

ABSTRACT
Autophagy is a conserved adaptive cellular pathway essential to maintain a variety of physiological functions. Core components of this machinery are the six human Atg8 orthologs that initiate formation of appropriate protein complexes. While these proteins are routinely used as indicators of autophagic flux, it is presently not possible to discern their individual biological functions due to our inability to predict specific binding partners. In our attempts towards determining downstream effector functions, we developed a computational pipeline to define structural determinants of human Atg8 family members that dictate functional diversity. We found a clear evolutionary separation between human LC3 and GABARAP subfamilies and also defined a novel sequence motif responsible for their specificity. By analyzing known protein structures, we observed that functional modules or microclusters reveal a pattern of intramolecular network, including distinct hydrogen bonding of key residues (F52/Y49; a subset of HP2) that may directly modulate their interaction preferences. Multiple molecular dynamics simulations were performed to characterize how these proteins interact with a common protein binding partner, PLEKHM1. Our analysis showed remarkable differences in binding modes via intrinsic protein dynamics, with PLEKHM1-bound GABARAP complexes showing less fluctuations and higher number of contacts. We further mapped 373 genomic variations and demonstrated that distinct cancer-related mutations are likely to lead to significant structural changes. Our findings present a quantitative framework to establish factors underlying exquisite specificity of human Atg8 proteins, and thus facilitate the design of precise modulators.
Abbreviations: Atg: autophagy-related; ECs: evolutionary constraints; GABARAP: GABA type A receptor-associated protein; HsAtg8: human Atg8; HP: hydrophobic pocket; KBTBD6: kelch repeat and BTB domain containing 6; LIR: LC3-interacting region; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MD: molecular dynamics; HIV-1 Nef: human immunodeficiency virus type 1 negative regulatory factor; PLEKHM1: pleckstrin homology and RUN domain containing M1; RMSD: root mean square deviation; SQSTM1/p62: sequestosome 1; WDFY3/ALFY: WD repeat and FYVE domain containing 3
KEYWORDS: Autophagy, evolution, functional diversity, GABARAP, LC3, molecular dynamics, non-covalent interactions, orthologs
Introduction
The primary function of single Atg8 protein in yeast is to facilitate the cellular process of autophagy by forming double-membrane vesicles called autophagosomes [1-3]. In humans, six distinct Atg8 orthologs participate as core autophagic proteins, namely LC3A, LC3B, LC3C, GABARAP, GABARAPL1, and GABARAPL2/GATE16. Each member of this family displays conserved ubiquitin fold and relatively variable N-terminal helices [4]. All proteins undergo post-translational lipid modification that allows them to recruit other binding partners to phagophore membranes [5,6]. Canonically, all the binding partners which may include adaptors and receptors, interact via a conserved motif known as the LC3-interacting region (LIR) comprising of [W/Y/F]XX[L/I/V] motif [7,8]. These binding partners along with human Atg8 (HsAtg8) orthologs coordinate several key processes such as autophagosome initiation and formation [9], transport [10], elongation [11] and lysosomal fusion [12], vesicular trafficking [13], selective autophagy [14], tumor suppression [15] and many others [16]. However, it is unclear to what extent individual family members differ, and what are the exact molecular details that dictate these large biological differences.
A pivotal step in autophagy evolution, thus, appears to have been the transition from standalone yeast Atg8 to multi-protein family in humans. Much of our present understanding of human Atg8 orthologs comes from the LC3B protein, with most of its molecular properties extrapolated to the family as a whole. Previous functional studies of HsAtg8 members underscored 2 broad autophagic roles [6,17]. While the LC3 subfamily mediates elongation of phagophore membrane, GABARAP proteins are proposed to act at the later stage in sealing of the autophagosome [17]. A large and diverse class of receptor and adaptor proteins is known to bind non-specifically to HsAtg8 orthologs [18]. Interestingly, recent reports highlight interactions of proteins exclusively binding to individual human Atg8 orthologs, suggesting the presence of distinct molecular features [19–22]. Using extensive structure and functional analysis, recent studies identified GABARAP recognition sites that uniquely bind to PLEKHM1 [12,23]. In addition, WDFY3/ALFY [24], KBTBD6/KBTBD7 [25], and NBR1 (neighbor of BRCA1 gene 1) [26] also bind preferentially to GABARAP proteins. On the other hand, FYCO1 (FYVE and coiled-coil domain containing 1) and FKBP8 (FKBP prolyl isomerase 8) exhibited binding preference for LC3A/B and LC3A, respectively [27,28]. Discrete substrate binding partners for LC3 homologs were identified that utilized non-canonical LIR motifs. For example, CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2) is a crucial receptor involved in anti-bacterial autophagy that binds specifically to LC3C via a non-canonical CLIR motif [29]. Utilizing atypical LIR motif, TAX1BP1 shows preference to both LC3C and LC3B [30]. Therefore, general principles underlying molecular recognition preferences of HsAtg8 proteins are not clear and what drives this specificity at large is missing.
In this computational study, we undertook four strategies to obtain selectivity factors responsible to discern human Atg8-family proteins: (i) evolutionary relationships amongst the species, and between proteins, (ii) characterization of molecular features within defined regions of the protein and how they are connected, (iii) local recognition differences within binding interface, and lastly (iv) mapping genomic variations across HsAtg8 orthologs. We pointed out how evolutionary and molecular constraints classified HsAtg8 proteins and proposed a novel sequence recognition motif that discriminates the two broad subfamilies. We then give examples of characteristic structural features in each HsAtg8 protein that directly contribute to their binding differences. By taking advantage of the experimentally resolved crystal structures of HsAtg8 family members and PLEKHM1, we identified factors that lead to preferential substrate recognition. Lastly, we compiled known missense variations in all HsAtg8 orthologs and mapped cancer-related mutations. Our results have important implications in understanding how topologically identical HsAtg8 proteins accomplish distinct functional roles by a repertoire of specific recognition motifs.
Results
Evolutionary and sequence relationships between Atg8 homologs
Aiming to understand Atg8 protein family evolution, we initially constructed a relationship between Atg8 homologs and their occurrence in 20 representative species ranging from fungi, plants to higher vertebrates. The number of proteins within each species varies significantly, with considerable expansion across the higher eukaryotes (Figure 1(a)). While yeast has a single Atg8 protein, most multicellular organisms have more than five protein family members. To obtain finer details of evolutionary features, we performed phylogenetic analysis of Atg8 sequences, as shown in Figure 1(b). Distinct protein specific clusters (Atg8, LC3A, LC3B, LC3C, GABARAP, GABARAPL1, and GABARAPL2/GATE16) were identified. Interestingly, the root of the tree originated from a protist, Entamoeba histolytica, constituting two isoforms of Atg8 (Atg8A and Atg8B). While LC3A-LC3B and GABARAP-GABARAPL1 originated from the same node, LC3C and GABARAPL2 proteins branched into separate clades. In addition, the species with two Atg8 proteins, for example honeybee and C. elegans, showed individual clustering with LC3 and GABARAP families, suggesting evolutionary significance of the two broad Atg8 subfamilies.
Figure 1.
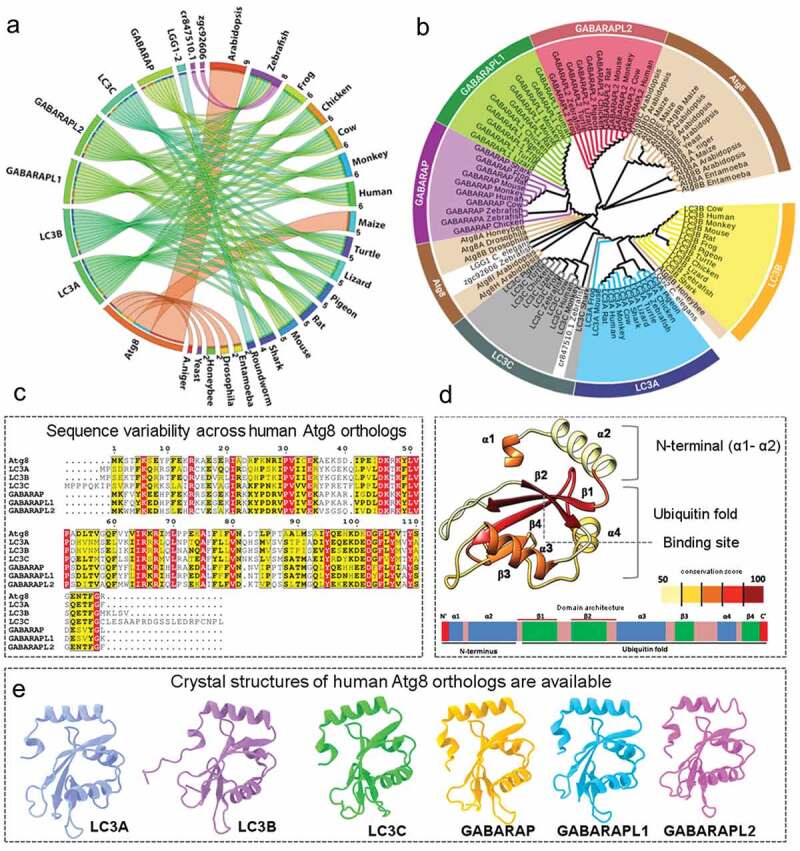
Evolutionary, sequence and structural overview of human Atg8 orthologs. (a) A Circos plot illustrating connection between Atg8 orthologs (left half) and their number in 20 representative species (right half). Different species are individually colored and the width of the ribbon is proportional to number of proteins present. (b) The phylogenetic tree of Atg8 homologs is constructed using maximum likelihood method with 500 bootstrap iterations. Proteins and branches are coloured according to protein name where Atg8, LC3A, LC3B, LC3C, GABARAP, GABARAPL1 and GABARAPL2 are colored in brown, blue, yellow, gray, purple, green and pink, respectively. Other taxa not belonging to any of the above categories are coloured in black. (c) Multiple sequence alignment of human Atg8 orthologs showing sequence conservation with identical (red) and similar residues (yellow and bold). (d) The sequence conservation score averaged according to secondary structural elements (lower bar) is mapped onto the LC3B structure. The low to high intensity of the color bar indicates sequence conservation. (e) The 6 experimentally resolved structures of 6 human Atg8 orthologs are illustrated and individually colored.
