

ABSTRACT
Obesity is associated with changes in the immune system that significantly hinder its ability to mount efficient immune responses. Previous studies have reported a dysregulation of immune responses caused by lipid challenge; however, the mechanisms underlying that dysregulation are still not completely understood. Autophagy is an essential catabolic process through which cellular components are degraded by the lysosomal machinery. In T cells, autophagy is an actively regulated process necessary to sustain homeostasis and activation. Here, we report that CD4+ T cell responses are inhibited when cells are challenged with increasing concentrations of fatty acids. Furthermore, analysis of T cells from diet-induced obese mice confirms that high lipid load inhibits activation-induced responses in T cells. We have found that autophagy is inhibited in CD4+ T cells exposed in vitro or in vivo to lipid stress, which causes decreased autophagosome formation and degradation. Supporting that inhibition of autophagy caused by high lipid load is a key mechanism that accounts for the effects on T cell function of lipid stress, we found that ATG7 (autophagy-related 7)-deficient T cells, unable to activate autophagy, did not show additional inhibitory effects on their responses to activation when subjected to lipid challenge. Our results indicate, thus, that increased lipid load can dysregulate autophagy and cause defective T cell responses, and suggest that inhibition of autophagy may underlie some of the characteristic obesity-associated defects in the T cell compartment.
Abbreviations: ACTB: actin, beta; ATG: autophagy-related; CDKN1B: cyclin-dependent kinase inhibitor 1B; HFD: high-fat diet; IFNG: interferon gamma; IL: interleukin; MAPK1/ERK2: mitogen-activated protein kinase 1; MAPK3/ERK1: mitogen-activated protein kinase 3; MAPK8/JNK: mitogen-activated protein kinase 8; LC3-I: non-conjugated form of MAP1LC3B; LC3-II: phosphatidylethanolamine-conjugated form of MAP1LC3B; MAP1LC3B: microtubule-associated protein 1 light chain 3 beta; MS: mass spectrometry; MTOR: mechanistic target of rapamycin kinase; NFATC2: nuclear factor of activated T cells, cytoplasmic, calcineurin dependent 2; NLRP3: NLR family, pyrin domain containing 3; OA: oleic acid; PI: propidium iodide; ROS: reactive oxygen species; STAT5A: signal transducer and activator of transcription 5A; TCR: T cell receptor; TH1: T helper cell type 1
KEYWORDS: Fatty acid, lipid stress, macroautophagy, obesity, T cell
Introduction
Obesity-associated changes in the immune system can affect both innate and adaptive immunity, dramatically impairing the immune response against infection [1–5]. Similarly, genetic mouse models of obesity or mice fed a high fat diet display reduced proliferative responses, altered cytokine secretion and reduced ability to respond to immunizations and clear infections [6–13]. It has been shown that lipids, such as fatty acids and cholesterol, are potent modulators of T cell function in laboratory animals and cell cultures, suggesting that high lipid load might directly contribute to immune dysfunction in obesity [14–18]. A series of mechanisms have been proposed to account for the effects of fatty acids on T cell activation, including decreased calcium signaling, reduced protein kinase C theta activation or altered cytoskeleton rearrangement [18–22]. In many cases, those changes appear to derive from the incorporation of those lipids into the plasma membrane, which causes modifications of the dynamics of lipid rafts and hinders recruitment of signaling intermediates [21–24]. The molecular and cellular mechanisms through which lipids may affect T cell function are likely affecting numerous processes that are involved in the regulation of the T cell response to antigen and remain to be fully elucidated.
Macroautophagy (hereafter referred as autophagy) is a highly conserved catabolic cellular process that has emerged as a crucial regulator of many cell functions [25,26]. This process involves the sequestration of cellular materials inside double membrane vesicles (autophagosomes) and their degradation upon fusion with lysosomes. Although it was first described as a survival mechanism induced upon nutrient starvation conditions, it was soon evident that autophagy constitutes an essential process of degradation and recycling, that participates in the regulation of programs of cell differentiation and function, homeostasis and quality control, and response to stress [26–28], and whose dysregulation participates in the pathogenesis of many diseases [29].
In T cells, autophagy controls organelle homeostasis [30,31], but it also participates in the regulation of T cell survival and proliferation [32–34]. Furthermore, autophagy may also help provide T cell with the bio-energetic requirements for activation [35], and can also regulate T cell activation through the controlled degradation of selective proteins that participate in specific signaling pathways activated downstream of the T cell receptor (TCR), including BCL10 and CDKN1B (cyclin dependent kinase inhibitor 1B) [34,36]. Degradation of cargo and sequestration inside autophagosomes appear to be essential to fully sustain the functions of autophagy in T cells, as cells lacking the small GTPase RAB7A, which promotes fusion between autophagosomes and lysosomes, show also impaired cell proliferation following TCR engagement [37].
Several reports support that autophagy is regulated by intra- and extracellular lipid levels [38–40]. The specific mechanisms of how lipids influence autophagy are still not completely understood. Upregulation of autophagy has been reported in response to increase levels of fatty acids in several cell types [40–42], whereas a number of reports have also shown inhibition of autophagy upon high lipid concentrations [39,43–45]. These results may suggest the existence of a lipid threshold below which autophagy is activated to respond to lipid stress, but above which autophagy can be negatively affected as a result of lipotoxicity.
In this work, we show evidence that in vitro and in vivo lipid challenge negatively affects the induction of activation-induced autophagy in T helper cells, which contributes to the inhibition of T cell responses observed in CD4+ T cells exposed to increasing concentrations of fatty acids or isolated from diet-induced obese mice. Our data supports that increased lipid load can dysregulate autophagy and cause defective T cell responses; and suggest that inhibition of autophagy may underlie some of the obesity-associated functional defects in the T cell compartment.
Results
In vitro lipid challenge downregulates CD4+ effector T cell function
Several studies have clearly established a direct correlation between obesity and inefficient immune responses [46]. Though many factors likely contribute to hinder immune function in obese individuals, we analyzed whether increased lipid challenge could directly have a negative effect on T cell function. As a model of increased lipid load in vitro, we determined the consequences for effector T cell function of exposure to increasing concentrations of fatty acids. After incubation of C57BL/6J CD4+ T helper cells type 1 (TH1) cells for 48 h with different concentrations of oleic acid (OA), cells were stimulated with anti-CD3 and anti-CD28 antibodies and proliferation and cytokine expression were assessed. These experiments revealed a dose-dependent inhibition of activation-induced proliferative capacity and cytokine production, in cells challenged with increasing concentrations of OA ranging from 0.25 mM to 0.75 mM (Figure 1(a–c)), that was not a consequence of increased cell death (Figure 1(d)). Similar results were obtained using T cells from Balb/c mice, excluding a strain-specific effect (Figure 1(e–g))
Figure 1.

OA-challenge inhibits activation-induced proliferation and cytokine secretion in mouse TH1 cells. CD4+ T cells isolated from C57Bl/6J mice were polarized into TH1 cells then incubated for 48 h in the presence of different concentrations of OA. Proliferation (a) and secretion of IL2 and IFNG (b and c) were assessed after 24 h of stimulation with plate bound anti-CD3 and anti-CD28 antibodies. Bars show mean+SEM from 11 independent experiments (*P < 0.05; **P < 0.01; ***P < 0.001. ANOVA). (d) Levels of live cells were measured by FACS (PI and ANXA5-FITC staining) in control and OA-challenged resting (Rest) and stimulated (Act) T cells. Bars represent average percentages of cell death from the total T cell population from 3 independent experiments. (e–g) Experiments as described in A-C were performed using T cells isolated from Balb/C mice. Bars show mean+SEM from 4 independent experiments (*P < 0.05; **P < 0.01. ANOVA).
In vitro lipid challenge inhibits activation-induced autophagy
Given the central role that autophagy plays in the regulation of programs that control T cells activation and differentiation [47] and the fact that when other cell types are challenged by exposure to high concentrations of lipids, autophagy activity is negatively affected [39,45], we explored the possibility that the lipid challenge-induced effect on T cell responses could correlate with an inhibition of autophagy in those cells. When we measured autophagy activity in TH1 cells exposed to increasing concentration of OA, assessing autophagy flux by either measuring turnover of the phosphatidylethanolamine-conjugated form of MAP1LC3B (microtubule-associated protein 1 light chain 3 beta) (LC3-II) by immunoblot or of LC3-II+ vesicles by immunofluorescence, we observed that cells challenged with OA showed reduced autophagy flux in a dose-dependent manner (Figure 2(a,b)), which correlated with the dose-dependent decrease of activation-induced responses we had observed (Figure 1)
To further characterize the effect of lipid challenge on activation-induced autophagy and determine if autophagosome formation was being impaired, we analyzed changes in LC3-II in T cells that were activated for 20 h and then incubated with ammonium chloride and leupeptin for 1, 2 or 3 h. Differences in the rate of accumulation of LC3-II after 1 or 3 h of incubation with lysosomal protease inhibitors provide information on the rate of autophagosome formation. Our analysis showed a dose-dependent reduction in the rate of accumulation of LC3-II in cells exposed to OA, supporting that lipid challenge was causing an inhibition of autophagosome formation in T cells (Figure 2(c)).
