

Abstract
Background
Cerivastatin was the most potent statin until it was withdrawn from the market due to a number of fatalities due to rhabdomyolysis, however, the dose‐related magnitude of effect of cerivastatin on blood lipids is not known.
Objectives
Primary objective
To quantify the effects of various doses of cerivastatin on the surrogate markers: LDL cholesterol, total cholesterol, HDL cholesterol and triglycerides in children and adults with and without cardiovascular disease.
The aim of this review is to examine the pharmacology of cerivastatin by characterizing the dose‐related effect and variability of the effect of cerivastatin on surrogate markers.
Secondary objectives To quantify the effect of various doses of cerivastatin compared to placebo on withdrawals due to adverse effects. To compare the relative potency of cerivastatin with respect to fluvastatin, atorvastatin and rosuvastatin for LDL cholesterol, total cholesterol, HDL cholesterol and triglycerides.
Search methods
The Cochrane Hypertension Information Specialist searched the following databases for RCTs up to March 2019: CENTRAL (2019, Issue 3), Ovid MEDLINE, Ovid Embase, the WHO International Clinical Trials Registry Platform, and ClinicalTrials.gov.We also searched the European Patent Office, FDA.gov, and ProQuest Dissertations & Theses, and contacted authors of relevant papers regarding further published and unpublished work. The searches had no language restrictions.
Selection criteria
RCTs and controlled before‐and‐after studies evaluating the dose response of different fixed doses of cerivastatin on blood lipids over a duration of three to 12 weeks in participants of any age with and without cardiovascular disease.
Data collection and analysis
Two review authors independently assessed eligibility criteria for trials to be included and extracted data. We entered data from RCTs and controlled before‐and‐after studies into Review Manager 5 as continuous and generic inverse variance data respectively. We collected information on withdrawals due to adverse effects from the RCTs. We assessed all trials using the 'Risk of bias' tool under the categories of sequence generation, allocation concealment, blinding, incomplete outcome data, selective reporting, and other potential biases.
Main results
Fifty trials (19 RCTs and 31 before‐and‐after studies) evaluated the dose‐related efficacy of cerivastatin in 12,877 participants who had their LDL cholesterol measured. The participants were of any age with and without cardiovascular disease and the trials studied cerivastatin effects within a treatment period of three to 12 weeks. Cerivastatin 0.025 mg/day to 0.8 mg/day caused LDL cholesterol decreases of 11.0% to 40.8%, total cholesterol decreases of 8.0% to 28.8% and triglyceride decreases of 9.0% to 21.4%. We judged the certainty of evidence for these effects to be high. Log dose‐response data over doses of 2.5 mg to 80 mg revealed strong linear dose‐related effects on LDL cholesterol, total cholesterol and triglycerides.
When compared to fluvastatin, atorvastatin and rosuvastatin, cerivastatin was about 250‐fold more potent than fluvastatin, 20‐fold more potent than atorvastatin and 5.5‐fold more potent than rosuvastatin at reducing LDL cholesterol; 233‐fold more potent than fluvastatin, 18‐fold more potent than atorvastatin and six‐fold more potent than rosuvastatin at reducing total cholesterol; and 125‐fold more potent than fluvastatin, 11‐fold more potent than atorvastatin and 13‐fold more potent than rosuvastatin at reducing triglycerides. There was no dose‐related effect of cerivastatin on HDL cholesterol, but overall cerivastatin increased HDL cholesterol by 5%. There was a high risk of bias for the outcome withdrawals due to adverse effects, but a low risk of bias for the lipid measurements. Withdrawals due to adverse effects were not different between cerivastatin and placebo in 11 of 19 of these short‐term trials (risk ratio 1.09, 95% confidence interval 0.68 to 1.74).
Authors' conclusions
The LDL cholesterol, total cholesterol, and triglyceride lowering effect of cerivastatin was linearly dependent on dose. Cerivastatin log dose‐response data were linear over the commonly prescribed dose range. Based on an informal comparison with fluvastatin, atorvastatin and rosuvastatin, cerivastatin was about 250‐fold more potent than fluvastatin, 20‐fold more potent than atorvastatin and 5.5‐fold more potent than rosuvastatin in reducing LDL cholesterol, and 233‐fold greater potency than fluvastatin, 18‐fold greater potency than atorvastatin and six‐fold greater potency than rosuvastatin at reducing total cholesterol. This review did not provide a good estimate of the incidence of harms associated with cerivastatin because of the short duration of the trials and the lack of reporting of adverse effects in 42% of the RCTs.
Plain language summary
Cerivastatin for lowering lipids
Review question
How different doses of cerivastatin affect fats in our blood.
Background
Cerivastatin is a very strong cholesterol‐lowering drug. We don't know how its dose size affects the amount of fats in our blood.
Search date
We looked at research up to March 2019.
Study characteristics
We looked for high quality randomised trials (RCTs) and before‐and‐after studies with cerivastatin in different dose sizes . The trials were between three and twelve weeks long. People of any age and gender, either with or without heart disease were in these trials.
Participants could be of any age and gender, with or without cardiovascular disease.
Key results
We found fifty trials with 13,018 participants who had their lipid levels measured. 12,877 participants had their LDL cholesterol measured.
People taking 0.025 to 0.8 mg of cerivastatin per day lowered their LDL cholesterol by 12% to 42%. The higher the dose, the lower the levels of three measures of cholesterol. HDL cholesterol increased by 5%.
For lowering LDL cholesterol, cerivastatin is 250‐times stronger than fluvastatin, 20‐times stronger than atorvastatin and 5.5 times stronger than rosuvastatin.
Only 11 of the 19 RCTs reported the number of people who dropped out of the studies because of adverse effects. The level of drop outs due to adverse effects were similar in the people who took cerivastatin and placebo.
Certainty of the evidence
There is a high level of trust around the results for total cholesterol and LDL cholesterol and very low to moderate for triglycerides. We have a low level of trust in the evidence around drop outs because many (8 out of 19 trials) did not report drop outs due to adverse effects.
Summary of findings
Background
Description of the condition
Cardiovascular disease is a major cause of death and disability in the developed world, accounting for more than one‐third of total deaths (Kreatsoulas 2010). In the USA, cardiovascular disease causes one in three reported deaths each year (CDC 2011; Roger 2011). Existing evidence shows a weak association between adverse cardiovascular events and blood concentrations of low‐density lipoprotein (LDL) cholesterol in adults (Grundy 2004). The current recommended treatment for secondary prevention of adverse cardiovascular events consists of diet and lifestyle changes plus drug therapy with the drug class widely known as ’statins’. In addition statins are commonly prescribed to lower cholesterol with the intent of reducing adverse cardiovascular events in primary prevention patients.
Description of the intervention
Cerivastatin is a synthetic statin and the most potent statin that has been marketed. However, it was withdrawn from the market in 2001, four years after its launch, due to a higher occurrence of rhabdomyolysis (breakdown of muscle fibres), including fatal cases (Furberg 2001), than other available statins. Before it was withdrawn, cerivastatin was prescribed to prevent adverse cardiovascular events and to lower total cholesterol and LDL cholesterol. Cerivastatin is rapidly absorbed, reaching peak plasma concentration within two to three hours and has a short half‐life, two to three hours. Cerivastatin is metabolised by cytochromes P‐450 2C8 and P‐450 3A4 to desmethylcerivastatin (M‐1) and its hydroxy metabolite (M‐23), which are also active (Muck 2000; Plosker 2000). Statins as a class have been shown in individual randomised controlled trials (RCTs) and systematic reviews of RCTs to reduce mortality and major vascular events in people with occlusive vascular disease (CTT 2005).
How the intervention might work
Cerivastatin acts in the liver by inhibiting an enzyme early in the pathway for cholesterol synthesis, 3‐hydroxy‐3‐methyl‐glutarylcoenzyme A reductase (HMG CoA reductase). This enzyme irreversibly converts HMG CoA to mevalonate (Moghadasian 1999). This reaction is the third step in a sequence of reactions resulting in the production of many compounds including cholesterol and its circulating blood derivatives, LDL cholesterol and very low‐density lipoprotein (VLDL) cholesterol (Gaw 2000). The prevailing hypothesis is that statins reduce mortality and morbidity in people with occlusive vascular disease by reducing liver production of cholesterol and thus causing a reduction in LDL cholesterol that increases the risk of atherogenesis. However, the HMG CoA reductase enzyme is also responsible for the production of ubiquinone (co‐enzyme Q10), heme A, vitamin D, steroid hormones and many other compounds. It remains possible that the beneficial effects of statins are due to actions other than the reduction of circulating cholesterol. These other actions have been referred to as the pleiotropic effects of statins (Liao 2005).
Why it is important to do this review
Statins are the most widely prescribed class of drugs in the world. Prescribing of statins is increasing, as are average prescribed doses. At the present time, clinicians have only an approximate sense of the different potency of the different statins. Previous systematic reviews have assessed the effect of statins on serum lipids (Bandolier 2004; Edwards 2003; Law 2003; Ward 2007). They have demonstrated that different statins have different potencies in terms of lipid lowering and that higher doses of statins cause greater lowering of serum lipids than lower doses (Kellick 1997; Schaefer 2004; Schectman 1996). However, a systematic assessment of the potency, dose‐response relationship, and variability of effect has only been published for atorvastatin (Adams 2015), rosuvastatin (Adams 2014) and fluvastatin (Adams 2018). These reviews showed that rosuvastatin is about three times more potent than atorvastatin and about 46‐fold more potent than fluvastatin in lowering LDL cholesterol. In addition, the slope of the dose‐response relationship was similar for those three statins. It is possible that, in addition to a difference in potency, the slope of the dose‐response or the variability of response is different for cerivastatin. Cerivastatin is not currently available as it was withdrawn from the market in 2001 but it is essential to determine the dose‐response relationship of cerivastatin as it may provide a clue as to why it was more toxic to muscle than the other statins (Psaty 2004). At the present time, the reason for cerivastatin’s increased toxicity is unknown. Statin‐induced myopathy, is common to all statins, and limits the use of statins in many people. Knowledge of the effects of statins on blood lipids can help us to use them more effectively. We will use the percentage reduction from baseline of the following surrogate markers to describe the dose‐response relationship of the effect of cerivastatin: total cholesterol, LDL cholesterol, high‐density lipoprotein (HDL) cholesterol, and triglycerides (Boekholdt 2012). We will use the results of this review to compare cerivastatin with rosuvastatin, atorvastatin and fluvastatin. Subsequent reviews of other drugs in the class (i.e. lovastatin, pravastatin, simvastatin, and pitavastatin) will also be done, in order to compare the results of all the statins.
Objectives
Primary objective
To quantify the effects of various doses of cerivastatin on the surrogate markers: LDL cholesterol, total cholesterol, HDL cholesterol and triglycerides in children and adults with and without cardiovascular disease.
The aim of this review is to examine the pharmacology of cerivastatin by characterizing the dose‐related effect and variability of the effect of cerivastatin on surrogate markers.
Secondary objectives
To quantify the variability of the effect of various doses of cerivastatin on withdrawals due to adverse effects. To quantify the relative potency of cerivastatin with respect to fluvastatin, atorvastatin and rosuvastatin for LDL cholesterol, total cholesterol, HDL cholesterol and triglycerides.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised, placebo‐controlled trials (RCTs) and controlled before‐and‐after studies. We included before‐and‐after studies because it has been shown that there is no placebo effect of statins on lipid parameters. Therefore a placebo control is not essential (Tsang 2002). We included data from cross‐over trials if the trial authors reported data for the initial treatment period versus parallel treatment groups, followed by an adequate washout period before crossing over to the other active treatments, and if they reported data in a similar manner during all treatment periods.
Types of participants
Participants could be of any age, with and without cardiovascular disease. They could have normal lipid parameters or any type of hyperlipidaemia or dyslipidaemia. We included participants with various comorbid conditions, including type 2 diabetes mellitus, hypertension, metabolic syndrome, chronic renal failure or cardiovascular disease.
Types of interventions
Cerivastatin had to be administered at a constant daily dose defined as a single dose per day compared to placebo or alone defined as a single cerivastatin dose per day for a period of three to 12 weeks. We have chosen this administration time window to allow at least three weeks for a steady‐state effect of cerivastatin to occur and to keep it short enough to minimise participants dropping out.
We included trials where cerivastatin was administered at any time during the day. Trials required a washout baseline dietary stabilisation period of at least three weeks, where all previous lipid‐altering medication was withdrawn. This baseline phase ensured that participants followed a standard lipid‐regulating diet and helped to stabilise baseline lipid values prior to treatment. In trials where participants were not receiving lipid‐altering medications or dietary supplements before receiving the test drug, we did not require washout baseline dietary stabilisation periods.
Types of outcome measures
The doses of cerivastatin that we studied were 0.05 mg/day, 0.1 mg/day, 0.2 mg/day, 0.3 mg/day, 0.4 mg/day and 0.8 mg/day.
Lipid parameters: for the RCTs, we present the mean percentage change from baseline for different doses of cerivastatin minus the mean percentage change from baseline with placebo for LDL cholesterol, total cholesterol, HDL cholesterol and triglycerides. For the before‐and‐after studies we present the mean percentage change from baseline of different doses of cerivastatin. We combined data from RCTs and before‐and‐after studies because it was shown that there was a lack of difference in the mean differences between the two types of trials (Tsang 2002). We used only data from RCTs for triglycerides because there was a difference in the mean differences between the two types of trials (Tsang 2002) and it was judged that RCT data provided a better efficacy estimate.
Primary outcomes
LDL cholesterol
Secondary outcomes
Total cholesterol
HDL cholesterol
Triglycerides
End‐of‐treatment variability (standard deviation (SD)) and coefficient of variation of LDL cholesterol measurements for each dose of cerivastatin. It is important to know whether cerivastatin has an effect on the variability of lipid measures and ultimately to compare this with the effect of other statins.
Withdrawals due to adverse effects limited to RCTs.
Search methods for identification of studies
Electronic searches
The Cochrane Hypertension Information Specialist searched the following databases without language, publication year or publication status restrictions:
the Cochrane Central Register of Controlled Trials (CENTRAL, 2019, Issue 2) via the Cochrane Register of Studies (CRS‐Web; searched 17 March 2019);
MEDLINE Ovid (from 1946 onwards), MEDLINE Ovid Epub Ahead of Print, and MEDLINE Ovid In‐Process & Other Non‐Indexed Citations (searched 17 March 2019);
Embase Ovid (from 1974 onwards; searched 17 March 2019);
ClinicalTrials.gov (www.clinicaltrials.gov; searched 17 March 2019);
World Health Organization International Clinical Trials Registry Platform (apps.who.int/trialsearch; searched 17 March 2019);
Epistemonikos (www.epistemonikos.org; searched 17 March 2019).
The Information Specialist modelled subject strategies for databases on the search strategy designed for MEDLINE. We present search strategies for major databases in Appendix 1.
Searching other resources
The Cochrane Hypertension Information Specialist searched the Cochrane Database of Systematic Reviews (CDSR) via Wiley and the Database of Abstracts of Reviews of Effects (DARE) via Wiley for related reviews so that we could scan their reference lists for additional trials.
We checked the bibliographies of included trials and any relevant systematic reviews identified for further references to relevant trials.
We contacted experts/organisations in the field to obtain additional information on relevant trials.
We contacted original authors for clarification and further data if trial reports were unclear.
We included grey literature by searching these other resources:
ProQuest Dissertations and Theses (search.proquest.com/pqdtft/)
US Food and Drug Administration (www.fda.gov/)
European Patent Office (worldwide.espacenet.com
We used the following keywords to search the grey literature resources: cerivastatin, baycol, lipobay, BAY w 6228, "BAY w 6228".
Data collection and analysis
Selection of studies
Initial selection of RCTs and before‐and‐after studies involved retrieving and reading the titles and abstracts of each paper found from the electronic search databases or bibliographic citations (see Figure 1 for PRISMA flow diagram (Moher 2009)). Two review authors (SA and NT) analysed the full‐text papers independently, to decide on the trials to be included. We resolved disagreements by recourse to a third review author (JMW). Two review authors (SA and NT) independently extracted the appropriate data from each of the included trials.
1.

0Cerivastatin review flow diagram
Data extraction and management
We extracted the mean percentage change directly from the data, or we calculated it from the baseline and endpoint values using the calculation found in Appendix 2. We added the calculated data to the 'Data and analyses' section of the review. If the calculated data differed from the given data by more than 10%, we did not include the data in the review. We extracted standard deviations (SDs) and standard errors (SEs) from the report or calculated them when possible using the calculation in Appendix 3. We entered data from RCTs and controlled before‐and‐after studies into Review Manager 5 (Review Manager 2014) as continuous and generic inverse variance data, respectively.
Assessment of risk of bias in included studies
We assessed all RCTs and before‐and‐after studies using the Cochrane 'Risk of bias' tool under the categories of random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data, selective reporting, and other potential biases. We produced 'Risk of bias' tables' as outlined in the Cochrane Handbook for Systematic Reviews of Interventions, Chapter 8 (Higgins 2011). Controlled before‐and‐after studies should be scored “High risk” and “Low grading” compared to RCTs. Having only one trial group makes this feature at high risk of bias, as the risk of bias for random sequence generation and allocation concealment were high for the before‐and‐after studies but other features in this trial design might be at lower risk, there may be unidentified differences between the intervention and control groups that may affect changes in the outcome measure. We appreciate that blinding of participants and personnel and blinding of outcome assessment are inappropriate for before‐and‐after studies and that this is a limitation. However, because the lipid parameter measurements are unlikely to be influenced by lack of blinding and were measured in a remote laboratory, we considered them unlikely to be affected by the trial design. We were able to use the Cochrane 'Risk of bias' tool for the controlled before‐and‐after studies because there was a lack of difference in the mean differences between the two type of trials (Tsang 2002).
Measures of treatment effect
We analysed the treatment effects as mean difference for each dose in the RCTs and generic inverse variance for each dose in the before‐and‐after controlled studies separately. In the event that the mean effects from the two trial designs were not statistically different, we re‐analysed all efficacy trial data using the generic inverse variance to determine the overall weighted treatment effects and their 95% confidence intervals (CIs) for LDL cholesterol, total cholesterol, HDL cholesterol, and triglycerides.
Unit of analysis issues
The unit of analysis is the mean values for the people completing the trial. We expect follow‐up to be reasonably high for these short‐term trials. The data however represent treatment efficacy and not real world effectiveness of cerivastatin on these lipid parameters.
Dealing with missing data
When data were missing, we requested them from the trial authors. The most common type of value that was not reported was the SD of the change.
In the case of a missing SD for the change in lipid parameters, we imputed the SD using the following hierarchy (listed from high to low preference).
SD calculated either from the T statistics corresponding to the exact P value reported or from the 95% CI of the mean difference between treatment groups.
Average weighted standard deviation of the change from other trials in the review (Furukawa 2006).
Because it is common for the SD to be miscalculated and in order not to overweight trials where it is inaccurately calculated and lower than expected, when SD values were less than 40% of the average weighted SDs, we used the imputed value by the method of Furukawa (Furukawa 2006).
Assessment of heterogeneity
The Chi2 test to identify heterogeneity was not appropriate because it has low power when there are few trials but has excessive power to detect clinically unimportant heterogeneity when there are many trials (Higgins 2002). The I2 is a better statistic. I2 calculates between‐trial variance/(between trial variance + within trial variance). This measures the proportion of total variation in the estimate of the treatment effect that is due to heterogeneity between trials. This statistic is also independent of the number of trials in the analysis (Higgins 2002).
We assessed the I2 statistic as moderate heterogeneity when it was 30% to 50% and high heterogeneity when greater than 50%.
Assessment of reporting biases
We assessed for publication bias using funnel plots, as outlined in chapter 13 of the Cochrane Handbook for Systematic Reviews of Interventions (Page 2019), when there were ten trials or more examining the same outcome (dose).
Data synthesis
We entered all RCTs into Review Manager 5 (Review Manager 2014), as mean difference using a fixed‐effect model to determine the weighted treatment effect and 95% CIs for LDL cholesterol, total cholesterol, HDL cholesterol, and triglycerides. We entered all controlled before‐and‐after studies as generic inverse variance fixed‐effect model data to determine the weighted treatment effect. If the effect in the RCTs was not statistically significantly different from the before‐and‐after studies, we entered all trials for each dose as fixed‐effect model generic inverse variance to determine the best overall weighted treatment effect for each dose.
We recorded data of each trial and dose in GraphPad Prism 4, to yield a weighted, least squares analysis, based on the inverse of the square of the SE for each lipid parameter, to generate weighted log dose‐response curves. We entered the number of participants in RCTs, who prematurely withdrew due to at least one adverse effect in Review Manager 5 (Review Manager 2014), as dichotomous data for each dose and all combined doses of cerivastatin.
We determined the relative potency of cerivastatin with respect to fluvastatin, atorvastatin and rosuvastatin as the ratio of the milligram (mg) amount of cerivastatin to the mg amount of fluvastatin or atorvastatin or rosuvastatin needed to produce the same specified effect. We calculated these values from the log dose‐response curves of cerivastatin, fluvastatin, atorvastatin and rosuvastatin for LDL cholesterol, total cholesterol, and triglycerides. We estimated the relative potencies from these dose ratios.
Subgroup analysis and investigation of heterogeneity
The main subgroup analyses were the different doses of cerivastatin. We assessed heterogeneity using I² statistic (Higgins 2002). If the I² statistic value was 50% or higher, we attempted to identify possible causes for this by carrying out a number of planned sensitivity analyses, provided there were sufficient numbers of trials (see below).
We analysed subgroups based on the following factors.
RCTs versus before‐and‐after studies (described above)
Men versus women
Morning administration time versus evening administration time as defined by the cut offs 6:00 am to noon and 6:00 pm to midnight
Bayer‐funded versus non‐Bayer‐funded trials
Sensitivity analysis
We conducted sensitivity analyses to assess the effect of different comorbidities, such as familial hyperlipidaemia, on the treatment effect. We compared the treatment effects as generic inverse variance between trials whose participants were reported to have type IIa or familial hypercholesterolaemia versus trials whose participants were not reported to have genetic hypercholesterolaemia. We excluded trials from this analysis if the participants in a trial included both familial and non‐familial hypercholesterolaemia. We conducted sensitivity analyses to assess the effect of different methods of dosing, such as twice daily versus single dose, on the treatment effect.
We analysed RCTs and before‐and‐after studies separately in the data and analysis section.
Summary of findings and assessment of the certainty of the evidence
We used the GRADE approach to assess the certainty of the supporting evidence behind each estimate of treatment effect (Schünemann 2019a; Schünemann 2019b). We presented key findings of the review, including a summary of the amount of data, the magnitude of the effect size and the overall certainty of the evidence, in Table 1, Table 2, Table 3 and Table 4. We did not summarize the findings on HDL cholesterol in a summary of findings table because cerivastatin doses ranging from 0.025 mg/day to 0.8 mg/day had no dose‐related effect on HDL cholesterol.
Summary of findings for the main comparison. Low‐density lipoprotein (LDL) cholesterol‐lowering efficacy of cerivastatin.
| Low‐density lipoprotein (LDL) cholesterol lowering efficacy of cerivastatin | ||||||
|
Patient or population: participants with normal or abnormal lipid profiles Settings: clinics or hospitals Intervention: different fixed doses of cerivastatin Comparison: placebo or baseline | ||||||
| Cerivastatin dose |
Anticipated absolute effects mmol/L (95%CI) |
Percentage change from baseline (95% CI) | № of participants (trials) | Certainty of the evidence (GRADE) | Comments | |
| LDL‐cholesterol before exposure to cerivastatina | LDL‐cholesterol after exposure to cerivastatin | |||||
| 0.05 mg/d | 5.08 (4.75 to 5.41) |
4.27 (4.21 to 4.33) |
‐16.0 (‐17.2 to ‐14.7) |
1811 (5) | ⊕⊕⊕⊕ High | Effect predicted from log dose‐response equation is −16.9%. Randomised and before‐and‐after design not different P = 0.46 |
| 0.1 mg/d | 5.01 (4.72 to 5.31) |
3.85 (3.81 to 3.90) |
‐23.1 (‐24.0 to ‐22.2) |
2327 (11) | ⊕⊕⊕⊕ High | Effect predicted from log dose‐response equation is −22.9%. Randomised and before‐and‐after design not different P = 0.13 |
| 0.2 mg/d | 5.09 (4.72 to 5.45) |
3.68 (3.64 to 3.73) |
‐27.6 (‐28.5 to ‐26.6) |
2498 (15) | ⊕⊕⊕⊕ High | Effect predicted from log dose‐response equation is −28.8%. Randomised and before‐and‐after design not different P = 0.07 |
| 0.3 mg/d | 5.09 (4.74 to 5.43) |
3.50 3.46 to 3.54) |
‐31.2 (‐32.0 to ‐30.5) |
3020 (19) | ⊕⊕⊕⊕ High | Effect predicted from log dose‐response equation is −32.3%. Randomised and before‐and‐after design not different P = 0.73 |
| 0.4 mg/d | 5.00 (4.69 to 5.32) |
3.28 (3.24 to 3.32) |
‐34.5 (‐35.3 to ‐33.7) |
3080 (13) | ⊕⊕⊕⊕ High | Effect predicted from log dose‐response equation is −34.8%. Randomised and before‐and‐after design not different P = 0.84 |
| 0.8 mg/d | 4.91 (4.55 to 5.27) |
2.84 (2.80 to 2.88) |
‐42.2 (‐43.1 to ‐41.3) |
2560 (6) | ⊕⊕⊕⊕ High | Effect predicted from log dose‐response equation is −40.8%. Randomised and before‐and‐after design not different P = 0.31 |
| CI: confidence interval; LDL: low‐density lipoprotein | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
aMean baseline values.
Summary of findings 2. Total cholesterol‐lowering efficacy of cerivastatin.
| Total cholesterol lowering efficacy of cerivastatin | ||||||
|
Patient or population: participants with normal or abnormal lipid profiles Settings: clinics or hospitals Intervention: different fixed doses of cerivastatin Comparison: placebo or baseline | ||||||
| Cerivastatin dose |
Anticipated absolute effects mmol/L (95%CI) |
Percentage change from baseline (95% CI) | № of participants (trials) | Certainty of the evidence (GRADE) | Comments | |
| Total cholesterol before exposure to cerivastatina | Total cholesterol after exposure to cerivastatin | |||||
| 0.05 mg/d | 7.14 (6.80 to 7.49) |
6.32 (6.22 to 6.43) |
‐11.5 (‐12.9 to ‐10.0) |
569 (3) | ⊕⊕⊕⊕ High | Effect predicted from log dose‐response equation is −12.2%. Randomised and before‐and‐after design not different P = 0.87 |
| 0.1 mg/d | 7.10 (6.80 to 7.39) |
5.91 (5.86 to 5.96) |
‐16.8 (‐17.5 to ‐16.1) |
2114 (10) | ⊕⊕⊕⊕ High | Effect predicted from log dose‐response equation is −16.3%. Randomised and before‐and‐after design not different P = 0.90 |
| 0.2 mg/d | 7.16 (6.80 to 7.52) |
5.73 (5.68 to 5.78) |
‐20.0 (‐20.7 to ‐19.3) |
1953 (14) |
⊕⊕⊕⊕ High | Effect predicted from log dose‐response equation is −20.5%. Randomised and before‐and‐after design was different P = 0.002 |
| 0.3 mg/d | 7.19 (6.85 to 7.54) |
5.58 (5.54 to 5.62) |
‐22.4 (‐23.0 to ‐21.8) |
2567 (17) |
⊕⊕⊕⊕ High | Effect predicted from log dose‐response equation is −22.9%. Randomised and before‐and‐after design not different P = 0.055 |
| 0.4 mg/d | 7.16 (6.88 to 7.44) |
5.41 (5.36 to 5.46) |
‐24.5 (‐25.2 to ‐23.8) |
2715 (10) |
⊕⊕⊕⊕ High | Effect predicted from log dose‐response equation is −24.7%. Randomised and before‐and‐after design was different P = 0.03 |
| 0.8 mg/d | 7.01 (6.66 to 7.36) |
4.91 (4.84 to 4.98) |
‐29.95 (‐31.0 to ‐28.9) |
1938 (5) |
⊕⊕⊕⊕ High | Effect predicted from log dose‐response equation is −28.8%. Randomised and before‐and‐after design not different P = 0.40 |
| CI: confidence interval | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
aMean baseline values.
Summary of findings 3. Triglyceride‐lowering efficacy of cerivastatin.
| Triglyceride‐lowering efficacy of cerivastatin | ||||||
|
Patient or population: participants with normal or abnormal lipid profiles Settings: clinics or hospitals Intervention: different fixed doses of cerivastatin Comparison: placebo | ||||||
| Cerivastatin dose |
Anticipated absolute effects mmol/L (95%CI) |
Mean difference from placebo (95% CI) | № of participants (trials) | Certainty of the evidence (GRADE) | Comments | |
| Triglycerides before exposure to cerivastatina | Triglycerides after exposure to cerivastatin | |||||
| 0.05 mg/d | 1.85 (1.70 to 1.99) |
1.62 (1.53 to 1.71) |
‐12.5 (‐17.4 to ‐7.6) |
504 (2) |
⊕⊝⊝⊝ Very lowb,c | Effect predicted from log dose‐response equation is −11.5% |
| 0.1 mg/d | 1.85 (1.73 to 1.98) |
1.55 (1.47 to 1.63) |
‐16.1 (‐20.4 to ‐11.9) |
731 (4) |
⊕⊕⊕⊝ Moderatec | Effect predicted from log dose‐response equation is −14.0% |
| 0.2 mg/d | 1.81 (1.68 to 1.94) |
1.52 (1.44 to 1.59) |
‐16.2 (‐20.3 to ‐12.0) |
780 (5) |
⊕⊕⊕⊝ Moderatec | Effect predicted from log dose‐response equation is −16.4% |
| 0.3 mg/d | 1.85 (1.73 to 1.97) |
1.49 (1.42 to 1.55) |
‐19.6 (‐23.2 to ‐16.0) |
1303 (8) |
⊕⊕⊕⊝ Moderatec | Effect predicted from log dose‐response equation is −17.9% |
| 0.4 mg/d | 1.88 (1.76 to 2.00) |
1.55 (1.50 to 1.60) |
‐17.6 (‐20.4 to ‐14.9) |
1969 (6) |
⊕⊕⊕⊝ Moderatec | Effect predicted from log dose‐response equation is −18.9% |
| 0.8 mg/d | 1.90 (1.77 to 2.04) |
1.50 (1.43 to 1.56) |
‐21.2 (‐24.5 to ‐18.0) |
1880 (4) |
⊕⊕⊕⊝ Moderatec | Effect predicted from log dose‐response equation is −20.0% |
| CI: confidence interval | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
a. Mean baseline values. b. Downgraded two levels due to small number of trials. c. Downgraded one level due to wide confidence intervals.
We used only data from RCTs for triglycerides because there was a difference in the mean differences between the two types of trials and it was judged that RCT data provided a better estimate of the true effect.
