

ABSTRACT
Chronic mitochondrial stress is associated with major neurodegenerative diseases; and thus, the recovery of those mitochondria constitutes a critical step of energy maintenance in early stages of neurodegeneration. Our recent study provides the first lines of evidence showing that the MUL1-MFN2 pathway acts as an early checkpoint to maintain mitochondrial integrity by regulating mitochondrial morphology and interplay with the endoplasmic reticulum (ER). This mechanism ensures that degradation through mitophagy is restrained in neurons under early stress conditions. MUL1 deficiency increases MFN2 activity, triggering the first phase of mitochondrial hyperfusion and acting as an antagonist of ER-mitochondria (ER-Mito) tethering. Reduced ER-Mito interplay enhances the cytoplasmic Ca2+ load that induces the DNM1L/Drp1-dependent second phase of mitochondrial fragmentation and mitophagy. Our study provides new mechanistic insights into neuronal mitochondrial maintenance under stress conditions. Identifying this pathway is particularly relevant because chronic mitochondrial dysfunction and altered ER-Mito contacts have been reported in major neurodegenerative diseases.
KEYWORDS: DNM1L, endoplasmic reticulum, ER-Mitochondrial contact, Mfn2, mitochondrial ubiquitin ligase, mitophagy, MUL1, neuronal mitochondria, PRKN
PRKN/parkin-mediated mitophagy is a key pathway for globally eliminating damaged mitochondria in many non-neuronal cell types. Neurons are post-mitotic cells that survive for the lifetime of the organism. Under chronic stress and pathological conditions, it is more advantageous to simply allow the recovery of stressed mitochondria rather than activate mitophagy to eliminate them, which can be fatal to neurons. Our previous studies revealed that PRKN-mediated mitophagy is observed only in a small portion of mature neurons and occurs much more slowly than in non-neuronal cells, arguing for unique mechanisms that maintain and/or recover neuronal mitochondrial integrity and thus energy homeostasis. In this study, we aimed to support this assumption by addressing 3 fundamental questions: Can neurons drive the recovery of chronically stressed mitochondria before PRKN-mediated mitophagy is activated? Is mitophagy the last resort for mitochondrial quality control after recovery mechanisms have failed? If this is the case, does MUL1 (mitochondrial ubiquitin ligase activator of NFKB 1) play an early role in maintaining mitochondrial integrity before the cytosolic ubiquitin ligase PRKN is recruited to stressed mitochondria for mitophagy in neurons [1]? Addressing these questions is highly relevant to major neurodegenerative diseases that are associated with chronic mitochondrial stress.
MUL1 is a multifunctional mitochondrial membrane protein; one of its roles depends upon its E3 ubiquitin ligase activity that can ubiquitinate and degrade MFN2. We hypothesize that MUL1 acts as the first-line of surveillance to maintain neuronal mitochondrial integrity; MUL1 deficiency facilitates recruitment of PRKN to stressed mitochondria. To study the mitochondrial response to mild stress, we used a 100-nM dose of the respiratory complex III inhibitor antimycin A (AA), a 400-times lower dose than widely used in the literature. We reasoned that this mild stress is more relevant to chronic mitochondrial dysfunction in aging-associated neurodegenerative diseases. We established a MUL1-deficient neuronal system by knocking down MUL1 or expressing its loss-of-function mutants. Neurons with MUL1 loss of function display enhanced PRKN/parkin recruitment to fragmented mitochondria and increased mitophagy in response to mild mitochondrial stress. In contrast, overexpressing MUL1 suppresses PRKN translocation and mitophagy. More intriguingly, MUL1-deficient neurons trigger a mitochondrial biphasic transition from hyperfusion at 10–11 day in vitro (DIV10-11) to fragmentation at DIV14-15. This transient hyperfusion phase is an early stress response to maintain bioenergetic capacity. Only those fragmented mitochondria that are associated with loss of membrane potential and ATP production capacity are subjected to mitophagic clearance. In contrast to HeLa cells where PRKN-mediated mitophagy is rapidly activated upon mitochondrial depolarization, our study highlights a unique checkpoint for neuronal mitochondrial quality control. After this pathway has failed, PRKN is then recruited to fragmentated mitochondria to initiate mitophagic clearance. This is consistent with the previous study showing that the loss of both Mul1 and Prkn aggravates mitochondrial damage and induces neuronal degeneration.
