

ABSTRACT
One of the most significant challenges of inflammatory bowel disease (IBD) research is to understand how alterations in the symbiotic relationship between the genetic composition of the host and the intestinal microbiota, under impact of specific environmental factors, lead to chronic intestinal inflammation. Genome-wide association studies, followed by functional studies, have identified a role for numerous autophagy genes in IBD, especially in Crohn disease. Studies using in vitro and in vivo models, in addition to human clinical studies have revealed that autophagy is pivotal for intestinal homeostasis maintenance, gut ecology regulation, appropriate intestinal immune responses and anti-microbial protection. This review describes the latest researches on the mechanisms by which dysfunctional autophagy leads to disrupted intestinal epithelial function, gut dysbiosis, defect in anti-microbial peptide secretion by Paneth cells, endoplasmic reticulum stress response and aberrant immune responses to pathogenic bacteria. A better understanding of the role of autophagy in IBD pathogenesis may provide better sub-classification of IBD phenotypes and novel approaches for disease management.
Abbreviations: AIEC: adherent-invasive Escherichia coli; AMPK: AMP-activated protein kinase; ATF6: activating transcription factor 6; ATG: autophagy related; Atg16l1[ΔIEC] mice: mice with Atg16l1 depletion specifically in intestinal epithelial cells; Atg16l1[HM] mice: mice hypomorphic for Atg16l1 expression; BCL2: B cell leukemia/lymphoma 2; BECN1: beclin 1, autophagy related; CALCOCO2: calcium binding and coiled-coil domain 2; CASP: caspase; CD: Crohn disease; CGAS: cyclic GMP-AMP synthase; CHUK/IKKA: conserved helix-loop-helix ubiquitous kinase; CLDN2: claudin 2; DAPK1: death associated protein kinase 1; DCs: dendritic cells; DSS: dextran sulfate sodium; EIF2A: eukaryotic translation initiation factor 2A; EIF2AK: eukaryotic translation initiation factor 2 alpha kinase; ER: endoplasmic reticulum; ERBIN: Erbb2 interacting protein; ERN1/IRE1A: ER to nucleus signaling 1; FNBP1L: formin binding protein 1-like; FOXP3: forkhead box P3; GPR65: G-protein coupled receptor 65; GSK3B: glycogen synthase kinase 3 beta; IBD: inflammatory bowel disease; IECs: intestinal epithelial cells; IFN: interferon; IL: interleukin; IL10R: interleukin 10 receptor; IRGM: immunity related GTPase M; ISC: intestinal stem cell; LAMP1: lysosomal-associated membrane protein 1; LAP: LC3-associated phagocytosis; MAP1LC3B: microtubule-associated protein 1 light chain 3 beta; LPS: lipopolysaccharide; LRRK2: leucine-rich repeat kinase 2; MAPK: mitogen-activated protein kinase; MHC: major histocompatibility complex; MIF: macrophage migration inhibitory factor; MIR/miRNA: microRNA; MTMR3: myotubularin related protein 3; MTOR: mechanistic target of rapamycin kinase; MYD88: myeloid differentiation primary response gene 88; NLRP3: NLR family, pyrin domain containing 3; NOD2: nucleotide-binding oligomerization domain containing 2; NPC: Niemann-Pick disease type C; NPC1: NPC intracellular cholesterol transporter 1; OMVs: outer membrane vesicles; OPTN: optineurin; PI3K: phosphoinositide 3-kinase; PRR: pattern-recognition receptor; PTPN2: protein tyrosine phosphatase, non-receptor type 2; PTPN22: protein tyrosine phosphatase, non-receptor type 22 (lymphoid); PYCARD/ASC: PYD and CARD domain containing; RAB2A: RAB2A, member RAS oncogene family; RELA: v-rel reticuloendotheliosis viral oncogene homolog A (avian); RIPK2: receptor (TNFRSF)-interacting serine-threonine kinase 2; ROS: reactive oxygen species; SNPs: single nucleotide polymorphisms; SQSTM1: sequestosome 1; TAX1BP1: Tax1 binding protein 1; Th: T helper 1; TIRAP/TRIF: toll-interleukin 1 receptor (TIR) domain-containing adaptor protein; TLR: toll-like receptor; TMEM173/STING: transmembrane protein 173; TMEM59: transmembrane protein 59; TNF/TNFA: tumor necrosis factor; Treg: regulatory T; TREM1: triggering receptor expressed on myeloid cells 1; UC: ulcerative colitis; ULK1: unc-51 like autophagy activating kinase 1; WT: wild-type; XBP1: X-box binding protein 1; XIAP: X-linked inhibitor of apoptosis.
KEYWORDS: Autophagy, immune responses, inflammatory bowel diseases, intestinal homeostasis, intestinal microbiota, microbial infection
Introduction
The etiology of inflammatory bowel diseases (IBD), including Crohn disease (CD) and ulcerative colitis (UC), has involved environmental factors, infectious agents and genetic susceptibility, leading to abnormal mucosal immune response against the intestinal microbiota [1]. Of the genetic factors implicated in IBD etiology, variants in the autophagy-related genes have been identified. Autophagy is a process conserved during evolution in eukaryotes, by which the cytoplasmic materials are degraded inside the lysosome [2]. Three distinct forms of autophagy have been described, including microautophagy, chaperone-mediated autophagy and macroautophagy [3]. Macroautophagy is the most studied form, and here we use the term autophagy to refer to macroautophagy if not otherwise mentioned. Autophagy was initially characterized as a nonspecific process induced under starvation conditions to recycle building blocks to compensate for the lack of nutrients, and thus was so-called non-selective bulk autophagy. Later, it has been evidenced that autophagy can be induced in non-starved cells to degrade specific substrates, such as aggregated proteins, damaged mitochondria or invading pathogens, which is known as selective autophagy [4]. Autophagy plays a key role in maintaining intestinal homeostasis, in regulating the interaction between gut microbiota and innate and adaptive immunity, and in host defense against intestinal pathogens [3]. A dysfunction of autophagy is associated with several human pathologies including IBD [2].
The pioneer evidence for the implication of autophagy in IBD etiology came from genome-wide association studies, which revealed single nucleotide polymorphisms (SNPs) in the autophagy-associated genes as susceptibility factors for CD. Most evidence for the association between these genetic variants and IBD etiology has come from functional studies using the ATG16L1 (autophagy related 16 like 1)T300A variant [2,5]. Pioneer studies showed that human cells having the ATG16L1T300A variant exhibit impaired autophagy-mediated clearance of intracellular bacteria and increased pro-inflammatory cytokine production [5]. Paneth cells from CD patients homozygous for the ATG16L1T300A allele, or from mice hypomorphic for Atg16l1 expression (Atg16l1[HM] mice), mice with Atg16l1 depletion specifically in intestinal epithelial cells (Atg16l1[ΔIEC] mice) or knock-in mice expressing the ATG16L1T300A variant exhibit structural and functional aberrances with decreased expression of antimicrobial peptides [2,5]. The ATG16L1T300A variant also leads to defects in autophagy induction, bacterial trafficking and antigen processing and presentation in dendritic cells (DCs) [2,5]. Together, these studies suggest a role for ATG16L1 in the control of intestinal epithelial homeostasis and inflammatory immune responses. The potential mechanisms linking the ATG16L1T300A variant to defective autophagy were recently discovered. Indeed, ATG16L1T300A protein exhibits enhanced CASP3 (caspase 3)-dependent degradation in response to stress signals [6]. The ATG16L1T300A variant also influences the ability of ATG16L1 to interact with TMEM59 (transmembrane protein 59), which engages ATG16L1 to induce autophagy in response to bacterial infection [7]. The interaction between ATG16L1 and NOD2 (nucleotide-binding oligomerization domain containing 2), encoded by the first CD susceptibility gene, reinforces the importance of autophagy in CD pathogenesis. Indeed, the CD-associated NOD2L1007fsinsC frameshift mutation leads to impaired autophagy-mediated intracellular bacterial clearance, due to a defect in recruiting ATG16L1 to the plasma membrane at the bacterial entry site, and fails to promote major histocompatibility complex (MHC) II-mediated antigen presentation by DCs [2,5]. Further studies have shown a link between the CD-associated susceptibility gene IRGM (immunity related GTPase M) and defect in autophagy-mediated clearance of CD-associated adherent-invasive Escherichia coli (AIEC), as well as the association between granulomas, one of the microscopic hallmarks of CD, and the genetic variants in the autophagy-associated genes ATG4A, ATG2A, FNBP1L (formin binding protein 1 like) and ATG4D [2,5]. However, the pathological relevance of the CD-associated SNPs in other autophagy-associated genes, such as ULK1 (unc-51 like autophagy activating kinase 1), LRRK2 (leucine rich repeat kinase 2), PTPN2 (protein tyrosine phosphatase non-receptor type 2) and CALCOCO2 (calcium binding and coiled-coil domain 2), has remained mostly unknown [2,5].
Efforts have been continuously made to explore the molecular mechanisms by which any shortcoming in autophagy could lead to disrupted intestinal epithelial function, gut dysbiosis, defect in antimicrobial peptide secretion by Paneth cells, endoplasmic reticulum (ER) stress response in enterocytes and aberrant immune responses to pathogenic bacteria, which are hallmarks of IBD pathogenesis. The latest important findings will be discussed in this review.
Autophagy and intestinal homeostasis
Autophagy and intestinal epithelial barrier function
The intestinal epithelium, which separates the luminal content from the mucosal immune system, is an important defensive line to protect the homeostasis of the gut microbiota and minimize intestinal inflammatory responses [8]. Increased intestinal permeability has been reported in IBD, which is associated with abnormal expression of tight junction proteins [9]. In 2015, autophagy was reported for the first time to regulate intestinal barrier function via inducing lysosomal degradation of the tight junction protein CLDN2 (claudin 2), thus decreasing epithelial permeability [10]. Increased CLDN2 level and tight junction defects in intestinal epithelial Caco-2 monolayers treated with the pro-inflammatory cytokine TNF/TNFA (tumor necrosis factor) partly arises from the inhibition of autophagy-mediated CLDN2 degradation [11] (Figure 1A). It was recently shown that defects in mitochondria and ER functions induce intestinal permeability, promoting E. coli internalization and transcytosis across the epithelium, and these are counteracted by selective autophagy-mediated elimination of intracellular bacteria, which is so-called xenophagy [12]. Moreover, the trans-epithelial permeability instigated by mitochondrial dysfunction is amplified by NOD2 knock-down in intestinal epithelial T84 cells [13]. These studies together showed a role for autophagy in protection of epithelial barrier function.
Figure 1.
