

ABSTRACT
Inhibition of host macroautophagy/autophagy is one of the strategies used by several intracellular pathogens, including H. pylori, to escape killing. Here we discuss our recent work that revealed the novel mechanism by which the vacuolating cytotoxin A (VacA) produced by H. pylori inhibits lysosomal and autophagic killing. We discovered that VacA impairs the activity of the lysosomal calcium channel MCOLN1/TRPML1 leading to the formation of enlarged, dysfunctional lysosomes and autophagosomes that serve as an intracellular niche, which allows the bacteria to escape eradication therapy.
KEYWORDS: Autophagy, autophagosomes, H. pylori, lysosomes, TRPML1, VacA, xenophagy
Autophagy was originally described as a response to glucagon, which represented a type of cellular stress, nutrient deprivation, and subsequently to other types of stress such as organellar damage. However, it is now well established that autophagy is also used as a defensive mechanism to battle the infection of host cells by intracellular pathogens. This kind of autophagy, named xenophagy, involves the recognition and delivery of intracellular bacteria to phagophores for subsequent degradation upon fusion with lysosomes. As a consequence, deficiency in xenophagy results in increased susceptibility to infection diseases. It is not surprising then that several pathogens subvert the autophagic pathway and manipulate this process at the molecular level as a strategy to establish a persistent infection. Many intracellular pathogens inhibit the signaling pathways that lead to autophagy induction or mask themselves with host proteins to escape autophagy recognition. Other bacteria block the fusion of autophagosomes with lysosomes or hijack autophagy components to promote their own intracellular growth. We recently discovered a novel mechanism by which Helicobacter pylori exploits the host cell autophagy machinery to generate a protective intracellular reservoir [1].
Vacuolating cytotoxin (VacA) is one of the most important virulence factors of H. pylori. This multifunctional secreted toxin inserts into the host plasma membrane where it oligomerizes to form a chloride-selective membrane channel. In vitro studies have shown that VacA generates large bacterium-containing vacuoles in infected cells through the fusion of late endocytic compartments. VacA also induces autophagy, but prolonged exposure to the toxin disrupts autophagosome maturation, causing bacterium-containing defective autophagosomes to accumulate within infected cells. Both vacuoles and autophagosomes acquire the lysosomal marker LAMP1 and acidic pH indicating that they fuse with lysosomes. However, these compartments are not degradative because they lack lysosomal hydrolases such as CTSD (cathepsin D). Given that VacA causes the formation of these dysfunctional vacuoles, we reasoned that these compartments might constitute an intracellular niche that confers H. pylori survival in vivo.
Because H. pylori is generally considered to be an extracellular, noninvasive bacterium, we initially established that VacA also generates an intracellular bacterial reservoir in vivo. By infecting mice with H. pylori for 8 weeks, followed by conventional eradication therapy with omeprazole, clarithromycin and metronidazole, we found that H. pylori infection is completely eliminated unless the bacterium expresses toxigenic VacA. In this case, intracellular and intravacuolar H. pylori are still detected by immunohistochemistry in the majority of the stomachs. Furthermore, these intracellular bacteria recolonized the gastric glands by 8 weeks after eradication therapy. Thus, VacA generates an intracellular niche that protects H. pylori from antibiotic eradication therapy allowing persistence and recolonization. This intracellular niche may also contribute to bacterial evasion of the host immune response.
Deciphering the mechanism of action of VacA should lead to the identification of potential therapeutic targets for H. pylori infection. We previously established that VacA causes the accumulation of enlarged dysfunctional lysosomes, which are unable to degrade both the bacteria taken up by the endolysosomal pathway and bacteria present within autophagosomes. A key player for efficient lysosomal functioning and cargo degradation is the lysosomal calcium channel MCOLN1. MCOLN1 mediates the transient and localized lysosomal calcium release that controls trafficking events between lysosomes and autophagosomes, late endosomes and the plasma membrane. Notably, deficiency or inhibition of MCOLN1 mimics the toxic effects of VacA at the cellular level, with the presence of enlarged endolysosomal compartments and impaired autophagy. Furthermore, loss-of-function mutations in Mcoln1 cause a lysosomal storage disorder called type IV mucolipidosis (ML4). ML4 patients and Mcoln1-deficient mice display a gastric phenotype that resembles H. pylori infection, including parietal cells defects with mislocalized and decreased expression of proton-pumps, hypochlorhydria and hypergastrinemia. Because MCOLN1 deficiency or inhibition recapitulates VacA effects, we hypothesized that TRPML1 is a downstream target of VacA.
