

ABSTRACT
Ferroptosis is a recently recognized form of regulated cell death that is characterized by lipid peroxidation. However, the molecular mechanisms regulating ferroptosis are largely unknown. In this study, we report that the RNA-binding protein ELAVL1/HuR plays a crucial role in regulating ferroptosis in liver fibrosis. Upon exposure to ferroptosis-inducing compounds, ELAVL1 protein expression was remarkably increased through the inhibition of the ubiquitin-proteasome pathway. ELAVL1 siRNA led to ferroptosis resistance, whereas ELAVL1 plasmid contributed to classical ferroptotic events. Interestingly, upregulated ELAVL1 expression also appeared to increase autophagosome generation and macroautophagic/autophagic flux, which was the underlying mechanism for ELAVL1-enhanced ferroptosis. Autophagy depletion completely impaired ELAVL1-mediated ferroptotic events, whereas autophagy induction showed a synergistic effect with ELAVL1. Importantly, ELAVL1 promoted autophagy activation via binding to the AU-rich elements within the F3 of the 3ʹ-untranslated region of BECN1/Beclin1 mRNA. The internal deletion of the F3 region abrogated the ELAVL1-mediated BECN1 mRNA stability, and, in turn, prevented ELAVL1-enhanced ferroptosis. In mice, treatment with sorafenib alleviated murine liver fibrosis by inducing hepatic stellate cell (HSC) ferroptosis. HSC-specific knockdown of ELAVL1 impaired sorafenib-induced HSC ferroptosis in murine liver fibrosis. Noteworthy, we retrospectively analyzed the effect of sorafenib on HSC ferroptosis in advanced fibrotic patients with hepatocellular carcinoma receiving sorafenib monotherapy. Attractively, ELAVL1 upregulation, ferritinophagy activation, and ferroptosis induction occurred in primary human HSCs from the collected human liver tissue. Overall, these results reveal novel molecular mechanisms and signaling pathways of ferroptosis, and also identify ELAVL1-autophagy-dependent ferroptosis as a potential target for the treatment of liver fibrosis.
Abbreviations: ACTA2/alpha-SMA: actin, alpha 2, smooth muscle, aorta; ACTB/beta-actin: actin beta; ARE: AU-rich element; ATG: autophagy related; BDL: bile duct ligation; BECN1: beclin 1; BSO: buthionine sulfoximine; COL1A1: collagen type I alpha 1 chain; ELAVL1/HuR: ELAV like RNA binding protein 1; FDA: fluorescein diacetate; FTH1: ferritin heavy chain 1; GOT1/AST: glutamic-oxaloacetic transaminase 1; GPT/ALT: glutamic–pyruvic transaminase; GPX4: glutathione peroxidase 4; GSH: glutathione; HCC: hepatocellular carcinoma; HSC: hepatic stellate cell; LCM: laser capture microdissection; MAP1LC3B: microtubule associated protein 1 light chain 3 beta; MDA: malondialdehydep; NCOA4: nuclear receptor coactivator 4; PTGS2: prostaglandin-endoperoxide synthase 2; ROS: reactive oxygen species; SQSTM1/p62: sequestosome 1; TBIL: total bilirubin; TEM: transmission electron microscopy; TGFB1: trasforming growth factor beta 1; UTR: untranslated region; VA-Lip-ELAVL1-siRNA: vitamin A-coupled liposomes carrying ELAVL1-siRNA.
KEYWORDS: Autophagy, ELAVL1, ferritinophagy, ferroptosis, hepatic stellate cell, liver fibrosis, therapeutic target
Introduction
Liver fibrosis is a reversible pathophysiological process correlated with intense repair and cicatrization mechanisms, and its end-stage cirrhosis is responsible for high morbidity and mortality worldwide [1–3]. The transdifferentiation of hepatic stellate cells (HSCs) into contractile, matrix-producing myofibroblasts is a central event in liver fibrosis [4,5]. Therefore, the targeting of HSCs, including inhibition of the fibrogenic function [6] and induction of apoptosis [7], necroptosis [8], pyroptosis [9], and senescence [10], are proposed therapeutic approaches to reverse liver fibrosis. We previously reported that autophagy regulates turnover of lipid droplets via a reactive oxygen species (ROS)-RAB25-dependent mechanism during HSC activation, and inhibition of autophagy reverses the lipocyte phenotype of activated HSCs [11]. Moreover, our recent studies revealed that senescent activated HSCs can reduce the secretion of extracellular matrix components, enhance immune surveillance, and facilitate the reversion of fibrosis [12]. In the current study, we aimed to evaluate the role of ferroptosis in liver fibrosis and to further elucidate the underlying mechanisms.
Ferroptosis is a newly discovered type of regulated cell death that differs from traditional apoptosis and necrosis, and results from iron-dependent lipid peroxide accumulation [13,14]. Ferroptosis can be triggered by physiological conditions (e.g., high extracellular glutamate) or small molecules (e.g., sorafenib, erastin, sulfasalazine) that block system X c−-mediated cystine import [15]. Moreover, ferroptosis can also be induced by genetic deletion of the glutathione-dependent antioxidant enzyme GPX4 (glutathione peroxidase 4), or by treatment with small molecules that trigger GPX4 degradation (e.g., FIN56) or covalently inhibit GPX4 function (e.g., RSL3) [16]. Ferroptosis is regulated by several genes, for example, iron metabolism genes TFRC (transferrin receptor) and IREB2 (iron responsive element binding protein 2), glutaminolysis-regulating genes SLC38A1 (solute carrier family 38 member 1) and GLS2 (glutaminase 2), and the pentose phosphate pathway gene G6PD (glucose-6-phosphate dehydrogenase) [17]. Interest in ferroptosis as a natural tumor-suppressing process has been spurred by the discovery that tumor-suppressor proteins RB1 (RB transcriptional corepressor 1) and TP53 (tumor protein p53) can activate ferroptosis [18,19]. More importantly, ferroptosis is recently reported as an autophagic cell death process (a process known as ferritinophagy) [20,21]. Ferritinophagy is mediated by NCOA4 (nuclear receptor coactivator 4), an autophagy cargo receptor that binds FTH1 (ferritin heavy chain 1) at the phagophore and is delivered into the lysosome for degradation to release iron for systemic physiological requirements [22]. We previously reported that autophagosome generation and autophagic flux are increased via a ROS-MAPK8/JNK1 (mitogen-activated protein kinase 8)-dependent mechanism during HSC activation [23]. Attractively, whether autophagy activation contributes to HSC ferroptosis by degradation of ferritin is worth further study.
To respond adequately to oxidative stress, mammalian cells elicit rapid and tightly controlled changes in gene expression patterns. Besides alterations in the subsets of transcribed genes, 2 posttranscriptional processes prominently influence the oxidant-triggered gene expression programs: mRNA stabilization and degradation [24]. Although the mechanisms underlying the regulation of ferroptosis have attracted extensive concern of numerous scholars and its acute regulation by iron-sensing signaling pathways is well described, the longer-term posttranscriptional regulation of ferroptosis is a relatively unexplored area [25]. The ubiquitous RNA-binding protein ELAVL1/HuR (ELAV like RNA binding protein 1) is one of the best-studied regulators of cytoplasmic mRNA fate [26]. Mechanistically, ELAVL1 regulates mRNA cargos that typically contain AU-rich elements (AREs) in the 3ʹ-untranslated region (UTR) [26]. Whereas ELAVL1 is normally localized to the nucleus, the stabilization of the ARE-containing mRNAs in response to stress conditions correlates with the translocation of ELAVL1 to the cytoplasm [26]. Currently, a wide range of mRNAs such as FOS (Fos proto-oncogene, AP-1 transcription factor subunit), CDKN1A (cyclin dependent kinase inhibitor 1A), CCNA2 (cyclin A2), CCNB1 (cyclin B1), CCND1 (cyclin D1), NOS2 (nitric oxide synthase 2), VEGFA (vascular endothelial growth factor A), TNF (tumor necrosis factor), BCL2 (BCL2, apoptosis regulator), IL3 (interleukin 3), and HIF1A (hypoxia inducible factor 1 subunit alpha) have been identified to bind to ELAVL1 [27–29]. Through these posttranscriptional influences on specific target mRNAs, ELAVL1 can alter the cellular response to oxidative stress, autophagy, proliferation, differentiation, apoptosis, senescence, and immune stimuli [27–29]. Interestingly, studying the posttranscriptional regulation mechanism of ferroptosis by ELAVL1 in liver fibrosis will provide a brand new perspective to reveal the pathological mechanism and find the effective diagnostic signs and therapeutic targets in iron overload diseases.
In the present study and for the first time, we elucidated novel molecular mechanisms and signaling pathways of ferroptosis in liver fibrosis. We found that erastin- or sorafenib-induced ELAVL1 upregulation promoted BECN1/Beclin1 production via binding to the AREs in the 3ʹ-UTR of BECN1 mRNA, thus triggering autophagy activation, promoting autophagic ferritin degradation, and eventually leading to iron-dependent ferroptosis. Our results indicate that ELAVL1 is a critical and novel posttranscriptional regulator of ferroptosis in liver fibrosis.
Results
RNA-binding protein ELAVL1 expression is increased during HSC ferroptosis
Previous studies have confirmed that the induction of ferroptosis by preclinical (e.g., erastin) and clinical (e.g., sorafenib) drugs facilitates the selective elimination of several tumor cells and represents an emerging anticancer strategy [30,31]. However, the functional contribution of erastin and sorafenib to non-cancer pathologies has not been fully understood. In the present study, we initially investigated whether erastin, buthionine sulfoximine (BSO), and sorafenib could induce HSC ferroptosis. Interestingly, we found that erastin-, BSO-, and sorafenib-mediated growth inhibition in human (HSC-LX2) and rat (HSC-T6) HSC lines was blocked by ferrostatin-1 (a potent ferroptosis inhibitor) but not ZVAD-FMK (a potent apoptosis inhibitor) and necrosulfonamide (a potent necroptosis inhibitor) (Figure 1(a)). Moreover, 3 different cell permeablization assays including trypan blue exclusion (Figure S1a), fluorescein diacetate (FDA) staining (Figure S1b), and calcein-AM-propidium iodide (PI) double staining (Figure S1c) showed that erastin treatment resulted in a drastic reduction in live cells and an increase in the dead cells compared with the untreated group, whereas ferrostain-1, but not ZVAD-FMK and necrosulfonamide, completely diminished the promoting effect of erastin on cell death (Figure S1a-c). It is well known that redox-active iron accumulation, GSH (glutathione) depletion, and lipid peroxidation are 3 critical events in ferroptosis [32]. Therefore, we detected the level of intracellular redox-active iron, GSH, and MDA (malondialdehyde) in HSC-LX2 and HSC-T6 cells treated with these ferroptosis stimulants. As expected, ferroptotic events, including GSH depletion (Figure S2a and b), redox-active iron accumulation (Figure S2c and d), and lipid peroxidation (Figure 1(b)), were significantly triggered following treatment with erastin, BSO, and sorafenib. Attractively, ferrostain-1, but not ZVAD-FMK and necrosulfonamide, completely abolished GSH depletion, MDA production, and redox-active iron accumulation in the induction of ferroptosis (Figure 1(b)) (Figure S2a-d). Similar to results from cancer research, these data indicate that erastin, BSO, and sorafenib can induce HSC ferroptosis in vitro.
Figure 1.
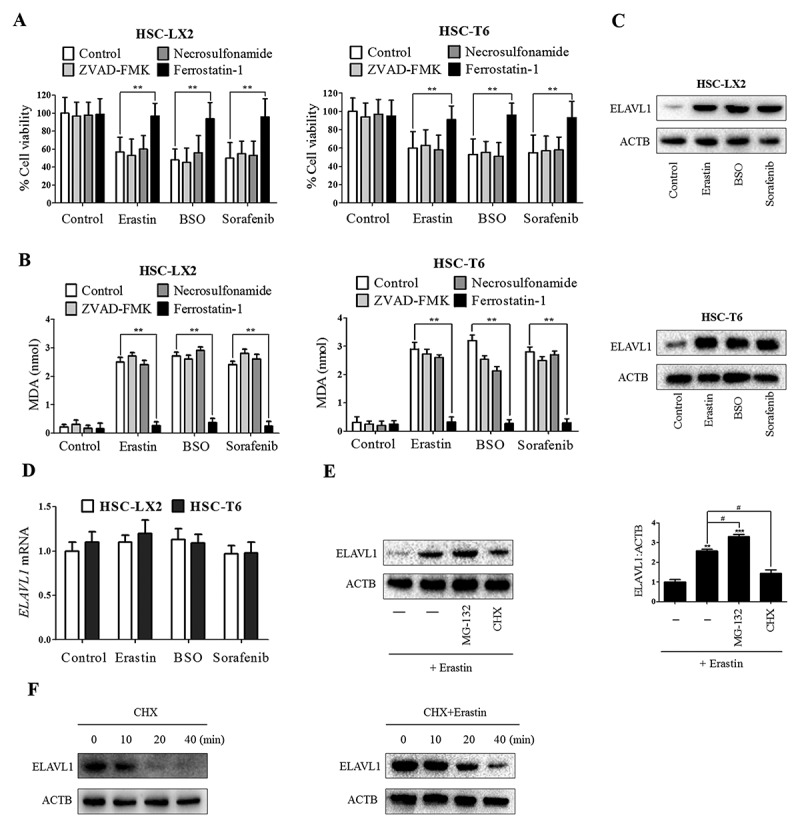
RNA-binding protein ELAVL1 expression is increased during HSC ferroptosis. HSC-LX2 and HSC-T6 cells were treated with erastin (10 μM), BSO (200 μM), and sorafenib (5 μM) with or without the indicated inhibitors (ZVAD-FMK, 10 μM; ferrostatin-1, 1 μM; necrosulfonamide, 0.5 μM) for 24 h. (a) Cell viability and (b) MDA levels were assayed (n = 3 in every group, **, p < 0.01). HSC-LX2 and HSC-T6 cells were treated with erastin (10 μM), BSO (200 μM), and sorafenib (10 μM) for 24 h. (c) ELAVL1 protein and (d) mRNA levels were determined (n = 3 in every group). (e) HSC-LX2 cells were treated with erastin (10 μM) with or without MG-132 (5 μM) or cycloheximide (CHX, 20 μg/ml) for 24 h, and ELAVL1 protein level was assayed (n = 3 in every group, **, p < 0.01, ***, p < 0.001, #, p < 0.05). (f) HSC-LX2 cells were treated with CHX (20 μg/ml) for 1 h or treated with erastin (10 μM) for 4 h followed by CHX (20 μg/ml) for 1 h. ELAVL1 protein level was determined at the indicated time points (n = 3 in every group).