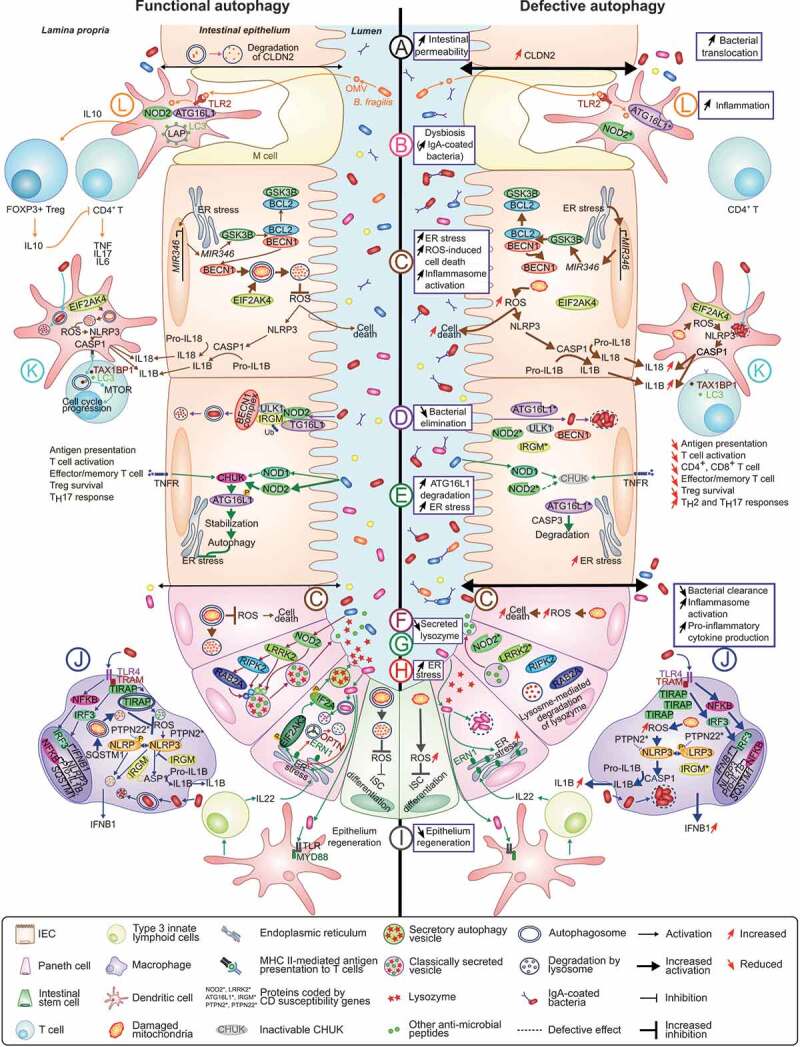
Role of autophagy in the maintenance of intestinal homeostasis and potential mechanisms by which defective autophagy may contribute to CD development. At the intestinal epithelium: (A) Autophagy modulates epithelial barrier function via lysosome-mediated degradation of CLDN2. Dysfunctional autophagy leads to increased CLDN2 level associated with increased intestinal permeability [10,11]. (B) Defective autophagy leads to intestinal dysbiosis and increased IgA-coated bacterial amount [26,27,32]. (C) By promoting mitochondrial homeostasis, autophagy protects IECs from cell death and prevents loss of Paneth cells [15,16]. MIR346, induced under ER stress, enhances GSK3B translation, favoring the dissociation between BCL2 and BECN1. This consequently activates mitophagy, thus reducing ROS level [41]. ROS-mediated NLRP3 inflammasome activation, which leads to CASP1 activation and subsequently IL18 and IL1B production, is also inhibited by EIF2AK4-induced mitophagy upon amino acid starvation [63]. Dysfunctional autophagy leads to accumulation of damaged mitochondria and ROS, increasing inflammasome activation and inflammation [63] and aggravating ROS-induced cell death [41]. (D) NOD2 recruits ATG16L1 to the plasma membrane at the bacterial entry site, initiating autophagy. Association of IRGM with NOD2 promotes IRGM ubiquitination and the assembly of the core autophagy machinery, promoting xenophagy [76]. Autophagy-associated risk variants induce defective autophagy and impaired intracellular bacterial clearance. (E) Stimulation of NOD and TNF receptors in IECs activates CHUK, which phosphorylates ATG16L1, leading to its stabilization, thus preventing ER stress during inflammation. Inactivation of CHUK fails to stabilize ATG16L1, which is consequently degraded by CASP3, increasing ER stress [47]. (F) In Paneth cells, NOD2 activation in response to commensal bacteria leads to recruitment of LRRK2, RIPK2 and RAB2A to dense core vesicles, a process required for sorting and secretion of lysozyme and other antimicrobial peptides. Dysfunctional NOD2 or LRRK2 result in lysosome-mediated degradation of lysozyme [21,22]. (G) During infection of Paneth cells with invasive bacteria, ER-Golgi secretion pathway is impaired, lysozyme is secreted via secretory autophagy. This process requires ER stress-mediated EIF2AK3-EIF2A activation in Paneth cells and activation of TLR-MYD88 in DCs that promotes IL22 secretion by type 3 innate lymphoid cells. Paneth cells with defective autophagy fails to secrete lysozyme via secretory autophagy [23]. (H) During ER stress in Paneth cells, ERN1 is recruited to autophagosomes via its interaction with OPTN, thus being degraded by autophagy. Impaired clearance of ERN1 aggregates during ER stress due to defective autophagy leads to increased ER stress and spontaneous CD-like transmural ileitis in mice [44]. (I) In intestinal stem cells, autophagy limits ROS accumulation that inhibits their differentiation, allowing epithelium regeneration. Defective autophagy leads to ROS accumulation and impaired epithelium regeneration [57]. (J) In macrophages, in response to TLR4 activation, which drives TIRAP-dependent inflammation, autophagy is activated to control TIRAP turnover and to limit production of IFNB1. Defective autophagy leads to TIRAP accumulation and subsequently increased IFNB1 production [83]. In response to TLR4 activation, NFKB activates expression of NLRP3, pro-IL1B and SQSTM1 [67]. SQSTM1 promotes mitophagy to prevent NLRP3 inflammasome activation, thus inhibiting IL1B production [67]. IRGM also limits NLRP3 inflammasome activation by preventing its assembly and by mediating selective autophagic degradation of the inflammasome components [68]. Defective autophagy leads to accumulation of dysfunctional mitochondria and ROS, enhancing NLRP3 inflammasome activation and subsequently IL1B production. (K) In DCs, autophagy degrades intracellular pathogens and participates in the presentation of antigens to T cells to induce adaptive immune responses. EIF2AK4-induced mitophagy inhibits ROS-mediated NLRP3 inflammasome activation, decreasing IL18 and IL1B production [63]. Defective autophagy leads to impaired bacterial elimination and antigen presentation, impairing T cell activation [92]. T cell activation is supported by the autophagic receptor TAX1BP1 that binds to LC3 and induces autophagy, providing critical amino acids that activates MTORC1 complex and induces metabolic transition of activated T cells [99]. An alteration of autophagy impairs T cell metabolic transition and proliferation, leading to decreased numbers of CD4+ and CD8+ T cells, impaired memory CD8+ T cell development, decreased Treg cell survival and increased Th2 and Th17 responses [91,93–99]. (L) Bacteroides fragilis from gut microbiota secrete immunomodulatory molecules through OMVs that are recognized by DCs via TLR2, activating LAP through NOD2 and ATG16L1 and inducing FOXP3+ Treg cells, which produce IL10, thus limiting CD4+ T cell-mediated inflammatory responses [101,102]. DCs having the autophagy-related risk variants fail to induce IL10 production by FOXP3+ Treg cells in response to B. fragilis-derived OMVs [101,102].
Autophagy and epithelial cell death
Epithelial apoptosis, especially TNF-induced apoptosis, has been associated with IBD pathogenesis [14]. Recently, it has been evidenced that autophagy can modulate cytokine-induced programmed cell death in intestinal epithelium, limiting intestinal inflammation [15,16]. Using Atg16l1ΔIEC mice exhibiting chronic colitis triggered by the intestinal opportunistic pathogen Helicobacter hepaticus, autophagy was demonstrated to protect IECs from TNF-induced apoptosis, allowing the maintenance of intestinal barrier integrity [16]. Atg16l1ΔIEC mice infected with murine norovirus and then treated with dextran sulfate sodium (DSS) exhibit exacerbated pathological score and increased non-apoptotic epithelial cell death compared to control mice [15]. Moreover, the viability in response to TNF treatment of intestinal organoids carrying the CD-associated ATG16L1T300A variant is decreased compared to those carrying the wild-type (WT) ATG16L1 allele [15]. It has been reported that anti-TNF antibodies, a major biological therapy for IBD, can induce regulatory macrophages resembling to M2 type macrophages implicated in limiting inflammation [17]. It was recently reported that anti-TNF-induced macrophages present an increased level of autophagy compared to IFNG (interferon gamma)-induced macrophages, and that the induction of macrophages by anti-TNF is impaired in healthy subjects carrying the ATG16L1T300A variant compared with those having the WT ATG16L1 allele [17]. Moreover, M2 phenotype and viability of anti-TNF-induced macrophages are dependent on the autophagy-related protein CTSS (cathepsin S) [17]. These results suggested that a functional autophagy is required for an optimal response to anti-TNF therapy in IBD.
A link between autophagy and ERBIN (Erbb2 interacting protein), a protein required for the polarity of epithelial cells and implicated in intestinal inflammation, was recently discovered [18]. ERBIN expression is decreased in the colons of UC patients, or of mice with DSS-induced colitis or mice deficient in Il10 (interleukin 10), and Erbin deficiency in mice leads to increased susceptibility to DSS-induced colitis due to excessive activation of autophagy leading to autophagy cell death [18]. Intraperitoneal injection of chloroquine, an autophagy inhibitor, attenuates excessive inflammatory response in DSS-treated erbin-/- mice [18]. Using in vitro and in vivo models of neonatal necrotizing enterocolitis, an inflammatory intestinal disease of new-born infants, it was reported that TNF induces autophagy via the MAPK1/ERK2 (mitogen-activated protein kinase 1)-MAPK3/ERK1 pathway to suppress cell proliferation and promote apoptosis, which might consequently favor disease development [19]. Even if these studies diverged in the precise mechanism of cell death, they all suggested that defective autophagy may lead to an excessive level of epithelial cell death, favoring IBD development.
