPD1 and the non-mitochondrial protein COPB1. (e) Quantification of FLAG-RAB10:HSPD1 in the mitochondrial fraction (n = 3). * P < 0.001 compared with DMSO. (f,g) Untransfected fibroblasts from Ctrl1 were treated with DMSO or Val. for 14 h, followed by western blotting of subcellular fractions for endogenous RAB10, HSPD1 and COPB1. (g) Quantification of endogenous RAB10:HSPD1 in the mitochondrial fraction (n = 10). * P < 0.001 compared with DMSO. (h) Ctrl1 fibroblasts were transfected with Myc-tagged RAB10 and HA-tagged PRKN. After 24 h cells were treated with DMSO or Val. (1 µM) for 3 h and immunostained for HSPD1, Myc and HA. White arrows indicate examples of mitochondria without PRKN or RAB10 signal. Arrowheads indicate examples of mitochondria that colocalize with PRKN and RAB10 puncta. The graph depicts the relative intensity of each channel over the drawn lines shown in the merged image. A.U., arbitrary units. (i,j) Ctrl1 fibroblasts were transfected with FLAG-tagged wild-type (WT) or ΔCC RAB10 WT. After 24 h cells were treated with DMSO or Val. (1 µM) for 24 h and immunostained for FLAG and HSPD1. Nuclei were stained with TOTO-3. Arrows in (i) indicate examples of RAB10 puncta that colocalize with mitochondria. (j) Quantification of the percentage of cells with colocalization of FLAG-RAB10 and HSPD1 (n = 4). * P < 0.001 compared with all other conditions. Scale bars: 10 µm.
Membrane association of RAB proteins is ensured by posttranslational modification of C-terminal cysteine residues with lipophilic geranylgeranyl groups that act as hydrophobic membrane anchors [7]. To assess the role of C-terminal geranylgeranylation in RAB10 recruitment to mitochondria, we deleted the 2 C-terminal cysteine residues of RAB10 (C199 and C200). This ΔCC RAB10 construct accumulated substantially less on depolarized mitochondria than full-length RAB10 (Figure 7(i,j)) despite similar expression levels (Figure S21), suggesting that C-terminal prenylation is important for RAB10 recruitment to mitochondria. Interestingly, overexpression of ΔCC RAB10 did not rescue mitochondrial recruitment of OPTN in the LRRK2 mutant fibroblasts, in contrast to wild-type RAB10 overexpression (Figure 5(k,l)). Thus, rescue of mitochondrial OPTN recruitment in LRRK2 mutant cells required RAB10 that was capable of being tethered to the membrane via its C-terminal lipid anchor.
RAB10 interacts with OPTN, colocalizes with OPTN on depolarized mitochondria and promotes mitochondrial OPTN recruitment in wild-type cells
Interestingly, the mitochondrial RAB10 puncta after valinomycin treatment of wild-type fibroblasts strongly colocalized with OPTN (Figure 8(a,b)). Moreover, siRNA-mediated knockdown of RAB10 reduced OPTN recruitment to depolarized mitochondria in wild-type cells, indicating that RAB10 is required for optimal OPTN accumulation on damaged mitochondria (Figure 8(c,d)).
Figure 8.
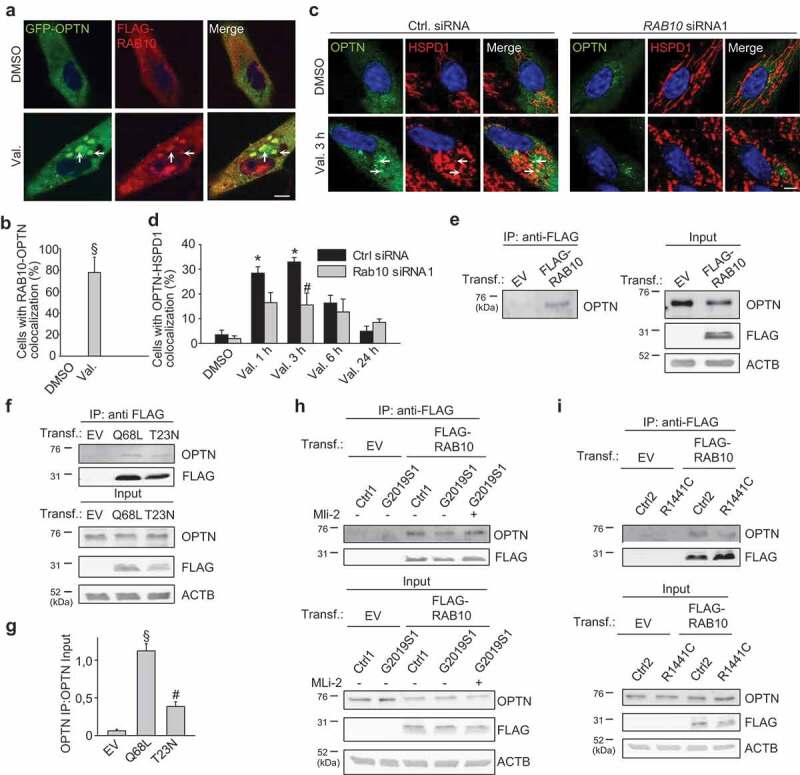
RAB10 colocalizes with OPTN on depolarized mitochondria, promotes mitochondrial OPTN recruitment and binds to OPTN in wild-type cells. (a,b) Fibroblasts from Ctrl1 were transfected with GFP-tagged OPTN and FLAG-RAB10, treated with DMSO or Val. (1 µM) for 3 h and immunostained for FLAG. Arrows in (a) indicate examples of colocalization of GFP-OPTN and FLAG-RAB10. Scale bars: 10 µm. (b) Quantification of the percentage of cells with colocalization of GFP-OPTN and FLAG-RAB10 (n = 3). § P < 0.005 compared with DMSO. (c,d) Ctrl1 fibroblasts were transfected with GFP-tagged OPTN and either control siRNA or RAB10 siRNA1. After 72 h cells were treated with DMSO of valinomycin (Val., 1 µM) for 3 h. and immunostained for HSPD1. Arrows in (c) indicate examples of colocalization of GFP-OPTN and HSPD1. Scale bars: 10 µm. (d) Quantification of the percentage of cells with colocalization of GFP-OPTN and HSPD1 (n = 3). * P < 0.001 compared with the ctrl siRNA DMSO condition. # P < 0.05 compared with the ctrl siRNA Val. 3 h condition. (e) Control fibroblasts were transfected (Transf.) with empty vector (EV) or FLAG-tagged RAB10, as indicated. After coimmunoprecipitation (Co-IP) with anti-FLAG beads, the immunoprecipitate (IP) and input samples were analyzed by SDS-PAGE and western using the indicated antibodies. (f,g) Fibroblasts were transfected with EV or FLAG-tagged Q68L or T23N RAB10, as indicated. After Co-IP with anti-FLAG beads, the IP and input samples were analyzed by SDS-PAGE and western. (g) Quantification of the ratio of OPTN in the IP over OPTN in the input (n = 3). § P < 0.005 compared with all other conditions. # P < 0.05 compared with EV. (h,i) Ctrl1 and G2019S fibroblasts (h) and Ctrl2 and R1441C fibroblasts (i) were transfected with EV or FLAG-tagged RAB10. In (h) cells were treated or not with MLi-2 (150 nM) for 24 h. After Co-IP with anti-FLAG beads, the IP and input samples were analyzed by SDS-PAGE and western.
