

ABSTRACT
The initiation of macroautophagy/autophagy is tightly regulated by the upstream ULK1 kinase complex, which affects many downstream factors including the PtdIns3K complex. The phosphorylation of the right position at the right time on downstream molecules is governed by proper complex formation. One component of the ULK1 complex, ATG101, known as an accessory protein, is a stabilizer of ATG13 in cells. The WF finger region of ATG101 plays an important role in the recruitment of WIPI1 (WD repeat domain, phosphoinositide interacting protein 1) and ZFYVE1 (zinc finger FYVE-type containing 1). Here, we report that the C-terminal region identified in the structure of the human ATG101-ATG13HORMA complex is responsible for the binding of the PtdIns3K complex. This region adopts a β-strand conformation in free ATG101, but either an α-helix or random coil in our ATG101-ATG13HORMA complex, which protrudes from the core and interacts with other molecules. The C-terminal deletion of ATG101 shows a significant defect in the interaction with PtdIns3K components and subsequently impairs autophagosome formation. This result clearly presents an additional role of ATG101 for bridging the ULK1 and PtdIns3K complexes in the mammalian autophagy process.
Abbreviations: ATG: autophagy related; BECN1: beclin 1; GFP: green fluorescent protein; HORMA: Hop1p/Rev7p/MAD2; HsATG13HORMA: HORMA domain of ATG13 from Homo sapiens; KO: knockout; MAD2: mitotic arrest deficient 2 like 1; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; PIK3C3/VPS34: phosphatidylinositol 3-kinase catalytic subunit type 3; PIK3R4/VPS15: phosphoinositide-3-kinase regulatory subunit 4; PtdIns3K: phosphatidylinositol 3-kinase; RB1CC1/FIP200: RB1 inducible coiled-coil 1; SAXS: small-angle X-ray scattering; ScAtg13HORMA: HORMA domain of Atg13 from Sccharomyces cerevisiae; SEC-SAXS: size-exclusion chromatography with small-angle X-ray scattering; SpAtg13HORMA: HORMA domain of Atg13 from Schizosaccharomyces pombe; SQSTM1/p62: sequestosome 1; ULK1: unc51-like autophagy activating kinase 1; UVRAG: UV radiation resistance associated; WIPI1: WD repeat domain: phosphoinositide interacting 1; ZFYVE1/DFCP1: zinc finger FYVE-type containing 1
KEYWORDS: ATG13, ATG101, C-terminal region, PtdIns3K complex, structure, ULK1 complex
Introduction
Autophagy is an evolutionarily conserved catabolic pathway by which cytoplasmic materials are delivered to the lysosome in animal cells or the vacuole in yeast for cellular homeostasis [1,2]. It is a selective and regulated process for engulfing specific cargoes, such as mitochondria, ribosomes, peroxisomes, protein aggregates, and even invading pathogens into autophagosomes [1,3–7]. Autophagy is increasingly being seen as important because of its linkage to various human diseases including cell death and cancer, aging, immunity, infectious diseases, and neurodegenerative diseases [8–14]. Autophagic processes are tightly regulated by several steps involving multi-protein complexes such as the ULK1 (unc51-like autophagy activating kinase 1) complex (Atg1 complex in yeast), PtdIns3K (phosphatidylinositol 3-kinase) complex, ATG9-containing vesicles, WIPI (WD repeat domain, phosphoinositide interacting) proteins (Atg2-Atg18 complex in yeast), and two ubiquitin-like conjugation systems [13,15]. Among them, 2 upstream complexes, ULK1 and PtdIns3K, are very complicated and attract lots of attention for understanding the initiation of autophagy [16].
The ULK1 complex is composed of 4 proteins: ULK1 kinase, RB1CC1/FIP200 (RB1 inducible coiled-coil 1), ATG13, and ATG101 [17,18]. Although most of the core autophagy proteins are quite conserved among various species, the Atg1 complex, the budding yeast counterpart of ULK1 complex, has different components such as Atg1 kinase, Atg11, Atg13, Atg17, Atg29, and Atg31 [19]. The ULK1 complex is regulated by mTORC1 (mechanistic target of rapamycin complex 1) and AMPK (AMP-activated protein kinase) in an opposite manner [20]. ULK1 kinase is a serine/threonine protein kinase that phosphorylates other autophagic molecules including ATG13 and RB1CC1 in the ULK1 complex [20,21] as well as ATG14/ATG14L, BECN1 (beclin 1), PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3), and AMBRA1 (autophagy and beclin1 regulator 1) in the PtdIns3K complex [22,23]. PtdIns3K is a lipid kinase for producing PtdIns3P (phosphatidylinositol 3-phosphate) and is divided into at least 2 distinct complexes, PtdIns3K-C1 and PtdIns3K-C2 [16,24]. Both complexes share the catalytic subunit PIK3C3, putative protein kinase PIK3R4/VPS15 (phosphoinositide-3-kinase regulatory subunit 4), and BECN1, while PtdIns3K-C1 contains ATG14 for directing the complex to the phagophore assembly site (PAS) [25], and PtdIns3K-C2 contains UVRAG (UV radiation resistance associated) for directing endosome and autophagosome maturation [26].
There is increasing evidence that the above 2 upstream autophagy complexes communicate with each other for autophagy regulation. The ULK1 complex is required for puncta formation of the PtdIns3K-C1 complex [27]. ULK1 kinase induces autophagy by phosphorylating BECN1 and activating PIK3C3 lipid kinase [28]. Furthermore, the ULK1 kinase phosphorylates serine 29 of ATG14 for autophagosome formation [29]. In Saccharomyces cerevisiae, the N-terminal HORMA (Hop1p/Rev7p/MAD2 proteins) domain of Atg13 is involved in recruiting Atg14 to the endoplasmic reticulum [30,31]. More recently, the HORMA domain of mammalian ATG13 (ATG13HORMA) was found to bind to the central region (residues 201 to 395) of ATG14 in the PtdIns3K-C1 complex [29].
Besides the differences between the components of the mammalian ULK1 and budding yeast Atg1 complex, ATG13 (Atg13 in budding yeast) in particular is quite dissimilar in size as well as in binding partners [15,32,33]. The HORMA domain in Atg13 from S. cerevisiae (ScAtg13HORMA) forms a monomer with a unique β-sheet cap domain [31], whereas mammalian ATG13HORMA forms a heterodimer with ATG101 [34,35]. The cap domain is lacking in mammals, but ATG101 is lacking in budding yeast. The ScAtg13HORMA and mammalian ATG13HORMA resemble the closed conformation of MAD2L1 (mitotic arrest deficient 2 like 1; C-MAD2) whereas the HORMA-fold binding partner, ATG101, resembles the open conformation of MAD2L1 (O-MAD2). Therefore, the structure of the heterodimeric ATG101-ATG13HORMA complex is similar to that of MAD2 conformational dimer [36,37].
Independently, we have also determined the structure of the full-length ATG101 and ATG13HORMA complex from Homo sapiens (HsATG101- ATG13HORMA) and identified a unique feature of the C-terminal region of ATG101. Previous structures of Atg101-Atg13HORMA from Schizosaccharomyces pombe (SpAtg101-Atg13HORMA) showed no electron density at the C-terminal region [35], and that of HsATG101-ATG13HORMA was determined using a C-terminal deletion construct of ATG101 [34]. Very intriguingly, the C-terminal region in the structure of free ATG101 from humans shows a β-strand conformation [38], which is completely different from our structure. In this paper, we have analyzed the conformation of the C-terminal region of ATG101 in detail and dissected the functional role of this C-terminal region. Our results show that the C-terminal region of ATG101 mediates the interaction between the ULK1 and PtdIns3K complexes, whereas the previously-studied WF finger of ATG101 does not. Besides the stabilization of ATG13, these findings highlight another function of ATG101 in the ULK1 complex that affects autophagy in cells.
Results
Helical conformation of the C-terminal region of human ATG101
When we determined the structure of the HsATG101-ATG13HORMA complex (Figure 1(a)), the structures of the SpAtg101-Atg103HORMA complex, the HsATG101 (with C-terminal deleted construct)-ATG13HORMA complex, and free HsATG101 were suddenly reported [34,35,38]. We used a bacterial co-expression vector including ATG13HORMA (residues from 1 to 190) and full-length ATG101. Crystals of the ATG101-ATG13HORMA complex were obtained in 4 different space groups (Table 1). All of our structures and those of others were analyzed and the structural features of 2 HORMA domains were essentially very similar (Figures 1(b) and S1). Two regions, the WF finger and the C-terminal segment of ATG101, showed noticeable differences (Figure 1(b)). The WF motif exists in 2 conformations, open (out) and closed (in), depending on the contacts with neighboring ATG13HORMA molecules as shown in previous papers [34,35]. This WF finger is critical for recruiting downstream factors to the PAS [35]. In our HsATG101-ATG13HORMA structure, it is involved in contact with a neighboring molecule and is thus also in an open conformation (Figures 1(a) and S2).