Figure 2.
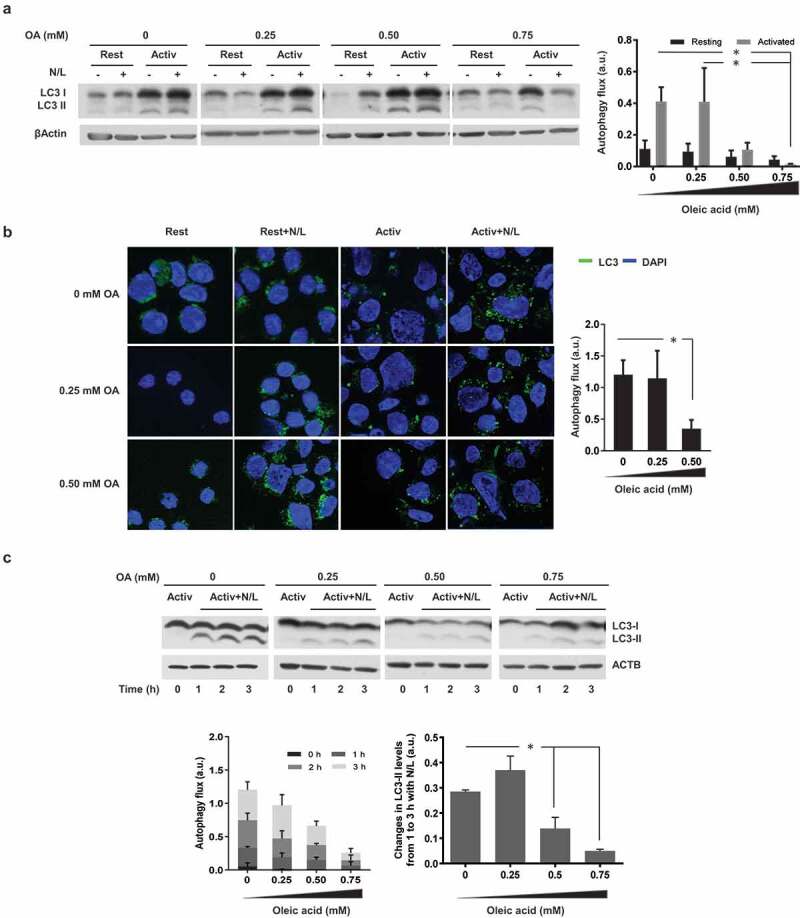
OA-challenge inhibits activation-induced macroautophagy activation. Mouse TH1 cells were incubated in the presence of different concentrations of OA for 48 h. Cells were then either left resting (Rest) or stimulated (Activ) with plate bound anti-CD3 and anti-CD28 antibodies for 24 h. NH4Cl and leupeptin (N/L) were added for the last 3 h of the 24-h stimulation to assess autophagy flux measured by (a) immunoblot (LC3-II degradation) or (b) immunofluorescence (LC3+ vesicle turnover). ACTB was used as loading control for normalization of values. Bar graphs represent mean+SEM of autophagy flux, measured as the difference between the intensity of the LC3-II band in cells activated in the presence or absence of N/L from 5 independent experiments or as the difference between the number of LC3+ puncta in cells cultured in the presence or absence of N/L from 10 fields per sample containing 30 cells per field (*P < 0.05. ANOVA). (c) Murine TH1 cells were incubated for 48 h in the presence or absence of different concentrations of OA and then activated with plate-bound anti-CD3 and anti-CD28 antibodies for 24 h. N/L were added for the last 1, 2 or 3 h of the 24-h activation period. Bar graphs represent mean+SEM of autophagy flux from 3 independent experiments, measured as the difference between the intensity of the LC3-II band in cells cultured in the presence or absence of N/L for different periods of time or the difference between LC3-II levels in cells incubated for 1 or 3 h with N/L (*P < 0.05; ANOVA).
In order to better understand how lipid challenge may affect T cells, we assessed if OA would incorporate into the cell membranes and lipids stores and alter their composition in T cells. When lipid extracts from control and OA-treated T cells were analyzed by thin layer chromatography, OA-treated cells showed an overall increase content of cholesterol, phosphatidylethanolamine and phosphatidylcholine (Figure 3(a)). We also subjected those lipid extracts to mass spectrometry (MS) analysis to confirm that the extra load of OA that was being used to challenge T cells would result in increased incorporation of OA into cell lipids and caused alteration of their normal distribution. For almost all classes of lipids identified (including cholesterol esters, triacylglycerol, phosphatidylcholine, phosphatidylethanolamine, phosphatidylinositol and phosphatidylglycerol) an increase of species that contained OA and a concomitant relative decrease in the presence of species containing other FA could be observed in OA-treated cells in a dose dependent manner (Figure 3(b,c)). These results confirmed that OA incorporated into the lipids of challenged T cells.
Figure 3.
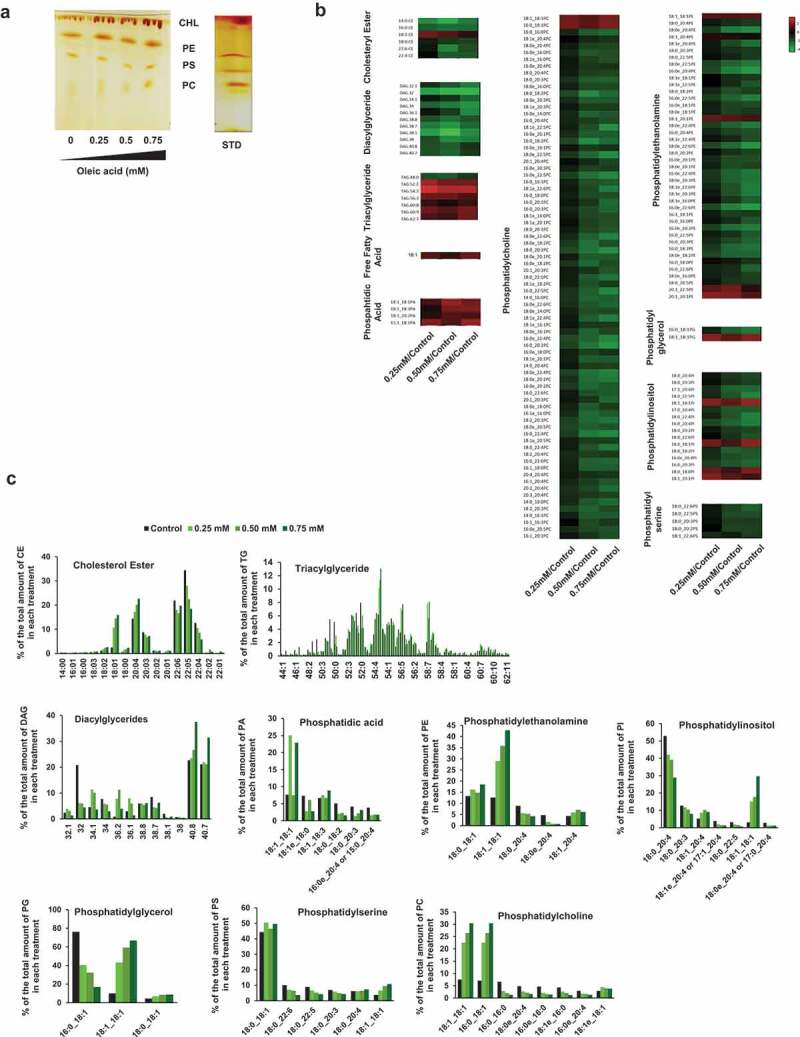
Qualitative and quantitative analysis of lipid extracts from murine and human CD4 + T cells challenged with oleic acid. (a) Thin layer chromatography of cholesterol (CHL), phosphatidylethanolamine (PE), phosphatidylserine (PS) and phosphatidylcholine (PC) of lipids extracted from murine CD4+ TH1 cells challenged with different doses of Oleic Acid. Dark-brown spots corresponding to the lipids of interest were identified using the corresponding lipid standards. (b) Heat map showing the comparative lipid profile of cells treated with increasing concentrations of OA. The three columns represent the normalized values of the individual lipid species. The color bar represents the log2 value of the ratio of each lipid species over the levels found on control untreated cells. Only changes that are greater than-2 fold are highlighted in the heat map. Relative increases and decreases are shown in red and green, respectively (n = 2). (c) Lipid distribution (expressed as relative percentage of each lipid species over the total amount of each lipid class) of control untreated cells and cells treated with increasing concentrations of oleic acid. CE, cholesterol ester; TG, triacyl glyceride; DAG, diacylglyceride; PA, phosphatidic acid; PE, phosphatidylethanolamine; PI, phosphatidylinositol; PG, phosphatidylglycerol; PS, phosphatidylserine; PC, phosphatidylcholine.