The colocalization of RAB10 and OPTN on depolarized mitochondria and the facilitating effect of RAB10 on mitochondrial recruitment of OPTN suggested that the two proteins might physically interact. Moreover, OPTN was previously reported to interact with RAB8A and RAB8B, the two RAB GTPases with the strongest sequence similarity to RAB10 [36]. We expressed FLAG-tagged RAB10 in control fibroblasts and found that it co-immunoprecipitated with endogenous OPTN (Figure 8(e)). The interaction already occurred in basal conditions and only marginally increased after valinomycin treatment, suggesting that a pre-existing RAB10-OPTN complex is recruited to depolarized mitochondria: after 9 h treatment with valinomycin (1 μM), the amount of OPTN co-immunoprecipitated with FLAG-RAB10, divided by the amount of OPTN present in the input, was 117 ± 5.0% of the value after DMSO treatment (n = 3; P = 0.04).
Next, we generated Q68L (GTP-locked, constitutively active) and T23N (GDP-locked, constitutively inactive) mutant RAB10 constructs [37]. Co-immunoprecipitation (Co-IP) experiments showed that GTP-locked RAB10 interacted more with OPTN than GDP-locked RAB10, indicating that RAB10 activity promotes binding with OPTN (Figure 8(f,g)). This is again reminiscent of the interaction between OPTN and RAB8, which is also enhanced by RAB8 activation [36].
RAB10 interaction with OPTN and accumulation on depolarized mitochondria are impaired in patients with LRRK2 mutations
To assess the effect of LRRK2-dependent RAB10 phosphorylation on the RAB10-OPTN interaction, we performed parallel Co-IP experiments in wild-type, G2019S mutant and R1441C mutant fibroblasts (Figure 8(h,i)). Significantly less OPTN co-immunoprecipitated with RAB10 in the G2019S and R1441C mutant cells than in control cells (Figure 8(h,i)). In G2019S and R1441C fibroblasts, the amount of OPTN co-immunoprecipitated with FLAG-RAB10, divided by the amount of OPTN present in the input, was 35.8 ± 10.4% (n = 3; P = 0.004) and 32.6 ± 4.9% (n = 3; P < 0.001), respectively, of the values in control cells. The interaction increased after treatment with MLi-2 (Figure 8(h)), further supporting the idea that T73 phosphorylation of RAB10 impairs the interaction with OPTN.
In G2019S and R1441C LRRK2 mutant fibroblasts, RAB10 accumulation on depolarized mitochondria was substantially reduced, as shown by immunocytochemistry and mitochondrial fractionation followed by western blotting (Figure 9(a-j); Figure S6C; Figure S22). Interestingly, RAB10 accumulation on depolarized mitochondria was also largely abrogated in fibroblasts from PD patients with PINK1 and PRKN mutations (Figure 9(k-t)). Thus, recruitment of RAB10 to depolarized mitochondria requires intact function of PINK1 and PRKN and is inhibited by PD-causing LRRK2 mutations.
Figure 9.
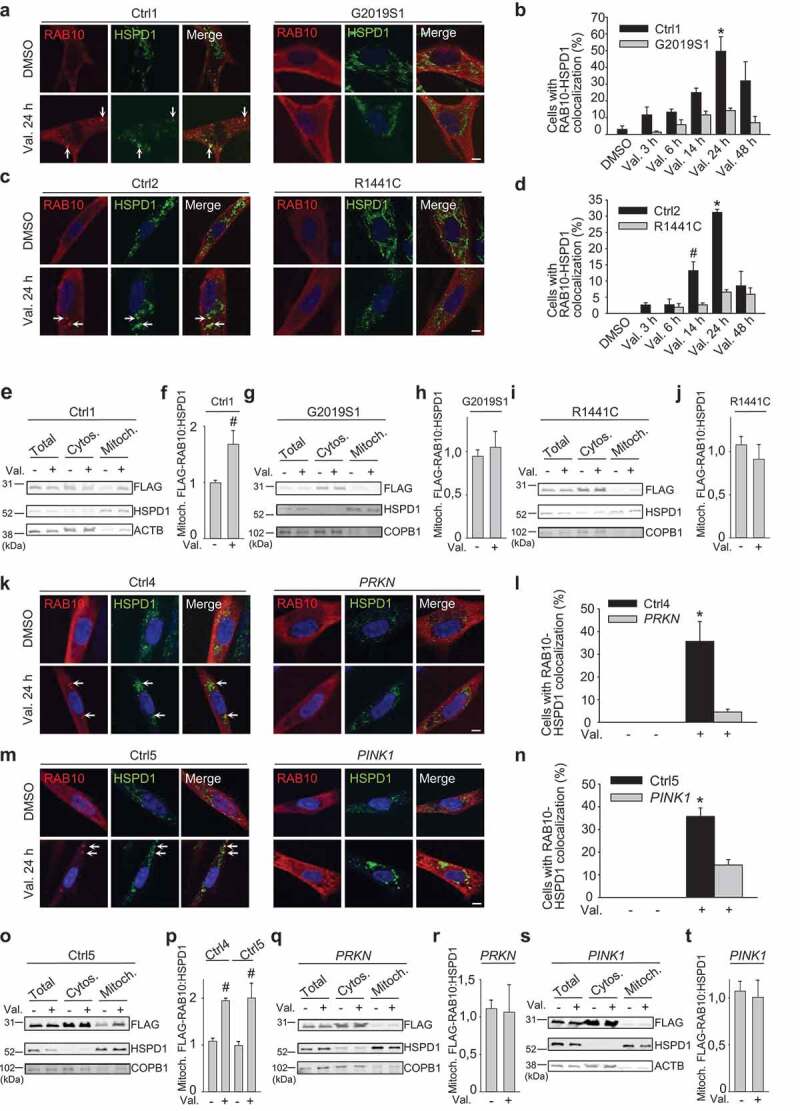
RAB10 accumulation on depolarized mitochondria is impaired in patient fibroblasts with LRRK2 mutations. (a-d) Control and LRRK2 mutant fibroblasts were transfected with FLAG-RAB10, treated with DMSO or valinomycin (Val., 1 µM) for the indicated time periods and immunostained for FLAG and HSPD1. Nuclei were stained with TOTO-3. Arrows in (a) and (c) indicate puncta with colocalization of FLAG-RAB10 and HSPD1. (b,d) Quantification of the percentage of cells with colocalization of FLAG-RAB10 and HSPD1 (n = 3). * P < 0.005, # P < 0.05 compared with DMSO in the same subject. (e-j) Ctrl1 and LRRK2 mutant fibroblasts transfected with FLAG-RAB10 were treated with DMSO or Val. for 14 h, followed by subcellular fractionation and western blotting of total, cytosolic (Cytos.) and mitochondrial (Mitoch.) fractions for FLAG, HSPD1 and the non-mitochondrial proteins ACTB or COPB1. (f,h,j) Quantification of FLAG-RAB10:HSPD1 in the mitochondrial fraction (n ≥ 3). # P < 0.05 compared with DMSO. (k-n) Fibroblasts from controls (Ctrl4, Ctrl5), a PD patient with compound heterozygous PRKN mutations and a PD patient with homozygous PINK1 mutations were transfected with FLAG-RAB10, treated with DMSO or valinomycin (Val., 1 µM) for 24 h and immunostained for FLAG and HSPD1. Arrows in (k) and (m) indicate puncta with colocalization of FLAG-RAB10 and HSPD1. (l,n) Quantification of the percentage of cells with colocalization of FLAG-RAB10 and HSPD1 (n = 3). * P < 0.005 compared with all other conditions. (o-t) Ctrl4, Ctrl5, PRKN mutant and PINK1 mutant fibroblasts transfected with FLAG-RAB10 were treated with DMSO or Val. for 14 h, followed by western blotting of subcellular fractions for FLAG, HSPD1 and ACTB or COPB1. (p,r,t) Quantification of FLAG-RAB10:HSPD1 in the mitochondrial fraction (n = 3–8). # P < 0.05 compared with DMSO in the same subject. Scale bars: 10 µm.
