

ABSTRACT
Sunitinib, a multikinase inhibitor approved for a number of cancer indications has a low response rate. Identifying mechanisms of resistance could lead to rational combination regimens that could improve clinical outcomes. Here we report that resistance to sunitinib therapy was driven by autophagic degradation of TP53/p53. Deletion of ATG7 or ATG5 suppressed TP53 degradation, as did knockdown of SQSTM1/p62. Mechanistically, the transport of TP53 from the nucleus to the cytoplasm was essential for the sunitinib-induced autophagic degradation of TP53 and did not require TP53 nuclear export signals (NESs). Moreover, TP53 degradation was achieved by the transport of its nuclear binding target, HMGB1, which shifted TP53 from the nucleus to the cytoplasm. The inhibition of HMGB1 sensitized cancer cells to sunitinib. Importantly, sunitinib induced the degradation of all TP53 proteins, except for TP53 proteins with mutations in the interaction domain of TP53 with HMGB1 (amino acids 313 to 352). In conclusion, our data identify an alternative HMGB1-mediated TP53 protein turnover mechanism that participates in the resistance of sunitinib and suggest HMGB1 as a potential therapeutic target for improving clinical outcomes of sunitinib.
KEYWORDS: Autophagy-lysosomal degradation, HMGB1, nuclear export, sunitinib, TP53
Introduction
Sunitinib is an oral small molecule multikinase inhibitor that targets PDGFR, KIT/cKIT, FLT1/VEGFR1, and KDR/VEGFR2. However, sunitinib has only been approved for the treatment of metastatic renal cell carcinoma (mRCC), Gleevec-resistant gastrointestinal stromal tumors (GISTs) and primitive neurotodermal tumor (PNET) [1]. Most of the sunitinib clinical trials performed to treat other common and malignant solid cancers, including colon and liver cancers, have failed because of poor patient response rates [2]. Evidence suggests that sunitinib fails to produce durable clinical responses in most mRCC and GIST patients; their tumors begin to regrow and the disease progresses after a short period of clinical benefit that is typically measured in months [3]. These findings suggest the existence of primary and acquired drug resistance mechanisms. The underlying mechanism of the resistance of sunitinib is largely unknown. Hence, it is necessary to investigate the biological basis and identify novel targets for overcoming sunitinib resistance.
The activation of macroautophagy/autophagy reduces the anti-cancer effect of sunitinib in numerous cancer cells [4,5]. However, a clinical trial of hydroxychloroquine (HCQ) and sunitinib in advanced solid tumors that failed to progress might have resulted from the insufficient anti-cancer activity and the intolerable side effects that prohibited further dose escalation [6]. Thus, new strategies to target sunitinib-induced autophagy are needed. As autophagy is a catabolic membrane trafficking process that degrades a variety of cellular constituents [7], it is possible that some tumor suppressors may be degraded by autophagy during treatment with sunitinib.
The master tumor suppressor TP53 is under exquisitely fine regulation and is a transcription factor that regulates the expression of thousands of genes that control apoptosis, necroptosis, ferroptosis, cell cycle arrest, metabolism and fertility [8,9]. TP53 is crucial for triggering efficient cell death in cancer cells upon cellular stress evoked by chemotherapy or radiation [9]. The loss of function (mutation) or abnormal degradation of wild-type (WT) TP53 is a pivotal aspect of tumor formation in different human cancers [10]. Although the proteasome-dependent degradation of WT TP53 is established, autophagy-lysosome degradation is the other pathway for controlling cellular protein stability [11], but little is known of the role of autophagy in degrading WT TP53. Numerous proteins are degraded through both proteasome-dependent and autophagy-lysosome pathways [11], implying that autophagy may be involved in WT TP53 protein turnover.
In this study, we observed that sunitinib induced the autophagic degradation of WT TP53 in multiple cancer cell lines, which was closely related to the resistance of sunitinib. Mechanistically, the nucleus-to-cytoplasm shift was essential for sunitinib induced-autophagic degradation of WT TP53 and did not require TP53 nuclear export signals (NESs). Moreover, sunitinib-induced TP53 degradation was achieved by the transport of its nuclear binding target, HMGB1, which shifted TP53 from the nucleus to the cytoplasm. The inhibition of HMGB1 sensitized cancer cells to sunitinib. Of note, sunitinib also induced TP53 degradation, unless there was a mutation in the 313 to 352 amino acid region of TP53, which is the interaction domain of TP53 with HMGB1. In summary, our study identified an alternative HMGB1-mediated TP53 protein turnover mechanism that affected the levels of TP53 under sunitinib treatment; therefore, the underlying mechanisms of sunitinib resistance in cancer therapy have been elucidated.