Disruption of autophagy in CD4 + T cells affects organelle homeostasis and the regulation of T cell metabolism [30,31,35]. To determine if the inhibitory effect of lipid challenge on autophagy would also result in reduced mitochondria turnover, we assessed mitochondrial content in cells treated with OA. Flow cytometry and fluorescence microscopy analyses using the mitochondrial dye Mitotracker Green confirmed increase accumulation of mitochondria in OA-treated T cells compared with control untreated T cells (Figure S1(a,b)). Likely as a consequence of the decreased mitochondrial turnover, OA-treated cells also generated increased levels of reactive oxygen species (ROS) following activation (Figure S2(a)). We also evaluated ATP generation in response to activation, which was markedly reduced in cells treated with OA (Figure S2(b)).
Diet-induced obesity negatively affects murine CD4+ T cell function
Having characterized the effects on CD4+T helper cell activation of an in vitro lipid challenge, we analyzed if in vivo, increased lipid intake would also result in inhibition of CD4+ T cell responses. For that purpose, we used a model of diet-induced obesity, were mice were fed for 16–20 weeks with a high-fat diet (HFD), in which fat provided 60% of the total calorie intake. HFD-fed mice gained approximately 40% more weight than mice fed with a control diet and showed increased levels of non-esterified fatty acids in serum (Figure 4(a,b)). CD4+ T cells isolated from HFD-fed mice showed a significant decrease in activation-induced proliferation and IL2 (interleukin 2) production when compared with cells from mice fed a control diet (Figure 4(c,d)). IFNG (interferon gamma) secretion was also reduced, though this difference did not reach statistical significance (Figure 4(e)). As we had seen in cells challenged in vitro with OA, we did not observe any significant difference in the levels of cell death between T cells obtained from HFD-fed mice and mice fed with a control diet (Figure 3(f)), supporting that decreased activation responded to an effect of HFD on the ability of T cells to engage activation-induced responses. In vivo immunization with ovalbumin in HFD-fed mice, resulted, however, in a biased type 1 response (Figure S3(a)), possibly due to the ability of obesity to potentiate type 1-like immune responses [48–50]. To circumvent that effect, which may be the result of chronic exposure to adipose tissue inflammation in obesity, and try assess in vivo the effect of high circulating lipids on T cell activation during an immunization, we utilized an adoptive transfer strategy. Ovalbumin323-339-peptide specific CD4 + T cells were isolated from OT-II mice and transferred into hosts that had been fed for 16 weeks with control diet or HFD. We then analyzed CD4+ T cell responses to ovalbumin peptide in cells isolated from those mice 7 days post-immunization. In this setting, cells transferred to HFD-fed mice showed markedly reduced responses when compared to cells from control diet-fed mice (Figure S3(b)).
Figure 5.

HFD-fed mice show reduced TCR-induced autophagy activation. CD4+ T cells isolated from HFD or control diet (CD) fed mice were either left resting (Rest) stimulated (Activ) with plate bound anti-CD3 and anti-CD28 antibodies for 24 h. NH4Cl and leupeptin (N/L) were added for the last 3 h of the 24-h stimulation to assess autophagy flux measured by (a) immunofluorescence (LC3+ vesicle turnover) or (b) immunoblot (LC3-II degradation). ACTB was used as loading control for normalization of values. Bar graphs represent mean+SEM of autophagy flux, measured as the difference between the number of LC3+ puncta in cells cultured in the presence or absence of N/L from 5 independent experiments or the difference between the intensity of the LC3-II band in cells activated in the presence or absence of N/L from 4 independent experiments (*P < 0.05; **P < 0.01; ***P < 0.001; ns: not significant. ANOVA or two-tailed t-test, respectively).
Figure 4.

High fat diet-induced obesity inhibits activation-induced proliferation and cytokine secretion in CD4+ T cells. (a) Final weight comparison of C57BL/6J female mice fed for a period of 16–20 weeks with a control diet (CD) or a high-fat diet (HFD). Graphs represent the mean final weight of 30 mice corresponding to 8 independent diets (**P < 0.01; Two-tailed t-test). (b) Serum non-esterified fatty acids levels (NEFA) of CD-fed mice and HFD-fed mice. Graph shows the mean serum fatty acid levels at the time of sacrifice from 20 mice representing 6 independent experiments (**P < 0.01. Two-tailed t-test. (c–e) CD4+ T cells from CD-fed and HFD-fed mice were left resting (Rest) or stimulated (Activ) for 24 h with plate bound anti-CD3 and anti-CD28 antibodies. Proliferation (c) and cytokine secretion (d–e) were measured. Graph shows mean from 26 mice representing 8 independent experiments (**P < 0.01; ***P < 0.001; ns: not significant. ANOVA). (f) Levels of live cells determined by FACS (Propidium iodide and ANXA5-FITC staining) in CD4+ T cells isolated from control and HFD-fed mice assessed in resting or activated cells. Bars indicate mean percentages of live cells in the total CD4 + T cell population from 6 mice corresponding to 2 independent experiments.
Diet-induced obesity negative effect on murine CD4+ T cell function correlates with a down-regulation of autophagy activation
To determine if, as observed in the in vitro experiments, reduced T cell function upon lipid dietary challenges in vivo would correlate with decreased autophagy activity, we compared the levels of autophagy flux in CD4+ T cells isolated from mice fed either a control diet or a HFD. CD4+ T cells isolated from HFD-fed mice failed to efficiently up-regulate autophagy activity in response to TCR engagement when compared to cells from mice that were fed a control diet (Figure 5).
To better understand the mechanisms by which autophagy was being affected in T cells in mice fed a HFD, we analyzed by electron microscopy the abundance of autophagic vesicles in those cells. Confirming a defect in activation-induced autophagy, following stimulation with anti-CD3 and anti-CD28 antibodies, CD4+ T cells from HFD-fed mice had a reduced number of autophagic vesicles when compared with cells form mice fed a control diet (Figure 6(a,b)). Interestingly, this decrease appeared to be mostly a consequence of a marked reduction in the number of autolysosomes (Figure 6), supporting that HFD could result in an inhibition of autophagosome-lysosome fusion. Similar results have been reported in hepatocytes subjected to high lipid load, where these defects in the fusion of autophagosomes and lysosomes [45], have been attributed to changes in the lipid composition of the membranes of these organelles. As we had seen in cells challenged in vitro with OA, the decrease in autophagy observed CD4 + T cells form HFD-fed mice also resulted in increased mitochondrial content when compared with cells fed with a control diet (Figure S1(c)).
Figure 6.
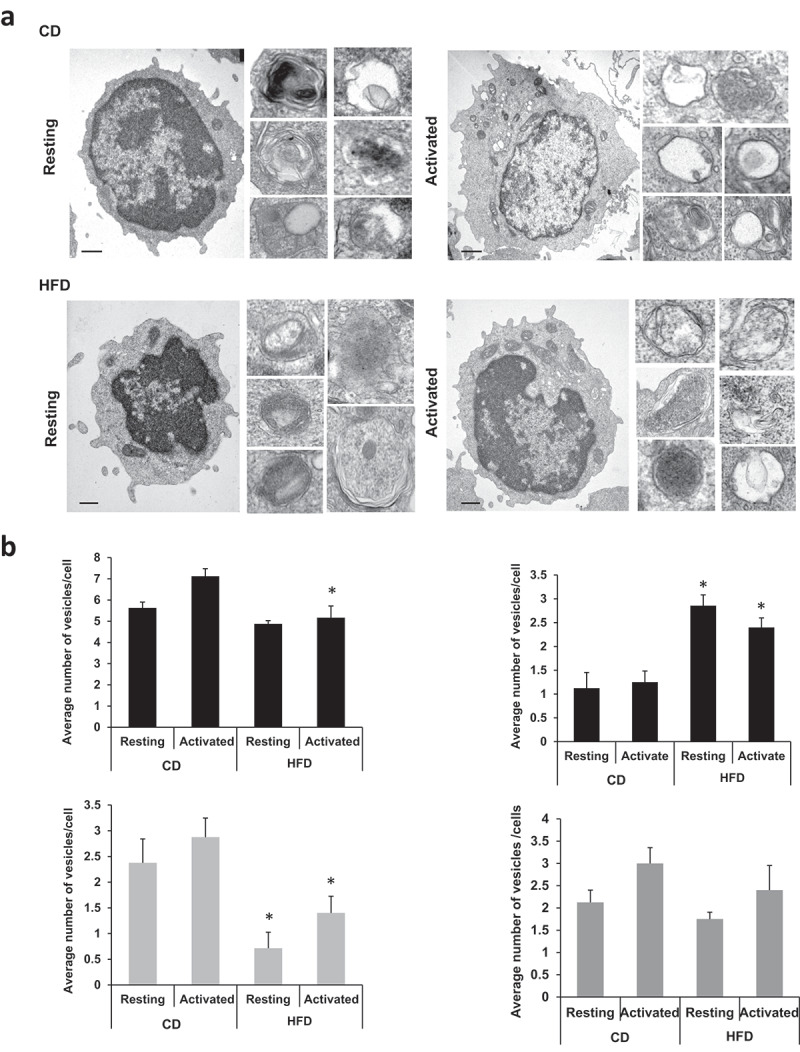
Ultrastructural analysis of the autophagic compartments in resting and activated CD4+ T cells in control diet (CD) and HFD-fed mice.CD4+ T cells isolated from CD- or HFD-fed C57BL/6J mice we left resting or stimulated for 24 h with plate bound anti-CD3 and anti-CD28 antibodies. Samples were processed for electron microscopy analysis. (a) Lower magnification fields show representative cells and higher magnification fields show individual autophagic vesicles. Bar: 1 µm. (b) Morphometric analysis was performed in 15–20 micrographies as the ones shown in panel a, corresponding to cells from two different experiments. The number of autophagic vacuoles, autophagosomes (APG), autolysosomes (AUL) and lysosomes (Lys) per cell are shown. Bars show mean+SEM. Differences between CD and HFD are shown (*P < 0.05. ANOVA).