Importantly, LRRK2 knockdown with two different siRNAs and LRRK2 kinase inhibition with LRRK2-IN-1 and PF-06447475 each rescued the mitochondrial accumulation of RAB10 (Figure 10) and OPTN (Figure S23) in valinomycin-treated G2019S or R1441C mutant fibroblasts, in parallel with the rescue of the mitophagy defect (Figure 2).
Figure 10.
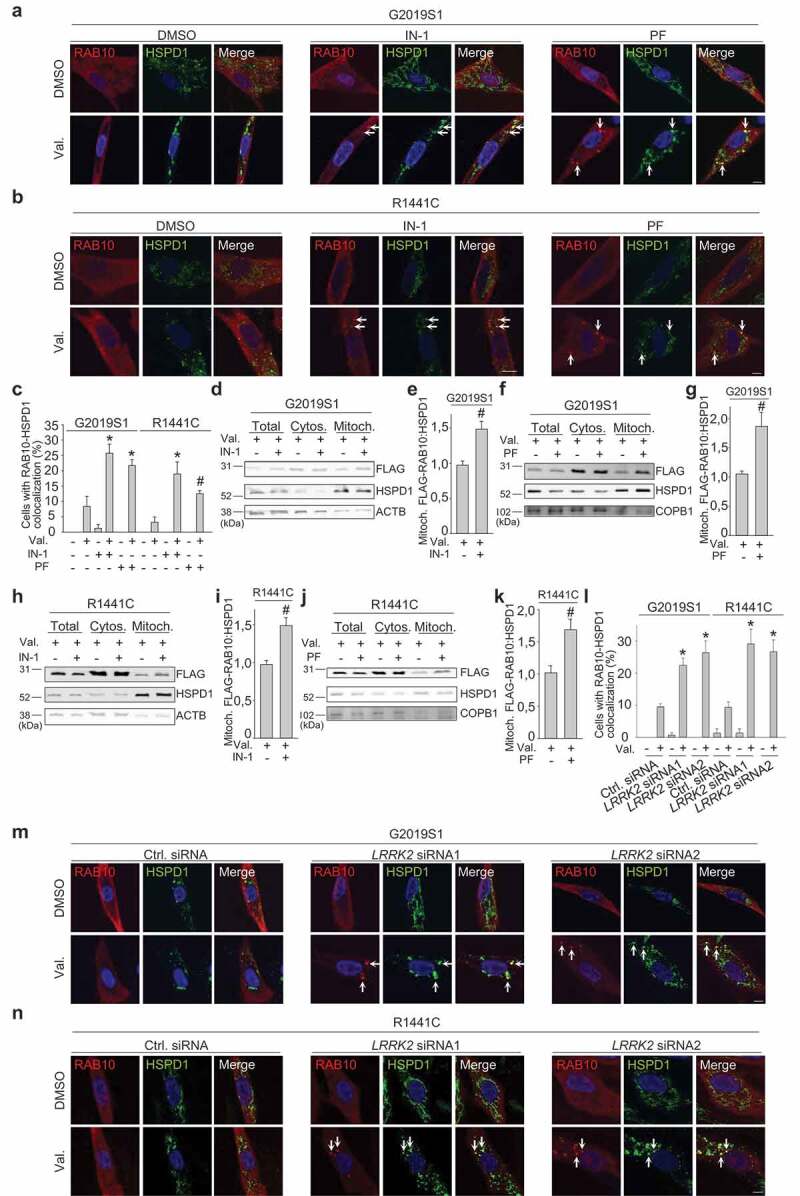
LRRK2 kinase inhibitors and LRRK2 knockdown rescue accumulation of RAB10 on depolarized mitochondria in patient fibroblasts with LRRK2 mutations. (a-c) G2019S and R1441C mutant fibroblasts were transfected with FLAG-RAB10. After pre-treatment for 24 h with DMSO or the LRRK2 kinase inhibitors LRRK2-IN-1 (IN-1, 1 µM) or PF-06447475 (PF, 0.5 µM), cells were treated for 24 h with DMSO or valinomycin (Val., 1 µM) in the continued presence or absence of the LRRK2 kinase inhibitors, and immunostained for FLAG and HSPD1. Arrows in (a) and (b) indicate puncta where FLAG-RAB10 and HSPD1 colocalize. (c) Quantification of the percentage of cells with colocalization of FLAG-RAB10 and HSPD1 (n = 3). * P < 0.005 and # P < 0.05 compared with Val. alone in the same subject. (D-K) G2019S and R1441C mutant fibroblasts were transfected with FLAG-RAB10. After pre-treatment for 24 h with DMSO, LRRK2-IN-1 (1 µM) or PF-06447475 (0.5 µM), cells were treated for 14 h with DMSO or Val. (1 µM) in the continued presence or absence of the LRRK2 kinase inhibitors, followed by western blotting of subcellular fractions for FLAG, HSPD1 and the non-mitochondrial proteins ACTB or COPB1. (e,g,i,k) Quantification of FLAG-RAB10:HSPD1 in the mitochondrial fraction (n = 3–5). # P < 0.05 compared with Val. alone. (l-n) G2019S and R1441C mutant fibroblasts were transfected with FLAG-RAB10 and control or LRRK2 siRNA1 or 2. After 72 h cells were treated with DMSO or Val. for 24 h and immunostained for FLAG and HSPD1. Arrows in (m) and (n) indicate puncta where FLAG-RAB10 and HSPD1 colocalize. (l) Quantification (n = 3). * P < 0.005 compared with Val. in control siRNA in the same subject.