A well-studied RNA-binding protein, ELAVL1, regulates oxidative stress through stabilizing and/or facilitating the translation of target mRNAs [26]. In the current study, we hypothesized that ELAVL1 could play a pivotal role in regulating ferroptosis. To test this hypothesis, we next analyzed protein and mRNA expression levels of ELAVL1 during HSC ferroptosis. Attractively, treatment with ferroptosis-inducing compounds significantly increased ELAVL1 protein expression (Figure 1(c)) but not its mRNA expression (Figure 1(d)), indicating that induction of ELAVL1 expression in ferroptosis occurred in a transcription-independent manner. To further determine the transcription-independent mechanism in ELAVL1 protein induction during ferroptosis, we used the chemical protein synthesis inhibitor cycloheximide (CHX) to inhibit ELAVL1 protein synthesis, and employed a selective 26S proteosomal inhibitor, MG-132, to block ELAVL1 protein degradation. As a result, we found that CHX limited the increased ELAVL1 protein level by erastin in HSC-LX2 cells, whereas MG-132 augmented the protein expression of ELAVL1 (Figure 1(e)). Last, we evaluated the effects of erastin on ELAVL1 protein stability. Results from western blot revealed that erastin treatment prolonged the half-life of ELAVL1 (Figure 1(f)). Taken together, these results show that ELAVL1 protein is stabilized in ferroptosis through the inhibition of ELAVL1 degradation.
Increased ELAVL1 expression contributes to HSC ferroptosis
To determine whether upregulated ELAVL1 is directly involved in the induction of ferroptosis, HSC-LX2 cells were pre-treated with ELAVL1 siRNA or ELAVL1 plasmid, followed by erastin and sorafenib treatment. Western blot analysis confirmed that ELAVL1 siRNA significantly decreased, whereas ELAVL1 plasmid markedly upregulated, ELAVL1 protein level in HSC-LX2 cells (Figure 2(a)). Then, cell viability evaluation was preformed to measure their effects on ferroptosis in both HSC-LX2 and HSC-T6 cells that were already exposed to erastin and sorafenib. As expected, siRNA-mediated knockdown of ELAVL1 abolished erastin- and sorafenib-induced growth inhibition, whereas pretreatment with ELAVL1 plasmid showed a synergistic effect with ferroptosis stimulants in human and rat HSC cells (Figure 2(b)). Besides, several ferroptotic events, including GSH depletion, lipid ROS production, lipid peroxidation, and iron accumulation, were evaluated following the ELAVL1 siRNA or ELAVL1 plasmid treatment. The results showed that ELAVL1 plasmid aggravated erastin- or sorafenib-induced GSH depletion, lipid ROS generation, MDA production, and redox-active iron accumulation (Figure 2(c)). In contrast, knockdown of ELAVL1 by RNA interference reversed lipid ROS accumulation, MDA production, and redox-active iron overload in ferroptosis (Figure 2(c)). Attractively, several ferroptosis inhibitors (ferrostatin-1 and liproxstatin-1) significantly reversed erastin- or sorafenib-induced growth inhibition in the presence of ELAVL1 plasmid (Figure 2(d)). However, ZVADFMK (a pan-caspase inhibitor), necrostatin-1 (a potent necroptosis inhibitor that targets RIPK1 [receptor interacting serine/threonine kinase 1]), and necrosulfonamide (a potent necroptosis inhibitor that targets MLKL [mixed lineage kinase domain like pseudokinase]) did not remarkably reverse this process (Figure 2(d)). Collectively, these findings suggest that increased ELAVL1 expression contributes to ferroptosis, but not apoptosis and necroptosis, following erastin and sorafenib treatment.
Figure 2.
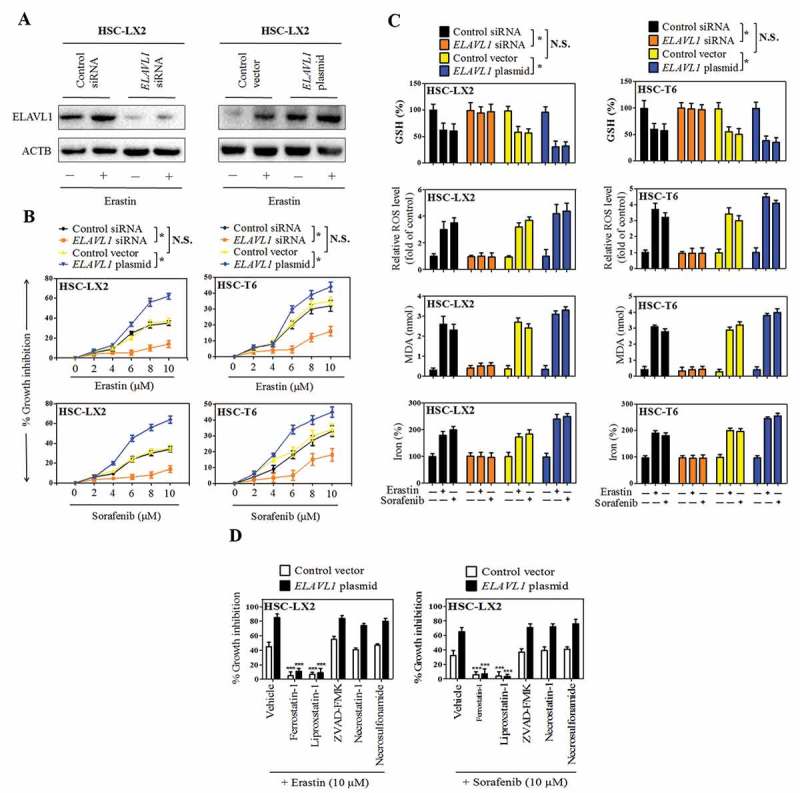
Increased ELAVL1 expression contributes to HSC ferroptosis. The indicated HSC cells were stably transfected with ELAVL1 siRNA or ELAVL1 plasmid, and then were treated with erastin (0–10 μM) or sorafenib (0–10 μM) for 24 h. (a) The transfection efficiency was confirmed by western blot analysis (n = 3 in every group). (b) Cell viability was assayed (n = 3 in every group, *, p < 0.05, N.S., not significant). (c) GSH, ROS, MDA, and iron levels were assayed (n = 3 in every group, *, p < 0.05, N.S., not significant). (d) The indicated ELAVL1-overexpressing HSC cells were treated with erastin (10 μM) and sorafenib (10 μM) with or without the indicated inhibitors (ferrostatin-1, 1 μM; liproxstatin-1, 100 nM; ZVAD-FMK,10 μM; necrostatin-1, 10 μM; necrosulfonamide, 0.5 μM) for 24 h, and cell viability was assayed (n = 3 in every group, ***, p < 0.001).
Enhanced ferroptosis by ELAVL1 is associated with autophagy activation
Several lines of evidence indicate that ferroptosis is an autophagic cell death process [20–22]. In the present study, we assumed that autophagy-lysosome pathways could have a pivotal role in ELAVL1-mediated ferroptosis. To evaluate this assumption, we initially examined the generation of autophagosomes after stable transfection of ELAVL1 siRNA or ELAVL1 plasmid. As expected, results from western blot analysis showed that ELAVL1 silencing significantly abrogated erastin-induced conversion of LC3-I to LC3-II, whereas ELAVL1 overexpression markedly enhanced LC3-II conversion in both HSC-LX2 and HSC-T6 cells (Figure 3(a,c)). To further confirm the role of autophagy in ELAVL1-enhanced ferroptosis, 9 important autophagy-related (ATG) genes were detected by western blot and real-time PCR analysis in ELAVL1 siRNA- or ELAVL1 plasmid-treated cells. The results revealed that ELAVL1 overexpression remarkably increased the expression of BECN1 without large differences in ATG3, ATG4A, ATG5, ATG7, ATG9, ATG12, and ATG14 levels (Figure 3(b)) (Figure S3a), although the expression of ATG5 was slightly increased in 2 HSC lines. In contrast, ELAVL1 silencing significantly inhibited the expression of BECN1, but did not obviously affect other ATG protein levels (Figure 3(b)) (Figure S3a). Given that BECN1 is one of the key regulatory proteins in LC3-II conversion and autophagosome generation [33], we hypothesized that ELAVL1 in ferroptosis promoted autophagy activation mainly by inducing BECN1. To further determine this mechanism during ferroptosis, immunofluorescent staining was performed on ELAVL1 siRNA- or ELAVL1 plasmid-treated HSCs, using a BECN1-specific antibody. The results showed that the expression of BECN1 was downregulated in ELAVL1-deficient cells, but was elevated in stable ELAVL1-expressing cells (Figure 3(c)).
Figure 3.
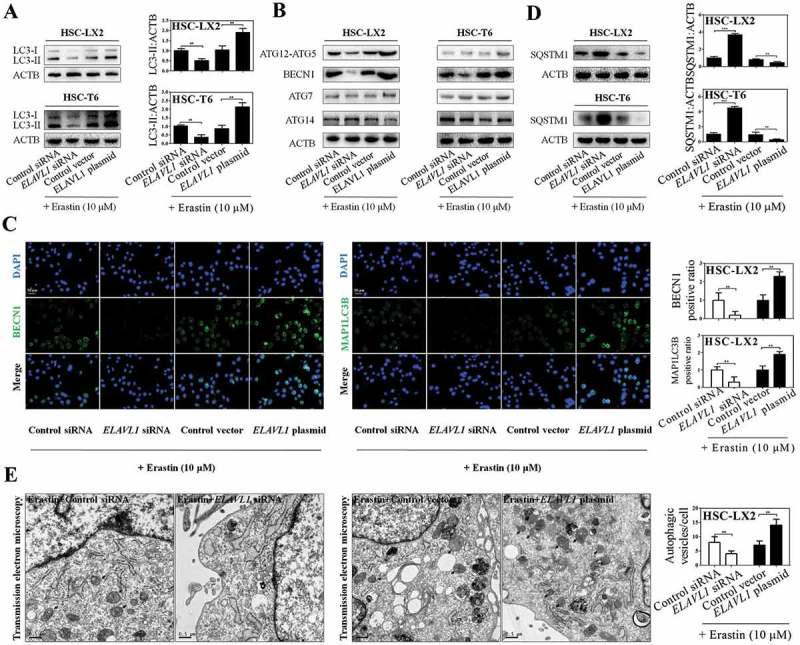
Enhanced ferroptosis by ELAVL1 is associated with autophagy activation. The indicated HSC cells were stably transfected with ELAVL1 siRNA or ELAVL1 plasmid, and then were treated with erastin (10 μM) for 24 h. (a) LC3-I/II protein expression was determined by western blot analysis (n = 3 in every group, **, p < 0.01). (b) 5 autophagy-related genes (ATG12-ATG5, BECN1, ATG7, and ATG14) were assayed by western blot analysis (n = 3 in every group). (c) BECN1 and MAP1LC3B protein expression were determined by immunofluorescence analysis. Scale bars: 50 μm. Representative photographs were shown (n = 3 in every group, **, p < 0.01). (d) SQSTM1 protein expression was assayed by western blot analysis (n = 3 in every group, **, p < 0.01, ***, p < 0.001). (e) HSC-LX2 cells were stably transfected with ELAVL1 siRNA or ELAVL1 plasmid, and then were treated with erastin (10 μM) for 24 h. Autophagosomes and autolysosomes were determined by transmission electron microscopy analysis. Scale bars: 0.5 μm. Representative photographs were shown (n = 3 in every group, **, p < 0.01).
Next, we employed 4 different methods to assess autophagic flux in ELAVL1 siRNA- or ELAVL1 plasmid-treated HSCs, respectively. First, autophagy-specific substrate SQSTM1/p62 (sequestosome 1) was detected in ELAVL1-overexpressing or -deficient HSCs with erastin treatment. SQSTM1 accumulates when autophagy is inhibited, and is decreased when there is autophagic flux [34]. As expected, western blot analysis revealed that ELAVL1 plasmid inhibited the protein expression of SQSTM1, whereas ELAVL1 siRNA augmented the SQSTM1 protein level in HSC-LX2 and HSC-T6 cells (Figure 3(d)). Second, long-lived protein degradation was detected to indicate autophagic flux because the rate was a key functional index of autophagic flux [34]. The longevity protein degradation rate reflected that pretreatment with ELAVL1 plasmid promoted autophagic flux, whereas ELAVL1 siRNA treatment showed the opposite effect during ferroptosis (Figure S3b). Third, we observed an increase in LC3-II level in HSC-LX2 cells which were stably transfected with ELAVL1 plasmid followed by chloroquine treatment compared with cells which were treated with ELAVL1 plasmid alone, suggesting that autophagic flux was increased in ELAVL1-overexpressing HSCs (Figure S3c). Last, transmission electron microscopy (TEM) was used to observe autophagy [34]. As a result, control siRNA- or control vector-treated HSC-LX2 cells displayed a certain number of autophagic vesicles (8 ± 4 vesicles/cell) in the cytoplasm during ferroptosis. Interestingly, the presence of autophagic vesicles was decreased (4 ± 2 vesicles/cell) after ELAVL1 siRNA treatment, whereas pretreatment with ELAVL1 plasmid resulted in a remarkable increase of autophagic vacuoles (14 ± 3 vesicles/cell), representing an intensified autophagy (Figure 3(e)). Overall, these data support the hypothesis that ELAVL1 accumulation during ferroptosis triggers autophagy activation.