At sequence level, the human Atg8 family members are also remarkably diverse (Figure 1(c)). While the overall sequence identity is highest between LC3A-LC3B (83%), and GABARAP-GABARAPL1 (87%), other proteins share relatively low pair wise sequence identity (Figure S1). Conversely, experimentally determined structures of 6 human Atg8 orthologs reveal similar fold, with a global RMSD of 0.96 Å (Figure 1(d,e)). To rank local structural differences, we averaged residue-wise conservation score of secondary structural elements. The structural fold consists of ubiquitin fold with four-stranded central β-sheet core (β1-β4), two α-helices (α3-α4), and relatively variable N-terminal α-helices (α1-α2). We observed that canonical binding site residing within β2 sheet possess highly similar residues (>90%), and other elements varied from 50% to 96%, with the two helices of N-terminus, β1-β2 loop and β2-α3 loop exhibiting high variation. Thus, our sequence based analysis confirms that human Atg8 orthologs showed clear evolutionary separation between two broad subfamilies (LC3 and GABARAP), and the extent of sequence variation is distributed across the protein fold.
Robust classification between LC3 and GABARAP subfamily reveals distinct co-evolved sites and recognition motifs
Using a promising computational approach of residue-covariation analysis, evolutionary constraints (ECs) can be exploited to infer correlations between amino acid at different sequence locations [31]. We utilized EVcouplings method to extract ECs for 6 human Atg8 orthologs, where high-ranking ECs are representative of strong evolutionary constraints and indicate functionally important interactions [32]. We compared the top 30 co-evolved residues within subfamilies, which were classified according to common residue pairs i.e., unique, common in at least 2 members, and common across all three proteins (Figure 2). Comparative analysis of these co-variation residues within GABARAP subfamily showed a large number of common co-evolutionary contacts (21 common ECs), and 4 unique ECs in each protein. The results indicated that individual GABARAP members have less propensity to acquire alternate functions. By contrast, the trend was opposite in LC3 family members, which showed only four common ECs within three subfamily members: S115(121)-L44(50), M111(117)-L81(87), G120(126)-Y113(119) and R70(76)-D48(54). See Methods for residue numbering pattern. Interestingly, LC3C displayed the most number of unique co-evolved residues, mostly harbored between N-terminal helices and ubiquitin fold.
Figure 2.
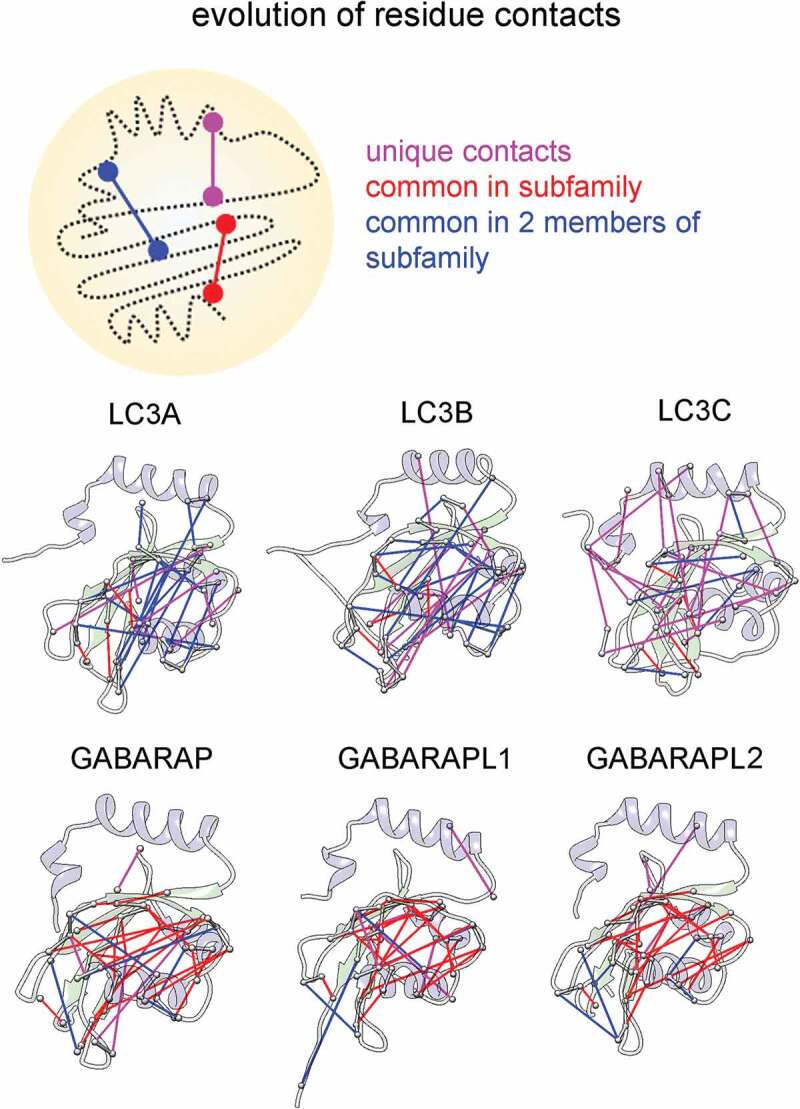
Co-evolution differentiating the broad LC3 and GABARAP subfamilies. Schematic diagram showing comparison of co-evolved contacts within subfamilies (top). We compared and mapped the top 30 co-evolved residue pairs onto the structures of all HsAtg8 orthologs and classified them as unique contacts (present only in individual member of subfamily), common in all members of subfamily and common only in 2 members of subfamily. The three categories are marked in magenta, red and blue, respectively.
To further robustly classify LC3 and GABARAP subfamilies; we utilized hidden Markov models (HMM) to obtain sequence-based recognition motifs (see Methods). We identified a distinct cluster of residues that are exclusive to either GABARAP or LC3 subfamily (Figure S2). The recognition sites were randomly distributed across the structure. Surprisingly, most of the residues were seen outside the binding pocket and only a few residues in LC3 (K30, I31, F52) and GABARAP (Y49, D54) conferred binding specificity. These results provided subfamily specific recognition motifs and co-evolved residues between subfamilies suggested that LC3 members with fewer coupled sites tend to evolve faster than the GABARAP proteins.
Changes in the terminal regions of LC3 and GABARAP subfamilies due to molecular surface properties
One of the important features that characterize protein function and drive molecular interactions is protein surface area accessible to solvent. To quantitatively detect conformational differences and how molecular surfaces evolve in human Atg8 proteins, we performed extensive microsecond-timescale molecular dynamics (MD) simulations (Figure S3). To characterize discrete molecular surfaces, we decomposed the protein into four distinct functional modules or microclusters: a) highly variable N-terminal helical sub-domain, b) binding region, c) membrane binding region [5], and d) C-terminus (Figure 3(a)).
As shown in Figure 3(b), the distribution of the accessible surface area in the N-terminus was found to be a distinguishing factor in LC3 subfamily, with LC3C displaying lowest surface area in the N-terminus (~27 nm2). Interestingly, the differences in molecular surfaces arise from unique residues, i.e., variable amino acids at corresponding amino-acid locations (Figure S4). On the other hand, we identified lack of difference within C-terminus in LC3 family, while GABARAP proteins showed diverse distribution, with GABARAP (~37.8 nm2), GABARAPL1 (~39.7 nm2), and GABARAPL2 (~36.7 nm2) displaying distinct accessible surfaces. The values of surface area within the binding and membrane binding region were close, except for GABARAPL2 in binding region (~37 nm2) and LC3C in membrane binding region (~35 nm2). These differences in LC3C were contributed by residues belonging to α3 and α3-β3 loop. This data suggests that molecular surface differences within LC3 and GABARAP subfamilies were limited to terminal regions and well-established binding pockets in HsAtg8 members may have no significant effect on the surface properties of the proteins.
Figure 3.
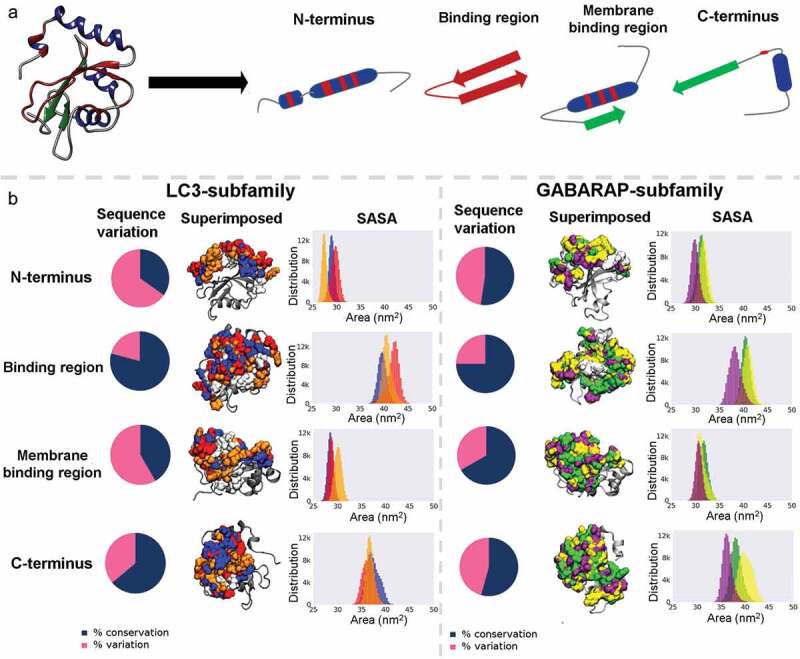
Residue based microclusters differentiate termini uniquely. (a) Schematic representation of four microclusters (functional modules) i.e., N-terminus, binding region, membrane binding region, and the C-terminus. The helices and sheets are colored in blue and green, respectively and the binding region is shown in maroon color. (b) Analysis of surface accessible surface area in LC3 and GABARAP subfamilies is shown. The first column shows pie chart illustrating sequence variation in percentage for each microcluster, with percentage of conserved and varied residues shown in blue and pink, respectively. The protein snapshots shows the superimposed molecular surface of three subfamily members where LC3A, LC3B, LC3C, GABARAP, GABARAPL1 and GABARAPL2/GATE16 proteins are colored in blue, red, orange, green, yellow and purple, respectively. The common surface in each microcluster is shown in white color. The last column shows the distribution of accessible surface area calculated from MD simulations and the color legends in the histograms refer to the same protein, as explained above.