Figure 1.
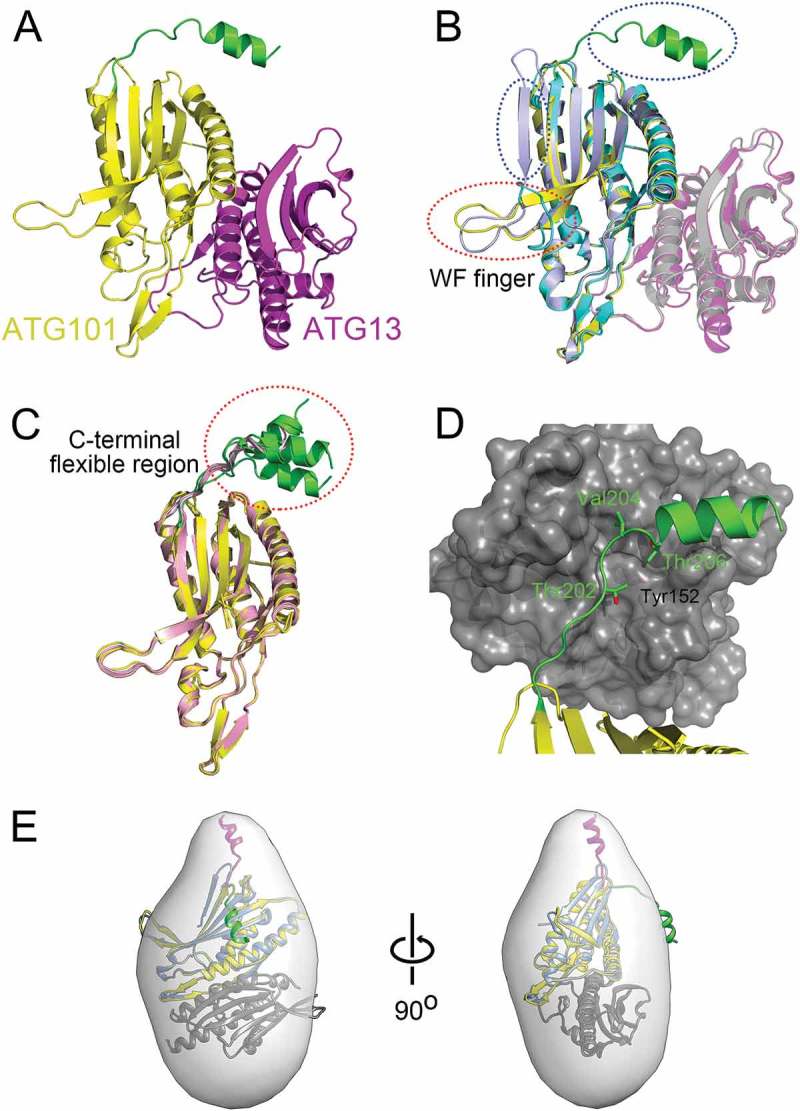
Structure of ATG101-ATG13HORMA complex. (a) Our HsATG101-ATG13HORMA complex showing the C-terminal helix of ATG101. ATG101 and ATG13HORMA are colored yellow and magenta, respectively. The unique C-terminal region of ATG101 is highlighted as green. (b) Superposition of HsATG101 structures showing clear differences in 2 regions, the WF-finger and the C-terminal region, which are highlighted by red and blue dotted circles, respectively. Our HsATG101-ATG13HORMA structure is colored as panel (A). The previous HsATG101-ATG13HORMA structure (C-terminal deleted version of HsATG101; PDB ID: 5C50) is colored slate and gray for ATG101 and ATG13, respectively. Free HsATG101 (β-sheet conformation of C-terminal region; PDB ID: 4WZG) is colored sky blue. (c) Superposition of all our structures of HsATG101 showing the flexibility of the C-terminal region. The HsATG101 with a helical C-terminal region is colored as panel (A) and that with random coil colored pink. (d) A close-up view of the interaction between the C-terminal region of HsATG101 and neighboring symmetry-equivalent ATG13 molecule shown in dark gray surface. The exposed aliphatic carbon atoms of Thr202, Val204, and Thr206 in ATG101 are stabilized by hydrophobic residues including Tyr152 in neighboring ATG13 in crystalline packing. (e) Our crystal structure of ATG101-ATG13HORMA and a modeled complex with the C-terminal β-sheet structure of ATG101 were fitted into the low-resolution molecular envelope generated from SAXS data. The C-terminal flexible α-helix of ATG101 colored green shows discrepancy for the SAXS molecular envelope and the simple rigid-body motion of the region colored magenta fits well into the SAXS envelope.
Table 1.
Data collection and refinement statistics.
| SeMet SAD | Form 1 | Form 2 | Form 3 | Form 4 | |
|---|---|---|---|---|---|
| Data collectiona | |||||
| Beamline | PAL 5C | PAL 5C | PF-AR NE3A | SP8 BL26B2 | PF BL17A |
| Space group | C2 | C2 | P2 | P21212 | C2221 |
| PDB ID | 5XUY | 5XV4 | 5XV3 | 5XV6 | |
| Cell dimensions | |||||
| a, b, c (Å) | 88.72, 124.83, 100.58 | 87.99, 125.18, 100.76 | 101.29, 125.36, 174.62 | 86.78, 101.12, 125.26 | 84.02, 123.49, 99.80 |
| α, β, γ (°) | 90, 102.0, 90 | 90, 102.08, 90 | 90, 95.69, 90 | 90, 90, 90 | 90, 90, 90 |
| Wavelength (Å) | 0.9795 (peak) | 0.9795 | 1.0000 | 1.0000 | 0.9800 |
| Resolution (Å) | 44.26–2.50 (2.59–2.50) | 31.46–2.20 (2.28–2.20) | 35.78–2.95 (3.05–2.95) | 38.59–2.55 (2.64–2.55) | 49.90–2.46 (2.54–2.46) |
| Total reflections | 149,964 (12,706) | 214,304 (20,001) | 317,829 (28,447) | 299,740 (27,032) | 136,168 (13,295) |
| Unique reflections | 36,369 (3,240) | 53,557 (5,224) | 87,500 (8,161) | 36,310 (3,286) | 19,209 (1,854) |
| Rmerge (%) | 9.62 (51.4) | 6.20 (62.86) | 8.87 (54.6) | 9.15 (91.91) | 9.02 (82.9) |
| I/σI | 16.52 (3.22) | 19.85 (3.40) | 9.21 (2.10) | 17.03 (2.55) | 18.67 (2.23) |
| Completeness (%) | 98.26 (87.78) | 98.73 (97.38) | 95.33 (88.14) | 99.12 (91.46) | 99.34 (97.48) |
| Multiplicity | 4.1 (3.9) | 4.0 (3.8) | 3.6 (3.5) | 8.3 (8.2) | 7.1 (7.2) |
| Wilson B-factor (Å2) | 47.13 | 52.96 | 58.91 | 55.56 | 64.22 |
| FOM before/after DM | 0.35/0.63 | ||||
| Refinement | |||||
| Resolution (Å) | 31.46–2.20 (2.28–2.20) | 35.78–2.95 (3.05–2.95) | 38.59–2.55 (2.64–2.55) | 49.90–2.46 (2.54–2.46) | |
| Rwork (%) | 23.72 (46.04) | 25.25 (35.45) | 23.21 (31.91) | 22.47 (30.25) | |
| Rfree (%) | 27.51 (48.72) | 30.30 (40.35) | 28.27 (34.50) | 27.17 (35.13) | |
| No. atoms | 6,049 | 23,827 | 5,900 | 3,017 | |
| Protein | 5,938 | 23,827 | 5,823 | 3,017 | |
| Water/PEG | 101/0 | 0/0 | 70/7 | 0/0 | |
| B-factors (Å2) | 79.5 | 68.8 | 83.0 | 77.5 | |
| Protein | 79.6 | 68.8 | 83.1 | 77.5 | |
| Water/PEG | 73.0/- | -/- | 72.4/85.2 | -/- | |
| R.m.s. deviations | |||||
| Bond lengths (Å) | 0.005 | 0.006 | 0.004 | 0.009 | |
| Bond angles (°) | 1.10 | 1.12 | 0.78 | 1.11 | |
| Ramachandran plot | |||||
| Favored (%) | 97 | 95.6 | 97 | 97 | |
| Outliers (%) | 0.13 | 0 | 0.13 | 0 |
a Values in parentheses are for the highest-resolution shell.