Disruption of autophagy by BECN1 siRNA impairs ELAVL1-enhanced ferroptosis
To investigate whether autophagy activation mediates ELAVL1-enhanced ferroptosis, we used BECN1 siRNA to block autophagosome formation and employed BECN1 plasmid to induce autophagy. Western blot analysis confirmed that BECN1 siRNA not only reduced BECN1 levels but also significantly decreased LC3-II conversion (Figure 4(a)). Next, we first measured cell viability when BECN1 was knocked down by specific siRNA. Indeed, siRNA-mediated knockdown of BECN1 led to resistance to erastin- and sorafenib-induced growth inhibition in human and rat HSC cells (Figure 4(b)). In contrast, upregulation of BECN1 expression by BECN1 plasmid significantly promoted erastin- and sorafenib-induced growth inhibition in both HSC-LX2 and HSC-T6 cells (Figure 4(b)). Interestingly, ELAVL1 plasmid exhibited superimposed effects on erastin- and sorafenib-induced growth inhibition in the presence of BECN1 plasmid. However, ELAVL1 overexpression did not dramatically promote growth inhibition by erastin and sorafenib in the presence of BECN1 knockdown (Figure 4(b)), indicating that ELAVL1 accumulation contributes to ferroptosis via an autophagy-dependent mechanism.
Figure 4.
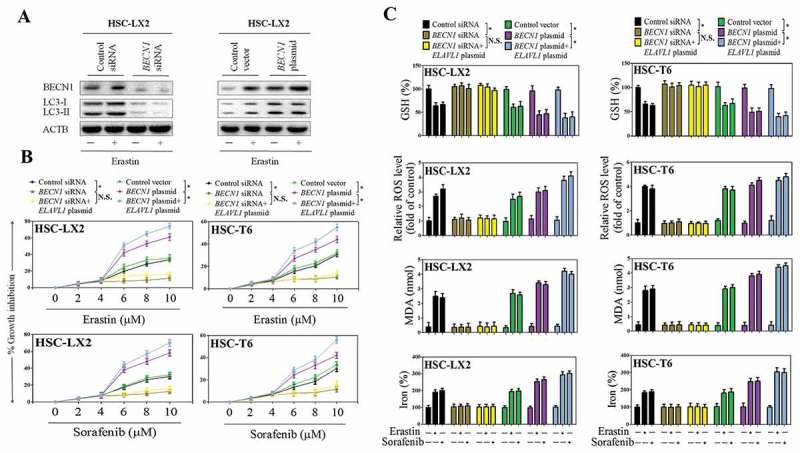
Disruption of autophagy by BECN1 siRNA impairs ELAVL1-enhanced ferroptosis. The indicated HSC cells were stably transfected with BECN1 siRNA or BECN1 plasmid, and then were treated with erastin (10 μM) for 24 h. (a) BECN1 and LC3-I/II protein expression was determined by western blot analysis. The indicated HSC cells were stably transfected with BECN1 siRNA, BECN1 plasmid, BECN1 siRNA+ ELAVL1 plasmid, or BECN1 plasmid+ ELAVL1 plasmid, and then were treated with erastin (0–10 μM) or sorafenib (0–10 μM) for 24 h. (b) Cell viability was assayed (n = 3 in every group, *, p < 0.05, N.S., not significant). (c) GSH, ROS, MDA, and iron levels were assayed (n = 3 in every group, *, p < 0.05, N.S., not significant).
Subsequently, several typical ferroptotic events including GSH depletion, lipid ROS production, lipid peroxidation, and iron accumulation were all investigated in HSC-LX2 and HSC-T6 cells co-treated with BECN1 siRNA and ELAVL1 plasmid. Similarly, pretreatment with BECN1 siRNA completely abolished erastin- and sorafenib-induced GSH depletion (Figure 4(c)), lipid ROS accumulation (Figure 4(c)), MDA production (Figure 4(c)), and redox-active iron accumulation (Figure 4(c)) in the absence or presence of ELAVL1 plasmid. In contrast, there appeared to be increases in ferroptotic events in cells co-treated with BECN1 plasmid and ELAVL1 plasmid compared with BECN1 plasmid alone (Figure 4(c)). Attractively, recent studies indicated that increased autophagy can degrade FTH1/ferritin to increase iron levels resulting in oxidative injury by the Fenton reaction [20,21]. Results from western blot of cell lysates showed that suppression of BECN1 expression by RNAi upregulated the expression of FTH1 during ferroptosis, but ELAVL1 plasmid did not significantly eliminate the increased FTH1 expression in the presence of BECN1 knockdown (Figure S4a and b). In contrast, pretreatment with BECN1 plasmid remarkably inhibited the expression of FTH1, following erastin treatment, and ELAVL1 plasmid showed a synergistic effect with the BECN1 plasmid (Figure S4c and d). Taken together, these results indicate that BECN1-dependent activation of autophagy is required for ELAVL1 to promote ferroptosis in vitro.
ELAVL1 promotes autophagy activation and BECN1 mRNA stability via binding to the AU-rich elements
Accumulating evidence indicate that ELAVL1 promotes mRNA stability and translation via binding to AREs in the 3ʹ-UTR of short-lived human transcripts [26–29]. Therefore, we first used computational methods to search AREs in the BECN1 mRNA sequence. Interestingly, a detailed primary sequence analysis of BECN1 mRNA revealed the presence of AREs in the 3ʹ-UTR, 3 of which (AUUUA) represent the canonical class I sequence recognized by the RNA-binding ELAVL1 protein (Figure 5(a)). Next, we examined whether ELAVL1 bound to the BECN1 mRNA in HSC-LX2 cells by performing RNP IP assays using anti-ELAVL1 antibody under conditions that preserved RNP integrity. As shown in Figure 5(b), the BECN1 PCR products were highly enriched in ELAVL1 samples compared with control IgG samples. The enrichment of GAPDH PCR product was treated as a negative control, and the amplification of PCR products (a nonspecific contaminating housekeeping transcript of ELAVL1) served to monitor the evenness of sample input as reported previously [35]. Furthermore, we measured the binding between ELAVL1 and BECN1 mRNA using biotinylated transcripts that spanned the BECN1 mRNA 5ʹ-UTR, coding region (CR), or 3ʹ-UTR. The interaction between the biotinylated BECN1 transcripts and ELAVL1 was examined by biotin affinity isolation followed by western blot analysis after incubation with cytoplasmic lysates. The results from mRNA-protein precipitation revealed that ELAVL1 could readily interact with the BECN1 3ʹ-UTR, whereas the BECN1 5ʹ-UTR and CR transcripts did not bind to ELAVL1 (Figure 5(c)). In contrast, none of the BECN1 partial transcripts (5ʹ-UTR, CR, or 3ʹ-UTR) were found to interact with ACTB (actin beta), included here as a negative control (Figure 5(c)).
Figure 5.
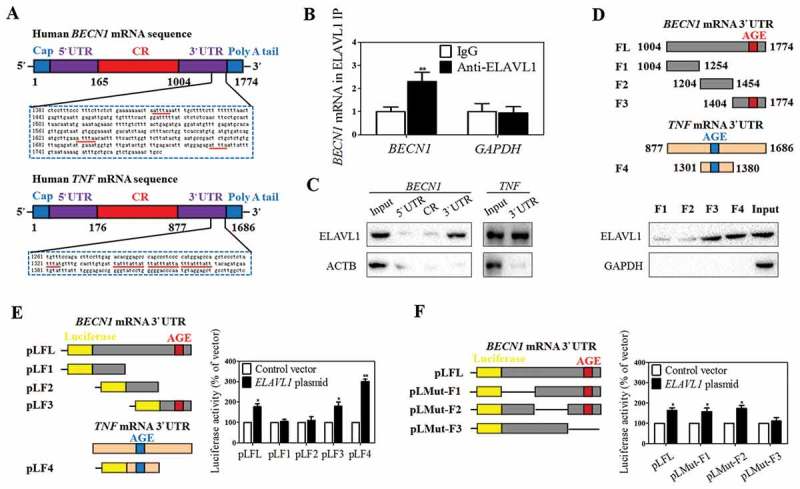
ELAVL1 promotes autophagy activation and BECN1 mRNA stability via binding to the AU-rich elements. (a) The predicted hits of the ELAVL1 signature motif in human BECN1 and TNF mRNA 3ʹ-UTR were assayed. (b) Association of endogenous ELAVL1 with endogenous BECN1 mRNA was measured by real-time PCR after ribonucleoprotein immunoprecipitation (RNP IP) (n = 3 in every group, **, p < 0.01). (c) mRNA affinity isolation assay was performed with biotinylated transcripts of the BECN1 mRNA 5ʹ-UTR, coding region (CR), 3ʹ-UTR or the TNF mRNA 3ʹ-UTR. ACTB served as a negative control (n = 3 in every group). (d) ELAVL1 binding to different fractions of the 3ʹ-UTR of BECN1 mRNA was determined by mRNA affinity isolation assay (n = 3 in every group). (e) The constructs of reporter that expressed chimeric RNA containing luciferase and the BECN1 mRNA 3ʹ-UTR are illustrated schematically. Activities of various luciferase reporters treated as indicated was determined (n = 3 in every group, *, p < 0.05, **, p < 0.01). (f) The deletion of specific ELAVL1-binding sites in the BECN1 3ʹ-UTR is illustrated. The luciferase reporter activity after ELAVL1 overexpression was assayed (n = 3 in every group, *, p < 0.05).
In order to further determine the specific ELAVL1-binding regions in the BECN1 3ʹ-UTR, various partial biotinylated transcripts spanning the BECN1 3ʹ-UTR were prepared (FL: spanning positions 1004–1774, F1: spanning positions 1004–1254, F2: spanning positions 1204–1454, F3: spanning positions 1404–1774). Moreover, an ARE-bearing probe, a portion of TNF 3ʹ-UTR (F4: spanning positions 1301–1380), was employed as the positive control of the experiment because it contains well characterized ELAVL1-target sequences (Figure 5(d)). As expected, F3 of the BECN1 3ʹ-UTR and F4 of the TNF 3ʹ-UTR, but not F1 or F2 of the BECN1 3ʹ-UTR, bound and precipitated ELAVL1 (Figure 5(d)). Although there was detectable binding of ELAVL1 to fragments F1 and F2, the signals were much weaker. These results suggested that ELAVL1 interacts with the BECN1 mRNA via specific RNA segments within the F3 of the 3ʹ-UTR.
To further confirm the mechanism, the constructs of reporter that expressed chimeric RNA containing luciferase and the BECN1 3ʹ-UTR were generated (Figure 5(e)). The results showed that ectopic ELAVL1 overexpression markedly increased luciferase activity when HSC-LX2 cells were transfected with the pLFL, pLF3, or pLF4 (Figure 5(e)). Consistent with their much weaker ELAVL1-binding capability, ELAVL1 exhibited no obvious effect on pLF1 and pLF2 (Figure 5(e)). More importantly, to further define the role of F3 regions in regulating BECN1 mRNA stability, the F3 region was deleted from pLFL for reverse validation. Indeed, the internal deletion of the F3 (not F1 and F2) region abrogated the ELAVL1-mediated upregulation of luciferase activity (Figure 5(f)). Collectively, these findings suggest that ELAVL1 promotes BECN1 mRNA stability via binding to the AREs within the F3 of the 3ʹ-UTR.
HSC-specific knockdown of ELAVL1 impairs sorafenib-induced HSC ferroptosis in murine liver fibrosis
To elucidate whether suppression of ELAVL1 impairs ferroptosis in vivo, a classical murine liver fibrosis model induced by bile duct ligation (BDL) was established [36]. Of note, we also generated vitamin A-coupled liposomes carrying ELAVL1 siRNA (VA-Lip-ELAVL1-siRNA) to knock down ELAVL1 expression in HSCs [37]. First and foremost, we evaluated the impact of HSC-specific ELAVL1 knockdown on liver fibrosis in vivo. Macroscopic examination showed that morphological changes pathologically occurred in livers of BDL-treated mice compared with the normal livers, but sorafenib treatment significantly improved the pathological changes in livers (Figure 6(a,b). Interestingly, the improvement of sorafenib on liver fibrosis was completely abrogated by VA-Lip-ELAVL1-siRNA (Figure 6(a,b)). Moreover, liver fibrosis was further demonstrated by histological analyses. Hematoxylin and eosin (H&E), Masson, and picro-Sirius red staining showed that intraperitoneal injection of sorafenib for 2 weeks significantly alleviated hepatic fibrosis characterized by decreased collagen deposition, whereas livers derived from mice treated with sorafenib in combination with VA-Lip-ELAVL1-siRNA exhibited severe fibrotic pathology compared with the mice treated with sorafenib alone (Figure 6(a,b)). Furthermore, immunohistochemistry revealed that sorafenib treatment markedly reduced the expression of ACTA2 (actin, alpha 2, smooth muscle, aorta) and COL1A1 (collagen type I alpha 1 chain), whereas VA-Lip-ELAVL1-siRNA almost completely abolished the inhibition of sorafenib on ACTA2 and COL1A1 expression (Figure 6(c)). Besides, real-time PCR also showed that sorafenib caused a decrease in ACTA2 and COL1A1 mRNA expression, whereas pretreatment with VA-Lip-ELAVL1-siRNA abrogated the effects of sorafenib on ACTA2 and COL1A1 mRNA expression (Figure S5a and b).
Figure 6.
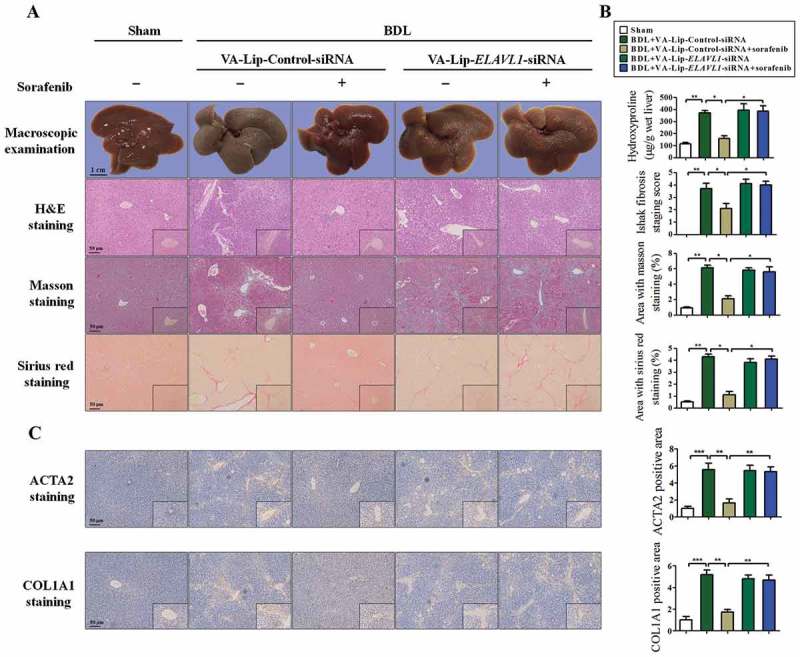
Sorafenib treatment alleviates murine liver fibrosis by inducing HSC ferroptosis. Fifty mice were randomly divided into 5 groups of 10 animals each with comparable mean body weight. Mice of 5 groups were treated with Sham, BDL+ VA-Lip-Control-siRNA, BDL+ VA-Lip-Control-siRNA+ sorafenib, BDL+ VA- Lip-ELAVL1-siRNA, or BDL+ VA-Lip-ELAVL1-siRNA+ sorafenib. (a and b) The pathological changes of the livers were observed by macroscopic examination. Scale bars: 1 cm. Representative photographs were shown. Thin sections (4 μm) were stained with H&E, Sirius Red, and Masson for histopathological study. The liver fibrosis stage was assessed by Ishak scale. Liver hydroxyproline level was determined using the Hydroxyproline Assay Kit (n = 6 in every group, *, p < 0.05, **, p < 0.01). (c) Immunohistochemical staining of ACTA2 and COL1A1 was determined. Scale bars: 50 μm. Representative photographs were shown (n = 6 in every group, *, p < 0.05, **, p < 0.01, ***, p < 0.001).