Hydrogen bonding network reveals a unique footprint for each protein
Next, we set out to compare how microclusters connect to each other. The extent of non-covalent bonding such as hydrogen bonds may potentially change the molecular recognition preferences [33,34]. We calculated and tabulated the number of hydrogen bonds between microclusters for each protein in a network diagram (Figure S5). Inspection of the obtained network shows significant changes, as protein intramolecular hydrogen bonding discriminated protein family members rather distinctly, with number and residue pairs unique to each protein.
Investigation of the obtained hydrogen bonding network revealed two interesting patterns. Firstly, we identified key binding residues that formed a unique pattern of hydrogen bonds (Figure 4(a)). For instance, the essential binding residue of GABARAP family Y49 was found to be involved in forming H-bonds with K66 and R67, while, the corresponding residue F52 of LC3 subfamily did not display any hydrogen bonds. Moreover, conserved residues (I67/64 and I23/21) within the binding region also displayed altered numbers of hydrogen bonds and residue pairs in each Atg8 ortholog. The complete list of unique hydrogen bonds formed by binding residues is shown in Table S1. Further, the substrate binding interaction is via two larger pockets, previously reported as hydrophobic pocket-1 (HP1), between α2 and β2, and hydrophobic pocket-2 (HP2), which lies between β2 and α3 [7,8]. Our analysis of residues lining the hydrophobic pockets showed no major changes within HP1, however, residues involved in HP2 showed remarkable differences. We observed a higher prevalence of hydrogen bonding in GABARAP proteins compared to LC3, with 21 and 19 hydrogen bonds, respectively, indicating tighter packing within hydrophobic pocket-2 (Figure 4(b)). These results imply that residues at recognition positions of HsAtg8 family members exhibit unique specificities that may directly modulate their interaction preferences.
Figure 4.
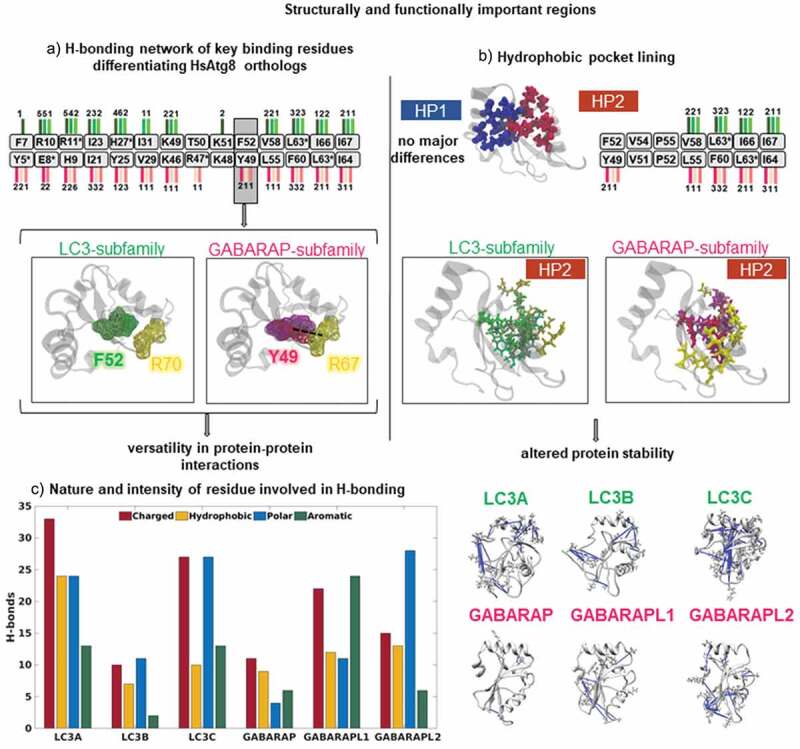
Hydrogen bonding network revealed differences in functionally important regions. (a) Hydrogen bonding pattern showing the key binding residues (in boxes) distinguishing LC3 and GABARAP subfamilies. The number of hydrogen bonds in LC3 and GABARAP subfamily are marked by lines colored in green and pink shades, respectively. The snapshot represents differences in the critical binding residue (F52/Y49) in LC3 and GABARAP subfamily. The dotted line represents distinct hydrogen bond with R67 in the GABARAP subfamily, however, the bond is absent in LC3 subfamily. (b) Differences in residues lining the hydrophobic pockets (HP1 and HP2), colored in blue and maroon, respectively. We observed no major differences in HP1. The number of hydrogen bonds in HP2 contacts shown in LC3 (green) and GABARAP (pink) subfamily markedly differ. The snapshot displays the hydrogen bonds of HP2 in both subfamilies, where the interacting residues are colored in yellow. (c) Bar plot showing number of unique H-bonds formed by polar, charged, aromatic and hydrophobic residues in each HsAtg8 ortholog. In addition, the snapshots highlight the distribution of polar residues involved in hydrogen bonds (in blue) in each protein structure.
The second remarkable property in the H-bonding was the nature of participating residues such as charged, hydrophobic, polar or aromatic. For instance, charged residues might contribute to higher stability due to stronger interactions as compared to a hydrogen bond between small hydrophobic residues like glycine or alanine. As shown in Figure 4(c), distribution of residue types showed drastic differences. In particular, the contribution of polar residues in forming hydrogen bonds was highest in GABARAPL2/GATE16, followed by LC3C, and was found to be the least in GABARAP. Further, we also noted that these residues were present on distinct surface sites, suggesting that the bonding pattern may restrict protein’s open and closed conformation [35]. Overall, our assessment of hydrogen bonding between microclusters showed significant differences in key recognition positions such as F52/Y49 and residues lining HP2 changes indicated precise molecular mechanisms underlying their recognition specificity.
Molecular details of PLEKHM1 and human Atg8 protein complexes reveal GABARAP proteins as stable interacting partner
Based on sequence, evolutionary and molecular surface properties of individual HsAtg8 proteins, we previously concluded that two subfamilies differ on the basis of sequence and evolution properties and non-covalent interactions that connect interacting microclusters uniquely identifies each family member. In the coming sections, we address an open question in autophagy that how human Atg8 protein complexes achieve binding specificity. Taking advantage from the high-resolution crystal structures of bound HsAtg8 proteins with LIR-containing PLEKHM1 peptide [12,23], we performed μs-timescale MD simulations of 6 additional peptide-bound protein complexes.
As shown in Figure 5(a), binding pocket measurements revealed that both LC3 and GABARAP subfamilies make similar utilization of deep binding pockets. By comparing the conformational dynamics between starting and final structures derived from MD simulations of the bound PLEKHM1 complexes, LC3 and GABARAP subfamilies showed marked differences in binding surfaces (Figure 5(b); Figure S6). The LC3 members had slightly loose binding with expanded accessible surfaces (37–42 nm2), as compared to tighter and smaller binding of GABARAP subfamily (33–35 nm2) with PLEKHM1. Also, the protein structural changes as a function of time indicated higher mobility in LC3A/B proteins (0.4 nm), as compared to GABARAP proteins (~0.27 nm), as shown in Figure S6. Interestingly, the bound PLEKHM1 peptide exhibited similar structural alterations, with RMSD values ranging from 0.2 to 0.4 nm.
Figure 5.
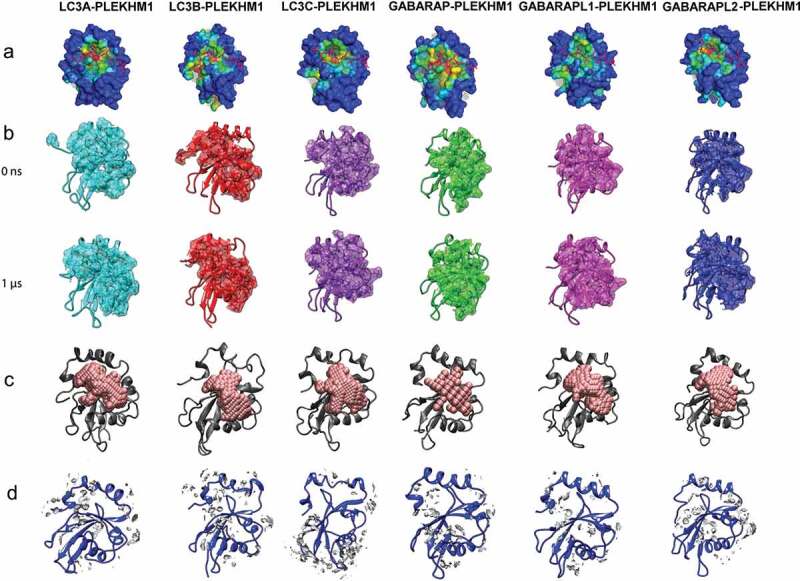
Protein dynamics of human Atg8 orthologs in PLEKHM1-bound state. (a) Binding pocket analysis with surface color ranging from blue (shallow) to red (deep) based on pocket depth, as calculated by Ghecom [89]. Pockets volumes are represented as grey spheres. Peptides are represented as sticks in pink. (b) The initial (0 ns) and last (1 μs) structures displaying the molecular binding surfaces. (c) The volume of binding site as calculated by POVME software for all the HsAtg8 orthologs is depicted through pink beads, and (d) Average water density around 3 Å of binding site, with occupancy of water density shown in silver color.