Summary of findings 4. All doses of cerivastatin compared to placebo for withdrawal due to adverse events.
| Withdrawals due to adverse events due to cerivastatin | ||||||
| Patient or population: participants with normal or abnormal lipid profiles Setting: clinics or hospitals Intervention: all doses of cerivastatin Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (trials) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with all doses of cerivastatin | |||||
| Withdrawals due to adverse events Follow‐up: range 3 weeks to 12 weeks | Trial population | RR 1.09 (0.68 to 1.74) | 6570 (11 RCTs) | ⊕⊝⊝⊝ Very lowa,b,c | ||
| 16 per 1000 | 17 per 1000 (11 to 28) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
a. Downgraded due to short duration of trials (3 to 12 weeks). b. Downgraded due to wide confidence interval. c. Downgraded due to selective reporting bias: only 11 out of 19 trials reported withdrawals due to adverse effects
Results
Description of studies
This review included 50 trials involving 14,149 intention‐to‐treat participants, of whom 13,018 (92%) participants had at least one lipid parameter measured and of whom 12,877 (91%) had LDL cholesterol reported. There were 31 before‐and‐after studies and 19 randomised, double‐blind, placebo‐controlled trials (RCTs). The number of placebo and cerivastatin participants were 1923 and 11,095 respectively. The number of male and female participants reported in 45 of the 50 trials were 7257 and 5669 respectively. Participants could be of any age. There were three familial hypercholesterolaemia trials and 23 non‐familial hypercholesterolaemia trials.
Results of the search
Database searching identified 2728 citations and 471 other resource citations giving a total of 3199 records. After the duplicates were removed, 2868 records remained. The number of irrelevant records was 2775. From these remaining records, we obtained 93 as full‐text articles, which we assessed for eligibility. We excluded 21 trials with reasons. The final number of included trials was 50 (Figure 1).
Included studies
Seventy‐two citations to 50 trials met the inclusion criteria and had extractable data to evaluate the dose‐related lipid‐lowering effect of cerivastatin. Each included trial is summarised in the Characteristics of included studies table. The publication languages of the 50 included trials were 36 (72%) English, 11 (22%) Japanese, one (2%) Russian, Chinese and Korean. All the RCTs were randomised double‐blind trials. Trials evaluating the lipid‐altering efficacy of cerivastatin were first published in 1992 and continued to be published until 2005 (Figure 2).
2.

Number of included trials according to publication year
The baseline mean (range) lipid parameters were as follows: total cholesterol, 7.22 mmol/L (5.41 mmol/L to 9.59 mmol/L), 279 mg/dL (209 mg/dL to 371 mg/dL); LDL cholesterol, 5.11 mmol/L (3.47 mmol/L to 7.63 mmol/L), 197 mg/dL (134 mg/dL to 295 mg/dL); HDL cholesterol, 1.28 mmol/L (0.57 mmol/L to 1.8 mmol/L), 49 mg/dL (22 mg/dL to 70 mg/dL) and triglycerides 1.90 mmol/L (1.05 mmol/L to 3.50 mmol/L), 168 mg/dL (93 mg/dL to 310 mg/dL). Trials were available for the dose range of 0.025 mg to 0.8 mg cerivastatin daily and were sufficient to generate dose‐response regression lines for LDL cholesterol, total cholesterol and triglycerides (Figure 3; Figure 4; Figure 5).
3.
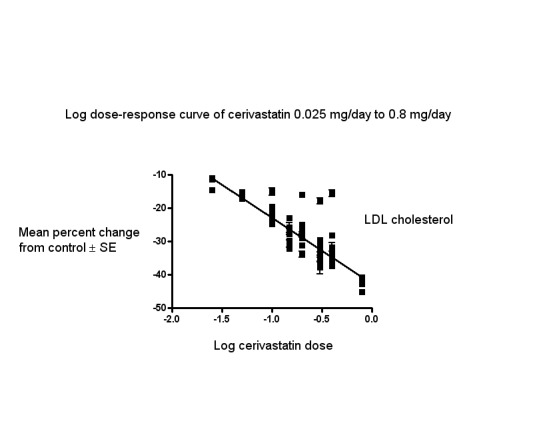
Log dose cerivastatin response curve for LDL cholesterol
Values represent the results of each trial for each dose comparison. The standard error bars cannot be seen because they all lie within the points
4.
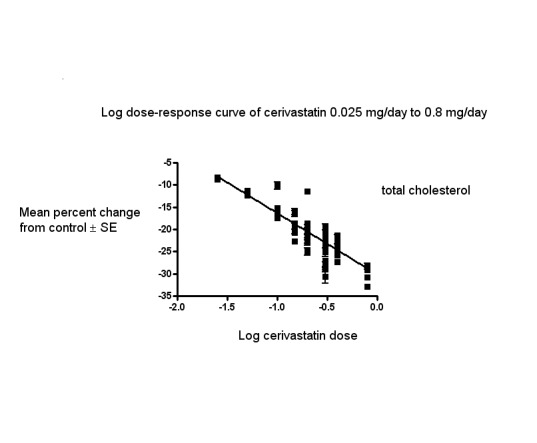
Log dose cerivastatin response curve for total cholesterol
Values represent the results of each trial for each dose comparison. The standard error bars cannot be seen because they all lie within the points
5.

Log dose cerivastatin response curve for triglycerides
Values represent the results of each trial for each dose comparison. The standard error bars cannot be seen because they all lie within the points
Excluded studies
We excluded 21 trials because they did not meet the inclusion criteria. Reasons for exclusion included confounding, inappropriate dosing, pooled data, attrition bias if more than 25% participants were not included in the efficacy analysis, inappropriate outcomes such as median percentage change from baseline or absolute change from baseline that could not be converted to percentage change from baseline, inadequate dietary baseline stabilisation period, and combined data for all cross‐over periods. We excluded trials in which participants were receiving drugs that affect blood lipid level concentrations: immunosuppressants (cyclosporine), protease inhibitors (ritonavir and indinavir), food supplements (fish oils), fibrates (gemfibrozil, fenofibrate and clofibrate), bile acid sequestrants (cholestyramine, colestipol, colesevelam), cholesterol absorption inhibitors (ezetimibe), vitamins (niacin) and the anti‐oxidant drug, probucol. The reasons for excluding each trial are listed in the Characteristics of excluded studies table.
Risk of bias in included studies
We judged random sequence generation bias to be high in the 31 before‐and‐after studies. The 19 RCTs did not report the method of sequence generation and so we judged them to be at unclear risk of bias.
Allocation
We judged allocation concealment bias to be high in the 31 before‐and‐after studies. The 19 RCTs did not report allocation concealment, and so we judged them to be at unclear risk of bias.
Blinding
We judged the risk of bias due to blinding of participants and personnel (performance bias) for lipid parameters to be low for all the trials as lipid parameter measurements are unlikely influenced by lack of blinding.
We judged the risk of bias for blinding of outcome assessment (detection bias) for lipids to be low for all the trials as lipid parameters were measured in a remote laboratory.
Of the 19 RCTs, we judged two (10.5%) at low risk of detection bias and seven (36.8%) at high risk of detection bias for withdrawals due to adverse effects.
Incomplete outcome data
Incomplete outcome reporting leading to attrition bias was not a problem in this review as few participants were lost to follow‐up. From 14,123 intention‐to‐treat participants, the LDL cholesterol was reported in 12,877 (91.2%).
Selective reporting
Out of 50 trials, 49 (98%) reported the primary lipid outcome LDL cholesterol, thus selection bias was not a potential source of bias for this outcome.
Out of 19 RCTs, only 11 (58%) reported withdrawals due to adverse effects. The trials that did not report could have deliberately not done so because withdrawals due to adverse effects were increased. Therefore, we judged selective reporting bias to be an important source of bias for this outcome. See 'Risk of bias' tables in Characteristics of included studies, and for the overall risk of bias, see (Figure 6).
6.
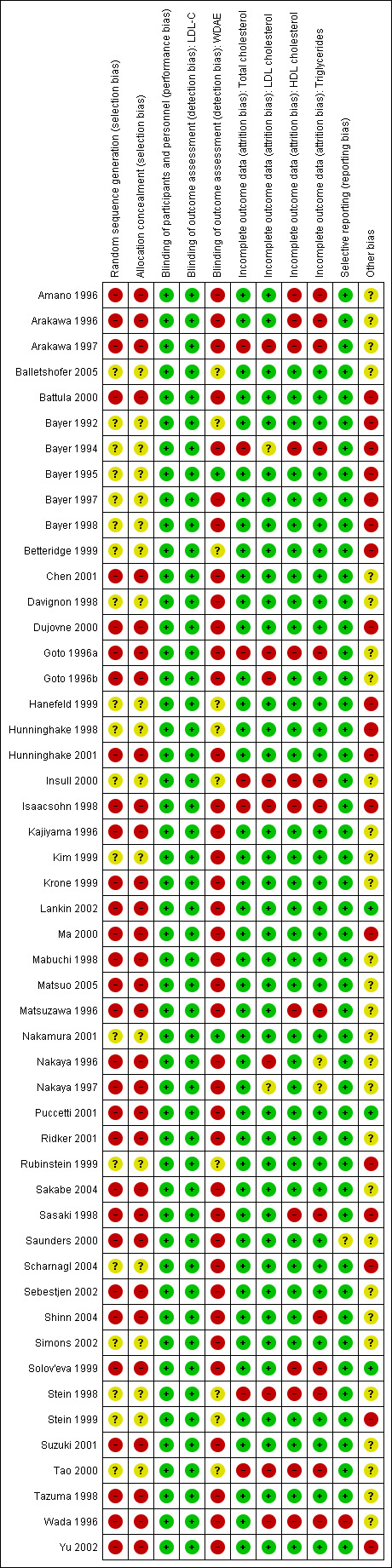
'Risk of bias' summary: review authors' judgements about each 'Risk of bias' item for each included trial
Other potential sources of bias
The main other potential source of bias is industry funding. Out of the 50 trials, 19 (38%) reported funding by industry, three (6%) reported partial funding by industry and government, three (6%) reported no industry funding and 25 (50%) trials did not report the source of funding. Out of 19 industry‐funded trials, 18 (94.7%) were funded by Bayer, marketers of cerivastatin and one (5.3%) was funded by another pharmaceutical company. The Bayer‐funded trials might be biased in favour of cerivastatin and may be expected to overestimate the treatment effect, while trials funded by rival pharmaceutical companies might be biased against cerivastatin and be expected to underestimate the treatment effect. In trials where the source of funding was not reported, bias could be for or against cerivastatin. Bayer‐funded versus non‐Bayer‐funded LDL cholesterol efficacy data were available for the doses of 0.2 mg/day and 0.3 mg/day. We analysed these data separately using the generic inverse variance fixed‐effect model in Review Manager 5. The sensitivity analysis revealed that the lipid‐lowering efficacy of cerivastatin in Bayer‐funded versus non‐Bayer funded trials was not different for the doses analysed; 0.20 mg/day (−27.6% versus −33.9%; P = 0.07) and 0.30 mg/day (−31.05% versus −30.2%; P = 0.52). We assessed publication bias by reviewing the funnel plots for all lipid outcomes with 10 or more trials. None of these funnel plots suggested publication bias.
Laboratories not connected to the trial personnel or participants determined lipids in the blood samples, therefore we judged the overall risk of bias to be low for both the RCTs and for the before‐and‐after studies (see Figure 6).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4
See: Table 1, Table 2, Table 3, and Table 4 for the LDL cholesterol lowering, total cholesterol lowering, triglyceride lowering efficacy of cerivastatin and withdrawal due to adverse effects for all trials. For relative potencies of cerivastatin with respect to fluvastatin, atorvastatin and rosuvastatin for LDL cholesterol, total cholesterol, triglycerides and HDL cholesterol; see Comparison of the effect with other statins subsection in the discussion section.
Overall efficacy of cerivastatin
We entered values from all data describing the efficacy of cerivastatin to lower the lipid parameters from placebo RCTs and before‐and‐after studies from the Data and analyses section as generic inverse variance data separately into GraphPad Prism 4 to yield log dose‐response curves for placebo and before‐and‐after studies. To compare slope results of RCTs versus before‐and‐after studies, we performed t‐tests from the formula T = (placebo slope‐before and after slope)/SQRT(SE2placebo slope+SE2before and after slope) from the slopes and standard errors of the curves for LDL cholesterol, total cholesterol, HDL cholesterol and triglycerides. The results showed that there were no differences between RCTs and before‐and‐after studies for LDL cholesterol: P = 0.944; total cholesterol: P = 0.054; HDL cholesterol: P = 0.157; and triglycerides P = 0.14. This demonstrates that the two trial designs provide similar estimates of the lipid‐lowering efficacy of cerivastatin.
In addition, we performed two‐tailed, one‐sample t‐tests from the RCTs to test for the difference between placebo mean effects and zero. The results of these tests demonstrated that the placebo means were not different from zero except for HDL cholesterol and the triglycerides: LDL cholesterol: 0.133 (95% CI −1.494 to 1.760) P = 0.866; total cholesterol: 0.342 (95% CI −1.019 to 1.703) P = 0.603; HDL cholesterol 1.892 (95% CI 0.328 to 6.456) P = 0.0205; and triglycerides: 3.825 (95%CI 0.435 to 7.215) P = 0.0296. The evidence of lack of a placebo effect provided further justification for combining all the trials to determine the overall efficacy.
Validation for combining the results from the two trial designs has been previously shown in the atorvastatin, rosuvastatin and fluvastatin reviews (Adams 2014; Adams 2015; Adams 2018).
We combined the results from the two trial designs by entering all data into Review Manager 5 using the generic inverse variance model outside of this review (data and analysis are not shown). The mean parameters from this analysis are summarised in Table 5. We combined the results from the two trial designs because the mean treatment effects were not statistically different between RCTs and before‐and‐after studies.
1. Cerivastatin overall efficacy.
| Cerivastatin dose (mg/d) | 0.025 | 0.05 | 0.1 | 0.15] | 0.2 | 0.3 | 0.4 | 0.8 |
|
Mean percentage change from control of LDL‐C (95%CI) |
‐12.2 (‐13.6 to ‐10.8) |
‐16.0 (‐17.2 to ‐14.7) |
‐23.1 (‐24.0 to ‐22.2) |
‐28.5 (‐29.6 to ‐27.4) |
‐27.55 (‐28.5 to ‐26.6) |
‐31.2 (‐32.0 to ‐30.5) |
‐34.5 (‐35.3 to ‐33.7) |
‐42.2 (‐43.1 to ‐41.3) |
|
Mean percentage change from control of total cholesterol (95%CI) |
‐8.4 (‐10.1 to ‐6.6) |
‐11.5 (‐12.9 to ‐10.0) |
‐16.8 (‐17.5 to ‐16.1) |
‐19.5 (‐20.3 to ‐18.7) |
‐20.0 (‐20.7 to ‐19.3) |
‐22.4 (‐23.0 to ‐21.8) |
‐24.5 (‐25.2 to ‐23.8) |
‐29.95 (‐31.0 to ‐28.9) |
|
Mean difference from placebo of triglycerides (95%CI) |
‐10.2 (‐12.5 to ‐8.0) |
‐9.8 (‐14.1 to −5.5) |
‐10.4 (‐13.0 to −7.75) |
‐5.4 (‐11.8 to ‐1.05) |
‐12.2 (‐14.6 to ‐9.7) |
‐12.9 (‐14.6 to ‐11.2) |
‐14.5 (‐16.5 to ‐12.6) |
‐21.2 (‐24.5 to ‐18.0) |
| CI: confidence interval; LDL‐C: low‐density lipoprotein cholesterol |
Primary outcome
LDL cholesterol
In total, 49 out of 50 (98%) trials and 12,877 out of 14,149 (91%) participants contributed to the LDL cholesterol data analysis.
The effect of different doses of cerivastatin on LDL cholesterol are shown in the Data and analyses section (Analysis 1.1; Analysis 2.1; Analysis 2.5; Analysis 3.1; Analysis 3.5; Analysis 4.1; Analysis 4.5; Analysis 5.1; Analysis 5.5; Analysis 6.1; Analysis 6.5; Analysis 7.1; Analysis 7.5; Analysis 8.1; Analysis 8.5). The analysis for LDL cholesterol yielded the log dose‐response straight‐line equation, y = −19.85 log(x) − 42.71. This equation provides the best estimate of the mean reductions in LDL cholesterol from baseline for cerivastatin doses ranging from 0.025 mg/day to 0.8 mg/day as it uses all the available data. Using this formula the calculated reductions in LDL cholesterol for doses of 0.025 mg per day to 0.8 mg per day was from 11.0% to 40.8%. For every two‐fold dose increase there was a 6.01% (95% CI 5.61 to 6.40) percentage decrease in LDL cholesterol (Figure 3).
1.1. Analysis.
Comparison 1 0.025 mg, Outcome 1 LDL‐cholesterol RCTs.
2.1. Analysis.
Comparison 2 0.05 mg, Outcome 1 LDL‐cholesterol RCTs.
2.5. Analysis.
Comparison 2 0.05 mg, Outcome 5 LDL‐cholesterol non‐RCTs.
3.1. Analysis.
Comparison 3 0.10 mg, Outcome 1 LDL‐cholesterol RCTs.
3.5. Analysis.
Comparison 3 0.10 mg, Outcome 5 LDL‐cholesterol non‐RCTs.
4.1. Analysis.
Comparison 4 0.15 mg, Outcome 1 LDL‐cholesterol RCTs.
4.5. Analysis.
Comparison 4 0.15 mg, Outcome 5 LDL‐cholesterol non‐RCTs.
5.1. Analysis.
Comparison 5 0.20 mg, Outcome 1 LDL‐cholesterol RCTs.
5.5. Analysis.
Comparison 5 0.20 mg, Outcome 5 LDL‐cholesterol non‐RCTs.
6.1. Analysis.
Comparison 6 0.30 mg, Outcome 1 LDL‐cholesterol RCTs.
6.5. Analysis.
Comparison 6 0.30 mg, Outcome 5 LDL‐cholesterol non‐RCTs.
7.1. Analysis.
Comparison 7 0.40 mg, Outcome 1 LDL‐cholesterol RCTs.
7.5. Analysis.
Comparison 7 0.40 mg, Outcome 5 LDL‐cholesterol non‐RCTs.
8.1. Analysis.
Comparison 8 0.80 mg, Outcome 1 LDL‐cholesterol RCTs.
8.5. Analysis.
Comparison 8 0.80 mg, Outcome 5 LDL‐cholesterol non‐RCTs.
Secondary outcomes
Total cholesterol
In total, 47 out of 50 (94%) trials and 10,365 out of 14,149 (73.3%) participants contributed to the total cholesterol data analysis. The effect of different doses of cerivastatin on total cholesterol are shown in the Data and analyses section (Analysis 1.2; Analysis 2.2; Analysis 2.6; Analysis 3.2; Analysis 3.6; Analysis 4.2; Analysis 4.6; Analysis 5.2; Analysis 5.6; Analysis 6.2; Analysis 6.6; Analysis 7.2; Analysis 7.6; Analysis 8.2; Analysis 8.6). The analysis for total cholesterol yielded the log dose‐response straight‐line equation, y = −13.83 log(x) − 30.15. This equation provides the best estimate of the mean reductions in total cholesterol from baseline for cerivastatin doses ranging from 0.025 mg/day to 0.8 mg/day as it uses all the available data. Using this formula, the calculated reductions in total cholesterol for doses of 0.025 mg per day to 0.8 mg per day was from 8.0% to 28.8%. For every two‐fold dose increase there was a 4.16% (95% CI 3.80 to 4.52) percentage decrease in total cholesterol (Figure 4).
1.2. Analysis.
Comparison 1 0.025 mg, Outcome 2 Total cholesterol RCTs.
2.2. Analysis.
Comparison 2 0.05 mg, Outcome 2 Total cholesterol RCTs.
2.6. Analysis.
Comparison 2 0.05 mg, Outcome 6 Total cholesterol non‐RCTs.
3.2. Analysis.
Comparison 3 0.10 mg, Outcome 2 Total cholesterol RCTs.
3.6. Analysis.
Comparison 3 0.10 mg, Outcome 6 Total cholesterol non‐RCTs.
4.2. Analysis.
Comparison 4 0.15 mg, Outcome 2 Total cholesterol RCTs.
4.6. Analysis.
Comparison 4 0.15 mg, Outcome 6 Total cholesterol non‐RCTs.
5.2. Analysis.
Comparison 5 0.20 mg, Outcome 2 Total cholesterol RCTs.
5.6. Analysis.
Comparison 5 0.20 mg, Outcome 6 Total cholesterol non‐RCTs.
6.2. Analysis.
Comparison 6 0.30 mg, Outcome 2 Total cholesterol RCTs.
6.6. Analysis.
Comparison 6 0.30 mg, Outcome 6 Total cholesterol non‐RCTs.
7.2. Analysis.
Comparison 7 0.40 mg, Outcome 2 Total cholesterol RCTs.
7.6. Analysis.
Comparison 7 0.40 mg, Outcome 6 Total cholesterol non‐RCTs.
8.2. Analysis.
Comparison 8 0.80 mg, Outcome 2 Total cholesterol RCTs.
8.6. Analysis.
Comparison 8 0.80 mg, Outcome 6 Total cholesterol non‐RCTs.
HDL cholesterol
In total, 39 out of 50 (78%) trials and 10,881 out of 14,149 (76.9%) participants contributed to the HDL cholesterol data analysis. The effect of different doses of cerivastatin on HDL cholesterol are shown in the Data and analyses section (Analysis 1.3; Analysis 2.3; Analysis 2.7; Analysis 3.3; Analysis 3.7; Analysis 4.3; Analysis 4.7; Analysis 5.3; Analysis 5.7; Analysis 6.3; Analysis 6.7; Analysis 7.3; Analysis 7.7; Analysis 8.3; Analysis 8.7). The GraphPad Prism 4 analysis showed that cerivastatin doses ranging from 0.025 mg/day to 0.8 mg/day had no dose‐related effect on HDL cholesterol. All doses of cerivastatin caused a small increase in HDL cholesterol. When we pooled all trials and doses using generic inverse variance the magnitude of the increase was 5.01% (95% CI 4.64 to 5.38).
1.3. Analysis.
Comparison 1 0.025 mg, Outcome 3 HDL‐cholesterol RCTs.
2.3. Analysis.
Comparison 2 0.05 mg, Outcome 3 HDL‐cholesterol RCTs.
2.7. Analysis.
Comparison 2 0.05 mg, Outcome 7 HDL‐cholesterol non‐RCTs.
3.3. Analysis.
Comparison 3 0.10 mg, Outcome 3 HDL‐cholesterol RCTs.
3.7. Analysis.
Comparison 3 0.10 mg, Outcome 7 HDL‐cholesterol non‐RCTs.
4.3. Analysis.
Comparison 4 0.15 mg, Outcome 3 HDL‐cholesterol RCTs.
4.7. Analysis.
Comparison 4 0.15 mg, Outcome 7 HDL‐cholesterol non‐RCTs.
5.3. Analysis.
Comparison 5 0.20 mg, Outcome 3 HDL‐cholesterol RCTs.
5.7. Analysis.
Comparison 5 0.20 mg, Outcome 7 HDL‐cholesterol non‐RCTs.
6.3. Analysis.
Comparison 6 0.30 mg, Outcome 3 HDL‐cholesterol RCTs.
6.7. Analysis.
Comparison 6 0.30 mg, Outcome 7 HDL‐cholesterol non‐RCTs.
7.3. Analysis.
Comparison 7 0.40 mg, Outcome 3 HDL‐cholesterol RCTs.
7.7. Analysis.
Comparison 7 0.40 mg, Outcome 7 HDL‐cholesterol non‐RCTs.
8.3. Analysis.
Comparison 8 0.80 mg, Outcome 3 HDL‐cholesterol RCTs.
8.7. Analysis.
Comparison 8 0.80 mg, Outcome 7 HDL‐cholesterol non‐RCTs.
Triglycerides
In total 15 out of 50 (30%) trials and 7831 out of 14,149 (55.3%) participants contributed to the triglyceride data analysis. The effect of different doses of cerivastatin on triglycerides are shown in the Data and analyses section (Analysis 1.4; Analysis 2.4; Analysis 2.8; Analysis 3.4; Analysis 3.8; Analysis 4.4; Analysis 4.8; Analysis 5.4; Analysis 5.8; Analysis 6.4; Analysis 6.8; Analysis 7.4;Analysis 7.8; Analysis 8.4). The analysis for triglycerides yielded the log dose‐response straight‐line equation, y = −8.24 log(x) − 22.19. This equation provides the best estimate of the mean reductions in triglycerides from baseline for cerivastatin doses ranging from 0.025 mg/day to 0.8 mg/day as it uses all the RCT data. Using this formula, the calculated reductions in total triglycerides for doses of 0.025 to 0.8 mg per day was from 9.0% to 21.4%. For every two‐fold dose increase there was a 2.48% (95% CI 1.57 to 3.39) percentage decrease in triglycerides (Figure 5).
1.4. Analysis.
Comparison 1 0.025 mg, Outcome 4 Triglycerides RCTs.
2.4. Analysis.
Comparison 2 0.05 mg, Outcome 4 Triglycerides RCTs.
2.8. Analysis.
Comparison 2 0.05 mg, Outcome 8 Triglycerides non‐RCTs.
3.4. Analysis.
Comparison 3 0.10 mg, Outcome 4 Triglycerides RCTs.
3.8. Analysis.
Comparison 3 0.10 mg, Outcome 8 Triglycerides non‐RCTs.
4.4. Analysis.
Comparison 4 0.15 mg, Outcome 4 Triglycerides RCTs.
4.8. Analysis.
Comparison 4 0.15 mg, Outcome 8 Triglycerides non‐RCTs.
5.4. Analysis.
Comparison 5 0.20 mg, Outcome 4 Triglycerides RCTs.
5.8. Analysis.
Comparison 5 0.20 mg, Outcome 8 Triglycerides non‐RCTs.
6.4. Analysis.
Comparison 6 0.30 mg, Outcome 4 Triglycerides RCTs.
6.8. Analysis.
Comparison 6 0.30 mg, Outcome 8 Triglycerides non‐RCTs.
7.4. Analysis.
Comparison 7 0.40 mg, Outcome 4 Triglycerides RCTs.
7.8. Analysis.
Comparison 7 0.40 mg, Outcome 8 Triglycerides non‐RCTs.
8.4. Analysis.
Comparison 8 0.80 mg, Outcome 4 Triglycerides RCTs.
End‐of‐treatment variability
There were not enough data to compare the effect of cerivastatin on the variability of blood lipids as a co‐efficient of variance as only three trials provided appropriate data.
Withdrawal data
Eleven (57.9%) of the 19 RCTs reported withdrawals due to adverse effects during the three‐ to 12‐week treatment period. In three trials no participant discontinued treatment due to adverse effects or died during the trial, therefore risk reduction was not estimable. There was no cerivastatin dose‐response relationship for withdrawals due to adverse effects. The effect of different doses of cerivastatin on withdrawal due to adverse effects are shown in the Data and analyses section (Analysis 1.5; Analysis 2.9; Analysis 3.9; Analysis 4.9; Analysis 5.9; Analysis 6.9; Analysis 7.9; Analysis 8.8 ). Withdrawals due to adverse effects were not different between cerivastatin and placebo for any of the cerivastatin doses. A pooled estimate for all doses compared to placebo showed a risk ratio (RR) of 1.09 (95% CI 0.68 to 1.74) for withdrawals due to adverse effects in these short‐term trials (Analysis 9.1).
1.5. Analysis.
Comparison 1 0.025 mg, Outcome 5 WDAEs.
2.9. Analysis.
Comparison 2 0.05 mg, Outcome 9 WDAEs.
3.9. Analysis.
Comparison 3 0.10 mg, Outcome 9 WDAEs.
4.9. Analysis.
Comparison 4 0.15 mg, Outcome 9 WDAEs.
5.9. Analysis.
Comparison 5 0.20 mg, Outcome 9 WDAEs.
6.9. Analysis.
Comparison 6 0.30 mg, Outcome 9 WDAEs.
7.9. Analysis.
Comparison 7 0.40 mg, Outcome 9 WDAEs.
8.8. Analysis.
Comparison 8 0.80 mg, Outcome 8 WDAEs.
9.1. Analysis.
Comparison 9 All doses of cerivastatin vs placebo, Outcome 1 WDAEs.
Subgroup analyses
Male versus female participant data were available for the 0.4 and 0.8 mg/day doses. We analysed these data separately for LDL cholesterol lowering efficacy using the generic inverse variance fixed‐effect model in Review Manager 5 outside of this review. The subgroup analysis revealed that the efficacy of cerivastatin was greater in female than in male participants. The efficacy for the 0.4 mg/day dose (male versus female participants) was −29.75 versus −43.15; P < 0.0001; and for the 0.8 mg/day dose (male versus female participants) was −38.9 versus ‐47.5; P = 0.0006.
A comparison of morning administration time versus evening administration time was not possible because no trial provided the appropriate data. Data for twice‐daily administration versus single dose administration were available for the dose 0.2 mg/day. We compared these data for LDL cholesterol lowering efficacy. The percentage reductions in twice‐daily versus single‐dose regimens showed no difference: 0.2 mg/day −27.00% (95% CI −33.76 to −20.24) versus −31.40% (95% CI −37.38 to −25.42); P = 0.34.
Sensitivity analyses
Familial versus non‐familial hypercholesterolaemia participant data were available for the doses 0.2 mg/day, 0.3 mg/day and 0.4 mg/day. We analysed these data separately for LDL cholesterol lowering efficacy using the generic inverse variance fixed‐effect model in Review Manager 5. The efficacy of cerivastatin were not consistently different in one direction for familial participants versus non‐familial participants: 0.20 mg/day −21.69 (95% CI −25.50 to −17.89) versus −27.14 (95% CI −28.60 to −25.68) P = 0.009; 0.30 mg/day −34.0 (95% CI −43.45 to −24.55) versus −32.2 (95% CI −33.5 to −30.9) P = 0.71; and 0.40 mg/day −15.60 (95% CI −22.95 to −8.25) versus −35.1(95% CI −36.4 to −33.8) P < 0.00001.
Discussion
Summary of main results
Daily cerivastatin intake is effective at lowering LDL cholesterol concentrations and does so in a predictable, dose‐related manner. The 'Summary of findings' table documents that cerivastatin lowers LDL cholesterol by 16% at 0.05 mg/day and by 42.2% at 0.8 mg/day (Table 1). These moderate reductions reflect a reduction in synthesis of cholesterol by the liver and indicate that liver HMG CoA reductase is being inhibited by up to two‐fifths over this dose range. This has significant implications beyond circulating LDL cholesterol, as LDL cholesterol is only one of many important biochemical products that are produced by the HMG CoA reductase pathway. Those other products, including co‐enzyme Q10, heme A, vitamin D, steroid hormones and many other compounds, are also likely to be reduced by about two‐fifths with the 0.8 mg dose of cerivastatin. It is important to recognise that the long‐term consequences of reduction of these products is presently unknown.
In the data and analysis section it can be seen that there are more trials and data with the before‐and‐after design than from RCTs. For the doses where there is a large number of trials and participants, it can be seen that estimates of the effect of cerivastatin on the lipid parameters are similar with the two different trial designs. This, plus the demonstration that the placebo effect was not different from zero, justified using generic inverse variance to pool and display the combined estimates in Table 1. In addition we entered all trial data into GraphPad Prism 4 to calculate the regression lines shown in Figure 3; Figure 4 and Figure 5. The overall efficacy results from GraphPad Prism 4 provide the best estimate of the treatment effect, because they are based on a regression line calculated from all the data for all the doses. The estimates of the average treatment effect from the regression lines are similar to the mean value for all the data for each dose (see Table 1).