The most striking feature of our structure is the helical conformation of the C-terminal region of ATG101. The C-terminal region in the SpAtg101 was missing in the structure (Figures S1 and S3) [35] and previous deuterium uptake data of HsATG101 showed that the C-terminal region is completely unstructured [34]. Furthermore, it showed even β-strand conformation in free ATG101 (Figures 1(b) and S4A) [38]. Most of our structures crystallized in 4 different crystal forms showed an α-helical conformation of the C-terminal region, which protrudes from the core HORMA domain (Figure 1(c)). Interestingly, the secondary structure prediction made by using PSIPRED server [39] matches perfectly with our structure (Figure S4B).
The C-terminal region of ATG101 is versatile and may participate in protein-protein interactions
The open conformation of the hydrophobic WF finger (Trp110-Pro111-Phe112 residues in the human sequence) region of ATG101 is stabilized by the hydrophobic region of the neighboring protein (Figure S2). Similarly, the hydrophobic C-terminal region, especially right before the C-terminal helix, (Thr202, Val204, and Thr206) makes contact with a neighboring molecule (Figures 1(d) and S2). There are in total 15 different ATG101 structures in 4 different crystal forms and all structures showed the clear electron density for this C-terminal segment showing either α-helices or random coils (Figures 1(c) and S3) and thus this region is very flexible. As described, this region in free HsATG101 is in the β-strand conformation allowing it to make a central β-sheet with neighboring strands (Figure 1(b)). However, it is not clear whether the β-strand conformation of the C-terminal region in free HsATG101 turns into an α-helix upon complex formation with ATG13HORMA. The C-terminal region of free HsATG101 also forms an intermolecular β-sheet with symmetry equivalent HsATG101 molecule in the crystal lattice [38].
To determine the structure of this region in solution, size-exclusion chromatography with small-angle X-ray scattering (SEC-SAXS) was performed (Figure S5). The measured molecular weight (MW) of the HsATG101-ATG13HORMA complex was 48.3 kDa and it is well-matched with the theoretical MW of a 1:1 heterodimer complex (Table S1). Furthermore, our crystal structure fitted well into the small-angle X-ray scattering (SAXS) envelope except for the C-terminal region of ATG101 (Figure 1(e)). However, the extra molecular envelope is easily occupied by a simple rigid-body motion of the C-terminal flexible region. Therefore, our crystal structure and SAXS data suggest that the C-terminal region of ATG101 is versatile and undergoes a large conformational change. As previous researchers noted regarding the flexible WF finger, it is tempting to speculate that this protruding C-terminal hydrophobic segment may be involved in protein-protein interactions, and thus we generated the C-terminal deletion construct of ATG101 (ATG101ΔC) for further analysis.
The C-terminal region of ATG101 interacts with the PtdIns3K complex
There are several lines of evidence that the ULK1 complex interacts with the PtdIns3K complex [21]. In S. cerevisiae, ScAtg13 (without Atg101) recruits the PtdIns3K complex mediated by Atg14 [31]. Recently, it has been reported that HsATG13HORMA interacts with the middle region (residues 201 to 395) of ATG14; however, this study did not consider the tight binding partner, ATG101 [29]. As found in our structural study, the C-terminal region of ATG101 protrudes from the core HORMA domain and seems to have the perfect properties to interact with other molecules (Figure 1(d)). Therefore, we decided to find out the role of this region in the interaction with the PtdIns3K complex. First, we examined the binding of ATG101 or ATG101ΔC with different PtdIns3K complex components including ATG14, UVRAG, BECN1, and PIK3C3 in over-expression conditions (Figure 2(a-d)). Compared with full-length ATG101, there is a binding defect between ATG101ΔC and PtdIns3K complex components, suggesting that the C-terminal region of ATG101 plays a role in interaction with the PtdIns3K complex. In the cases of ATG14 and UVRAG, there is almost no binding if the C-terminal region of ATG101 is removed (Figure 2(a,b)). The other components, BECN1 and PIK3C3, also show much-reduced binding (Figure 2(b,c)). The PIK3R4 was only stably expressed in the presence of PIK3C3 (data not shown). The PtdIns3K complex exists as at least 2 forms, PtdIns3K-C1 and PtdIns3K-C2, depending on the presence of ATG14 or UVRAG with a tight core of PIK3C3, PIK3R4, and BECN1. The deletion of the C-terminal region of ATG101 causes a defect in binding to both ATG14 and UVRAG (Figure 2(a,b)), and thus it suggests that it is involved in the interaction with both PtdIns3K-C1 and PtdIns3K-C2 complexes.
Figure 2.
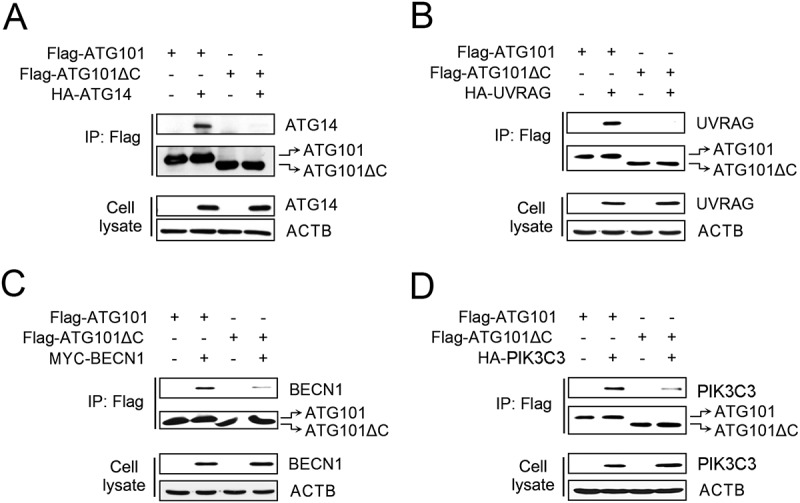
The C-terminal flexible region of ATG101 is important to the interaction with PtdIns3K complex components. To determine the role of the C-terminal region of ATG101 on PtdIns3K complex binding, either full-length wild-type Flag-ATG101 or C-terminal deletion Flag-ATG101 mutant (Flag-ATG101ΔC) were co-transfected with plasmids encoding PtdIns3K complex subunits (HA-ATG14 [A], HA-UVRAG [B], Myc-BECN1 [C], and HA-PIK3C3 [D]) into HEK293 cells as indicated. Flag-ATG101s were immunoprecipitated with anti-Flag antibody coupled with protein G-Sepharose and the immunoprecipitates were examined by western blots using the indicated antibodies. ACTB was used as a loading control.
Direct interaction between the C-terminal region of ATG101 and the PtdIns3K complex
The dissection of the interaction between the ULK1 and PtdIns3K complexes is extremely complicated because there is a mixture between direct and indirect interactions, and there might be multivalent interactions, which means a particular protein binds simultaneously to several different partners in different complexes. Furthermore, some proteins exist as a component of a larger complex as well as a free form in the cells. Unfortunately, ATG13 forms a tight complex with ATG101 and also interacts with ATG14 independently [29], which makes for a complicated explanation. Therefore, we performed the same immunoprecipitation experiments, with the addition of co-transfecting ATG13 (Figure S6). When we co-transfected ATG101ΔC and ATG13, the amount of precipitated ATG14, UVRAG, BECN1, and PIK3C3 was significantly decreased compared with the full-length ATG101 (Figure S6), which is a consistent result with endogenous ATG13 (Figure 2(a-c)).
To further dissect molecular interactions mediated by the C-terminal region of ATG101, we generated the constructs with only the C-terminal region of ATG101 fused with GST (GST- ATG101 C197) and only the N-terminal region of ATG101 fused with GST (GST-ATG101N196), and then checked in vitro binding with an immunoprecipitation assay (Figure 3(a)). The purified PtdIns3K immune complex was mixed with bacterially-expressed ATG101 constructs for assessing direct interaction. Two different PtdIns3K complexes, PIK3C3-BECN1 and PIK3C3-BECN1-ATG14 were tested and there was a binding between both complexes with full-length ATG101. Furthermore, only the C-terminal region of ATG101 was able to interact with both complexes, however, the interaction was enhanced in the presence of ATG14 (Figure 3(b)). These results indicate that the C-terminal region of ATG101 directly interacts with the PtdIns3K complex, and the interaction might be mediated by ATG14 or with a higher affinity state of PtdIns3K complex in the presence of ATG14. Based on our immunoprecipitation assay, the interaction must be very weak, which is one of the multivalent interactions between the ULK1 and PtdIns3K complexes. The C-terminal region of ATG101 plays a role in the direct interaction with the PtdIns3K complex no matter which molecules are involved.