Importantly, the primary hepatocytes, macrophages, and HSCs were isolated from fibrotic livers of C57BL/6 mice treated with BDL and/or sorafenib, and the effect of sorafenib on ferroptosis in isolated primary hepatocytes, macrophages, and HSCs was investigated. Interestingly, sorafenib could not increase the expression of the ferroptosis marker PTGS2 (prostaglandin-endoperoxide synthase 2) in isolated primary hepatocytes and macrophages, but could upregulate the expression of PTGS2 in isolated primary HSCs (Figure S6a). Moreover, treatment with sorafenib could not trigger redox-active iron overload (Figure S6b), lipid ROS accumulation (Figure S6c), and MDA production (Figure S6d) in isolated primary hepatocytes and macrophages. By contrast, ferroptotic events in isolated primary HSCs, including redox-active iron overload (Figure S6b), lipid ROS accumulation (Figure S6c), and lipid peroxidation (Figure S6d), were significantly triggered following treatment with sorafenib.
To further investigate the effect of HSC-specific ELAVL1 knockdown on ferritinophagy and ferroptosis in vivo, primary HSCs were also isolated from fibrotic livers of C57BL/6 mice treated with sorafenib+VA-Lip-ELAVL1-siRNA or sorafenib+VA-Lip-Control-siRNA, and total RNAs were extracted for sequencing (RNA-Seq). Using >1.4-fold change for upregulation and <0.65-fold change for downregulation as cutoffs, 347 mRNAs were upregulated and 421 mRNAs were downregulated in ELAVL1-silenced primary HSCs as shown in the heat map (Figure 7(a)) and volcano plot (Figure 7(b)). It is noteworthy that some of the known autophagy-related genes and ferroptosis-related genes, such as MAP1LC3B/LC3B (0.21 fold), BECN1 (0.28 fold), SQSTM1 (2.36 fold), PTGS2 (0.32 fold), and FTH1 (2.86 fold), were found among these genes (Figure 7(c)). Interestingly, consistent with in vitro results, the expression of other autophagy-related genes including ATG3 (0.89 fold), ATG4 (1.12 fold), ATG5 (0.91 fold), ATG7 (0.98 fold), ATG9 (1.03 fold), ATG12 (1.05 fold), and ATG14 (0.96 fold) did not change significantly in the primary mouse HSCs infected with VA-Lip-ELAVL1-siRNA (Figure 7(c)).
Figure 7.
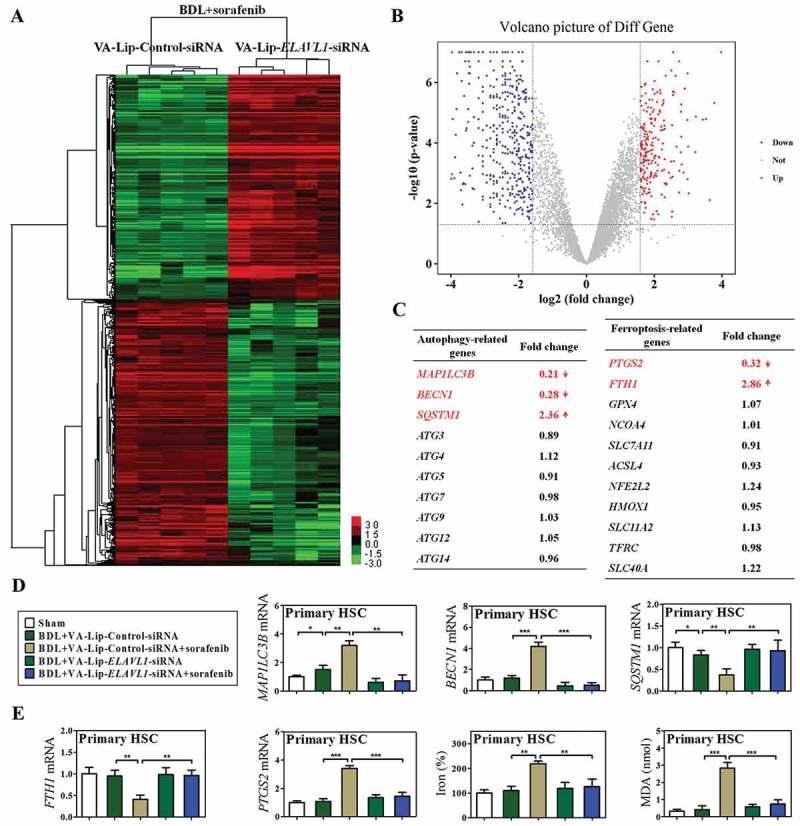
HSC-specific knockdown of ELAVL1 impairs sorafenib-induced HSC ferroptosis in murine liver fibrosis. Fifty mice were randomly divided into 5 groups of 10 animals each with comparable mean body weight. Mice of 5 groups were treated with Sham, BDL+VA-Lip-Control-siRNA, BDL+VA-Lip-Control-siRNA+sorafenib, BDL+VA-Lip-ELAVL1-siRNA, or BDL+VA-Lip-ELAVL1-siRNA+sorafenib. Primary HSCs were further isolated from BDL+VA-Lip-Control-siRNA+sorafenib or BDL+VA-Lip-ELAVL1-siRNA+sorafenib treated mice, and total RNAs were extracted for RNA-Seq. (a) Microarray heat map demonstrates clustering of isolated primary HSCs. Hierarchical cluster analysis of significantly differentially expressed mRNAs: bright green, underexpression; gray, no change; bright red, overexpression (BDL+VA-Lip-Control-siRNA+sorafenib, n = 5; BDL+VA-Lip-ELAVL1-siRNA+sorafenib, n = 5). (b) Volcano plot demonstrates clustering of isolated primary HSCs. Hierarchical cluster analysis of significantly differentially expressed mRNAs: bright blue, underexpression; gray, no change; bright red, overexpression (BDL+VA-Lip-ELAVL1-siRNA, n = 5; BDL+VA-Lip-Control-siRNA+sorafenib, n = 5). (c) Fold changes of autophagy-related genes and ferroptosis-related genes were identified. (d) The mRNA expression of autophagy markers MAP1LC3B/LC3B, BECN1, and SQSTM1 was determined by real-time PCR in isolated primary HSCs (n = 6 in every group, *, p < 0.05, **, p < 0.01, ***, p < 0.001). (e) Ferroptosis markers FTH1 and PTGS2 mRNA expression and ferroptotic events including iron accumulation and lipid peroxidation were determined in isolated primary HSCs (n = 6 in every group, **, p < 0.01, ***, p < 0.001).
Additionally, these findings were further confirmed by real-time PCR. As expected, the results showed that sorafenib treatment significantly upregulated the expression of MAP1LC3B/LC3B and downregulated the expression of SQSTM1, whereas VA-Lip-ELAVL1-siRNA remarkably suppressed the ability of sorafenib in the induction of autophagosome generation and autophagic flux (Figure 7(d)). The above in vitro experiments suggested that ELAVL1 accumulation triggered autophagy activation via a BECN1-dependent mechanism. In agreement with this finding, ELAVL1 knockdown in HSCs by VA-Lip-ELAVL1-siRNA apparently abrogated sorafenib-induced upregulation of BECN1 mRNA expression (Figure 7(d)). As the main iron storage protein, FTH1 plays a central role in the maintenance of cellular iron balance [38]. Attractively, sorafenib treatment significantly decreased the expression of the ferritinophagy substrate FTH1, whereas VA-Lip-ELAVL1-siRNA markedly reversed the inhibitory effect of sorafenib on FTH1 (Figure 7(e)). More importantly, the HSC ferroptosis marker PTGS2 was significantly upregulated after sorafenib treatment, showing that HSCs undergo a ferroptosis process in vivo as well (Figure 7(e)). Interestingly, VA-Lip-ELAVL1-siRNA completely abolished sorafenib-induced HSC ferroptosis with decreased ferroptotic events including redox-active iron reduction (Figure 7(e)), GSH generation (Figure S5c), lipid ROS clearance (Figure S5d), and MDA elimination (Figure 7(e)). Taken together, these results show that suppression of ELAVL1 impairs sorafenib-induced HSC ferroptosis in murine liver fibrosis.
ELAVL1 upregulation, ferritinophagy activation, and ferroptosis induction occur in human HSCs from fibrotic patients with HCC receiving sorafenib monotherapy
To evaluate the potential mechanism clinically, we retrospectively analyzed consecutive advanced fibrotic patients with hepatocellular carcinoma (HCC) who were treated with sorafenib monotherapy in the Second Affiliated Hospital of Southeast University (Nanjing, China) between February 2013 and June 2017. Data on patient characteristics (Table 1) were collected retrospectively from all patients by reviewing their electronic medical records. Importantly, among the enrolled patients, 5 patients underwent liver biopsy for accurate diagnosis of liver fibrosis and HCC before sorafenib treatment, while 5 patients received curative hepatectomy for the treatment of liver fibrosis and HCC after sorafenib monotherapy. We picked out the above eligible samples from our hospital preserved samples for the subsequent laboratory experiments according to the inclusion criteria (Figure S7). Blood samples were collected for the analyses of liver functions including GPT/ALT (alanine aminotransferase), GOT1/AST (aspartate aminotransferase), and TBIL (total bilirubin). Moreover, primary human HSCs were isolated from the collected liver tissue by laser capture microdissection (LCM) as described previously [39–41], and total RNAs were extracted for RNA-Seq.
Table 1.
Patient characteristics.
| Characteristics | Patients |
|---|---|
| Number of patients, n | 28 |
| Gender (male/female), n (%) | 15 (53.6%)/13 (46.4%) |
| Age (years), mean±SD; min.-max. | 56.8 ± 12.6; 27–85 |
| BMI (kg/m2), mean±SD; min.-max. | 27.7 ± 5.2; 17.2–28.6 |
| Child-Pugh grade (A/B/C), n (%) | 7 (25.0%)/11 (39.3%)/10 (35.7%) |
| MELD score, mean±SD; min.-max. | 10.5 ± 2.8; 7–8 |
| ASA class (I/II/III/IV), n (%) | 0 (0%)/9 (32.1%)/17 (60.7%)/2 (7.2%) |
| Preoperative serum GPT/ALT level (IU/L), mean±SD | 135.7 ± 12.4 |
| Preoperative serum GOT1/AST level (IU/L), mean±SD | 213.4 ± 18.0 |
| Preoperative serum ALPL level (IU/L), mean±SD | 386.3 ± 38.2 |
| Preoperative serum TBIL level (μmol/L), mean±SD | 46.8 ± 14.4 |
| Preoperative serum GGT1 level (IU/L), mean±SD | 85.6 ± 18.7 |
| Preoperative serum ALB level (g/L), mean±SD | 23.7 ± 5.3 |
| Preoperative serum IGH level (g/L), mean±SD | 54.6 ± 7.4 |
| Origin of cirrhosis, n (%) | |
| HBV infection | 8 (28.6%) |
| HCV infection | 2 (7.1%) |
| Alcohol consumption | 7 (25.0%) |
| HBV infection plus alcohol consumption | 4 (14.3%) |
| NAFLD/NASH | 6 (21.4%) |
| Hereditary hemochromatosis | 1 (3.6%) |
Abbreviations: BMI, body mass index; MELD, model for end-stage liver disease; ASA, American Society of Anesthesiologists; GPT/ALT, glutamic–pyruvic transaminase; GOT1/AST, glutamic-oxaloacetic transaminase 1; ALPL, alkaline phosphatase; TBIL, total bilirubin; GGT1, gamma-glutamyltransferase 1; ALB, albumin; IGH, immunoglobulin heavy; HBV, hepatitis B virus; HCV, hepatitis C virus; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis. Quantitative values were expressed as means with standard deviations or medians with range, while categorical values were expressed as numbers with percentages.
The data revealed that 1024 mRNAs were upregulated and 831 mRNAs were downregulated in sorafenib-treated human HSCs as shown in the heat map (Figure 8(a)) and volcano plot (Figure 8(b)). As expected, some established fibrosis-related genes, such as ACTA2 (0.44 fold), COL1A1 (0.37 fold), COL1A2 (0.58 fold), FN1 (fibronectin 1) (0.61 fold), and MMP2 (matrix metallopeptidase 2) (0.53 fold), were found among these downregulated genes (Figure 8(c)). Moreover, several well-known autophagy-related genes, such as BECN1 (5.79 fold), MAP1LC3B/LC3B (4.38 fold), ATG4 (1.98 fold), ATG5 (1.87 fold), ATG10 (1.81 fold), ATG12 (2.14 fold), and ATG16L1 (2.39 fold), were found among these upregulated genes (Figure 8(c)). Attractively, some recognized ferroptosis-related genes, such as ELAVL1 (3.85 fold), PTGS2 (6.04 fold), SLC7A11 (solute carrier family 7 member 11) (2.68 fold), ACSL4 (acyl-CoA synthetase long chain family member 4) (2.95 fold), NFE2L2 (nuclear factor, erythroid 2 like 2) (2.09 fold), HMOX1 (heme oxygenase 1) (1.94 fold), SLC11A2 (solute carrier family 11 member 2) (1.78 fold), TFRC (1.93 fold), SLC40A1 (solute carrier family 40 member 1) (2.05 fold), and TP53 (2.31 fold), were found among these upregulated genes (Figure 8(c)).