In this review we established, using regression analysis, that there was a correlation between the baseline value and cerivastatin effect on LDL cholesterol when the effect was expressed as absolute change from baseline (P = 0.0028). There was little correlation between the baseline value and the cerivastatin effect when the effect was expressed as percentage reduction from baseline (P = 0.0467). This finding provides support for the fact that systematic reviews reporting the effect of statins on absolute changes in lipid parameters are problematic and potentially misleading.
What is the effect of cerivastatin on the end‐of‐treatment variability?
We could not assess end‐of‐treatment variabilities of cerivastatin and placebo because most trials did not report this outcome and there were not enough data.
Does cerivastatin increase withdrawals due to adverse effects?
Of 19 RCTs, 11 (57.9%) reported withdrawals due to adverse effects. This analysis represented only 6570 participants, 5370 of whom received cerivastatin and 1200 of whom received placebo. The pooled estimate for all doses provided a risk ratio (RR) of 1.09 (95% CI 0.68 to 1.74), demonstrating uncertainty, but the possibility of an increase in adverse effects even in these short‐term trials. As eight (42.1%) of 19 RCTs did not report withdrawals due to adverse effects, risk of selective reporting bias for this outcome is high, and the null effect may be a result of that bias. Furthermore, this analysis was limited to trials of three to 12 weeks’ duration and thus does not reflect adverse effects of cerivastatin that occur after intake of longer duration. Risk of participant selection bias is also high in these trials, as many of the participants studied probably were known to tolerate statins at baseline.
Overall completeness and applicability of evidence
This review included 50 trials with 13,018 out of 14,123 intention‐to‐treat participants, of whom 12,877 participants had their LDL cholesterol reported. As such it provided us with robust evidence of the dose‐related, lipid‐lowering effects of cerivastatin. It was unknown when we did the review whether the time of cerivastatin administration is important with respect to lipid lowering. Unfortunately, there were no trials comparing morning and evening administration. A sensitivity analysis comparing twice‐daily versus single‐dose regimen data was available for the dose of 0.20 mg/day. The percentage reductions in twice‐daily versus single‐dose regimens showed no difference. We therefore felt justified in combining data from both dosing regimens. Subsequently a Cochrane Review has attempted to answer this question and concluded that statin lipid‐lowering effect is the same for morning and evening administration (Izquiero‐Palomares 2016).
Practitioners can use this evidence to calculate the expected effect of doses of cerivastatin commonly utilised in society. It is unlikely that further research will change these estimates appreciably. However, there was a fair amount of heterogeneity in many of the estimates and it is possible that this was due to differences in the populations being studied (e.g. gender or genetic differences; Thompson 2005). To explore this, where it was possible, we compared the effect of cerivastatin in male and female participants plus in participants with familial and non‐familial hypercholesterolaemia. A subgroup analysis comparing male versus female participant data was available for the doses 0.4 mg/day and 0.8 mg/day and suggested that efficacy of cerivastatin was greater in women than in men. A greater effect in women than men could be because women, on average ,weigh less than men. This finding corroborates this subgroup analysis in the atorvastatin and rosuvastatin reviews, which also showed a larger effect in female than male participants (Adams 2014; Adams 2015). There was no statistically significant difference in the effect in male and female participants for fluvastatin (Adams 2018).
Familial versus non‐familial hypercholesterolaemia participant data were available for the cerivastatin doses of 0.20 mg/day, 0.30 mg/day and 0.40 mg/day. We analysed these data separately for LDL cholesterol lowering efficacy using the generic inverse variance fixed‐effect model in Review Manager 5. The percentage reductions were not consistently in one direction for familial participants versus non‐familial participants (see Effects of interventions). This finding is consistent with what was found in the rosuvastatin review (Adams 2014). However, it is not consistent with the findings in the atorvastatin and fluvastatin reviews, where the LDL‐lowering effect was less in participants with familial hypercholesterolaemia (Adams 2015; Adams 2018).
The profound and relatively consistent effect of cerivastatin on lipid parameters shown in this review is probably appreciated by clinicians who treat patients with these drugs. Investigators involved in placebo‐controlled RCTs are likely to know whether participants are taking or not taking statins. Knowledge of the lipid parameters almost certainly leads to loss of blinding in statin RCTs. The present review calls attention to that problem, and efforts to prevent this loss of blinding are needed in future statin RCTs (Higgins 2011).
Quality of the evidence
The summary of all ’Risk of bias’ tools for the lipid effects suggests a high risk of bias (Figure 6). However, the lipid parameter outcomes are probably relatively resistant to bias. If anything, a high risk of bias would lead to an overestimate of the lipid‐lowering effects rather than an underestimate. However, because of the objectivity of the measurement of the lipid parameters by independent laboratories we think that the estimates of effects are reasonably accurate. This view is strengthened by the fact that we could not show evidence of funding bias. Comparing Bayer‐funded trials with non‐Bayer‐funded trials did not show any difference. Furthermore, a review of funnel plots did not suggest evidence of publication bias.
Low risk of bias is not true for the harm outcome, withdrawals due to adverse effects. Eleven (57.9%) of the 19 RCTs reported withdrawals due to adverse effects. There is therefore a high risk of selective reporting bias for this outcome and this, combined with the high risk of other biases, means that we cannot be confident that the finding of no increase in withdrawals due to adverse effects is accurate (Table 4).
Potential biases in the review process
Combining the RCTs with the before‐and‐after studies is a limitation of the review. We have explained why the increased risk of bias associated with the before‐and‐after design is less in this instance because the lipid parameters were measured in remote independent laboratories. Another limitation of this review is that many trials did not report standard deviations for the lipid‐lowering effects. In those trials we imputed the standard deviation of the percentage change from baseline of the blood lipid parameters as the average of this parameter from trials that reported it. These values were determined by the method of (Furukawa 2006). Such imputation might weight some trials more or less; however, this has been shown in other reviews not to have much effect on the magnitude of the effect estimate (Heran 2008; Musini 2014). Another limitation is that in this review few trials were available to demonstrate the lipid‐lowering effect of cerivastatin at doses of less than 0.025 mg/day and more than 0.8 mg/day. We did not downgrade the certainty of evidence due to heterogeneity of LDL cholesterol because the confidence intervals for the pooled result estimates were narrow.
Agreements and disagreements with other studies or reviews
The best estimate of the mean percentage reduction in LDL cholesterol for any dose of cerivastatin can be calculated from our log dose‐response equation. Using this equation y = −19.85 log(x) − 42.71, a cerivastatin dose of 0.4 mg/day reduces LDL cholesterol by an average of 34.8%. This is similar to the estimate of 36% reduction in LDL cholesterol in 527 participants in Edwards 2003.
Comparison of the effect with other statins
The greatest value in doing this type of review is the ability to compare cerivastatin to other statins. At present we can compare it to fluvastatin, atorvastatin and rosuvastatin, which have been reviewed using the same protocol. The most important finding in this review is that the slope of the dose‐response effect for cerivastatin on LDL cholesterol, total cholesterol and triglycerides is not different from the slopes of the dose‐response curves for atorvastatin (Adams 2015), rosuvastatin (Adams 2014), and fluvastatin (Adams 2018). This provides some confirmation that the four statins are all causing lipid lowering by a similar mechanism. However, it also demonstrates that cerivastatin is more potent than the other three drugs: for lowering LDL cholesterol; cerivastatin is 250‐fold more potent than fluvastatin, 20‐fold more potent than atorvastatin and 5.5‐fold more potent than rosuvastatin, for lowering total cholesterol; cerivastatin is 233‐fold more potent than fluvastatin, 18‐fold more potent than atorvastatin and six‐fold more potent than rosuvastatin and for lowering triglycerides cerivastatin is 125‐fold more potent than fluvastatin, 11‐fold more potent than atorvastatin and 13‐fold more potent than rosuvastatin. Relative potencies could not be determined for HDL cholesterol. When we compare cerivastatin 0.3 mg/day, which reduces LDL cholesterol by 32.3% on average with the other statins, the dose of fluvastatin, atorvastatin and rosuvastatin to achieve the same reduction in LDL cholesterol is 80 mg/day, 6 mg/day and 1.7 mg/day respectively.
When cerivastatin in the recommended dose range is compared to the other statins for their effect to lower LDL cholesterol it is more than fluvastatin and less than atorvastatin and rosuvastatin.
Cerivastatin 0.1 mg to 0.8 mg (23% to 41%) decrease in LDL cholesterol
Fluvastatin 20 mg to 80 mg (21 to 33%) decrease in LDL cholesterol
Atorvastatin 10 mg to 80 mg (37% to 52%) decrease in LDL cholesterol
Rosuvastatin 5 mg to 40 mg (41% to 55%) decrease in LDL cholesterol
Does this review provide an explanation as to why cerivastatin caused more cases of rhabdomyolysis than other statins?
The answer to this question is unfortunately no. We thought that the slope of the dose‐response effect for cerivastatin might be greater, thus leading to more dose‐related toxicity of cerivastatin, but in effect, the dose‐response slopes are similar for cerivastatin when compared to the other statins. Another possibility was that the lipid‐lowering effect for the recommended doses might be greater for cerivastatin. The review showed the range of LDL lipid lowering at 28% to 42% for cerivastatin to be more than fluvastatin at 20% to 35%, but it is substantially less than atorvastatin at 37% to 52% and rosuvastatin at 39% to 55%. Thus, the reason why cerivastatin caused a higher incidence of rhabdomyolysis than atorvastatin, leading it to be removed from the market, remains a mystery. It is certainly worth further study, as muscle toxicity associated with statins remains a problem with the long‐term use of these drugs. Muscle symptoms occur in up to 11% of patients taking high‐dose statins (Bruckert 2005). Furthermore research suggests that muscle toxicity sometimes leading to rhabdomyolysis is not a rare, idiopathic adverse effect. In fact, measurable muscle toxicity occurs in people taking statins without symptoms and likely occurs in most if not all people taking statins (Draeger 2006; Mohaupt 2009). Statin muscle toxicity has also been demonstrated in people in heavily exercised muscles (Urso 2005). A number of researchers have tried to study statin muscle toxicity in isolated muscle preparations (Jaskiewicz 2018; Jaskiewicz 2019; Kaufmann 2006; McTaggart 2003; Morikawa 2005; Nishimoto 2003; Sakamoto 2013; Yamazaki 2006). Unfortunately the exact mechanism of toxicity remains unexplained. In addition, nobody has been able to explain why cerivastatin was more toxic in the clinical setting. Further research is critical, as it could lead to ways that this serious and sometimes fatal toxicity of statin therapy could be avoided.
Authors' conclusions
Implications for practice.
Cerivastatin 0.025 mg/day to 0.8 mg/day causes a linear dose‐response reduction in the percentage change from control of LDL cholesterol, total cholesterol and triglycerides, but not for HDL cholesterol. Manufacturer‐recommended cerivastatin doses of 0.2 mg/day to 0.8 mg/day resulted in a range of 28% to 42% decrease of LDL cholesterol. From the slope of the lines for every two‐fold dose increase, there was a 6.01%, 4.16%, and 2.48% decrease in LDL cholesterol, total cholesterol and triglycerides, respectively.
To determine the relative potency of cerivastatin with respect to atorvastatin, rosuvastatin and fluvastatin, we determined the ratio of the mg amount of cerivastatin to the mg amount of atorvastatin, rosuvastatin or fluvastatin needed to produce the same effect. We calculated these values from the log dose‐response curves of cerivastatin, fluvastatin, atorvastatin and rosuvastatin for LDL cholesterol and total cholesterol. We determined that cerivastatin was about 250‐fold more potent than fluvastatin, 20‐fold more potent than atorvastatin and 5.5‐fold more potent than rosuvastatin in reducing LDL cholesterol.
We are uncertain about the risk of withdrawal due to adverse events from all doses of cerivastatin as compared to placebo (RR 1.09; 95% CI 0.68 to 1.74). The evidence for this outcome is very low certainty and thus it cannot be considered reliable.
Implication of these findings
Cerivastatin is much more potent than fluvastatin, atorvastatin and rosuvastatin but in the recommended dose range it lowered LDL more than fluvastatin but substantially less that atorvastatin and rosuvastatin.
Implications for research.
Since cerivastatin is no longer on the market, it is unlikely that any more clinical trials will be conducted. More basic research into why cerivastatin caused a higher incidence of rhabdomyolysis is needed.
Acknowledgements
The review authors would like to acknowledge assistance provided by staff of the Cochrane Hypertension.
Appendices
Appendix 1. Search Strategies
Database: Cochrane Central Register of Controlled Trials (CENTRAL; 2019, Issue 3) via Cochrane Register of Studies (CRS‐Web) Search Date: 18 March 2019 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ #1 cerivastatin AND CENTRAL:TARGET #2 baycol AND CENTRAL:TARGET #3 certa AND CENTRAL:TARGET #4 lipobay AND CENTRAL:TARGET #5 rivastatin AND CENTRAL:TARGET #6 #1 OR #2 OR #3 OR #4 OR #5 AND CENTRAL:TARGET ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Database: Ovid MEDLINE(R) and Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, Daily and Versions(R) <1946 to March 15, 2019> Search Date: 17 March 2019 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 cerivastatin.mp. 2 baycol.mp. 3 certa.mp. 4 lipobay.mp. 5 rivastatin.mp. 6 or/1‐5 7 animals/ not (humans/ and animals/) 8 6 not 7 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Database: Embase <1974 to 2019 March 15> Search Date: 17 March 2019 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 cerivastatin.mp. 2 baycol.mp. 3 certa.mp. 4 lipobay.mp. 5 rivastatin.mp. 6 or/1‐5 7 cholesterol$.mp. 8 (HDL or LDL).mp. 9 lipoprotein?.mp. 10 lipid$.mp. 11 triglyceride$.mp. 12 triacylglycerol.mp. 13 or/7‐12 14 6 and 13 15 (exp animal/ or animal.hw. or nonhuman/) not (exp human/ or human cell/ or (human or humans).ti.) 16 14 not 15
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Database: Cochrane Database of Systematic Reviews (CDSR) via Wiley Search Date: 17 March 2019 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ #1 All Text cerivastatin OR baycol OR certa OR lipobay OR rivastatin ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Database: ClinicalTrials.gov Search Date: 17 March 2019 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Other terms: cerivastatin OR baycol OR certa OR lipobay OR rivastatin ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Database: WHO International Clinical Trials Registry Platform (ICTRP) Search Date: 17 March 2019 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ cerivastatin OR baycol OR certa OR lipobay OR rivastatin ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Database: Epistemonikos Search Date: 17 March 2019 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ cerivastatin OR baycol OR lipobay OR rivastatin Publication type: Systematic Review
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
Database: ISI Clarivate Web of Science ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ TS = (cerivastatin or baycol or lipoby or "BAY w 6228" or BAY w 6228) NOT TS =(animal*) ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Database: BIOSIS Previews ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1. (cerivastatin or baycol or lipobay) AND Taxa Notes=(humans) 2. ("BAY w 6228" or BAY w 6228) AND Taxa Notes=(humans) 3. (#1 or #2) ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Database: International Pharmaceutical Abstracts (IPA) ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1. cerivastatin.af 2. baycol.af 3. lipobay.af 4. "BAY w 6228".af 5. BAY w 6228.af 6. 1 or 2 or 3 or 4 or 5 7. limit 6 to human ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Database: ProQuest Dissertations & Theses ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Cerivastatin or baycol or lipobay or "BAY w 6228" or BAY w 6228 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Database: SciFinder Scholar ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Cerivastatin or baycol or lipobay or BAY w 6228 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Database: Bayer.com ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Cerivastatin or baycol or lipobay or BAY w 6228 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Database: metaRegister (mRCT) ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Cerivastatin or baycol or lipobay or BAY w 6228
Appendix 2. Mean percentage change
[(Endpoint‐Baseline)/Baseline]*100
Appendix 3. Extracted standard deviations (SDs) and standard errors (SEs)
SE = |MD/t|
SD = (√n)*SE
SD = √n(upper confidence limit ‐ lower confidence limit)/2t
Data and analyses
Comparison 1. 0.025 mg.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 LDL‐cholesterol RCTs | 3 | 1100 | Mean Difference (IV, Fixed, 95% CI) | ‐12.65 [‐14.32, ‐10.97] |
| 2 Total cholesterol RCTs | 2 | 510 | Mean Difference (IV, Fixed, 95% CI) | ‐8.37 [‐10.13, ‐6.62] |
| 3 HDL‐cholesterol RCTs | 2 | 510 | Mean Difference (IV, Fixed, 95% CI) | 0.89 [‐1.49, 3.27] |
| 4 Triglycerides RCTs | 2 | 510 | Mean Difference (IV, Fixed, 95% CI) | ‐6.34 [‐11.27, ‐1.40] |
| 5 WDAEs | 2 | 562 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.46 [0.42, 5.06] |
Comparison 2. 0.05 mg.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 LDL‐cholesterol RCTs | 4 | 1750 | Mean Difference (IV, Fixed, 95% CI) | ‐15.76 [‐17.12, ‐14.41] |
| 2 Total cholesterol RCTs | 2 | 504 | Mean Difference (IV, Fixed, 95% CI) | ‐11.56 [‐13.32, ‐9.80] |
| 3 HDL‐cholesterol RCTs | 2 | 504 | Mean Difference (IV, Fixed, 95% CI) | 3.21 [0.73, 5.69] |
| 4 Triglycerides RCTs | 2 | 504 | Mean Difference (IV, Fixed, 95% CI) | ‐12.52 [‐17.45, ‐7.59] |
| 5 LDL‐cholesterol non‐RCTs | 1 | 61 | Mean Difference (Fixed, 95% CI) | ‐17.1 [‐20.36, ‐13.84] |
| 6 Total cholesterol non‐RCTs | 1 | 65 | Mean Difference (Fixed, 95% CI) | ‐11.3 [‐13.85, ‐8.75] |
| 7 HDL‐cholesterol non‐RCTs | 1 | 65 | Mean Difference (Fixed, 95% CI) | 2.2 [‐1.69, 6.09] |
| 8 Triglycerides non‐RCTs | 1 | 63 | Mean Difference (Fixed, 95% CI) | ‐0.5 [‐9.56, 8.56] |
| 9 WDAEs | 2 | 558 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.34, 4.47] |
Comparison 3. 0.10 mg.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 LDL‐cholesterol RCTs | 7 | 1908 | Mean Difference (IV, Fixed, 95% CI) | ‐22.40 [‐23.71, ‐21.10] |
| 2 Total cholesterol RCTs | 6 | 1677 | Mean Difference (IV, Fixed, 95% CI) | ‐16.72 [‐17.76, ‐15.68] |
| 3 HDL‐cholesterol RCTs | 6 | 1675 | Mean Difference (IV, Fixed, 95% CI) | 4.49 [3.04, 5.94] |
| 4 Triglycerides RCTs | 4 | 731 | Mean Difference (IV, Fixed, 95% CI) | ‐16.17 [‐20.46, ‐11.89] |
| 5 LDL‐cholesterol non‐RCTs | 4 | 419 | Mean Difference (Fixed, 95% CI) | ‐23.84 [‐25.14, ‐22.54] |
| 6 Total cholesterol non‐RCTs | 4 | 437 | Mean Difference (Fixed, 95% CI) | ‐16.81 [‐17.74, ‐15.87] |
| 7 HDL‐cholesterol non‐RCTs | 4 | 437 | Mean Difference (Fixed, 95% CI) | 3.20 [1.85, 4.56] |
| 8 Triglycerides non‐RCTs | 4 | 426 | Mean Difference (Fixed, 95% CI) | ‐6.87 [‐10.18, ‐3.56] |
| 9 WDAEs | 4 | 942 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.40, 2.76] |
Comparison 4. 0.15 mg.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 LDL‐cholesterol RCTs | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐23.05 [‐30.64, ‐15.46] |
| 2 Total cholesterol RCTs | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐19.1 [‐25.17, ‐13.03] |
| 3 HDL‐cholesterol RCTs | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 37.2 [29.10, 45.30] |
| 4 Triglycerides RCTs | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐17.9 [‐33.84, ‐1.96] |
| 5 LDL‐cholesterol non‐RCTs | 10 | 483 | Mean Difference (Fixed, 95% CI) | ‐28.63 [‐29.74, ‐27.51] |
| 6 Total cholesterol non‐RCTs | 11 | 555 | Mean Difference (Fixed, 95% CI) | ‐19.51 [‐20.36, ‐18.66] |
| 7 HDL‐cholesterol non‐RCTs | 3 | 94 | Mean Difference (Fixed, 95% CI) | 1.03 [‐2.20, 4.26] |
| 8 Triglycerides non‐RCTs | 3 | 92 | Mean Difference (Fixed, 95% CI) | ‐2.82 [‐9.86, 4.23] |
| 9 WDAEs | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 5. 0.20 mg.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 LDL‐cholesterol RCTs | 7 | 2140 | Mean Difference (IV, Fixed, 95% CI) | ‐28.37 [‐29.66, ‐27.07] |
| 2 Total cholesterol RCTs | 6 | 1589 | Mean Difference (IV, Fixed, 95% CI) | ‐21.17 [‐22.24, ‐20.11] |
| 3 HDL‐cholesterol RCTs | 6 | 1584 | Mean Difference (IV, Fixed, 95% CI) | 4.69 [3.21, 6.18] |
| 4 Triglycerides RCTs | 5 | 780 | Mean Difference (IV, Fixed, 95% CI) | ‐16.21 [‐20.39, ‐12.02] |
| 5 LDL‐cholesterol non‐RCTs | 8 | 358 | Mean Difference (Fixed, 95% CI) | ‐26.65 [‐28.01, ‐25.29] |
| 6 Total cholesterol non‐RCTs | 8 | 364 | Mean Difference (Fixed, 95% CI) | ‐18.91 [‐19.86, ‐17.96] |
| 7 HDL‐cholesterol non‐RCTs | 7 | 349 | Mean Difference (Fixed, 95% CI) | 6.14 [4.71, 7.58] |
| 8 Triglycerides non‐RCTs | 6 | 328 | Mean Difference (Fixed, 95% CI) | ‐10.43 [‐13.70, ‐7.15] |
| 9 WDAEs | 4 | 1102 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.31, 2.37] |
Comparison 6. 0.30 mg.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 LDL‐cholesterol RCTs | 10 | 2327 | Mean Difference (IV, Fixed, 95% CI) | ‐31.33 [‐32.55, ‐30.12] |
| 2 Total cholesterol RCTs | 8 | 1874 | Mean Difference (IV, Fixed, 95% CI) | ‐23.25 [‐24.30, ‐22.19] |
| 3 HDL‐cholesterol RCTs | 8 | 1931 | Mean Difference (IV, Fixed, 95% CI) | 6.39 [4.96, 7.81] |
| 4 Triglycerides RCTs | 8 | 1303 | Mean Difference (IV, Fixed, 95% CI) | ‐19.59 [‐23.15, ‐16.04] |
| 5 LDL‐cholesterol non‐RCTs | 9 | 693 | Mean Difference (Fixed, 95% CI) | ‐31.06 [‐32.03, ‐30.08] |
| 6 Total cholesterol non‐RCTs | 9 | 693 | Mean Difference (Fixed, 95% CI) | ‐21.98 [‐22.74, ‐21.23] |
| 7 HDL‐cholesterol non‐RCTs | 7 | 632 | Mean Difference (Fixed, 95% CI) | 4.59 [3.55, 5.62] |
| 8 Triglycerides non‐RCTs | 6 | 592 | Mean Difference (Fixed, 95% CI) | ‐10.86 [‐12.82, ‐8.89] |
| 9 WDAEs | 5 | 665 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.31 [0.38, 4.52] |
Comparison 7. 0.40 mg.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 LDL‐cholesterol RCTs | 8 | 2531 | Mean Difference (IV, Fixed, 95% CI) | ‐34.61 [‐35.76, ‐33.46] |
| 2 Total cholesterol RCTs | 7 | 2344 | Mean Difference (IV, Fixed, 95% CI) | ‐25.29 [‐26.26, ‐24.31] |
| 3 HDL‐cholesterol RCTs | 8 | 2551 | Mean Difference (IV, Fixed, 95% CI) | 4.85 [3.65, 6.04] |
| 4 Triglycerides RCTs | 6 | 1969 | Mean Difference (IV, Fixed, 95% CI) | ‐17.63 [‐20.39, ‐14.87] |
| 5 LDL‐cholesterol non‐RCTs | 5 | 549 | Mean Difference (Fixed, 95% CI) | ‐34.45 [‐35.54, ‐33.35] |
| 6 Total cholesterol non‐RCTs | 3 | 371 | Mean Difference (Fixed, 95% CI) | ‐23.73 [‐24.69, ‐22.77] |
| 7 HDL‐cholesterol non‐RCTs | 4 | 533 | Mean Difference (Fixed, 95% CI) | 6.31 [5.35, 7.26] |
| 8 Triglycerides non‐RCTs | 1 | 244 | Mean Difference (Fixed, 95% CI) | ‐11.4 [‐14.15, ‐8.65] |
| 9 WDAEs | 2 | 603 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.95 [0.55, 6.87] |
Comparison 8. 0.80 mg.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 LDL‐cholesterol RCTs | 4 | 1879 | Mean Difference (IV, Fixed, 95% CI) | ‐41.55 [‐43.05, ‐40.06] |
| 2 Total cholesterol RCTs | 4 | 1880 | Mean Difference (IV, Fixed, 95% CI) | ‐30.17 [‐31.33, ‐29.01] |
| 3 HDL‐cholesterol RCTs | 4 | 1880 | Mean Difference (IV, Fixed, 95% CI) | 5.67 [4.23, 7.10] |
| 4 Triglycerides RCTs | 4 | 1880 | Mean Difference (IV, Fixed, 95% CI) | ‐21.23 [‐24.46, ‐18.01] |
| 5 LDL‐cholesterol non‐RCTs | 2 | 681 | Mean Difference (Fixed, 95% CI) | ‐42.52 [‐43.63, ‐41.42] |
| 6 Total cholesterol non‐RCTs | 1 | 58 | Mean Difference (Fixed, 95% CI) | ‐29.1 [‐31.34, ‐26.86] |
| 7 HDL‐cholesterol non‐RCTs | 2 | 681 | Mean Difference (Fixed, 95% CI) | 8.49 [7.33, 9.65] |
| 8 WDAEs | 2 | 435 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.04 [0.52, 8.05] |
Comparison 9. All doses of cerivastatin vs placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 WDAEs | 11 | 6570 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.68, 1.74] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Amano 1996.
| Methods | 4‐week placebo run‐in period 12‐week before‐and‐after study |
|
| Participants | 11 men and women with type IIa and IIb hyperlipidaemia and type 2 diabetes mellitus age > 20 years old TC ≥ 220 mg/dL (5.69 mmol/L) Exclusion criteria: secondary hypercholesterolaemia, FH, statin hypersensitivity, difficulty evaluating drug efficacy, Cerivastatin 0.15 mg/d baseline TC: 5.88 mmol/L (227 mg/dL) Cerivastatin 0.15 mg/d baseline LDL‐C: 3.54 mmol/L (137 mg/dL) |
|
| Interventions | Cerivastatin 0.15 mg/d evening dosing | |
| Outcomes | Percentage change from baseline at 4‐12 weeks of blood TC and LDL‐C | |
| Source of funding | Unknown | |
| Notes | HDL‐C and TG were not included in the efficacy analysis because the given values and the calculated values differed by > 10% | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | High risk | Not included in the efficacy analysis because the given values and the calculated values differed by > 10% |
| Incomplete outcome data (attrition bias) Triglycerides | High risk | Not included in the efficacy analysis because the given values and the calculated values differed by > 10% |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Arakawa 1996.
| Methods | 4‐week placebo washout period 12‐week before‐and‐after study |
|
| Participants | 73 men and women with type IIa and IIb hyperlipidaemia aged 20‐64 years old, hypertension, mild diabetes, obesity, cholelithiasis TC ≥ 260 mg/dL (6.72 mmol/L) TG < 400 mg/dL (4.52 mmol/L) Exclusion criteria: secondary hyperlipidaemia, hypothyroidism, Cushings syndrome, obstructive gallbladder disease, SLE, nephrotic syndrome, poorly controlled diabetes, severe hypertension, alcohol abuse, drug‐induced hyperlipidaemia, dietary treatment for obesity, heart, brain, kidney, liver diseases, MI within 3 months, cerebrovascular disorder, statin hypersensitivity, pregnancy potential and lactation, those participants considered inappropriate by the investigator Cerivastatin 0.05 mg/d baseline TC: 7.30 mmol/L (282 mg/dL) Cerivastatin 0.15 mg/d baseline TC: 7.79 mmol/L (301 mg/dL) Cerivastatin 0.15 mg/d baseline LDL‐C: 5.66 mmol/L (219 mg/dL) Cerivastatin 0.3 mg/d baseline TC: 7.98 mmol/L (309 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 5.76 mmol/L (223 mg/dL) |
|
| Interventions | Cerivastatin 0.15 mg/d evening dosing Cerivastatin 0.3 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 4‐12 weeks of blood TC and LDL‐C | |
| Source of funding | Unknown | |
| Notes | HDL‐C and TG data were excluded because the given data and the calculated values differed by > 10% | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis for the cerivastatin 0.15 mg/d group for TC [(40‐39)/40]*100 = 2.5% participants were not included in the efficacy analysis for the cerivastatin 0.3 mg/d group |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | [(33‐28)/33]*100 = 15.2% participants were not included in the efficacy analysis for the cerivastatin 0.15 mg/d group [(40‐39)/40]*100 = 2.5% participants were not included in the efficacy analysis for the cerivastatin 0.3 mg/d group [(73‐67)/73]*100 = 8.2% participants were not included in the efficacy analysis for combined doses |
| Incomplete outcome data (attrition bias) HDL cholesterol | High risk | Not included in the efficacy analysis because the given values and the calculated values differed by > 10% |
| Incomplete outcome data (attrition bias) Triglycerides | High risk | Not included in the efficacy analysis because the given values and the calculated values differed by > 10% |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Arakawa 1997.
| Methods | 4‐week washout period 12‐week before‐and‐after study |
|
| Participants | 58 men and women with type IIa and IIb hyperlipidaemia aged 20‐64 years old TC ≥ 260 mg/dL (6.72 mmol/L) TG ≤ 400 mg/dL (4.52 mmol/L), moderate hypertension, mild diabetes mellitus, obesity, cholelithiasis Exclusion criteria: secondary hyperlipidaemia Cerivastatin 0.15 mg/d baseline TC: 7.92 mmol/L (306 mg/dL) Cerivastatin 0.15 mg/d baseline LDL‐C: 5.74 mmol/L (222 mg/dL) |
|
| Interventions | Cerivastatin 0.15 mg/d evening dosing | |
| Outcomes | Percentage change from baseline at 12 weeks of blood TC and LDL‐C | |
| Source of funding | Unknown | |
| Notes | HDL‐C and TG data were excluded because the given data and the calculated values differed by > 10% | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | High risk | [(108‐56)/108]*100 = 48.1% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | High risk | [(108‐53)/108]*100 = 50.9% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | High risk | Not included in the efficacy analysis because the given values and the calculated values differed by > 10% |
| Incomplete outcome data (attrition bias) Triglycerides | High risk | Not included in the efficacy analysis because the given values and the calculated values differed by > 10% |
| Selective reporting (reporting bias) | Low risk | LD‐C outcome was reported |
| Other bias | Unclear risk | Source of funding was not reported |
Balletshofer 2005.