Figure 3.
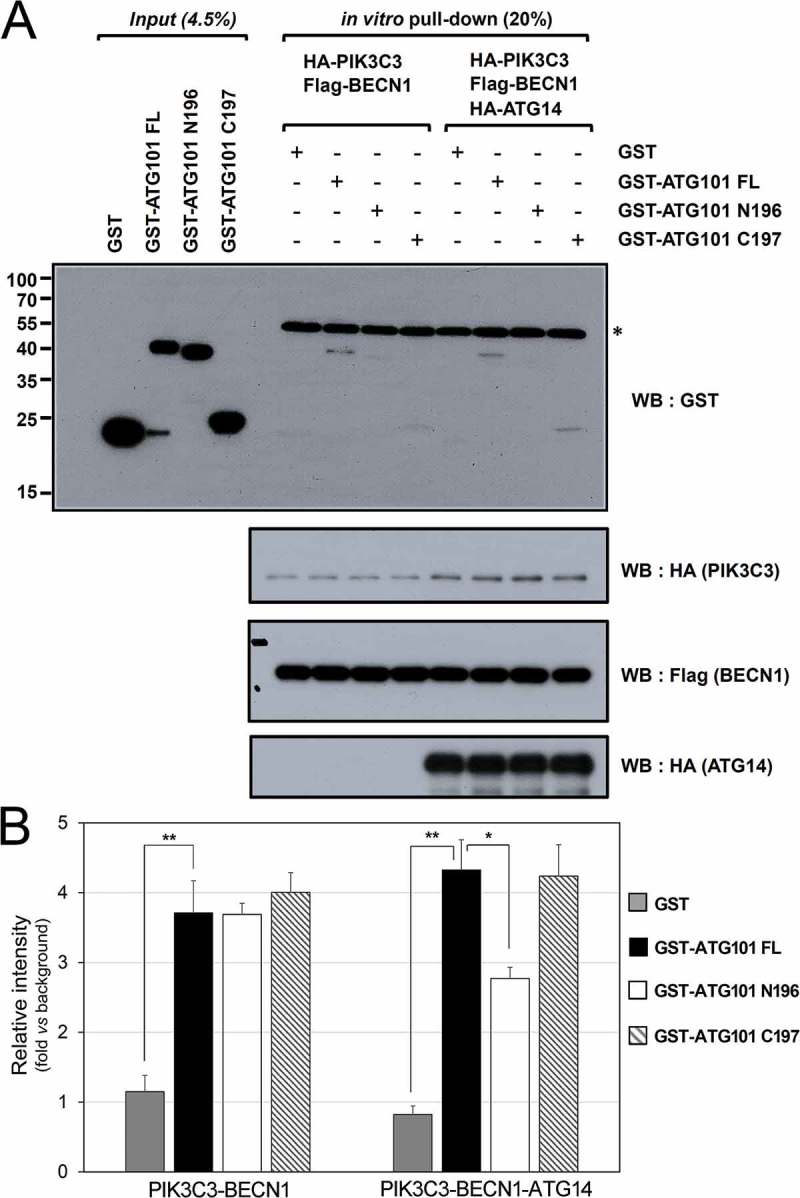
ATG101 directly interacts with the PtdIns3K complex through its C terminus. GST proteins (4.5%) used in the binding assay and 20% of final ATG101-PtdIns3K complex binding product in vitro were subjected to the analysis as described in Materials and Methods. *, non-specific band (presumably IgG heavy-chain). (B) Quantification data for the interaction between ATG101 and PtdIns3K complex in presence or absence of HA-ATG14. Fold changes are represented by an assay background as a reference (mean ± S.D.; n = 3; **, P < 0.01; *, P < 0.05).
The WF finger region of ATG101 does not bind to the PtdIns3K complex
Previously, the WF finger region of ATG101 has been proposed to bind downstream factors in the autophagy pathway [35]. Clearly, it plays a critical role in autophagy based on the study using a triple W110A/P111A/F112A mutant; however, the details of the mechanism are not clear and we have checked the binding of this region to PtdIns3K complex. Initially, we mutated both Trp110 and Phe112 residues of ATG101 to alanine (APA mutant) because the proline residue may play a role in the maintenance of a particular conformation, and the interactions between the APA mutant and each of the PtdIns3K complex components, ATG14, UVRAG, BECN1, and PIK3C3, were monitored. Interestingly, there was no effect on the band intensity of the PtdIns3K complex components using either the WT or APA mutants (Figure 4). The side chain of alanine is small compared with tryptophan and phenylalanine residues but still hydrophobic; therefore, we generated another mutant, SPS, which mutates the hydrophobic Trp110 and Phe112 residues to polar serine residues. The immunoprecipitation result using an SPS mutant is basically the same as that obtained using an APA mutant (Figure 4). Therefore, we conclude that this WF finger region shows no binding affinity to the PtdIns3K complex.
Figure 4.
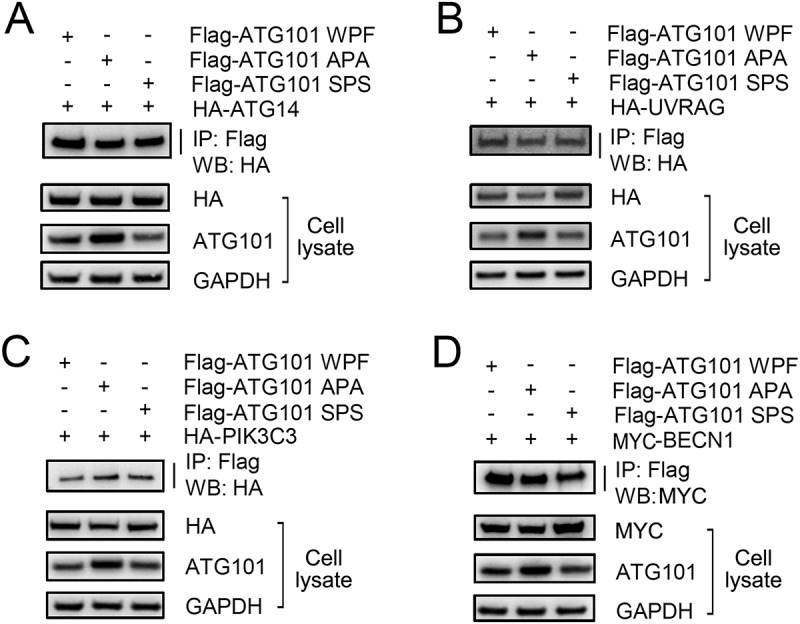
The WF finger motif of ATG101 is not responsible for binding to PtdIns3K complex. To determine the function of WF motif of ATG101, full-length wild-type Flag-ATG101 (Flag-ATG101-WPF) or full-length Flag-ATG101 mutants (Flag-ATG101 APA, Flag-ATG101 SPS) were co-transfected with PtdIns3K complex components (HA-ATG14 [A], HA-UVRAG [B], HA-PIK3C3 [C], and myc-BECN1 [D]) into HEK293 cells. For western blotting, cells were harvested and immunoblotted with the indicated antibodies. GAPDH was used as a loading control.
Effect of the C-terminal region of ATG101 on autophagy
Since the C-terminal region of ATG101 interacts with the PtdIns3K complex (Figure 3), the next obvious question is whether this interaction affects autophagy in cells. Therefore, we performed the standard cell-based autophagy assays using several ATG101 mutants. First, we generated an ATG101 knockout (KO) cell line and then transfected full-length ATG101, ATG101ΔC, and the WF finger APA mutant. The previous report showed that the WF finger (AAA triple mutant) mutant caused a significant reduction of autophagosome formation, which is monitored by MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) turnover, SQSTM1/p62 (sequestosome 1) accumulation, and a GFP (green fluorescent protein)-LC3 puncta assay [35]. LC3 turnover is slightly impaired by ATG101 deletion, but restored by the addition of full-length wild-type ATG101; however, the restoration was affected when the APA mutant was introduced (Figure 5(a)). This result shows that our APA mutant also behaved similarly to previous AAA triple mutants [35], although the effect of LC3 turnover and accumulation of SQSTM1 are not prominent due to the induction of basal autophagy in the ATG101 KO MIA PaCa-2 human pancreatic cancer cell line (Figure 5(a)). Interestingly, the ATG101ΔC mutant behaved much obviously to patterns of LC3 turnover autophagy in the ATG101 KO HeLa cell line (Figure S7), suggesting that the C-terminal region of ATG101 plays an important role in autophagy.