By using lysosomal calcium indicators and measuring MCOLN1-mediated calcium release we showed that, consistent with an impairment in MCOLN1 activity, VacA treatment causes calcium to accumulate in lysosomes. Thus, having established that VacA disrupts MCOLN1 function, we investigated whether increasing MCOLN1 activity by the administration of the MCOLN1 small-molecule agonist ML-SA1 could reverse the toxic effects of VacA. By performing live imaging studies, we showed that ML-SA1 induces the tubulation and fragmentation of the VacA-generated vacuoles into lysosome-sized compartments. ML-SA1 administration restores VacA-disrupted vesicular trafficking, leading to the proper localization and functioning of retrograde trafficking sorting receptors and reestablishing the delivery of CTSD to lysosomes. Further biochemical and immunofluorescence assays indicated that indeed the reactivation of MCOLN1 by ML-SA1 leads to the reformation of functional, degradative lysosomes. As a consequence, ML-SA1 restores the autophagy pathway, as shown by a reduction in both VacA-mediated autophagosome accumulation (LC3 puncta) and LC3-II protein levels. Most importantly, gentamycin protection assays revealed that reactivation of MCOLN1 by ML-SA1 culminates in efficient clearance of intracellular H. pylori.
Pathogens use toxins or effectors to escape host-mediated killing. Our data elucidated a novel mechanism by which H. pylori VacA promotes intracellular bacterial survival. VacA interferes with the activity of a key regulator of lysosomal function, the calcium channel MCOLN1, thereby impairing MCOLN1-mediated cellular responses that promote lysosomal and autophagic degradation of invading pathogens (Figure 1). Further work remains to be done to understand the precise mechanism by which VacA interferes with MCOLN1 activity. However, the observations that MCOLN1 activation reverses the detrimental effects of VacA, restores endolysosomal and autophagic bacterial killing and eliminates the intracellular H. pylori niche, provide the first evidence that MCOLN1 could represent a therapeutic target for fighting infection.
Figure 1.
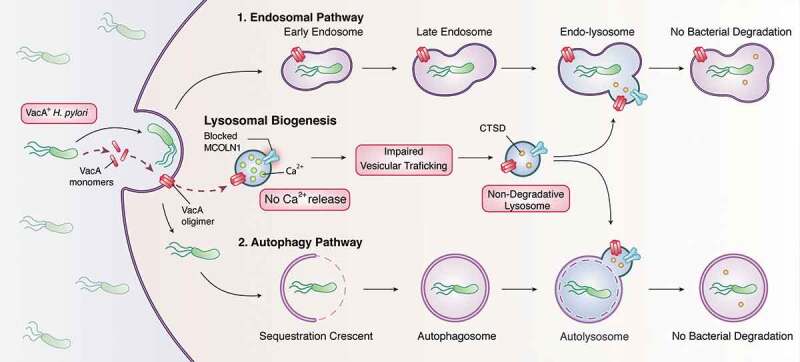
VacA mechanism of action. H. pylori secretes VacA toxin that, upon insertion and oligomerization in the plasma membrane, impairs the activity of the lysosomal calcium channel MCOLN1. Blocking MCOLN1 disrupts vesicular retrograde trafficking affecting the delivery of CTSD to lysosomes, leading to non-degradative lysosomes. These dysfunctional lysosomes are unable to degrade (1) the bacteria taken up by the endosomal pathway or (2) the intracellular bacteria that trigger autophagy. Dysfunctional endolysosomes and autophagosomes serve as an intracellular niche that confers H. pylori survival.
Funding Statement
This work was supported by the Canadian Institute of Health Research (CIHR) and the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition Foundation (NASPGHAN).
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Capurro MI, Greenfield LK, Prashar A, et al. VacA generates a protective intracellular reservoir for Helicobacter pylori that is eliminated by activation of the lysosomal calcium channel TRPML1. Nat Microbiol. 2019. August;4(8):1411–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]