| Methods | 4‐week run‐in period 12‐week RCT |
|
| Participants | 58 men and women with type 2 diabetes aged 35‐75 years old LDL‐C 100 mg/dL (2.57 mmol/L)‐190 mg/dL (4.91 mmol/L) Exclusion criteria: patients under therapy with heparin, known thrombophilia, neoplasia, unstable angina, known intolerance or hypersensitivity to statins, major organ system failure, transplantation, organic brain syndrome, clinically manifest hypothyroidism, pregnancy or lactation period, women of childbearing potential not using adequate methods of contraception, myopathy (CK > 3 times ULN), impaired hepatic function (SGOT or SGPT > 2 times ULN), SCr > 2 mg/dL, intake of drugs influencing endothelial function if a dose change (inc. start or withdrawal) occurred within the last 12 weeks prior to the screening visit, intake of statins within the last 8 weeks prior to the screening visit, drug or alcohol abuse, reversal of a normal sleep/wake cycle (e.g. patients on nightshift) Placebo baseline TC: 6.0 mmol/L (232.1 mg/dL) Placebo baseline LDL‐C: 3.9 mmol/L (150.8 mg/dL) Placebo baseline HDL‐C: 1.1 mmol/L (42.5 mg/dL) Placebo baseline TG: 2.3 mmol/L (203.7 mg/dL) Cerivastatin 0.2 mg/d baseline TC: 5.9 mmol/L (228.2 mg/dL) Cerivastatin 0.2 mg/d baseline LDL‐C: 3.8 mmol/L (146.9 mg/dL) Cerivastatin 0.2 mg/d baseline HDL‐C: 1.3 mmol/L (50.3 mg/dL) Cerivastatin 0.2 mg/d baseline TG: 2.0 mmol/L (177.1 mg/dL) Cerivastatin 0.8 mg/d baseline TC: 5.4 mmol/L (208.8 mg/dL) Cerivastatin 0.8 mg/d baseline LDL‐C: 3.5 mmol/L (135.3 mg/dL) Cerivastatin 0.8 mg/d baseline HDL‐C: 1.2 mmol/L (46.4 mg/dL) Cerivastatin 0.8 mg/d baseline TG: 1.8 mmol/L (159.4 mg/dL) |
|
| Interventions | Placebo Cerivastatin 0.2 mg/d Cerivastatin 0.8 mg/d |
|
| Outcomes | Percentage change from baseline at 12 weeks of plasma TC, LDL‐C, HDL‐C, TG and WDAEs | |
| Source of funding | Unknown | |
| Notes | SDs were imputed by the method of Furukawa 2006 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of sufficient blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | LDL‐C was measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | Unclear risk | Blinding method was not described |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Battula 2000.
| Methods | 4‐week run‐in period 4‐week before‐and‐after study |
|
| Participants | 8 men and women with type 2 diabetes age 40‐70 years LDL‐C > 213 mg/dL (5.52 mmol/L), TG > 133 mg/dL (1.5 mmol/L) Exclusion criteria: evidence of hepatic or renal disease, unstable hypertension, proliferative retinopathy, unstable angina, or recent MI (within 3 months) or patients with FH or those using any lipid‐lowering agent in the previous 3 months Cerivastatin 0.3 mg/d baseline TC: 6.2 mmol/L (239.8 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 4.5 mmol/L (174 mg/dL) Cerivastatin 0.3 mg/d baseline HDL‐C: 1.1 mmol/L (42.5 mg/dL) Cerivastatin 0.3 mg/d baseline TG: 2.4 mmol/L (212.6 mg/dL) |
|
| Interventions | Cerivastatin 0.3 mg/d | |
| Outcomes | Percentage change from baseline at 4 weeks of plasma TC, LDL‐C, HDL‐C and TG | |
| Source of funding | Bayer UK | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | LDL‐C was measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Trial supported by Bayer UK, data may support bias for cerivastatin |
Bayer 1992.
| Methods | No washout required because participants were not receiving any lipid‐lowering medications within 4 weeks of the trial; 6 months for probucol 1‐month, RCT |
|
| Participants | 36 men and women with primary hypercholesterolaemia aged 18‐75 years LDL‐C ≥ 130 mg/dL (3.36 mmol/L) and TG ≤ 350 mg/dL (4.02 mmol/L) Exclusion criteria: homozygous FH, CVD or cerebrovascular disease, TIA, uncontrolled hypertension, diabetes mellitus, clinically significant eye disease, malignancy, psychosis, chronic liver disease, prior exposure to cerivastatin nor HMG CoA reductase inhibitor hypersensitivity, drug or alcohol abuse, child bearing potential, patients who had taken another investigational drug within 30 d of trial, concurrent use of corticosteroids, erythromycin, oral anticoagulants, hypoglycaemic agents, digoxin, androgens, immunosuppressants or cimetidine and significant laboratory abnormalities Placebo baseline TC: 6.90 mmol/L (267 mg/dL) Placebo baseline LDL‐C: 4.84 mmol/L (187 mg/dL) Placebo baseline HDL‐C: 1.22 mmol/L (47 mg/dL) Placebo baseline TG: 1.89 mmol/L (167 mg/dL) Cerivastatin 0.3 mg/d baseline TC: 6.90 mmol/L (267 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 4.68 mmol/L (181 mg/dL) Cerivastatin 0.3 mg/d baseline HDL‐C: 1.29 mmol/L (50 mg/dL) Cerivastatin 0.3 mg/d baseline TG: 1.99 mmol/L (176 mg/dL) |
|
| Interventions | Placebo evening dosing Cerivastatin 0.3 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 4 weeks of blood TC, LDL‐C, HDL‐C, TG, and WDAEs | |
| Source of funding | Bayer | |
| Notes | SDs were imputed by the method of Furukawa 2006 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind: placebo and cerivastatin tablets were all identical in appearance |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | Unclear risk | Method of blinding for WDAEs was not reported |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis for the placebo group [(24‐23)/24]*100 = 4.2% participants were not included in the efficacy analysis for the cerivastatin 0.3 mg/d group |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis for the placebo group [(24‐23)/24]*100 = 4.2% participants were not included in the efficacy analysis for the cerivastatin 0.3 mg/d group |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis for the placebo group [(24‐23)/24]*100 = 4.2% participants were not included in the efficacy analysis for the cerivastatin 0.3 mg/d group |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis for the placebo group [(24‐23)/24]*100 = 4.2% participants were not included in the efficacy analysis for the cerivastatin 0.3 mg/d group |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Bayer funded the trial, data may support bias for cerivastatin |
Bayer 1994.
| Methods | 10‐week run‐in washout period 24‐week, RCT |
|
| Participants | 785 men and women with primary hypercholesterolaemia both familial and non‐familial LDL‐C ≥ 160 mg/dL (4.12 mmol/L) and TG ≤ 350 mg/dL (4.02 mmol/L) Exclusion criteria: MI, unstable angina, stroke, TIA, uncontrolled hypertension within the last 3 months, CABG or PTCA within the previous 6 months diabetes mellitus, clinically significant eye disease, muscular or neuromuscular disease, CK level ≥ 3 times the ULN, significant infection, malignancy, or psychosis, GI disorders that could affect drug absorption, chronic liver disease, serum lambda amylase > 1.2 times ULN, current use of immunosuppressants, corticosteroids, oral anticoagulants, androgens, or erythromycin, concomitant treatment with other hypolipidaemic drugs, probucol within 6 months, child bearing potential, drug or alcohol abuse, HMG CoA reductase inhibitor hypersensitivity, reversal of normal sleep/wake cycle and treatment with an investigational drug within 30 d of randomisation Placebo baseline LDL‐C: 5.15 mmol/L (199 mg/dL) Cerivastatin 0.05 mg/d baseline LDL‐C: 5.20 mmol/L (189 mg/dL) Cerivastatin 0.1 mg/d baseline LDL‐C: 5.07 mmol/L (196 mg/dL) Cerivastatin 0.2 mg/d baseline LDL‐C: 5.08 mmol/L (196 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 4.99 mmol/L (193 mg/dL) |
|
| Interventions | Placebo evening dosing Cerivastatin 0.05 mg/d evening dosing Cerivastatin 0.1 mg/d evening dosing Cerivastatin 0.2 mg/d evening dosing Cerivastatin 0.3 mg/d evening dosing Lovastatin 40 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 4‐12 weeks of plasma LDL‐C | |
| Source of funding | Bayer | |
| Notes | Lovastatin 40 mg/d group was not analysed SDs were imputed by the method of Furukawa 2006 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind lipid parameter measurements unlikely influenced by lack of sufficient blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | LDL‐C was measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No WDAE data reported for the 3‐12‐week period |
| Incomplete outcome data (attrition bias) Total cholesterol | High risk | No 3‐12‐week data |
| Incomplete outcome data (attrition bias) LDL cholesterol | Unclear risk | [(154‐139)/154]*100 = 9.7% participants were not included in the efficacy analysis for the placebo group [(159‐145)/159]*100 = 8.8% participants were not included in the efficacy analysis for the cerivastatin 0.05 mg/d group [(157‐136)/157]*100 = 13.4% participants were not included in the efficacy analysis for the cerivastatin 0.1 mg/d group [(159‐138)/159]*100 = 13.2% participants were not included in the efficacy analysis for the cerivastatin 0.2 mg/d group [(156‐137)/156]*100 = 12.2% participants were not included in the efficacy analysis for the cerivastatin 0.3 mg/d group [(785‐695)/785]*100 = 11.5 % participants were not included in the efficacy analysis for all doses |
| Incomplete outcome data (attrition bias) HDL cholesterol | High risk | No 3‐12‐week data |
| Incomplete outcome data (attrition bias) Triglycerides | High risk | No 3‐12‐week data |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Bayer funded the trial, data may support bias for cerivastatin |
Bayer 1995.
| Methods | 10‐week run‐in washout period 6‐week RCT |
|
| Participants | 55 men and women with heterozygous FH aged 21‐75 years LDL‐C > 5.0 mmol/L (193 mg/dL), TG ≤ 4.02 mmol/L (356 mg/dL), hypertensive patients on a stable dose of beta‐blockers or diuretics and patients on thyroid replacement therapy TSH was ≤ 7.5mu/L with T4 within normal range, postmenopausal women on HRT Exclusion criteria: homozygous FH, MI, stroke, PTCA or coronary bypass surgery within the previous 6 months, congestive heart failure grade 3 or 4, significant cardiac arrhythmias, uncontrolled diabetes mellitus, endocrine disease, significant renal disease, respiratory disease, hepatic dysfunction, significant eye disease, neuromuscular disease, infections that may interfere with the trial, cancer within 5 years, women of child bearing potential, drug or alcohol abuse, night shift workers, mental disorders, HIV positive, pancreatitis, use of immunosuppressants, corticosteroids, androgens, erythromycin, niacin, psyllium, fish oil and excess bran and therapy with any other investigational drug within 30 d prior to the screening visit Placebo baseline TC: 9.00 mmol/L (348 mg/dL) Placebo baseline LDL‐C: 7.24 mmol/L (280 mg/dL) Placebo baseline HDL‐C: 1.19 mmol/L (46 mg/dL) Placebo baseline TG: 1.24 mmol/L (110 mg/dL) Cerivastatin 0.2 mg/d baseline TC: 9.08 mmol/L (351 mg/dL) Cerivastatin 0.2 mg/d baseline LDL‐C: 7.27 mmol/L (281 mg/dL) Cerivastatin 0.2 mg/d baseline HDL‐C: 1.16 mmol/L (45 mg/dL) Cerivastatin 0.2 mg/d baseline TG: 1.39 mmol/L (123 mg/dL) Cerivastatin 0.3 mg/d baseline TC: 9.59 mmol/L (371 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 7.63 mmol/L (295 mg/dL) Cerivastatin 0.3 mg/d baseline HDL‐C: 1.19 mmol/L (46 mg/dL) Cerivastatin 0.3 mg/d baseline TG: 1.68 mmol/L (149 mg/dL) |
|
| Interventions | Placebo evening dosing Cerivastatin 0.2 mg/d evening dosing Cerivastatin 0.3 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 6 weeks of plasma TC, LDL‐C, HDL‐C, TG and WDAEs | |
| Source of funding | Bayer | |
| Notes | SDs were imputed by the method of Furukawa 2006 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind fashion lipid parameter measurements unlikely influenced by lack of sufficient blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | Low risk | No participants withdrew due to adverse events |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Bayer funded the trial, data may support bias for cerivastatin |
Bayer 1997.
| Methods | 10‐week run‐in washout period 8‐week RCT |
|
| Participants | 1170 men and women with primary hypercholesterolaemia aged 21‐75 years; participants with no atherosclerotic disease LDL‐C > 160 mg/dL (4.14 mmol/L) participants with atherosclerotic disease LDL‐C > 130 mg/dL (3.36 mmol/L) participants with ≥ 2 cardiovascular risk factors plasma HDL‐C < 35 mg/dL (0.9 mmol/L) and LDL‐C > 130 mg/dL (3.36 mmol/L) plasma TG < 400 mg/dL (4.52 mmol/L) Exclusion criteria: MI, stroke, TIA, unstable angina, PTCA or coronary bypass surgery within the previous 6 months, for hypertensive patients change in diuretic of beta‐blocker therapy, diabetes mellitus, endocrine diseases except hypothyroidism, significant renal disease, respiratory disease, active liver disease, HMG CoA reductase hypersensitivity, unstable eye disease, cancer except skin cancer, or psychosis, women of child bearing potential, drug or alcohol abuse, night shift workers, GI tract absorption impairment, significant laboratory abnormalities, use of immunosuppressants, corticosteroids, androgens, erythromycin, probucol within 6 months, H2 blockers other than cimetidine, azole antifungals, niacin, psyllium, fish oil and excess bran, treatment with cerivastatin for > 10 d within 6 months of visit 1 and therapy with any other investigational drug within 30 d prior to the screening visit Placebo baseline TC: 6.90 mmol/L (267 mg/dL) Placebo baseline LDL‐C: 4.77 mmol/L (184 mg/dL) Placebo baseline HDL‐C: 1.24 mmol/L (48 mg/dL) Placebo baseline TG: 1.89 mmol/L (167 mg/dL) Cerivastatin 0.4 mg/d baseline TC: 7.14 mmol/L (276 mg/dL) Cerivastatin 0.4 mg/d baseline LDL‐C: 4.91 mmol/L (190 mg/dL) Cerivastatin 0.4 mg/d baseline HDL‐C: 1.27 mmol/L (49 mg/dL) Cerivastatin 0.4 mg/d baseline TG: 2.04 mmol/L (181 mg/dL) Cerivastatin 0.8 mg/d baseline TC: 7.11 mmol/L (275 mg/dL) Cerivastatin 0.8 mg/d baseline LDL‐C: 4.89 mmol/L (189 mg/dL) Cerivastatin 0.8 mg/d baseline HDL‐C: 1.27 mmol/L (49 mg/dL) Cerivastatin 0.8 mg/d baseline TG: 2.11 mmol/L (187 mg/dL) |
|
| Interventions | Placebo Cerivastatin 0.4 mg/d Cerivastatin 0.8 mg/d |
|
| Outcomes | Percentage change from baseline at 8 weeks of serum TC, LDL‐C, HDL‐C, TG | |
| Source of funding | Bayer | |
| Notes | SDs were imputed by the method of Furukawa 2006 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind lipid parameter measurements unlikely influenced by lack of adequate blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No WDAEs were reported for the 8‐week period of interest |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | [(199‐197)/199]*100 = 1% participants were not included in the efficacy analysis for the placebo group [(195‐193)/195]*100 = 1% participants were not included in the efficacy analysis for the cerivastatin 0.4 mg/d group [(776‐770)/776]*100 = 0.8% participants were not included in the efficacy analysis for the cerivastatin 0.8 mg/d group |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | [(199‐197)/199]*100 = 1% participants were not included in the efficacy analysis for the placebo group [(195‐193)/195]*100 = 1% participants were not included in the efficacy analysis for the cerivastatin 0.4 mg/d group [(776‐770)/776]*100 = 0.8% participants were not included in the efficacy analysis for the cerivastatin 0.8 mg/d group |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | [(199‐197)/199]*100 = 1% participants were not included in the efficacy analysis for the placebo group [(195‐193)/195]*100 = 1% participants were not included in the efficacy analysis for the cerivastatin 0.4 mg/d group [(776‐770)/776]*100 = 0.8% participants were not included in the efficacy analysis for the cerivastatin 0.8 mg/d group |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | [(199‐197)/199]*100 = 1% participants were not included in the efficacy analysis for the placebo group [(195‐193)/195]*100 = 1% participants were not included in the efficacy analysis for the cerivastatin 0.4 mg/d group [(776‐770)/776]*100 = 0.8% participants were not included in the efficacy analysis for the cerivastatin 0.8 mg/d group |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Bayer funded the trial, data may support bias for cerivastatin |
Bayer 1998.
| Methods | 10‐week baseline washout period 6 months for probucol 24‐week RCT |
|
| Participants | 908 men and women aged 18‐75 with documented primary hypercholesterolaemia Exclusion criteria: weight > 140% ideal body weight, homozygous FH, cancer except squamous or basal cell skin cancer or psychosis, women of childbearing potential, night shift workers, drug or alcohol abuse, MI, stroke, TIA, unstable angina, CABG and PTCA within 6 months of trial, uncontrolled hypertension within 3 months of trial, patients with hypertension who had a change in diuretic or beta blocker therapy within 3 months of trial, diabetes mellitus or other endocrine disorders, significant eye disease, active hepatic disease, GI disorders that could affect drug absorption, HMG CoA reductase inhibitor hypersensitivity, renal dysfunction, current use of corticosteroids, erythromycin, all macrolide antibiotics, rifampin, androgens, immunosuppressants, ketoconazole and itraconazole, treatment with cerivastatin within 6 months of trial and therapy with another investigational product within 30 d Placebo baseline TC: 7.10 mmol/L (275 mg/dL) Placebo baseline LDL‐C: 4.94 mmol/L (191 mg/dL) Placebo baseline HDL‐C: 1.25 mmol/L (48 mg/dL) Placebo baseline TG: 1.99 mmol/L (176 mg/dL) Cerivastatin 0.3 mg/d baseline TC: 7.15 mmol/L (276 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 4.96 mmol/L (192 mg/dL) Cerivastatin 0.3 mg/d baseline HDL‐C: 1.26 mmol/L (49 mg/dL) Cerivastatin 0.3 mg/d baseline TG: 2.11 mmol/L (187 mg/dL) Cerivastatin 0.4 mg/d baseline TC: 7.05 mmol/L (273 mg/dL) Cerivastatin 0.4 mg/d baseline LDL‐C: 4.84 mmol/L (187 mg/dL) Cerivastatin 0.4 mg/d baseline HDL‐C: 1.27 mmol/L (49 mg/dL) Cerivastatin 0.4 mg/d baseline TG: 2.08 mmol/L (184 mg/dL) |
|
| Interventions | Placebo Cerivastatin 0.3 mg/d Cerivastatin 0.4 mg/d |
|
| Outcomes | Percentage change from baseline at 8 weeks of blood TC, LDL‐C, HDL‐C and TG | |
| Source of funding | Bayer | |
| Notes | SDs were imputed by the method of Furukawa 2006 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind lipid parameter measurements unlikely influenced by lack of adequate blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No WDAE data for the 8‐week period |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Bayer funded the trial, data may support bias for cerivastatin |
Betteridge 1999.
| Methods | 10‐week run‐in period 12‐week RCT |
|
| Participants | 978 men and women aged 21‐75 years with uncomplicated primary hypercholesterolaemia LDL‐C ≥ 160 mg/dL (4.12 mmol/L) and TG ≤ 350 mg/dL (4.02 mmol/L) Exclusion criteria: homozygous FH, MI, stroke or bypass surgery within the last 6 months, diabetes mellitus, significant renal hepatic muscular or neuromuscular or eye abnormalities Placebo baseline TC: 7.75 mmol/L (300 mg/dL) Placebo baseline LDL‐C: 5.64 mmol/L (218 mg/dL) Placebo baseline HDL‐C: 1.39 mmol/L (54 mg/dL) Placebo baseline TG: 1.57 mmol/L (139 mg/dL) Cerivastatin 0.025 mg/d baseline TC: 7.71 mmol/L (298 mg/dL) Cerivastatin 0.025 mg/d baseline LDL‐C: 5.63 mmol/L (218 mg/dL) Cerivastatin 0.025 mg/d baseline HDL‐C: 1.35 mmol/L (52 mg/dL) Cerivastatin 0.025 mg/d baseline TG: 1.59 mmol/L (141 mg/dL) Cerivastatin 0.05 mg/d baseline TC: 7.62 mmol/L (295 mg/dL) Cerivastatin 0.05 mg/d baseline LDL‐C: 5.55 mmol/L (215 mg/dL) Cerivastatin 0.05 mg/d baseline HDL‐C: 1.38 mmol/L (53 mg/dL) Cerivastatin 0.05 mg/d baseline TG: 1.53 mmol/L (136 mg/dL) Cerivastatin 0.1 mg/d baseline TC: 7.56 mmol/L (292 mg/dL) Cerivastatin 0.1 mg/d baseline LDL‐C: 5.49 mmol/L (212 mg/dL) Cerivastatin 0.1 mg/d baseline HDL‐C: 1.33 mmol/L (51 mg/dL) Cerivastatin 0.1 mg/d baseline TG: 1.61 mmol/L (143 mg/dL) Cerivastatin 0.2 mg/d baseline TC: 7.64 mmol/L (295 mg/dL) Cerivastatin 0.2 mg/d baseline LDL‐C: 5.58 mmol/L (216 mg/dL) Cerivastatin 0.2 mg/d baseline HDL‐C: 1.34 mmol/L (52 mg/dL) Cerivastatin 0.2 mg/d baseline TG: 1.58 mmol/L (140 mg/dL) |
|
| Interventions | Placebo evening dosing Cerivastatin 0.025 mg/d evening dosing Cerivastatin 0.05 mg/d evening dosing Cerivastatin 0.1 mg/d evening dosing Cerivastatin 0.2 mg/d evening dosing Simvastatin 20 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 12 weeks of plasma TC, LDL‐C, HDL‐C, TG and WDAEs | |
| Source of funding | Bayer AG | |
| Notes | Simvastatin 20 mg/d group was not analysed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind cerivastatin and placebo tablets were all identical in appearance |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | Unclear risk | Blinding method was not described |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Bayer AG funded the trial, data may support bias for cerivastatin |
Chen 2001.
| Methods | No washout required because no participants were receiving lipid‐lowering agents before the trial 4‐week before‐and‐after study |
|
| Participants | 20 elderly men and women with hyperlipidaemia TC > 240 mg/dL (6.24 mmol/L) LDL‐C > 160 mg/dL (4.16 mmol/L) for those without coronary risk factors TC > 220 mg/dL (5.72 mmol/L) LDL‐C > 140 mg/dL (3.64 mmol/L) for those with coronary risk factors TC > 200 mg/dL (5.20 mmol/L) LDL‐C > 120 mg/dL (3.20 mmol/L) for those with coronary heart disease or atherosclerotic disease Exclusion criteria: serious cardiovascular and cerebrovascular disease within 6 months, severe trauma or major surgery, severe liver and kidney dysfunction, taking other drugs that affect on blood lipids, nephrotic syndrome, hypothyroidism, uncontrolled diabetes and pregnant and lactating women Cerivastatin 0.3 mg/d baseline TC: 6.65 mmol/L (257 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 3.64 mmol/L (141 mg/dL) Cerivastatin 0.3 mg/d baseline HDL‐C: 1.06 mmol/L (41 mg/dL) Cerivastatin 0.3 mg/d baseline TG: 2.15 mmol/L (190 mg/dL) |
|
| Interventions | Cerivastatin 0.3 mg/d evening dosing Simvastatin 20 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 4 weeks of serum TC, LDL‐C, HDL‐C and TG | |
| Source of funding | Unknown | |
| Notes | Simvastatin 20 mg/d group was not analysed SDs were imputed by the method of Furukawa 2006 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | LDL‐C was measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Davignon 1998.
| Methods | 10‐week run‐in/washout period 8‐week RCT |
|
| Participants | 349 men and women aged from 21‐75 years old with primary hypercholesterolaemia (LDL‐C ≥ 190 mg/dL; TG ≤ 350 mg/dL, or LDL‐C ≥ 160 mg/dL; TG ≤ 350 mg/dL in the presence of additional CAD risk factors Exclusion criteria: none reported No baseline data provided |
|
| Interventions | Placebo evening dosing Cerivastatin 0.3 mg/d evening dosing Cerivastatin 0.4 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 8 weeks of plasma LDL‐C, HDL‐C and TG | |
| Source of funding | Unknown | |
| Notes | Only European phase IIB higher‐dose trial was analysed. US phase shows 6‐ and 12‐month data, and Canadian phase shows 32‐week data only SDs were imputed by the method of Furukawa 2006 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of sufficient blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | WDAEs data were not reported |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Dujovne 2000.
| Methods | 6‐8‐week run‐in phase 8‐week before‐and‐after study |
|
| Participants | 479 men and women with type IIa or IIb hypercholesterolaemia age 18‐75 years old TG < 350 mg/dL (3.95 mmol/L) Exclusion criteria: history of MI, angina, stroke, recent TIA, recent coronary revascularisation, uncontrolled hypertension or hypothyroidism, type 2 diabetes, chronic liver disease, renal dysfunction, alcohol or drug abuse, CPK values are 3 x ULN Cerivastatin 0.3 mg/d baseline TC: 6.7 mmol/L (259 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 4.49 mmol/L (174 mg/dL) Cerivastatin 0.3 mg/d baseline HDL‐C: 1.3 mmol/L (50 mg/dL) Cerivastatin 0.3 mg/d baseline TG: 1.99 mmol/L (176 mg/dL) Cerivastatin 0.4 mg/d baseline TC: 6.75 mmol/L (261 mg/dL) Cerivastatin 0.4 mg/d baseline LDL‐C: 4.55 mmol/L (176 mg/dL) Cerivastatin 0.4 mg/d baseline HDL‐C: 1.3 mmol/L (50 mg/dL) Cerivastatin 0.4 mg/d baseline TG: 1.98 mmol/L (175 mg/dL) |
|
| Interventions | Cerivastatin 0.3 mg/d evening dosing Cerivastatin 0.4 mg/d evening dosing Pravastatin 20 mg/d evening dosing Pravastatin 40 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 8 weeks of serum TC, LDL‐C, HDL‐C and TG | |
| Source of funding | Bayer | |
| Notes | Pravastatin 20 mg/d and pravastatin 40 mg/d groups were not analysed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Bayer funded the trial, data may support bias for cerivastatin |
Goto 1996a.
| Methods | 4‐week washout period 12‐week before‐and‐after study |
|
| Participants | 163 men and women with type IIa and IIb hyperlipidaemia aged 20‐70 years old were randomised to receive cerivastatin 162 men and women with type IIa and IIb hyperlipidaemia aged 20‐70 years old were randomised to receive pravastatin TC ≥ 220 mg/dL (5.69 mmol/L), moderate hypertension, mild diabetes, obese, cholelithiasis Exclusion criteria: secondary hyperlipidaemia, hypothyroidism, Cushings syndrome, obstructive gallbladder disease, SLE, uncontrolled diabetes, severe hypertension, alcoholism, various hormonal agents, drugs that interfere with lipid metabolism, severe brain disease, liver and kidney dysfunction, diet therapy for obesity, pancreatic disease, cerebrovascular disease, MI within 3 months of trial, drug hypersensitivity, potential for pregnancy and lactation Cerivastatin 0.15 mg/d baseline TC: 7.12 mmol/L (275 mg/dL) Cerivastatin 0.15 mg/d baseline LDL‐C: 4.98 mmol/L (193 mg/dL) |
|
| Interventions | Cerivastatin 0.15 mg/d evening dosing Pravastatin 10 mg/d |
|
| Outcomes | Percentage change from baseline at 8 weeks of blood TC and LDL‐C | |
| Source of funding | Unknown | |
| Notes | Pravastatin 10 mg/d was not analysed HDL‐C and TG data were excluded because the given data and the calculated values differed by > 10% |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | High risk | [(163‐137)/163]*100 = 16.0% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | High risk | [(163‐130)/163]*100 = 20.2% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | High risk | Excluded because the given data and the calculated values differed by > 10% |
| Incomplete outcome data (attrition bias) Triglycerides | High risk | Excluded because the given data and the calculated values differed by > 10% |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Goto 1996b.