Effects of lipid challenge on T cells are reversible
Having established that function and autophagy activity on CD4+T cells were inhibited by in vitro or in vivo lipid challenges, we determined whether those effects were permanent or could be reversed. We proceeded to assess the effect of culturing previously OA-challenged T cells in media with no added OA for different periods of time. TH1 were cultured in the presence 0.75 mM OA for 48h and allowed to recover for different periods of time (0, 1 or 2 days) in media with no added OA. When cytokine secretion in response to re-stimulation were analyzed, a clear pattern of restored responses was observed when cells were allowed to recover for longer periods of time (Figure 7(a)). Restoration of T cell responses correlated with a recovery of autophagy activity in activated T cells (Figure 7(b)). Similar recovery of T cell responses was observed when CD4 + T cells isolated from HFD-fed mice were cultured for 7 days in media without addition of extra fatty acids (Figure 7(c)). To determine if this recovery of the effects of high lipid load on autophagy could also be achieved in vivo, mice that were fed HFD for 16 weeks were either left on HFD or returned to a control diet for an additional 12 weeks. Within a month of withdrawing the HFD, mice restored their weight to levels similar to those of mice in a control diet and maintained it for the rest of the experiment (Figure 7(d)). Supporting our in vitro results, whereas CD4+ T cells from mice fed with HFD displayed a significantly lower activation-induced autophagy flux, cells from mice that were switched from HFD to a control diet showed levels of autophagy similar to those found in control mice (Figure 7(e)).
Figure 7.

Recovery of cell function and autophagy activity in CD4+ T cells exposed to lipid challenge after removal of excess lipid load. (a) Murine TH1 cells were cultured in normal media (Ctrl) or in the presence of 0.75 mM OA for 48 h followed by recovery in excess OA-free media for 2 days (2d rec), 1 day (1d rec) or without any recovery time (No rec). Cells were then stimulated with antiCD3 and antiCD28 antibodies for 24 h and cytokine production measured by ELISA. Bars show mean+ SEM from 3 independent experiment. (*P < 0.05; ns. not significant. ANOVA). (b) Autophagy flux was measured on those cells after NH4Cl and leupeptin (N/L) were added for the last 3 h of the 24-h stimulation. Levels of LC3-II were measured by immunoblot using anti-LC3 antibodies. ACTB was used as loading control for normalization of values. Bar graphs represent mean+SEM of quantification of activation induced autophagy flux from 2 independent experiments. (c) Murine CD4 + T cells were isolated from C57BL/6J mice that were fed with either CD or HFD for 16 weeks. IL2 production was measured in resting T cells (Rest) and in cell activated with plate bound anti-CD3 and anti-CD28 antibodies for 24 h (Act) by ELISA immediately after isolation (Day 0) or following 7 days of culture in media without excess fatty acids (Day 7). Bars represent mean+SEM from 5 different mice per condition. (*P < 0.05. Two-tailed t-test). (d) Body weight (mean±SEM) curves of C57BL/6J mice that were fed with either CD or HFD for 28 weeks, and mice fed with HFD for 16 weeks and switched to CD for 12 more weeks. Weight was measured every week. (e) CD4+ T cells were isolated from those mice and were either left resting or stimulated with plate bound anti-CD3 and anti-CD28 antibodies for 24 h. To measure autophagy flux, NH4Cl and leupeptin (N/L) were added for the last 3 h of the 24-h stimulation period. Levels of LC3-II in the presence or absence of inhibitors were determined by immunoblot using anti-LC3 antibodies on whole cell lysates. ACTB was used as loading control for normalization of values. Bar graph represent mean+SEM of autophagy flux (measured as the difference between the intensity of the LC3-II band in cells cultured in the presence or absence of N/L) from 5 mice per condition, (*P < 0.05, **P < 0.01, ns = not significant. Two-tailed t-test).
Lipid challenge fails to further inhibit T cell function in autophagy-deficient CD4+ T cells
We first determined if exposure to increasing concentration to OA might have any effect on TCR-mediated signaling that could explain the inhibition of activation-induced responses observed in those cells. We did not detect any significant differences in TCR internalization, phosphorylation of MAPK1/ERK2 (mitogen-activated protein kinase 1)-MAPK3/ERK1 and MAPK8/JNK (mitogen-activated protein kinase 8) or dephosphorylation of the transcription factor NFATC2 (nuclear factor of activated T cells, cytoplasmic, calcineurin dependent 2) between control cells and cells exposed to 0.25 mM or 0.5 mM of OA, though a small decreased of those events could be observed in cells treated with 0.75 mM OA (Figures 8(a,b) and S2). Similar analyses performed in CD4+ T cells isolated from mice fed with either HFD or a control diet also fail to detect any significant difference in the extent of activation-induced internalization of the TCR complex or in the levels of phosphorylation of MAPK1, MAPK3 and MAPK8 (Figure 8(c,d)). We found, however, a small decrease in the levels of MTOR (mechanistic target of rapamycin kinase) phosphorylation and a significant reduction of STAT5A (signal transducer and activator of transcription 5A) phosphorylation when cells were exposed to OA concentrations of 0.5 mM or 0.75 mM (Figure S4), which might respond to the reported direct effect of inhibition of autophagy on MTOR activation in T cells or to the reduced ability of T cells to produced IL2 in conditions where activation of autophagy is hindered [35,51].
Figure 8.
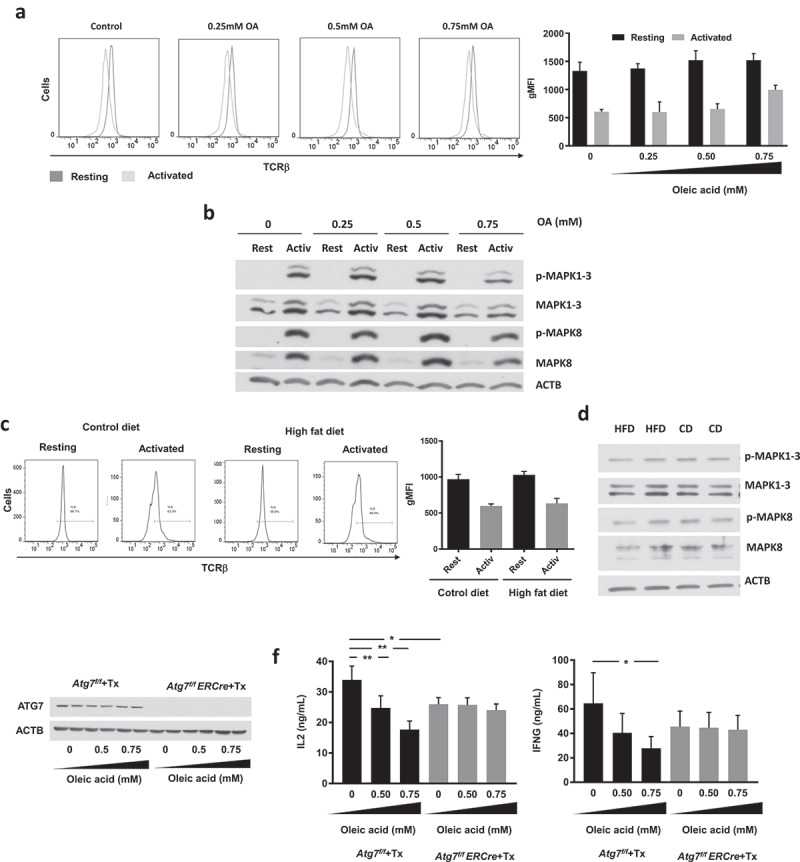
Decreased activation-induced responses to lipid challenge respond to the inhibition of autophagy in T cells. C57BL/6J CD4+ TH1 cells were cultured in the presence or absence of different concentrations of OA for 48 h. (a) Cells were then left resting or stimulated with plate-bound antiCD3 and antiCD28 antibodies for 24 h. Levels of TCR present in the plasma membrane were measured by FACS using fluorochrome-coupled anti-TCR beta chain antibodies. Bar graph represents mean+SEM of the geometrical mean fluorescence intensity (gMFI) from T cells from 2 independent experiments. (b) Cells were stimulated with plate-bound anti-CD3 and anti-CD28 antibodies for 1 h. MAPK1-MAPK3 and MAPK8 phosphorylation were determined by immunoblot on whole cell lysates. ACTB was used as loading control for normalization of values. (c,d) Similar experiments as in a-b were performed in CD4 + T cells isolated from control diet (CD) or high-fat diet (HFD) fed mice. (e) CD4 + T cells from Cre-ERT2-atg7flox/flox or Cre-ERT2-atg7+/+ mice were polarized into TH1 cells. Cells were treated with 3.5 μM hydroxy-tamoxifen for 24 h and grown in TH1 polarizing condition for an additional 4 days. Whole cell lysates from these samples were analyzed for ATG7 levels by immunoblot 48 h after treatment with oleic acid. (f) CD4 + T cells from Cre-ERT2-atg7flox/flox or Cre-ERT2-atg7+/+ mice were treated with 3.5μM hydroxytamoxifen to delete Atg7 as described. Cells were then incubated for 48 h in in the presence of absence of different concentrations of OA. After washing away the OA, cells were stimulated for 24 h with plate-bound anti-CD3 and anti-CD28 antibodies and IL2 and IFNG secretion determined by ELISA. Bars show mean+SEM from 3–5 different mice (*P < 0.05; **P < 0.01. ANOVA).