Autophagy and paneth cell homeostasis and function
Autophagy is critical for the development, maintenance and functions of Paneth cells. Under physiological conditions, antimicrobial peptides are synthesized in the ER and transported to the Golgi, where they are packed into immature secretory dense core vesicles in Paneth cells [20]. Then, immature dense core vesicles undergo a maturation process, during which the peptides that are destined to extracellular secretion are retained into dense core vesicles, while the others are directed to the endosomal-lysosomal pathway to be degraded. Recent studies have highlighted the mechanism by which lysozyme is sorted into dense core vesicles in Paneth cells, which requires the small GTPase RAB2A (RAB2A, member RAS oncogene family), NOD2, LRRK2 and the adaptor protein RIPK2 (receptor [TNFRSF]-interacting serine-threonine kinase 2) encoded by CD susceptibility genes [21,22]. Mechanistically, NOD2 is activated by symbiotic bacteria-derived muramyl dipeptide, which in turn activates the LRRK2-RIPK2-RAB2A pathway. This allows the recruitment of NOD2, LRRK2, RIPK2 and RAB2A to dense core vesicles, which is required for lysozyme sorting [21,22] (Figure 1F). Dysfunctional NOD2 or LRRK2 results in lysosomal degradation of lysozyme [21,22]. The role of autophagy in maintaining Paneth cell-mediated lysozyme secretion during invasive pathogen infection, which induces ER stress and damages in Golgi apparatus, was recently discovered [23]. Indeed, to counteract Golgi apparatus damages induced by Salmonella Typhimurium infection, Paneth cells can bypass the ER-Golgi secretion pathway and redirect lysozyme through an autophagy-based alternative secretion pathway [23] (Figure 1G). Activation of secretory autophagy in Paneth cells requires bacteria-induced ER stress through the EIF2AK3 (eukaryotic translation initiation factor 2 alpha kinase 3)-EIF2A (eukaryotic translation initiation factor 2 alpha) pathway [23]. In addition, it requires activation of MYD88 (myeloid differentiation primary response gene 88) in DCs and IL22 secretion by type 3 innate lymphoid cells [23]. Importantly, knock-in mice expressing the ATG16L1T300A variant exhibit disruption of both ER-Golgi secretion pathway and secretory autophagy in Paneth cells in response to invasive bacterial infection [23] (Figure 1G). These results supported a role for autophagy in lysozyme secretion during bacterial infection, and suggested how the CD-associated variants in autophagy genes could lead to aberrant lysozyme packaging and secretion that characterize dysfunctional Paneth cells in CD patients. Furthermore, using Atg16l1[ΔIEC] mice and a virally-triggered mouse model of IBD, it was reported that by promoting mitochondrial homeostasis, autophagy is essential to prevent Paneth cell loss and exaggerated cell death, thus contributing to intestinal barrier maintenance [15] (Figure 1C).
The autophagy-associated protein IRGM and its murine ortholog IRGM1 have also a role in Paneth cell morphology and function. Indeed, irgm1-/- mice display marked Paneth cell alterations, including abnormal secretory granule development, decreased expression of selected antimicrobial peptides and increased susceptibility to DSS-induced colitis [24]. Moreover, in contrast to conventionally raised irgm1-/- mice, specific-pathogen-free irgm1-/- mice present only a slight increase in susceptibility to DSS-induced inflammation and minimal abnormalities in Paneth cell morphology and antimicrobial peptide production [25]. These mice also present a profound difference in the gut microbiota composition [25]. These suggest that environmental conditions, by modifying the gut microbiota composition, strongly impact the effect of genetic susceptibility on Paneth cells, and subsequently on epithelial functions and susceptibility to intestinal inflammation.
Together, these studies show that autophagy is a key regulator of Paneth cell homeostasis, and that autophagy dysfunction might be implicated in the abnormality of Paneth cells observed in CD.
Autophagy and gut microbiota
Autophagy has a role in the control of gut microbiota composition, and dysfunctional autophagy has been linked with intestinal dysbiosis (Figure 1B). Indeed, mice with colonic epithelial cell-specific Atg7 deficiency show altered fecal microbiota composition with an increase in total bacteria and an enrichment in Clostridum leptum, Eubacterium cylindroides and Bacteroides fragilis compared to WT control mice [26]. Furthermore, mice with colonic epithelial cell-specific Atg7 deficiency exhibit increased bacterial burden in the colonic epithelium compared with WT mice under the steady state and following DSS-induced colitis [26]. Similarly, compared with WT mice, Atg5[ΔIEC] mice exhibit altered gut microbiota composition with reduced diversity, as observed in CD, and a decreased number of the bacteria implicated in the control of inflammatory responses, such as those from the Lachnospiraceae and Ruminococcaceae families, of which the abundance is decreased in CD [27]. Conversely, Atg5[ΔIEC] mice exhibit an increased number of pro-inflammatory bacteria, such as those from the Pasteurellaceae family, of which the abundance is increased in CD [27]. Moreover, Atg5[ΔIEC] mice exhibit decreased abundance of Akkermansia muciniphila, as shown in IBD, and increased abundance of Candidatus Athromitus, segmented filamentous bacteria able to induce the differentiation of naïve CD4+ T cells towards Th (T helper) 1 and Th17 cells implicated in IBD pathogenesis [27]. Recently, changes in the fecal microbiota composition of knock-in mice expressing the ATG16L1T300A variant compared to WT mice at steady state with an increase in the order Bacteroidales was reported [28]. During DSS-induced colitis, the knock-in mice display altered fecal microbial composition associated with a decrease in the phylum Firmicutes and an increase in the phyla Bacteroidetes, Proteobacteria and Cyanobacteria, compared to WT mice [28]. Gnotobiotically-housed knock-in mice expressing the ATG16L1T300A variant that received stool from a patient with active CD, but not from one with active UC, exhibit a difference in the fecal microbiota composition, with an increase in Bacteroides abundance, compared to WT mice [28]. This is associated with increased Th17 and Th1 cells in the lamina propria of the colon and the ileum without the development of intestinal inflammation [28]. Together, these results revealed that the ATG16L1T300A risk variant contributes to gut dysbiosis and dysregulated immune response prior to disease symptoms. However, how host genotype or particular SNPs influence gut microbiota composition or function is still not fully understood. Or in other words, cause-consequence mechanisms behind the link between human host genetics and the gut microbiota are still unclear.
Recently, our group showed that deficiency of the amino acid deprivation sensor EIF2AK4 (eukaryotic translation initiation factor 2 alpha kinase 4) in mice, which leads to impaired autophagy-mediated clearance of CD-associated AIEC and consequently enhanced inflammation [29], induces changes in the fecal microbiota composition upon AIEC colonization but not under the steady state [30]. This suggested that in some conditions, defective autophagy alone does not trigger intestinal dysbiosis but its combination with an infectious agent can do so. It was reported that the IBD-associated risk variants, including those in NOD2, ATG16L1 and IRGM genes, can influence the fecal microbiota composition of healthy individuals, characterized by a decrease in Roseburia spp., an acetate-to-butyrate converter that produces butyrate, a short-chain fatty acid having an important role in IEC homeostasis [31]. Zhang and co-authors also reported an increased amount of IgA-coated bacteria, which were identified as IBD-promoting microbes [32], in the feces of mice with myeloid cell-specific Atg16l1 deficiency and in both non-IBD and CD patients carrying the ATG16L1T300A variant [33] (Figure 1B). Furthermore, CD patients homozygous for the ATG16L1T300A variant exhibit a higher abundance of the pathosymbiont groups, including Enterobacteriaceae (mostly Escherichia coli), Bacteroidaceae (mostly Bacteroides fragilis) and Fusobacteriaceae, in the inflamed ileum compared with the patients homozygous for the WT ATG16L1 allele [34].
Autophagy in host cells can be influenced by the gut microbiota composition. It was shown that fecal microbiota transplantation induces an increase in autophagy-related protein levels in the intestinal mucosa of piglets, suggesting that intestinal microbiota modulation may influence mucosal autophagy [35]. Bacterial metabolites have a major effect on energy homeostasis and also on autophagy in colonic epithelial cells [36]. In particular, germ-free mice exhibit altered energy metabolism, leading to enhanced autophagy in colonic epithelial cells compared to conventionally raised control mice [36]. Colonization of germ-free mice with the microbiota derived from conventionally raised mice or with Butyrivibrio fibriosolvens, a butyrate-producing bacterial strain, restores autophagy in colonic epithelial cells to normal levels [36].
We and others have shown that abnormal AIEC colonization suppresses autophagy by modulating microRNA (MIR/miRNA) levels in host cells [37,38]. Lu and co-authors revealed that MIR106B and MIR93 levels are increased in various human cell lines upon AIEC infection, inhibiting ATG16L1 expression and impairing autophagy-mediated AIEC elimination [38]. We also showed in vitro and in vivo that AIEC induce an upregulation of Mir30c and Mir130a levels in IECs, leading to inhibition of ATG5 and ATG16L1 expression, respectively, and to impaired autophagy-mediated clearance of intracellular AIEC [37]. This consequently leads to abnormal intracellular replication of AIEC and enhanced AIEC-induced inflammation [37]. We also observed an inverse correlation between the levels of MIR30C and MIR130A and those of ATG5 and ATG16L1 mRNAs in the ileal mucosa of CD patients compared to control subjects or UC patients. Although the presence of AIEC in these human biopsies was not defined, this result suggested that AIEC might reduce autophagy to favor their own colonization in CD.
Together, these data highlighted how autophagy in host cells regulates intestinal microbial communities and limits the expansion of pathobionts, and how the pathobionts have adapted to subvert this process to evade host cells.
Autophagy and ER stress
Autophagy, unfolded protein response and ER stress are interconnected at many levels [39]. ER is the primary site for folding and quality control of proteins, and any perturbation that alters ER homeostasis can cause misfolded protein accumulation, leading to activation of unfolded protein response, autophagic response or apoptosis [39]. In response to extended ER stress, unfolded protein response activation induces Ca2+ release from the ER, which in turn activates the key regulator of autophagy AMPK (AMP-activated protein kinase) [40]. It was reported that ER stress activates ATF6 (activating transcription factor 6)-DAPK1 (death-associated protein kinase 1) signaling in IECs, enhancing autophagic killing of intracellular bacteria [12]. Recently, it was shown that ER stress upregulates MIR346 level in both epithelial and squamous cell lines, leading to enhanced translation of GSK3B (glycogen synthase kinase 3 beta) mRNA and consequently increased GSK3B protein level. This favors the dissociation between BCL2 (B-cell lymphoma 2) and BECN1 (beclin 1), thus promoting mitophagy, a selective degradation of mitochondria by autophagy, and reducing levels of intracellular reactive oxygen species (ROS) [41] (Figure 1C).
Autophagy can reduce ER stress in various inflammatory and immune diseases [42]. In a mouse model of DSS-induced colitis, pharmacological or genetical inhibition of TREM1 (triggering receptor expressed on myeloid cells 1), known to amplify inflammation in several diseases, enhances both macroautophagy and chaperone-mediated autophagy by inducing MTOR (mechanistic target of rapamycin kinase) deregulation, thus compensating the unfolded protein response to reduce ER stress [43]. This also re-establishes gut microbiota homeostasis, thus preventing colonic inflammation and attenuating experimental colitis severity [43]. Dysfunctional autophagy leads to increased ER stress in murine IECs [44–46]. A correlation between the presence of the ATG16L1T300A variant and ER stress in Paneth cells and increased AIEC colonization in patients with quiescent CD was reported [46]. Atg16l1ΔIEC mice develop spontaneously CD-like transmural ileitis, and this is mediated via impaired clearance of the aggregates of the ER stress sensor ERN1/IRE1A (ER to nucleus signaling 1) during ER stress in Paneth cells [44]. ERN1 accumulation was also observed in intestinal epithelial crypts of healthy individuals and CD patients homozygous for the ATG16L1T300A allele [44]. Mechanistically, in mouse IECs experiencing ER stress, when autophagy is defective, ERN1 is not recruited to autophagosomes, which is mediated via its interaction with the autophagy receptor OPTN (optineurin), and thus is not degraded by autophagy [44] (Figure 1G). Together, these results showed that the impaired clearance of ERN1 aggregates during ER stress due to defective autophagy may predispose to IBD [44].