To obtain a microscopic view, we also computed the dynamics of binding pocket volume that allowed us to understand the evolution of protein conformational space (Figure 5(c)). The PLEKHM1 was found to occupy deeper protein volume and bind tightly with GABARAP proteins, with GABARAP protein exhibiting maximum pocket volume (516 Å3), followed by GABARAPL1 (484 Å3) and GABARAPL2/GATE16 displaying the least (445 Å3). Despite being highly similar to LC3A (525 Å3) in terms of sequence (83% identical) and structure, LC3B displayed reduced binding site volume (375 Å3). Since the pocket volume was found to be different amongst HsAtg8 orthologs, we hypothesized that internal protein cavities might also accommodate water molecules differently. Leveraging atomistic scale trajectories of explicit-solvent, the water densities around the binding site were mapped (Figure 5(d)). Consistent with the protein volume changes, quantitative estimation of water molecules around the binding site showed GABARAP to be least hydrated, while water density was found to be highest in LC3A (Figure S6). From protein dynamics of six complexes, we identified that GABARAP proteins exhibit stable interactions with low structural fluctuations, occupying smaller and tighter binding surface with PLEKHM1 rather than expanded and hydrated pockets observed in LC3 subfamily.
Structural signatures of LIR motif within binding pocket differs
We further focused on local residue-based interactions that contribute to specificity. Table S2 lists the binding residues at the corresponding amino acid locations in HsAtg8 orthologs, along with binding strength, computed from molecular simulations. Most of the binding core belonged to conserved amino acids, and unique residues constituted only 18.5% of the binding site. In particular, five residues in GABARAP subfamily G18, K20, R/K47, D54, F/W62 contribute to PLEKHM1 specificity. Although specific unique residues of GABARAP proteins such as G18 and K20 displayed strong interactions with PLEKHM1, corresponding cluster of residues within α2 of LC3 family members participated negligibly. In addition, Figure 6 showed the detailed atomistic interaction map of PLEKHM1 with HsAtg8 orthologs. The core LIR motif ‘WVNV’ in PLEKHM1 showed stronger binding (~5 to 15 contacts) with both LC3 and GABARAP family members. Interestingly, N at X2 position displayed higher number of contacts with residues V51 and P52 of GABARAP proteins, while the corresponding residues in LC3 subfamily displayed weaker or no binding. In addition, conserved binding residue D48/D45 displayed major differences in terms of interaction strength. The interaction of D48 with PLEKHM1 was strong in LC3A and LC3B (92% and 70% occupancy), but relatively weak in LC3C (41%) and GABARAPL2/GATE16 (2%). Similarly, key residue R70 was observed to show differential binding ability, especially in GABARAP, whereby additional interactions were observed with Q639 and Y640 of PLEKHM1. These results indicated that both unique and conserved residues contribute to specific LIR binding in GABARAP-PLEKHM1 complexes. Five variable binding residues displayed strong interactions in GABARAP subfamily and N of WVNV motif distinguished between LC3 and GABARAP subfamilies.
Figure 6.
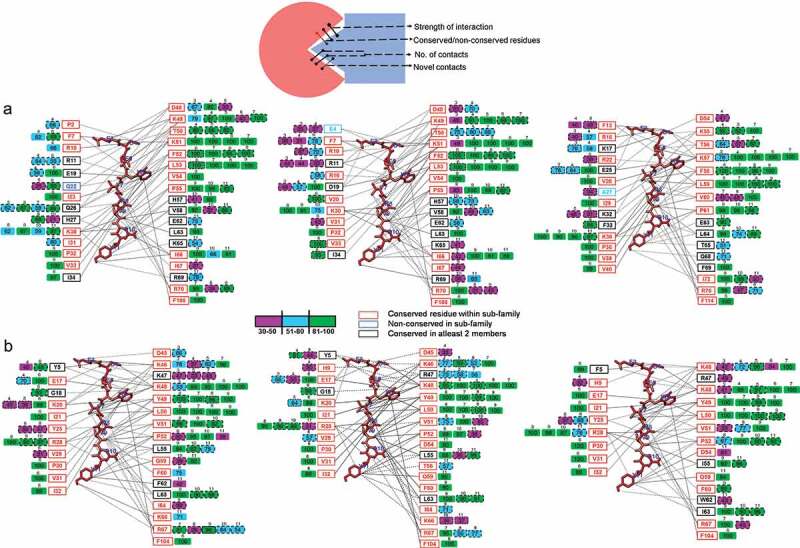
Schematic view of the binding properties of (a) LC3-subfamily and (b) GABARAP-subfamily in the PLEKHM1 bound complex. All binding residues of six HsAtg8 proteins within 0.5 nm of PLEKHM1 are considered, which is illustrated in three dimensional representations. The PLEKHM1 residues, 632–640 are numbered 3–11 in all HsAtg8 complexes. The residue boxes marked next to the PLEKHM1 peptide (shown in red as line representation) depict conservedness. The conserved (red), variable (blue), and residues conserved in at least 2 members of subfamily (black) are highlighted to show uniqueness of the interaction. The next column with the filled boxes are colored according to the strength of interaction with occupancy 30–50%, 51–80%, and >80% colored in magenta, blue, and green, respectively. The interacting PLEKHM1 residue position is marked above the filled box and the novel contacts are shown as dashed lines. The interactions of proteins are shown in black bold lines, and the dashed black outline shows distinct novel interactions.
Specific charged interactions also play a critical distinguishing factor
We have also investigated the interactions and position of charged residues which have been previously found to be critical in binding [36,37]. As shown in Figure 7(a), K49 (55,46/47) and K51 (57,48) form salt-bridges in both the subfamilies. However, LC3 members formed two additional salt-bridges localized at the N-terminus (R10 and R11 with E632 and D633 of PLEKHM1), which were found to be absent in GABARAP subfamily. In kinetic trajectories, the frequency of K51 was highest throughout most of the molecular simulations. In contrast, other salt-bridges displayed relatively lower interaction strength (Figure 7(b)). Moreover, in PLEKHM1-bound complexes, the residues forming ionic interactions were found to be surface exposed, as shown in Figure S7. We also performed in silico alanine scanning and saturation mutagenesis to understand the effect of amino acid mutations on protein stability and protein-protein affinity. Our mutagenesis analysis on each binding site residues also showed that charge differences in the binding region of the LC3 and GABARAP structures seem to play an important role in binding (Figure S8). In addition, F/Y mutation in β2 distinguished well between LC3 and GABARAP subfamily with 83% accuracy (Figure S9). To summarize these observations, significant differences in binding modes also originated from the charged residues and K51 forms salt bridge interactions ubiquitously in all proteins.
Figure 7.
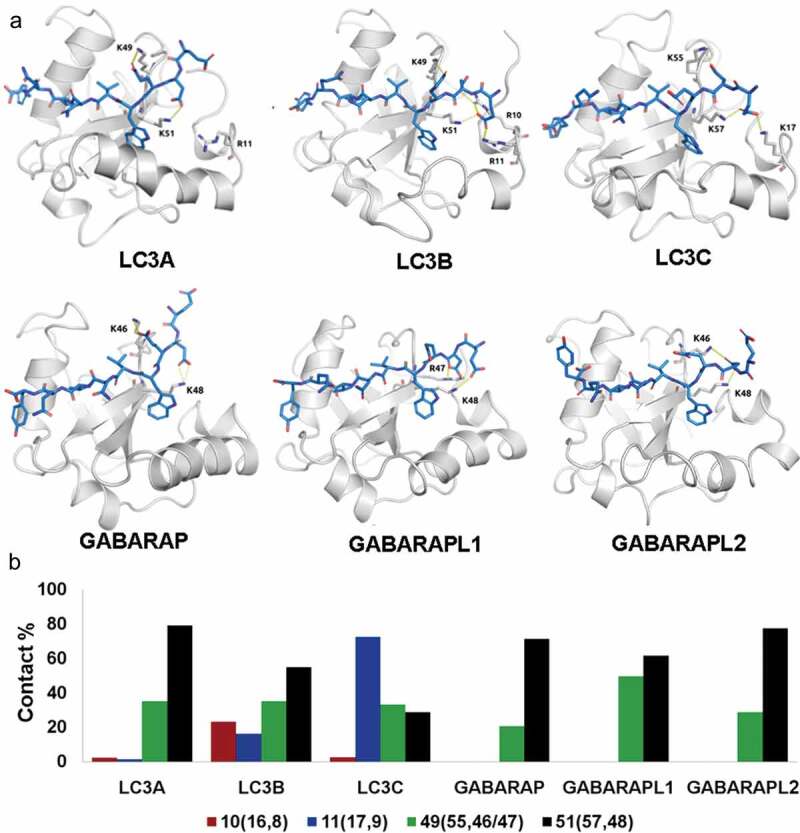
Ionic interactions formed by human Atg8 orthologs at protein-protein interface. (a) The residues forming ionic interaction are colored in blue and white representing PLEKHM1 and Atg8 orthologs, respectively. The bonds are shown by dashed lines. (b) The bar plot displaying the frequency of salt-bridge contacts in MD simulations, with 100% representing the presence of contact throughout the entire trajectory. The ionic contacts made by the human Atg8 orthologs are shown as a function of residue positions marked below. The residue numbers are according to LC3A and LC3B while the numbers in brackets (x,y) indicate that of LC3C and GABARAP proteins, respectively.