Next, we examined the mechanism(s) behind the biphasic change in mitochondrial morphology in MUL1-deficient neurons. As a mitochondrial E3 ligase, MUL1 regulates MFN levels by ubiquitinating and degrading Marf/Mfn in Drosophila. We demonstrate that of the 2 mammalian MFN isoforms, MFN2, but not MFN1, mediates MUL1-deficient phenotypes; elevated MFN2 activity alone is sufficient to induce MUL1-deficient phenotypes. MFN2 is involved in multiple pathways in addition to mitochondrial fusion. Overexpressing MFN2 induces mitochondrial dysfunction and cell death. MFN2 is also involved in the regulation of ER-Mito contacts in non-neuronal cells. Our study in mature cortical neurons further reveals an antagonistic role of MFN2 in maintaining ER-Mito contacts. Using 3D stimulated emission depletion nanoscopy (3D-STED) and immuno-TEM ultrastructural analysis combined with imaging of mitochondria Ca2+ uptake, our study consistently supports a role for MUL1 in maintaining ER-Mito contacts by reducing MFN2 levels. Enhancing ER-Mito contacts by expressing an ER-Mito anchoring protein, RMDN3/PTPIP51, in MUL1-deficient neurons rescues mitochondrial fragmentation and reduces PRKN-mediated mitophagy. Thus, maintaining ER-Mito contacts in mature neurons is critical to suppress mitochondrial fragmentation and mitophagy. This notion is consistent with a recent report showing that impaired ER-Mito contact increases PRKN-mediated mitophagy upon acute mitochondrial depolarization.
Our study further shows that prolonged stress impairs mitochondrial functions and Ca2+ homeostasis. Elevated cytoplasmic Ca2+ load due to reduced ER-Mito coupling activates DNM1L/Drp1 through PPP3/calcineurin, thus triggering mitochondrial fragmentation. We provided 6 lines of evidence to support this mechanistic model: (1) mitochondrial association of DNM1L is increased at DIV13, a time point just before fragmentation occurs; (2) mitochondria are locked in the hyperfusion state when expressing catalytically inactive mutant DNM1LK38A; (3) cytoplasmic Ca2+ load is elevated in MUL1-deficient neurons; (4) DNM1L is activated through Ca2+-PPP3 when ER-Mito contacts are impaired; (5) expressing an ER-Mito anchoring protein rescues mitochondrial fragmentation; and (6) we further confirmed this mechanistic model by applying 2 blockers of PPP3 activity, which arrest stressed mitochondria in the hyperfusion state in MUL1-depleted neurons. Thus, our study reveals a new mechanistic pathway that links impaired ER-Mito contacts to DNM1L-dependent mitochondrial fragmentation, a process necessary for PRKN-mediated mitophagy.