Moreover, stimulation of NOD and TNF receptors in IECs in a mouse model of DSS-induced colitis leads to ATG16L1 phosphorylation specifically at serine 218 by CHUK/IKKA (conserved helix-loop-helix ubiquitous kinase), thus protecting ATG16L1 from CASP3-dependent cleavage and limiting ER stress activation [47] (Figure 1E). However, in mice expressing inactivable CHUK specifically in IECs, ATG16L1 degradation is enhanced as observed with the ATG16L1T300A protein, leading to increased ER stress response [47]. This consequently increases CASP12 level, resulting in CASP1 activity inhibition and decreased secretion of the cytoprotective IL18, thus delaying epithelial regeneration and enhancing inflammation [47]. This study shows a novel mechanism by which CHUK prevents ER stress and inflammation via limiting degradation of the CD-associated ATG16L1T300A protein.
ER stress and defective autophagy can act in synergy to promote IBD development [45]. Indeed, mice deficient in both Atg16l1 and Xbp1 (X-box binding protein 1) specifically in IECs (Atg16l1[ΔIEC]Xbp1[ΔIEC]) develop more severe colitis compared to Atg16l1[ΔIEC] or Xbp1[ΔIEC] mice [45]. However, all Atg16l1[ΔIEC], Xbp1[ΔIEC] and Atg16l1[ΔIEC]Xbp1[ΔIEC] mice exhibit increased accumulation and hyper-activation of ERN1 [45], which seems to be due to defective ERN1 degradation by autophagy [44]. Primary IECs from CD patients carrying several risk variants implicated in both autophagy and ER stress, but not in autophagy alone, exhibit significantly higher ER stress response after ER stress induction but not at the basal level, indicating that the genetic predispositions in both pathways should be considered when looking at functional level [48].
Autophagy and mitochondrial ROS secretion
Autophagy, especially mitophagy, by eliminating damaged or superfluous mitochondria, plays a major role in limiting ROS accumulation. Mutations in the autophagy-related genes or autophagy deficiency have an impact on ROS levels via the impaired elimination of dysfunctional mitochondria in several cell types [49,50]. Murine macrophages expressing dysfunctional ATG16L1 produce higher amount of ROS following lipopolysaccharide (LPS) stimulation compared to WT macrophages [51]. Consistently, ROS production by Atg16l1-deficient murine DCs is increased compared to WT DCs either at baseline or upon Salmonella Typhimurium infection, and this is associated with an increased number of mitochondria probably due to impaired mitophagy [52]. In regulating ROS level, autophagy has an impact on cytokine secretion [53]. Indeed, in human intestinal epithelial T84 cells depleted in NOD2 or ATG16L1 gene, disruption of mitochondrial functions leads to increased IL8 production in response to commensal E. coli compared to WT cells [54]. Furthermore, autophagy deficiency increases secretion of the pro-inflammatory cytokine MIF (macrophage migration inhibitory factor) by macrophages both basally and in response to bacterial LPS in a mitochondrial ROS-dependent manner [55]. Macrophages depleted in the autophagy gene Map1lc3b (microtubule-associated protein 1 light chain 3B) exhibit dysfunctional mitochondria, associated with increased ROS production, leading to enhanced IL1B and IL18 secretion in response to LPS [56].
Autophagy and ROS are involved in intestinal stem cell (ISC) maintenance [57] and IEC defense against intracellular bacteria [13] (Figure 1I). Indeed, Atg5[ΔIEC] mice exhibit a fewer number of ISCs, associated with higher ROS level and defective ISC-associated intestinal recovery after irradiation, compared to WT mice [57]. Treatment of Atg5[ΔIEC] mice with an antioxidant rescues the defect in ISC-associated intestinal recovery, suggesting that autophagy, by reducing excessive ROS level, allows the maintenance of ISCs and intestinal regeneration [57].
Finally, in modulating autophagy, intestinal microbiota may influence ROS production. Indeed, trimethylamine-N-oxide, a choline-derived metabolite produced by gut microbiota, via inhibiting ATG16L1, SQSTM1 (sequestosome 1) and LC3-II, increases ROS production in a dose and time dependent manner, thus explaining its involvement in IBD [58].
Autophagy and inflammasome activation
Previous studies showed that dysfunctional autophagy leads to abnormal inflammasome activation, which is associated with increased CASP1 activity, elevated IL1B and IL18 production and higher susceptibility to experimental intestinal inflammation in mice [59,60]. Recently, it was shown that mice with macrophage-specific Atg5 depletion or knock-in mice expressing the ATG16L1T300A variant exhibit increased inflammasome-mediated IL1B secretion compared to their controls, leading to more severe endotoxin-induced uveitis, an eye disease frequently developed by CD patients [61]. Murine macrophages deficient in the anti-inflammatory cytokine lL10 exhibit impaired mitophagy together with damaged mitochondrial accumulation, increased ROS production and aberrant inflammasome activation upon LPS stimulation [62]. Macrophages from il10-/- mice or IBD patients with a null mutation in the IL10R (interleukin 10 receptor) gene exhibit damaged mitochondrial accumulation, associated with dysregulated NLRP3 (NLR family pyrin domain containing 3) inflammasome activation and elevated IL1B production [62]. These results suggested that IL10 prevents the development of intestinal inflammation, at least in part, via promoting mitophagy to limit inflammasome activation and inflammatory responses [62]. Autophagy-mediated inhibition of inflammasome activation was also reported as a novel mechanism underlying the role of EIF2AK4 in controlling intestinal inflammation [63]. Indeed, upon amino acid starvation, EIF2AK4 activation exerts a protective effect on DSS-induced colitis in mice by inducing autophagy, thus reducing ROS-dependent inflammasome activation in ITGAX/CD11c+ antigen-presenting cells and IECs and inhibiting intestinal inflammation [63] (Figure 1K). Eif2ak4 deficiency specifically in CD11c+ antigen-presenting cells or IECs in mice results in impaired autophagy activation, leading to increased mitochondrial ROS level and subsequently to increased susceptibility to DSS-induced colitis [63] (Figure 1K). Recently, the CD susceptibility genes PTPN2 (protein tyrosine phosphatase non-receptor type 2) and PTPN22 have been implicated in the regulation of autophagy and inflammasome activities [64,65]. It has been reported that a loss-of-function variant in PTPN2 gene is a risk factor for CD, while a gain-of-function variant in PTPN22 gene is associated with reduced risk of CD [66]. PTPN22 dephosphorylates NLRP3 inflammasome to limit the sequestration of NLRP3 into autophagosomes to be degraded in autolysosomes [64] (Figure 1J). In human macrophages and murine bone marrow-derived DCs, loss of PTPN22 leads to enhanced NLRP3 phosphorylation and subsequent NLRP3 degradation, thus decreasing IL1B secretion [64]. Loss of functional autophagy counteracts the reduction of NLRP3 activation observed in PTPN22-deficient cells, leading to increased IL1B secretion [64] (Figure 1J). PTPN2, involved in autophagy regulation, was shown to regulate inflammasome activation and IL1B secretion by limiting the phosphorylation of the adaptor molecule ASC (apoptosis-associated speck-like protein containing a CARD), which is required for inflammasome assembly. In mice, loss of Ptpn2 in myeloid cells results in increased susceptibility to DSS-induced colitis due to increased inflammasome assembly and elevated IL1B production [65] (Figure 1J). Autophagy and inflammasome activation are recently linked via the key inflammatory regulator NFKB (nuclear factor of kappa light polypeptide gene enhancer in B cells) [67]. Indeed, NFKB activates expression of the selective autophagy receptor SQSTM1, promoting the mitophagy-mediated elimination of damaged mitochondria that secrete NLRP3-activating signals, thus inhibiting inflammasome activation and IL1B production [67] (Figure 1J). Recently, Mehto et al. showed for the first time that IRGM/IRGM1 mediates its anti-inflammatory activity by suppressing the activation of NLRP3 inflammasomes via at least two distinct mechanisms: (i) IRGM/IRGM1 physically interacts with NLRP3 inflammasome components and prevents inflammasome assembly, and (ii) IRGM/IRGM1 interacts with SQSTM1 and mediates SQSTM1-dependent selective autophagic degradation of NLRP3 inflammasome components [68] (Figure 1J). Using IRGM-depleted human THP-1 macrophages and irgm1-/- mice, it was shown that IRGM/IRGM1, by restricting inflammasome activity, protects from inflammation-induced cell death and suppresses DSS-induced colitis [68]. This work importantly showed a direct role of IRGM in suppressing inflammation and suggested that the CD-associated IRGM risk variant fails to do so, thus exacerbating inflammasome activity and the subsequent inflammation.
Whereas most of the studies on the cross-talk between autophagy and inflammasome have reported that autophagy inhibits inflammasome activation to prevent excessive inflammation, ROS and inflammasome can be involved in the regulation of autophagy activation. Indeed, NLRP3 was identified as a novel binding partner of the autophagy inhibitor MTOR [69]. Under inflammatory conditions, NLRP3 inflammasome binds to and promotes MTOR phosphorylation, inhibiting autophagy and impairing the autophagy-mediated elimination of pro-inflammatory mediators and consequently exacerbating inflammation [69]. Hypoxia, an important environmental factor that influences IBD development, can counteract intestinal inflammation in a mouse model of DSS-induced colitis or in il10-/- mice by reducing NLRP3-MTOR binding, thus activating the autophagy-mediated degradation of NFKB signaling mediators and reducing expression of pro-inflammatory genes [69]. However, it was reported that during Legionella pneumophila infection, inflammasome activation promotes autophagy, thus facilitating bacterial clearance [70]. Furthermore, in response to intracellular pathogens, CASP4 is activated, leading to inflammasome activation, thus positively regulating autophagosome biogenesis and trafficking to lysosomes in macrophages [71]. This consequently increases xenophagy-mediated pathogen elimination [71].
Therapies involving the modulation of autophagy and NLRP3 inflammasome to alleviate IBD have been largely studies. Administration of andrographolide, a herbal extract isolated from a Southeast Asian plant, to mice with DSS-induced colitis enhances mitophagy in peritoneal macrophages, leading to NLRP3 inflammasome inhibition and subsequently amelioration of colitis [72]. Agonist-mediated activation of cannabinoid receptor 2, a G protein-coupled receptor mainly located on immune cells and has been used to treat IBD, increases autophagy, which in turn inhibits NLRP3 inflammasome activation and attenuates DSS-induced colitis in mice [73]. Conversely, inhibition of NRLP3 inflammasome could be an efficient strategy to promote autophagy activation. Administration of Ginsenoside Rd, an anti-inflammatory molecule, was reported to inactivate NLRP3 inflammasome, thus inducing SQSTM1-driven mitophagy, leading to amelioration of DSS-induced colitis in mice [74].
Together, these studies showed a mutual regulation between autophagy and inflammasome, and raised the possibility to use NLRP3 inhibitors that induce autophagy or autophagy activators that inhibit inflammasome as potential therapeutic molecules for IBD treatment.