Mutations in human Atg8 orthologs implicated in cancer
Here, we extracted genomic variations in HsAtg8 orthologs from publicly available datasets that assemble genome sequences of healthy and diseased individuals (See methods). This approach identified 215 and 158 nonsynonymous mutations in LC3 and GABARAP subfamily, respectively. While LC3C showed the largest number of 95 mutations, GABARAP protein exhibited the lowest count of 45 mutations. We mapped these missense variants onto HsAtg8 functional modules, including N-terminus, binding region, membrane binding, and C-terminus (Figure 8(a)). The mutations were found to be distributed across the protein length, with majority of them located in the C-terminus where the protein pre-processing steps involving enzymatic activity by ATG4 may be perturbed [38].
Out of 373 wide set of annotated nonsynonymous mutations in 6 proteins, 174 were classified as disease-related. The functional impact of these variants was calculated by employing mutation prediction algorithms to classify them as high, medium and low/neutral impact (Figure 8(b)). Surprisingly, 50% of mutations were cataloged to have medium to high impact. In addition, human Atg8 orthologs were found to be associated with various diseases including some cancers [39], therefore, we investigated the clinical significance of these amino acid changes. In total, 43 cancer-associated mutations were found in the binding pocket region (Table S3). Interestingly, the occurrence of amino acid change at R70/76/67 position was found in all six proteins. The mutations with high allele frequencies are displayed in Figure 8(c), with endometrial cancer as the most prevalent type of cancer associated with mutations in all HsAtg8 orthologs. In addition, high occurrence of LC3A mutations were found in bladder cancer patients, LC3B was found to be associated with various lung cancers, and LC3C was related to prostate and skin cancer. The mutations in GABARAP subfamily were linked with thymic cancer in GABARAP, stomach cancer in GABARAPL1 and breast cancer in GABARAPL2/GATE16. In summary, compilation of 174 clinically relevant mutations out of 373 genomic variations was performed, along with their structural mapping on microclusters which provides a rich resource of information to deduce variability of HsAtg8 proteins in the disease context.
Figure 8.
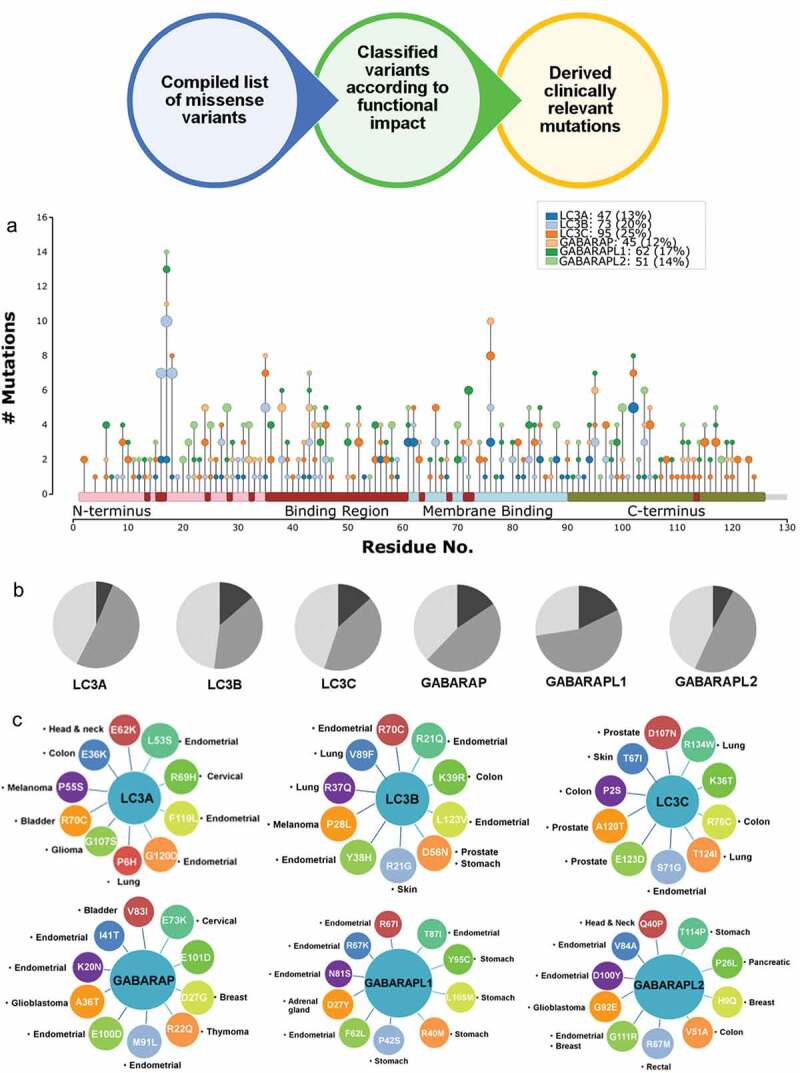
Functional and clinical impact of mutations in HsAtg8 orthologs. Schematic representing the workflow of mutation analysis. (a) The Needle plot demonstrates variations mapped on microclusters in human Atg8 orthologs. The residue numbering of all the HsAtg8 orthologs has been modified according to LC3C. (b) Pie chart displaying the functional impact of variations with high, medium and low impact colored in dark grey, grey and light grey, respectively. (c) A circular network representation of top 10 mutations associated with various cancers in each HsAtg8 ortholog.
Discussion
During the course of evolution, many prominent protein families contain functionally overlapping orthologs that have structurally similar domains. Here, we assessed the six human Atg8 protein family members that have evolved from a single yeast protein, Atg8. By systematically analyzing the experimentally available protein structures, we attributed precise regions that contribute to functional similarities and dissimilarities across the human Atg8 family. Figure 9 provides highlights of our work based on quantitative information on how the repertoire of HsAtg8 orthologs acquires different structural modulations. Our analysis distinguished the family members on the basis of four putative mechanisms that are described in detail below.
Figure 9.
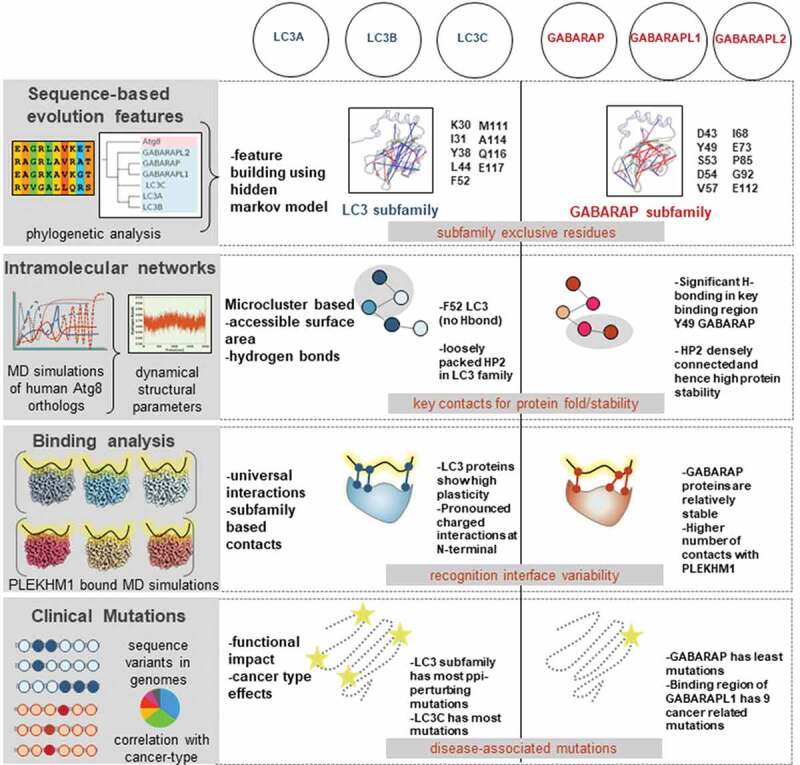
Schematic representation revealing highlights of this study to discern LC3 and GABARAP subfamilies on the basis of molecular signatures. The distinct subfamilies are shown in two columns, with four major highlights depicted in rows. Overall, we identified selectivity determinants using evolutionary relationships (sequence motifs), intramolecular networks (H-bond pattern in binding region and HP2), binding analysis (PLEKHM1 binds more stably with GABARAP), and finally clinical mutations (specific disease-related sites). We propose that these measurements highlight how human Atg8 orthologs achieve selectivity via distinct structural modulations.
Role of evolutionary constraints
The expansion of one Atg8 in yeast to several in mammals and other species is a consequence of 68 gene duplication events [40] which forms the basis of evolution of species and creation of most of the gene families in higher eukaryotes. As a result of various transposition, translocation and recombination events during the course of evolution, all the six human paralogs were found to have different chromosomal locations with LC3A, LC3B, LC3C, GABARAP, GABARAPL1 and GABARAPL2/GATE16 located on chromosome 20, 16, 1, 17, 2 and 16, respectively [16].
Our phylogenetic analysis revealed a clear evolutionary separation between LC3 and GABARAP subfamilies, and also identified a sequence-based recognition motif that distinguishes Atg8 homologs (Figures 1 and 2). Previous reports have also demonstrated evolution-based sorting of Atg8 into LC3/GABARAP subfamilies in lower eukaryotes. For example, two Atg8 orthologs in C. elegans, LGG-1 and LGG-2 were found to show structural and functional similarity to GABARAP and LC3 subfamily, respectively [41,42]. However, in many plants, there can be more than 6 Atg8 isoforms [43]. As shown in our phylogenetic tree, nine Arabidopsis proteins were grouped with Atg8 cluster into two groups (Atg8H-I and Atg8A-G), with latter dividing further into two subgroups. In a recent study, potato Atg8 isoforms were also proposed to bind to a distinct set of proteins [44]. The recognition motif for subfamilies, identified in our study, thus also provides a valuable resource for the autophagic community to decode variability within the multi-member Atg8 family.