Figure 9.
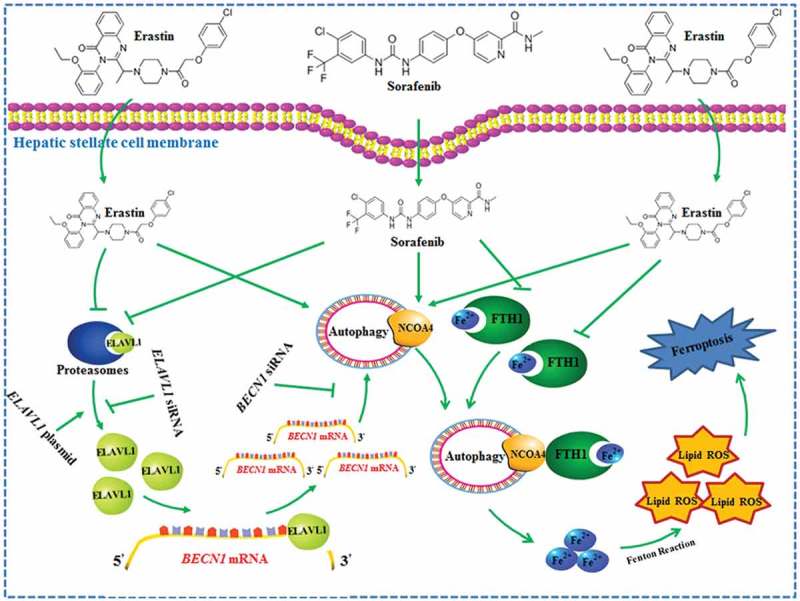
The RNA-binding protein ELAVL1 regulates ferroptosis by inducing ferritinophagy activation in HSCs. Ferroptosis-inducing compounds increase ELAVL1 expression through the inhibition of the ubiquitin-proteasome pathway. Increased ELAVL1 can bind to the AREs of the BECN1 mRNA 3ʹ-UTR, trigger autophagy activation, promote autophagic ferritin degradation, and in turn, lead to HSC ferroptosis.
Figure 8.
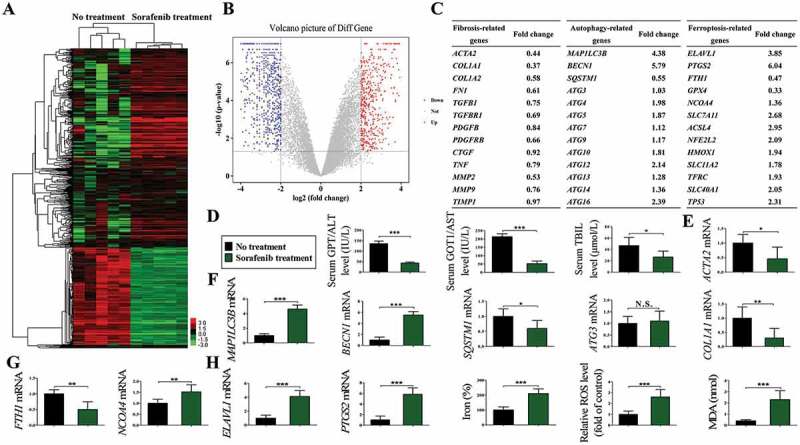
ELAVL1 upregulation, ferritinophagy activation, and ferroptosis induction occur in primary human HSCs from fibrotic patients with HCC receiving sorafenib monotherapy. Primary human HSCs were isolated from the collected liver tissue by laser capture microdissection (LCM), and total RNAs were extracted for RNA-Seq. (a) Microarray heat map demonstrates clustering of isolated primary HSCs before and after sorafenib treatment. Hierarchical cluster analysis of differentially expressed mRNAs: bright green, underexpression; gray, no change; bright red, overexpression (No treatment, n = 5; sorafenib treatment, n = 5). (b) Volcano plot demonstrates clustering of isolated primary HSCs before and after sorafenib treatment. Hierarchical cluster analysis of differentially expressed mRNAs: bright blue, underexpression; gray, no change; bright red, overexpression (No treatment, n = 5; sorafenib treatment, n = 5). (c) Fold changes of liver fibrosis-related genes, autophagy-related genes, and ferroptosis-related genes were identified. (d) Blood samples were collected for the analyses of liver functions including GPT/ALT, GOT1/AST, and TBIL (total bilirubin) (n = 28 in every group, *, p < 0.05, ***, p < 0.001). (e) The mRNA expression of liver fibrosis markers ACTA2 and COL1A1 was determined by real-time PCR in sorafenib-treated liver biopsies and untreated controls (n = 5 in every group, *, p < 0.05, **, p < 0.01). (f and g) The mRNA expression of ferritinophagy markers MAP1LC3B/LC3B, BECN1, SQSTM1, ATG3, FTH1, and NCOA4 was determined by real-time PCR in isolated primary HSCs before and after sorafenib treatment (n = 5 in every group, *, p < 0.05, **, p < 0.01, ***, p < 0.001, N.S., not significant). (h) Ferroptosis markers ELAVL1 and PTGS2 mRNA expression and ferroptotic events including iron accumulation, lipid ROS production, and lipid peroxidation were all determined in isolated primary HSCs before and after sorafenib treatment (No treatment, n = 5; sorafenib treatment, n = 5; *, p < 0.05, ***, p < 0.001).
Of note, these results were further confirmed by real-time PCR analysis and biochemical assay. First and foremost, liver function was assessed by measurement of GPT/ALT, GOT1/AST, and TBIL. The liver function tests prior to sorafenib treatment were as follows: GPT/ALT, 135.7 ± 12.4 international units per liter (IU/L) (normal: 4–36 IU/L); GOT1/AST, 213.4 ± 18.0 IU/L (normal: 13–39 IU/L); TBIL, 46.8 ± 14.4 μmol/L (normal: 2.0–20.0 μmol/L) (Figure 8(d)). After sorafenib treatment, liver function tests were as follows: GPT/ALT, 43.6 ± 4.4 IU/L; GOT1/AST, 52.4 ± 15.8 IU/L; TBIL, 26.67 ± 10.4 μmol/L (Figure 8(d)). These results suggest that treatment with the ferroptosis inducer sorafenib significantly improves liver function in patients with liver fibrosis. Second, we evaluated the effect of sorafenib on liver fibrosis. Real-time PCR analysis indicated that the mRNA expression of ACTA2 and COL1A1 was decreased in sorafenib-treated liver biopsies compared to untreated controls (Figure 8(e)). Third, we investigated the changes of ferritinophagy in primary human HSCs before and after sorafenib treatment. The data showed that the expression of MAP1LC3B/LC3B and BECN1 was remarkably upregulated after sorafenib treatment, suggesting an increase of autophagosome generation (Figure 8(f)). Consistent with this finding, the expression of ferritinophagy substrates SQSTM1 and FTH1 was markedly decreased in primary human HSCs from fibrotic patients treated with sorafenib, indicating that autophagic flux was increased (Figure 8(f,g)). Interestingly, the cargo receptor NCOA4 mediates autophagic ferritin degradation [22]. Our results showed that sorafenib treatment remarkably upregulated NCOA4 expression, which suggested an increase of ferritinophagy in primary human HSCs (Figure 8(g)). Fourth, the induction of ferroptosis was determined in primary human HSCs before and after sorafenib treatment. As expected, real-time PCR analysis indicated that ELAVL1 mRNA expression was increased by above 4 fold in primary human HSCs from fibrotic patients treated with sorafenib (Figure 8(h)). Moreover, ferroptotic events, including PTGS2 upregulation (Figure 8(h)), redox-active iron accumulation (Figure 8(h)), ROS generation (Figure 8(h)), and lipid peroxidation (Figure 8(h)), were significantly triggered following treatment with sorafenib in primary human HSCs. Collectively, these findings suggest that 3 critical events including ELAVL1 upregulation, ferritinophagy activation, and ferroptosis induction occur in primary human HSCs from fibrotic patients receiving sorafenib monotherapy.
Discussion
Ferroptosis is a novel cell death mechanism that is characterized morphologically by cell volume shrinkage and increased mitochondrial membrane density without typical apoptotic and necrotic manifestations [42–44]. An improved understanding of the role of ferroptosis in cancer has provided a new opportunity for diagnosis and therapeutic intervention. Alvarez et al. [45] recently demonstrated that NFS1 (NFS1, cysteine desulfurase) undergoes positive selection in lung tumors and protects cells from ferroptosis. Moreover, Hao et al. [46] revealed that CDO1 (cysteine dioxygenase type 1) mediates erastin-induced ferroptosis in human gastric cancer cells. Furthermore, Chen et al. [47] reported that inhibition of ATF4 (activating transcription factor 4) is a valid target for diminishing tumor growth and vasculature via sensitizing tumor cells for ferroptosis. However, the functional contribution of ferroptosis to non-cancer pathologies has not been fully understood.
In the present study, we reported that erastin and sorafenib can induce HSC ferroptosis in both human (HSC-LX2) and rat (HSC-T6) HSC lines, and also can trigger the accumulation of ferroptotic HSCs in murine fibrotic liver. Moreover, the induction of HSC ferroptosis remarkably alleviated liver fibrosis characterized by decreased collagen deposition. To our knowledge, this is the first report that the ferroptosis of activated HSCs provides a brake on the fibrogenic response to damage by limiting the expansion of the cell type responsible for producing the fibrotic scar. Importantly, we should also take into account the possibility that ferroptosis-inducing agents may have deleterious effects on hepatocytes and macrophages. Recently, Wang et al. [44] reported that the ferroptosis stimulant ferric citrate potently induces ferroptosis in murine primary hepatocytes and bone marrow-derived macrophages. Interestingly, unlike ferric citrate, we found that the ferroptosis-inducing agent sorafenib can not trigger redox-active iron overload, lipid ROS accumulation, and MDA production in primary hepatocytes and macrophages.
In fact, a large number of papers have shown that sorafenib has protective effects on hepatocytes, and alleviates the pathological state of hepatic fibrosis [48–50]. Interestingly, Chen et al. reported that sorafenib inhibits TGFB1 (transforming growth factor beta 1)-mediated epithelial-mesenchymal transition and apoptosis in mouse hepatocytes [48]. Moreover, they further present in vitro evidence that sorafenib ameliorates the proapoptotic and profibrotic effects of TGFB1 in hepatocytes, suggesting that this drug exerts a protective effect on hepatocytes and has therapeutic potential for the treatment of liver fibrosis [48]. Furthermore, Deng et al. [49] also demonstrated that sorafenib significantly attenuates chronic liver injury and fibrosis, including reduction in liver inflammation and histopathology as well as decreased expression of liver fibrosis-related genes, including ACTA2, COL1A1, matrix metallopeptidases, and TIMP1 (TIMP metallopeptidase inhibitor 1). More importantly, Wang et al. [50] showed new insights into the antifibrotic effects of sorafenib on HSCs and liver fibrosis. Sorafenib treatment attenuates liver fibrosis and is associated with a significant decrease in intrahepatic fibrogenesis, hydroxyproline accumulation, and collagen deposition [50]. Overall, although these studies did not directly determine the impact of sorafenib on hepatocyte ferroptosis, these results support the idea that sorafenib can not induce hepatocyte death, but protect hepatocytes from apoptosis, which could contribute to reversion of hepatic fibrosis.
Importantly, our study identified the RNA-binding protein ELAVL1 as a novel posttranscriptional regulator in the facilitation of erastin-induced HSC ferroptosis. ELAVL1 is a ubiquitously expressed member of the embryonic lethal abnormal vision (ELAV)-like/Hu-protein family of RNA-binding proteins [26]. ELAVL1 selectively binds to target mRNAs bearing specific sequence elements, often U- and AU-rich, and generally found in the mRNA 3ʹ-UTR, and plays a critical role in their posttranscriptional regulation [26]. Yu et al. [51] recently showed that the RNA-binding protein ELAVL1 promotes bladder cancer progression by competitively binding to the long noncoding HOTAIR with MIR1-1HG. Similarly, Xu et al. [52] found that downregulation of ELAVL1 inhibits the progression of esophageal cancer through IL18. Moreover, Badawi et al. [53] demonstrated that silencing of the mRNA-binding protein ELAVL1 increases the sensitivity of colorectal cancer cells to ionizing radiation through upregulation of CASP2 (caspase2).
Despite its pro-tumorigenic properties, some studies also reported anti-tumorigenic effects of ELAVL1, with a negative impact on cell growth and inflammatory responses, which indicates that ELAVL1 function largely depends on the cellular context and microenvironmental conditions [54–56]. Gubin et al. [54] reported that overexpression of the RNA binding protein ELAVL1 impairs tumor growth in triple-negative breast cancer associated with deficient angiogenesis. Furthermore, Katsanou et al. [55] identified ELAVL1 as a negative posttranscriptional modulator in inflammation. Besides, Yiakouvaki et al. [56] revealed that myeloid cell expression of the RNA-binding protein ELAVL1 protects mice from pathological inflammation and colorectal carcinogenesis.
In the current study, we showed that ELAVL1 protein expression was remarkably increased through the inhibition of the ubiquitin-proteasome pathway after cultured HSCs were exposed to ferroptosis-inducing compounds (erastin, sorafenib, and BOS). ELAVL1 silencing led to ferroptosis resistance, whereas ELAVL1 overexpression contributed to HSC ferroptosis characterized by redox-active iron accumulation, GSH depletion, and lipid peroxidation. Similarly, HSC-specific knockdown of ELAVL1 by VA-Lip-ELAVL1-siRNA also impaired sorafenib-induced HSC ferroptosis in murine liver fibrosis. Although more experiments are needed to determine the exact role of ELAVL1 in ferroptosis, our results indicate a novel function of ELAVL1 in addition to controlling inflammation, apoptosis, proliferation, and senescence.
