

ABSTRACT
Like other biological processes, macroautophagy/autophagy must be tightly controlled for maintenance of cellular homeostasis and for proper response to changing cellular conditions. To gain insights into the regulation of autophagy, we recently conducted a genome-wide CRISPR-Cas9 knockout screen using cells expressing endogenous LC3B tagged with GFP-mCherry as a reporter. This approach allowed us to identify the ubiquitin-activating enzyme UBA6 and the hybrid ubiquitin-conjugating enzyme/ubiquitin ligase BIRC6 as novel autophagy regulators. We found that these enzymes cooperate to mediate monoubiquitination and proteasomal degradation of LC3B, thus limiting the pool of LC3B available for autophagy. Depletion of UBA6 or BIRC6 increased the level of cytosolic LC3B, enhancing the degradation of autophagy adaptors and the clearance of intracellular proteins aggregates. This finding could be the basis for the development of pharmacological inhibitors of UBA6 or BIRC6 for the treatment of protein aggregation disorders. Recent work by another group showed that BIRC6 itself is subject to ubiquitination and proteasomal degradation, highlighting the existence of a complex regulatory network for the control of LC3B levels.
KEYWORDS: Apollon, BIRC6, BRUCE, LC3, UBA6, Ubiquitin Proteasome Pathway
By now it is likely that most, if not all, core components of the autophagy machinery have been identified, mainly from genetic screens originally performed in yeast. It is thus pertinent to ask if there is any value in conducting additional genome-wide screens for autophagy in mammalian cells, now that they have become feasible through the use of CRISPR-Cas9 knockout (KO) technology. Judging from several such screens reported over the past year, the answer appears to be a definite “yes”, particularly when it comes to the identification of regulatory, accessory, or mammalian- or cell-type-specific factors. A case in point is the recent discovery by 3 independent groups of the role of the endoplasmic reticulum (ER) transmembrane protein TMEM41B as a novel regulator of autophagosome biogenesis in mammals.
We conducted our own CRISPR-Cas9 KO autophagy screen using the human H4 neuroglioma cell line gene-edited to express endogenous MAP1LC3B (microtubule associated protein 1 light chain 3 beta; herein abbreviated LC3B) tagged with a tandem of the fluorescent proteins GFP and mCherry (H4-tfLC3B cells) [1]. As originally developed in the laboratory of Tamotsu Yoshimori (Osaka University, Japan), the GFP-mCherry-LC3B reporter protein has been widely used as an indicator of autophagy, based on the property of GFP, but not mCherry, fluorescence to be quenched at the acidic pH of autolysosomes. Defects in autophagy are thus manifested by an increased GFP:mCherry ratio. Taking advantage of this property, we mutagenized H4-tfLC3B cells using a lentiviral whole-genome, single-guide RNA (sgRNA) CRISPR-Cas9 KO library, and cells displaying high GFP:mCherry ratios were collected by fluorescence-activated cell sorting (FACS). The sgRNAs responsible for this phenotype were identified by next-generation sequencing and analysis using the MAGeCK algorithm. Among the many candidates found in this screen, we chose to focus on 2 ubiquitination-related proteins, UBA6 (ubiquitin like modifier activating enzyme 6) and BIRC6/BRUCE/Apollon (baculoviral IAP repeat containing 6), because their roles in autophagy had not been previously demonstrated or were unclear. UBA6 is one of 8 ubiquitin-activating (E1) enzymes encoded in the human genome, and it was reported by others to activate both ubiquitin and the ubiquitin-like protein UBD/FAT10 (ubiquitin D). BIRC6 is a hybrid enzyme that combines ubiquitin-conjugating (E2) and ubiquitin-ligase (E3) activities, and in previous studies was shown to inhibit apoptosis by catalyzing the ubiquitination and proteasomal degradation of DIABLO/SMAC (diablo IAP-binding mitochondrial protein) and CASP9 (caspase 9) (Figure 1).
Figure 1.
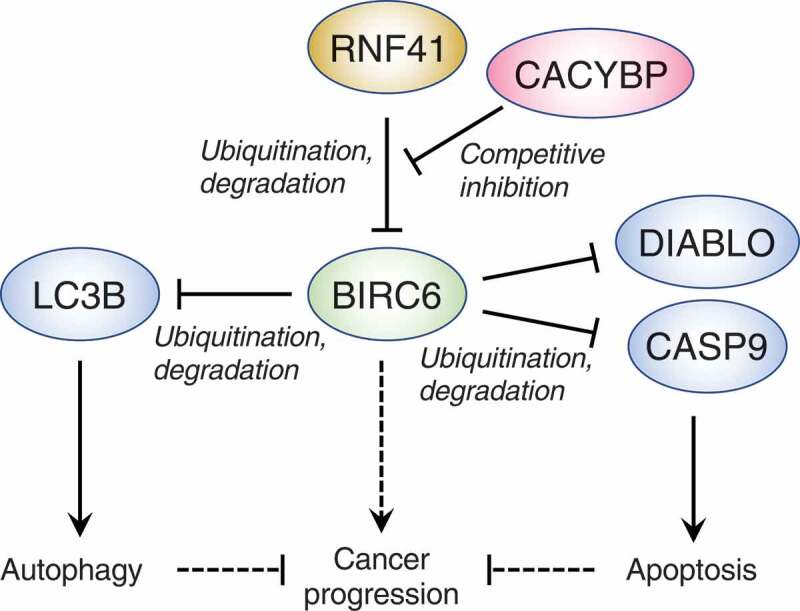
Schematic representation of the roles of BIRC6 in regulating autophagy and apoptosis. BIRC6 is a non-canonical, hybrid ubiquitin-conjugating enzyme (E2)-ubiquitin ligase (E3) that, in conjunction with the ubiquitin-activating enzyme (E1) UBA6, mediates ubiquitination and proteasomal degradation of LC3B, thus reducing LC3B-dependent autophagy. BIRC6 also promotes ubiquitination and proteasomal degradation of DIABLO and CASP9, inhibiting apoptosis. BIRC6 itself is targeted for ubiquitination and proteasomal degradation by another E3, RNF41, a process that is inhibited by competitive binding of CACYBP to RNF41. BIRC6 expression is elevated in some cancers, but it is currently unknown if this elevation promotes cancer progression and, if so, whether the inhibitory effects of BIRC6 on autophagy and apoptosis contribute to progression. Blunt and pointed arrows denote negative and positive effects, respectively. Dashed arrows indicate hypothetical effects.