Having shown that under the conditions used in our experiments, lipid challenge did not appear to have a direct effect on early TCR signaling, we determined if the impairment in autophagy caused by high lipid load in T cells could be the mechanism responsible for the effect of lipids on T cell function. We assessed if exposure to OA would have any functional consequences in autophagy deficient T cells. If autophagy were the target of lipid challenge that accounted to reduced T cell responses, we would not expect autophagy deficient cells to show any additional inhibitory effect when exposed to lipid challenge. On the other hand, if reduced activation-induced responses were the consequence of the effect of lipids on other processes, autophagy-deficient cells would still be affected by exposure to high lipid concentrations. We used the Cre-ERT2-atg7flox/flox inducible system in order to generate a conditional somatic mutant mouse that allowed us to perform differentiation of effector T cells and then induce deletion of the essential autophagy gene Atg7 using tamoxifen. Tamoxifen induced a practically complete deletion of Atg7 in Cre-ERT2-atg7flox/flox TH1 cells, which resulted in only residual levels of ATG7 detected by immunoblot (Figure 8(e)). Supporting that autophagy may be a major target of lipid challenge responsible for its effects on T cell function, Atg7-deleted cells, which exhibited decreased T cell function due to their inability to activate autophagy [35], were, however, insensitive to challenge with 0.5 mM or 0.75 mM OA (Figure 8(f)).
Lipid challenge decreases T cell responses and inhibits autophagy in human CD4+ T cells
To confirm that the inhibitory effect of lipid challenge on autophagy and T cell function i also occurred in human CD4+ T cells, we analyzed the responses to OA exposure of CD4+ T cells isolated from peripheral human blood. Incubation with OA resulted in a dose-dependent decrease on the proliferative capacity and impaired cytokine secretion of activated human CD4+ T cells (Figure 9(a)). As we had seen in murine T cells, this effect was not a consequence of defective TCR signaling, as we detected no significant differences in TCR internalization or MAPK1-MAPK3 phosphorylation in OA-treated cells (Figure 9(b,c)). However, a marked dose-dependent decrease in activation-induced autophagy was detected in human T cells challenged with increasing concentrations of OA (Figure 9(d)).
Figure 9.

OA-challenge inhibits activation-induced proliferation, cytokine secretion and autophagy in human CD4+ T cells. (a) Human CD4+ T cells were incubated for 48 h in in the absence (Control) or presence of different concentrations of oleic acid. Proliferation and secretion of IL2 and IFNG were assessed after stimulation with anti-CD3 and anti-CD28 antibodies. Bars show mean+ SEM from 4 independent experiments. (b) Levels of TCR present in the plasma membrane were measured by FACS using fluorochrome-coupled anti-TCR antibodies . (c) MAPK1-MAPK3 phosphorylation was determined by immunoblot on whole cell lysates. ACTB was used as loading control for normalization of values. (d) Human CD4 + T cells were incubated in the presence or absence (Control) of different concentrations of oleic acid. Cells were then either left resting (Rest) stimulated (Activ) with plate bound anti-CD3 and anti-CD28 antibodies for 24 h and autophagy flux measured by LC3-II immunoblot. NH4Cl and leupeptin (N/L) were added for the last 3 h of the 24-h stimulation to assess LC3-II turnover. ACTB was used as loading control for normalization of values. Bar graphs represent mean+SEM of autophagy flux (measured as the difference between the intensity of the LC3-II band in cells activated in the presence or absence of lysosomal hydrolases inhibitors) from 6 independent experiments.
Discussion
It is well established that increased lipid load has a negative effect on immune function. Although the effects of lipids likely extend to different immune cell populations [9,52,53], many reports highlight a clear dysfunction of the T cell compartment. In vitro challenge with lipids or obesity can cause disturbed responses to antigen stimulation that result in increased susceptibility to infection [9,14,54]. The molecular mechanisms that may account for altered T cells responses upon exposure to lipid stress have remained so far not fully characterized. Our data show that the negative effect of lipid challenge on autophagy is, at least in part, responsible for the decreased responses observed in fatty-acid challenged CD4+ T cells or in T cells from obese mice, and support that altered autophagy constitutes a major mechanism behind T cell dysfunction in cells subjected to lipid challenge.
Autophagy has emerged as a crucial regulator of immune cell function [55], and dysregulation of autophagy has been linked to numerous immune and inflammatory pathologies [56–59]. In T cells, autophagy has been shown to modulate T cell homeostasis, survival and proliferation; activation-induced responses, including modulation of metabolism; and programs of differentiation that include generation of memory T cell populations, differentiation of T helper cells and maintenance of the regulatory T cell phenotype and function [30–36,47,51,60–68]. It is not surprising thus that inhibition of autophagy has a profound effect on T cell activation [33,35,51]. Here, we report that mouse and human CD4+ T cells exposed to increasing concentrations of oleic acid or T cells isolated from obese mice show a clear inhibition of their ability to induce autophagy in response to TCR engagement, which correlates with a defect in proliferation and cytokine production. Levels of fatty acids used in our in vitro experiments are of similar magnitude to the levels we measured in serum from diet-induced obese mice and to those reported in some pathological conditions [18,69], though fatty acid levels have been shown to rise even beyond those values under certain stress or disease conditions [70–72]. Under the conditions we tested, lipid challenge did not appear to affect TCR-induced signaling, and the effect of lipid stress on T cell function responded mainly to the downregulation of autophagy in those cells. Indeed, when we analyzed the effects of excess oleic acid in ATG7-deficient cells, we failed to detect any additive inhibitory effect on T cell responses caused by that lipid challenge. Interestingly, the inhibition of cytokine expression caused by exposure to oleic acid resulted in levels of activation-induced cytokine secretion similar to those found in autophagy-deficient T cells. Providing a connection between lipid challenge, autophagy and the immune response, it has also been reported that in the context of HFD, saturated fatty acids, such as palmitate, induce the activation of the NLRP3 (NLR family, pyrin domain containing 3) inflammasome in macrophages, which results in the secretion of the proinflammatory cytokines IL1B and IL18 and the downregulation of autophagy [73]. Given the role that autophagy may play in the regulation of the differentiation and function of many other cells in the immune system [55], it is reasonable to speculate that defective autophagy induced by lipid stress may alter the immune response through a more global dysregulation involving altered function of many cell types.
In many of our analyses, we saw a possible positive effect of low concentrations of oleic acid that turned inhibitory as the amount of fatty acid was increased. This suggest the existence of a threshold effect whereby activation or inhibition of autophagy would depend on the intensity of the lipid challenge received by the cells. Induction of autophagy by low concentrations of fatty acids has been reported in cancer cells, due to different mechanisms of activation depending on the nature- unsaturated or saturated- of the fatty acid [40]. Interestingly, studies in hepatocytes found a similar threshold effect as the one we have seen in T cells, where low amounts of fatty acids stimulated autophagy, but higher concentrations inhibited it [39,45]. Several studies have also examined the responses of T cells challenged with lipids and found that lipid challenge can have a positive or a negative effect on proliferation of T cells [17,24,74]. The difference might lie on the particular species of lipids used, the length of time they are present and the concentration at which they are employed [39,40,45,50,75,76]. Though fatty acid metabolism, which may be fueled by uptake of fatty acids by activated T cells, has been shown to be required to ensure adequate T cell activation [75], our data show, however, that chronic exposure to high levels of fatty acids may cause instead lipid-induced stress, which can target autophagy. Inhibition of autophagy under those conditions would cause an inhibitory effect on T cell proliferation and cytokine production.