Autophagy and innate and adaptive immune responses
The role of autophagy in maintaining intestinal immune homeostasis has been largely investigated. Previous studies have shown that autophagy is required for intracellular bacterial handling, inhibiting pro-inflammatory cytokine production in IECs and immune cells, and that the CD-associated variants in the autophagy-associated genes fail to do so [2,5] (Figure 1D and J). Recently, it was shown that primary Atg16l1-deficient murine macrophages and primary human macrophages harboring the ATG16L1T300A variant exhibit increased ROS production, impaired mitophagy and diminished fusion of Salmonella Typhimurium-containing vesicles with LAMP1 (lysosomal-associated membrane protein 1)-labeled lysosomes, leading to impaired bacterial clearance and reduced MHC class II antigen processing [33]. Mice with Atg16l1 deficiency in myeloid cells show exacerbated DSS-induced colitis with increased ratio of pro-inflammatory to anti-inflammatory macrophages, enhanced pro-inflammatory cytokine production, and elevated IgA-coated intestinal bacterial numbers [33]. Using bone marrow-derived macrophages from transgenic mice overexpressing Lrrk2 infected with Mycobacterium leprae, LRRK2 was shown to bind to BECN1 to induce BECN1 degradation, thus inhibiting autophagy [75]. Moreover, increased LRRK2 expression, as given by the CD-associated risk variant in LRRK2 gene, leads to enhanced NFKB activation induced by CLEC7A/Dectin-1, a recently discovered pattern-recognition receptor (PRR), and consequently excessive pro-inflammatory cytokine secretion [75]. LRRK2 inhibitors decrease CLEC7A-induced TNF production by mouse DCs and ameliorate DSS-induced colitis in control and Lrrk2 transgenic mice, and inhibit TNF production by DCs from CD patients [75]. Although the implication of IRGM in xenophagy-mediated intracellular bacterial restriction and regulation of inflammation has been documented, the exact underlying molecular mechanism was just recently revealed. Indeed, IRGM functions as a key player in the formation and activation of the autophagy initiation complex, including ULK1 and BECN1, while at the same time physically couples to NOD2 and ATG16L1, and this is crucial for the antimicrobial and anti-inflammatory functions of autophagy [76] (Figure 1D).
Early onset of CD was recently associated with Niemann-Pick disease type C (NPC), a neurodegenerative lysosomal lipid storage disorder, in which the NPC1 (NPC intracellular cholesterol transporter 1) gene involved in lipid transport is mutated [77]. Using peripheral blood monocyte-derived macrophages from patients having the NPC1 gene mutation, it was shown that this mutation is linked to impaired autophagy due to a defective autophagosome formation. This consequently abolishes NOD2-mediated handling of intracellular bacteria, and this is similar to the effects given by the CD-associated variants in NOD2 or XIAP (X-linked inhibitor of apoptosis) gene [77]. XIAP binds and ubiquitinates the adaptor protein RIPK2 following muramyl dipeptide-mediated NOD2 activation, thus activating NFKB signaling pathway and cytokine production [77]. In contrast to NOD2 and XIAP variants, NPC1 variant does not influence NOD2-RIPK2-XIAP-dependent cytokine production [77]. The defect in bacterial clearance given by the NPC1 variant can be overcome by pharmacological activation of autophagy [77]. Recently, it was revealed that inhibitors of apoptosis proteins, such as XIAP, are required for the fusion of autophagosome with lysosome during autophagy [78]. Inhibition or loss of function of inhibitor of apoptosis proteins results in impaired autophagic flux, responsible for defective mitophagy and impaired bacterial clearance, and increased secretion of TNF and IL1B, which may lead to CD development [78]. In addition to an efficient fusion of autophagosome with lysosome, a functional lysosome is also required to guarantee pathogen elimination. It was recently shown that GPR65 (G protein-coupled receptor 65), a H+-sensing G protein-coupled receptor encoded by an IBD susceptibility gene, plays a role in maintaining lysosomal pH and thus lysosomal function, preserving autophagy and pathogen defense [79]. Epithelial cells or lymphoblasts from IBD patients carrying the GPR65/I231L missense variant exhibit aberrant lysosomal acidification, which results in lysosomal dysfunction, and impaired autophagy-mediated intracellular bacterial clearance [79]. Mice with Gpr65 deficiency are more susceptible than WT mice to Citrobacter rodentium-induced intestinal disease, as shown by higher levels of C. rodentium in the caecum, colon and feces and more severe colonic inflammation with enhanced infiltration of T cells [79].
However, other studies have reported that autophagy deficiency increases resistance to infection with C. rodentium [80,81]. Marchiando et al. reported that compared to WT mice, Atg16l1[HM] mice are more resistant to C. rodentium infection, displayed by reduced number of C. rodentium associated to the colon and caecum, increased survival, decreased weight loss and diarrheal disease and protection from colonic inflammation [80]. Mechanistically, this protection is due to enhanced immune response in Atg16l1[HM] mice and is dependent on monocytes and NOD2 [80]. Similarly, Atg16l1[HM] mice are protected against C. rodentium infection via enhanced IFN-I response to the gut microbiota [81]. The same results were obtained in atg4b–/– and lc3b–/– mice, but not in mice deficient in Atg16l1 specifically in myeloid cells, indicating that the autophagy-mediated protection against C. rodentium requires myeloid cells [81]. These studies suggested that autophagy-associated gene variants may have conferred the host a protection against certain pathogen infections.
Recently, an unexpected role of ATG16L1 in regulating IL22 signaling in the intestinal epithelium via modulating CGAS (cyclic GMP-AMP synthase)-TMEM173/STING (stimulator of interferon genes protein)-IFN-I pathway was revealed [82]. Aden et al. showed that IL22 stimulation results in transient ER stress, thus activating IFN-I signaling via CGAS-TMEM173/STING in intestinal organoids, and this is increased in those derived from Atg16l1[ΔIEC] mice [82]. This consequently exacerbates TNF production and TNF-induced cell death in Atg16l1-deficient intestinal organoids. Systemic treatment of Atg16l1[ΔIEC] mice with IL22 aggravates intestinal inflammation and causes excessive necroptotic intestinal cell death, and these effects are ameliorated by inhibiting IFN-I signaling [82]. This study suggested that IL22 signals together with autophagy-associated risk factors may contribute to tissue damage and inflammation in CD. IFN-I responses and cytokine secretion may be also regulated by selective autophagy-mediated control of the turnover of the innate adaptor TIRAP/TRIF (toll-interleukin 1 receptor [TIR] domain-containing adaptor protein) in response to TLR (toll-like receptor) activation [83] (Figure 1J). Indeed, the autophagy receptors SQSTM1 and TAX1BP1 (Tax1 binding protein 1) target TIRAP for degradation by selective autophagy, and that Atg16l1-deficient murine bone marrow-derived macrophages or DCs exhibit TIRAP accumulation, driving TIRAP-dependent inflammatory signaling upon TLR stimulation with increased IFNB1 and IL1B production [83] (Figure 1J). Consistently, elevated production of IFNB1 and IL1B upon LPS treatment by human macrophages carrying the ATG16L1T300A allele compared to those harboring the WT ATG16L1 allele was observed [83].
Another IBD-associated MTMR3 (myotubularin-related protein 3) risk allele was shown to amplify the PRR-induced innate immune response by decreasing autophagy [84]. Indeed, myeloid cells from healthy individuals carrying the IBD-associated polymorphism in the MTMR3 gene exhibit increased MTMR3 expression, leading to decreased PRR-induced autophagy by reducing cellular levels of phosphatidylinositol-3-phosphate, which are associated with autophagy initiation and maturation. This consequently leads to increased PRR-induced CASP1 activation and cytokine secretion [84].
Selective autophagy receptors, such as OPTN, are required for an appropriate regulation of cytokine response. Indeed, bone marrow-derived macrophages from Optn-deficient mice present a reduced ability to secrete TNF and IL6 upon stimulation with heat-killed E. coli, due to a mis-trafficking of these cytokines to lysosomes [85]. Optn-deficient mice are more susceptible to C. rodentium-induced colitis, associated with greater colitis score, reduced TNF level in the serum and impaired neutrophil recruitment to the site of infection compared to WT mice [85]. Transcriptomic analysis of CD and control macrophages revealed that the gene encoding OPTN is under-expressed in approximately 10% of CD patients, and this is associated with decreased OPTN protein level and reduced cytokine secretion by macrophages upon bacterial infection [86]. Moreover, neutrophils can be the target of CD-associated AIEC, which invade and replicate within these cells [87]. Indeed, AIEC reside in non-acidic autolysosomes without being degraded, suggesting that AIEC either delay or prevent the full maturation of autolysosomes [87]. This is correlated with the induction of autophagic cell death and exacerbated IL8 production [87].
It was recently shown that ATG16L1 can act as an important player in innate immune defense by limiting pathogen dissemination during infection via an autophagy-independent mechanism. Indeed, ATG16L1 and its binding partners ATG5 and ATG12 are required for the repair of plasma membrane damage caused by bacterial pore-forming toxins, thus maintaining cell viability [88]. ATG16L1 was also found to restrict cell-to-cell spread of Listeria monocytogenes by limiting plasma membrane damage mediated by listeriolysin O, a pore-forming toxin expressed by the bacterium [88]. Importantly, expression of the CD-associated ATG16L1T300A risk allele leads to a defect in plasma membrane damage repair upon L. monocytogenes infection and increased cell-to-cell spread by the bacterium [88]. Thus, it should be kept in mind that the autophagy-related genetic variants can contribute to IBD pathogenesis via autophagy-independent pathways or mechanisms.
In addition to its role in innate immunity, autophagy has been implicated in adaptive immunity. After the degradation of intracellular pathogens in autolysosomes, the produced antigens are presented by MHC class II molecules to T cells to induce adaptive immune responses, a process that can be promoted by autophagy [89]. Infection of DCs by AIEC activates autophagy, thus contributing to the elimination of intracellular AIEC and to generation of MHC class II antigen-specific CD4+ T cell response [90]. Furthermore, autophagy negatively modulates adaptive responses by destabilizing the immunologic synapse, which is the DC-T cell contact site [91]. Depletion of Atg16l1 or Irgm1 in DCs in mice results in hyperstable DC-T cell interaction, increased T cell activation, and activation of a Th17 response [91]. DCs from CD patients carrying the ATG16L1T300A allele have a similar hyperstable DC-T cell interaction [91]. This suggests that the IBD-associated genetic variants in autophagy-associated genes might lead to increased adaptive immunity, enhancing disease development.
Several studies have revealed that T cells are highly sensitive to alterations of autophagy process [92]. Indeed, murine models with T-cell-specific defective autophagy exhibit decreased numbers of CD4+ and CD8+ T cells and alteration of T cell proliferation, revealing the key role of autophagy in T cell homeostasis [93–95] (Figure 1K). Moreover, autophagy substantially contributes to effector CD8+ T cell survival and memory formation [96–98] (Figure 1K). It was recently demonstrated that autophagy mediated by the autophagy receptor TAX1BP1 is implicated in the control of cellular metabolism enabling T cell activation, revealing the crucial relationship between immune activation, autophagy and metabolism of immune cells [99] (Figure 1K). Th cells play a critical role in intestinal immune responses, and their number and behavior vary during IBD [100]. Autophagy is directly involved in favoring regulatory T (Treg) cell survival and in restricting pro-inflammatory Th2 cell expansion, and T cell-specific autophagy inhibition leads to changes in the balance between different Th cell types in the intestine [95]. Mice lacking Atg16l1 in CD4+ T cells develop spontaneously intestinal inflammation characterized by Th2 responses against dietary and gut microbiota antigens and by a loss of Treg cells, suggesting that autophagy plays a role in promoting the survival of Treg cells, limiting mucosal Th2 cell expansion and thus limiting Th2 type inflammation [95] (Figure 1K). Mice deficient in Eif2ak4 specifically in antigen-presenting cells or IECs exhibit exacerbated Th17 responses and intestinal inflammation, and this is mechanistically due to impaired autophagy, enhanced inflammasome activation and IL1B production [63] (Figure 1K).