| Methods | 4‐week placebo run‐in washout period 12‐week before‐and‐after study |
|
| Participants | 294 men and women with type IIa, type IIb and type IV hypercholesterolaemia aged 20‐70 years old with moderate hypertension, mild diabetes mellitus, obese, cholelithiasis, TC ≥ 220 mg/dL (5.69 mmol/L) and TG ≤ 400 mg/dL (4.52 mmol/L) Exclusion criteria: hypothyroidism, Cushings syndrome, obstructive gallbladder disease, SLE, nephrosis, poorly controlled diabetes, severe hypertension, alcohol abuse, drug induced hyperlipidaemia, diet therapy for obesity, secondary hyperlipidaemia, severe heart, brain, kidney, liver diseases, MI within 3 months, cerebrovascular disorder, statin hypersensitivity, pregnancy potential and lactation Cerivastatin 0.05 mg/d baseline TC: 7.30 mmol/L (282 mg/dL) Cerivastatin 0.05 mg/d baseline LDL‐C: 5.22 mmol/L (202 mg/dL) Cerivastatin 0.05 mg/d baseline HDL‐C: 1.31 mmol/L (51 mg/dL) Cerivastatin 0.05 mg/d baseline TG: 1.86 mmol/L (165 mg/dL) Cerivastatin 0.10 mg/d baseline TC: 7.21 mmol/L (279 mg/dL) Cerivastatin 0.10 mg/d baseline LDL‐C: 5.20 mmol/L (201 mg/dL) Cerivastatin 0.10 mg/d baseline HDL‐C: 1.23 mmol/L (48 mg/dL) Cerivastatin 0.10 mg/d baseline TG: 1.94 mmol/L (172 mg/dL) Cerivastatin 0.15 mg/d baseline TC: 7.13 mmol/L (276 mg/dL) Cerivastatin 0.15 mg/d baseline LDL‐C: 5.04 mmol/L (195 mg/dL) Cerivastatin 0.15 mg/d baseline HDL‐C: 1.39 mmol/L (54 mg/dL) Cerivastatin 0.15 mg/d baseline TG: 1.60 mmol/L (142 mg/dL) Cerivastatin 0.20 mg/d baseline TC: 7.25 mmol/L (280 mg/dL) Cerivastatin 0.20 mg/d baseline LDL‐C: 5.11 mmol/L (198 mg/dL) Cerivastatin 0.20 mg/d baseline HDL‐C: 1.40 mmol/L (54 mg/dL) Cerivastatin 0.20 mg/d baseline TG: 1.84 mmol/L (163 mg/dL) |
|
| Interventions | Cerivastatin 0.05 mg/d evening dosing Cerivastatin 0.1 mg/d evening dosing Cerivastatin 0.15 mg/d evening dosing Cerivastatin 0.2 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 4‐12 weeks of blood TC, LDL‐C, HDL‐C and TG | |
| Source of funding | Unknown | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | [(71‐65)/71]*100 = 8.5% participants were not included in the efficacy analysis for the cerivastatin 0.05 mg/d group for total cholesterol [(79‐73)/79]*100 = 7.6% participants were not included in the efficacy analysis for the cerivastatin 0.1 mg/d group for total cholesterol [(70‐67)/70]*100 = 4.3% participants were not included in the efficacy analysis for the cerivastatin 0.15 mg/d group for total cholesterol [(74‐70)/74]*100 = 5.4% participants were not included in the efficacy analysis for the cerivastatin 0.2 mg/d group for total cholesterol |
| Incomplete outcome data (attrition bias) LDL cholesterol | High risk | [(71‐61)/71]*100 = 14.1% participants were not included in the efficacy analysis for the cerivastatin 0.05 mg/d group [(79‐66)/79]*100 = 16.5% participants were not included in the efficacy analysis for the cerivastatin 0.1 mg/d group [(70‐63)/70]*100 = 10.0% participants were not included in the efficacy analysis for the cerivastatin 0.15 mg/d group [(74‐64)/74]*100 = 13.5% participants were not included in the efficacy analysis for the cerivastatin 0.2 mg/d group [(294‐254)/294]*100 = 13.6% participants were not included in the efficacy analysis for all doses |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | [(71‐65)/71]*100 = 8.5% participants were not included in the efficacy analysis for the cerivastatin 0.05 mg/d group [(79‐73)/79]*100 = 7.6% participants were not included in the efficacy analysis for the cerivastatin 0.1 mg/d group [(70‐67)/70]*100 = 4.3% participants were not included in the efficacy analysis for the cerivastatin 0.15 mg/d group [(74‐70)/74]*100 = 5.4% participants were not included in the efficacy analysis for the cerivastatin 0.2 mg/d group |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | [(71‐63)/71]*100 = 11.3% participants were not included in the efficacy analysis for the cerivastatin 0.05 mg/d group [(79‐70)/79]*100 = 8.9% participants were not included in the efficacy analysis for the cerivastatin 0.1 mg/d group [(70‐65)/70]*100 = 7.1% participants were not included in the efficacy analysis for the cerivastatin 0.15 mg/d group [(74‐69)/74]*100 = 6.8% participants were not included in the efficacy analysis for the cerivastatin 0.2 mg/d group |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Hanefeld 1999.
| Methods | 4‐week optional washout /dietary stabilisation 6‐week placebo run‐in period 8‐week RCT |
|
| Participants | 349 men and women with hypercholesterolaemia aged 18‐75 years old LDL‐C level of ≥ 190 mg/dL (≥ 4.90 mmol/L) or ≥ 160 mg/dL (≥ 4.12 mmol/L) if associated with one or more of the following risk factors: male sex; family history of premature coronary heart disease (definite MI or sudden death < 55 years of age in a parent or sibling); current cigarette smoker (> 10 cigarettes/d); hypertension; coronary disease; low HDL‐C concentration < 35 mg/dL (< 0.9 mmol/L), history of definite cerebrovascular or occlusive PVD, obesity, TG ≤ 350 mg/dL (≤ 3.99 mmol/L) Exclusion criteria: established contraindications for statin intake; unstable CVD including severe hypertension, MI within the past 6 months, cerebrovascular events, congestive heart failure (NYHA class 3 or 4), cardiac arrhythmias; diabetes mellitus, hypothyroidism; HIV‐positive; malignancy; hepatic or renal disorders; history of pancreatitis; known muscular or neuromuscular disease; GI disease (e.g. Crohri's disease) which could result in impaired absorption of the trial drug; clinically significant ophthalmic abnormalities; nightshift workers (reversal of normal sleep/wake cycle); concomitant medication affecting lipid levels or known to interact with statins including immunosuppressants, erythromycin, vitamin tablets containing > 50 mg/d niacin, regular therapeutic use of psyllium, fish oil or excess bran for lipid‐lowering purposes, corticosteroids (including inhalation formulation) and androgens history of hypersensitivity to HMG CoA reductase inhibitors; CK > 3 times ULN and/or hepatic transaminases (ALT, AST) > 1.5 times ULN; treatment with any other investigational drug within 30 d prior to screening Placebo baseline TC: 8.12 mmol/L (314 mg/dL) Placebo baseline LDL‐C: 5.99 mmol/L (232 mg/dL) Placebo baseline HDL‐C: 1.42 mmol/L (55 mg/dL) Placebo baseline TG: 1.56 mmol/L (138 mg/dL) Cerivastatin 0.3 mg/d baseline TC: 7.92 mmol/L (306 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 5.77 mmol/L (223 mg/dL) Cerivastatin 0.3 mg/d baseline HDL‐C: 1.42 mmol/L (55 mg/dL) Cerivastatin 0.3 mg/d baseline TG: 1.61 mmol/L (143 mg/dL) Cerivastatin 0.4 mg/d baseline TC: 7.77 mmol/L (300 mg/dL) Cerivastatin 0.4 mg/d baseline LDL‐C: 5.64 mmol/L (218 mg/dL) Cerivastatin 0.4 mg/d baseline HDL‐C: 1.39 mmol/L (54 mg/dL) Cerivastatin 0.4 mg/d baseline TG: 1.63 mmol/L (144 mg/dL) |
|
| Interventions | Placebo Cerivastatin 0.3 mg/d Cerivastatin 0.4 mg/d |
|
| Outcomes | Percentage change from baseline at 8 weeks of serum TC, LDL‐C, HDL‐C, TG and WDAEs | |
| Source of funding | Unknown | |
| Notes | SDs were imputed by the method of Furukawa 2006 except for LDL‐C | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind tablets were identical in appearance |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | Unclear risk | Blinding method was not described |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Bayer helped with the statistical analysis |
Hunninghake 1998.
| Methods | 10‐week dietary run‐in period 4‐week RCT |
|
| Participants | 647 men and women with primary hypercholesterolaemia age 21‐65 years were randomised to cerivastatin 33 men and women with primary hypercholesterolaemia age 21‐65 years were randomised to lovastatin 31 men and women with primary hypercholesterolaemia age 21‐65 years were randomised to simvastatin LDL‐C > 160 mg/dL (4.14 mmol/L) or < 250 mg/dL (6.465 mmol/L), TG < 350 mg/dL (3.95 mmol/L) Exclusion criteria: cardiovascular or cerebrovascular disease, diabetes mellitus, renal or respiratory disease, homozygous FH, clinically significant eye disease, muscular or neuromuscular disease, child bearing potential, CK > 3 x ULN significant infection, malignancy or psychosis, GI tract disease, liver dysfunction, pancreatitis, use of corticosteroids, erythromycin, oral anticoagulants, beta‐blockers, diuretics, digitalis, H2 blockers, androgens, progestins, estrogens or other lipid‐lowering drugs, drug or alcohol use, history of statin hypersensitivity, patients with reversal of the normal sleep/wake cycle, anyone treated with an investigational drug within 30 d of randomisation Placebo baseline TC: 7.40 mmol/L (286 mg/dL) Placebo baseline LDL‐C: 5.21 mmol/L (201 mg/dL) Placebo baseline HDL‐C: 1.38 mmol/L (53 mg/dL) Placebo baseline TG: 1.76 mmol/L (156 mg/dL) Cerivastatin 0.025 mg/d baseline TC: 7.20 mmol/L (278 mg/dL) Cerivastatin 0.025 mg/d baseline LDL‐C: 5.12 mmol/L (198 mg/dL) Cerivastatin 0.025 mg/d baseline HDL‐C: 1.25 mmol/L (48 mg/dL) Cerivastatin 0.025 mg/d baseline TG: 1.82 mmol/L (161 mg/dL) Cerivastatin 0.05 mg/d baseline TC: 7.22 mmol/L (279 mg/dL) Cerivastatin 0.05 mg/d baseline LDL‐C: 5.08 mmol/L (196 mg/dL) Cerivastatin 0.05 mg/d baseline HDL‐C: 1.22 mmol/L (47 mg/dL) Cerivastatin 0.05 mg/d baseline TG: 2.01 mmol/L (178 mg/dL) Cerivastatin 0.1 mg/d baseline TC: 7.27 mmol/L (281 mg/dL) Cerivastatin 0.1 mg/d baseline LDL‐C: 5.15 mmol/L (199 mg/dL) Cerivastatin 0.1 mg/d baseline HDL‐C: 1.21 mmol/L (47 mg/dL) Cerivastatin 0.1 mg/d baseline TG: 2.01 mmol/L (178 mg/dL) Cerivastatin 0.2 mg/d baseline TC: 7.20 mmol/L (278 mg/dL) Cerivastatin 0.2 mg/d baseline LDL‐C: 5.11 mmol/L (198 mg/dL) Cerivastatin 0.2 mg/d baseline HDL‐C: 1.25 mmol/L (48 mg/dL) Cerivastatin 0.2 mg/d baseline TG: 1.84 mmol/L (163 mg/dL) |
|
| Interventions | Placebo Cerivastatin 0.025 mg/d Cerivastatin 0.05 mg/d Cerivastatin 0.1 mg/d Cerivastatin 0.2 mg/d; 0.1 mg twice a day or 0.2 mg evening dosing Lovastatin 40 mg/d Simvastatin 20 mg/d |
|
| Outcomes | Percentage change from baseline at 4 weeks of plasma TC, LDL‐C, HDL‐C, TG and WDAEs | |
| Source of funding | Bayer | |
| Notes | Lovastatin 40 mg/d and simvastatin 20 mg/d groups were not analysed SDs were imputed by the method of Furukawa 2006 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind Trial drug was packaged in brown glass bottles; cerivastatin and placebo tablets were all identical in appearance |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | Unclear risk | Blinding method was not described |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | [(115‐110)/115]*100 = 4.3% participants were not included in the efficacy analysis for the placebo group [(68‐65)/68]*100 = 4.4% participants were not included in the efficacy analysis for the 0.025 mg/d group [(69‐65)/68]*100 = 5.9% participants were not included in the efficacy analysis for the 0.05 mg/d group [(68‐64)/68]*100 = 5.9% participants were not included in the efficacy analysis for the 0.1 mg/d group [(340‐325)/340]*100 = 4.4% participants were not included in the efficacy analysis for the 0.2 mg/d group |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | [(115‐110)/115]*100 = 4.3% participants were not included in the efficacy analysis for the placebo group [(68‐65)/68]*100 = 4.4% participants were not included in the efficacy analysis for the 0.025 mg/d group [(69‐65)/68]*100 = 5.9% participants were not included in the efficacy analysis for the 0.05 mg/d group [(68‐64)/68]*100 = 5.9% participants were not included in the efficacy analysis for the 0.1 mg/d group [(340‐325)/340]*100 = 4.4% participants were not included in the efficacy analysis for the 0.2 mg/d group |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | [(115‐110)/115]*100 = 4.3% participants were not included in the efficacy analysis for the placebo group [(68‐65)/68]*100 = 4.4% participants were not included in the efficacy analysis for the 0.025 mg/d group [(69‐65)/68]*100 = 5.9% participants were not included in the efficacy analysis for the 0.05 mg/d group [(68‐64)/68]*100 = 5.9% participants were not included in the efficacy analysis for the 0.1 mg/d group [(340‐325)/340]*100 = 4.4% participants were not included in the efficacy analysis for the 0.2 mg/d group |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | [(115‐110)/115]*100 = 4.3% participants were not included in the efficacy analysis for the placebo group [(68‐65)/68]*100 = 4.4% participants were not included in the efficacy analysis for the 0.025 mg/d group [(69‐65)/68]*100 = 5.9% participants were not included in the efficacy analysis for the 0.05 mg/d group [(68‐64)/68]*100 = 5.9% participants were not included in the efficacy analysis for the 0.1 mg/d group [(340‐325)/340]*100 = 4.4% participants were not included in the efficacy analysis for the 0.2 mg/d group |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Bayer funded the trial, data may support bias for cerivastatin |
Hunninghake 2001.
| Methods | 4‐week washout period 6‐week before‐and‐after study |
|
| Participants | 107 men and women with primary hypercholesterolaemia age 18‐80 years old LDL‐C ≥ 160 mg/dL (4.14 mmol/L) and TG ≤ 400 mg/dL (4.52 mmol/L) Exclusion criteria: pregnancy or breast feeding, hyperlipoproteinaemia secondary to uncontrolled primary hypothyroidism, nephrotic syndrome or renal dysfunction; diabetes mellitus type 1 or uncontrolled diabetes mellitus type 2; active liver disease or hepatic dysfunction; CK levels > 3 x ULN; uncontrolled hypertension (systolic BP ≥140 mm Hg or diastolic BP ≥ 90 mm Hg); current or recent history of alcohol abuse; unreliability as a trial participant based on the investigator’s prior knowledge of the patient, such as inability or unwillingness to adhere to a lipid‐lowering diet; participation in another clinical trial within the 30‐d period before consideration for entry into this trial; known hypersensitivity to HMG CoA reductase inhibitors; MI, coronary angioplasty, CABG, or severe or unstable angina within 3 months before screening; significant abnormalities that the investigator believed could compromise the patient’s safety in participating in the trial; and use of any drugs known to affect lipid levels, immunosuppressive agents, drugs associated with rhabdomyolysis in combination with HMG CoA reductase inhibitors (e.g. cyclosporine and erythromycin), or mibefradil dihydrochloride Cerivastatin 0.3 mg/d baseline TC: 7.4 mmol/L (286 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 5.22 mmol/L (202 mg/dL) Cerivastatin 0.3 mg/d baseline HDL‐C: 1.28 mmol/L (49.5 mg/dL) Cerivastatin 0.3 mg/d baseline TG: 1.96 mmol/L (174 mg/dL) |
|
| Interventions | Cerivastatin 0.3 mg/d evening dosing Atorvastatin 10 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 6 weeks of blood TC, LDL‐C, HDL‐C, and TG | |
| Source of funding | Pfizer Inc | |
| Notes | Atorvastatin group was not analysed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | [(107‐106)/107]*100 = 0.9% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | [(107‐106)/107]*100 = 0.9% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | [(107‐106)/107]*100 = 0.9% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | [(107‐106)/107]*100 = 0.9% participants were not included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Pfizer Inc funded the trial data may support bias against cerivastatin |
Insull 2000.
| Methods | 10‐week dietary washout period 8‐week RCT |
|
| Participants | 1170 men and women with hypercholesterolaemia age 18‐75 years LDL‐C was to be ≥ 160 mg/dL for those without definite atherosclerotic disease and < cardiovascular risk factors or ≥ 130 mg/dL for those with definite atherosclerotic disease or ≥ 2 cardiovascular risk factors Exclusion criteria: MI, unstable angina, stroke, TIA, or uncontrolled hypertension within 3 months of enrolment; a coronary revascularisation procedure within 6 months of enrolment; a change in diuretic or β‐blocker therapy for hypertension within 2 months of enrolment; diabetes mellitus; other endocrine disorders (except for hypothyroidism on stable replacement therapy with TSH 140% of ideal; homozygous FH; history of malignancy (except squamous or basal cell skin cancer), psychosis or GI disorders that might impair absorption of trial medications; night‐shift work that reverses the normal sleep/wake cycle; pregnancy/ breast‐feeding/childbearing potential; ingestion of other lipid‐lowering substances (including fish oil, psyllium, bran or niacin > 100 mg 4 times daily); and hypersensitivity to HMG CoA reductase inhibitors, concomitant use of corticosteroids (except low‐dose inhaled agents for asthma), macrolide antibiotics, azole antifungals (including ketoconazole and itraconazole), androgens, oral anticoagulants, rifampin, immunosuppressants or H2‐antagonists (except cimetidine); use of another investigational product within 30 d of enrolment; or use of cerivastatin (for > 10 d) or probucol within 6 months of enrolment Placebo baseline TC: 6.88 mmol/L (266 mg/dL) Placebo baseline LDL‐C: 4.74 mmol/L (183 mg/dL) Placebo baseline HDL‐C: 1.25 mmol/L (48 mg/dL) Placebo baseline TG: 1.95 mmol/L (173 mg/dL) Cerivastatin 0.4 mg/d baseline TC: 7.15 mmol/L (276 mg/dL) Cerivastatin 0.4 mg/d baseline LDL‐C: 4.94 mmol/L (191 mg/dL) Cerivastatin 0.4 mg/d baseline HDL‐C: 1.26 mmol/L (49 mg/dL) Cerivastatin 0.4 mg/d baseline TG: 2.07 mmol/L (183 mg/dL) Cerivastatin 0.8 mg/d baseline TC: 7.10 mmol/L (275 mg/dL) Cerivastatin 0.8 mg/d baseline LDL‐C: 4.89 mmol/L (189 mg/dL) Cerivastatin 0.8 mg/d baseline HDL‐C: 1.27 mmol/L (49 mg/dL) Cerivastatin 0.8 mg/d baseline TG: 2.06 mmol/L (182 mg/dL) |
|
| Interventions | Placebo evening dosing Cerivastatin 0.4 mg/d evening dosing Cerivastatin 0.8 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 8 weeks of plasma TC, LDL‐C, HDL‐C, TG and WDAEs | |
| Source of funding | Bayer and SmithKline Beecham | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind lipid parameter measurements unlikely influenced by lack of sufficient blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | Unclear risk | Blinding method was not described |
| Incomplete outcome data (attrition bias) Total cholesterol | High risk | [(199 ‐177)/199]*100 = 11.1% participants were not included in the efficacy analysis for the placebo group [(195 ‐164)/195]*100 = 15.9% participants were not included in the efficacy analysis for the cerivastatin 0.4 mg/d group [(776 ‐ 656)/776]*100 = 15.5% participants were not included in the efficacy analysis for the cerivastatin 0.8 mg/d group |
| Incomplete outcome data (attrition bias) LDL cholesterol | High risk | [(199 ‐177)/199]*100 = 11.1% participants were not included in the efficacy analysis for the placebo group [(195 ‐164)/195]*100 = 15.9% participants were not included in the efficacy analysis for the cerivastatin 0.4 mg/d group [(776 ‐ 656)/776]*100 = 15.5% participants were not included in the efficacy analysis for the cerivastatin 0.8 mg/d group |
| Incomplete outcome data (attrition bias) HDL cholesterol | High risk | [(199 ‐177)/199]*100 = 11.1% participants were not included in the efficacy analysis for the placebo group [(195 ‐164)/195]*100 = 15.9% participants were not included in the efficacy analysis for the cerivastatin 0.4 mg/d group [(776 ‐ 656)/776]*100 = 15.5% participants were not included in the efficacy analysis for the cerivastatin 0.8 mg/d group |
| Incomplete outcome data (attrition bias) Triglycerides | High risk | [(199 ‐177)/199]*100 = 11.1% participants were not included in the efficacy analysis for the placebo group [(195 ‐164)/195]*100 = 15.9% participants were not included in the efficacy analysis for the cerivastatin 0.4 mg/d group [(776 ‐ 656)/776]*100 = 15.5% participants were not included in the efficacy analysis for the cerivastatin 0.8 mg/d group |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Bayer and SmithKline Beecham funded the trial |
Isaacsohn 1998.
| Methods | 10‐week washout period 12‐week before‐and‐after study |
|
| Participants | 200 men and women aged 18‐75 years with documented primary hypercholesterolaemia LDL‐C ≥ 157.5 mg/dL (4.07 mmol/L) or ≥ 130 mg/dL (3.36 mmol/L) with documented CAD of ≥ 2 cardiovascular risk factors plasma TG ≤ 400 mg/dL (4.52 mmol/L) have a food rating score ≤ 15 Exclusion criteria: clinically active CVD, hypertension with alterations in diuretic or beta blocker therapy within 2 months of entry, uncontrolled diabetes mellitus or other endocrine abnormalities and uncontrolled hypothyroidism, ophthalmic abnormalities, cancer other than basil cell or squamous cell carcinoma, psychosis, hepatic dysfunction, weight 140% ideal body weight, statin hypersensitivity, significant GI tract disorders, child bearing potential homozygous FH, renal dysfunction, current use of other medications that would interfere with the trial, treatment with other hypolipidaemic drugs within 10 weeks of entry, drug or alcohol abuse, night shift workers, therapy with another investigational product within 30 d, other medical conditions which might interfere with the trial Cerivastatin 0.2 mg/d baseline TC: 6.94 mmol/L (268 mg/dL) Cerivastatin 0.2 mg/d baseline LDL‐C: 4.74 mmol/L (183 mg/dL) Cerivastatin 0.2 mg/d baseline HDL‐C: 1.24 mmol/L (48 mg/dL) Cerivastatin 0.2 mg/d baseline TG: 2.03 mmol/L (180 mg/dL) |
|
| Interventions | Fluvastatin 20 mg/d for 0‐6 weeks evening dosing Fluvastatin 40 mg/d for 6‐12 weeks evening dosing Cerivastatin 0.2 mg/d for 0‐6 weeks evening dosing Cerivastatin 0.3 mg/d for 6‐12 weeks evening dosing |
|
| Outcomes | Percentage change from baseline at 6 weeks of plasma TC, LDL‐C, HDL‐C, and TG | |
| Source of funding | Novartis | |
| Notes | Fluvastatin 20 mg/d for 0‐6 weeks Fluvastatin 40 mg/d for 6‐12 weeks Cerivastatin 0.3 mg/d for 6‐12 weeks Fluvastatin groups were not included in the efficacy analysis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | High risk | [(200‐174)/200]*100 = 13% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | High risk | [(200‐174)/200]*100 = 13% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | High risk | [(200‐174)/200]*100 = 13% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | High risk | [(200‐174)/200]*100 = 13% participants were not included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Novartis funded the trial, data may support bias against cerivastatin |
Kajiyama 1996.
| Methods | 4‐week run‐in stabilisation period 12‐week before‐and‐after study |
|
| Participants | 21 men and women with type IIa or IIb hyperlipidaemia aged 20‐70 years old TC ≥ 220 mg/dL (5.7 mmol/L) and TG < 400 mg/dL (4.52 mmol/L) Exclusion criteria: severe organ disease, history of stroke, MI history, child bearing potential, statin hypersensitivity Cerivastatin 0.2 mg/d baseline TC: 7.029 mmol/L (272 mg/dL) Cerivastatin 0.2 mg/d baseline LDL‐C: 5.0168 mmol/L (194 mg/dL) Cerivastatin 0.2 mg/d baseline HDL‐C: 1.3395 mmol/L (52 mg/dL) Cerivastatin 0.2 mg/d baseline TG: 1.47 mmol/L (130 mg/dL) |
|
| Interventions | Cerivastatin 0.2 mg/d evening dosing | |
| Outcomes | Percentage change from baseline at 4‐12 weeks of serum TC, LDL‐C, HDL‐C, and TG | |
| Source of funding | Unknown | |
| Notes | SDs were imputed by the method of Furukawa 2006 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | [(21 ‐ 20)/21]*100 = 4.8% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | [(21 ‐ 20)/21]*100 = 4.8% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | [(21 ‐ 20)/21]*100 = 4.8% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | [(21 ‐ 20)/21]*100 = 4.8% participants were not included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Kim 1999.
| Methods | 4‐week placebo run‐in period 6‐week RCT Allocations via block randomisation in groups of 6 |
|
| Participants | 32 men and women with primary hypercholesterolaemia aged 18‐75 years. Average LDL‐C measured during diet‐stabilisation and placebo run‐in stage had to be > 160 mg/dL (4.14 mmol/L) with definite personal history of coronary heart disease and each measurement had to be not > ± 12% from the average (> 130 mg/dL (3.36 mmol/L) for participants with > 2 risk factors for CAD or CVD) fasting plasma TG below 350 mg/dL (3.95 mmol/L) for all participants, consent received from all participants During period A, included participants had to have diet satisfied by dietician (i.e. meeting dietician recommendations). Compliance rate to placebo during period A was between 80%‐120%; 26 did not finish placebo run‐in period A; 47 participants were randomised Exclusion criteria: women of childbearing age but not on IUD or OCP; pregnant or breastfeeding; participants with history of MI, unstable angina, stroke, TIA, and uncontrolled hypertension within 3 months of starting trial; CABG or PCI within 6 months of starting trial. Regardless of whether they're on treatment or not, if fasting glucose is > 140 mg/dL or having other endocrine disease, or someone with systolic BP > 180 mmHg or diastolic BP > 110 mmHg; clinically known to have cataract or malignant tumour or psychiatric condition or chronic seizures or “homogenous hypercholesterolaemia” (homogenous family history?). BMI > 30, SCr > 2 mg/dL, AST/ALT/amylase > 1.5 ULN or CK > 3, chronic or acute infection, if require regular care visits or compliance expected to be affected, if alcohol or drug abuse in medical history (> 14 drinks/week for alcohol use), if occupational shift work requiring working overnight, bodybuilders or weightlifters, people on other cholesterol medications are excluded; if they stop the statin 4 weeks before or fibrates 8 weeks before or probucol 6 months before, then they can be included in the trial; people with sensitivities to statins, anyone involved with any other trial within 30 d of starting this trial Placebo baseline TC: 6.42 mmol/L (248 mg/dL) Placebo baseline LDL‐C: 4.36 mmol/L (169 mg/dL) Placebo baseline HDL‐C: 1.26 mmol/L (49 mg/dL) Placebo baseline TG: 1.77 mmol/L (157 mg/dL) Cerivastatin 0.1 mg/d baseline TC: 6.97 mmol/L (270 mg/dL) Cerivastatin 0.1 mg/d baseline LDL‐C: 4.86 mmol/L (188 mg/dL) Cerivastatin 0.1 mg/d baseline HDL‐C: 1.19 mmol/L (46 mg/dL) Cerivastatin 0.1 mg/d baseline TG: 2.01 mmol/L (178 mg/dL) Cerivastatin 0.3 mg/d baseline TC: 6.77 mmol/L (262 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 4.59 mmol/L (177 mg/dL) Cerivastatin 0.3 mg/d baseline HDL‐C: 1.31 mmol/L (51 mg/dL) Cerivastatin 0.3 mg/d baseline TG: 1.90 mmol/L (168 mg/dL) |
|
| Interventions | Placebo evening dosing Cerivastatin 0.1 mg/d evening dosing Cerivastatin 0.3 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 4‐6 weeks of blood TC, LDL‐C, HDL‐C, and TG | |
| Source of funding | Unknown | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of sufficient blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | WDAEs were not reported |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding was not reported |
Krone 1999.
| Methods | 10‐week run‐in period 18‐week before‐and‐after study |
|
| Participants | 225 men and women with primary hypercholesterolaemia aged between 18‐75 years old LDL‐C ≥ 160 mg/dL (4.14 mmol/L) or LDL‐C ≥ 130 mg/dL (3.36 mmol/L) in patients with proven CHD or ≥ 2 risk factors Exclusion criteria: MI, unstable angina, stroke, TIA or uncontrolled hypertension within the previous 3 months; diabetes mellitus or other endocrine problems obesity, homozygous FH, hepatic or renal dysfunction and concomitant treatment with other hypolipidaemic drugs Cerivastatin 0.1 mg/d baseline TC: 6.96 mmol/L (269 mg/dL) Cerivastatin 0.1 mg/d baseline LDL‐C: 4.97 mmol/L (192 mg/dL) Cerivastatin 0.1 mg/d baseline HDL‐C: 1.36 mmol/L (53 mg/dL) Cerivastatin 0.1 mg/d baseline TG: 1.39 mmol/L (123 mg/dL) |
|
| Interventions | Cerivastatin 0.1 mg/d for 0‐6 weeks evening dosing Cerivastatin 0.2 mg/d for 6‐12 weeks evening dosing Cerivastatin 0.4 mg/d for 12‐18 weeks evening dosing Pravastatin 10 mg/d for 0‐6 weeks evening dosing Pravastatin 20 mg/d for 6‐12 weeks evening dosing Pravastatin 40 mg/d for 12‐18 weeks evening dosing |
|
| Outcomes | Percentage change from baseline at 6 weeks of blood TC, LDL‐C, HDL‐C, and TG | |
| Source of funding | Unknown | |
| Notes | Cerivastatin 0.2 mg/d for 6‐12 weeks Cerivastatin 0.4 mg/d for 12‐18 weeks Pravastatin 10 mg/d for 0‐6 weeks Pravastatin 20 mg/d for 6‐12 weeks Pravastatin 40 mg/d for 12‐18 weeks Pravastatin groups were not analysed |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | [(225 ‐ 219)/225]*100 = 2.7% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | [(225 ‐ 219)/225]*100 = 2.7% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | [(225 ‐ 219)/225]*100 = 2.7% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | [(225 ‐ 219)/225]*100 = 2.7% participants were not included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Lankin 2002.
| Methods | 12‐week washout/8‐week dietary period 12‐week before‐and‐after controlled study |
|
| Participants | 32 men with chronic CHD and primary type IIa hyperlipoproteinaemia 16 received cerivastatin 16 received probucol TC > 7.4 mmol/L (286.2 mg/dL) Exclusion criteria: none reported Cerivastatin 0.4 mg/d baseline LDL‐C: 5.27 mmol/L (203.8 mg/dL) |
|
| Interventions | Cerivastatin 0.4 mg/d Probucol 250 mg/d |
|
| Outcomes | Percentage change from baseline at 4, 8 and 12 weeks of plasma LDL‐C | |
| Source of funding | Russian Foundation for Basic Research | |
| Notes | Probucol 250 mg/d data not analysed SDs were imputed by the method of Furukawa 2006 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | LDL‐C was measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Low risk | Work supported by Russian Foundation for Basic Research |
Ma 2000.