Interestingly, several lines of evidence indicate a vital relationship between ferroptosis and autophagy [20–22]. Autophagy is identified as an upstream mechanism in the induction of ferroptosis by regulating cellular iron homeostasis and cellular ROS generation [20,21]. Upon induction of ferroptosis, autophagy is activated, leading to degradation of the cellular iron stock protein ferritin, and thus an increase of cellular labile iron level via an NCOA4-mediated autophagy pathway [22]. Gao et al. [20] recently demonstrated that ferroptosis is an autophagic cell death process. Using RNAi screening coupled with subsequent genetic analysis, they identified multiple autophagy-related genes as positive regulators of ferroptosis [20]. Ferroptosis induction leads to autophagy activation and consequent degradation of ferritin and the ferritinophagy cargo receptor NCOA4 [20]. Consistent with this observation, inhibition of ferritinophagy by blockage of autophagy or knockdown of NCOA4 abrogates the accumulation of ferroptosis-associated cellular labile iron and ROS, as well as eventual ferroptotic cell death [20]. Furthermore, Hou et al. [21] indicated that autophagy contributes to ferroptosis by degradation of ferritin in fibroblasts and cancer cells. Knockout or knockdown of ATG5 or ATG7 limits erastin-induced ferroptosis with decreased intracellular ferrous iron levels and lipid peroxidation [21]. In addition, Latunde-Dada et al. [22] further revealed that ferritinophagy is the autophagic degradation of the iron-storage ferritin protein that maintains homeostasis during iron depletion. The pathway is mediated, during low iron levels, by NCOA4, an autophagy cargo receptor that binds FTH1 at the phagophore and delivers it into the lysosome for degradation to release iron for systemic physiological requirements [22]. Initiation of ferroptosis activates ferritinophagy in a process whereby ferritin catabolism increases the labile iron pool, which promotes ROS accumulation that derives ferroptosis [22].
Consistent with previous studies, we showed that the upregulated ELAVL1 expression appeared to increase autophagosome generation and autophagic flux, which was the underlying mechanism for ELAVL1-enhanced ferroptosis. Disruption of autophagy by BECN1 siRNA impaired ELAVL1-enhanced ferroptosis. Although our data suggested a direct connection between autophagy and ELAVL1-enhanced HSC ferroptosis, we could not eliminate other effects that may mediate the promoting effect. Of note, we should also take full account of the profibrotic properties of autophagy during HSC activation [57–59]. Wu et al. [57] recently reported that quercetin prevents liver fibrosis by inhibiting HSC activation and reducing autophagy via the TGFB1-SMAD and phosphoinositide 3-kinase-AKT1 pathways. Moreover, Thoen et al. [58] showed that HSC activation is followed by an increased autophagic flux and its inhibition can partially inhibit the HSC myofibroblastic transition. Furthermore, Hernández-Gea et al. [59] demonstrated that autophagy fuels tissue fibrogenesis. Inhibition of autophagy by pharmacological antagonism or ATG5 knockdown in mice also results in attenuation of fibrogenesis, as well as increased lipid content in HSCs [59].
Overall, autophagy is a novel target playing dual roles in human diseases including liver fibrosis. Autophagy may help cells to live through stress conditions and attenuate inflammation, leading to fibrosis reduction. Autophagy is also involved in collagen degradation, which may contribute to fibrosis attenuation. However, autophagy fuels HSCs to be activated and promote fibrosis. The effect of autophagy on liver fibrosis is complex and still controversial. With a better understanding of the effects of autophagy on hepatic fibrosis, autophagy may have potential as a target of antifibrotic therapy.
BECN1 is an evolutionarily conserved protein that functions in autophagy in a wide variety of species [60]. In mammalian cells, BECN1 functions as part of a class III phosphatidylinositol 3-kinase, thus participating in the generation of phosphatidylinositol-3-phosphate, which is of critical significance in autophagy modulation [60]. Huang et al. [61] recently demonstrated that the APLN (apelin) active peptide APLN13 induces autophagy in hepatoma HepG2 cells through a MAPK1/ERK2 signaling pathway-dependent upregulation of BECN1. Moreover, Shrivastava et al. [62] reported that hepatitis C virus upregulates BECN1 for induction of autophagy and activates MTOR signaling. Similarly, Ma et al. [63] found that DACT1 (disheveled binding antagonist of beta catenin 1) promotes autophagy by enhancing BECN1-PIK3C3/VPS34-ATG14 complex formation. Consistent with previous studies, we showed that autophagy activation by ELAVL1 during HSC ferroptosis may occur in a BECN1-dependent mechanism in both human and rat HSCs. Further research revealed that ELAVL1 promotes BECN1 mRNA stability via binding to the AREs within the 3ʹ-UTR. For the first time, we found 3 canonical class I sequences (AUUUA) present in the 3ʹ-UTR of human BECN1 mRNA. Moreover, the results from mRNA-protein precipitation revealed that ELAVL1 readily interacts with the BECN1 3ʹ-UTR, but the BECN1 5ʹ-UTR and CR transcripts did not bind to ELAVL1. Besides, various partial biotinylated transcripts spanning the BECN1 3ʹ-UTR were prepared (FL: spanning positions 1004–1774, F1: spanning positions 1004–1254, F2: spanning positions 1204–1454, F3: spanning positions 1404–1774). As expected, F3 of the BECN1 3ʹ-UTR and F4 of the TNF 3ʹ-UTR (they contain AUUUA sequences), but not F1 or F2 of the BECN1 3ʹ-UTR, bound and precipitated ELAVL1. Consistent with this finding, the internal deletion of the F3 (but not F1 and F2) region abrogated the ELAVL1-mediated upregulation of luciferase activity. These data presented here demonstrate that ELAVL1 promoted BECN1 mRNA stability via binding to the AREs within the F3 of the 3ʹ-UTR, which was the fundamental cause of autophagy activation and subsequent ferroptosis enhancement.
Attractively, we retrospectively analyzed consecutive advanced fibrotic patients with HCC who were treated with sorafenib monotherapy in the Second Affiliated Hospital of Southeast University between February 2013 and June 2017. Interestingly, ELAVL1 upregulation, ferritinophagy activation, and ferroptosis induction occurred in primary human HSCs from the collected human liver tissue. However, our study has several limitations that should be mentioned. First, potential bias in the selection of samples is inherent to the retrospective nature. The most suitable clinical samples are fibrotic patients who were treated with sorafenib monotherapy. However, the oral multikinase inhibitor sorafenib has been clinically approved for the treatment of HCC. Therefore, fibrotic patients with HCC were included in the retrospective cohort study. Importantly, to minimize the risk of selection bias, primary human HSCs were isolated from the collected liver tissue samples for detection of ferroptosis markers. Given that HSCs are the most principal cellular players promoting synthesis and deposition of extracellular matrix proteins in liver fibrosis, it is reasonable that the results of isolated HSCs are mainly related to liver fibrosis. Second, the number of patients subjected to liver biopsy was relatively small. Liver biopsy remains the gold standard in the diagnosis of several liver disorders. However, many patients did not have the physical conditions required for the operation, and were also mentally reluctant to undergo surgery. Third, this was a single-center retrospective study rather than a nationwide investigation, which limits the generalizability of our data. The results obtained from this study need to be verified in a multicenter study with a large sample size.
In conclusion, the interaction of ELAVL1 and BECN1 mRNA serves as a key molecular event that triggers autophagy activation, promotes autophagic ferritin degradation, and in turn, leads to ferroptosis (Figure 9). Understanding the molecular mechanisms and signaling pathways of ferroptosis may provide new diagnostic and therapeutic approaches to regulate HSC survival and death in liver fibrosis.
Materials and methods
Human liver specimens
We retrospectively analyzed consecutive advanced fibrotic patients with HCC who were treated with sorafenib (Nexavar; Bayer Healthcare Pharmaceuticals, Leverkusen, Germany) monotherapy in the Second Affiliated Hospital of Southeast University between February 2013 and June 2017. The diagnosis of liver fibrosis and HCC was based on the criteria of the American Association for the Study of Liver Diseases (AASLD) [64]. An initial sorafenib dosage of 400 mg was administered orally twice daily, after breakfast and dinner [65]. Subsequently, discontinuations and dosage reductions of sorafenib were based on tolerance [65]. Treatment was continued until clinical disease progression or unacceptable drug-related toxicity occurred, or upon withdrawal of consent [65]. Informed consent in writing was obtained from patients. This study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki Principles, and was approved by the review committee of the Second Affiliated Hospital of Southeast University.
The total number of advanced fibrotic patients with HCC in the Second Affiliated Hospital of Southeast University between February 2013 and June 2017 were 136. Among these patients, 72 patients were treated with sorafenib monotherapy or polytherapy, whereas 64 patients were treated with other treatment modality but not sorafenib treatment. Importantly, among the above 72 patients, 26 patients were excluded due to combined use of other drug treatment; 5 patients were excluded due to post-liver transplantation usage of sorafenib; 2 patients were excluded due to adjuvant usage of sorafenib after curative surgery; and 11 patients were excluded because they did not follow up after their first prescriptions. Therefore, 28 advanced fibrotic patients with HCC who had undergone sorafenib monotherapy were assigned to the study. For detailed patients’ enrollment, please refer to Figure S7.
Data on patient characteristics were collected retrospectively from all patients by reviewing their electronic medical records. Patient records were scanned for the following information: gender, age, body mass index (BMI), Child-Pugh grade, model for end-stage liver disease (MELD) score, American Society of Anesthesiologists (ASA) class, serum GPT/ALT, GOT1/AST, ALPL (alkaline phosphatase), TBIL, GGT1 (gamma-glutamyltransferase 1), ALB (albumin), and IGH (immunoglobulin heavy) levels, etiology of liver cirrhosis, treatment and outcome data. For detailed patient characteristics, please refer to Table 1. Noteworthy, among the enrolled 28 patients, 5 patients underwent liver biopsy for accurate diagnosis of liver fibrosis and HCC before sorafenib treatment, while 5 patients received curative hepatectomy for the treatment of liver fibrosis and HCC after sorafenib monotherapy. We picked out the above eligible samples from our hospital preserved samples for the subsequent laboratory experiments. Blood samples were collected for the analyses of liver functions including GPT/ALT, GOT1/AST, and TBIL. Moreover, primary human HSCs were also isolated from the collected liver tissue by laser capture microdissection (LCM) as described previously [39–41], and total RNAs were extracted for RNA-Seq and real-time PCR.
Liver function assessment
Liver function was assessed by measurement of GPT/ALT, GOT1/AST, and TBIL using an Integrated Chemistry System Dimension® RxL Max® (Siemens Healthcare Diagnostics, Erlangen, Germany).
Primary human HSC isolation and characterization
Primary human HSCs were isolated from the collected human liver tissue by laser capture microdissection (LCM) as described previously [39–41]. Briefly, frozen human liver tissue was sectioned at 10 μm with a cryostat and stained with immunofluorescence for DES (desmin) (Abcam, ab15200). Once the tissue was stained, the DES-positive cells were located using specialized microdissection software. Then, a specific CapSure® LCM Cap (Thermo Fisher Scientific, LCM0211) was placed on the section. Pulsing the laser through the cap caused the thermoplastic film to form a thin protrusion that bridges the gap between the cap and tissue and adheres to the DES-positive cells. Lifting of the cap could remove the target cells attached to the cap. Purification and characterization of the obtained human HSCs were confirmed by detection of ACTA2 and PDGFRB (platelet derived growth factor receptor beta) [39–41]. Moreover, total RNAs were extracted from the isolated human HSCs for RNA-Seq and real-time PCR.
Animals and experimental design
All experimental procedures were approved by the institutional and local committee on the care and use of animals of Nanjing University of Chinese Medicine (Nanjing, China), and all animals received humane care according to the National Institutes of Health (USA) guidelines. Eight-week-old male C57BL/6 mice were purchased from the Experimental Animal Center of Yangzhou University (Yangzhou, China). Fifty mice were randomly divided into 5 groups of 10 animals each with comparable mean body weight. Mice of 5 groups were treated with Sham, BDL+VA-Lip-Control- siRNA, BDL+VA-Lip-Control-siRNA+sorafenib, BDL+VA-Lip-ELAVL1-siRNA, or BDL+VA-Lip-ELAVL1-siRNA+sorafenib. Mice were anesthetized with isoflurane. A midline laparotomy was performed, and the common bile duct was ligated close to the liver hilus immediately below the bifurcation with 3–0 surgical silk and cut between the ligatures as described previously [37]. Controls underwent a sham operation that consisted of exposure, but not ligation, of the common bile duct. VA-Lip-Control-siRNA and VA-Lip-ELAVL1-siRNA (0.75 mg/kg) were administrated intravenously 3 times a week for 2 weeks after the BDL operation [37]. Sorafenib (10 mg/kg, once every other day) was suspended in sterile phosphate-buffered saline (PBS; Sigma, P5368) and given by intraperitoneal injection for 2 weeks after the BDL operation [37]. The livers were collected 2 weeks after surgery under general anesthesia. A small portion of the liver was removed for histopathological and immunohistochemical studies by fixation with 10% formalin and subsequent embedment with paraffin. The remaining liver was cut in pieces and rapidly frozen with liquid nitrogen for extraction of total RNA and hepatic proteins. Moreover, primary mouse HSCs were further isolated from mouse liver and total RNAs were extracted for RNA-Seq and real-time PCR.
Histological analysis and immunohistochemistry
The specimen was sequentially fixed in 10% formalin for 2 days, transferred to ethanol of different concentration and embedded in paraffin in preparation for histopathological analysis. Thin sections (4 μm) were stained with H&E, Sirius Red, and Masson for histopathological study. H&E, Sirius Red, and Masson-stained areas from 10 random fields were quantified with ImageJ software (NIH, Bethesda, MD, USA). The liver fibrosis stage was assessed by Ishak scale [66]. According to the above results, 3 sections were chosen from each group for immunohistochemistry analysis. Briefly, sections prepared on slides were first submitted to antigen retrieval by incubation in citrate buffer (pH 6.0) for 5 min at 108°C and pretreated with 3% H2O2 in PBS for 15 min at room temperature followed by washing with PBS. Slides were subsequently incubated in normal goat serum (EMD Millipore, NI02) for 20 min to block the nonspecific immunoreactivity. Next, the slides were treated with primary antibody ACTA2 (Abcam, ab5694), or COL1A1 (Abcam, ab34710) overnight at 4°C. In addition, tissue sections were processed omitting the primary antibody as the negative control. The slides were incubated with secondary antibody (horseradish peroxidase-conjugated anti-rabbit IgG) (Abcam, ab6728) and the reaction products were visualized using diaminobenzidine (Cell Signaling Technology, 8059) and monitored by microscopy. ACTA2- and COL1A1-stained areas from 10 random fields were quantified with ImageJ software.