Recently, it was reported that a functional autophagy is required for the sensing of microbiota-derived protective signals to initiate an efficient immune response and to inhibit intestinal inflammation [101]. Indeed, the commensal bacterium Bacteroides fragilis secretes the immunomodulatory molecule polysaccharide A through outer membrane vesicles (OMVs), which is then sensed by DCs through TLR2 [102], resulting in enhanced conversion of naïve CD4+ T cells into FOXP3+ (forkhead box P3) Treg cells that produce IL10 and subsequently protection of mice from experimental colitis [102]. OMVs require ATG16L1 and NOD2 to activate the LC3-associated phagocytosis (LAP), a non-canonical autophagy pathway specifically activated by microbial ligands, in murine bone marrow-derived DCs [101] (Figure 1L). Upon oral administration of OMVs, although WT mice are protected from 2,4-dinitrobenzenesulfonic acid-induced colitis, mice deficient in Atg16l1 specifically in CD11+ DCs exhibit increased weight loss and elevated mortality similarly to those that did not receive OMVs [101]. Moreover, the production of IL10 by CD4+FOXP3+ Treg cells is significantly reduced in mice deficient in Atg16l1 in CD11+ DCs versus WT mice [101]. Murine bone marrow-derived DCs deficient in Nod2 are also defective in inducing IL10 production by FOXP3+ Treg cells in response to B. fragilis-derived OMVs during in vitro co-culture [101]. Finally, in contrast to those carrying the WT alleles, monocyte-derived DCs from CD patients or healthy controls homozygous for the ATG16L1T300A variant pulsed with B. fragilis OMVs and co-cultured with syngeneic CD4+ T cells are unable to induce IL10 production by FOXP3+ Treg cells. These findings suggest that IBD-associated genetic variants may lead to defective sensing of protective signals from the microbiota, favoring the disease development [101] (Figure 1L).
Collectively, these studies highlighted the molecular mechanisms contributing to the modulation of innate and adaptive immune cell function by autophagy, thus influencing the outcome immune responses.
Future directions and conclusion
As discussed in this review, defective autophagy may have a strong impact on the course of IBD, via disruption of intestinal homeostasis, affecting gut microbiota composition, impairing intracellular bacterial clearance and amplifying intestinal inflammation. Thus, efforts have been made to develop IBD treatments based on modulating autophagy.
Treatment with the rapamycin analogue everolimus, an inducer of autophagy, ameliorates CD-like ileitis in il10-/- mice [103] and improves CD symptoms [104,105]. Two case reports revealed an improvement of CD symptoms in two patients treated with everolimus and another rapamycin analogue named sirolimus by enhancing autophagy level [104,105]. However, in these contexts, the beneficial effects of rapamycin analogues may be given by the autophagy-independent immunosuppressive effects. The use of autophagy modulators as a therapy remains challenging given the low pharmacological specificity for their targets, the absence of specificity for a particular cell type and the autophagy-independent effects.
Thus, strategies to modulate specifically autophagy with no toxic effects should be developed to establish an efficient therapy for IBD treatment. Interestingly, some compounds derived from dietary products, such as vitamin D and curcumin, have been shown to induce autophagy [106,107]. Vitamin D deficiency is a critical factor in the pathology of IBD, and it was recently shown that treatment of IECs with vitamin D3 leads to increased autophagy activation [108]. This suggested that a vitamin D complementation in IBD patients may palliate their deficiency and increase autophagy level, leading to beneficial effects. Similarly, it was reported that glutamine enhances autophagy in IECs both under basal and stress-induced conditions by regulating MTOR and MAPK/p38 pathways, thus limiting stress-induced cellular apoptosis [109]. Several studies have recently revealed that curcumin treatment, which has been shown to ameliorate pathology in animal models of IBD and in clinical studies [110], promotes autophagy in vitro and in vivo via diverse molecular mechanisms [111–113]. Andrographolide, a herbal extract, enhances mitophagy in macrophages, inhibiting NLRP3 inflammasome and ameliorating DSS-induced colitis [72]. Another herbal extract called celastrol extracted from the root of Tripterygium wilfordii, a traditional Chinese medicine, enhances autophagy by inhibiting the PI3K (phosphoinositide 3-kinase)-AKT-MTOR signaling pathway in colonic tissues of il10-/- mice, thus limiting colonic inflammation [114]. These outcomes suggested that nutritional complementation of IBD patients with natural products that induce autophagy may alleviate the disease, and further investigations using large patient cohorts are needed to confirm this effect.
Given the multifactorial etiology of IBD and the fact that the underlying risk factors and pathophysiology being specific to each patient, the development of efficient personalized approaches for disease treatment appears essential. The induction of autophagy in CD patients having autophagy-associated genetic defects to restore functional antimicrobial peptide secretion and antimicrobial activity in Paneth cells and to activate an efficient immune response could be an attractive therapeutic concept [115]. Gene therapy is also a potential therapeutic strategy to relieve, but also cure, IBD. Several miRNAs have been reported to suppress autophagy by inhibiting expression of autophagy-associated genes [106]. In this regard, we showed in vitro and in vivo that inhibition of Mir30c and Mir130a in IECs abolished the AIEC-induced autophagy inhibition, reestablishing an efficient AIEC clearance and inhibiting intestinal inflammation [37]. This suggested that targeting these miRNAs could be a strategy to treat specifically CD patients with abnormal AIEC colonization. The use of an autophagy-inducing peptide termed ‘Tat-BECN1’, which can be internalized and induce autophagy in various human cell types, has been also proposed to treat human diseases with defective autophagy [116]. However, the use of autophagy-inducing peptide or miRNA-based therapy to treat IBD remains problematic and requires the development of gut-specific delivery systems as well as demands further assessment on the effectiveness and security.
In conclusion, understanding the molecular mechanisms behind the impact of IBD-associated autophagy alleles and their interactions with other etiology factors required for disease manifestation, including the resident microbiota and environmental triggers, is crucial when developing new therapeutic strategies. It should be kept in mind that the IBD-associated autophagy gene variants are also present in healthy individuals, and their presence alone is not sufficient to induce IBD. Thus, the development of multiple pathway-integrated treatments, depending on each patient subset, may be a successful strategy to provide a cure for IBD. In particular, vectorized therapies to target and modulate autophagy specifically in IBD patients with defective autophagy should be highly considered.
Funding Statement
This work was supported by the Ministère de la Recherche et de la Technologie, Inserm (UMR1071) and INRA (USC 2018), the Agence Nationale de la Recherche of the French government through the program “Investissements d’Avenir” (16-IDEX-0001 CAP 20-25) (to H.N.) and the European Union FP7 People Marie Curie International Incoming Fellowship (to H.N.).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Carrière J, Darfeuille-Michaud A, Nguyen HTT.. Infectious etiopathogenesis of crohn’s disease. World J Gastroenterol. [Internet] 2014;20:12102–12117. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25232246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nguyen HTT, Lapaquette P, Bringer M-A, et al. Autophagy and crohn’s disease. J Innate Immun. [Internet] 2013;5:434–443. Available from: https://www.karger.com/Article/FullText/345129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mizushima N. A brief history of autophagy from cell biology to physiology and disease [Internet]. Nat Cell Bio. 2018;20:521–527. Available from: http://www.nature.com/articles/s41556-018-0092-5 [DOI] [PubMed] [Google Scholar]
- [4].Kim S, Eun H, Jo E-K. Roles of autophagy-related genes in the pathogenesis of inflammatory bowel disease. Cells. [Internet] 2019;8:77. Available from: http://www.mdpi.com/2073-4409/8/1/77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lapaquette P, Thi Thu Nguyen H, Faure M. L’autophagie garante de l’immunité et de l’inflammation. médecine/sciences. [Internet] 2017;33:305–311. Available from: http://www.medecinesciences.org/10.1051/medsci/20173303018 [DOI] [PubMed] [Google Scholar]
- [6].Murthy A, Li Y, Peng I, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. [Internet] 2014;506:456–462. [DOI] [PubMed] [Google Scholar]
- [7].Boada-Romero E, Serramito-Gómez I, Sacristán MP, et al. The T300A Crohn’s disease risk polymorphism impairs function of the WD40 domain of ATG16L1. Nat Commun. [Internet] 2016;7:11821. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27273576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol. [Internet] 2014;15:19–33. [DOI] [PubMed] [Google Scholar]
- [9].Michielan A, D’Incà R. Host-microbiome interaction in Crohn’s disease: A familiar or familial issue? World J Gastrointest Pathophysiol. [Internet] 2015;6:159–168. Available from: http://www.wjgnet.com/2150-5330/full/v6/i4/159.htm [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nighot PK, Hu C-A-A, Ma TY. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. J Biol Chem [Internet]. 2015;290:7234–7246. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25616664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang C, Yan J, Xiao Y, et al. Inhibition of autophagic degradation process contributes to claudin-2 expression increase and epithelial tight junction dysfunction in tnf-α treated cell monolayers. Int J Mol Sci [Internet]. 2017;18:157. Available from: http://www.mdpi.com/1422-0067/18/1/157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lopes F, Keita ÅV, Saxena A, et al. ER-stress mobilization of death-associated protein kinase-1-dependent xenophagy counteracts mitochondria stress-induced epithelial barrier dysfunction. J Biol Chem [Internet]. 2018;293:3073–3087. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29317503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Saxena A, Lopes F, Poon KKH, et al. Absence of the NOD2 protein renders epithelia more susceptible to barrier dysfunction due to mitochondrial dysfunction. Am J Physiol Liver Physiol [Internet]. 2017;313:G26–38. Available from: http://ajpgi.physiology.org/lookup/doi/10.1152/ajpgi.00070.2017 [DOI] [PubMed] [Google Scholar]
- [14].Blander JM. Death in the intestinal epithelium-basic biology and implications for inflammatory bowel disease. Febs J [Internet]. 2016;283:2720–2730. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27250564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med. [Internet] 2017:jem.20170558. DOI: 10.1084/jem.20170558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pott J, Kabat AM, Maloy KJ. Intestinal epithelial cell autophagy is required to protect against TNF-induced apoptosis during chronic colitis in mice. Cell Host Microbe. [Internet] 2018;1–12. DOI: 10.1016/j.chom.2017.12.017 [DOI] [PubMed] [Google Scholar]