In summary, our study reveals MUL1 acts at an early checkpoint to protect neuronal mitochondria from rapid mitophagic clearance under mild stress conditions, thus maintaining energy supply and ensuring neuron survival. MUL1 deficiency leads to increased MFN2 activity that plays 2 parallel roles: inducing mitochondrial hyperfusion and impairing ER-Mito interplay, the latter of which disturbs mitochondrial bioenergetics and Ca2+ homeostasis. Elevated cytoplasmic Ca2+ activates DNM1L through PPP3, triggering mitochondrial fragmentation and PRKN-mediated mitophagy. Consistently, stabilizing ER-Mito contacts by overexpressing an anchoring protein partially rescues MUL1-deficient phenotypes, whereas overexpressing MFN2 mimics MUL1-deficient phenotypes. By regulating mitochondrial morphology and ER-Mito contacts, the MUL1-MFN2 pathway plays a critical role in maintaining neuronal mitochondrial integrity and restraining PRKN-mediated mitophagy (Figure 1). Thus, our study reveals new mechanistic insights into neuronal mitochondrial maintenance under stress conditions; our findings add to the emerging evidence demonstrating important differences in how neurons respond to chronic and mild mitochondrial damage. In addition, our study provides an explanation of (1) why mitophagy in mature neurons is observed only in a small proportion of mitochondria under chronic stress, and (2) why prkn or pink1 knockout mice show only subtle changes in mitochondrial integrity. This MUL1-MFN2 pathway is particularly relevant to chronic mitochondrial dysfunction and altered ER-Mito interplay reported in major neurodegenerative diseases including Alzheimer and Parkinson diseases, amyotrophic lateral sclerosis, and hereditary spastic paraplegia.
Figure 1.
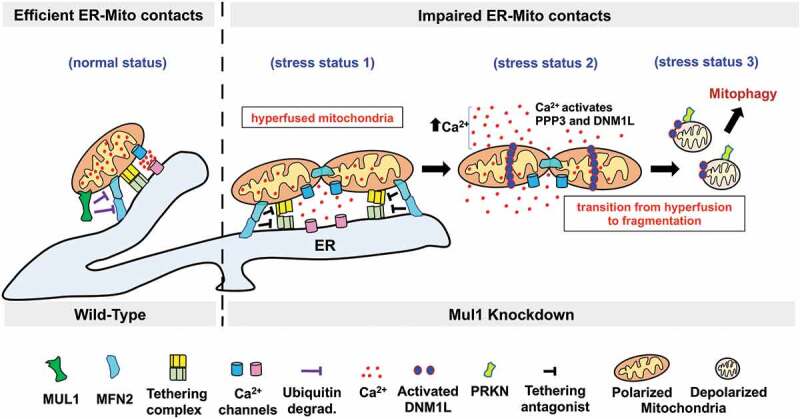
Schematic illustration of the MUL1-MFN2 pathway in restraining mitophagy in mature neurons. MUL1 is a mitochondrial E3 ubiquitin ligase that binds and ubiquitinates MFN2, leading to MFN2 degradation, thus maintaining mitochondrial integrity (normal status). MUL1 deficiency results in the first phase of mitochondrial hyperfusion due to an increase in MFN2 activity (stress status 1). Dysregulation of MFN2 activity acts in parallel as an antagonist of ER-Mito tethering, thus impairing ER-Mito coupling, disturbing mitochondrial bioenergetics, and reducing mitochondrial Ca2+ uptake from the ER. An increase in cytoplasmic Ca2+ load upregulates PPP3 phosphatase activity, which activates and recruits DNM1L onto stressed mitochondria (stress status 2), triggering a mitochondrial morphology transition from hyperfusion to fragmentation. Those fragmented mitochondria lose their membrane potential and ATP production capacity and recruit PRKN for mitophagic clearance (stress status 3). Thus, the MUL1-MFN2 pathway serves a dual role: maintain mitochondrial morphology and ER-Mito contacts, thereby contributing to allow the recovery of stressed mitochondria at early stages of chronic pathological conditions. Failure of the MUL1-MFN2 pathway activates mitophagy to eliminate damaged mitochondria.
Funding Statement
The work was supported by the Intramural Research Program of NINDS, NIH ZIA NS003029 and ZIA NS002946 (Z-H. Sheng) and NINDS Competitive Fellowship (R. Puri).
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Puri R, Cheng XT, Lin MY, et al. Mul1 restrains Parkin-mediated mitophagy in mature neurons by maintaining ER-mitochondrial contacts. Nat Commun. 2019;10(1):3645. [DOI] [PMC free article] [PubMed] [Google Scholar]