| Methods | 10‐week washout run‐in period 8‐week before‐and‐after study |
|
| Participants | 174 men and women with combined (type IIb) dyslipidaemia age 18‐80 years old received cerivastatin LDL‐C ≥ 130 mg/dL (3.4 mmol/L) or ≥ 100 mg/dL (2.6 mmol/L) if the patient had ≥ 2 of the following CAD risk factors: male > 45 years old; female > 55 years old, or postmenopausal and not on HRT; family history of premature CAD in a 1st degree relative; smoking ≥ 1 cigarettes/d; hypertension (systolic BP > 140 mmHg, diastolic BP > 90 mmHg, or antihypertensive therapy); TG ≥ 200 mg/dL (2.8 mmol/L) to ≤ 800 mg/dL (9 mmol/L) Exclusion criteria: women of childbearing potential who were not using adequate contraception, who were breast feeding, or who had a positive pregnancy test; unstable weight (variation of > 3 kg during the last 4 weeks of the run‐in); type 1 diabetes; type 2 diabetes (WHO classification) unless this was controlled by diet and/or oral treatment and/or fixed‐dose insulin (HbA1c ≥ 8.5% for 90 d before enrolment and fasting glucose < 10 mmol/L at randomisation); severe hypertension (diastolic BP ≥ 110 mmHg and/or systolic BP ≥ 180 mmHg on ≥ 3 consecutive occasions); MI, cerebrovascular accident, PTCA or CABG within the 3 months prior to the enrolment visit; congestive heart failure (NYHA class III or IV); significant arrhythmia or conduction disturbances; hypothyroidism (TSH > 7.5 mU/L or TSH > 5 mU/L and ≤ 7.5 mU/L plus total T4 < 7 ug/dL); any malignant tumour requiring treatment in the past 5 years (except squamous or basal cell skin cancer); known significant renal impairment (SCr ≥ 2.0 mg/dL) or known nephrotic syndrome; known liver disease and/or elevated serum transaminase (AST, ALT) > 1.5 x ULN and/or hepatosplenomegaly; known muscular or neuromuscular disease and/or serum CK > 3 x ULN if not otherwise explainable (e.g. Crohn's disease) which could result in impaired absorption of the trial drug; drug or alcohol abuse; concomitant treatment with immunosuppressants, rifampin, macrolide antibiotics, H2 blockers, azoles, corticosteroids or androgens; concomitant therapy for hyperlipidaemia; treatment with any other investigational drug (including either atorvastatin or cerivastatin) within 30 d prior to enrolment. Cerivastatin 0.4 mg/d baseline TC: 7.01 mmol/L (270.9 mg/dL) Cerivastatin 0.4 mg/d baseline LDL‐C: 4.35 mmol/L (168.4 mg/dL) Cerivastatin 0.4 mg/d baseline HDL‐C: 0.96 mmol/L (37.2 mg/dL) Cerivastatin 0.8 mg/d baseline TC: 6.65 mmol/L (257.0 mg/dL) Cerivastatin 0.8 mg/d baseline LDL‐C: 4.02 mmol/L (155.6 mg/dL) Cerivastatin 0.8 mg/d baseline HDL‐C: 1.04 mmol/L (40.3 mg/dL) |
|
| Interventions | Cerivastatin 0.4 mg/d evening dosing Cerivastatin 0.8 mg/d evening dosing Atorvastatin 10 mg/d evening dosing Atorvastatin 20 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 8 weeks of blood TC, LDL‐C and HDL‐C | |
| Source of funding | Bayer | |
| Notes | Atorvastatin 10 mg/d and atorvastatin 20 mg/d groups were not analysed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Bayer funded the trial, data may support bias for cerivastatin |
Mabuchi 1998.
| Methods | 4‐week dietary period 4‐week before‐and‐after study |
|
| Participants | 20 men and women with heterozygous FH aged from 33‐70 years old received cerivastatin Exclusion criteria: none reported Cerivastatin 0.2 mg/d baseline TC: 8.78 mmol/L (339.6 mg/dL) Cerivastatin 0.2 mg/d baseline LDL‐C: 6.92 mmol/L (267.6 mg/dL) Cerivastatin 0.2 mg/d baseline HDL‐C: 0.99 mmol/L (38.3 mg/dL) |
|
| Interventions | Cerivastatin 0.2 mg/d Cerivastatin 0.2 mg/d + cholestyramine 8g/d Cerivastatin 0.2 mg/d + procubol 1g/d |
|
| Outcomes | Percentage change from baseline at 4 weeks of plasma TC, LDL‐C and HDL‐C | |
| Source of funding | Unknown | |
| Notes | Cerivastatin 0.2 mg/d associated with cholestyramine 8 g/d or procubol 1 g/d started after 4 weeks, data were not analysed. SDs were imputed by the method of Furukawa 2006 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Matsuo 2005.
| Methods | 1‐month run‐in period 4‐week before‐and‐after study |
|
| Participants | 10 men and women with hypercholesterolaemia mean age 63 years TC ≥ 220 mg/dL (5.70 mmol/L) Exclusion criteria: none reported Cerivastatin 0.15 mg/d baseline TC: 6.47 mmol/L (250 mg/dL) Cerivastatin 0.15 mg/d baseline LDL‐C: 4.30 mmol/L (166 mg/dL) Cerivastatin 0.15 mg/d baseline HDL‐C: 1.29 mmol/L (50 mg/dL) Cerivastatin 0.15 mg/d baseline TG: 1.81 mmol/L (160 mg/dL) |
|
| Interventions | Cerivastatin 0.15 mg/d | |
| Outcomes | Percentage change from baseline at 4 weeks of serum TC, LDL‐C, HDL‐C and TG | |
| Source of funding | Unknown | |
| Notes | SDs were imputed by the method of Furukawa 2006 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Matsuzawa 1996.
| Methods | 4‐week run‐in period 12‐week before‐and‐after study |
|
| Participants | 117 men and women with type IIa or IIb hyperlipidaemia aged 23‐80 years old TC > 220 mg/dL (5.69 mmol/L) on 2 occasions Exclusion criteria: secondary hypercholesterolaemia, people with severe disabilities, statin allergy, hyperthyroidism, Cushing syndrome, obstructive bile duct disease, lupus, nephrosis, uncontrolled diabetes, severe hypertension, alcoholism, hormone therapy and dieting obese patients Cerivastatin 0.15 mg/d baseline TC: 6.94 mmol/L (268 mg/dL) Cerivastatin 0.15 mg/d baseline LDL‐C: 4.83 mmol/L (187 mg/dL) Cerivastatin 0.15 mg/d baseline HDL‐C: 1.29 mmol/L (50 mg/dL) Cerivastatin 0.15 mg/d baseline TG: 1.88 mmol/L (167 mg/dL) |
|
| Interventions | Cerivastatin 0.15 mg/d evening dosing | |
| Outcomes | Percentage change from baseline at 4‐12 weeks of blood TC and LDL‐C | |
| Source of funding | Unknown | |
| Notes | HDL‐C and TG data were excluded because the given data and the calculated values differed by > 10% | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | [(117‐111)/117]*100 = 5.1% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | [(117‐107)/117]*100 = 8.5% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | High risk | Excluded because the given data and the calculated values differed by > 10% |
| Incomplete outcome data (attrition bias) Triglycerides | High risk | Excluded because the given data and the calculated values differed by > 10% |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Nakamura 2001.
| Methods | 3‐month placebo run‐in period 6‐month RCT |
|
| Participants | 60 men and women with type 2 diabetes, microalbuminuria and dyslipidaemia mean age 56.5 years Exclusion criteria: none reported Placebo baseline TC: 6.67 mmol/L (258 mg/dL) Placebo baseline LDL‐C: 5.43 mmol/L (210 mg/dL) Placebo baseline HDL‐C: 0.62 mmol/L (24 mg/dL) Placebo baseline TG: 2.23 mmol/L (198 mg/dL) Cerivastatin 0.15 mg/d baseline TC: 6.77 mmol/L (262 mg/dL) Cerivastatin 0.15 mg/d baseline LDL‐C: 5.38 mmol/L (208 mg/dL) Cerivastatin 0.15 mg/d baseline HDL‐C: 0.57 mmol/L (22 mg/dL) Cerivastatin 0.15 mg/d baseline TG: 2.28 mmol/L (202 mg/dL) |
|
| Interventions | Placebo Cerivastatin 0.15 mg/d |
|
| Outcomes | Percentage change from baseline at 12 weeks of plasma TC, LDL‐C, HDL‐C, TG and WDAEs | |
| Source of funding | Unknown | |
| Notes | 3‐6 month treatment period was not included in the efficacy and safety analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of sufficient blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | Low risk | No adverse events during the trial |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy and safety analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy and safety analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy and safety analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy and safety analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Nakaya 1996.
| Methods | 4‐week run‐in period 8‐week before‐and‐after study |
|
| Participants | 82 men and women aged ≥ 20 years with type IIa and IIb hyperlipidaemia with informed consent TC ≥ 220 mg/dL (5.69 mmol/L) and TG ≤ 400 mg/dL (4.52 mmol/L) Exclusion criteria: secondary hyperlipidaemia, hypothyroidism, Cushing's syndrome, SLE, nephrosis, alcohol abuse, drug‐induced hyperlipidaemia, diet therapy for obesity, poorly controlled diabetes mellitus Cerivastatin 0.1 mg/d baseline TC: 7.19 mmol/L (278 mg/dL) Cerivastatin 0.1 mg/d baseline LDL‐C: 5.10 mmol/L (197 mg/dL) Cerivastatin 0.1 mg/d baseline HDL‐C: 1.42 mmol/L (54.9 mg/dL) Cerivastatin 0.1 mg/d baseline TG: 1.69 mmol/L (150 mg/dL) |
|
| Interventions | Cerivastatin 0.1 mg/d for 0‐8 weeks evening dosing Cerivastatin 0.15 mg/d for 8‐24 weeks evening dosing Cerivastatin 0.15‐0.3 mg/d for 24‐60 weeks evening dosing |
|
| Outcomes | Percentage change from baseline at 4‐8 weeks of plasma TC, LDL‐C, HDL‐C and TG | |
| Source of funding | Unknown | |
| Notes | Cerivastatin 0.15 mg/d for 8‐24 weeks Cerivastatin 0.15‐0.3 mg/d for 24‐60‐week periods were not included in the efficacy analysis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | [(82‐78)/82]*100 = 4.9% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | High risk | [(82‐70)/82]*100 = 14.6% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | [(82‐78)/82]*100 = 4.9% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Unclear risk | [(82‐74)/82]*100 = 9.8% participants were not included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Nakaya 1997.
| Methods | 4‐week washout period 8‐week before‐and‐after study |
|
| Participants | 70 men and women with type IIa and IIb hyperlipidaemia 2 groups: young aged 20‐64 years; old aged > 65 years TC ≥ 220 mg/dL (2.48 mmol/L) TG < 400 mg/dL (4.52 mmol/L) Exclusion criteria: secondary hyperlipidaemia, hypothyroidism, Cushing's syndrome, gallbladder disease, SLE, nephrosis, alcohol and drug abuse Cerivastatin 0.1 mg/d baseline TC: 7.31 mmol/L (263 mg/dL) Cerivastatin 0.1 mg/d baseline LDL‐C: 4.7 mmol/L (182 mg/dL) Cerivastatin 0.1 mg/d baseline HDL‐C: 1.42 mmol/L (55 mg/dL) Cerivastatin 0.1 mg/d baseline TG: 1.66 mmol/L (147 mg/dL) |
|
| Interventions | Cerivastatin 0.1 mg/d for 8 weeks evening dosing Cerivastatin 0.15 mg/d titrated dose from 8 to 60 weeks |
|
| Outcomes | Percentage change from baseline at 4 weeks of serum TC, LDL‐C, HDL‐C and TG | |
| Source of funding | Unknown | |
| Notes | Cerivastatin 0.15 mg/d titrated dose was not included in the efficacy analysis because there was no washout between the 0.1 mg/d dose and the 0.15 mg/d dose | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | [(70‐67)/70]*100 = 4.3% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Unclear risk | [(70‐62)/70]*100 = 11.4% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | [(70‐67)/70]*100 = 4.3% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Unclear risk | [(70‐63)/70]*100 = 10% participants were not included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding was not reported |
Puccetti 2001.
| Methods | 6‐week dietary period 6‐week before‐and‐after study |
|
| Participants | 25 men and women with primary hypercholesterolaemia mean age 48.2 received cerivastatin TC 6.93 mmol/L (268 mg/dL), HDL‐C 1.25 mmol/L (48.3 mg/dL) and TG 1.15 mmol/L (102 mg/dL) Exclusion criteria: history of cardiovascular events or current hypertension, diabetes, or liver, renal, thyroid, infectious, immunological or malignant diseases. None of the participants had a personal or family history of deep vein thrombosis or a tendency to bleed, an none of them were taking hypolipidaemic, antiplatelet, anticoagulant or profibrinolytic drugs Cerivastatin 0.2 mg/d baseline TC: 6.62 mmol/L (256 mg/dL) Cerivastatin 0.2 mg/d baseline LDL‐C: 4.89 mmol/L (189.1 mg/dL) Cerivastatin 0.2 mg/d baseline HDL‐C: 1.24 mmol/L (47.9 mg/dL) Cerivastatin 0.2 mg/d baseline TG: 1.05 mmol/L (93 mg/dL) |
|
| Interventions | Cerivastatin 0.2 mg/d Atorvastatin 10 mg/d Simvastatin 20 mg/d Pravastatin 20 mg/d Fluvastatin 20 mg/d |
|
| Outcomes | Percentage change from baseline at 6 weeks of plasma TC, LDL‐C, HDL‐C and TG | |
| Source of funding | University of Siena | |
| Notes | Atorvastatin 10 mg/d, simvastatin 20 mg/d, pravastatin 20 mg/d and fluvastatin 20 mg/d data were not analysed. SDs were imputed by the method of Furukawa 2006 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Low risk | Work funded by University of Siena |
Ridker 2001.
| Methods | 10‐week run‐in period 8‐week before‐and‐after study |
|
| Participants | 785 men and women with primary hypercholesterolaemia age 24‐76 years Exclusion criteria: acute vascular events within the past 3 months, revascularisation within the past 6 months, diabetes, or active liver disease Cerivastatin 0.4 mg/d baseline LDL‐C: 4.98 mmol/L (193 mg/dL) Cerivastatin 0.4 mg/d baseline HDL‐C: 1.26 mmol/L (49 mg/dL) Cerivastatin 0.8 mg/d baseline LDL‐C: 4.97 mmol/L (192 mg/dL) Cerivastatin 0.8 mg/d baseline HDL‐C: 1.27 mmol/L (49 mg/dL) |
|
| Interventions | Cerivastatin 0.4 mg/d evening dosing Cerivastatin 0.8 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 4 weeks of plasma LDL‐C and HDL‐C | |
| Source of funding | Government grant and Bayer Inc | |
| Notes | SDs were imputed by the method of Furukawa 2006 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Trial was funded by a government grant and Bayer Inc |
Rubinstein 1999.
| Methods | 10‐week dietary/washout period 12‐week RCT |
|
| Participants | 265 men and women with type 2 diabetes age 30‐80 years LDL‐C > 3.35 mmol/L (130 mg/dL), TG < 4.56 mmol/L (400 mg/dL) Exclusion criteria: patients with any significant medical or surgical events within 6 months before enrolment, were known to have renal impairment (creatinine > 177 mmol/L (2.0 mg/dL)), hepatic impairment (AST or ALT > 2 x ULN) or any other serious disease, suffered from any condition that necessitated insulin treatment, or had a BMI > 32 kg/m2 Placebo baseline TC: 6.39 mmol/L (247.1 mg/dL) Placebo baseline LDL‐C: 4.29 mmol/L (165.9 mg/dL) Placebo baseline HDL‐C: 1.14 mmol/L (44 mg/dL) Placebo baseline TG: 2.09 mmol/L (185.1 mg/dL) Cerivastatin 0.1 mg/d baseline TC: 6.45 mmol/L (249.4 mg/dL) Cerivastatin 0.1 mg/d baseline LDL‐C: 4.34 mmol/L (167.8 mg/dL) Cerivastatin 0.1 mg/d baseline HDL‐C: 1.14 mmol/L (44 mg/dL) Cerivastatin 0.1 mg/d baseline TG: 2.09 mmol/L (185.1 mg/dL) Cerivastatin 0.3 mg/d baseline TC: 6.34 mmol/L (245.2 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 4.24 mmol/L (164 mg/dL) Cerivastatin 0.3 mg/d baseline HDL‐C: 1.14 mmol/L (44 mg/dL) Cerivastatin 0.3 mg/d baseline TG: 2.09 mmol/L (185.1 mg/dL) |
|
| Interventions | Placebo evening dosing Cerivastatin 0.1 mg/d evening dosing Cerivastatin 0.3 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 4 weeks of plasma TC, LDL‐C, HDL‐C, TG and WDAEs | |
| Source of funding | Bayer | |
| Notes | SDs were imputed by the method of Furukawa 2006 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of sufficient blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | Unclear risk | Blinding method was not described |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | [(51 ‐45)/51]*100 = 11.8% participants were not included in the efficacy analysis for the placebo group [(107‐101)/107]*100 = 5.6% participants were not included in the efficacy analysis for the cerivastatin 0.1 mg/d group [(107‐106)/107]*100 = 0.9% participants were not included in the efficacy analysis for the cerivastatin 0.3 mg/d group [(265‐252)/265]*100 = 4.9% participants were not included in the efficacy analysis for all doses |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | [(51 ‐45)/51]*100 = 11.8% participants were not included in the efficacy analysis for the placebo group [(107‐101)/107]*100 = 5.6% participants were not included in the efficacy analysis for the cerivastatin 0.1 mg/d group [(107‐106)/107]*100 = 0.9% participants were not included in the efficacy analysis for the cerivastatin 0.3 mg/d group [(265‐252)/265]*100 = 4.9% participants were not included in the efficacy analysis for all doses |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | [(51 ‐45)/51]*100 = 11.8% participants were not included in the efficacy analysis for the placebo group [(107‐101)/107]*100 = 5.6% participants were not included in the efficacy analysis for the cerivastatin 0.1 mg/d group [(107‐106)/107]*100 = 0.9% participants were not included in the efficacy analysis for the cerivastatin 0.3 mg/d group [(265‐252)/265]*100 = 4.9% participants were not included in the efficacy analysis for all doses |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | [(51 ‐45)/51]*100 = 11.8% participants were not included in the efficacy analysis for the placebo group [(107‐101)/107]*100 = 5.6% participants were not included in the efficacy analysis for the cerivastatin 0.1 mg/d group [(107‐106)/107]*100 = 0.9% participants were not included in the efficacy analysis for the cerivastatin 0.3 mg/d group [(265‐252)/265]*100 = 4.9% participants were not included in the efficacy analysis for all doses |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Bayer funded the trial, data may support bias for cerivastatin |
Sakabe 2004.
| Methods | No washout required because participants were not receiving lipid‐lowering medications 12‐week before‐and‐after study |
|
| Participants | 17 men and women LDL‐C ≥ 160 mg/dL (4.14 mmol/L) and TG ≤ 400 mg/dL (4.52 mmol/L) Exclusion criteria: patients who smoked, had diabetes, hypertension, previous vascular events and revascularisation, stable CAD, or active liver disease Cerivastatin 0.15 mg/d baseline TC: 6.70 mmol/L (259.1 mg/dL) Cerivastatin 0.15 mg/d baseline LDL‐C: 4.40 mmol/L (170.1 mg/dL) Cerivastatin 0.15 mg/d baseline HDL‐C: 1.58 mmol/L (61.1 mg/dL) Cerivastatin 0.15 mg/d baseline TG: 1.56 mmol/L (138.2 mg/dL) |
|
| Interventions | Cerivastatin 0.15 mg/d evening dosing Atorvastatin 10 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 12 weeks of plasma TC, LDL‐C, HDL‐C and TG | |
| Source of funding | Unknown | |
| Notes | Atorvastatin 10 mg/d data not analysed SDs were imputed by the method of Furukawa 2006 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | LDL‐C was measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Sasaki 1998.
| Methods | 4‐week placebo run‐in period 12‐week before‐and‐after study |
|
| Participants | 73 men and women with severe primary hypercholesterolaemia aged 20‐64 years TC ≥ 260 mg/dL (3.36 mmol/L) and TG ≤ 400 mg/dL (4.52 mmol/L) Exclusion criteria: secondary hyperlipidaemia, poorly controlled diabetes mellitus or severe hypertension, or a history of alcoholism or heavy drinking, obese patients on diet therapy for weight reduction, women who were pregnant or hoped to become pregnant, and those with any clinically critical condition Cerivastatin 0.15 mg/d baseline TC: 7.79 mmol/L (301 mg/dL) Cerivastatin 0.15 mg/d baseline LDL‐C: 5.66 mmol/L (219 mg/dL) Cerivastatin 0.3 mg/d baseline TC: 7.98 mmol/L (309 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 5.76 mmol/L (223 mg/dL) Cerivastatin 0.3 mg/d baseline HDL‐C: 1.46 mmol/L (56 mg/dL) |
|
| Interventions | Cerivastatin 0.15 mg/d evening dosing Cerivastatin 0.3 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 4‐12 weeks of serum TC, LDL‐C and HDL‐C | |
| Source of funding | Bayer Yakuhin | |
| Notes | HDL‐C data for the 0.15 mg/d group not used because the calculated value and the given value differed by > 10% TG data for both groups not used because the calculated values and the given values differed by > 10% |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | High risk | Excluded because the calculated values and the given values differed by > 10% for the 0.15 mg/d group All participants were included in the efficacy analysis for the 0.3 mg/d group [(73 ‐40)/73]*100 = 45.2% participants were not included in the efficacy analysis for all doses |
| Incomplete outcome data (attrition bias) Triglycerides | High risk | Excluded because the calculated values and the given values differed by > 10% |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Bayer Yakuhin funded the trial, data may support bias for cerivastatin |
Saunders 2000.
| Methods | 6‐8‐week run‐in period 8‐week before‐and‐after study |
|
| Participants | 221 men and women aged 18‐75 years with type IIa or IIb hypercholesterolaemia TG < 400 mg/dL (4.52 mmol/L) LDL‐C > 160 mg/dL (4.14 mmol/L) for patients with 0‐1 risk factor, > 130 mg/dL (3.36 mmol/L) for those with > 2 risk factors, or > 100 mg/dL (2.59 mmol/L) for those with documented CHD or PVD Exclusion criteria: MI (within the previous year), unstable angina, angina at rest, stroke or recent TIAs, recent coronary revascularisation, uncontrolled hypertension or hypothyroidism, diabetes, chronic liver disease, renal dysfunction, or drug or alcohol abuse, CK levels > 3 x ULN Cerivastatin 0.3 mg/d baseline TC: 6.84 mmol/L (264 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 4.63 mmol/L (179 mg/dL) Cerivastatin 0.3 mg/d baseline HDL‐C: 1.32 mmol/L (51 mg/dL) Cerivastatin 0.3 mg/d baseline TG: 1.96 mmol/L (174 mg/dL) |
|
| Interventions | Cerivastatin 0.3 mg/d evening dosing Pravastatin 20 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 8 weeks of blood TC, LDL‐C, HDL‐C and TG | |
| Source of funding | Unknown | |
| Notes | Pravastatin 20 mg/d group was not included in the efficacy analysis SDs were imputed by the method of Furukawa 2006 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | [(221‐202)/221]*100 = 8.6% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | [(221‐202)/221]*100 = 8.6% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | [(221‐202)/221]*100 = 8.6% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | [(221‐202)/221]*100 = 8.6% participants were not included in the efficacy analysis |
| Selective reporting (reporting bias) | Unclear risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Scharnagl 2004.
| Methods | 10‐week run‐in period 12‐week RCT |
|
| Participants | 69 men and women age < 75 years old LDL‐C > 130 mg/dL (3.36 mmol/L), TG > 100 mg/dL (1.13 mmol/L) Exclusion criteria: TG ≥ 600 mg/dL (6.77 mmol/L), LDL‐C ≥ 300 mg/dL (7.76 mmol/L); diabetes mellitus requiring treatment, HRT, renal disease liver disease, acute coronary syndromes or stroke within 3 months of trial, severe hypertension, heart failure, arrhythmia, thyroid diseases, cancer, nephrotic syndrome, pancreatitis, myopathy, GI disease, depression, alcohol or drug abuse, statin hypersensitivity; participation in other clinical trials within 2 months of enrolment, use of other lipid‐lowering drugs or supplements, beta blockers, diuretics, androgens, immunosuppressants, vitamins Placebo baseline TC: 6.88 mmol/L (266 mg/dL) Placebo baseline LDL‐C: 3.67 mmol/L (142 mg/dL) Placebo baseline HDL‐C: 1.11 mmol/L (43 mg/dL) Cerivastatin 0.4 mg/d baseline TC: 6.59 mmol/L (255 mg/dL) Cerivastatin 0.4 mg/d baseline LDL‐C: 3.62 mmol/L (140 mg/dL) Cerivastatin 0.4 mg/d baseline HDL‐C: 1.11 mmol/L (43 mg/dL) |
|
| Interventions | Placebo Cerivastatin 0.4 mg/d |
|
| Outcomes | Percentage change from baseline at 6‐12 weeks of serum TC, LDL‐C and HDL‐C | |
| Source of funding | Bayer | |
| Notes | SDs were imputed by the method of Furukawa 2006 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind cerivastatin and placebo capsules were all identical in appearance |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | LDL‐C was measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | WDAEs were not reported |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Bayer funded the trial, data may support bias for cerivastatin |
Sebestjen 2002.
| Methods | 6‐week run‐in period 12‐week before‐and‐after study |
|
| Participants | 38 men with combined hyperlipidaemia 19 received cerivastatin 19 received fenofibrate TC > 6.0 mmol/L (232 mg/dL), LDL‐C > 4.0 mmol/L (155 mg/dL) and TG is between 2.2‐4.6 mmol/L (195‐407 mg/dL) Exclusion criteria: none reported Cerivastatin 0.2 mg/d baseline TC: 6.8 mmol/L (263 mg/dL) Cerivastatin 0.2 mg/d baseline LDL‐C: 4.2 mmol/L (162 mg/dL) Cerivastatin 0.2 mg/d baseline HDL‐C: 0.96 mmol/L (37 mg/dL) Cerivastatin 0.2 mg/d baseline TG: 3.5 mmol/L (310 mg/dL) |
|
| Interventions | Cerivastatin 0.2mg/d for 0‐6 weeks Cerivastatin 0.2‐0.4 mg/d for 6‐12 weeks Fenofibrate 250 mg/d for 0‐12 weeks |
|
| Outcomes | Percentage change from baseline at 8 weeks of blood TC, LDL‐C, HDL‐C and TG | |
| Source of funding | Unknown | |
| Notes | Cerivastatin 0.2‐0.4 mg/d for 6‐12 weeks Fenofibrate 250 mg/d for 0‐12 weeks groups were not analysed |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Shinn 2004.
| Methods | No washout was required since no participants received lipid medications before the trial 6‐week before‐and‐after study |
|
| Participants | 11 men and women with diabetes and hypercholesterolaemia Exclusion criteria: patients taking gemfibrozil Cerivastatin 0.4 mg/d baseline TC: 5.8 mmol/L (224.3 mg/dL) Cerivastatin 0.4 mg/d baseline LDL‐C: 3.75 mmol/L (145.0 mg/dL) Cerivastatin 0.4 mg/d baseline HDL‐C: 1.27 mmol/L (49.1 mg/dL) |
|
| Interventions | Cerivastatin 0.4 mg/d | |
| Outcomes | Percentage change from baseline at 8 weeks of plasma TC, LDL‐C and HDL‐C | |
| Source of funding | Unknown | |
| Notes | TG were median percentage change and not analysed SDs were imputed by the method of Furukawa 2006 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | High risk | TG were median percentage change and were not included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Simons 2002.
| Methods | 4‐week dietary/washout period 4‐week RCT |
|
| Participants | 152 men and women with primary hypercholesterolaemia aged ≥ 18 years LDL‐C ≥ 97 mg/dL (2.5 mmol/L), TG ≤ 400 mg/dL (4.5 mmol/L) Exclusion criteria: none reported Placebo baseline TC: 7.66 mmol/L (296.2 mg/dL) Placebo baseline LDL‐C: 5.42 mmol/L (209.6 mg/dL) Placebo baseline HDL‐C: 1.36 mmol/L (52.6 mg/dL) Placebo baseline TG: 1.97 mmol/L (174.5 mg/dL) Cerivastatin 0.4 mg/d baseline TC: 7.50 mmol/L (290 mg/dL) Cerivastatin 0.4 mg/d baseline LDL‐C: 5.22 mmol/L (201.9 mg/dL) Cerivastatin 0.4 mg/d baseline HDL‐C: 1.39 mmol/L (53.7 mg/dL) Cerivastatin 0.4 mg/d baseline TG: 1.97 mmol/L (174.5 mg/dL) |
|
| Interventions | Placebo + regular margarine evening dosing Placebo + sterol‐ester margarine evening dosing Cerivastatin 0.4 mg/d + regular margarine evening dosing Cerivastatin 0.4 mg/d + sterol‐ester margarine evening dosing |
|
| Outcomes | Percentage change from baseline at 4 weeks of plasma TC, LDL‐C, HDL‐C, TG and WDAEs | |
| Source of funding | Bayer | |
| Notes | SDs were imputed by the method of Furukawa 2006 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind Cerivastatin and placebo tablets were all identical in appearance |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | WDAEs were not reported |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C was reported |
| Other bias | Unclear risk | Bayer and Unilever funded the trial |
Solov'eva 1999.
| Methods | 1‐month washout period 3‐month before‐and‐after study |
|
| Participants | 15 men aged 21‐64 years with type IIa and IIb hypercholesterolaemia Exclusion criteria: secondary hyperlipidaemia, thyroid dysfunction, diabetes, endocrine disorders, chronic kidney disease, liver disease, cancer Cerivastatin 0.2 mg/d baseline TC: 8.07 mmol/L (312 mg/dL) Cerivastatin 0.2 mg/d baseline LDL‐C: 6.18 mmol/L (239 mg/dL) |
|
| Interventions | Cerivastatin 0.2 mg/d | |
| Outcomes | Percentage change from baseline at 1‐3 months of blood TC and LDL‐C | |
| Source of funding | Russian Ministry of Health | |
| Notes | HDL‐C and TG were not included in the efficacy analysis because the given values and the calculated values differed by > 10% | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | High risk | Excluded because the calculated values and the given values differed by > 10% |
| Incomplete outcome data (attrition bias) Triglycerides | High risk | Excluded because the calculated values and the given values differed by > 10% |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Low risk | Government grant |
Stein 1998.