Formation of autophagosomes and autophagosome-lysosome fusion events may be both altered in cells subjected to lipid challenge. Incorporation of fatty acids into cell lipids may change the distribution of different lipid species in cell structures and account for the changes in generation of autophagosome membrane and/or fusion events. It this sense, it has been shown that autophagosomes and lysosomes isolated from hepatocytes of mice fed a high fat diet present marked changes in their membrane lipid composition, which result in decreased ability of autophagosomes to fuse with lysosomes [45]. Though not explored in this study, we need to consider that lipid stress may also affect other forms of autophagy, which also control T cell function. Chaperone-mediated autophagy is also inhibited in hepatocytes from mice fed a high fat diet, which causes changes in the lysosomal membrane lipid composition that prevent the efficient assembly of the translocation complex required for substrate transport into the lysosomal lumen for degradation [77]. This same form of autophagy has been shown to regulate cell levels of key signaling molecules downstream of the TCR to regulate T cell activation [78]. Whether chaperone-mediated autophagy is also altered in T cells subjected to lipid stress remains to be determined.
Our data support that changes in T cell autophagy are restored after mice that have been fed with a HFD are reverted to a control diet or grown for one week ex vivo in media without added fatty acids, which highlights that negative effects of obesity and lipid challenge in T cells can be reversed by removing the source of stress. Interestingly, the gradual deterioration of the immune system associated with aging may be considered somewhat similar to the effects of obesity on the immune system. Whether dyslipidemia associated with the metabolic syndrome of aging contributes also to T cell malfunction remains to be determined. However, it is important to note that autophagy is also inhibited in T cells from old individuals [79] and that changes in lysosomal lipid membrane composition induced by high fat diet have been found to be similar to those present in lysosomes in aged cells [77].
This study defines, thus, reduced autophagy as a major mediator of the inhibitory effects of lipid challenge on T cell activation, and supports that targeting autophagy may help improve immune function in situations where increased lipid load may otherwise thwart T cell responses.
Material and methods
Mice
C57BL/6J, BalB/cJ and B6.129-Gt(ROSA)25Sortm1(cre/ERT2)Tyj/J mice were purchased from Jackson Laboratory (Bar Harbor, ME). Cre-ERT2-atg7flox/flox- mice were generated by crossing B6.129-Gt(ROSA)25Sortm1(cre/ERT2)Tyj/J mice with atg7flox/flox mice [80]. Mice were housed in a selective pathogen-free barrier facility. All procedures were reviewed and approved by The Albert Einstein College of Medicine’s Institutional Animal Care and Use Committee, confirming to accepted standards of humane animal care.
Animal diets
Four-week-old C57BL/6J mice were fed control chow diet (21.6% calories from fat; Lab Supply, Inc., Picolab Mouse Diet 20) for 2 weeks before starting any especial diet. To study diet induced obesity, mice were randomly separated in two groups of animals each with similar average body weight. Animals were then fed either a control diet (21.6% of calories provided by fat) or a HFD (60% calories from fat; OpenSource DIETS, DIO Series Diet HFD) for a total period of 16 weeks. Fresh food was provided every 7 days and food intake as well as body weight were recorded weekly.
Isolation and culture of CD4+ T cells
Mouse CD4+ T cells were isolated by positive selection using anti-CD4-coupled magnetic beds (Dynabeads® mouse CD4; Fisher Scientific, 11145D). To differentiate TH1 cells, purified CD4+ T cells were activated with plate-bound anti-CD3 (0.25 μg/ml; eBioscience, 16-0031-85, clone 145-2C11) and anti-CD28 (0.25 μg/ml; eBioscience, 16-0281-82, clone 37.51) antibodies in T cell media (DMEM [HyClone, SH30243.01] supplemented with 10% fetal bovine serum, 50 U/ml penicillin and 50 μg/ml streptomycin [Mediatech Inc., 30-001-Cl], 4 mM glutamax [Fisher Scientific, 35050061], 1X nonessential amino-acids [Lonza, BW13-114E], 1X MEM Eagle vitamin mixture [Lonza, 13-607C], 50 μM 2-mercaptoethanol [Fisher Scientific, BP176], 660 μM L-arginine [Acros Organics, AC10500], 270 μM L-asparagine [Acros Organics, AC175271000], 6 μM folic acid [Fisher Scientific, BP251910], 10 mM HEPES [Lonza, 17-737E], 1 mM sodium pyruvate [Life Technologies, 11360-070]) in the presence of 10 μg/ml anti-mouse IL4 (clone 11B11; BioXcell, BE0001-1) and recombinant mouse IL12 (eBioscience, 14812180). After 24 h, recombinant human IL2 (10 U/ml; Biological Resources Branch NCI) was added. TH1 cells were differentiated for 6 days. To delete the Atg7 gene, CD4+ T cells from Cre-ERT2-atg7flox/fox-mice were treated with hydroxytamoxifen (3.5 μM; Sigma-Aldrich, H7904) for 48 h. Human CD4+ T cells were isolated from anonymous donor leukopaks obtained from the New York Blood Center, following a protocol approved under the guidelines of the Institutional Review Board of the Albert Einstein College of Medicine. Human primary CD4+ T cells were isolated from peripheral blood mononuclear cells obtained in a density gradient generated using Ficoll-plaque plus (GE Healthcare Fisher, 17-1440-02), using a CD4 + T cell isolation Kit (Miltenyi Biotec, 130-104-453). Where stated, cells were activated with Dynabeads Human T-Activator CD3:CD28 beads (Thermo Fisher Scientific, 11131D). Human T cells were grown in RPMI medium supplemented with 10% fetal calf serum, 10 mM HEPES and 2 mM L-glutamax.
Fatty acid preparation and use
OA:bovine serum albumin was purchased from (Sigma-Aldrich, O3008). T cells were cultured for 48 h at the desired concentration of OA before being profusely washed in PBS (Fischer scientific, BP243820) prior to stimulation to analyze T cell activation and autophagy.
T cell function assessment
CD4+ T cells were activated with activator beads (human cells) or plate-bound 0.25 μg/ml anti-mouse CD3 and 0.25 μg/ml anti-mouse CD28 antibody for 24 h., before the addition of 10 µM 5-bromo-2‘-deoxyuridine (BrdU) for a 12-h labeling. Detection of BrdU incorporation was measured according to the manufacturer’s instructions using the BrdU Labeling and Detection Kit III (Sigma-Aldrich, 11444611001). To measure cytokine production cells were activated for 24 h. Supernatants were harvested and assessed for IL2 or IFNG presence using sandwich ELISA (for IL2, anti-mouse IL2 [eBioscience, 14-7022-85] and biotinylated anti-mouse IL2 [eBioscience 13-7021-81] or anti-human IL2 [BD Bioscience, 555051] and biotinylated mouse anti-human IL2 [BD Bioscience, 555040]; for IFNG: anti-mouse IFNG [eBioscience, 14-7313-85] and biotinylated anti-mouse IFNG [eBioscience 551506] or anti-human IFNG [BD Bioscience, 551221] and biotinylated anti-human IFNG [BD Bioscience, 554550]).
Immunoblot and immunofluorescence analyses
Total cell lysates were prepared using RIPA buffer (50 mM Tris-HCl, pH 7.7, 150 mM NaCl, 1% [v:v] NP40 [AmericanBio, AB01425], 0.5% sodium deoxycholate [Sigma-Aldrich, 30970], and 0.1% SDS, protease Inhibitor cocktail [Sigma-Aldrich, 11697498001], 2 mM phenylmethylsulfonyl fluoride [Sigma-Aldrich, 10837091001], 1 mM 1,4-dithiothreitol [American Bioanalytical, AB00490) and phosphatase inhibitor cocktail [Sigma-Aldrich, P2850]), resolved using sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. The following primary antibodies were used to detect proteins of interest: anti-LC3 (human, mouse; MBL, PM036), anti-ACTB (human, mouse; Abcam, Ab6276), anti-MAPK8 (human, mouse; Cell Signaling Technology, 9252), anti-phospho-MAPK8 (Thr183-Tyr185; human, mouse; Cell Signaling Technology, 9255), anti-phospho-MAPK1-MAPK3 (Thr202-Tyr204; human, mouse. Cell Signaling Technology, 9101), anti-MAPK1/3 (human, mouse; Cell Signaling Technology, 9102). Band intensities were quantified using ImageJ (National Institute of Health, Bethesda, MD, USA). Values of each lane were normalized to its corresponding ACTB. To assess autophagy flux extracts were prepared from T cells that were left resting or activated for 20 h before adding 200 mM ammonium chloride and 100 µm leupeptin (Fisher Scientific, BP26625) for 3 h. We subtracted the normalized LC3-II densitometric value from the sample without lysosomal inhibitors from the value of the sample incubated with inhibitors to obtain a value that represents the amount of LC3-II degraded by autophagy during the time the inhibitors are present. By comparing the flux in activated cells versus resting cells in each condition, we assessed TCR-induced autophagy activation. Human or murine CD4+ T cells (1–2 × 105) were deposited in glass slides using a cytospin centrifuge (Thermo Scientific). Cells were fixed in paraformaldehyde 4% and permeabilized using Triton X-100 (Sigma-Aldrich, T8787). Anti-MAP1LC3B antibody (MBL, PM036; 1:500) was used to detect MAP1LC3B+ structures. Images were acquired using a fluorescence microscope (Carl Zeiss). Autophagy flux was calculated by subtracting the number MAP1LC3B+ puncta in cells left resting or activated and then incubated without lysosomal inhibitors for 3 h from the values in cells incubated with lysosomal inhibitors. Comparing the flux of activated versus resting cells gave us a quantitative measure of the TCR-induced activation of autophagy.