Figure 5.
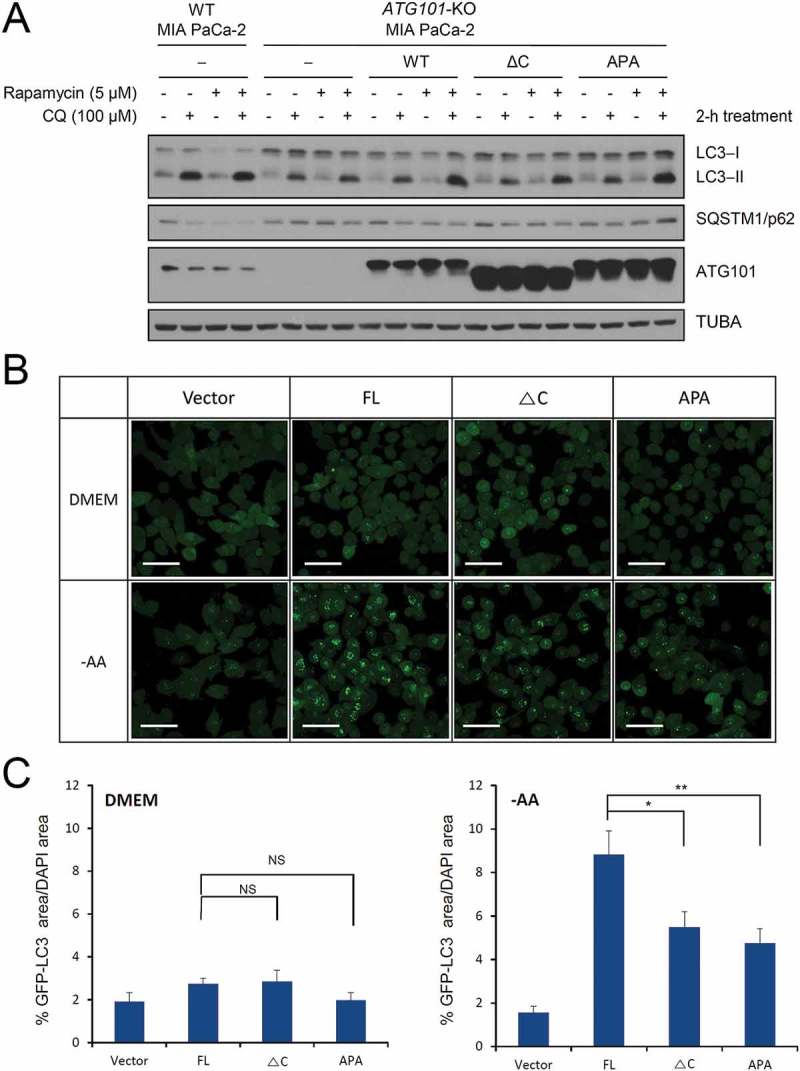
The role of the C-terminal region of ATG101 in the autophagy pathway. Autophagy defect shown in ATG101 knockout cells is restored by the expression of full-length ATG101 but not ATG101ΔC. (a) ATG101-knockout MIA PaCa-2 cells were transfected by a full-length wild-type of ATG101 (WT), a C-terminal deletion mutant of ATG101 (ΔC), and full-length ATG101 APA mutant (APA), respectively, and incubated for 48 h. After replacement with complete media containing 5 μM rapamycin (Sigma, R8781) with or without 100 μM chloroquine (CQ; Sigma, C6628), cells were further incubated for 2 h. For western blotting, cells were harvested and immunoblotted for ATG101, LC3, SQSTM1, and TUBA. (b) GFP-LC3 puncta were analyzed by live imaging using fluorescence microscopy after 2 h incubation of either complete (DMEM) or amino acid-depleted (-AA) media. Scale bar: 50 μm. (c) Quantification data for GFP-LC3 puncta area are expressed as a percentage of the DAPI area within the cell. NS, *, and ** mean P > 0.05, P < 0.05, and P < 0.01, respectively.
To further examine the immunoprecipitation data of ATG101 and its mutants, various constructs were generated for the monitoring of autophagosome formation using GFP-tagged LC3, a well-known autophagy marker [6,33,40,41]. Autophagy was induced by nutrient-starvation, and punctate GFP-LC3 fluorescent signals indicating the recruitment of LC3 protein to autophagosomes were detected in the cytosol (Figure 5(b)). The GFP-LC3 puncta were markedly decreased in the ATG101 KO cell line in both complete DMEM and amino acid (AA)-depleted media. However, autophagosome formation was restored by the addition of full-length wild-type ATG101, especially in AA-depleted autophagy induction media (Figure 5(b)). In autophagy induction media, overexpression of both ATG101ΔC and APA mutants cannot fully restore the GFP puncta in the cells, indicating that LC3 recruitment to the autophagosomes is defective. Quantification of the fluorescence data indicated that a significant reduction was shown by both APA and ATG101ΔC mutants, but the APA mutant seems to have a slightly severe defect on autophagy (Figure 5(c)). Although the functional output is similar, the mechanism is different. As a previous study suggested, the WF finger region of ATG101 affects autophagy by the other downstream factors whereas the flexible C-terminal region of ATG101 affects the PtdIns3K complex (Figure 6).
Figure 6.
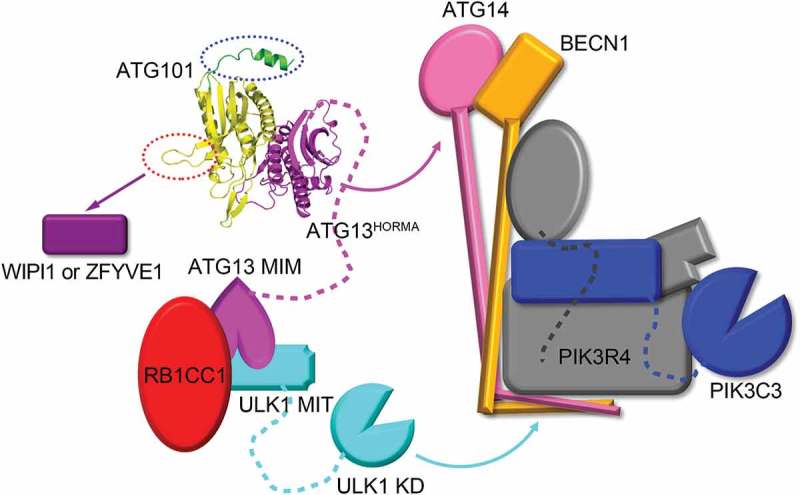
A model of the function of ATG101 in the ULK1 complex. The interaction between ATG13HORMA and ATG14 is indicated with the magenta curved arrow and the ULK1 kinase phosphorylates several components of PtdIns3K complex indicated by the cyan curved arrow. The C-terminal region of ATG101 (green ribbon) that binds to PtdIns3K complexes (C1 and C2) and is indicated by the blue dotted oval. A WF finger of ATG101 (red dotted oval) cannot interact with the PtdIns3K complex but affects WIPI1 or ZFYVE1 function indicated by the purple arrow. KD, kinase domain; MIT, microtubule interacting and transport; MIM, MIT-interacting motif; RB1CC1, RB1 inducible coiled-coil 1; WIPI1, WD repeat domain, phosphoinositide interacting 1; ZFYVE1, zinc finger FYVE-type containing 1.