Based on the characteristics of our screen, we initially expected that KO of UBA6 or BIRC6 would impair some step of the autophagic process, impeding the delivery of GFP-mCherry-LC3B to autolysosomes and thus preventing the quenching of GFP. Paradoxically, we found that KO of these genes does not reduce autophagic flux, but rather enhances it under conditions of nutrient deprivation, protein synthesis inhibition or formation of intracellular protein aggregates. What was the explanation for this apparent paradox? As it turned out, we found that UBA6 and BIRC6 cooperate to mediate monoubiquitination and proteasomal degradation of the cytosolic, non-lipidated form of LC3B (LC3B-I) (Figure 1). UBA6 and BIRC6 play a similar role for the LC3A and LC3C, but not GABARAP, GABARAPL1, and GABARAPL2, members of the Atg8 family of autophagy proteins. Thus, everything we say about LC3B, probably applies to LC3A and LC3C as well. KO of UBA6 or BIRC6 raises the levels of LC3B-I without altering the levels of the membrane-bound, phosphatidylethanolamine-conjugated form of LC3B (LC3B-II) under basal culture conditions. The larger pool of LC3B-I in the UBA6- or BIRC6-KO cells actually enables increased autophagic flux, as manifested by increased production of LC3B-II from LC3B-I when cells are starved of serum and amino acids. In addition, UBA6- or BIRC6-KO cells exhibit faster degradation of the autophagic receptors SQSTM1 and NBR1 upon inhibition of protein synthesis with cycloheximide. Finally, UBA6 or BIRC6 KO delay the accumulation of puromycin-induced aggresome-like induced structures in H4 cells, and BIRC6 knockdown (KD) decreases the accumulation of aggregates of mutant SNCA/α-synuclein in the axon of rat hippocampal neurons. These effects of UBA6 or BIRC6 depletion are suppressed by incubation with the vacuolar-type H+-ATPase inhibitor bafilomycin A1 or KD of the autophagy protein ATG7, which restore aggregate accumulation in both cell types. These results indicated that the levels of LC3B-I are rate-limiting, and that UBA6 and BIRC6 contribute to keeping them in check, most likely to prevent excessive autophagy. The finding that depletion of UBA6 and BIRC6 decreases the accumulation of intracellular protein aggregates could be the basis for the development of pharmacological inhibitors to treat protein aggregation diseases such as various neurodegenerative disorders.
The importance of controlling the levels of LC3B-I for the maintenance of cellular homeostasis is further underscored by the recent discovery by Xiao-Bo Qiu (Beijing Normal University, China) and colleagues that levels of BIRC6 itself are subject to regulation through ubiquitination and proteasomal degradation mediated by the ubiquitin-ligase RNF41 (ring finger protein 41) (Figure 1). The picture gets even more complex, as another protein named CACYBP (calcyclin binding protein), inhibits the ubiquitination of BIRC6 by competitive binding to RNF41 (Figure 1). Accordingly, depletion of CACYBP reduces the levels of LC3B-I, inhibiting the autophagic clearance of cytoplasmic proteins aggregates and damaged mitochondria.
The expression of BIRC6 is elevated in cell lines and tissues from some prostate, liver and colorectal cancers, suggesting that this increase may be beneficial to the cancer cells. This benefit may stem from the role of BIRC6 in targeting the apoptotic factors DIABLO and CASP9 for proteasomal degradation, thus promoting cancer cell survival. The relationship of autophagy to cancer is more complex and variable in different types of tumors. The role of BIRC6 in limiting autophagy could prevent autophagic cell death. However, some cancer cells are dependent on autophagy, so increased levels of BIRC6 would be detrimental to their survival. These uncertainties emphasize the need for further research on the role of BIRC6 as a negative regulator of both apoptosis and autophagy in normal and cancer cells.
Funding Statement
Work in the authors’ laboratory is supported by the Intramural Program of NICHD, NIH [ZIA HD001607].
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Jia R, Bonifacino JS.. Negative regulation of autophagy by UBA6-BIRC6-mediated ubiquitination of LC3. eLife. 2019;8. PMID:31692446. doi: 10.7554/eLife.50034. [DOI] [PMC free article] [PubMed] [Google Scholar]