Flow cytometry
T cells were pre-incubated with Fc block (anti-CD16:CD32; Invitrogen, 14016185) and then incubated with fluorochrome-coupled antibodies anti-TCR (BD Biosciences, MR9-4 or T10B9.1A-3). Data were acquired by flow cytometry using a DXP10 FACSCalibur (Becton Dickinson) and analyzed with FLowJo software (Tree Star). Apoptosis levels were assessed with ANXA5-FITC and propidium iodide (PI) (eBioscience, BMS500FI). Live cells are negative for both stainings.
Electron microscopy
CD4+ T cells were fixed in 2.5% glutaraldehyde in 100 mM sodium cacodylate, pH 7.43, and were post-fixed in 1% osmium tetroxide in 100 mM sodium cacodylate, pH 7.43, followed by 1% uranyl acetate. After ethanol dehydration and embedment in LX112 resin (LADD Research Industries, 21210), ultrathin sections were cut on a Reichert Ultracut E and were stained with uranyl acetate followed by lead citrate. All grids were viewed on a JEOL 100CX II transmission electron microscope at 80kV. Morphometric analysis was done with ImageJ software (National Institutes of Health) in 10–20 different micrographs for each condition after ‘thresholding’. Autophagic vacuoles were identified by visual inspection of the micrographs as previously described [81]. Autophagic vacuoles (vesicles <0.5 μm) were identified as autophagosomes when they met the following criteria: i. double membranes (complete or at least partially visible); ii. absence of ribosomes attached to the cytosolic side of the membrane; and iii. luminal density similar to the cytosol and/or identifiable organelles or regions of organelles in the lumen. Vesicles of similar size but with a single membrane (or less than 40% of the membrane visible as a double membrane) with luminal density lower than the surrounding cytosol, or multiple single membrane-limited vesicles containing light or dense amorphous material were classified as autolysosomes.
Lipidomic analysis
Extraction of lipids from cells was performed by solubilizing cell pellets in 100 μl of water to which 375 μl of chloroform:methanol:12 N HCl (2:4:0.1, v:v) was added. After thorough mixing, 125 μl of chloroform was further added, and the solution vortexed for 30 s followed by the addition of another 125 μl of water. Suspensions were centrifuged at 2,000 × g. The lower chloroform layer was recovered and transferred to a glass tube for evaporation in a vacuum centrifuge. The lipid film was dissolved in 50 μl of 1:1:0.3 chloroform:methanol:water and subjected to further analysis by thin layer chromatography and nano-liquid chromatography tandem MS (MS/MS). For qualitative thin layer chromatography analysis, the lipid extracts were spotted on silica-coated plates, and developed in a closed jar with chloroform:methanol:water (65:25:1), supplemented with 2% phosphoric acid to enable identification of major phospholipids and cholesterol. The thin layer chromatography plate was air-dried and developed with iodine vapors. The dark-brown spots corresponding to the lipids of interest were identified using the corresponding lipid standards. For nano-liquid chromatography MS/MS, lipid extracts were diluted 1 to 10 in 1:1:0.3 (v:v:v) chloroform:methanol:water containing 25 mM piperidine. Each sample (10 μl) was loaded into a 2 μm static nanospray emitter. A high-resolution mass spectrometer was used in negative ionization mode for targeted and untargeted MS/MS. Data were collected with the Xcalibur package (ThermoFinnigan). The MS Analysis Tool provided by the Resources at the LIPID MAPS website (www.lipidmaps.org) was used to identify lipid species from MS/MS fragmentation profiles.
Statistical analysis
GraphPad Instat software (GraphPad Prism) was used for statistical analyses. Differences between multiple groups were analyzed by ANOVA with Tukey’s post-test. Comparisons of data pairs were analyzed with a t-test. Data is presented as mean and standard deviation (SD) or standard error of the mean (SEM).
Funding Statement
This work was supported by National Institutes of Health grant AG031782 (AMC, FM and LS), and T32GM007491 (CR), the Hisrchl-Caulier Trust (FM) and the Glenn Foundation for Biomedical Research (AMC and FM), and a fellowship from the American Federation for Aging Research (IG-R). This study was also supported by research cores funded by National Institutes of Health grants to the Albert Einstein Institute for Aging Research (AG038072) and Cancer Center (CA013330).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Jain S, Chaves SS.. Obesity and influenza. Clin Infect Dis. 2011;53:422–424. [DOI] [PubMed] [Google Scholar]
- [2].Harrison LM, Balan KV, Babu US. Dietary fatty acids and immune response to food-borne bacterial infections. Nutrients. 2013;5:1801–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Anderson M, Fritsche KL. (n-3) Fatty acids and infectious disease resistance. J Nutr. 2002;132:3566–3576. [DOI] [PubMed] [Google Scholar]
- [4].Chandra RK, Kutty KM. Immunocompetence in obesity. Acta Paediatr Scand. 1980;69:25–30. [DOI] [PubMed] [Google Scholar]
- [5].Amar S, Zhou Q, Shaik-Dasthagirisaheb Y, et al. Diet-induced obesity in mice causes changes in immune responses and bone loss manifested by bacterial challenge. Proc Natl Acad Sci U S A. 2007;104:20466–20471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mito N, Hosoda T, Kato C, et al. Change of cytokine balance in diet-induced obese mice. Metabolism. 2000;49:1295–1300. [DOI] [PubMed] [Google Scholar]
- [7].Verwaerde C, Delanoye A, Macia L, et al. Influence of high-fat feeding on both naive and antigen-experienced T-cell immune response in DO10.11 mice. Scand J Immunol. 2006;64:457–466. [DOI] [PubMed] [Google Scholar]
- [8].Papathanassoglou E, El-Haschimi K, Li XC, et al. Leptin receptor expression and signaling in lymphocytes: kinetics during lymphocyte activation, role in lymphocyte survival, and response to high fat diet in mice. J Immunol. 2006;176:7745–7752. [DOI] [PubMed] [Google Scholar]
- [9].Strandberg L, Verdrengh M, Enge M, et al. Mice chronically fed high-fat diet have increased mortality and disturbed immune response in sepsis. PLoS One. 2009;4:e7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Karlsson EA, Sheridan PA, Beck MA. Diet-induced obesity impairs the T cell memory response to influenza virus infection. J Immunol. 2010;184:3127–3133. [DOI] [PubMed] [Google Scholar]
- [11].Paich HA, Sheridan PA, Handy J, et al. Overweight and obese adult humans have a defective cellular immune response to pandemic H1N1 influenza A virus. Obesity. 2013;21:2377–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sheridan PA, Paich HA, Handy J, et al. Obesity is associated with impaired immune response to influenza vaccination in humans. Int J Obes (Lond). 2012;36:1072–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Smith AG, Sheridan PA, Harp JB, et al. Diet-induced obese mice have increased mortality and altered immune responses when infected with influenza virus. J Nutr. 2007;137:1236–1243. [DOI] [PubMed] [Google Scholar]
- [14].Edelman R. Obesity: does it modulate infectious disease and immunity? Prog Clin Biol Res. 1981;67:327–337. [PubMed] [Google Scholar]
- [15].Zurier RB, Rossetti RG, Seiler CM, et al. Human peripheral blood T lymphocyte proliferation after activation of the T cell receptor: effects of unsaturated fatty acids. Prostaglandins Leukot Essent Fatty Acids. 1999;60:371–375. [DOI] [PubMed] [Google Scholar]
- [16].Gorjao R, Cury-Boaventura MF, de Lima TM, et al. Regulation of human lymphocyte proliferation by fatty acids. Cell Biochem Funct. 2007;25:305–315. [DOI] [PubMed] [Google Scholar]
- [17].Ioan-Facsinay A, Kwekkeboom JC, Westhoff S, et al. Adipocyte-derived lipids modulate CD4+ T-cell function. Eur J Immunol. 2013;43:1578–1587. [DOI] [PubMed] [Google Scholar]
- [18].Stulnig TM, Berger M, Roden M, et al. Elevated serum free fatty acid concentrations inhibit T lymphocyte signaling. FASEB J. 2000;14:939–947. [DOI] [PubMed] [Google Scholar]
- [19].Costabile M, Hii CS, Melino M, et al. The immunomodulatory effects of novel beta-oxa, beta-thia, and gamma-thia polyunsaturated fatty acids on human T lymphocyte proliferation, cytokine production, and activation of protein kinase C and MAPKs. J Immunol. 2005;174:233–243. [DOI] [PubMed] [Google Scholar]
- [20].Fan YY, Ly LH, Barhoumi R, et al. Dietary docosahexaenoic acid suppresses T cell protein kinase C theta lipid raft recruitment and IL-2 production. J Immunol. 2004;173:6151–6160. [DOI] [PubMed] [Google Scholar]
- [21].Stulnig TM, Berger M, Sigmund T, et al. Polyunsaturated fatty acids inhibit T cell signal transduction by modification of detergent-insoluble membrane domains. J Cell Biol. 1998;143:637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yog R, Barhoumi R, McMurray DN, et al. n-3 polyunsaturated fatty acids suppress mitochondrial translocation to the immunologic synapse and modulate calcium signaling in T cells. J Immunol. 2010;184:5865–5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kim W, Fan YY, Barhoumi R, et al. n-3 polyunsaturated fatty acids suppress the localization and activation of signaling proteins at the immunological synapse in murine CD4+ T cells by affecting lipid raft formation. J Immunol. 2008;181:6236–6243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zeyda M, Staffler G, Horejsi V, et al. LAT displacement from lipid rafts as a molecular mechanism for the inhibition of T cell signaling by polyunsaturated fatty acids. J Biol Chem. 2002;277:28418–28423. [DOI] [PubMed] [Google Scholar]