Discussion
Although the cellular output of autophagy is a relatively simple destruction of cargo molecules, the molecular details of the autophagy pathway are very complicated and include multiple steps from initiation (or induction) to the complete destruction inside lysosomes [2,42,43]. A variety of autophagy molecules are involved in each step and even more, multi-protein complexes are responsible for the specific molecular events. Among them, the initiation of autophagy is triggered by the ULK1 complex (ULK1, RB1CC1, ATG13, and ATG101) [16]. The enzymatic activity of ULK1 kinase is critical, however, the modes of regulation are diverse, depending on the different complex formations. For phosphorylation of different substrates by ULK1, kinase must be a consequence of different modes of interaction between ULK1 complex and either the substrate itself or the complex-containing substrate [19]. Several components of the PtdIns3K complex are phosphorylated by the ULK1 complex and this event is critical for the downstream autophagy process. Previously, it has been reported that ATG13 binds to ATG14 and ULK1 subsequently phosphorylates serine 29 of ATG14 [29]. BECN1 and PIK3C3 are also known as ULK1 substrates [28], but the molecular contacts that regulate these phosphorylations are still unknown. ATG13 also binds the other molecules within the ULK1 complex such as the C-terminal MIM (MIT-interacting motif) of ATG13 interacts with the MIT (microtubule interacting and transport) domain of ULK1; the N-terminal HORMA domain of ATG13 binds the HORMA domain of ATG101. ATG13 forms a tight complex with ATG101, which resembles the heterodimeric HORMA domain interaction found in O-MAD2:C-MAD2 complex [34,36,44]. ATG101 was originally identified as a binding partner of ATG13 but not studied extensively [17,18]. Only recently, the physiological roles of ATG101 are starting to be revealed [15] and are now classified into 3 roles.
First, it has been reported that ATG101 controls the stability of ATG13 in cells [17,18]. Deletion of ATG101 causes a marked reduction of ATG13 and this earlier finding is now clearly explained by the structural and biochemical study of the ATG101-ATG13HORMA complex [34,35]. The structure of ATG101 is mainly a HORMA domain fold (Figure 1(a)) and the binding interface with ATG13 is large, thus ATG13 is not stable without an ATG101 molecule. This is not direct evidence of the instability of ATG13 in mammalian cells, but the HORMA domain of HsATG13 is only stably expressed in E. coli by the co-expression of ATG101 simultaneously, whereas that of Atg13 from budding yeast can be expressed in E. coli without a binding partner [31].
Second, Suzuki et al. identified the role of the WF finger region of SpAtg101 for interaction with downstream factors in the autophagy pathway [35]. They did not show the direct interactions, but ATG101 plays an important role in the recruitment of WIPI1 (WD repeat domain, phosphoinositide interacting 1) and ZFYVE1/DFCP1 (zinc finger FYVE-type containing 1) (Figure 6). Later, the conformational change of this WF finger region in HsATG101 was reported [34]. The hydrophobic residues are inward and this makes for a closed conformation of HsATG101. However, when they are outward, it makes for an open conformation for better interaction with downstream factors (Figure 1(b)). Very interestingly, our study shows that this WF finger region does not interact with the PtdIns3K complex at all (Figure 3).
Third, this PtdIns3K interaction is mediated by the C-terminal flexible region of ATG101 (Figure 2). As we noted, while we were preparing the manuscript, structural studies of ATG101-ATG13HORMA have been reported [34,35,38]; however, fortunately, we were able to get the complete structure of ATG101 including the C-terminal region (Figure 1(a)). Therefore, the role of the C-terminal region was further studied using the C-terminal deletion mutant of ATG101. Clearly, the deletion of this region causes a marked reduction of the binding to PtdIns3K components (Figure 2), and this interaction is directly mediated by the C-terminal region of ATG101 and is augmented in the presence of ATG14 (Figure 3). More importantly, this C-terminal deletion impairs autophagosome formation based on the GFP-LC3 puncta assay results (Figure 5(b,c)). It has been reported that the HORMA domain of ATG13 directly interacts with the middle region of ATG14 [29] and the newly identified interaction between the ULK1 and PtdIns3K complexes in this study might be important for correct autophagy induction signaling.
Therefore, taken together, this small 25-kDa ATG101 protein within a huge 1-MDa ULK1 complex plays critical roles as a signaling hub, such as stabilizing ATG13 by heterodimer formation, the interaction with the PtdIns3K complex via the C-terminal region, and the interaction with other downstream factors via the WF finger (Figure 6). As noted, a clear dissection of the interaction among the multi-protein complexes is not straightforward. The deletion of the C-terminal region of ATG101 significantly affects the binding to both ATG14 and UVRAG, which independently forms the different complexes with the PtdIns3K complex core, PtdIns3K-C1 and PtdIns3K-C2. Furthermore, no sequence homology between ATG14 and UVRAG is detected. The easier interpretation might be that the C-terminal region of ATG101 binds to the PtdIns3K complex core components (BECN1, PIK3R4, and PIK3C3), however, our immunoprecipitation data with the C-terminal region of ATG101 showed that the direct interaction to the PtdIns3K complex is augmented in the presence of ATG14. Therefore, another interpretation might be the conformational transition of the PtdIns3K complex core components to high affinity state in the presence of ATG14 or UVRAG. For a complete understanding of the communication between the ULK1 and PtdIns3K complexes, we must await the structural study of the whole complex by X-ray crystallography or, more likely, cryo-electron microscopy and strong supporting biochemical and cell biology data.
Materials and methods
Chemicals and reagents
Anti-Flag antibody was obtained from Sigma (F3165) and from Cell Signaling Technology (8146). Anti-HA was from Covance (MMS-101P) and from Cell Signaling Technology (3724). Horseradish peroxidase (HRP)-conjugated anti-Flag and anti-HA were purchased from Cell Signaling Technology (86,861 and 2999, respectively). Anti-MYC was from Covance (MMS-164P) and from Cell Signaling Technology (2276). Primary antibodies against ATG101, LC3A/B, and SQSTM1 were purchased from Cell Signaling Technology (13,492, 4108, and 5114, respectively). Primary antibodies against GFP and GAPDH were purchased from Santa Cruz Biotechnology (sc-9996 and sc-25,778, respectively), TUBA from Sigma-Aldrich (T6074), and ACTB from Bethyl Laboratories (A300-485A). The secondary antibodies, HRP-linked anti-rabbit and anti-mouse antibodies were from Bethyl Laboratories (A120-101P and A90-131P, respectively). Non-essential amino acids (11140076), essential amino acids (11130051), glutamine (25030081), HEPES (15630080), vitamin solution (11120052), sodium bicarbonate (A19875), DAPI (D3571), and Hoechst 33258 (H3569) were from Thermo Fisher Scientific.
Cloning, expression, and purification
DNAs encoding full-length ATG101 (residues 1–218) and ATG13HORMA (residues 1–190) were amplified from the cDNA of the human testis (Clontech, 637209). These were cloned into Escherichia coli expression vector pETDuet-1 (Novagen, 71146) and the resulting vector co-expresses the 6xHis-tag fused ATG13HORMA and full-length of ATG101 simultaneously. For crystallization, we designed a triple mutant K40A/K41A/E42A of ATG101 based on the suggestion of SERp server (http://services.mbi.ucla.edu/SER/) [45]. The mutation was introduced by PCR-mediated site-directed mutagenesis. The mutated vector was transformed into E. coli BL21(DE3) cells. The transformed cells were grown at 37°C to an OD600 of 0.6 and then induced by the addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG; Gold Biotechnology, 12481C100) at 18°C for 22 h. The cells were harvested by centrifugation at 8,671 × g for 20 min and resuspended in 50 mM Tris-HCl pH 8.0, 200 mM NaCl, 5 mM imidazole (Affymetrix, 17525), 1 mM tris(2-carboxyethyl)phosphine (TCEP; Gold Biotechnology, TCEP25). After sonication and centrifugation at 27,216 × g for 1 h, the supernatant was loaded onto a His-Trap column (GE healthcare, 17-5255-01) and then eluted with 200 ~ 300 mM imidazole. The eluted sample was further purified by ion-exchange column chromatography using 5 ml HiTrap Q Fast Flow (GE Healthcare, 17-5156-01). Finally, the ATG101 and ATG13HORMA protein complex was loaded onto a HiLoad 16/600 Superdex 75 pg column (GE Healthcare, 28-9893-33) pre-equilibrated with 20 mM HEPES (Sigma, H3375) pH 7.5, 200 mM NaCl, and 1 mM TCEP. Fractions were collected and concentrated to 10–13 mg/ml for crystallization by ultrafiltration using an Amicon Ultra-15 Centrifugal Filter Unit (Millipore, UFC903024).
Selenomethionine (SeMet)-labeled protein complex was expressed in B834(DE3) cells. SeMet-substituted ATG101-ATG13HORMA complex was purified in a similar manner to that described for the wild-type protein complex.