Non-covalent interactions and binding mode differ across HsAtg8 orthologs
We assessed the functional impact of inter-microcluster hydrogen bond network across all HsAtg8 orthologs that revealed significant differences in HP2 and in key binding residues, in particular, F52/Y49 residing in β2 (Figure 4). The primary distinction in a highly conserved β2 is F/Y, which distinguishes LIR binding in LC3 and GABARAP subfamily. The importance of these residues has been established by mutagenesis experiments whereby alanine mutant of F52 and Y49 abrogates LC3 binding to SQSTM1/p62 [36], GABARAP binding to NBR1 [26] and BNIP3L/Nix (BCL2 interacting protein 3 like) [21], respectively. In addition, other residues around HP1 and HP2 like Y25, V29, K46 and L55 in GABARAP proteins have been reported to display specific binding to WDFY3/ALFY, KBTBD6 and HIV-1 Nef [24,25,45]. In addition, HP1, being more conserved than HP2 in Atg8 protein family [7], displayed no major differences in H-bonding pattern. On the other hand, we observed more inter-microcluster H-bonds in HP2 of GABARAP proteins (21) than LC3 (19), indicating tighter packing in GABARAP proteins. Previous reports also suggested similar observations where two-dimensional (2D) 1H-15N-heteronuclear single quantum coherence (HSQC) spectra showed HP2 to be more affected than HP1 on HIV-1 Nef binding to GABARAP [45]. It has also been reported that KBTBD6 engages with the bulkier residues of HP2 in GABARAP proteins, thus, forming tight-complex contributing to high binding affinity [25].
Although it is not clear that all the non-covalent interactions may be specific [46,47], universally conserved positions in family members may contribute to overall stability with minimal backbone changes. In a parallel analysis, we observed major differences in conserved salt-bridge interactions (Figure 7). It is reported that the N-terminus of LC3-subfamily is basic in nature while GABARAP subfamily is acidic or neutral [48], and thus have been addressed to be critical for carrying out specific function [36,39]. Previous studies have shown that truncation of N-terminus in LC3 and not GABARAPL2/GATE16 abates SQSTM1/p62 binding. This was further verified by domain swap experiments where GABARAPL2/GATE16 chimera containing the LC3 N-terminus recruits SQSTM1/p62 to LC3 in a similar manner [37].
Recognition binding variability
In a high-throughput study, LC3/GABARAP subfamilies display interactome differences where around one-third of binding partners were found to be specific for LC3 subfamily, one-third for GABARAP and one-third were found to be common for both groups [18]. At the molecular level, many reports have identified unique binders to HsAtg8 orthologs [21,23–30,45]. We, therefore, surmised that molecular differences between bound complexes of human Atg8 orthologs would entail underlying mechanisms into their selectivity. Molecular dynamics simulations revealed underlying recognition variability, with GABARAP proteins showing less rigidity and tighter packing with PLEKHM1 (Figure 5). These findings were found to be in concordance with previous studies on PLEKHM1-bound complexes, with increased binding affinities in GABARAP subfamily of proteins [23]. At residue level, a more robust approach was constructed. The five unique residues (G18, K20, R/K47, D54, F/W62) contributed to specificity in GABARAP proteins. These findings agree with earlier reports, where D54 and F62 contribute to GABARAP specific binding with WDFY3/ALFY and HIV-1 Nef, respectively [24,45]. The residue R/K47 has been reported to display an important electrostatic interaction with E386 of ATG4B which was found to be conserved in GABARAP subfamily [49]. In terms of LIR motif, we found N of WVNV motif within binding pocket of PLEKHM1 contributed to stronger interactions with GABARAP proteins which was found to be in agreement with the previous reports [23]. Further, there are reports which suggest that the presence of Y25 dictates preferential binding of GABARAP proteins to KBTBD6, where it forms an H-bond with R670 of WVRV motif [25]. Contrary to this, we observed weaker or no interaction of Y25 with PLEKHM1 in GABARAP subfamily, in spite of similar LIR motif. These observations indicated that all HsAtg8 orthologs employ global and local conformational variability to bind to different protein partners.
Clinical impact of mutations in Atg8 orthologs
Although several reports have linked autophagic proteins to cancer, it remains unclear what are the genomic variations originating from HsAtg8 genes, and to what extent disease-related mutations are located on their functional domains [50,51]. We extracted and mapped 373 mutations in LC3 and GABARAP proteins from publicly available resources, and further narrowed down to 174 cancer-related mutations (Figure 8). Depending on the structural location, mutations may be linked with altered protein folding, stability and protein-protein interactions. In total, 43 cancer mutations were present in the binding region. A critical residue R70, involved in LIR binding, was found to be mutated in all proteins and also showed a higher prevalence in endometrial cancer patients. Another binding interface residue P55S in LC3A is found in melanoma patients that plays an important role in Atg13 interaction [52]. Additionally, it was observed that various mutations were located in the C-terminus that is critical for protein processing within Atg8 family members. For instance, LC3A-G120D [53], LC3B-G120R [54], LC3C-R76H [55] and GABARAP-G116W [56] are prominently linked with accumulation of the proform of each protein by potentially inhibiting the cleavage of HsAtg8 orthologs. While there is less evidence for GABARAP mutations involved in abolishing direct protein-protein interactions, the F62L mutation was reported to effect HIV-1 Nef binding to GABARAPL1 [45].
Towards future peptide design and binding modulators
Finally, given the indispensable biological nature of human Atg8 orthologs and its association with cancers of different tissue of origin, the design of specific modulators for each protein is an interesting subject to explore further. There are studies that report the design of peptides for closely related proteins which target specific transmembrane helices to modulate the activity of integrins [57]. A recent report by Stolz et al., demonstrated the use of engineered peptides as intracellular sensors specifically recognizing individual protein family members [58]. Thus at the atomistic level, our findings offer first step towards defining structure-based principles to discern Atg8 human family members.
Materials and methods
Phylogenetic analysis
For phylogenetic analysis, protein sequences of Atg8 orthologs from 20 eukaryotic species including unicellular protist (E. histolytica), fungi (A. niger, S. cerevisiae), plants (Arabidopsis, Maize), a nematode (C. elegans), insects (honeybee, Drosophila), fishes (shark, zebrafish), amphibian (frog), reptiles (lizard, turtle), birds (pigeon, chicken) and mammals (rat, mouse, cow, monkey and human) were selected. A total of 90 sequences were downloaded from UniProt database. Sequence alignment was carried out using MUSCLE [59] and phylogenetic tree construction was carried out using MEGA (Molecular Evolutionary Genetics Analysis) software [60]. Maximum likelihood (ML) method was used for phylogeny reconstruction using LG model. Tree robustness and reliability was assessed with 500 bootstrap replicates. Tree visualization was carried out using Evolview [61]. The coevolution analysis was carried out using the EVCoupling webserver [32].
Profile HMMs were generated for the Atg8 subfamilies (LC3 and GABARAP) using HMMER (v3.2.1) [62] for representative orthologous sequences of each subfamily (34 for LC3 and 31 for GABARAP). The sequences used to create the profiles are available as Supplementary Materials. Each profile was then used to search against the database of sequences for both subfamilies with e-values for the full sequences taken into account for classification purposes. Multiple sequence alignment using MAFFT (v7) [63] was performed per subfamily.
Starting structures and docking
The protein structures of all six human Atg8 orthologs (LC3A, LC3B, LC3C, GABARAP, GABARAPL1, and GABARAPL2/GATE16) were taken from the PDB Database (PDB-ID: 3WAL, 3VTU, 3WAM, 1KJT, 2R2Q, and 4CO7). The missing residues in the crystal structure at the N- and C-terminus were modeled as random coil using DS Visualizer [64] in accordance with the UniProt database. In addition, the bound structures of 5 Atg8 orthologs (LC3A, LC3B, LC3C, GABARAP, and GABARAPL1) bound to PLEKHM1 were taken from PDB (PDB-ID: 5DPR, 3X0W, 5DPW, 5DPS, and 5DPT). The crystal structure of GABARAPL2/GATE16 bound to PLEKHM1 was not available, and hence we reconstituted the binding co-ordinates by superimposing with other crystal structures. Further, in three of the bound complexes (LC3A, LC3B, and GABARAP), PLEKHM1 was found in the fused form. Therefore, the bound conformation of PLEKHM1 in LC3A and LC3B was generated from the LC3C structural details. Similarly, the bound conformation of PLEKHM1 in GABARAP and GABARAPL2/GATE16 was generated by superimposition with GABARAPL1 structure.
Molecular dynamics simulations
The MD simulations were performed using the program GROMACS [65], and the OPLS all-atom force field [66]. The water molecules were modeled with the TIP4P representation [67]. Periodic boundary conditions were used and long-range electrostatic interactions were treated with the Particle Mesh Ewald (PME) summation using grid spacing of 0.16 nm combined with a fourth-order cubic interpolation to deduce the potential and forces in-between grid points [68]. The real space cut-off distance was set to 1.0 nm and the van der Waals cut-off to 1.2 nm. The bond lengths were fixed [69] and a time step of 2 fs for numerical integration of the equations of motion was used. Coordinates were saved every 10 ps. 12 independent MD trajectories, each 1 μs long at 300 K were carried out for all the human Atg8 orthologs in unbound and bound form as starting structures. The protein was placed in a dodecahedral water box, large enough to contain protein and at least 1.0 nm of solvent on all sides. The starting structures were subjected to energy minimization using the steepest descent method. The simulations were subjected to Nose-Hoover T-coupling bath to maintain the exact temperature [70]. The structures were then subjected to Parrinello-Rahman barostat for pressure coupling at 1 bar [71], before the 1 μs production run were started. The details of simulation are given in Table S4.