- [17].Levin AD, Koelink PJ, Bloemendaal FM, et al. Autophagy contributes to the induction of Anti-TNF induced macrophages. J Crohns Colitis [Internet]. 2016;10:323–329. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26417049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shen T, Li S, Cai L, et al. Erbin exerts a protective effect against inflammatory bowel disease by suppressing autophagic cell death. Oncotarget [Internet]. 2018;9:12035–12049. Available from: http://www.oncotarget.com/fulltext/23925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yuan Y, Ding D, Zhang N, et al. TNF-α induces autophagy through ERK1/2 pathway to regulate apoptosis in neonatal necrotizing enterocolitis model cells IEC-6. Cell Cycle. [Internet] 2018;1. DOI: 10.1080/15384101.2018.1482150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Holly M, Smith J. Paneth cells during viral infection and pathogenesis. Viruses. [Internet] 2018;10:225. Available from: http://www.mdpi.com/1999-4915/10/5/225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang Q, Pan Y, Yan R, et al. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat Immunol. [Internet] 2015;1–12. DOI: 10.1038/ni.3233 [DOI] [PubMed] [Google Scholar]
- [22].Wang H, Zhang X, Zuo Z, et al. Rip2 is required for nod2-mediated lysozyme sorting in paneth cells. J Immunol. [Internet] 2017;1601583. Available from: http://www.jimmunol.org/lookup/doi/10.4049/jimmunol.1601583 [DOI] [PubMed] [Google Scholar]
- [23].Bel S, Pendse M, Wang Y, et al. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science. [Internet] 2017;357:1047–1052. Available from: http://www.sciencemag.org/lookup/doi/10.1126/science.aal4677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liu B, Gulati AS, Cantillana V, et al. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. [Internet] 2013;305:G573–G584. Available from: http://ajpgi.physiology.org/cgi/doi/10.1152/ajpgi.00071.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rogala AR, Schoenborn AA, Fee BE, et al. Environmental factors regulate Paneth cell phenotype and host susceptibility to intestinal inflammation in Irgm1-deficient mice. Dis Model Mech. [Internet] 2018;11:dmm031070. Available from: http://dmm.biologists.org/lookup/doi/10.1242/dmm.031070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tsuboi K, Nishitani M, Takakura A, et al. Autophagy protects against colitis by the maintenance of normal gut microflora and secretion of mucus. J Biol Chem. [Internet] 2015;290:20511–20526. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26149685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yang L, Liu C, Zhao W, et al. Impaired autophagy in intestinal epithelial cells alters gut microbiota and host immune responses. Appl Environ Microbiol. [Internet] 2018;84:AEM.00880–18. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30006408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lavoie S, Conway KL, Lassen KG, et al. The Crohn’s disease polymorphism, ATG16L1 T300A, alters the gut microbiota and enhances the local Th1/Th17 response. Elife. [Internet] 2019;8:1–28. Available from: https://elifesciences.org/articles/39982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bretin A, Carrière J, Dalmasso G, et al. Activation of the EIF2AK4-EIF2A/eIF2α-ATF4 pathway triggers autophagy response to Crohn disease-associated adherent-invasive Escherichia coli infection. Autophagy. [Internet] 2016;12:770–783. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bretin A, Lucas C, Larabi A, et al. AIEC infection triggers modification of gut microbiota composition in genetically predisposed mice, contributing to intestinal inflammation. Sci Rep. [Internet] 2018;8:12301. Available from: http://www.nature.com/articles/s41598-018-30055-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Imhann F, Vich Vila A, Bonder MJ, et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut. [Internet] 2018;67:108–119. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27802154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Palm NW, de Zoete MR, Cullen TW, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell. [Internet] 2014;158:1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang H, Zheng L, McGovern DPB, et al. Myeloid ATG16L1 facilitates host–bacteria interactions in maintaining intestinal homeostasis. J Immunol. [Internet] 2017;198:2133–2146. Available from: http://www.jimmunol.org/lookup/doi/10.4049/jimmunol.1601293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sadaghian Sadabad M, Regeling A, de Goffau MC, et al. The ATG16L1–T300A allele impairs clearance of pathosymbionts in the inflamed ileal mucosa of Crohn’s disease patients. Gut. [Internet] 2015;64:1546–1552. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25253126 [DOI] [PubMed] [Google Scholar]
- [35].Cheng S, Ma X, Geng S, et al. Fecal microbiota transplantation beneficially regulates intestinal mucosal autophagy and alleviates gut barrier injury. mSystems. [Internet] 2018;3:1–19. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30320222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Donohoe DR, Garge N, Zhang X, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. [Internet] 2011;13:517–526. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1550413111001434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nguyen HTT, Dalmasso G, Müller S, et al. Crohn’s disease-associated adherent invasive escherichia coli modulate levels of microRNAs in intestinal epithelial cells to reduce autophagy. Gastroenterology. [Internet] 2014;146:508–519. [DOI] [PubMed] [Google Scholar]
- [38].Lu C, Chen J, Xu HG, et al. MIR106B and MIR93 prevent removal of bacteria from epithelial cells by disrupting ATG16L1-mediated autophagy. Gastroenterology. [Internet] 2014;146:188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. [Internet] 2015;40:141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Høyer-Hansen M, Bastholm L, Szyniarowski P, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase Kinase-β, and Bcl-2. Mol Cell [Internet]. 2007;25:193–205. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1097276506008458 [DOI] [PubMed] [Google Scholar]
- [41].Guo J, Yang Z, Yang X, et al. miR-346 functions as a pro-survival factor under ER stress by activating mitophagy. Cancer Lett. [Internet] 2018;413:69–81. [DOI] [PubMed] [Google Scholar]
- [42].Cao SS. Endoplasmic reticulum stress and unfolded protein response in inflammatory bowel disease. Inflamm Bowel Dis. [Internet] 2015;21:636–644. Available from: https://academic.oup.com/ibdjournal/article/21/3/636-644/4602937 [DOI] [PubMed] [Google Scholar]
- [43].Kökten T, Gibot S, Lepage P, et al. TREM-1 inhibition restores impaired autophagy activity and reduces colitis in mice. J Crohns Colitis. [Internet] 2018;12:230–244. Available from: https://academic.oup.com/ecco-jcc/article/12/2/230/4159885 [DOI] [PubMed] [Google Scholar]
- [44].Tschurtschenthaler M, Adolph TE, Ashcroft JW, et al. Defective ATG16L1-mediated removal of IRE1α drives Crohn’s disease–like ileitis. J Exp Med. [Internet] 2017;214:401–422. Available from: http://www.jem.org/lookup/doi/10.1084/jem.20160791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Adolph TE, Tomczak MF, Niederreiter L, et al. Paneth cells as a site of origin for intestinal inflammation. Nature. [Internet] 2013;503:272–276. Available from: http://www.nature.com/doifinder/10.1038/nature12599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Deuring JJ, Fuhler GM, Konstantinov SR, et al. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn’s disease. Gut. [Internet] 2014;63:1081–1091. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23964099 [DOI] [PubMed] [Google Scholar]
- [47].Diamanti MA, Gupta J, Bennecke M, et al. IKKα controls ATG16L1 degradation to prevent ER stress during inflammation. J Exp Med. [Internet] 2017;214:423–437. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28082356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Vanhove W, Nys K, Arijs I, et al. Biopsy-derived intestinal epithelial cell cultures for pathway-based stratification of patients with inflammatory bowel disease. J Crohn’s Colitis. [Internet] 2018;12:178–187. Available from: http://academic.oup.com/ecco-jcc/article/12/2/178/4210679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tal MC, Sasai M, Lee HK, et al. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci. [Internet] 2009;106:2770–2775. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.0807694106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Billmann-Born S, Lipinski S, Böck J, et al. The complex interplay of NOD-like receptors and the autophagy machinery in the pathophysiology of Crohn disease. Eur J Cell Biol. [Internet] 2011;90:593–602. [DOI] [PubMed] [Google Scholar]
- [51].Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature. [Internet] 2008;456:264–268. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18849965 [DOI] [PubMed] [Google Scholar]
- [52].Zhang H, Zheng L, Chen J, et al. The protection role of Atg16l1 in CD11c + dendritic cells in murine colitis. Immunobiology. [Internet] 2017;222:831–841. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0889159115004717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med. [Internet] 2017;6:24. Available from: http://clintransmed.springeropen.com/articles/10.1186/s40169-017-0154-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Saxena A, Lopes F, McKay DM. Reduced intestinal epithelial mitochondrial function enhances in vitro interleukin-8 production in response to commensal escherichia coli. Inflamm Res. [Internet] 2018. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30030553 [DOI] [PubMed] [Google Scholar]
- [55].Lee JPW, Foote A, Fan H, et al. Loss of autophagy enhances MIF/macrophage migration inhibitory factor release by macrophages. Autophagy. [Internet] 2016;12:907–916. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Nakahira K, Haspel JA, Rathinam VAK, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. [Internet] 2011;12:222–230. Available from: http://www.nature.com/doifinder/10.1038/ni.1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Asano J, Sato T, Ichinose S, et al. Intrinsic autophagy is required for the maintenance of intestinal stem cells and for irradiation-induced intestinal regeneration. Cell Rep. [Internet] 2017;20:1050–1060. [DOI] [PubMed] [Google Scholar]
- [58].Yue C, Yang X, Li J, et al. Trimethylamine N-oxide prime NLRP3 inflammasome via inhibiting ATG16L1-induced autophagy in colonic epithelial cells. Biochem Biophys Res Commun. [Internet] 2017;490:541–551. [DOI] [PubMed] [Google Scholar]
- [59].Takahama M, Akira S, Saitoh T. Autophagy limits activation of the inflammasomes. Immunol Rev. [Internet] 2018;281:62–73. Available from. [DOI] [PubMed] [Google Scholar]
- [60].Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. [Internet] 2002;10:417–426. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12191486 [DOI] [PubMed] [Google Scholar]
- [61].Santeford A, Wiley LA, Park S, et al. Impaired autophagy in macrophages promotes inflammatory eye disease. Autophagy. [Internet] 2016;12:1876–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ip WKE, Hoshi N, Shouval DS, et al. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. [Internet] 2017;356:513–519. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28473584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ravindran R, Loebbermann J, Nakaya HI, et al. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature. [Internet] 2016;531:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Spalinger MR, Lang S, Gottier C, et al. PTPN22 regulates NLRP3-mediated IL1B secretion in an autophagy-dependent manner. Autophagy. [Internet] 2017;13:1590–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Spalinger MR, Manzini R, Hering L, et al. PTPN2 regulates inflammasome activation and controls onset of intestinal inflammation and colon cancer. Cell Rep. [Internet] 2018;22:1835–1848. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2211124718301013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Pike KA, Tremblay ML. Protein tyrosine phosphatases: regulators of CD4 T cells ininflammatory bowel disease. Front Immunol. [Internet] 2018;9:1–14. Available from: https://www.frontiersin.org/article/10.3389/fimmu.2018.02504/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. [Internet] 2016;164:896–910. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0092867415017213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Mehto S, Jena KK, Nath P, et al. The crohn’s disease risk factor IRGM limits NLRP3 inflammasome activation by impeding its assembly and by mediating its selective autophagy. Mol Cell. [Internet] 2019;73:429–445.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Cosin-Roger J, Simmen S, Melhem H, et al. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat Commun. [Internet] 2017;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Byrne BG, Dubuisson J-F, Joshi AD, et al. Inflammasome components coordinate autophagy and pyroptosis as macrophage responses to infection. MBio. [Internet] 2013;4:1–12. Available from: http://mbio.asm.org/cgi/doi/10.1128/mBio.00620-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Krause K, Caution K, Badr A, et al. CASP4/caspase-11 promotes autophagosome formation in response to bacterial infection. Autophagy. [Internet] 2018;1–15. Available from: https://www.tandfonline.com/doi/full/10.1080/15548627.2018.1491494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Guo W, Sun Y, Liu W, et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy. [Internet] 2014;10:972–985. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24879148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Shao B-Z, Wei W, Ke P, et al. Activating cannabinoid receptor 2 alleviates pathogenesis of experimental autoimmune encephalomyelitis via activation of autophagy and inhibiting NLRP3 inflammasome. CNS Neurosci Ther. [Internet] 2014;20:1021–1028. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Liu C, Wang J, Yang Y, et al. Ginsenoside Rd ameliorates colitis by inducing p62-driven mitophagy-mediated NLRP3 inflammasome inactivation in mice. Biochem Pharmacol. [Internet] 2018;155:366–379. [DOI] [PubMed] [Google Scholar]