| Methods | 10‐week washout period 8‐week RCT |
|
| Participants | 3113 men and women with primary hypercholesterolaemia type IIa and IIb Exclusion criteria: none reported Placebo baseline TC: 7.655 mmol/L (296 mg/dL) Placebo baseline LDL‐C: 5.472 mmol/L (212 mg/dL) Placebo baseline HDL‐C: 1.324 mmol/L (51 mg/dL) Cerivastatin 0.025 mg/d baseline LDL‐C: 5.617 mmol/L (217 mg/dL) Cerivastatin 0.05 mg/d baseline LDL‐C: 5.606 mmol/L (217 mg/dL) Cerivastatin 0.1 mg/d baseline TC: 7.605 mmol/L (294 mg/dL) Cerivastatin 0.1 mg/d baseline LDL‐C: 5.379 mmol/L (208 mg/dL) Cerivastatin 0.1 mg/d baseline HDL‐C: 1.249 mmol/L (48 mg/dL) Cerivastatin 0.2 mg/d baseline TC: 7.458 mmol/L (288 mg/dL) Cerivastatin 0.2 mg/d baseline LDL‐C: 5.263 mmol/L (204 mg/dL) Cerivastatin 0.2 mg/d baseline HDL‐C: 1.272 mmol/L (49 mg/dL) Cerivastatin 0.3 mg/d baseline TC: 7.634 mmol/L (295 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 5.345 mmol/L (207 mg/dL) Cerivastatin 0.3 mg/d baseline HDL‐C: 1.29 mmol/L (50 mg/dL) Cerivastatin 0.4 mg/d baseline TC: 7.717 mmol/L (298 mg/dL) Cerivastatin 0.4 mg/d baseline LDL‐C: 5.57 mmol/L (215 mg/dL) Cerivastatin 0.4 mg/d baseline HDL‐C: 1.402 mmol/L (54 mg/dL) |
|
| Interventions | Placebo Cerivastatin 0.025 mg/d Cerivastatin 0.05 mg/d Cerivastatin 0.1 mg/d Cerivastatin 0.2 mg/d Cerivastatin 0.3 mg/d Cerivastatin 0.4 mg/d |
|
| Outcomes | Percentage change from baseline at 8 weeks of plasma TC, LDL‐C, HDL‐C and WDAEs for all doses combined only | |
| Source of funding | Unknown | |
| Notes | Total number of participants for TG could not be determined from the 3 TG subgroups from figure 3 in the paper SDs were imputed by the method of Furukawa 2006 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of sufficient blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | Unclear risk | Blinding method was not described |
| Incomplete outcome data (attrition bias) Total cholesterol | High risk | [(520‐393)/520]*100 = 24.4% participants were not included in the efficacy analysis for the placebo group for total cholesterol All participants were included in the efficacy analysis for the 0.025 mg/d group All participants were included in the efficacy analysis for the 0.05 mg/d group [(487‐407)/487]*100 = 16.4% participants were not included in the efficacy analysis for the 0.1 mg/d group [(758‐416)/758]*100 = 45.1% participants were not included in the efficacy analysis for the 0.2 mg/d group [(408‐389)/408]*100 = 4.6% participants were not included in the efficacy analysis for the 0.3 mg/d group [(132‐122)/132]*100 = 7.6% participants were not included in the efficacy analysis for the 0.4 mg/d group [(3113‐1727)/3113]*100 = 44.5% participants were not included in the efficacy analysis for all doses |
| Incomplete outcome data (attrition bias) LDL cholesterol | High risk | [(520‐372)/520]*100 = 28.5% participants were not included in the efficacy analysis for the placebo group All participants were included in the efficacy analysis for the 0.025 mg/d group All participants were included in the efficacy analysis for the 0.05 mg/d group [(487‐384)/487]*100 = 21.1% participants were not included in the efficacy analysis for the 0.1 mg/d group [(758‐403)/758]*100 = 46.8% participants were not included in the efficacy analysis for the 0.2 mg/d group [(408‐376)/408]*100 = 7.8% participants were not included in the efficacy analysis for the 0.3 mg/d group [(132‐122)/132]*100 = 7.6% participants were not included in the efficacy analysis for the 0.4 mg/d group [(3113‐2465)/3113]*100 = 20.8% participants were not included in the efficacy analysis for all doses |
| Incomplete outcome data (attrition bias) HDL cholesterol | High risk | [(520‐391)/520]*100 = 24.8% participants were not included in the efficacy analysis for the placebo group All participants were included in the efficacy analysis for the 0.025 mg/d group All participants were included in the efficacy analysis for the 0.05 mg/d group [(487‐407)/487]*100 = 16.4% participants were not included in the efficacy analysis for the 0.1 mg/d group [(758‐413)/758]*100 = 45.5% participants were not included in the efficacy analysis for the 0.2 mg/d group [(408‐388)/408]*100 = 4.9% participants were not included in the efficacy analysis for the 0.3 mg/d group [(132‐122)/132]*100 = 7.6% participants were not included in the efficacy analysis for the 0.4 mg/d group [(3113‐1721)/3113]*100 = 44.7% participants were not included in the efficacy analysis for all doses |
| Incomplete outcome data (attrition bias) Triglycerides | High risk | No TG data included in the efficacy analysis because total number of subjects for the TG could not be determined |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding was not reported |
Stein 1999.
| Methods | 4‐week dietary run‐in period 4‐week RCT |
|
| Participants | 41 men and women with primary hypercholesterolaemia aged 18‐75 years LDL‐C ≥ 160 mg/dL (4.14 mmol/L) or ≥ 130 mg/dL (3.36 mmol/L) with CAD or ≥ cardiovascular risk factors TG ≤ 400 mg/dL) (4.52 mmol/L) Exclusion criteria: clinically active CVD or uncontrolled hypertension within 3 months of entry; CABG or PTCA 6 months before entry; hypertension with alterations in diuretic or β‐blocker therapy within 2 months of entry; uncontrolled diabetes mellitus or other endocrine abnormalities; unstable ophthalmic abnormalities (cataracts, or corrected visual acuity < 20/50); malignancy; psychosis; active liver disease, or unexplained persistent elevations of hepatic transaminases (> 1.1 x ULN), SCr ≥ 2 mg/dL, ≥ 1.5 x ULN, serum amylase > 1.2 x ULN; fasting serum glucose > 140 mg/dL; weight > 130% of ideal body weight; history of hypersensitivity to HMG CoA reductase inhibitors; nephrotic syndrome; or GI disorders. Disallowed concurrent medications included aspirin, corticosteroids, erythromycin, rifampin, androgens, immunosuppressants, or antidiabetic medications. Patients were excluded if they were concomitantly treated with other hypolipidaemic medication, had used probucol within the prior 6 months, or had a history of alcohol or substance abuse, pregnancy, lactation, child bearing potential and night shift workers Placebo baseline TC: 6.67 mmol/L (258 mg/dL) Placebo baseline LDL‐C: 4.55 mmol/L (176 mg/dL) Placebo baseline HDL‐C: 1.22 mmol/L (47 mg/dL) Placebo baseline TG: 2.03 mmol/L (180 mg/dL) Cerivastatin 0.8 mg/d baseline TC: 6.72 mmol/L (260 mg/dL) Cerivastatin 0.8 mg/d baseline LDL‐C: 4.50 mmol/L (174 mg/dL) Cerivastatin 0.8 mg/d baseline HDL‐C: 1.22 mmol/L (47 mg/dL) Cerivastatin 0.8 mg/d baseline TG: 2.36 mmol/L (209 mg/dL) |
|
| Interventions | Placebo evening dosing Cerivastatin 0.8 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 4 weeks of plasma TC, LDL‐C, HDL‐C, TG and WDAEs | |
| Source of funding | Bayer | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of sufficient blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | Unclear risk | Blinding method was not described |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Bayer funded the trial, data may support bias for cerivastatin |
Suzuki 2001.
| Methods | 4‐week washout period 12‐week before‐and‐after study |
|
| Participants | 53 men and women with hypercholesterolaemia aged 30‐79 years old LDL‐C ≥ 140 mg/dL (3.62 mmol/L) TG < 400 mg/dL (4.52 mmol/L) Exclusion criteria: none reported no baseline data reported |
|
| Interventions | Cerivastatin 0.15 mg/d Cerivastatin 0.3 mg/d Pravastatin 10 mg/d Simvastatin 5 mg/d Simvastatin 10 mg/d |
|
| Outcomes | Percentage change from baseline at 12 weeks of serum TC and LDL‐C | |
| Source of funding | Unknown | |
| Notes | Pravastatin 10 mg/d, simvastatin 5 mg/d, simvastatin 10 mg/d groups were not included in the efficacy analysis SD was imputed by the method of Furukawa 2006 for TC |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Tao 2000.
| Methods | 5‐week placebo run‐in period 8‐week RCT |
|
| Participants | 470 men and women with hypercholesterolaemia aged 18‐75 years LDL‐C ≥ 160 mg/dL (4.14 mmol/L) or ≥ 130 mg/dL (3.36 mmol/L) if the patient had ≥ 2 cardiovascular risk factors Exclusion criteria: DBP ≥ 115 mmHg, MI history, stroke, PTCA or CABG within the previous 6 months, congestive heart failure, heart rhythm problems, diabetes mellitus, hypothyroidism, cancer within 5 years, renal dysfunction, liver disease, history of pancreatitis, muscular or neuromuscular disease, GI malabsorption, drug or alcohol abuse or psychological instability, HIV positive, severe cataract, glaucoma, hypolipidaemic therapy, use of immunosuppressants, erythromycin niacin > 50 mg/d, corticosteroids, androgens or anticoagulants Placebo baseline TC: 6.5 mmol/L (251 mg/dL) Placebo baseline LDL‐C: 4.7 mmol/L (182 mg/dL) Placebo baseline HDL‐C: 1.0 mmol/L (39 mg/dL) Placebo baseline TG: 1.6 mmol/L (142 mg/dL) Cerivastatin 0.1 mg/d baseline TC: 6.5 mmol/L (251 mg/dL) Cerivastatin 0.1 mg/d baseline LDL‐C: 4.7 mmol/L (182 mg/dL) Cerivastatin 0.1 mg/d baseline HDL‐C: 1.09 mmol/L (42 mg/dL) Cerivastatin 0.1 mg/d baseline TG: 1.5 mmol/L (133 mg/dL) Cerivastatin 0.2 mg/d baseline TC: 6.5 mmol/L (251 mg/dL) Cerivastatin 0.2 mg/d baseline LDL‐C: 4.7 mmol/L (182 mg/dL) Cerivastatin 0.2 mg/d baseline HDL‐C: 1.1 mmol/L (42.5 mg/dL) Cerivastatin 0.2 mg/d baseline TG: 1.5 mmol/L (133 mg/dL) Cerivastatin 0.3 mg/d baseline TC: 6.6 mmol/L (255 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 4.8 mmol/L (186 mg/dL) Cerivastatin 0.3 mg/d baseline HDL‐C: 1.1 mmol/L (42.5 mg/dL) Cerivastatin 0.3 mg/d baseline TG: 1.5 mmol/L (133 mg/dL) |
|
| Interventions | Placebo evening dosing Cerivastatin 0.1 mg/d evening dosing Cerivastatin 0.2 mg/d evening dosing Cerivastatin 0.3 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 4 weeks of plasma TC, LDL‐C, HDL‐C, TG and WDAEs | |
| Source of funding | Unknown | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of sufficient blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | Unclear risk | Blinding method was not described |
| Incomplete outcome data (attrition bias) Total cholesterol | High risk | [(118 ‐100)/118]*100 = 15.2% participants were not included in the efficacy analysis for the placebo group [(119 ‐103)/119]*100 = 13.4% participants were not included in the efficacy analysis for the cerivastatin 0.1 mg/d group [(117 ‐ 101)/117]*100 = 13.7% participants were not included in the efficacy analysis for the cerivastatin 0.2 mg/d group [(116 ‐ 96)/116]*100 = 17.2% participants were not included in the efficacy analysis for the cerivastatin 0.3 mg/d group [(470 ‐ 400)/470]*100 = 14.9% participants were not included in the efficacy analysis for all doses |
| Incomplete outcome data (attrition bias) LDL cholesterol | High risk | [(118 ‐100)/118]*100 = 15.2% participants were not included in the efficacy analysis for the placebo group [(119 ‐103)/119]*100 = 13.4% participants were not included in the efficacy analysis for the cerivastatin 0.1 mg/d group [(117 ‐ 101)/117]*100 = 13.7% participants were not included in the efficacy analysis for the cerivastatin 0.2 mg/d group [(116 ‐ 96)/116]*100 = 17.2% participants were not included in the efficacy analysis for the cerivastatin 0.3 mg/d group [(470 ‐ 400)/470]*100 = 14.9% participants were not included in the efficacy analysis for all doses |
| Incomplete outcome data (attrition bias) HDL cholesterol | High risk | [(118 ‐100)/118]*100 = 15.2% participants were not included in the efficacy analysis for the placebo group [(119 ‐103)/119]*100 = 13.4% participants were not included in the efficacy analysis for the cerivastatin 0.1 mg/d group [(117 ‐ 101)/117]*100 = 13.7% participants were not included in the efficacy analysis for the cerivastatin 0.2 mg/d group [(116 ‐ 96)/116]*100 = 17.2% participants were not included in the efficacy analysis for the cerivastatin 0.3 mg/d group [(470 ‐ 400)/470]*100 = 14.9% participants were not included in the efficacy analysis for all doses |
| Incomplete outcome data (attrition bias) Triglycerides | High risk | [(118 ‐100)/118]*100 = 15.2% participants were not included in the efficacy analysis for the placebo group [(119 ‐103)/119]*100 = 13.4% participants were not included in the efficacy analysis for the cerivastatin 0.1 mg/d group [(117 ‐ 101)/117]*100 = 13.7% participants were not included in the efficacy analysis for the cerivastatin 0.2 mg/d group [(116 ‐ 96)/116]*100 = 17.2% participants were not included in the efficacy analysis for the cerivastatin 0.3 mg/d group [(470 ‐ 400)/470]*100 = 14.9% participants were not included in the efficacy analysis for all doses |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Source of funding not reported |
Tazuma 1998.
| Methods | 4‐6‐week placebo run‐in period 12‐week before‐and‐after study |
|
| Participants | 21 men and women with hypercholesterolaemia age 38‐77 years TC ≥ 220 mg/dL (5.69 mmol/L) TG ≤ 400 mg/dL (4.52 mmol/L) Exclusion criteria: none reported Cerivastatin 0.2 mg/d baseline TC: 7.03 mmol/L (272 mg/dL) Cerivastatin 0.2 mg/d baseline LDL‐C: 5.02 mmol/L (194 mg/dL) Cerivastatin 0.2 mg/d baseline HDL‐C: 1.34 mmol/L (52 mg/dL) Cerivastatin 0.2 mg/d baseline TG: 1.47 mmol/L (130 mg/dL) |
|
| Interventions | Cerivastatin 0.2 mg/d evening dosing | |
| Outcomes | Percentage change from baseline at 4‐12 weeks of serum TC, LDL‐C, HDL‐C and TG | |
| Source of funding | Bayer and government grant | |
| Notes | SDs were imputed by the method of Furukawa 2006 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | Unclear risk | Partially funded by Bayer and government grant |
Wada 1996.
| Methods | 4‐week washout period 8‐week before‐and‐after study |
|
| Participants | 19 men and women with type IIa and IIb hyperlipidaemia age ≥ 20 years TC ≥ 220 mg/dL (5.69 mmol/L) Exclusion criteria: statin hypersensitivity, hypothyroidism, Cushing's syndrome, obstructive gallbladder disease, SLE, nephrosis, HDL seborrhoea, drug‐induced hyperlipidaemia, diet therapy for obesity, secondary hyperlipidaemia, alcohol abuse Cerivastatin 0.15 mg/d baseline TC: 6.90 mmol/L (267 mg/dL) |
|
| Interventions | Cerivastatin 0.15 mg/d evening dosing | |
| Outcomes | Percentage change from baseline at 4‐8 weeks of blood TC | |
| Source of funding | Unknown | |
| Notes | LDL‐C, HDL‐C and TG were not included in the efficacy analysis because the given values and the calculated values differed by > 10% | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | [(19‐18)/19)*100 = 5.3% participants were not included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | High risk | Excluded because was not included in the efficacy analysis because the given values and the calculated values differed by > 10% |
| Incomplete outcome data (attrition bias) HDL cholesterol | High risk | Excluded because was not included in the efficacy analysis because the given values and the calculated values differed by > 10% |
| Incomplete outcome data (attrition bias) Triglycerides | High risk | Excluded because was not included in the efficacy analysis because the given values and the calculated values differed by > 10% |
| Selective reporting (reporting bias) | High risk | LDL‐C outcome was not reported |
| Other bias | Unclear risk | Source of funding not reported |
Yu 2002.
| Methods | 8‐week dietary and washout run‐in period 8‐week before‐and‐after study |
|
| Participants | 21 men and women with primary hypercholesterolaemia age 18‐75 years old LDL‐C > 160 mg/dL (4.14 mmol/L), TG < 350 mg/dL (3.95 mmol/L) Exclusion criteria: severe uncontrolled hypertension. diabetes mellitus, uncontrolled hypothyroidism, renal impairment (SCr > 2.0 mg/dL), known chronic liver disease or elevated serum transaminase levels (ALT/AST > 1.5 x ULN). MI, unstable angina, cerebral vascular accident, and TIA within 3 months before the trial; and CABG or PCTA within 6 months before the trial, concomitant treatment with corticosteroids, androgens, erythromycin, oral anticoagulants, or other lipid‐lowering agents was not permitted during the trial. Cerivastatin 0.3 mg/d baseline TC: 6.60 mmol/L (255.2 mg/dL) Cerivastatin 0.3 mg/d baseline LDL‐C: 4.63 mmol/L (179.1 mg/dL) Cerivastatin 0.3 mg/d baseline HDL‐C: 1.31 mmol/L (50.7 mg/dL) Cerivastatin 0.3 mg/d baseline TG: 1.43 mmol/L (126.5 mg/dL) |
|
| Interventions | Cerivastatin 0.3 mg/d evening dosing Lovastatin 20 mg/d evening dosing |
|
| Outcomes | Percentage change from baseline at 8 weeks of plasma TC, LDL‐C, HDL‐C and TG | |
| Source of funding | Bayer Taiwan Co | |
| Notes | Lovastatin 20 mg/d group was not analysed SDs were imputed by the method of Furukawa 2006 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Controlled before‐and‐after design |
| Allocation concealment (selection bias) | High risk | Controlled before‐and‐after design |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Lipid parameter measurements unlikely influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) LDL‐C | Low risk | Lipid parameters were measured in a remote laboratory |
| Blinding of outcome assessment (detection bias) WDAE | High risk | No comparison possible |
| Incomplete outcome data (attrition bias) Total cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) LDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) HDL cholesterol | Low risk | All participants were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) Triglycerides | Low risk | All participants were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | LDL‐C outcome was reported |
| Other bias | High risk | Trial supported by Bayer Taiwan Co. data may support bias for cerivastatin |
ALT: alanine transaminase; AST: aspartate transaminase; BMI: body mass index; BP: blood pressure; CABG: coronary artery bypass graft; CAD: coronary artery disease; CK: creatine kinase; CVD: cardiovascular disease; FH: familial hypercholesterolaemia; GI: gastrointestinal; HBA1c: haemoglobin A1c; HDL‐C: high‐density lipoprotein cholesterol; HMG CoA; 3‐hydroxy‐3‐methyl‐glutarylcoenzyme A; HRT: hormone‐replacement therapy; IUD: intrauterine device; LDL‐C: low‐density lipoprotein cholesterol; MI: myocardial infarction; NYHA: New York Heart Association; OCP: oral contraceptive pill; PCI: percutaneous coronary intervention; PTCA: percutaneous transluminal coronary angioplasty; PVD: peripheral vascular disease; RCT: randomised, double‐blind, placebo‐controlled trial; SCr: serum creatinine; SD: standard deviation; SGOT: serum glutamic oxaloacetic transaminase; SGPT: serum glutamic pyruvic transaminase; SLE: systemic lupus erythematosus; TC: total cholesterol; TG: triglyceride; TIA: transient ischaemic attack; TSH: thyroid‐stimulating hormone; ULN: upper limit of normal; WDAEs: withdrawals due to adverse effects; WHO: World Health Organization
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Anonymous 1998 | All libraries report lacking this supplement: article is not available |
| Bergstrom 2006 | Number of participants taking cerivastatin is not clear since the article focuses on HRT |
| Deighan 2001 | Data were combined for all cross‐over periods |
| Fegan 2005 | Postprandial data |
| Fleischmann 2004 | Data were combined for all cross‐over periods |
| Fujiwara 2000 | Participants were receiving simvastatin up to the time they received cerivastatin; simvastatin was not washed out within at least 3 weeks |
| Garcia 2002 | Confounding factor: immunosuppressant |
| Habib 2000 | Confounding factor: lipid‐lowering agents received during the 4‐week run‐in period |
| ISRCTN22144829 | No lipid data provided |
| Koizumi 1996 | Confounding factors: cholestyramine or probucol |
| Lauterbach 2001 | No lipid data provided |
| Leiter 1999 | Doses not specified |
| Leslie 2004 | Data were combined for all cross‐over periods |
| McPherson 2001 | Doses not specified |
| Morisaki 1996 | Confounding factors: cholestyramine or probucol |
| Paniagua 2002 | Data were combined for all cross‐over periods |
| Renders 2003 | Confounding factor: immunosuppressant tacrolimus |
| Schmage 2000 | ITT n values were not reported for the separate cerivastatin doses of 0.1, 0.2 and 0.3 mg/d. 1749 participants for 0.1‐0.3 mg/d cerivastatin and 651 participants for placebo |
| Tran 2005 | Data were combined for all cross‐over periods |
| Ural 2002 | Results are internally inconsistent |
| Wilmink 2001 | Postprandial data |
HRT: hormone replacement therapy; ITT: intention‐to‐treat
Differences between protocol and review
We excluded trials in which participants were receiving drugs that affect blood lipid level concentrations: for example, immunosuppressants such as cyclosporine; protease inhibitors such as ritonavir and indinavir; food supplements such as fish oils; fibrates such as gemfibrozil, fenofibrate and clofibrate; bile acid sequestrants such as cholestyramine, colestipol, colesevelam; the cholesterol absorption inhibitor ezetimibe; the vitamin niacin; and the anti‐oxidant drug probucol. These exclusion criteria were not mentioned in the protocol.
We conducted sensitivity analyses to assess the effect of different methods of dosing, such as twice daily versus single dose, on the treatment effect. This sensitivity analysis was not mentioned in the protocol.
The secondary objective to quantify the relative potency of cerivastatin with respect to fluvastatin, atorvastatin and rosuvastatin for LDL cholesterol, total cholesterol, and triglycerides was not mentioned in the protocol.
Contributions of authors
Stephen P Adams: contributed to the design of the protocol, screened the citations, assessed all trials for inclusion or exclusion, extracted the data, analysed the data and made contributions to the discussion. Nicholas Tiellet: assessed all trials for inclusion or exclusion, checked some data extraction.
Nima Alaeiilkhchi: checked some data extraction. James M Wright: interpreted the data, made contributions to the discussion and conclusions.
Sources of support
Internal sources
-
Department of Anesthesiology, Pharmacology & Therapeutics, University of BC, Canada.
Office space
External sources
-
BC Ministry of Health grant to the Therapeutics Initiative, Canada.
Salary support
Declarations of interest
Stephen P Adams: none known Nicholas Tiellet: none known Nima Alaeiilkhchi: none known James M Wright: none known
New
References
References to studies included in this review
Amano 1996 {published data only}
- Amano K. The clinical effect of novel HMG CoA reductase inhibitor, BAY w 6228, on hyperlipidemia in diabetic patients. Japanese Pharmacology and Therapeutics 1996;24 Suppl 9:253‐62. [EMBASE: 1996316447] [Google Scholar]
Arakawa 1996 {published data only}
- Arakawa K. A comparative and long term trial of a new HMG CoA reductase inhibitor, BAY w 6228 150 mug/day and 300 mug/day in patients with hyperlipidemia. Japanese Pharmacology and Therapeutics. Japan, 1996; Vol. 24 Suppl 9:147‐70. [EMBASE: 1996316441]
Arakawa 1997 {published data only}
- Arakawa K. Long‐term trial of a new HMG‐CoA reductase inhibitor, BAY w 6228 (cerivastatin) in patients with hyperlipidemia. Japanese Pharmacology and Therapeutics 1997;25(10):127‐43. [EMBASE: 1998035849] [Google Scholar]
Balletshofer 2005 {published data only}
- Balletshofer BM, Goebbel S, Rittig K, Enderle M, Schmolzer I, Wascher TC, et al. Intense cholesterol lowering therapy with a HMG‐CoA reductase inhibitor does not improve nitric oxide dependent endothelial function in type‐2‐diabetes‐‐a multicenter, randomised, double‐blind, three‐arm placebo‐controlled clinical trial. Experimental and Clinical Endocrinology & Diabetes 2005;113(6):324‐30. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Battula 2000 {published data only}
- Battula SB, Fitzsimons O, Moreno S, Owens D, Collins P, Johnson A, et al. Postprandial apolipoprotein B48‐and B100‐containing lipoproteins in type 2 diabetes: do statins have a specific effect on triglyceride metabolism?. Metabolism: Clinical and Experimental 2000;49(8):1049‐54. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Bayer 1992 {published data only}
- Bayer. A placebo‐controlled, double‐blind, parallel group 28 day study of the safety and tolerability of BAY w 6228 3000 ug once daily in patients with hypercholesterolemia. Study # D92‐010 (0123)/Report #1139 1998.
- Mullican WS. A placebo‐controlled, double‐blind, parallel group 28 day study of the safety and tolerability of BAY w 6228 3000 ug once daily in patients with hypercholesterolemia. Study 0123 (D92‐010) 1992.
Bayer 1994 {published data only}
- Bayer. A double‐blind dose ranging study of BAY w 6228 in doses of 50 ug, 100 ug, 200 ug. and 300 ug once daily compared to placebo and to lovastatin 40 mg once daily in patients with hypercholesterolemia. Study # D91‐031 (0124)/ Report # 1304 1998.
- Bayer. A double‐blind dose ranging study of BAY w 6228 in doses of 50 ug, 100 ug, 200 ug. and 300 ug once daily compared to placebo and to lovastatin 40 mg once daily in patients with hypercholesterolemia. Study D/X91‐031 1994.
Bayer 1995 {published data only}
- Bayer. Efficacy of cerivastatin in FH heterozygotes. Study 0139 1995.
- Marais AD. A randomized, double‐blind, parallel group comparison study to compare the efficacy of BAY w 6228 to placebo in the treatment of patients with familial hypercholesterolemia. Study #0139/Report #25062 1999.
Bayer 1997 {published data only}
- Bayer. Protocol BAY D97‐008. Study BAY D97‐008 1997.
Bayer 1998 {published data only}
- Bayer. NDA:20‐740/S002. Study D96‐008A 1998.
Betteridge 1999 {published data only}
- Bayer 1994. A multinational, multicentre, randomised, double‐blind, dose ranging study of BAY w 6228 in doses of 25 ug, 50 ug, 100 ug, 200 ug, once daily compared to placebo and simvastatin 20 mg once daily in patients with hypercholesterolemia. Bayer Study 120 1994. [Study 0120]
- Betteridge DJ. International multicentre comparison of cerivastatin with placebo and simvastatin for the treatment of patients with primary hypercholesterolaemia. International Cerivastatin Study Group. International Journal of Clinical Practice 1999;53(4):243‐50. [MEDLINE: ] [PubMed] [Google Scholar]
Chen 2001 {published data only}
- Chen Z, Zhang J. Clinical comparison of cerivastatin with simvastatin for aged hyperlipidemia. Chinese Pharmaceutical Journal 2001;36(4):276‐7. [EMBASE: 2001165218] [Google Scholar]
Davignon 1998 {published data only}
- Davignon J, Hanefeld M, Nakaya N, Hunninghake DB, Insull W Jr, Ose L. Clinical efficacy and safety of cerivastatin: summary of pivotal phase IIb/III studies. American Journal of Cardiology 1998;82(4B):32J‐9J. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Dujovne 2000 {published data only}
- Bayer. Double blind efficacy and safety study of two doses of Baycol (cerivastatin) and Pravachol (pravastatin) in the treatment of patients with primary hypercholesterolemia (Protocol 001). Cerivastatin Clinical Report Study 001 1998.
- Dujovne CA, Knopp R, Kwiterovich P, Hunninghake D, McBride TA, Poland M. Randomized comparison of the efficacy and safety of cerivastatin and pravastatin in 1,030 hypercholesterolemic patients. The Cerivastatin Study Group. Mayo Clinic Proceedings 2000;75(11):1124‐32. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Goto 1996a {published data only}
- Goto Y. Clinical evaluation of a new HMG CoA reductase inhibitor, BAY w 6228: a double blind comparative study with pravastatin in patients with hyperlipidemia. Japanese Pharmacology & Therapeutics 1996;24 Suppl 9:263‐96. [EMBASE: 1996316448] [Google Scholar]
Goto 1996b {published data only}
- Goto Y. Clinical efficacy of a new HMG‐CoA reductase inhibitor, BAY w 6228: a dose finding among four groups by the double blind method. Japanese Pharmacology and Therapeutics 1996;24 Suppl 9:87‐115. [EMBASE: 1996316439] [Google Scholar]
Hanefeld 1999 {published data only}
- Bayer. A multinational, multicentre, double‐blind, randomised study of BAY w 6228 in doses of 300 ug and 400 ug once daily compared to placebo in patients with primary hypercholesterolemia. Study #0149 / Report #25068 1995.
- Hanefeld M. Effectivity and tolerance of 300 microgram and 400 microgram cerivastatin per day in patients with primary hypercholesterolamia: a European multi‐centre study. Zeitschrift fur Kardiologie 1998;87 Suppl 5:15. [CENTRAL: CN‐00336724 UPDATE] [Google Scholar]
- Hanefeld M, Deslypere JP, Ose L, Durrington PN, Farnier M, Schmage N. Efficacy and safety of 300 micrograms and 400 micrograms cerivastatin once daily in patients with primary hypercholesterolaemia: a multicentre, randomized, double‐blind, placebo‐controlled study. Journal of International Medical Research 1999;27(3):115‐29. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Hunninghake 1998 {published data only}
- Balarac N, Betteridge DJ, Klӧr HU. A double‐blind pilot dose‐ranging study of BAY w 6228 in doses of 25 ug, 50 ug, 100 ug, and 200 ug once daily compared to placebo and to simvastatin 20 mg once daily in patients with hyperlipidemia. British Columbia Pharmacare Program 1992; Vol. Study #0110 / Report #23202:275‐82.
- Bayer. A double‐blind pilot dose ranging study of BAY w 6228 in doses of 25 ug, 50 ug, 100 ug, and 200 ug once daily compared to placebo and to lovastatin 40 mg once daily in patients with hypercholesterolemia. Study # D91‐012 (0109) / Report #1199 1991.
- Bayer. A double‐blind placebo‐controlled dose scheduling study of BAY w 6228 in doses of 100 ug twice daily with breakfast and dinner compared to 200 ug once daily with dinner or at bedtime in patients with hypercholesterolemia. D/X 91‐031 1993.
- Bayer. A double‐blind placebo‐controlled dose scheduling study of BAY w 6228 in doses of 100 ug twice daily with breakfast and dinner compared to 200 ug once daily with dinner or at bedtime in patients with hypercholesterolemia. Study #D91‐016 (0111) / Report #1215 1993.
- Hunninghake DB. Clinical efficacy of cerivastatin: phase IIa dose‐ranging and dose‐scheduling studies. American Journal of Cardiology 1998;82(4B):26J‐31J. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Stein E, Sprecher D, Allenby KS, Tosiello RL, Whalen E, Ripa SR. Cerivastatin, a new potent synthetic HMG Co‐A reductase inhibitor: effect of 0.2 mg daily in subjects with primary hypercholesterolemia. Journal of Cardiovascular Pharmacology and Therapeutics 1997;2(1):7‐16. [EMBASE: 1997110288] [DOI] [PubMed] [Google Scholar]
Hunninghake 2001 {published data only}
- Hunninghake D, Insull W, Knopp R, Davidson M, Lohrbauer L, Jones P, et al. Comparison of the efficacy and safety of atorvastatin versus cerivastatin in patients with hypercholesterolemia. Atherosclerosis 1999;144(1):28‐9. [CENTRAL: CN‐00445809] [DOI] [PubMed] [Google Scholar]
- Hunninghake D, Insull W, Knopp R, Davidson M, Lohrbauer L, Jones P, et al. Comparison of the efficacy of atorvastatin versus cerivastatin in primary hypercholesterolemia. American Journal of Cardiology 2001;88(6):635‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Insull 2000 {published data only}
- Insull W Jr, Isaacsohn J, Kwiterovich P, Ra P, Brazg R, Dujovne C, et al. Efficacy and safety of cerivastatin 0.8 mg in patients with hypercholesterolaemia: the pivotal placebo‐controlled clinical trial. Cerivastatin Study Group. Journal of International Medical Research 2000;28(2):47‐68. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Isaacsohn J, Insull W Jr, Stein E, Kwiterovich P, Patrick M A, Brazg R, et al. Long‐term efficacy and safety of cerivastatin 0.8 mg in patients with primary hypercholesterolemia. Clinical Cardiology 2001;24(9 Suppl):IV1‐9. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Isaacsohn 1998 {published data only}
- Isaacsohn J, Stein E, Orchard T, Weinstein R, Huh C, Whalen E, et al. Cerivastatin Study Group. Comparative efficacy, safety, and tolerability of cerivastatin a new HMG‐CoA reductase inhibitor, and fluvastatin in subjects with primary hypercholesterolemia. Drugs affecting lipid metabolism: abstract book. Milan, Italy: Milano Fondazione Giovanni Lorenzini, 1998; Vol. Lorenzini Foundation symposium, 273:50.