- [25].Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15:713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mizushima N. Physiological functions of autophagy. Curr Top Microbiol Immunol. 2009;335:71–84. [DOI] [PubMed] [Google Scholar]
- [27].Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ. 2005;12 Suppl 2:1535–1541. [DOI] [PubMed] [Google Scholar]
- [29].Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jia W, He YW. Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J Immunol. 2011;186:5313–5322. [DOI] [PubMed] [Google Scholar]
- [31].Pua HH, Guo J, Komatsu M, et al. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. 2009;182:4046–4055. [DOI] [PubMed] [Google Scholar]
- [32].Li C, Capan E, Zhao Y, et al. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol. 2006;177:5163–5168. [DOI] [PubMed] [Google Scholar]
- [33].Pua HH, Dzhagalov I, Chuck M, et al. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jia W, He MX, McLeod IX, et al. Autophagy regulates T lymphocyte proliferation through selective degradation of the cell-cycle inhibitor CDKN1B/p27Kip1. Autophagy. 2015;11:2335–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hubbard VM, Valdor R, Patel B, et al. Macroautophagy regulates energy metabolism during effector T cell activation. J Immunol. 2010;185:7349–7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Paul S, Kashyap AK, Jia W, et al. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-kappaB. Immunity. 2012;36:947–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Roy SG, Stevens MW, So L, et al. Reciprocal effects of rab7 deletion in activated and neglected T cells. Autophagy. 2013;9:1009–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kaushik S, Rodriguez-Navarro JA, Arias E, et al. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 2011;14:173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Niso-Santano M, Malik SA, Pietrocola F, et al. Unsaturated fatty acids induce non-canonical autophagy. Embo J. 2015;34:1025–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nowicki M, Serke H, Kosacka J, et al. Oxidized low-density lipoprotein (oxLDL) induces cell death in neuroblastoma and survival autophagy in schwannoma cells. Exp Mol Pathol. 2010;89:276–283. [DOI] [PubMed] [Google Scholar]
- [42].Tang Y, Chen Y, Jiang H, et al. Short-chain fatty acids induced autophagy serves as an adaptive strategy for retarding mitochondria-mediated apoptotic cell death. Cell Death Differ. 2011;18:602–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mei S, Ni HM, Manley S, et al. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther. 2011;339:487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Liu K, Zhao E, Ilyas G, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11:271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010;24:3052–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Andersen CJ, Murphy KE, Fernandez ML. Impact of obesity and metabolic syndrome on immunity. Adv Nutr. 2016;7:66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Botbol Y, Guerrero-Ros I, Macian F. Key roles of autophagy in regulating T-cell function. Eur J Immunol. 2016;46:1326–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jung MY, Kim HS, Hong HJ, et al. Adiponectin induces dendritic cell activation via PLCgamma/JNK/NF-kappaB pathways, leading to Th1 and Th17 polarization. J Immunol. 2012;188:2592–2601. [DOI] [PubMed] [Google Scholar]
- [49].Procaccini C, Jirillo E, Matarese G. Leptin as an immunomodulator. Mol Aspects Med. 2012;33:35–45. [DOI] [PubMed] [Google Scholar]
- [50].Haghikia A, Jorg S, Duscha A, et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity. 2015;43:817–829. [DOI] [PubMed] [Google Scholar]
- [51].Whang MI, Tavares RM, Benjamin DI, et al. The ubiquitin binding protein TAX1BP1 mediates autophagasome induction and the metabolic transition of activated T cells. Immunity. 2017;46:405–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Smith AG, Sheridan PA, Tseng RJ, et al. Selective impairment in dendritic cell function and altered antigen-specific CD8+ T-cell responses in diet-induced obese mice infected with influenza virus. Immunology. 2009;126:268–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].O‘Shea D, Cawood TJ, O‘Farrelly C, et al. Natural killer cells in obesity: impaired function and increased susceptibility to the effects of cigarette smoke. PLoS One. 2010;5:e8660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tanaka S, Inoue S, Isoda F, et al. Impaired immunity in obesity: suppressed but reversible lymphocyte responsiveness. Int J Obes Relat Metab Disord. 1993;17:631–636. [PubMed] [Google Scholar]
- [55].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Brest P, Lapaquette P, Souidi M, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn‘s disease. Nat Genet. 2011;43:242–245. [DOI] [PubMed] [Google Scholar]
- [57].Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Cadwell K, Patel KK, Maloney NS, et al. Virus-plus-susceptibility gene interaction determines Crohn‘s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Clarke AJ, Ellinghaus U, Cortini A, et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis. 2015;74:912–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Alissafi T, Banos A, Boon L, et al. Tregs restrain dendritic cell autophagy to ameliorate autoimmunity. J Clin Invest. 2017;127:2789–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kabat AM, Harrison OJ, Riffelmacher T, et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. Elife. 2016;5:e12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Marcel N, Sarin A. Notch1 regulated autophagy controls survival and suppressor activity of activated murine T-regulatory cells. Elife. 2016;5:e14023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Rivera Vargas T, Cai Z, Shen Y, et al. Selective degradation of PU.1 during autophagy represses the differentiation and antitumour activity of TH9 cells. Nat Commun. 2017;8:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kovacs JR, Li C, Yang Q, et al. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ. 2012;19:144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Puleston DJ, Zhang H, Powell TJ, et al. Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife. 2014;3:e03706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wei J, Long L, Yang K, et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol. 2016;17:277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Willinger T, Flavell RA. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc Natl Acad Sci USA. 2012;109:8670–8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Xu X, Araki K, Li S, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol. 2014;15:1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Brown RE, Steele RW, Marmer DJ, et al. Fatty acids and the inhibition of mitogen-induced lymphocyte transformation by leukemic serum. J Immunol. 1983;131:1011–1016. [PubMed] [Google Scholar]
- [70].Potter BJ, Sorrentino D, Berk PD. Mechanisms of cellular uptake of free fatty acids. Annu Rev Nutr. 1989;9:253–270. [DOI] [PubMed] [Google Scholar]
- [71].Yli-Jama P, Meyer HE, Ringstad J, et al. Serum free fatty acid pattern and risk of myocardial infarction: a case-control study. J Intern Med. 2002;251:19–28. [DOI] [PubMed] [Google Scholar]
- [72].Zhang J, Zhao Y, Xu C, et al. Association between serum free fatty acid levels and nonalcoholic fatty liver disease: a cross-sectional study. Sci Rep. 2014;4:5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wen H, Gris D, Lei Y, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Geyeregger R, Zeyda M, Zlabinger GJ, et al. Polyunsaturated fatty acids interfere with formation of the immunological synapse. J Leukoc Biol. 2005;77:680–688. [DOI] [PubMed] [Google Scholar]
- [75].Angela M, Endo Y, Asou HK, et al. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARgamma directs early activation of T cells. Nat Commun. 2016;7:13683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Mauro C, Smith J, Cucchi D, et al. Obesity-induced metabolic stress leads to biased effector memory CD4+ T cell differentiation via PI3K p110delta-Akt-mediated signals. Cell Metab. 2017;25:593–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Rodriguez-Navarro JA, Kaushik S, Koga H, et al. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2012;109:E705–E714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Valdor R, Mocholi E, Botbol Y, et al. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat Immunol. 2014;15:1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Raz Y, Guerrero-Ros I, Maier A, et al. Activation-induced autophagy is preserved in CD4+ T-cells in familial longevity. J Gerontol A Biol Sci Med Sci. 2017;72:1201–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3:181–206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.