Analysis of trajectories
Graphs were constructed using Graphing, Advanced Computation and Exploration (GRACE) program, version 5.1.22 and MATLAB [72]. All molecular images were generated using VMD [73], Pymol [74] and Chimera [75].
Protein-ligand contacts: The protein-peptide contacts across the trajectory were calculated using MDcons [76]. Two residues were considered to be in contact with at least two heavy atoms being at a distance <0.5 nm. The conservation rate (CR) for each inter-residue pair was evaluated across the trajectory run which is calculated using the equation:
| (1) |
where nckl is the total number of frames when residue k and l of protein A and B are in contact. N is the total no. of frames analyzed. All the contacts with conservation rate ≥0.3 were retained for further analysis.
LC3-interacting region (LIR) residues in HsAtg8 orthologs: The binding site for LIR included residues of HsAtg8 orthologs interacting with PLEKHM1 within 0.5 nm distance.
Novel contacts were defined as interactions that were newly formed during the simulations, as compared to the first 50 ns of the run length of PLEKHM1 bound HsAtg8 orthologs.
Microclusters decomposition
On the basis of protein architecture, we decomposed each structure into four distinct functional modules or microclusters. (i) The first two α-helices along with α2-β1 loop constitute the N-terminal helical sub-domain which is known to be variable in all Atg8 orthologs [39]. (ii) The binding region constitutes residues from conserved β-sheets (β1-β2), β1-β2 loop and residues from two hydrophobic pockets (HP1 and HP2), responsible for interaction with the autophagy receptors [27,39,77]. In addition, few N-terminal residues like F7, R10, R11, H27 [28,78,79] and some residues from α3 (L63, I66, I67, R70) are also known to be involved in binding [80]. (iii) Further, we have previously shown that lipidated LC3 is attached to the membrane via α3 and β3 secondary structural elements, referred here as membrane binding segment [5]. (iv) The rest of the protein beyond β3 constitutes the C-terminus.
Conserved and variable residues
Variable residues were identified from the alignment of LC3 and GABARAP subfamily, where a residue is said to be variable if it is different in all the three subfamily members.
Inter-segment H-bonding calculations
The inter-segment protein hydrogen-bonds were calculated using Gromacs module, where the intra-segment H-bonds and main-chain H-bonds involved in forming the secondary structural elements were ignored. The residue numbers in LC3C were modified to match the numbering pattern of LC3A and LC3B.
Binding site volume
The binding site volume in all the bound-conformations was computed as a function of time using POVME 2.0 [81,82].
Salt-bridge calculations
The salt-bridge interactions were calculated using Salt Bridges Plugin in VMD [73].
Residue numbering
The residue numbering is different for LC3 subfamily. For comparison, the canonical numbering refers to LC3A and LC3B and the residue number in the bracket corresponds to LC3C protein. To compare LC3 and GABARAP residue locations, X/Y notion was used to present LC3/GABARAP proteins.
Average water density
Average water density was calculated in Volmap plugin in VMD [73]. The map type was selected to be density with mass as weights. The average water density was calculated for the entire length of simulation run of the complex trajectories. Final results were visualized in Chimera [75].
Mutation studies
Missense variations in all the Atg8 orthologs were compiled from Ensembl [40] and cBioPortal [83] resulting in a total of 373 variations. We further classified the variants according to their functional impact into high, medium and low impact variations using Mutation Assessor [84]. The diseased mutations implicated in cancer were compiled from cBioPortal, where only the missense mutations were taken into account.
In silico scanning mutagenesis
The effects of each mutation on protein folding and stability was assessed using mCSM-Stability [85], SDM [86] and DUET [87]. The effects of each mutation on the binding affinity for its partners were assessed using mCSM-PPI [85]. They represent a class of novel machine-learning methods that extract patterns from graph representations of the three dimensional residue environment structure in order to quantitatively predict the effects of missense mutations on protein stability [85,86] and protein-protein interactions [85,88]. For the in silico saturation scanning mutagenesis, the predicted changes in Gibb’s free energy were averaged at each residue position for the 19 possible mutations.
Funding Statement
This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico; Jack Brockhoff Foundation [JBF 4186, 2016]; Newton Fund RCUK-CONFAP Grant awarded by the Medical Research Council (MRC) and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) [MR/M026302/1]; IRR/FIOCRUZ Minas; Department of Biotechnology, Ministry of Science and Technology (IN); INSPIRE Faculty fellowship, Department of Science and Technology, Government of India (IN) [LSBM-45]; NHMRC CJ Martin Fellowship [APP1072476]; Science and Engineering Research Board [PDF/2017/000905].
Acknowledgments
LT and NJ acknowledge the support from CSIR-IGIB for infrastructure and CSIR-4PI for supercomputing facilities. We also thank Ms. Saman Fatihi for helping us with the analysis and extracting cancer-related mutations. LT acknowledges the support from DST-INSPIRE Faculty funded research grant from Department of Science and Technology, India. LT also thanks DST-SERB Early Career Award [ECR/2017/000768] from Department of Science and Technology. RSG is a J.C. Bose Fellow of the Department of Science and Technology, Government of India. NJ is thankful to DBT-RA and DST-NPDF Fellowships. DEVP and DBA were funded by Newton Fund RCUK-CONFAP Grant awarded by The Medical Research Council (MRC) and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG), DBA acknowledges support of NHMRC CJ Martin Fellowship (APP1072476) and Jack Brockhoff Foundation (JBF 4186, 2016) (DBA), DEVP was further supported by Instituto René Rachou (IRR/FIOCRUZ Minas) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Hurley JH, Young LN.. Mechanisms of autophagy initiation. Annu Rev Biochem. 2017;86:225–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nakatogawa H, Ichimura Y, Ohsumi Y.. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130(1):165–178. [DOI] [PubMed] [Google Scholar]
- [3].Tsukada M, Ohsumi Y. Isolation and characterisation of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333(1–2):169–174. [DOI] [PubMed] [Google Scholar]
- [4].Paz Y, Elazar Z, Fass D. Structure of GATE-16, membrane transport modulator and mammalian ortholog of autophagocytosis factor Aut7p. J Biol Chem. 2000;275(33):25445–25450. [DOI] [PubMed] [Google Scholar]
- [5].Thukral L, Sengupta D, Ramkumar A, et al. The molecular mechanism underlying recruitment and insertion of Lipid-Anchored LC3 protein into membranes. Biophys J. 2015;109(10):2067–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Weidberg H, Shpilka T, Shvets E, et al. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell. 2011;20(4):444–454. [DOI] [PubMed] [Google Scholar]
- [7].Noda NN, Ohsumi Y, Inagaki F. Atg8-family interacting motif crucial for selective autophagy. FEBS Lett. 2010;584(7):1379–1385. [DOI] [PubMed] [Google Scholar]
- [8].Birgisdottir ÅB, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci. 2013;126(Pt 15):3237–3247. [DOI] [PubMed] [Google Scholar]
- [9].Kraft C, Kijanska M, Kalie E, et al. Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. Embo J. 2012;31(18):3691–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pankiv S, Alemu EA, Brech A, et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010;188(2):253–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Xie Z, Nair U, Klionsky DJ. Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell. 2008;19(8):3290–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].McEwan DG, Popovic D, Gubas A, et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell. 2015;57(1):39–54. [DOI] [PubMed] [Google Scholar]
- [13].Jacob TC, Moss SJ, Jurd R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci. 2008;9(5):331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7(3):279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Klebig C, Seitz S, Arnold W, et al. Characterisation of {gamma}-aminobutyric acid type A receptor-associated protein, a novel tumor suppressor, showing reduced expression in breast cancer. Cancer Res. 2005;65(2):394–400. [PubMed] [Google Scholar]
- [16].Schaaf MB, Keulers TG, Vooijs MA, et al. LC3/GABARAP family proteins: autophagy-(un)related functions. FASEB J. 2016;30(12):3961–3978. [DOI] [PubMed] [Google Scholar]
- [17].Weidberg H, Shvets E, Shpilka T, et al. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. Embo J. 2010;29(11):1792–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Behrends C, Sowa ME, Gygi SP, et al. Network organization of the human autophagy system. Nature. 2010;466(7302):68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Koukourakis MI, Kalamida D, Giatromanolaki A, et al. Autophagosome proteins LC3A, LC3B and LC3C have distinct subcellular distribution kinetics and expression in cancer cell lines. PLoS One. 2015;10(9):e0137675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang H, Sun HQ, Zhu X, et al. GABARAPs regulate PI4P-dependent autophagosome: lysosome fusion. Proc Natl Acad Sci U S A. 2015;112(22):7015–7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Novak I, Kirkin V, McEwan DG, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11(1):45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol. 2012;22(1):R29–R34. [DOI] [PubMed] [Google Scholar]
- [23].Rogov VV, Stolz A, Ravichandran AC, et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep. 2017;18(8):1382–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lystad AH, Ichimura Y, Takagi K, et al. Structural determinants in GABARAP required for the selective binding and recruitment of ALFY to LC3B-positive structures. EMBO Rep. 2014;15(5):557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Genau HM, Huber J, Baschieri F. CUL3-KBTBD6/KBTBD7 ubiquitin ligase cooperates with GABARAP proteins to spatially restrict TIAM1-RAC1 signaling. Mol Cell. 2015;57(6):995–1010. [DOI] [PubMed] [Google Scholar]
- [26].Rozenknop A, Rogov VV, Rogova NY, et al. Characterisation of the interaction of GABARAPL-1 with the LIR motif of NBR1. J Mol Biol. 2011;410(3):477–487. [DOI] [PubMed] [Google Scholar]