Kajiyama 1996 {published data only}
- Kajiyama G. Effects of BAY w 6228 on biliary lipid metabolism in patients with hyperlipidemia. Japanese Pharmacology and Therapeutics 1996;24(Suppl 9):235‐43. [EMBASE: 1996316445] [Google Scholar]
Kim 1999 {published data only}
- Kim S, Kim DJ, Hahm JR, Kim BJ, Chung JH, Min YK, et al. Cholesterol lowering effect of cerivastatin in Korean patients with primary hypercholesterolemia. Journal of Korean Society of Endocrinology 1999;14(4):729‐38. [CENTRAL: CN‐01045329 UPDATE] [Google Scholar]
Krone 1999 {published data only}
- Krone W, Schmage N, Breuer HW. Comparison of long‐term efficacy and safety of cerivastatin versus pravastatin in primary hypercholesterolaemia. Journal of Drug Assessment 1999;2(Pt 4):327‐40. [CENTRAL: CN‐00281951 UPDATE] [Google Scholar]
Lankin 2002 {published data only}
- Lankin VZ, Tikhaze AK, Konovalova GG, Tutunov VS, Medvedeva NV, Kotkina TI, et al. Intensification of free radical oxidation of low‐density lipoproteins in the plasma of patients with ischemic heart disease receiving beta‐hydroxy‐beta‐methylglutaryl‐coenzyme A reductase inhibitor cerivastatin and inhibition of low‐density lipoprotein peroxidation with antioxidant probucol. Bulletin of Experimental Biology & Medicine 2002;134(1):39‐42. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ma 2000 {published data only}
- Ma P, Hegele R, Yale JF, Schwartz B. CAVEAT: A randomised, double‐blind, parallel group evaluation of cerivastatin 0.4 mg and 0.8 mg compared to atorvastatin 10 mg and 20 mg once daily in patients with combined (type IIb) dyslipidaemia. British Journal of Cardiology 2000;7(12):780‐6. [EMBASE: 2001009668] [Google Scholar]
- Ma, Hegele R, Yale JF, Schwartz B. CAVEAT: a comparison of cerivastatin 0.4 mg and 0.8mg with atorvastatin 10 mg and 20 mg in patients with combined (type IIB) dyslipidaemia. Atherosclerosis Supplements 2001;2(2):48. [MEDLINE: ] [Google Scholar]
Mabuchi 1998 {published data only}
- Mabuchi H, Koizumi J, Kajinami K. Clinical efficacy and safety of cerivastatin in the treatment of heterozygous familial hypercholesterolemia. American Journal of Cardiology 1998;82(4B):52J‐5J. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Matsuo 2005 {published data only}
- Matsuo T, Iwade K, Hirata N, Yamashita M, Ikegami H, Tanaka N, et al. Improvement of arterial stiffness by the antioxidant and anti‐inflammatory effects of short‐term statin therapy in patients with hypercholesterolemia. Heart and Vessels 2005;20(1):8‐12. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Matsuzawa 1996 {published data only}
- Matsuzawa Y. Clinical evaluation of BAY w 6228, a new HMG CoA reductase inhibitor, in the long term treatment of hyperlipidemia: effect on serum lipids and steroid hormones. Japanese Pharmacology and Therapeutics 1996;24(Suppl 9):117‐45. [EMBASE: 1996316440] [Google Scholar]
Nakamura 2001 {published data only}
- Nakamura T, Ushiyama C, Hirokawa K, Osada S, Shimada N, Koide H. Effect of cerivastatin on urinary albumin excretion and plasma endothelin‐1 concentrations in type 2 diabetes patients with microalbuminuria and dyslipidemia. American Journal of Nephrology 2001;21(6):449‐54. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Nakaya 1996 {published data only}
- Nakaya N. Clinical study of Bay w 6228, a new HMG CoA reductase inhibitor, in the long term treatment of hyperlipidemia: comparison between the elderly and younger subjects. Japanese Pharmacology and Therapeutics 1996;24(SUPPL. 9):171‐95. [EMBASE: 1996316442] [Google Scholar]
Nakaya 1997 {published data only}
- Nakaya N. Clinical study of BAY w 6228, a new HMG‐CoA reductase inhibitor, in the long‐term treatment of hyperlipidemia: comparison between the elderly and younger subjects (report II). Japanese Pharmacology and Therapeutics 1997;25(10):145‐69. [EMBASE: 1998035850] [Google Scholar]
Puccetti 2001 {published data only}
- Puccetti L, Bruni F, Bova G, Cercignani M, Palazzuoli A, Console E, et al. Effect of diet and treatment with statins on platelet‐dependent thrombin generation in hypercholesterolemic subjects. Nutrition Metabolism & Cardiovascular Diseases 2001;11(6):378‐87. [MEDLINE: ] [PubMed] [Google Scholar]
Ridker 2001 {published data only}
- Ridker PM, Rifai N, Lowenthal SP. Rapid reduction in C‐reactive protein with cerivastatin among 785 patients with primary hypercholesterolemia. Circulation 2001;103(9):1191‐3. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Rubinstein 1999 {published data only}
- Rubinstein A, Maritz FJ, Soule SG, Markel A, Chajek‐Shaul T, Maislos M, et al. Efficacy and safety of cerivastatin for type 2 diabetes and hypercholesterolaemia. Hyperlipidaemia in Diabetes Mellitus investigators. Journal of Cardiovascular Risk 1999;6(6):399‐403. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sakabe 2004 {published data only}
- Sakabe K, Fukuda N, Wakayama K, Nada T, Shinohara H, Tamura Y. Time course differences for statin‐induced pleiotropic effects in hypercholesterolemic patients. International Journal of Cardiology 2004;94(1):111‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sasaki 1998 {published data only}
- Sasaki J, Arakawa K, Yamamoto K, Kobori S, Ageta M, Kono S. A comparative long‐term trial of sodium cerivastatin, a new HMG‐CoA reductase inhibitor, in patients presenting with primary hypercholesterolemia [Essai comparatif a long terme de la cerivastatine sodique, un nouvel inhibiteur de l'HMG‐CoA reductase, chez des patients presentant une hypercholesterolemie primaire]. Revue de Medecine Interne 1999;20(Suppl 3):393s‐8s. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Sasaki J, Arakawa K, Yamamoto K, Kobori S, Ageta M, Kono S. A long‐term comparative trial of cerivastatin sodium, a new HMG‐CoA reductase inhibitor, in patients with primary hypercholesterolemia. Clinical Therapeutics 1998;20(3):539‐48. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Saunders 2000 {published data only}
- Saunders E, Ferdinand K, Yellen LG, Tonkon MJ, Krug‐Gourley S, Poland M. Efficacy and safety of cerivastatin and pravastatin in the treatment of primary hypercholesterolemia. Journal of the National Medical Association 2000;92(7):319‐26. [MEDLINE: ] [PMC free article] [PubMed] [Google Scholar]
Scharnagl 2004 {published data only}
- Scharnagl H, Stojakovic T, Winkler K, Rosinger S, Marz W, Boehm BO. The HMG‐CoA reductase inhibitor cerivastatin lowers advanced glycation end products in patients with type 2 diabetes. Experimental and Clinical Endocrinology & Diabetes 2007;115(6):372‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Scharnagl H, Winkler K, Mantz S, Baumstark MW, Wieland H, Marz W. Inhibition of HMG‐CoA reductase with cerivastatin lowers dense low density lipoproteins in patients with elevated fasting glucose, impaired glucose tolerance and type 2 diabetes mellitus. Experimental and Clinical Endocrinology & Diabetes 2004;112(5):269‐77. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sebestjen 2002 {published data only}
- Sebestjen M, Keber I, Zegura B, Simcic S, Bozic M, Fressart MM, et al. Statin and fibrate treatment of combined hyperlipidemia: the effects on some novel risk factors. Thrombosis and Haemostasis 2004;92(5):1129‐35. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Sebestjen M, Zegura B, Keber I. Both cerivastatin and fenofibrate improve arterial vasoreactivity in patients with combined hyperlipidaemia. Journal of Internal Medicine 2002;251(1):77‐85. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Shinn 2004 {published data only}
- Shinn AH. Evaluating the effects of HMG ‐CoA reductase inhibitors on C‐reactive protein, butyrylcholinesterase, and lipids [PhD thesis]. Stockton, California (USA): University of the Pacific, 2004. [ProQuest Document ID: 305146858] [Google Scholar]
Simons 2002 {published data only}
- Simons LA. Additive effect of plant sterol‐ester margarine and cerivastatin in lowering low‐density lipoprotein cholesterol in primary hypercholesterolemia. American Journal of Cardiology 2002;90(7):737‐40. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Solov'eva 1999 {published data only}
- Solov'eva E, Rozhkova TA, Tvorogova MG, Kukharchuk VV. Cerivastatin‐a new synthetic 3‐hydroxy‐3‐methylglutaryl (HMG) inhibitor: effect of 0,2 mg dose in patients with primary hyperlipidemias [Tserivastatin‐novyi sinteticheskii ingibitor reduktazy kofermenta A 3‐gidroksi‐3‐metilgliutarila: effekt dozy 0,2 mg u bol'nykh s pervichnymi giperlipidemiiami]. Terapevticheskii Arkhiv 1999;71(8):30‐4. [MEDLINE: ] [PubMed] [Google Scholar]
Stein 1998 {published data only}
- Stein E. Cerivastatin in primary hyperlipidemia‐‐a multicenter analysis of efficacy and safety. Atherosclerosis 1998;139(Suppl 1):S15‐22. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Stein E. Cerivastatin in primary hyperlipidemia: a multicenter analysis of efficacy and safety. American Journal of Cardiology 1998;82(4B):40J‐6J. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Stein 1999 {published data only}
- Stein E, Isaacsohn J, Stoltz R, Mazzu A, Liu MC, Lane C, et al. Pharmacodynamics, safety, tolerability, and pharmacokinetics of the 0.8‐mg dose of cerivastatin in patients with primary hypercholesterolemia. American Journal of Cardiology 1999;83(10):1433‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Suzuki 2001 {published data only}
- Suzuki K. Randomized comparison of the efficacy between cerivastatin and simvastatin in hypercholesterolemia. Therapeutic Research 2001;22(5):1105‐11. [EMBASE: 2002094763] [Google Scholar]
Tao 2000 {published data only}
- Tao P, Wu XS, Yu XJ, Zhu J, Tao SQ. Efficacy and safety of cerivastatin 0.1 mg, 0.2 mg and 0.3 mg in Chinese patients with primary hypercholesterolaemia: a multicentre, randomised, double‐blind, placebo‐controlled study. Journal of Clinical Research 2000;3:29‐40. [EMBASE: 2000231116] [Google Scholar]
- Wang YQ, Jia YH, Tao P. Efficacy and safety of cerivastatin in the treatment of primary hypercholesterolemia: a multicentre, randomized, double‐blind study. Chinese Journal of Cardiology 2000;28(4):259‐63. [CENTRAL: CN‐00361391 UPDATE] [Google Scholar]
Tazuma 1998 {published data only}
- Tazuma S, Yamashita G, Ochi H, Miura H, Kajihara T, Hattori Y, et al. Effects of cerivastatin sodium, a new HMG‐CoA reductase inhibitor, on biliary lipid metabolism in patients with hypercholesterolemia. Clinical Therapeutics 1998;20(3):477‐85. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wada 1996 {published data only}
- Wada H. Effects of BAY w 6228 on coagulation and fibrinolytic systems in patients with hyperlipidemia. Japanese Pharmacology and Therapeutics 1996;24(Suppl 9):243‐52. [EMBASE: 1996316446] [Google Scholar]
Yu 2002 {published data only}
- Yu WC, Chen CH, Tsao HM, Ding YA. A randomized, double‐blind comparison of cerivastatin and lovastatin for treatment of primary hypercholesterolemia. Chung Hua i Hsueh Tsa Chih ‐ Chinese Medical Journal 2002;65(6):260‐7. [MEDLINE: ] [PubMed] [Google Scholar]
References to studies excluded from this review
Anonymous 1998 {published data only}
- Anonymous. Cerivastatine‐‐profile of a new lipid‐reducing drug. Internist 1998;39(1 Suppl Cerivastat):1‐8. [MEDLINE: ] [PubMed] [Google Scholar]
Bergstrom 2006 {published data only}
- Bergstrom H, Tonstad S. Combined hormone replacement therapy improves lipids but may increase leucocytes. Journal of Internal Medicine 2006;259(5):537‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Deighan 2001 {published data only}
- Deighan CJ, Caslake MJ, McConnell M, Boulton‐Jones JM, Packard CJ. Comparative effects of cerivastatin and fenofibrate on the atherogenic lipoprotein phenotype in proteinuric renal disease. Journal of the American Society of Nephrology 2001;12(2):341‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fegan 2005 {published data only}
- Fegan PG, Shore AC, Mawson D, Tooke JE, MacLeod KM. Microvascular endothelial function in subjects with type 2 diabetes and the effect of lipid‐lowering therapy. Diabetic Medicine 2005;22(12):1670‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fleischmann 2004 {published data only}
- Fleischmann EH, John S, Delles C, Schneider MP, Schmidt BM, Schmieder RE. The effect of statins on angiotensin II‐induced hemodynamic changes in young, mildly hypercholesterolemic men. American Journal of Hypertension 2004;17(12 Pt 1):1120‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fujiwara 2000 {published data only}
- Fujiwara A. Effect of cerivastatin on the hypercholesterolemia pretreated with simvastatin. Therapeutic Research 2000;21(2):373‐8. [EMBASE: 2000135385] [Google Scholar]
Garcia 2002 {published data only}
- Garcia I, Errasti P, Lavilla FJ, Ballester B, Manrique J, Rossich E, et al. Effects of cerivastatin in dyslipemia and other cardiovascular risk factors after renal transplantation. Transplantation Proceedings 2002;34(1):401‐2. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Habib 2000 {published data only}
- Habib G, Paillard F, Charpentier G, Angellier JF, Roux T, Portal JJ, et al. A multicenter, open‐label, randomized study comparing the efficacy of atorvastatin versus usual care in reducing refractory hypercholesterolemia in high‐risk patients to target levels. Current Therapeutic Research, Clinical and Experimental 2000;61(4):175‐90. [EMBASE: 2000177377] [Google Scholar]
ISRCTN22144829 {published data only}
- ISRCTN22144829. Lipids in Diabetes Study. doi.org/10.1186/ISRCTN22144829 2002.
Koizumi 1996 {published data only}
- Koizumi J. The effect of a new HMG CoA reductase inhibitor, BAY w 6228, in combination therapy with colestyramine or probucol for familial hypercholesterolemia (FH). Japanese Pharmacology and Therapeutics 1996;24(Suppl 9):197‐214. [EMBASE: 1996316443] [Google Scholar]
Lauterbach 2001 {published data only}
- Lauterbach KW, Binnen T, Evers T, Harnischmacher U, Ludwig D, Hanrath P, Krone W, Lehmacher W, Leys D, Neuhaus K‐L, Windler E. Primary prevention of stroke: RESPECT. European heart journal, supplement 2000;2(D):D51‐D53. [CENTRAL: CN‐00423870] [Google Scholar]
Leiter 1999 {published data only}
- Leiter LA, Hanna K. Efficacy and safety of cerivastatin in primary hypercholesterolemia: a long term comparative titration study with simvastatin. Canadian Journal of Cardiology 1999;15(5):545‐55. [MEDLINE: ] [PubMed] [Google Scholar]
Leslie 2004 {published data only}
- Leslie SJ, Spratt JC, Grieg L, Attina T, Denvir MA, Webb DJ. The effect of cerivastatin therapy on vascular responses to endothelin antagonists in humans. Journal of Cardiovascular Pharmacology 2004;44(Suppl 1):S410‐2. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
McPherson 2001 {published data only}
- McPherson R, Hanna K, Agro A, Braeken A, Canadian Cerivastatin Study Group. Cerivastatin versus branded pravastatin in the treatment of primary hypercholesterolemia in primary care practice in Canada: a one‐year, open‐label, randomized, comparative study of efficacy, safety, and cost‐effectiveness. Clinical Therapeutics 2001;23(9):1492‐507. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Morisaki 1996 {published data only}
- Morisaki N. Long term trial of a new HMG CoA reductase inhibitor, BAY w 6228 in patients with heterozygous familial hypercholesterolemia. Japanese Pharmacology and Therapeutics 1996;24(Suppl 9):215‐34. [EMBASE: 1996316444] [Google Scholar]
Paniagua 2002 {published data only}
- Paniagua JA, Lopez‐Miranda J, Escribano A, Berral FJ, Marin C, Bravo D, et al. Cerivastatin improves insulin sensitivity and insulin secretion in early‐state obese type 2 diabetes. Diabetes 2002;51(8):2596‐603. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Renders 2003 {published data only}
- Renders L, Haas CS, Liebelt J, Oberbarnscheidt M, Schocklmann HO, Kunzendorf U. Tacrolimus and cerivastatin pharmacokinetics and adverse effects after single and multiple dosing with cerivastatin in renal transplant recipients. British Journal of Clinical Pharmacology 2003;56(2):214‐9. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schmage 2000 {published data only}
- Schmage N, Cagatay M, Park SM, Lommerzheim A, Schopen U. Cerivastatin, a novel HMG‐CoA reductase inhibitor: efficacy and tolerability [Cerivastatin, ein neuer HMG‐CoA‐reduktase‐inhibitor: Wirksamkeit und vertraglichkeit]. Herz Kreislauf 2000;32(7‐8):242‐52. [EMBASE: 2000279263] [Google Scholar]
Tran 2005 {published data only}
- Tran D, Lowy A, Howes JB, Howes LG. Effects of cerivastatin on forearm vascular responses, blood pressure responsiveness and ambulatory blood pressure in type 2 diabetic men. Diabetes, Obesity & Metabolism 2005;7(3):273‐81. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ural 2002 {published data only}
- Ural AU, Yilmaz MI, Avcu F, Yalcin A. Treatment with cerivastatin in primary mixed hyperlipidemia induces changes in platelet aggregation and coagulation system components. International Journal of Hematology 2002;76(3):279‐83. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wilmink 2001 {published data only}
- Wilmink HW, Twickler MB, Banga JD, Dallinga‐Thie GM, Eeltink H, Erkelens DW, et al. Effect of statin versus fibrate on postprandial endothelial dysfunction: role of remnant‐like particles. Cardiovascular Research 2001;50(3):577‐82. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Additional references
Adams 2014
- Adams SP, Sekhon SS, Wright JM. Rosuvastatin for lowering lipids. Cochrane Database of Systematic Reviews 2014, Issue 11. [DOI: 10.1002/14651858.CD010254.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Adams 2015
- Adams SP, Tsang M, Wright JM. Atorvastatin for lowering lipids. Cochrane Database of Systematic Reviews 2015, Issue 3. [DOI: 10.1002/14651858.CD008226.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Adams 2018
- Adams SP, Sekhon SS, Wright JM, Tsang M. Fluvastatin for lowering lipids. Cochrane Database of Systematic Reviews 2018, Issue 3. [DOI: 10.1002/14651858.CD012282.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bandolier 2004
- Bandolier. Cholesterol lowering with statins. Bandolier 2004:121‐2.
Boekholdt 2012
- Boekholdt SM, Arsenault BJ, Mora S, Pedersen TR, LaRosaJC, Nestel PJ, et al. Association of LDL cholesterol, non‐HDL cholesterol, and apolipoprotein B levels with risk of cardiovascular events among patients treated with statins: a meta‐analysis. Journal of the American Medical Association 2012;307(12):1302–9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Bruckert 2005
- Bruckert E, Hayem G, Dejager S, Yau C, Begaud B. Mild to moderate muscular symptoms with high‐dosage statin therapy in hyperlipidemic patients‐‐the PRIMO study. Cardiovascular Drugs and Therapy / Sponsored by the International Society of Cardiovascular Pharmacotherapy 2005;19(6):403‐14. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
CDC 2011
- Centers for Disease Control and Prevention (CDC). Million hearts: strategies to reduce the prevalence of leading cardiovascular disease risk factors‐‐United States, 2011. MMWR ‐ Morbidity & Mortality Weekly Report 2011;60(36):1248‐51. [MEDLINE: ] [PubMed] [Google Scholar]
CTT 2005
- Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Cholesterol Treatment Trialists (CTT) Collaborators. Efficacy and safety of cholesterol‐lowering treatment: prospective meta‐analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005;366(9493):1267‐78. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Draeger 2006
- Draeger A, Monastyrskaya K, Mohaupt M, Hoppeler H, Savolainen H, Allemann C, et al. Statin therapy induces ultrastructural damage in skeletal muscle in patients without myalgia. Journal of Pathology 2006;210:94‐102. [DOI] [PubMed] [Google Scholar]
Edwards 2003
- Edwards JE, Moore RA. Statins in hypercholesterolaemia: a dose‐specific meta‐analysis of lipid changes in randomised, double blind trials. BMC Family Practice 2003;4:18. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Furberg 2001
- Furberg CD, Pitt B. Withdrawal of cerivastatin from the world market. Current Controlled Trials in Cardiovascular Medicine 2001;2(5):205‐7. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(1):7‐10. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Gaw 2000
- Gaw A, Packard CJ, Shepherd J. Statins: the HMG CoA Reductase Inhibitors in Perspective. London, England: Martin Dunitz Ltd, 2000. [1853174688] [Google Scholar]
GraphPad Prism 4 [Computer program]
- GraphPad Software Inc.. GraphPad Prism 4. Version Version 4.0. La Jolia: GraphPad Software Inc., 2003.
Grundy 2004
- Grundy SM, Cleeman JI, Merz CN, Brewer HB Jr, ClarkLT, Hunninghake DB, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. Journal of the American College of Cardiology 2004;44(3):720–32. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Heran 2008
- Heran BS, Wong MM, Heran IK, Wright JM. Blood pressure lowering efficacy of angiotensin converting enzyme (ACE) inhibitors for primary hypertension. Cochrane Database of Systematic Reviews 2008, Issue 4. [DOI: 10.1002/14651858.CD003823.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2002
- Higgins JP, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;21(11):1539‐58. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Altman DG, Sterne JA. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Izquiero‐Palomares 2016
- Izquierdo‐Palomares JM, Fernandez‐Tabera JM, Plana MN, Añino Alba A, Gómez Álvarez P, Fernandez‐Esteban I, et al. Chronotherapy versus conventional statins therapy for the treatment of hyperlipidaemia. Cochrane Database of Systematic Reviews 2016, Issue 11. [DOI: 10.1002/14651858.CD009462.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jaskiewicz 2018
- Jaskiewicz A, Pajak B, Litwiniuk A, Urbanska K, Orzechowski A. Geranylgeraniol prevents statin‐dependent myotoxicity in C2C12 muscle cells through RAP1 GTPase prenylation and cytoprotective autophagy. Oxidative Medicine and Cellular Longevity 2018;2018:6463807. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jaskiewicz 2019
- Jaskiewicz, Pajak B, Labieniec‐Watala M, Palma C, Orzechowski A. Diverse action of selected statins on skeletal muscle cells‐an attempt to explain the protective effect of geranylgeraniol (GGOH) in statin‐associated myopathy (SAM). Journal of Clinical Medicine 2019;8(5):694. [DOI: 10.3390/jcm8050694; MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kaufmann 2006
- Kaufmann P, Torok M, Zahno A, Waldhauser KM, Brecht K, Krahenbuhl S. Toxicity of statins on rat skeletal muscle mitochondria. Cellular and Molecular Life Sciences 2006;63(19‐20):2415‐25. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kellick 1997
- Kellick KA, Burns K, McAndrew E, Haberl E, Hook N, Ellis AK. Focus on atorvastatin: an HMG‐CoA reductase inhibitor for lowering both elevated LDL cholesterol and triglycerides in hypercholesterolemic patients. Formulary 1997;32(4):352‐63. [EMBASE: 1997129035] [Google Scholar]
Kreatsoulas 2010
- Kreatsoulas C, Anand SS. The impact of social determinants on cardiovascular disease. Canadian Journal of Cardiology 2010;26 Suppl C:8C–13C. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Law 2003
- Law MR, Wald NJ, Rudnicka AR. Quantifying effect of statins on low density lipoprotein cholesterol, ischaemic heart disease, and stroke: systematic review and meta‐analysis. BMJ 2003;326(7404):1423. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Liao 2005
- Liao JK, Laufs U. Pleiotropic effects of statins. Annual Review of Pharmacology and Toxicology 2005;45:89‐118. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
McTaggart 2003
- McTaggart F. Comparative pharmacology of rosuvastatin. Atherosclerosis. Supplements 2003;4(1):9‐14. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Moghadasian 1999
- Moghadasian MH. Clinical pharmacology of 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitors. Life Sciences 1999;65(13):1329–37. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Mohaupt 2009
- Mohaupt MG, Karas RH, Babiychuk EB, Sanchez‐Freire V, Monastyrskaya K, Iyer L, et al. Association between statin‐associated myopathy and skeletal muscle damage. Canadian Medical Association Journal 2009;181:E11‐E18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group (2009). Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA Statement. PLos Medicine 2009;6(7):e1000097. [DOI: 10.1371/journal.pmed1000097] [DOI] [PMC free article] [PubMed] [Google Scholar]
Morikawa 2005
- Morikawa S, Murakami T, Yamazaki H, Izumi A, Saito Y, Hamakubo T, et al. Analysis of the global RNA expression profiles of skeletal muscle cells treated with statins. Journal of Atherosclerosis and Thrombosis 2005;12(3):121‐31. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Muck 2000
- Mück W. Clinical pharmacokinetics of cerivastatin. Clinical Pharmacokinetics 2000;39(2):99‐116. [DOI: 10.2165/00003088-200039020-00002] [DOI] [PubMed] [Google Scholar]
Musini 2014
- Musini VM, Nazer M, Bassett K, Wright JM. Blood pressure‐lowering efficacy of monotherapy with thiazide diuretics for primary hypertension. Cochrane Database of Systematic Reviews 2014, Issue 5. [DOI: 10.1002/14651858.CD003824.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nishimoto 2003
- Nishimoto T, Tozawa R, Amano Y, Wada T, Imura Y, Sugiyama Y. Comparing myotoxic effects of squalene synthase inhibitor, T‐91485, and 3‐hydroxy‐3‐methylglutaryl coenzyme A (HMG‐CoA) reductase inhibitors in human myocytes. Biochemical Pharmacology 2003;66(11):2133‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Page 2019
- Page MJ, Higgins JPT, Sterne JAC. Chapter 13: Assessing risk of bias due to missing results in a synthesis. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.0 (updated July 2019).Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Plosker 2000
- Plosker GL, Dunn CI, Figgitt DP. Cerivastatin: a review of its pharmacological properties and therapeutic efficacy in the management of hypercholesterolaemia. Drugs 2000;60:1179. [DOI: 10.2165/00003495-200060050-00011] [DOI] [PubMed] [Google Scholar]
Psaty 2004
- Psaty BM, Furberg CD, Ray WA, Weiss NS. Potential for conflict of interest in the evaluation of suspected adverse drug reactions: use of cerivastatin and risk of rhabdomyolysis. Journal of the American Medical Association 2004;292(21):2622‐31. [DOI: 10.1001/jama.292.21.2622] [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Roger 2011
- Roger VL, Go AS, Lloyd‐Jones DM, Adams RJ, Berry JD, Brown TM, et al. Heart disease and stroke statistics‐2011 update: a report from the American Heart Association. Circulation 2011;123(4):e18–e209. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sakamoto 2013
- Sakamoto K, Kimura J. Mechanism of statin‐induced rhabdomyolysis. Journal of Pharmacological Sciences 2013;123(4):289‐94. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Schaefer 2004
- Schaefer EJ, McNamara JR, Tayler T, Daly JA, Gleason JL, Seman LJ, et al. Comparisons of effects of statins (atorvastatin, fluvastatin, lovastatin, pravastatin, and simvastatin) on fasting and postprandial lipoproteins in patients with coronary heart disease versus control subjects. American Journal of Cardiology 2004;93(1):31‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Schectman 1996
- Schectman G, Hiatt J. Dose‐response characteristics of cholesterol‐lowering drug therapies: implications for treatment. Annals of Internal Medicine 1996;125(12):990‐1000. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Schünemann 2019a
- Schünemann HJ, Higgins JPT, Vist GE, Glasziou P, Akl EA, Skoetz N, Guyatt GH. Chapter 14: Completing ‘Summary of findings’ tables and grading the certainty of the evidence. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Schünemann 2019b
- Schünemann HJ, Vist GE, Higgins JPT, Santesso N, Deeks JJ, Glasziou P, Akl EA, Guyatt GH. Chapter 15: Interpreting results and drawing conclusions. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Thompson 2005
- Thompson JF, Man M, Johnson KJ, Wood LS, Lira ME, Lloyd DB, et al. An association study of 43 SNPs in 16 candidate genes with atorvastatin response. Pharmacogenomics Journal 2005;5(6):352‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Tsang 2002
- Tsang MB, Adams SP, Jauca C, Wright JM. In some systematic reviews placebos may not be necessary: an example from a statin dose‐response study. 10th Cochrane Colloquium. Stavanger, 31 July‐3 August 2002; Vol. Abstracts:Poster 29.
Urso 2005
- Urso ML, Clarkson PM, Hittel D, Hoffman EP, Thompson PD. Changes in ubiquitin proteasome pathway gene expression in skeletal muscle with exercise and statins. Ateriosclerosis, Thrombosis, and Vascular Biology 2005;25:2560‐6. [DOI] [PubMed] [Google Scholar]
Ward 2007
- Ward S, Lloyd Jones M, Pandor A, Holmes M, Ara R, Ryan A, et al. A systematic review and economic evaluation of statins for the prevention of coronary events. Health Technology Assessment (Winchester, England) 2007;11(14):1‐160, iii‐iv. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Yamazaki 2006
- Yamazaki H, Suzuki M, Aoki T, Morikawa S, Maejima T, Sato F, et al. Influence of 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitors on ubiquinone levels in rat skeletal muscle and heart: relationship to cytotoxicity and inhibitory activity for cholesterol synthesis in human skeletal muscle cells. Journal of Atherosclerosis and Thrombosis 2006;13(6):295‐307. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]