Hydroxyproline assay
The hydroxyproline content in liver tissue was evaluated using a Hydroxyproline Assay Kit (Abcam, ab222941) according to the manufacturer’s instructions.
Primary mouse hepatocyte isolation and characterization
Primary mouse hepatocytes were isolated from mouse liver according to a reported protocol [67]. Briefly, the livers of the mice were first perfused in situ via the portal vein with Ca2+- and Mg2+-free Hanks’ balanced salt solution (HBSS, Sigma, H1641) supplemented with 0.5 mM EGTA (Sigma, E3889) and 25 mM HEPES (Sigma, H3375) at 37.8°C. Then, the buffer was replaced with 0.1% collagenase I solution (Sigma, C0130) in HBSS (containing 4 mM CaCl2, 0.8 mM MgSO4). After a few minutes of perfusion, the liver was excised rapidly from the body cavity and dispersed into cold HBSS. The cell suspension generated was filtered through a sterile 70-μm pore size nylon cell strainer (Sigma, CLS431751) and spun 3 times at 30 x g for 4 min. The pellets were suspended in Dulbecco’s modified essential medium (DMEM; Gibco, 12491–015) containing 10% fetal bovine serum (FBS; Gibco, 10099141) for primary hepatocyte culture. Purification and characterization of the obtained hepatocytes were confirmed by detection of ALB [67].
Primary mouse HSC isolation and characterization
Primary mouse HSCs were isolated from the mouse liver according to a previous protocol [11]. Briefly, the livers of the mice were first perfused in situ with DMEM-free containing 1 mg/ml collagenase IV (Vetec, V900893) and 2 mg/ml pronase (Roche, PRON-RO) following HBSS including 0.5 mM EDTA (Sigma, E6758). After a few minutes of perfusion, the livers were removed, and the digested hepatic cells were dispersed in DMEM-free. Next, DNase enzymes (Sigma, D4263) were added to prevent filamentous gelatinous material, and the undigested debris was removed through a filter. The filtrates were centrifuged at 50 x g in a centrifuge tube for 5 min at 4 °C. The supernatant was collected following gradient centrifugation with 25% Nycodenz (Sigma, D2158) to isolate primary HSCs. Cells were washed and plated on 60-mm diameter tissue culture dishes (Sigma, CLS430599). Purification and characterization of the obtained HSCs were confirmed by detection of ACTA2 and PDGFRB [11].
Primary mouse macrophage isolation and characterization
Primary mouse macrophages were isolated from mouse liver according to a reported protocol [67]. Briefly, the livers of the mice were first perfused in situ with Ca2+- and Mg2+-free HBSS containing 2.5 mM EGTA via portal vein and then they were perfused again with 0.05% collagenase I HBSS solution. Digested livers were dissected and then gently teased with forceps until they were in solution. The cell suspensions were filtered through a 70-μm nylon cell strainer. Nonparenchymal cells were separated from the hepatocytes by 1 cycle of differential centrifugation (300 x g for 5 min). The supernatant was centrifuged further (300 x g for 5 min and 2 cycles of 1200 x g for 5 min) to obtain nonparenchymal cells. The obtained nonparenchymal cells were resuspended in DMEM with 2 % FBS, and separated by centrifugation on a 25%-50% Percoll (Sigma, P4937) gradient. The macrophage fraction located at the interface of the 25%-50% Percoll layer was seeded in DMEM containing 10% FBS and 10 mM HEPES. Purification and characterization of the obtained macrophages were confirmed by detection of ADGRE1/F4/80 [67].
Cell culture conditions and drug treatment
HSC-LX2 (BNCC337957) and HSC-T6 (BNCC337976) cells were obtained from BeNa culture collection (Beijing, China). These HSC cells were cultured in DMEM with 10% FBS, 1% antibiotics, and maintained at 37°C in a humidified incubator of 5% CO2 and 95% air. Cell morphology was assessed using an inverted microscope with a Leica Qwin System (Leica, Germany). Erastin (Selleck Chemicals, S7242) and sorafenib (Selleck Chemicals, S7397) were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10 mM and stored in a dark-colored bottle at −20°C. The stock was diluted to the required concentration with DMSO when needed. Prior to the drug treatment, cells were grown to ~ 70% confluence, and then exposed to drug at different concentrations (0–10 μM) for different periods of time (0–24 h). Cells grown in a medium containing an equivalent amount of DMSO without drugs served as a control.
Reagents and antibodies
Ferrostatin-1 (S7243), liproxstatin-1 (S7699), and necrosulfonamide (S8251) were purchased from Selleck Chemicals. BSO (B2515), Z-VAD-FMK (V116), necrostatin-1 (N9037), MG-132 (M7449), CHX (C7698), chloroquine (PHR1258), DMSO (156914), and CCl4 (488488) were purchased from Sigma-Aldrich. Opti MEM medium (51985034) and trypsin-EDTA (25200114) were bought from Gibco BRL. Primary antibodies against ELAVL1 (ab200342), LC3-I/II (ab128025), MAP1LC3B (ab48394), ATG12-ATG5 (ab108327), BECN1 (ab227107), ATG7 (ab133528), ATG14 (ab139727), SQSTM1 (ab56416), ACTA2 (ab5694), and COL1A1 (ab34710) were purchased from Abcam Technology. Primary antibodies against FTH1 (3998), ACTB (4970), GAPDH (2118), anti-rabbit IgG (7054), and anti-mouse IgG (7076) were purchased from Cell Signaling Technology.
Plasmid construction and gene transfection
The ELAVL1 siRNA (sc-35619, sc-270446), BECN1 siRNA (sc-29797, sc-29798), and Control siRNA were purchased from Santa Cruz Biotechnology. The pCDNA3.1-ELAVL1 plasmid (NM_001419.2, NM_001108848.1), pCDNA3.1- BECN1 plasmid (NM_003766, NM_001034117.1), and Control vector were ordered from Hanbio (KW20170208RFF-LP01). The full length or indicated fragments of human BECN1 mRNA 3ʹ-UTR (FL: spanning positions 1004–1774, F1: spanning positions 1004–1254, F2: spanning positions 1204–1454, F3: spanning positions 1404–1774) and a portion of human TNF mRNA 3ʹ-UTR (F4: spanning positions 1301–1380) were amplified and inserted in pEGFP-C2 (Clontech, 6083–1). The resulting plasmid was verified by sequencing. Transfections were performed with Lipofectamine™ 3000 (Invitrogen, L3000008) according to the manufacturer’s instructions. VA-Lip-ELAVL1-siRNA and VA-Lip-Control-siRNA were prepared according to a reported method [37]. Briefly, siRNA of ELAVL1 (5ʹ-AAGAGGCAAUUACCAGUUUCA-3ʹ) was synthesized by Genscript (Nanjing, China). VA (5 mg) was added into DMSO (50 μl) to form VA solution. VA solution (280 nmol) and Lipotrust solution (0.14 μmol; Hokkaido System Science, LEO-01) were mixed by vortexing in a 1.5-ml tube at 25°C. Control-siRNA or ELAVL1-siRNA (12.24 nmol) was added into VA-Lip solution with stirring at 25°C. The VA-Lip-siRNA solution was filtered. Fractions were collected and the material trapped in the filter was reconstituted with PBS to achieve the desired dose for in vivo use.
Immunofluorescence analysis
Immunofluorescence staining with treated cells was performed as we previously reported [11]. 4ʹ,6-Diamidino-2-phenylindole (DAPI, Sigma, D9542) was used to stain the nucleus in HSCs. All the images were captured with the fluorescence microscope and representative images were shown. The ImageJ software was used to quantify the fluorescence intensity on the micrographs.
Cell viability analysis
Cell viability was evaluated with a CCK8 Cell Counting Kit (Beyotime Institute of Biotechnology, C0042) according to the manufacturer’s instructions. Briefly, HSC cells were plated in a 96-well plates (Sigma, CLS9898) and exposed to various concentrations of the cytotoxic compounds for the indicated times. The 10 μl CCK8 reagents were added to each well and incubated at 37°C in 5% CO2 for 4 h, and then the plates were measured at 450 nm using the Tecan Safire2 Multi-detection Microplate Reader (Morrisville, NC, USA).
Trypan blue staining
HSC-LX2 cells were cultured in 6-well plates (Sigma, CLS3516) for 24 h and then treated with vehicle, erastin, erastin+ ZVAD-FMK, erastin+ necrosulfonamide, and erastin+ ferrostatin-1. After the indicated treatment times, cell suspension and 0.4% trypan blue solution (Sigma, T8154) were mixed in 9:1 ratio. After 3 min, the counting plate containing the live cells (no cytoplasmic fluorescence) and dead cells (blue cytoplasmic fluorescence) were counted. The trypan blue-positive ratio from 10 random fields was quantified with ImageJ software.
Fluorescein diacetate staining
After the indicated treatment, HSC-LX2 cells were rinsed twice with sterile HBSS, incubated in 10 ml of FDA working solution (10 mg/mL; Sigma, F7378) at 37°C for 15 min, and then washed twice with HBSS. Subsequently, HSC-LX2 cells were observed and photos were taken using an inverted fluorescence microscope (Leica, Germany). FDA-positive ratio from 10 random fields was quantified with ImageJ software.
Calcein-AM/PI fluorescence staining
After the indicated treatment, HSC-LX2 cells were washed twice with PBS, and a solution of 2 μmol/L calcein-AM and 4.5 μmol/L propidium iodide (Sigma, 04511) was added to the culture wells, and incubated at 37°C in the dark for 20 min. Living cells (green cytoplasmic fluorescence) and dead cells (red nucleus) were observed in an inverted fluorescence microscope (Leica, Germany). Live:dead cell ratio from 10 random fields was quantified with ImageJ software.
Lipid peroxidation assay
The lipid peroxidation product MDA concentration in cell lysates was assessed using a Lipid Peroxidation Assay Kit (Abcam, ab118970) according to the manufacturer’s instructions.
Iron assay
The relative iron concentration in cell lysates was assessed using an Iron Assay Kit (Abcam, ab83366) according to the manufacturer’s instructions.
Glutathione assay
The relative GSH concentration in cell lysates was assessed using a Glutathione Assay Kit (Sigma, CS0260) according to the manufacturer’s instructions.
Reactive oxygen species assay
Intracellular ROS level was measured using an oxidation-sensitive fluorescent probe DCFH-DA (Sigma, D6883) according to the manufacturer’s instructions.
Transmission electron microscopy
HSC-LX2 cells were seeded onto 4-well Chambered Coverglass (Thermo Scientific, 155382) at a density of 2 × 104 cells/mL (14,000 cells/well). Images were acquired using the Olympus EM208S transmission electron microscope.
Western blot analysis
HSC cells were lysed using a mammalian lysis buffer (Sigma, MCL1) and immunoblotting was performed according to the manufacturer’s guidelines (Bio/Rad, Hercules, CA, USA). Densitometry analysis was performed using the ImageJ software.
RNA isolation and real-time PCR
Total RNA was isolated and qPCR performed using the QuantiTect SYBR Green PCR Kit (Qiagen, 204141) in accordance with the manufacturer’s instructions. ACTB levels were taken for normalization and fold change was calculated using 2−ddCt. Primer Sequence available on request.
RNA-Seq and computational analysis
The isolated primary HSCs were collected and lysed with Trizol reagent (Sigma, T9424). RNA (1 μg) was used for library preparation with TruSeq Stranded Total RNA with Ribo-Zero Gold kit (Illumina, MRZG12324). The sequenced reads were aligned to the mouse reference genome (GRCm38.p5) or the human reference genome (GRCh38.p10) using TopHat v1.4.1. Differential gene expression was performed with EdgeR (Empirical analysis of digital gene expression data in R) version 3.08. Adjusted P values were computed using the Benjamini-Hochburg method. We used >1.4-fold change for upregulation and <0.65-fold change for downregulation as cutoffs.
Luciferase reporter system construction
The construction of chimeric firefly luciferase reporter plasmids were generated as described previously [35]. Briefly, the full length or indicated fragments of 3ʹ-UTR of human BECN1 mRNA were subcloned into the pGL3-Luc plasmid (Promega, E1751) at the Xba1 site to generate the chimeric pGL3-Luc-BECN1-3ʹ-UTR (Luc-BECN1-3ʹ-UTR) reporter construct. The mutants of the BECN1 3ʹ-UTR were generated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, 200518) according to the manufacturer’s instructions. The sequence and orientation of the luciferase reporter were verified by sequencing and enzyme digestion. Primers for the luciferase reporter system constructs or for deleting the fragments are available on request. The luciferase reporter constructs were transfected in cells along with phRL-null (luciferase control reporter vector; Promega, E2231) to monitor transfection efficiencies as described [35]. Luciferase activity was measured using the Dual Luciferase Assay System (Promega, E1910).
RNP IP
The immunoprecipitation of endogenous ribonucleoprotein complexes was analyzed using Imprint® RNA Immunoprecipitation Kit (Sigma, RIP-12RXN) according to the manufacturer’s instructions.
GST affinity isolation
The synthesis of biotinylated transcripts and analysis of RNA binding proteins bound to biotinylated RNA were performed as described previously [37]. cDNA from HSC-LX2 cells was used as a template for PCR amplification of the 5ʹ-UTR, CR, and 3ʹ-UTR of BECN1. All sequences of oligonucleotides for preparation of various short RNA probes for mapping the BECN1 3ʹ-UTR were as follows: FL: spanning positions 1004–1774, F1: spanning positions 1004–1254, F2: spanning positions 1204–1454, F3: spanning positions 1404–1774. A portion of human TNF 3ʹ-UTR was as follows: F4: spanning positions 1301–1380. Approximately 8 × 106 cells were washed twice with PBS, harvested and lysed in Cell Lysis Buffer (Cell Signaling Technology, 9803) plus complete protease inhibitor cocktail (Sigma, P8340). Biotinylated transcripts (9 μg) were incubated with cytoplasmic lysates (120 μg) for 30 min at room temperature. Complexes were isolated with paramagnetic streptavidin-conjugated Dynabeads (Dynal, 11206D) and analyzed by western blot analysis.