To further test whether T73 phosphorylation of RAB10 impedes its interaction with OPTN, its translocation to depolarized mitochondria and mitophagy, we generated a phosphomimetic T73E mutant version of RAB10. Expression levels of transfected wild-type and T73E FLAG-tagged RAB10 were similar in control and LRRK2 mutant fibroblasts (Figure 11(a)). Co-IP experiments in control fibroblasts showed that RAB10T73E indeed interacted less well with OPTN than wild-type RAB10 (Figure 11(b)). The amount of OPTN that was co-immunoprecipitated with RAB10T73E, divided by the amount of OPTN in the input, was 38.5 ± 6.7% (n = 7; P < 0.001) of the value after Co-IP of OPTN with wild-type RAB10. Moreover, the T73E mutation impaired RAB10 translocation to depolarized mitochondria (Figure 11(c,d)), and overexpression of RAB10T73E failed to rescue valinomycin-induced mitophagy in G2019S and R1441C mutant cells, in contrast to overexpression of wild-type RAB10 (Figure 11(e-h)). We also made a non-phosphorylatable T73A version of RAB10, but protein levels of FLAG-RAB10T73A were much lower than those of wild-type FLAG-RAB10 and FLAG-RAB10T73E, despite extensive optimization of transfection conditions (Figure 11(a)). This made it difficult to perform Co-IP experiments with the T73A construct and to compare its subcellular distribution with that of wild-type RAB10 and RAB10T73E. However, despite its much lower abundance, RAB10T73A rescued the mitophagy defect of LRRK2 mutant fibroblasts as potently as wild-type RAB10, in contrast to the lack of rescue by RAB10T73E (Figure 11(e-h)). This further supported the conclusion that phosphorylation of RAB10 at T73 impairs its facilitating role in mitophagy.
Figure 11.
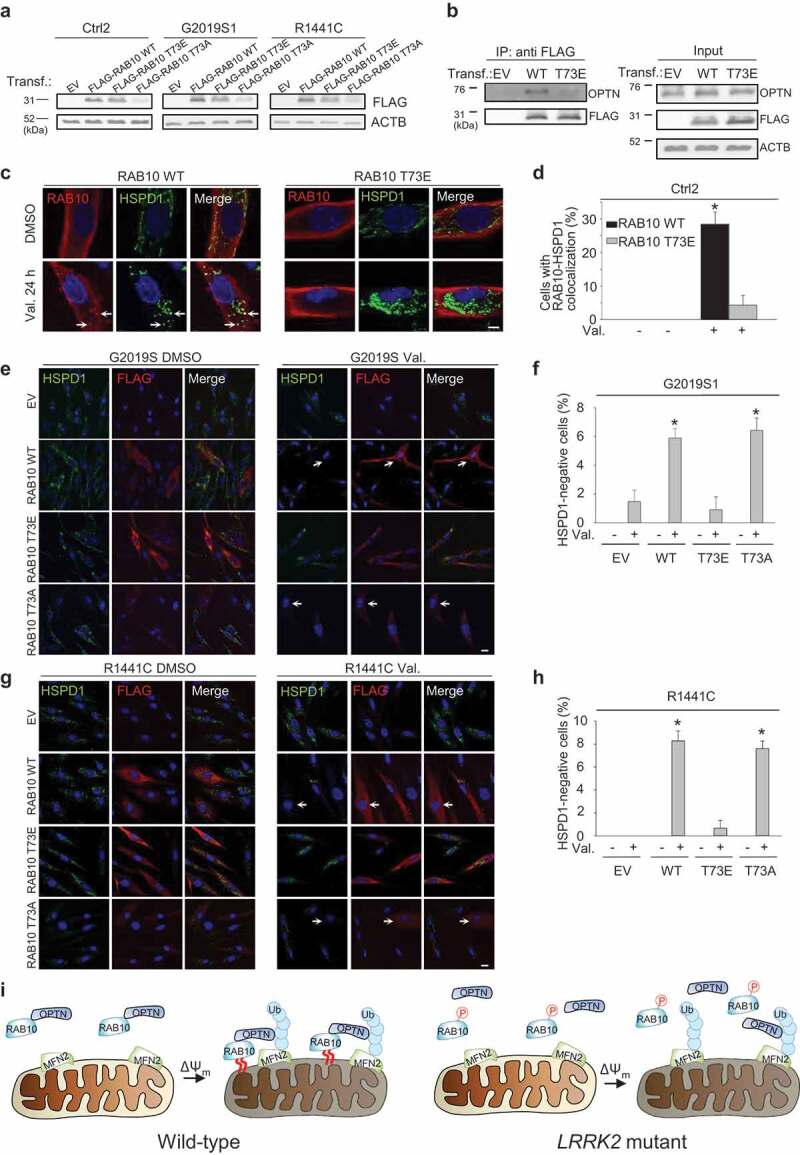
Phosphomimetic T73E RAB10 binds less with OPTN, accumulates less on depolarized mitochondria, and fails to rescue the mitophagy deficit of LRRK2 mutant cells. (a) Western blot for FLAG and ACTB in control (Ctrl2) and LRRK2 (G2019S1 and R1441C) mutant fibroblasts transfected with empty vector (EV), FLAG-tagged wild-type (WT) RAB10, FLAG-RAB10 with a phosphomimetic (T73E) mutation or FLAG-RAB10 with a non-phosphorylatable (T73A) mutation. (b) Control (Ctrl1) fibroblasts were transfected with EV, FLAG-tagged WT or T73E RAB10, as indicated. After Co-IP with anti-FLAG beads, the IP and input samples were analyzed by SDS-PAGE and western. (c,d) Control (Ctrl2) fibroblasts were transfected with FLAG-tagged WT or T73E RAB10 and treated with DMSO or valinomycin (Val., 1 µM) for 24 h. Fibroblasts were immunostained for FLAG and HSPD1. Nuclei were stained with TOTO-3 (blue). Arrows in (c) indicate puncta with colocalization of FLAG-RAB10 and HSPD1. (d) Quantification of the percentage of FLAG-positive cells with colocalization of FLAG-RAB10 and HSPD1 (n = 3). * P < 0.001 compared with all other conditions. (e-h) G2019S1 and R1441C fibroblasts were transfected with empty vector (EV), FLAG-tagged WT RAB10, T73E FLAG-RAB10 or T73A FLAG-RAB10, and treated with DMSO or Val. (1 µM) for 48 h, followed by immunostaining for FLAG and HSPD1. Nuclei were stained with TOTO-3 (blue). Arrows in (e) and (g) indicate cells without detectable HSPD1. (f,h) Quantification of the percentage of FLAG-positive cells with no detectable HSPD1 (n = 3). * P < 0.001 compared with all other conditions. Scale bars: 10 µm. (i) Model. OPTN forms a complex with RAB10 in basal conditions in WT cells. This complex translocates to mitochondria after mitochondrial depolarization. Optimal accumulation of OPTN on mitochondria requires both binding to mitochondrial ubiquitin (Ub) chains and interaction with RAB10 that is capable of insertion into the membrane via its C-terminal lipid anchor (indicated by red curved lines). In LRRK2 mutant cells, enhanced T73 phosphorylation of RAB10 impairs interaction with OPTN. Some OPTN recruitment to mitochondria is still possible via binding of OPTN to ubiquitin, but accumulation of OPTN on mitochondria is reduced because the interaction with membrane-inserted RAB10 is missing. P, phosphate. ΔΨm, mitochondrial depolarization.
Discussion
We show that the two most common PD-causing LRRK2 mutations (G2019S and R1441C) disrupt depolarization-induced mitophagy at a site downstream to the steps that are affected by PD-linked PINK1 and PRKN mutations. In PD patient cells expressing endogenous mutant LRRK2, PINK1 accumulation and PRKN-mediated MFN2 ubiquitination on depolarized mitochondria are preserved, but mitochondrial accumulation of the autophagy receptor OPTN is defective. Consistent with a specific detrimental effect of LRRK2 mutations on OPTN recruitment, starvation-induced autophagy, which does not require OPTN, is intact in LRRK2 mutant cells.