- [27].Olsvik HL, Lamark T, Takagi K, et al. FYCO1 contains a C-terminally extended, LC3A/B-preferring LC3-interacting Region (LIR) motif required for efficient maturation of autophagosomes during basal autophagy. J Biol Chem. 2015;290(49):29361–29374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bhujabal Z, Birgisdottir ÅB, Sjøttem E, et al. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017;18(6):947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].von Muhlinen N, Akutsu M, Ravenhill BJ, et al. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol Cell. 2012;48(3):329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tumbarello DA, Manna PT, Allen M, et al. The autophagy receptor TAX1BP1 and the molecular motor myosin vi are required for clearance of Salmonella typhimurium by autophagy. PLoS Pathog. 2015;11(10):e1005174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Echave J, Spielman SJ, Wilke CO. Causes of evolutionary rate variation among protein sites. Nat Rev Genet. 2016;17(2):109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Marks DS, Colwell LJ, Sheridan R, et al. Protein 3D structure computed from evolutionary sequence variation. PLoS One. 2011;6(12):e28766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Joachimiak LA, Kortemme T, Stoddard BL, et al. Computational design of a new hydrogen bond network and at least a 300-fold specificity switch at a protein-protein interface. J Mol Biol. 2006;361(1):195–208. [DOI] [PubMed] [Google Scholar]
- [34].Klumb LA, Chu V, Stayton PS. Energetic roles of hydrogen bonds at the ureido oxygen binding pocket in the streptavidin-biotin complex. Biochemistry. 1998;37(21):7657–7663. [DOI] [PubMed] [Google Scholar]
- [35].Wierenga RK, Noble ME, Postma JP, et al. The crystal structure of the “open” and the “closed” conformation of the flexible loop of trypanosomal triosephosphate isomerase. Proteins. 1991;10(1):33–49. [DOI] [PubMed] [Google Scholar]
- [36].Shvets E, Fass E, Scherz-Shouval R, et al. The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J Cell Sci. 2008;121(Pt 16):2685–2695. [DOI] [PubMed] [Google Scholar]
- [37].Shvets E, Abada A, Weidberg H, et al. Dissecting the involvement of LC3B and GATE-16 in p62 recruitment into autophagosomes. Autophagy. 2011;7(7):683–688. [DOI] [PubMed] [Google Scholar]
- [38].Satoo K, Noda NN, Kumeta H, et al. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. Embo J. 2009;28(9):1341–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lee YK, Lee JA. Role of the mammalian Atg8/LC3 family in autophagy: differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016;49(8):424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zerbino DR, Achuthan P, Akanni W, et al. Ensembl 2018. Nucleic Acids Res. 2018;46(D1):D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wu F, Watanabe Y, Guo XY, et al. Structural basis of the differential function of the two C. elegans Atg8 homologs, LGG-1 and LGG-2. Autophagy. 2015;60(6):914–929. [DOI] [PubMed] [Google Scholar]
- [42].Wu F, Wang P, Shen Y. Small differences make a big impact: structural insights into the differential function of the 2 Atg8 homologs in C. elegans. Autophagy. 2016;12(3):606–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sláviková S, Shy G, Yao Y, et al. The autophagy-associated Atg8 gene family operates both under favourable growth conditions and under starvation stresses in Arabidopsis plants. J Exp Bot. 2005;56(421):2839–2849. [DOI] [PubMed] [Google Scholar]
- [44].Zess EK, Cassandra Jensen C, Cruz-Mireles N, et al. N-terminal β-strand underpins biochemical specialization of an Atg8 isoform. bioRxiv 453563. doi: 10.1101/453563 [DOI] [PMC free article] [PubMed]
- [45].Boeske A, Schwarten M, Ma P. Direct binding to GABARAP family members is essential for HIV-1 Nef plasma membrane localization. Sci Rep. 2017;7(1):5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gallivan JP, Dougherty DA. A computational study of cation−π interactions vs salt bridges in aqueous media: implications for protein engineering. J Am Chem Soc. 2000;122(5):870–874. [Google Scholar]
- [47].Waldburger CD, Schildbach JF, Sauer RT. Are buried salt bridges important for protein stability and conformational specificity? Nat Struct Biol. 1995;2(2):122–128. [DOI] [PubMed] [Google Scholar]
- [48].Sugawara K, Suzuki NN, Fujioka Y, et al. The crystal structure of microtubule-associated protein light chain 3, a mammalian homologue of Saccharomyces cerevisiae Atg8. Genes Cells. 2004;9(7):611–618. [DOI] [PubMed] [Google Scholar]
- [49].Skytte Rasmussen M, Mouilleron S, Kumar Shrestha B, et al. ATG4B contains a C-terminal LIR motif important for binding and efficient cleavage of mammalian orthologs of yeast Atg8. Autophagy. 2017;13(5):834–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kar R, Singha PK, Venkatachalam MA, et al. A novel role for MAP1 LC3 in nonautophagic cytoplasmic vacuolation death of cancer cells. Oncogene. 2009;28(28):2556–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Scherz-Shouval R, Weidberg H, Gonen C, et al. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci U S A. 2010;107(43):18511–18516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Suzuki H, Tabata K, Morita E, et al. Structural basis of the autophagy-related LC3/Atg13 LIR complex: recognition and interaction mechanism. Structure. 2014;22(1):47–58. [DOI] [PubMed] [Google Scholar]
- [53].Costa JR, Prak K, Aldous S, et al. Autophagy gene expression profiling identifies a defective microtubule-associated protein light chain 3A mutant in cancer. Oncotarget. 2016;7(27):41203–41216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wang W, Chen Z, Billiar TR, et al. The carboxyl-terminal amino acids render pro-human LC3B migration similar to lipidated LC3B in SDS-PAGE. PLoS One. 2013;8(9):e74222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Liu C, Ma H, Wu J, et al. Arginine68 is an essential residue for the C-terminal cleavage of human Atg8 family proteins. BMC Cell Biol. 2013;14:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chakrama FZ, Seguin-Py S, Le Grand JN, et al. GABARAPL1 (GEC1) associates with autophagic vesicles. Autophagy. 2010;6(4):495–505. [DOI] [PubMed] [Google Scholar]
- [57].Yin H, Slusky JS, Berger BW, et al. Computational design of peptides that target transmembrane helices. Science. 2007;315(5820):1817–1822. [DOI] [PubMed] [Google Scholar]
- [58].Stolz A, Putyrski M, Kutle I, et al. Fluorescence-based Atg8 sensors monitor localization and function of LC3/GABARAP proteins. Embo J. 2017;36(4):549–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].He Z, Zhang H, Gao S, et al. Evolview v2: an online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 2016;44(W1):W236–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Eddy SR. Accelerated Profile HMM Searches. PLoS Comput Biol. 2011;7(10):e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Studio CU Discovery ”version 25”. San Diego: Accelrys Inc; 2009. [Google Scholar]
- [65].Van Der Spoel D, Lindahl E, Hess B, et al. Gromacs: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. [DOI] [PubMed] [Google Scholar]
- [66].Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118(45):11225–11236. [Google Scholar]
- [67].Jorgensen WL, Chandrasekhar J, Madura JD, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. [Google Scholar]
- [68].Darden T, York D, Pedersen L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092. [Google Scholar]
- [69].Hess B, Bekker H, Berendsen HJ, et al. LINCS: A linear constraint solver for molecular simulations. J Comput Chem. 1997;18(12):1463–1472. [Google Scholar]
- [70].Nośe S. A unified formulation of the constant temperature molecular dynamics methods. J Chem Phys. 1984;81(1):511–519. [Google Scholar]
- [71].Parrinello M, Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys. 1981;52(12):7182–7190. [Google Scholar]
- [72].MATLAB, 2012b STR. Natick, Massachusetts, United States: The MathWorks, Inc.; 2012. [Google Scholar]
- [73].Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–38, 27–28. [DOI] [PubMed] [Google Scholar]
- [74].The PyMOL molecular graphics system. Version 2.0. Schrödinger, LLC; 2010. [Google Scholar]
- [75].Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. [DOI] [PubMed] [Google Scholar]
- [76].Abdel-Azeim S, Chermak E, Vangone A, et al. MDcons: intermolecular contact maps as a tool to analyze the interface of protein complexes from molecular dynamics trajectories. BMC Bioinformatics. 2014;15 Suppl 5:S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Rogov V, Dötsch V, Johansen T, et al. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014;53(2):167–178. [DOI] [PubMed] [Google Scholar]
- [78].Lv M, Wang C, Li F, et al. Structural insights into the recognition of phosphorylated FUNDC1 by LC3B in mitophagy. Protein Cell. 2017;8(1):25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Ichimura Y, Kumanomidou T, Sou YS, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283(33):22847–22857. [DOI] [PubMed] [Google Scholar]
- [80].Noda NN, Kumeta H, Nakatogawa H, et al. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells. 2008;13(12):1211–1218. [DOI] [PubMed] [Google Scholar]
- [81].Durrant JD, de Oliveira CAF, McCammon JA. POVME: an algorithm for measuring binding-pocket volumes. J Mol Graph Model. 2011;29(5):773–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Durrant JD, Votapka L, Sørensen J, et al. POVME 2.0: an enhanced tool for determining pocket shape and volume characteristics. J Chem Theory Comput. 2014;10(11):5047–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: applications to cancer genomics. Nucleic Acids Res. 2011;39(17):e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Pires DE, Ascher DB, Blundell TL. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics. 2014;30(3):335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Pandurangan AP, Ochoa-Montaño B, Ascher DB, et al. SDM: a server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017;45(W1):W229–W235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Pires DE, Ascher DB, Blundell TL. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014;42(WebServer issue):W314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Pires DE, Ascher DB. mCSM-AB: a web server for predicting antibody-antigen affinity changes upon mutation with graph-based signatures. Nucleic Acids Res. 2016;44(W1):W469–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kawabata T. Detection of multiscale pockets on protein surfaces using mathematical morphology. Proteins. 2010;78(5):1195–1211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.