- [75].Takagawa T, Kitani A, Fuss I, et al. An increase in LRRK2 suppresses autophagy and enhances Dectin-1–induced immunity in a mouse model of colitis. Sci Transl Med. [Internet] 2018;10:eaan8162. Available from: http://stm.sciencemag.org/lookup/doi/10.1126/scitranslmed.aan8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Chauhan S, Mandell MA, Deretic V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol Cell. [Internet] 2015;58:507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Schwerd T, Pandey S, Yang H-T, et al. Impaired antibacterial autophagy links granulomatous intestinal inflammation in Niemann–pick disease type C1 and XIAP deficiency with NOD2 variants in Crohn’s disease. Gut. [Internet] 2017;66:1060–1073. Available from: http://gut.bmj.com/lookup/doi/10.1136/gutjnl-2015-310382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Gradzka S, Thomas OS, Kretz O, et al. Inhibitor of apoptosis proteins are required for effective fusion of autophagosomes with lysosomes. Cell Death Dis. [Internet] 2018;9:529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lassen KG, McKenzie CI, Mari M, et al. Genetic coding variant in GPR65 alters lysosomal pH and links lysosomal dysfunction with colitis risk. Immunity. [Internet] 2016;44:1392–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Marchiando AM, Ramanan D, Ding Y, et al. A deficiency in the autophagy gene Atg16L1 enhances resistance to enteric bacterial infection. Cell Host Microbe. [Internet] 2013;14:216–224. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23954160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Martin PK, Marchiando A, Xu R, et al. Autophagy proteins suppress protective type I interferon signalling in response to the murine gut microbiota. Nat Microbiol. [Internet] 2018;3:1131–1141. Available from: http://www.nature.com/articles/s41564-018-0229-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Aden K, Tran F, Ito G, et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS–STING. J Exp Med. [Internet] 2018;215:2868–2886. Available from: http://www.jem.org/lookup/doi/10.1084/jem.20171029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Samie M, Lim J, Verschueren E, et al. Selective autophagy of the adaptor TRIF regulates innate inflammatory signaling. Nat Immunol. [Internet] 2018;19:246–254. [DOI] [PubMed] [Google Scholar]
- [84].Lahiri A, Hedl M, Abraham C. MTMR3 risk allele enhances innate receptor-induced signaling and cytokines by decreasing autophagy and increasing caspase-1 activation. Proc Natl Acad Sci. [Internet] 2015;112:10461–10466. Available from: http://www.pnas.org/content/112/33/10461.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Chew TS, O’Shea NR, Sewell GW, et al. Optineurin deficiency in mice contributes to impaired cytokine secretion and neutrophil recruitment in bacteria-driven colitis. Dis Model Mech. [Internet] 2015;8:817–829. Available from: http://dmm.biologists.org/cgi/doi/10.1242/dmm.020362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Smith AM, Sewell GW, Levine AP, et al. Disruption of macrophage pro-inflammatory cytokine release in Crohn’s disease is associated with reduced optineurin expression in a subset of patients. Immunology. [Internet] 2015;144:45–55. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Chargui A, Cesaro A, Mimouna S, et al. Subversion of autophagy in adherent invasive Escherichia coli-infected neutrophils induces inflammation and cell death. PLoS One. [Internet] 2012;7:e51727. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23272151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Tan JMJ, Mellouk N, Osborne SE, et al. An ATG16L1-dependent pathway promotes plasma membrane repair and limits Listeria monocytogenes cell-to-cell spread. Nat Microbiol. [Internet] 2018;3:1472–1485. [DOI] [PubMed] [Google Scholar]
- [89].Dengjel J, Schoor O, Fischer R, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci. [Internet] 2005;102:7922–7927. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.0501190102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. [Internet] 2010;16:90–97. [DOI] [PubMed] [Google Scholar]
- [91].Wildenberg ME, Vos ACW, Wolfkamp SCS, et al. Autophagy attenuates the adaptive immune response by destabilizing the immunologic synapse. Gastroenterology. [Internet] 2012;142:1493–503.e6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22370477 [DOI] [PubMed] [Google Scholar]
- [92].Ma Y, Galluzzi L, Zitvogel L, et al. Autophagy and cellular immune responses. Immunity. [Internet] 2013;39:211–227. [DOI] [PubMed] [Google Scholar]
- [93].Jia W, He Y-W. Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J Immunol. [Internet] 2011;186:5313–5322. Available from: http://www.jimmunol.org/cgi/doi/10.4049/jimmunol.1002404 [DOI] [PubMed] [Google Scholar]
- [94].Kovacs JR, Li C, Yang Q, et al. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ. [Internet] 2012;19:144–152. Available from: http://www.nature.com/articles/cdd201178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kabat AM, Harrison OJ, Riffelmacher T, et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. Elife. [Internet] 2016;5:1–27. Available from: https://elifesciences.org/articles/12444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Puleston DJ, Zhang H, Powell TJ, et al. Autophagy is a critical regulator of memory CD8+ T cell formation. Elife. [Internet] 2014;3:1–21. Available from: https://elifesciences.org/articles/03706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Xu X, Araki K, Li S, et al. Autophagy is essential for effector CD8+ T cell survival and memory formation. Nat Immunol. [Internet] 2014;15:1152–1161. Available from: http://www.nature.com/doifinder/10.1038/ni.3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Schlie K, Westerback A, DeVorkin L, et al. Survival of effector CD8 + T cells during influenza infection is dependent on autophagy. J Immunol. [Internet] 2015;194:4277–4286. Available from: http://www.jimmunol.org/lookup/doi/10.4049/jimmunol.1402571 [DOI] [PubMed] [Google Scholar]
- [99].Whang MI, Tavares RM, Benjamin DI, et al. The ubiquitin binding protein TAX1BP1 mediates autophagosome induction and the metabolic transition of activated T cells. Immunity. [Internet] 2017;46:405–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Geremia A, Biancheri P, Allan P, et al. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev. [Internet] 2014;13:3–10. [DOI] [PubMed] [Google Scholar]
- [101].Chu H, Khosravi A, Kusumawardhani IP, et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science. [Internet] 2016;352:1116–1120. Available from: http://www.sciencemag.org/cgi/doi/10.1126/science.aad9948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Shen Y, Torchia MLG, Lawson GW, et al. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe. [Internet] 2012;12:509–520. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1931312812002752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Matsuda C, Ito T, Song J, et al. Therapeutic effect of a new immunosuppressive agent, everolimus, on interleukin-10 gene-deficient mice with colitis. Clin Exp Immunol. [Internet] 2007;148:348–359. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Massey DCO, Bredin F, Parkes M. Use of sirolimus (rapamycin) to treat refractory Crohn’s disease. Gut. [Internet] 2008;57:1294–1296. Available from: http://gut.bmj.com/cgi/doi/10.1136/gut.2008.157297 [DOI] [PubMed] [Google Scholar]
- [105].Dumortier J, Lapalus M-G, Guillaud O, et al. Everolimus for refractory Crohn’s disease: a case report. Inflamm Bowel Dis. [Internet] 2008;14:874–877. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18275074 [DOI] [PubMed] [Google Scholar]
- [106].El-Khider F, McDonald C. Links of autophagy dysfunction to inflammatory bowel disease onset. Dig Dis. [Internet] 2016;34:27–34. Available from: https://www.karger.com/Article/FullText/442921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Shakeri A, Cicero AFG, Panahi Y, et al. Curcumin: A naturally occurring autophagy modulator. J Cell Physiol. [Internet] 2018. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30239005 [DOI] [PubMed] [Google Scholar]
- [108].Wu S, Zhang Y, Lu R, et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut. [Internet] 2015;64:1082–1094. Available from: http://gut.bmj.com/lookup/doi/10.1136/gutjnl-2014-307436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Sakiyama T, Musch MW, Ropeleski MJ, et al. Glutamine increases autophagy under basal and stressed conditions in intestinal epithelial cells. Gastroenterology. [Internet] 2009;136:924–932.e2. Available from: http://linkinghub.elsevier.com/retrieve/pii/S001650850802177X [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Yanai H, Salomon N, Lahat A. Complementary therapies in inflammatory bowel diseases. Curr Gastroenterol Rep. [Internet] 2016;18:62. [DOI] [PubMed] [Google Scholar]
- [111].Li X, Feng K, Li J, et al. Curcumin inhibits apoptosis of chondrocytes through activation ERK1/2 signaling pathways induced autophagy. Nutrients. [Internet] 2017;9:414. Available from: http://www.mdpi.com/2072-6643/9/4/414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Guo S, Long M, Li X, et al. Curcumin activates autophagy and attenuates oxidative damage in EA.hy926 cells via the Akt/mTOR pathway. Mol Med Rep. [Internet] 2016;13:2187–2193. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26781771 [DOI] [PubMed] [Google Scholar]
- [113].Lee YJ, Kim N-Y, Suh Y-A, et al. Involvement of ROS in curcumin-induced autophagic cell death. Korean J Physiol Pharmacol. [Internet] 2011;15:1. Available from: https://synapse.koreamed.org/DOIx.php?id=10.4196/kjpp.2011.15.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Zhao J, Sun Y, Shi P, et al. Celastrol ameliorates experimental colitis in IL-10 deficient mice via the up-regulation of autophagy. Int Immunopharmacol. [Internet] 2015;26:221–228. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1567576915001356 [DOI] [PubMed] [Google Scholar]
- [115].Nys K, Agostinis P, Vermeire S. Autophagy: a new target or an old strategy for the treatment of Crohn’s disease? Nat Rev Gastroenterol Hepatol. [Internet] 2013;10:395–401. [DOI] [PubMed] [Google Scholar]
- [116].Shoji-Kawata S, Sumpter R, Leveno M, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature [Internet]. 2013;494:201–206. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23364696 [DOI] [PMC free article] [PubMed] [Google Scholar]