We show that the recently identified LRRK2 substrate RAB10 accumulates on depolarized mitochondria in wild-type cells in a manner dependent on its C-terminal cysteine residues. RAB10 interacts with OPTN, colocalizes with OPTN on depolarized mitochondria, promotes mitochondrial OPTN accumulation and facilitates mitophagy. In LRRK2 mutant cells, T73 phosphorylation of RAB10 is enhanced, while RAB10 interaction with OPTN, accumulation of RAB10 and OPTN on depolarized mitochondria, mitophagy and mitochondrial function are all impaired. These defects are rescued by LRRK2 knockdown and LRRK2 kinase inhibition.
Based on these data we can depict a model in which a subpopulation of OPTN forms a complex with RAB10 in basal conditions in wild-type cells (Figure 11(i)). This complex translocates to mitochondria after mitochondrial depolarization. Optimal accumulation of OPTN on mitochondria requires both binding to mitochondrial ubiquitin chains and interaction with RAB10 that is capable of insertion into the membrane via its C-terminal lipid anchor. In LRRK2 mutant cells, enhanced T73 phosphorylation of RAB10 impairs the interaction with OPTN. Some recruitment of OPTN to mitochondria is still possible (e.g. via binding of OPTN to ubiquitin), but accumulation of OPTN on mitochondria is reduced because the interaction with membrane-inserted RAB10 is lacking (Figure 11(i)).
Our data show that the kinetics of mitochondrial recruitment of OPTN and RAB10 are not identical, as the peak of mitochondrial accumulation occurs earlier for OPTN than for RAB10. Clearly, recruitment of OPTN to mitochondria does not depend exclusively on RAB10 but is multimodal, also relying on binding to ubiquitin on mitochondria [18] and on amplification by binding to TBK1 (TANK binding kinase 1) [38,39]. We show that interaction with prenylation-competent RAB10 is an additional mechanism that contributes to OPTN recruitment. These different mechanisms for OPTN recruitment likely have distinct but overlapping time courses. Binding of OPTN to ubiquitin is probably the earliest mechanism, while TBK1 and RAB10 rather amplify OPTN recruitment or stabilize OPTN on mitochondria. Our data show that RAB10 knockdown reduced OPTN recruitment but did not completely abolish it, which is consistent with the multimodal nature of OPTN recruitment.
Our finding that LRRK2 mutations as well as a phosphomimetic RAB10 mutation impaired the RAB10-OPTN interaction, suggests that T73 phosphorylation of RAB10 reduces its affinity for OPTN. It is possible that T73 phosphorylation of RAB10 has additional effects that further compromise mitochondrial accumulation of RAB10 and OPTN. For example, T73 phosphorylation of RAB10 by mutant LRRK2 decreases its affinity for several members of the RAB geranylgeranyl transferase complex [6], which could hamper C-terminal prenylation and membrane association of RAB10 in LRRK2 mutant cells.
Our data indicate that PINK1 and PRKN activity are also required for RAB10 recruitment to depolarized mitochondria, as mitochondrial accumulation of RAB10 was largely abrogated in cells from patients with PINK1 and PRKN mutations. It is likely that ubiquitin-binding proteins facilitate recruitment of RAB10 to mitochondria. Alternatively, RAB10 might selectively associate with patches of inner mitochondrial membrane that are exposed to the cytosol after PRKN- and proteasome-dependent OMM rupture [40].
The RAB GTPase-activating proteins TBC1D15 and TBC1D17 participate in PINK1- and PRKN-mediated mitophagy but not in starvation-induced autophagy [41,42]. Our data provide further evidence for the existence of RAB-based mechanisms that operate specifically in mitophagy and not in non-selective autophagy. Recently, RAB35 was found to recruit the selective autophagy receptor CALCOCO2 (calcium binding and coiled-coil domain 2) to damaged mitochondria in mitophagy [43]. Intriguingly, RAB35 is also a substrate of LRRK2 [32], but whether LRRK2 mutations impair the effects of RAB35 on CALCOCO2, in analogy to the disruption of RAB10-mediated OPTN recruitment, is not yet known.
We show that fibroblasts from LRRK2 mutation carriers have dysfunctional mitochondria, consistent with previous reports [44,45]. Interventions that corrected the mitophagy defect, such as RAB10 overexpression or LRRK2 kinase inhibition, also rescued the mitochondrial defects, suggesting that the mitochondrial dysfunction was a consequence of impaired mitophagy. It was previously noted that the pattern of changes in respiratory chain activity differed between fibroblasts from LRRK2 mutation carriers and those from PRKN and PINK1 mutation carriers [45]. This could be due to the fact that LRRK2 mutations affect mitochondrial quality control at a more distal site than PRKN and PINK1 mutations. As shown by our data, LRRK2 mutations impair mitophagy, but do not affect PRKN-mediated mitochondrial ubiquitination and therefore should not interfere with proteasomal degradation of damaged mitochondrial components or the mitochondria-derived vesicle pathway [16,46,47]. By contrast, PINK1 and PRKN mutations disrupt mitochondrial ubiquitination and may therefore have a more global, severe impact on mitochondrial quality control pathways. This may also explain why PINK1 and PRKN mutation carriers typically develop PD at a younger age than subjects with LRRK2 mutations [3].
We found a similar mitophagy defect in fibroblasts from two young non-manifesting G2019S mutation carriers as in the clinically manifesting carriers. The penetrance of this mutation is incomplete and age-dependent, and it is possible that the non-manifesting subjects will develop PD later in life. Assessment of fibroblasts of larger numbers of LRRK2 mutation carriers and longitudinal clinical follow-up will be necessary to determine whether impaired mitophagy in fibroblasts constitutes a consistent endophenotype of this mutation or only occurs in carriers who will eventually develop PD.
Previous studies on the effect of LRRK2 mutations on mitophagy have produced mixed results. Hsieh et al. [31] and Bonello et al. [48] also found impaired mitophagy in fibroblasts from patients with LRRK2 mutations, and Hsieh et al. [31] confirmed this in iPSC-derived dopaminergic neurons. However, two earlier studies [49,50] reported that mutant LRRK2 enhanced mitophagy. The latter two studies used other cell types than Hsieh et al. [31], Bonello et al. [48] and our group, and relied exclusively on overexpression of LRRK2. By contrast, Hsieh et al. [31], Bonello et al. [48] and we used cells that express endogenous wild-type and mutant LRRK2. Hsieh et al. [31] ascribed the observed mitophagy defect of LRRK2 mutant cells to impaired interaction of mutant LRRK2 with RHOT1 and delayed RHOT1 degradation and reported that this defect was not rescued by LRRK2 kinase inhibition. By contrast, Bonello et al. [48] found that LRRK2 kinase inhibition did rescue the mitophagy defect of LRRK2 mutant cells, in agreement with our findings. They suggested that the mitophagy defect was at least in part due to impaired recruitment of the mitochondrial fission-promoting protein DNM1L (also known as Drp1) to depolarized mitochondria, but the LRRK2 kinase substrate that interfered with DNM1L recruitment was not identified [48]. A limitation of our fibroblast experiments as well as those of Bonello et al. [48] and Hsieh et al. [31] is that the control fibroblasts were not isogenic. Therefore, unknown differences in genetic background between control and mutant fibroblasts may have contributed to the observed phenotypes and to some of the apparent discrepancies.