Long-lived proteins degradation analysis
The HSC-LX2 cells stably transfected with ELAVL1 siRNA or ELAVL1 plasmid were cultured in DMEM supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, glutamine, 10% FBS, and labeled with either L-arginine and L-lysine, L-[U-13C6,14N4] arginine and L-[2H4] lysine, or L-[U-13C6,15N4] arginine and L-[U-13C6,15N2] lysine (Cambridge Isotope Laboratories, 74–79-3; Sigma-Aldrich, 23128). After erastin treatment, the cells were scraped in cell scraping buffer (0.25M sucrose [Sigma-Aldrich, BP818], 1 mM sodium orthovanadate [Sigma, S6508], 5 mM NaF [Sigma-Aldrich, S7920], 5 mM β-glycerophosphate [Sigma, G5422], and protease inhibitor mixture [Roche, 04693132001]) and normalized by cell counting. Mixed cells were centrifuged for 5 min at 1500 x g and lysed in 6 M urea (Sigma, U5378), 2 M thiourea (Sigma-Aldrich, PHR1758), and 2% benzonase (Sigma, E1014) was added before samples were concentrated on spin tubes (Sigma, CLS8160). Protein mixtures were separated by SDS-PAGE (4–12% Bis-Tris Midi Protein Gels, NuPAGE; Invitrogen, WG1401A). Gel lanes were cut into 15 slices, and samples were in-gel digested, and resulting peptide mixtures were STAGE-tipped. Relative quantification and identification of peptides were analyzed by LC-MS/MS as described previously [12].
Calculations and statistics
Individual cell experiments and animal experiments were performed in duplicate or triplicate and repeated 3 times using matched controls, and the data were pooled. Data were expressed as mean ±standard error of the mean (SEM). Statistical analysis was performed using either Student’s t-test (two-group comparison) or one-way analysis of variance followed by Student-Newman-Keuls test (more than two groups). In the retrospective cohort study, quantitative values were expressed as means with standard deviations or medians with range, while categorical values were expressed as numbers with percentages. Categorical variables were analyzed using the chi-squared test or Fisher’s exact test as appropriate. Continuous variables were analyzed using the paired t-test or Wilcoxon tests as appropriate. In all analyses, a probability of less than 0.05 was considered to indicate statistical significance. All statistical analyses were performed using the SPSS program (version 20.0; IBM, Somers, NY, USA).
Funding Statement
This work was supported by the National Natural Science Foundation of China (NSFC) [81270514];National Natural Science Foundation of China (NSFC) [31571455];National Natural Science Foundation of China (NSFC) [31401210];Postgraduate Research & Practice Innovation Program of Jiangsu Province [KYCX17_1322];National Natural Science Foundation of China (NSFC) [81600483];the Open Project Program of Jiangsu Key Laboratory for Pharmacology and Safety Evaluation of Chinese Materia Medica [JKLPSE 201502];Project of the Priority Academic Program Development of Jiangsu Higher Education Institutions [PAPD];National Natural Science Foundation of China (NSFC) [31600653].
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81270514, 31571455, 31401210, 31600653, and 81600483), the Natural Science Foundation of Jiangsu Province (BK20140955), the Open Project Program of Jiangsu Key Laboratory for Pharmacology and Safety Evaluation of Chinese Materia Medica (JKLPSE 201502), the Project of the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX17_1322). Finally, I sincerely thank my wife (Mei Guo) for her support and encouragement in a difficult period. I shall love you as long as I have breath.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
Supplemental data for this article can be accessed here.
References
- [1].Ding BS, Cao Z, Lis R, et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].De Magalhaes Filho CD, Downes M, Evans R.. Bile acid analog intercepts liver fibrosis. Cell. 2016;166:789. [DOI] [PubMed] [Google Scholar]
- [3].Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen Y, Choi SS, Michelotti GA, et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology. 2012;143:1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shackel NA, Vadas MA, Gamble JR, et al. Beyond liver fibrosis: hepatic stellate cell senescence links obesity to liver cancer by way of the microbiome. Hepatology. 2014;59:2413–2415. [DOI] [PubMed] [Google Scholar]
- [6].Ceccarelli S, Panera N, Alisi A, et al. Hepatic stellate cell proliferation: a potential role of protein kinase R. Hepatology. 2011;54:1484–1485. [DOI] [PubMed] [Google Scholar]
- [7].Paik YH, Kim JK, Lee JI, et al. Celecoxib induces hepatic stellate cell apoptosis through inhibition of Akt activation and suppresses hepatic fibrosis in rats. Gut. 2009;58:1517–1527. [DOI] [PubMed] [Google Scholar]
- [8].Choi HS, Kang JW, Lee SM.. Melatonin attenuates carbon tetrachloride-induced liver fibrosis via inhibition of necroptosis. Transl Res. 2015;166:292–303. [DOI] [PubMed] [Google Scholar]
- [9].Wree A, Eguchi A, McGeough MD, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59:898–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Krizhanovsky V, Yon M, Dickins RA, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang Z, Zhao S, Yao Z, et al. Autophagy regulates turnover of lipid droplets via ROS-dependent Rab25 activation in hepatic stellate cell. Redox Biol. 2017;11:322–334. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [12].Zhang Z, Yao Z, Zhao S, et al. Interaction between autophagy and senescence is required for dihydroartemisinin to alleviate liver fibrosis. Cell Death Dis. 2017;8:e2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sun X, Ou Z, Chen R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Conrad M, Friedmann Angeli JP. Glutathione peroxidase 4 (Gpx4) and ferroptosis: what’s so special about it? Mol Cell Oncol. 2015;2:e995047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dixon SJ. Ferroptosis: bug or feature? Immunol Rev. 2017;277:150–157. [DOI] [PubMed] [Google Scholar]
- [18].Louandre C, Marcq I, Bouhlal H, et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015;356(2 Pt B):971–977. [DOI] [PubMed] [Google Scholar]
- [19].Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gao M, Monian P, Pan Q, et al. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Latunde-Dada GO. Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta. 2017;1861:1893–1900. [DOI] [PubMed] [Google Scholar]
- [23].Zhang Z, Guo M, Zhao S, et al. ROS-JNK1/2-dependent activation of autophagy is required for the induction of anti-inflammatory effect of dihydroartemisinin in liver fibrosis. Free Radic Biol Med. 2016;101:272–283. [DOI] [PubMed] [Google Scholar]
- [24].Dodson RE, Shapiro DJ. Regulation of pathways of mRNA destabilization and stabilization. Prog Nucleic Acid Res Mol Biol. 2002;72:129–164. [DOI] [PubMed] [Google Scholar]
- [25].Angeli JPF, Shah R, Pratt DA, et al. Ferroptosis inhibition: mechanisms and opportunities. Trends Pharmacol Sci. 2017;38:489–498. [DOI] [PubMed] [Google Scholar]
- [26].Chang SH, Hla T. Post-transcriptional gene regulation by HuR and microRNAs in angiogenesis. Curr Opin Hematol. 2014;21:235–240. [DOI] [PubMed] [Google Scholar]
- [27].Wang J, Guo Y, Chu H, et al. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int J Mol Sci. 2013;14:10015–100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zucal C, D’Agostino V, Loffredo R, et al. Targeting the multifaceted HuR protein, benefits and caveats. Curr Drug Targets. 2015;16:499–515. [DOI] [PubMed] [Google Scholar]
- [29].Srikantan S, Gorospe M. HuR function in disease. Front Biosci (Landmark Ed). 2012;17:189–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yu Y, Xie Y, Cao L, et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol. 2015;2:e1054549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lachaier E, Louandre C, Godin C, et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014;34:6417–6422. [PubMed] [Google Scholar]
- [32].Bertrand RL. Iron accumulation, glutathione depletion, and lipid peroxidation must occur simultaneously during ferroptosis and are mutually amplifying events. Med Hypotheses. 2017;101:69–74. [DOI] [PubMed] [Google Scholar]
- [33].Kang R, Zeh HJ, Lotze MT, et al. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yu TX, Gu BL, Yan JK, et al. CUGBP1 and HuR regulate E-cadherin translation by altering recruitment of E-cadherin mRNA to processing bodies and modulate epithelial barrier function. Am J Physiol Cell Physiol. 2016;310:C54–C65. [DOI] [PubMed] [Google Scholar]
- [36].Chen Q, Chen L, Kong D, et al. Dihydroartemisinin alleviates bile duct ligation-induced liver fibrosis and hepatic stellate cell activation by interfering with the PDGF-βR/ERK signaling pathway. Int Immunopharmacol. 2016;34:250–258. [DOI] [PubMed] [Google Scholar]
- [37].Wu X, Wu X, Ma Y, et al. CUG-binding protein 1 regulates HSC activation and liver fibrogenesis. Nat Commun. 2016;7:13498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wen X, Paine ML. Iron deposition and ferritin heavy chain (FTH) localization in rodent teeth. BMC Res Notes. 2013;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shang L, Hosseini M, Liu X, et al. Human hepatic stellate cell isolation and characterization. J Gastroenterol. 2018;53:6–17. [DOI] [PubMed] [Google Scholar]
- [40].McDaniel K, Huang L, Sato K, et al. The let-7/Lin28 axis regulates activation of hepatic stellate cells in alcoholic liver injury. J Biol Chem. 2017;292:11336–11347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].McDaniel K, Meng F, Wu N, et al. Forkhead box A2 regulates biliary heterogeneity and senescence during cholestatic liver injury in mice. Hepatology. 2017;65:544–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gao M, Jiang X. To eat or not to eat-the metabolic flavor of ferroptosis. Curr Opin Cell Biol. 2017;51:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wenzel SE, Tyurina YY, Zhao J, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171:628–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wang H, An P, Xie E, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66:449–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Alvarez SW, Sviderskiy VO, Terzi EM, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551:639–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hao S, Yu J, He W, et al. Cysteine dioxygenase 1 mediates erastin-induced ferroptosis in human gastric cancer cells. Neoplasia. 2017;19:1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chen D, Fan Z, Rauh M, et al. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene. 2017;36:5593–5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen YL, Lv J, Ye XL, et al. Sorafenib inhibits transforming growth factor β1-mediated epithelial-mesenchymal transition and apoptosis in mouse hepatocytes. Hepatology. 2011;53:1708–1718. [DOI] [PubMed] [Google Scholar]
- [49].Deng YR, Ma HD, Tsuneyama K, et al. STAT3-mediated attenuation of CCl4-induced mouse liver fibrosis by the protein kinase inhibitor sorafenib. J Autoimmun. 2013;46:25–34. [DOI] [PubMed] [Google Scholar]
- [50].Wang Y, Gao J, Zhang D, et al. New insights into the antifibrotic effects of sorafenib on hepatic stellate cells and liver fibrosis. J Hepatol. 2010;53:132–144. [DOI] [PubMed] [Google Scholar]
- [51].Yu D, Zhang C, Gui J. RNA-binding protein HuR promotes bladder cancer progression by competitively binding to the long noncoding HOTAIR with MIR-1. Onco Targets Ther. 2017;10:2609–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Xu X, Song C, Chen Z, et al. Downregulation of HuR inhibits the progression of esophageal cancer through interleukin-18. Cancer Res Treat. 2018;50:71–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Badawi A, Hehlgans S, Pfeilschifter J, et al. Silencing of the mRNA-binding protein HuR increases the sensitivity of colorectal cancer cells to ionizing radiation through upregulation of caspase-2. Cancer Lett. 2017;393:103–112. [DOI] [PubMed] [Google Scholar]
- [54].Gubin MM, Calaluce R, Davis JW, et al. Overexpression of the RNA binding protein HuR impairs tumor growth in triple negative breast cancer associated with deficient angiogenesis. Cell Cycle. 2010;9:3337–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Katsanou V, Papadaki O, Milatos S, et al. HuR as a negative posttranscriptional modulator in inflammation. Mol Cell. 2005;19:777–789. [DOI] [PubMed] [Google Scholar]
- [56].Yiakouvaki A, Dimitriou M, Karakasiliotis I, et al. Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. J Clin Invest. 2012;122:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wu L, Zhang Q, Mo W, et al. Quercetin prevents hepatic fibrosis by inhibiting hepatic stellate cell activation and reducing autophagy via the TGF-β1/Smads and PI3K/Akt pathways. Sci Rep. 2017;7:9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Thoen LF, Guimarães EL, Grunsven LA. Autophagy: a new player in hepatic stellate cell activation. Autophagy. 2012;8:126–128. [DOI] [PubMed] [Google Scholar]
- [59].Hernández-Gea V, Friedman SL. Autophagy fuels tissue fibrogenesis. Autophagy. 2012;8:849–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Huang J, Yang J, Shen Y, et al. HMGB1 mediates autophagy dysfunction via perturbing Beclin1-Vps34 complex in dopaminergic cell model. Front Mol Neurosci. 2017;10:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Huang Q, Liu X, Cao C, et al. Apelin-13 induces autophagy in hepatoma HepG2 cells through ERK1/2 signaling pathway-dependent upregulation of Beclin1. Oncol Lett. 2016;11:1051–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Shrivastava S, Bhanja Chowdhury J, Steele R, et al. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J Virol. 2012;86:8705–8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ma B, Cao W, Li W, et al. Dapper1 promotes autophagy by enhancing the Beclin1-Vps34-Atg14L complex formation. Cell Res. 2014;24:912–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Muratori L, Muratori P, Lanzoni G, et al. Application of the 2010 American Association for the study of liver diseases criteria of remission to a cohort of Italian patients with autoimmune hepatitis. Hepatology. 2010;52:1857–1858. [DOI] [PubMed] [Google Scholar]
- [65].Nagai H, Kanekawa T, Kobayashi K, et al. Changes of cytokines in patients with liver cirrhosis and advanced hepatocellular carcinoma treated by sorafenib. Cancer Chemother Pharmacol. 2014;73:223–229. [DOI] [PubMed] [Google Scholar]
- [66].Standish RA, Cholongitas E, Dhillon A, et al. An appraisal of the histopathological assessment of liver fibrosis. Gut. 2006;55:569–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Aparicio-Vergara M, Tencerova M, Morgantini C, et al. Isolation of Kupffer cells and hepatocytes from a single mouse liver. Methods Mol Biol. 2017;1639:161–171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.