Crystallization
Crystallization of wild-type ATG101-ATG13HORMA complex was performed with a hanging drop vapor diffusion method at 20°C. Initial crystals were grown with drops containing 1 μl of 10 mg/ml protein and 1 μl of reservoir solution (0.1 M Tris-HCl, pH 8.5 and 1.0 M [NH4]2HPO4) for 1 d which were then used for micro-seeds. The optimized crystal was grown with drops containing 1.2 μl of 12 mg/ml protein, 0.4 μl of micro-seed solution, and 0.8 μl of reservoir solution (0.1 M HEPES, pH 7.5, 0.2 M MgCl2, 6 ~ 8% [w:v] PEG 3,350 [Hampton Research, HR2-527]) for 2 d. The crystals were flash-frozen in liquid nitrogen with cryo-protectant that is above reservoir solution containing 30% (w:v) glycerol. Crystals of SeMet-labeled ATG101-ATG13HORMA complex were prepared in the same condition with native crystal.
A single-wavelength anomalous dispersion (SAD) and the highest resolution data were collected at the 5C beamline of Pohang Accelerator Laboratory, South Korea (Table 1). The data sets with different crystal forms were collected at the NE3A and the BL17A beamline of Photon Factory and at the BL26B2 beamline of SPring-8, Japan. All diffraction data were processed with HKL2000 software [46].
Structure determination and refinement
After 14 selenium sites were located and the initial phases calculated, density modification and automated model building were performed with AutoSol [47] and AutoBuild [48]. Models were rebuilt manually using COOT [49]. Refinement with non-crystallographic symmetry restrains was carried out using the Phenix.refine and REFMAC [50]. Statistics for the collected data and refinement are listed in Table 1.
Size-exclusion chromatography with small angle X-ray scattering (SEC-SAXS)
SAXS data were collected on the beamline BL-10C of Photon Factory, Tsukuba, Japan with a sample-to-detector distance of 2.0 m. The wavelength of X-ray was set to 1.488 Å. These data were collected using the SEC-SAXS technique, which was used to exclude the effect of protein aggregation. The protein complex (100 μl at 7.8 mg/ml) was injected on to a Superdex 200 Increase 10/300 GL (GE Healthcare, 28-9909-44) pre-equilibrated with 20 mM HEPES pH7.5, 200 mM NaCl, and 1 mM TCEP, and eluted at a flow rate of 0.05 ml/min. Scattering data were collected in 20 s exposures over the course of the elution. Details of the experimental parameters are shown in Table S1. Guinier analysis of each profile across the single elution peak showed a consistent radius of gyration (Rg) and a total of scatter profiles were averaged and background subtracted. Guinier analysis of data was performed using PRIMUS and indirect Fourier transform with GNOM to obtain the distance distribution function, P(r), and the maximum dimension of the scattering particle, Dmax [51]. Theoretical scattering curves were calculated from crystal structure atomic coordinates and compared with experimental scattering curves using CRYSOL. Ab initio modeling and averaging of these models were performed using DAMMIF and DAMAVER, respectively. Rigid body modeling of the crystallographic structure of dummy-atom models was computed using the Situs program suite [52].
Cell lines and culture conditions
HEK293 cells (ATCC, CRL-1573) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone, SH30243.01) medium containing 10% fetal bovine serum (Hyclone, SH30084.03) and 50 mg/ml penicillin/streptomycin (Gibco, 15070063). Transfection with polyethylenimine (PEI; Sigma, 408727) and Lipofectamine 2000 (Invitrogen, 11668019) was performed as described [41]. MIA PaCa-2 human pancreatic cancer cells (ATCC, CRL-1420) were maintained in DMEM. All cells were maintained in a humidified atmosphere of 5% (v:v) CO2 at 37°C in medium supplemented with 10% fetal bovine serum.
Stable cell lines
To generate ATG101 KO cells, pLentiCRISPRv2-based ATG101 CRISPR-Cas9 guide RNA expression plasmid (Gene Script, USA, U0448BI200-1) was transfected into HEK293T cells (ATCC, CRL-3216) in combination with the lentiviral packaging plasmids, pMDLG/pRRE (Addgene, 12251), pRSV-Rev (Addgene, 12253) and envelope plasmid, pMD2.G (Addgene, 12259) using Lipofectamine 2000. The lentivirus particles-containing culture medium was harvested at 24–72 h after transfection. MIA PaCa-2 cells were incubated with lentivirus-containing medium together with polybrene (Sigma, H9268). After 24–48 h incubation, the culture medium then was removed and replaced with complete DMEM media. After 1 d recombinant lentiviruses-infected cells were selected with puromycin (2 μg/ml; Gibco, A1113802)-containing culture medium. For isolation of single clone from ATG101 KO cells, cells were sorted into 96-well plates using flow cytometry and further incubated in culture media with puromycin (2 μg/ml) to reach confluency. Immunoblotting was performed to test the ATG101 KO.
Immunoblot and immunoprecipitation assay
To perform immunoprecipitation assay for MAP1LC3 turnover, several human cells expressing the following plasmids were prepared. The ATG101ΔC, APA, SPS, LHRR mutants of ATG101 were generated using the standard mutagenesis method. After 48 h transfection, cells were lysed with immunoprecipitation buffer containing 20 mM Tris-HCl pH 7.5, 500 mM NaCl, 0.5% (v:v) NP-40 (Sigma, I8896), 1 mM EDTA (Sigma, E4884), 1 mM PMSF (Roche, 20039220), 5 μg/ml leupeptin (Thermo Fisher Scientific, AMR-M180-1), and 2 mM Na3VO4 (Sigma, 450243). The lysates were mixed with antibodies overnight, and the immune complexes were then incubated with protein G-Sepharose beads (Thermo Fisher Scientific, 20399) for 2 h. After washing, beads were boiled with SDS sample buffer (100 mM Tris-HCl pH 6.8, 4% [w:v] sodium dodecyl sulfate [affymetrix, 75819], 0.2% [w:v] bromophenol blue [GOLDBIO, B-092–25], 20% [v:v] glycerol [affymetrix, 16374], and 200 mM dithiothreitol [GOLDBIO, DTT50]) for 5 min. The samples were loaded onto SDS-PAGE gels, transferred to a PVDF membrane (Invitrogen, IB24001), and incubated with primary antibody at 4°C overnight. After a further incubation with HRP-conjugated secondary antibody for 1 h at room temperature, immune-reactive bands were visualized using a Immobilon Forte Western HRP substrate solution (EMD Millipore, WBLUF0500).
To test a direct ATG101-PtdIns3K complex binding in vitro, 2 different PtdIns3K complexes (PIK3C3-BECN1 and PIK3C3-BECN1-ATG14) were first immuno-purified from the transfected HEK293 cells. The purified PtdIns3K immune complexes were then incubated with 8 μg of bacterial-purified GST-ATG101 full-length (FL), GST-ATG101 N-terminal fragment (N196; residues 1–196), and GST-ATG101 C-terminal fragment (C197; residues 197–218) as indicated, in which GST alone was used as a negative control for binding assay. After in vitro binding reaction, the immune complexes were extensively washed with mild lysis buffer (10 mM Tris-HCl, pH 7.5, 2 mM EDTA, 100 mM NaCl, 0.1% NP-40, 50 mM NaF [Sigma, 47072], 1 mM Na3VO4, and protease inhibitor cocktail [Roche, 05892791001]) and then ATG101 binding and the PtdIns3K immune complexes were analyzed by western blotting. To further confirm, after 2 d cells were lysed with mild lysis buffer containing protease inhibitors and then 200 μg of total cell lysates were added onto Flag antibody-coated immunoassay plates. The GST-ATG101 proteins (1 μg) were added and then the binding of GST-ATG101 proteins with PtdIns3K complex immobilized on immunoassay plate were determined by ELISA assay using GST antibody.
Fluorescence microscopy
Subcellular localization of GFP-LC3 was monitored by LSM780 confocal fluorescent microscope (Carl Zeiss, Germany). The levels of GFP-LC3 puncta were quantified as total puncta area per cell which was normalized by the area of the DAPI-stained nucleus of that cell using the image analysis software provided by microscope (Carl Zeiss, Germany).
Accession codes
Atomic coordinates and structure factor files have been deposited in the Protein Data Bank under the following accession codes: 5XUY, 5XV3, 5XV4, and 5XV6 for C2, P21212, P2, and C2221 crystal forms, respectively.
Funding Statement
This work was supported by National Research Foundation of Korea (NRF) grants from the Korean government (NRF-2016R1E1A1A01942623, BRL grant: No. 2015041919, and International Cooperation Program: No. 2015K2A2A6002008 to H.K.S.; NRF-2017R1A2B4011732 to H.C.).