Recent work has revealed considerable overlap in the molecular machinery regulating mitophagy and the selective autophagy of cytosol-invading bacteria (xenophagy). PRKN, OPTN, CALCOCO2 and TBK1 are all critically involved in both mitophagy and xenophagy [19,38,39,43,51,52], and polymorphisms in PRKN are associated with increased susceptibility to Mycobacterium leprae (M. leprae) infection [53]. Intriguingly, LRRK2 variants are also linked to susceptibility to M. leprae infection [54], and future studies will need to investigate the role of LRRK2 and RAB10 in xenophagy.
In addition to RAB10 LRRK2 phosphorylates several other RAB GTPases [6,32] that may perform distinct roles on different membranes. This may account for the pleiotropy of the reported cellular effects of LRRK2 mutations [5]. Nevertheless, our data connect LRRK2 with the PINK1- and PRKN-mediated mitophagy pathway via its substrate RAB10, and this convergence of LRRK2, PRKN and PINK1 on mitochondrial quality control further increases the likelihood that this pathway is fundamentally important in the pathogenesis of PD.
Materials and methods
Antibodies
The following primary antibodies were used for immunofluorescence (IF) or western blot (WB): mouse anti-FLAG (WB, 1:5000; IF, 1:1000; Sigma, F3165), rabbit anti-FLAG (WB, 1:1000; Sigma, F7425), mouse anti-HA (IF, 1:250; Covance, MMS-101R-200), goat anti-MYC (IF, 1:250; Bethyl, A190-104A), mouse anti-ACTB (WB, 1:5000; Sigma, A5441), goat anti-HSPD1 (IF, 1:200; WB, 1:1000; Santa Cruz Biotechnology, sc-1052), rabbit anti-HSPD1 (IF, 1:1000; WB, 1:1000; Abcam, ab53109), mouse anti-ATP5F1B (IF, 1:500; WB 1:500; Abcam, ab14730), mouse anti-MFN2 (WB, 1:1000; Abcam, ab56889), mouse anti-RHOT1 (WB, 1:1000; Novus Biologicals, H00055288-M01), mouse anti-LC3B (WB, 1:1000; Novus Biologicals, NB100-2220), mouse anti-LAMP1 (IF, 1:500; BD Biosciences, 555798), rabbit anti-RAB10 (WB, 1:1000; IF, 1:200; Cell Signaling Technology, 8127S), rabbit anti-RAB10 (phospho T73; WB, 1:500; Abcam, ab230261) [33], rabbit anti-RAB3A (WB, 1:1000; Abcam, ab3335), rabbit anti-PINK1 (WB, 1:1000; Novus Biologicals, BC100-494), rabbit anti-OPTN (IF, 1:200; WB, 1:1000; Abcam, ab23666), mouse anti-COPB1 (WB, 1:1000; Sigma, G6160), rabbit anti-LRRK2 (WB, 1:1000; Abcam, ab133474). Peroxidase-linked secondary antibodies for WB were from GE Healthcare (NA931, NA934) and Agilent (P044901-2). Secondary antibodies for IF were donkey anti-mouse and anti-rabbit Alexa Fluor-488 and −555, anti-sheep Alexa Fluor-555 and anti-goat Alexa Fluor-488 (Thermo Fisher, a21202, a31570, a21206, a31572, a21436, a11055).
Cdnas and sirnas
pCMV6-Entry vectors containing cDNAs for FLAG-tagged human RAB3A, RAB5B, RAB7A, RAB10, RAB12 and RAB29 and pCMV6-XL5 vector containing cDNA for untagged human RAB10 were purchased from Origene (RC203973, RC208529, RC201776, RC201464, RC211472, RC200431 and SC114472). GFP-tagged RAB10 was constructed by subcloning RAB10 in pCMV6-AC-GFP (Origene, PS100010). The mito-Keima construct (mt/mKeima/pIND[SP1]) was a gift from Dr. A. Miyawaki (RIKEN Brain Science Institute, Japan) [25], and the Keima construct (mKeima-Red-N1) was a gift from Dr. M. Davidson (Addgene, 54597). The GFP-LC3 construct was a gift from Dr. J. Debnath (Harvard Medical School, USA). The OPTN-EGFP was a gift from Dr. B. Yue (Addgene, 27052) [55]. The HA-PRKN construct was a gift from Dr. O. Corti (ICM, Paris) [56]. Empty vector was pcDNA3 (Invitrogen, V79020). The RAB10 Q68L, T23N, ΔCC, T73E and T73A mutant constructs were generated using the QuikChange Lightning Site- Directed Mutagenesis Kit (Agilent, 210518) with the following primers: 5‘- GGGATACAGCAGGCCTGGAGCGATTTCACAC -3‘ and 5‘- GTGTGAAATCGCTCCAGGCCTGCTGTATCCC -3‘ (for Q68L); 5‘- CCGGAGTGGGGAAGAACTGCGTCCTTTTTCG -3‘ and 5‘- CGAAAAAGGACGCAGTTCTTCCCCACTCCGG -3‘ (for T23N); 5‘-GCGTACGCGTTTTGCTCTTCCAGCCTGTC -3‘ and 5‘- GACAGGCTGGAAGAGCAAAACGCGTACGC -3‘ (for ΔCC); 5‘- CCAGGAGCGATTTCACGAAATCACAACCTCCTA-3‘ and 5‘- TAGGAGGTTGTGATTTCGTGAAATCGCTCCTGG-3‘ (for T73E); and 5‘-CCAGGAGCGATTTCACGCCATCACAACCTCCTA-3‘ and 5‘-TAGGAGGTTGTGATGGCGTGAAATCGCTCCTGG-3‘ (for T73A). Mutagenesis was verified by sequencing. LRRK2 siRNA1 (ID263837), siRNA2 (ID272780), siRNA3 (ID272779) and siRNA4 (ID272781) were from Thermo Fisher, and RAB10 siRNA1 was from Dharmacon (L-010823–00-0005). The sequence of RAB10 siRNA2, RAB3A siRNA1, RAB3A siRNA2 and control siRNA was 5ʹ-GGGTAATGCAGAAGTGATAGC-3ʹ, 5ʹ-ACCAACGAGGAATCCTTCAAT-3ʹ, 5ʹ-CGAGAAGATGTCCGAGTCGTT-3ʹ and 5′-AATTCTCCGAACGTGTCACGT-3′, respectively (Qiagen).
Fibroblast culture and transfection
Fibroblasts from PD patients and age-matched controls were obtained via skin biopsy from the medial aspect of the upper limb after written informed consent. All procedures were approved by the local ethics committee and were in accordance with the latest version of the World Medical Association Declaration of Helsinki. Demographic and clinical characteristics of the subjects are provided in Table EV1. Fibroblasts were grown as described [22] in DMEM F12 (Invitrogen, 31331093) supplemented with fetal bovine serum (10%; Greiner Bio-One, 10270106), non-essential amino acids (1%; Thermo Fisher, 11140035), penicillin (100 U/ml; Thermo Fisher, 15140112), streptomycin (100 µg/ml; Thermo Fisher, 15140112) and sodium bicarbonate (0.12%; Sigma, S8761) at 37°C in a 5% CO2 humidified atmosphere. Cultures were repeatedly tested for Mycoplasma infection and tests were always negative. Fibroblasts were transiently transfected with 50 nm siRNA or 3 µg cDNA using the Neon Transfection System (Invitrogen, MPK1096) according to the manufacturer’s instructions. Transfection efficiency for siRNAs was determined using the BLOCK-iT fluorescent oligo (Thermo Fisher, 13750062) and was 97.8 ± 0.2% for Control 1, 95.5 ± 1.8% for G2019S1, 100 ± 0% for Control 2 and 98,3 ± 0.4% for R1441C (n = 3). Transfection efficiency for the Keima cDNA was 43.6 ± 2.8% for Control 1, 45.5 ± 2.5% for G2019S1, 48.6 ± 3.4% for Control 2 and 41.0 ± 3.5% for R1441C (n = 3). Transfection efficiency for FLAG-RAB10 cDNA was 22.7 ± 0.6% for Control 1, 22.4 ± 1.1% for G2019S1, 20.0 ± 3.5% for Control 2, and 19.3 ± 2.0% for R1441C (n = 3). Experiments were performed at passage numbers 5–15. Within the same experiment control and mutant fibroblasts were used at equivalent passage numbers. Starvation-induced autophagy was triggered by incubation in Earle’s balanced salt solution (EBSS) (Sigma, E2888). Cells were treated with valinomycin, oligomycin, antimycin A, 3-methyladenine (Sigma, V3639, 495455, A8674, M9281), bafilomycin A1 (Abcam, ab120497), CCCP (Calbiochem, 215911), LRRK2-IN-1 (Merck, 438193), PF-06447475 and MLi-2 (Tocris, 5716, 5756).
Immunofluorescence, confocal microscopy and image analysis
Immunostaining of cultured cells was performed as described previously [12,22]. Confocal images with 1 µm slice thickness were acquired at room temperature with an Axioskop 2 microscope (LSM510 META; Carl Zeiss), a 63X NA 1.4 oil Plan Apochromat objective and an AxioCam HR camera (Carl Zeiss). Brightness and contrast were adjusted with NIH ImageJ software. Random images were captured and analyzed by an investigator blinded to genotype and experimental condition.
Western blot, mitochondrial isolation and co-ip
Western blot was performed as described [12,22]. In brief, fibroblasts were washed with ice-cold PBS, removed with a scraper and resuspended in PBS (350 mM NaCl, 2.7 mM KCl, 10.2 mM Na2PO4, 1.75 mM KH2PO4, pH 7.4) with 1% Triton X-100 (Sigma, 93,443). After solubilization on ice for 30 min, insoluble material was removed by centrifugation at 20 000g for 5 min. Protein concentrations were determined using Bio-Rad Protein assay. SDS loading dye was added to the samples, followed by denaturation at 99°C for 10 min, SDS-PAGE, blotting onto PVDF membranes and incubation with blocking solution, primary and secondary antibodies. Immunoreaction was visualized with ECL (Sigma, WBLUC, WBLUR) or ECL Prime (GE Healthcare, RPN2232) on a Fujifilm LAS-3000 Imager. The density of scanned signals was measured with UN-SCAN-IT gel 6.1 (Silk Scientific). Mitochondria were isolated as described [22]. For Co-IP, fibroblasts transfected with FLAG-tagged RAB10 constructs were washed with ice-cold PBS, removed with a scraper, and resuspended in lysis buffer (20 mM Tris-HCl, 500 mM NaCl and 1% Triton X-100, pH 7.4). After solubilization for 30 min at 4°C, insoluble material was removed by centrifugation at 20,000 g for 5 min. Equal protein amounts were incubated with anti-FLAG M2 Affinity Gel (Sigma, A2220) for 2 h at 4°C after which beads were washed 3 times with lysis buffer. Bound proteins were eluted in lysis buffer containing FLAG peptides (0.1 µg/µl; Sigma, F3290) for 30 min at 4°C, followed by western blot.
Live quantification of autophagy
Fibroblasts were transfected with mito-Keima or Keima for live imaging of mitophagy and starvation-induced autophagy, respectively. Transfected cells were analyzed using a Leica TCS SP5 II confocal microscope equipped with a 63x objective lens (HC PL APO 63x/1.4 CS2), a multi-argon laser (458, 476, 488 nm) and a He/Ne laser (543 nm). Keima and mito-Keima were imaged in 2 channels via 2 sequential excitations (458 nm, green; 543 nm, red) and using a 600- to 695-nm emission range. Ratio (543:458) images were created using the Ratio Plus plugin in ImageJ, and high (543:458) ratio areas were segmented. The parameter (high [543:458] ratio area:total cell area) was used as an index of autophagy and the parameter (high [543:458] ratio area:total mitochondrial area) as an index of mitophagy, as described [25]. LysoTracker Green DND-26 (Thermo Fisher, L7526) was imaged using a 488-nm excitation and a 495–550 nm emission filter.
Mitochondrial membrane potential and ROS production
Mitochondrial membrane potential and ROS production were assessed using TMRM (Thermo Fisher, T668) and MitoSOX Red (Thermo Fisher, M36008) imaging, respectively. Briefly, fibroblasts were plated in 12-well plates (at 5 × 104 cells per well) and allowed to attach overnight at 37°C. Following the required treatments, cells were pulsed with either TMRM (100 nM) or MitoSOX (2 µg/ml) diluted in cell media for 30 min at 37°C. Subsequently, cells were rinsed twice with PBS, trypsinized and collected by centrifugation (300 x g, 5 min). Samples were then re-suspended in FACs buffer (PBS containing 1% BSA [Sigma, A7030], 2% fetal bovine serum) and kept on ice for the remainder of the experiment. The fluorescent intensities of 10,000 cells per condition were acquired using an Attune flow cytometer (Life Technologies).
Statistics
Significance of differences was analyzed with two-tailed Student’s t-test for comparison between two groups and with one-way ANOVA and post hoc Holm – Sidak test for comparison between more than two groups (SigmaStat 3.5, Systat). Values and error bars represent mean ± SEM.
Supplementary Material
Funding Statement
This work was supported by the KU Leuven [Opening the Future Campaign]; Onderzoeksraad, KU Leuven [C16/15/073]; Onderzoeksraad, KU Leuven [GOA/13/017]; Onderzoeksraad, KU Leuven [OT/13/091]; Geneeskundige stichting Koningin Elisabeth [NA].