Acknowledgments
We thank the staff at beamline 5C and 11C, Pohang Accelerator Laboratory, Korea and beamline BL17A and NE3A, Photon Factory, Japan for their help with the X-ray data collection. This work was in part performed under the International Collaborative Research Program of Institute for Protein Research, Osaka University (ICR-17-05). Diffraction data were collected at the Osaka University beamline BL44XU at SPring-8 (Harima, Japan) (Proposal Nos. 2017A6775 and 2017B6775). We also thank the staff at beamline BL10C, Photon Factory, Japan and beamline 4C, Pohang Accelerator Laboratory, Korea for their help with the SAXS data collection. MIA PaCa-2 human pancreatic cancer cell line was kindly provided by Dr. Yun-Hee Kim, National Cancer Center, Korea.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data for this article are accessed here.
References
- [1].Klionsky DJ, Codogno P.. The mechanism and physiological function of macroautophagy. J Innate Immun. 2013;5(5):427–433. PMID: 23774579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Reggiori F, Klionsky DJ.. Autophagic processes in yeast: mechanism, machinery and regulation. Genetics. 2013;194(2):341–361. PMID: 23733851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zaffagnini G, Martens S. Mechanisms of selective autophagy. J Mol Biol. 2016;428(9Pt A):1714–1724. PMID: 26876603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kim BW, Kwon DH, Song HK. Structure biology of selective autophagy receptors. BMB Rep. 2016;49(2): 73–80. PMID: 26698872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Farre JC, Subramani S. Mechanistic insights into selective autophagy pathways: lessons from yeast. Nat Rev Mol Cell Biol. 2016;17(9):537–552. PMID: 27381245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kwon DH, Kim S, Jung YO, et al. The 1:2 complex between RavZ and LC3 reveals a mechanism for deconjugation of LC3 on the phagophore membrane. Autophagy. 2017;13(1):70–81. PMID: 27791457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kwon DH, Kim L, Kim BW, et al. A novel conformation of the LC3-interacting region motif revealed by the structure of a complex between LC3B and RavZ. Biochem Biophys Res Commun. 2017;490(3):1093–1099. PMID: 28668392. [DOI] [PubMed] [Google Scholar]
- [8].Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015;22(3):367–376. PMID: 25257169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34(7):856–880. PMID: 25712477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13(10):722–737. PMID: 24064518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Manzanillo PS, Ayres JS, Watson RO, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501(7468):512–516. PMID: 24005326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in parkinson’s disease. Neuron. 2015;85(2):257–273. PMID: 25611507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].RA Nixon. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983–997. PMID: 23921753. [DOI] [PubMed] [Google Scholar]
- [14].Kwon DH, Song HK. A structural view of xenophagy, a battle between host and microbes. Mol Cells. 2018;41(1):27–34. PMID: 29370690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Noda NN, Mizushima N. Atg101: not just an accessory subunit in the autophagy-initiation complex. Cell Struct Funct. 2016;41(1):13–20. PMID: 26754330. [DOI] [PubMed] [Google Scholar]
- [16].Hurley JH, Young LN. Mechanisms of autophagy initiation. Annu Rev Biochem. 2017;86:225–244. PMID: 28301741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hosokawa N, Sasaki T, Iemura S, et al. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy. 2009;5(7):973–979. PMID: 19597335. [DOI] [PubMed] [Google Scholar]
- [18].Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy. 2009;5(5): 649–662. PMID: 19287211. [DOI] [PubMed] [Google Scholar]
- [19].Noda NN, Fujioka Y. Atg1 family kinases in autophagy initiation. Cell Mol Life Sci. 2015;72(16):3083–3096. PMID: 25948417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lin MG, Hurley JH. Structure and function of the ULK1 complex in autophagy. Curr Opin Cell Biol. 2016;39:61–68. PMID: 26921696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Papinski D, Kraft C. Regulation of autophagy by signaling through the Atg1/ULK1 Complex. J Mol Biol. 2016;428(9):1725–1741. PMID: WOS:000376695000006. [DOI] [PubMed] [Google Scholar]
- [22].Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–461. PMID: 21205641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. PMID: 21258367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ma M, Liu JJ, Li Y, et al. Cryo-EM structure and biochemical analysis reveal the basis of the functional difference between human PI3KC3-C1 and -C2. Cell Res. 2017. PMID: 28731030. doi: 10.1038/cr.2017.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhong Y, Wang QJ, Li X, et al. Distinct regulation of autophagic activity by Atg14L and rubicon associated with beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11(4):468–476. PMID: 19270693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sun T, Li X, Zhang P, et al. Acetylation of Beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat Commun. 2015;6:7215 PMID: 26008601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6(6): 764–776. PMID: 20639694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741–750. PMID: 23685627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Park JM, Jung CH, Seo M, et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy. 2016;12(3):547–564. PMID: 27046250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jao CC, Ragusa MJ, Stanley RE, et al. What the N-terminal domain of Atg13 looks like and what it does: a HORMA fold required for PtdIns 3-kinase recruitment. Autophagy. 2013;9(7):1112–1114. PMID: 23670046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jao CC, Ragusa MJ, Stanley RE, et al. A HORMA domain in Atg13 mediates PI 3-kinase recruitment in autophagy. Proc Natl Acad Sci U S A. 2013;110(14):5486–5491. PMID: 23509291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Alers S, Wesselborg S, Stork B. ATG13: just a companion, or an executor of the autophagic program? Autophagy. 2014;10(6):944–956. PMID: 24879146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Popelka H, Klionsky DJ. The molecular mechanism of Atg13 function in autophagy induction: what is hidden behind the data? Autophagy. 2017;13(3):449–451. PMID: 28118060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Qi S, Kim DJ, Stjepanovic G, et al. Structure of the human Atg13-Atg101 HORMA heterodimer: an interaction hub within the ULK1 complex. Structure. 2015;23(10):1848–1857. PMID: 26299944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Suzuki H, Kaizuka T, Mizushima N, et al. Structure of the Atg101-Atg13 complex reveals essential roles of Atg101 in autophagy initiation. Nat Struct Mol Biol. 2015;22(7):572–580. PMID: 26030876. [DOI] [PubMed] [Google Scholar]
- [36].Mapelli M, Massimiliano L, Santaguida S, et al. The mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell. 2007;131(4):730–743. PMID: 18022367. [DOI] [PubMed] [Google Scholar]
- [37].Popelka H, Klionsky DJ. One step closer to understanding mammalian macroautophagy initiation: interplay of 2 HORMA architectures in the ULK1 complex. Autophagy. 2015;11(11):1953–1955. PMID: 26325630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Michel M, Schwarten M, Decker C, et al. The mammalian autophagy initiator complex contains 2 HORMA domain proteins. Autophagy. 2015;11(12):2300–2308. PMID: 26236954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Buchan DW, Minneci F, Nugent TC, et al. Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Res. 2013;41(Web Server issue):W349–W357. PMID: 23748958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–5728. PMID: 11060023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kim JH, Hong SB, Lee JK, et al. Insights into autophagosome maturation revealed by the structures of ATG5 with its interacting partners. Autophagy. 2015;11(1):75–87. PMID: 25484072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat Struct Mol Biol. 2014;21(4):336–345. PMID: 24699082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. PMID: 21801009. [DOI] [PubMed] [Google Scholar]
- [44].Suzuki SW, Yamamoto H, Oikawa Y, et al. Atg13 HORMA domain recruits Atg9 vesicles during autophagosome formation. Proc Natl Acad Sci U S A. 2015;112(11):3350–3355. PMID: 25737544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Goldschmidt L, Cooper DR, Derewenda ZS, et al. Toward rational protein crystallization: A Web server for the design of crystallizable protein variants. Protein Sci. 2007;16(8):1569–1576. PMID: 17656576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode In: Carter JCW, Sweet RM, editors. Methods Enzymol. Vol. 276 New York: Academic Press; 1997. p. 307–326. [DOI] [PubMed] [Google Scholar]
- [47].Terwilliger TC, Adams PD, Read RJ, et al. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr D Biol Crystallogr. 2009;65(Pt6):582–601. PMID: 19465773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr. 2008;64(Pt1):61–69. PMID: 18094468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt12 Pt 1):2126–2132. PMID: 15572765. [DOI] [PubMed] [Google Scholar]
- [50].Afonine PV, Grosse-Kunstleve RW, Echols N, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68(Pt4):352–367. PMID: 22505256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Petoukhov MV, Franke D, Shkumatov AV, et al. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Crystallogr. 2012;45(Pt2):342–350. PMID: 25484842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wriggers W, Chacon P. Using Situs for the registration of protein structures with low-resolution bead models from X-ray solution scattering. Journal of Applied Crystallography. 2001;34:773–776. PMID: WOS:000172233900016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.