Acknowledgments
T.C. is a Postdoctoral Fellow and W.V. a Senior Clinical Investigator of the Research Foundation Flanders (FWO). This work was supported by KU Leuven (GOA/13/017 and ‘Opening the Future’ campaign) and the Queen Elisabeth Medical Foundation. The P.V. lab is supported by KU Leuven (C16/15/073 and OT/13/091) and Interuniversity Attraction Poles (P7/13). We are grateful to A. Miyawaki, M. Davidson, J. Debnath, O. Corti and B. Yue for providing constructs.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Poewe W, Seppi K, Tanner CM, et al. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013. [DOI] [PubMed] [Google Scholar]
- [2].Kowal SL, Dall TM, Chakrabarti R, et al. The current and projected economic burden of Parkinson‘s disease in the United States. Mov Disord. 2013;28:311–318. [DOI] [PubMed] [Google Scholar]
- [3].Corti O, Lesage S, Brice A.. What genetics tells us about the causes and mechanisms of Parkinson‘s disease. Physiol Rev. 2011;91:1161–1218. [DOI] [PubMed] [Google Scholar]
- [4].Nalls MA, Pankratz N, Lill CM, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson‘s disease. Nat Genet. 2014;46:989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Martin I, Kim JW, Dawson VL, et al. LRRK2 pathobiology in Parkinson’s disease. J Neurochem. 2014;131:554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Steger M, Tonelli F, Ito G, et al. Phosphoproteomics reveals that Parkinson‘s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife. 2016;5:e12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhen Y, Stenmark H. Cellular functions of Rab GTPases at a glance. J Cell Sci. 2015;128:3171–3176. [DOI] [PubMed] [Google Scholar]
- [8].Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson‘s disease. Neuron. 2015;85:257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Narendra D, Tanaka A, Suen DF, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. [DOI] [PubMed] [Google Scholar]
- [11].Matsuda N, Sato S, Shiba K, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Van Humbeeck C, Cornelissen T, Hofkens H, et al. Parkin interacts with Ambra1 to induce mitophagy. J Neurosci. 2011;31:10249–10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kazlauskaite A, Kondapalli C, Gourlay R, et al. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014;460:127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kane LA, Lazarou M, Fogel AI, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Koyano F, Okatsu K, Kosako H, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. [DOI] [PubMed] [Google Scholar]
- [16].Chan NC, Salazar AM, Pham AH, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sarraf SA, Raman M, Guarani-Pereira V, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014;111:E4439–E4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cornelissen T, Vilain S, Vints K, et al. Deficiency of parkin and PINK1 impairs age-dependent mitophagy in Drosophila. eLife. 2018;7:e35878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018;20:1013–1022. [DOI] [PubMed] [Google Scholar]
- [22].Cornelissen T, Haddad D, Wauters F, et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet. 2014;23:5227–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Papkovskaia TD, Chau KY, Inesta-Vaquera F, et al. G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum Mol Genet. 2012;21:4201–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tashiro K, Shishido M, Fujimoto K, et al. Age-related disruption of autophagy in dermal fibroblasts modulates extracellular matrix components. Biochem Biophys Res Comm. 2014;443:167–172. [DOI] [PubMed] [Google Scholar]
- [25].Katayama H, Kogure T, Mizushima N, et al. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18:1042–1052. [DOI] [PubMed] [Google Scholar]
- [26].Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. [DOI] [PubMed] [Google Scholar]
- [27].Bravo-San Pedro JM, Niso-Santano M, Gomez-Sanchez R, et al. The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell Mol Life Sci. 2013;70:121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Manzoni C, Mamais A, Dihanich S, et al. Pathogenic Parkinson’s disease mutations across the functional domains of LRRK2 alter the autophagic/lysosomal response to starvation. Biochem Biophys Res Commun. 2013;441:862–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gegg ME, Cooper JM, Chau KY, et al. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang X, Winter D, Ashrafi G, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hsieh CH, Shaltouki A, Gonzalez AE, et al. Functional impairment in Miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson‘s disease. Cell Stem Cell. 2016;19:709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Steger M, Diez F, Dhekne HS, et al. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. eLife. 2017;6:e31012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lis P, Burel S, Steger M, et al. Development of phosphor-specific Rab protein antibodies to monitor in vivo activity of the LRRK2 Parkinson’s disease kinase. Biochem J. 2018;475:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gómez-Suaga P, Rivero-Ríos P, Fdez E, et al. LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum Mol Genet. 2014;23:6779–6796. [DOI] [PubMed] [Google Scholar]
- [35].Purlyte E, Dhekne HS, Sarhan AR, et al. Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. Embo J. 2018;37:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hattula K, Peränen J. FIP-2, a coiled-coil protein, links Huntington to Rab8 and modulates cellular morphogenesis. Curr Biol. 2000;10:1603–1606. [DOI] [PubMed] [Google Scholar]
- [37].English AR, Voeltz GK. Rab10 GTPase regulates ER dynamics and morphology. Nat Cell Biol. 2013;15:169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Heo JM, Ordureau A, Paulo JA, et al. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. 2015;60:7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Richter B, Sliter DA, Herhaus L, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 2016;113:4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wei Y, Chiang WC, Sumpter R, et al. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168:224–238.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yamano K, Fogel AI, Wang C, et al. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife. 2014;3:e01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yamano K, Wang C, Sarraf SA, et al. Endosomal Rab cycles regulate Parkin-mediated mitophagy. eLife. 2018;7:e31326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Minowa-Nozawa A, Nozawa T, Okamoto-Furuta K, et al. Rab35 GTPase recruits NPD52 to autophagy targets. Embo J. 2017;36:2790–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mortiboys H, Johansen KK, Aasly JO, et al. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology. 2010;75:2017–2020. [DOI] [PubMed] [Google Scholar]
- [45].Mortiboys H, Furmston R, Bronstad G, et al. UDCA exerts beneficial effect on mitochondrial dysfunction in LRRK2(G2019S) carriers and in vivo. Neurology. 2015;85:846–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Vincow ES, Merrihew G, Thomas RE, et al. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A. 2013;110:6400–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].McLelland GL, Soubannier V, Chen CX, et al. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. Embo J. 2014;33:282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bonello F, Hassoun SM, Mouton-Liger F, et al. LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: pathologic insights into Parkinson’s disease. Hum Mol Genet. 2019. DOI: 10.1093/hmg/ddz004. [DOI] [PubMed] [Google Scholar]
- [49].Cherra SJ, Steer E, Gusdon AM, et al. Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol. 2013;182:474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhu Y, Wang C, Yu M, et al. ULK1 and JNK are involved in mitophagy incurred by LRRK2 G2019S expression. Protein Cell. 2013;4:711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wild P, Farhan H, McEwan DG, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Manzanillo PS, Ayres JS, Watson RO, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501:512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mira MT, Alcaïs A, Nguyen VT, et al. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature. 2004;427:636–640. [DOI] [PubMed] [Google Scholar]
- [54].Zhang FR, Huang W, Chen SM, et al. Genomewide association study of leprosy. N Engl J Med. 2009;361:2609–2618. [DOI] [PubMed] [Google Scholar]
- [55].Park BC, Shen X, Samaraweera M, et al. Studies of optineurin, a glaucoma gene: golgi fragmentation and cell death from overexpression of wild-type and mutant optineurin in two ocular cell types. Am J Pathol. 2006;169:1976–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hampe C, Ardila-Osorio H, Fournier M, et al. Biochemical analysis of Parkinson‘s disease-causing variants of parkin, an E3 ubiquitin ligase with monoubiquitylation capacity. Hum Mol Genet. 2006;15:2059–2075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.