Results
Sunitinib-induced degradation of TP53 by autophagy is involved in its low anti-cancer activity
We began with examining TP53 expression levels after sunitinib treatment in multiple cell lines. TP53 protein levels were reduced by sunitinib treatment in HCT116 and RCC4 cells in a concentration- and time-dependent manner (Figure 1(a) and S1). To confirm these responses, we re-examined the effect of sunitinib treatment on TP53 expression in numerous cell lines. As shown in Figure S2 and 1(b), sunitinib induced a significant decrease in WT TP53 expression in all of the cells that we examined, including cancer cells lines, immortalized hepatocytes, mouse embryonic fibroblasts (MEFs) and primary colon cancer cells that expressed WT TP53. Next, we investigated the effect of sunitinib-induced TP53 decrease on the anti-cancer activity of sunitinib. A cell proliferation assay was performed and the data showed that cells overexpressing TP53 were more sensitive to sunitinib than control cells. This result is consistent with previous observations that TP53 is critical for the potent suppressive effect on cancer cell growth (Figure S3).
Figure 1.
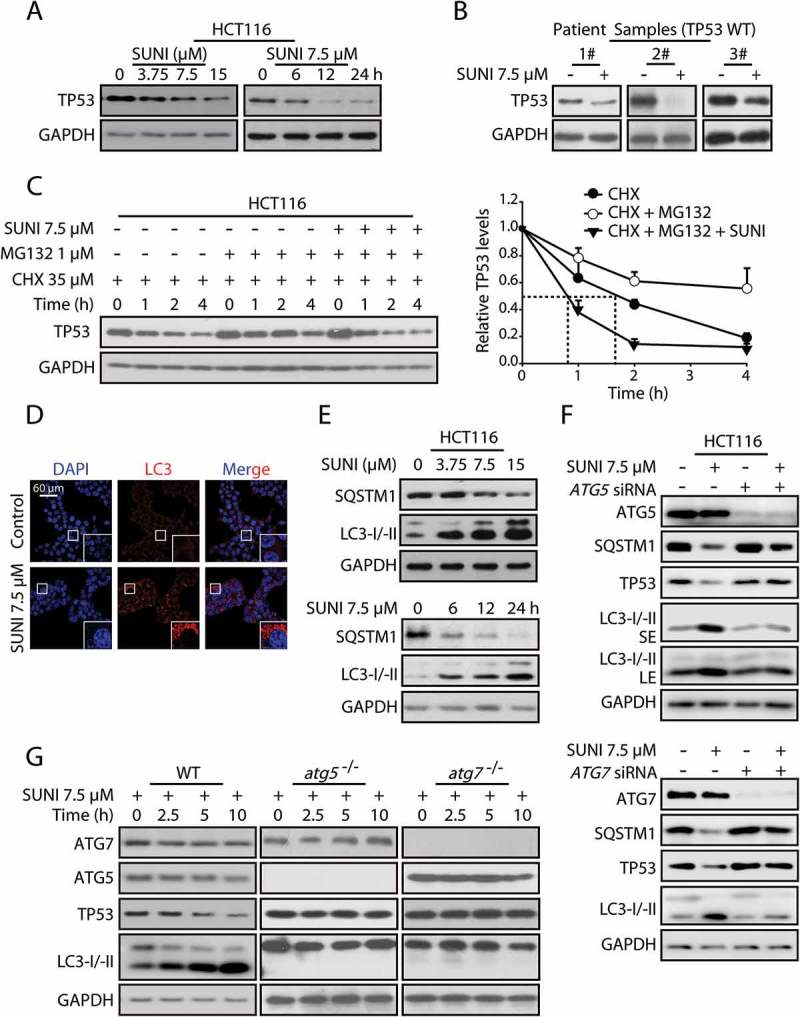
Sunitinib accelerates the degradation of TP53 protein via the autophagy-lysosome pathway. (a) Representative western blots show TP53 expression in untreated and sunitinib-treated HCT116 cells. The cells were treated for 6, 12 or 24 h with 7.5 µM sunitinib or for 24 h with 3.75, 7.5 or 15 µM sunitinib. (b) Representative western blots show TP53 expression in untreated and sunitinib-treated primary colon cancer cells. (c) HCT116 cells were treated with 35 µM CHX and 1 µM MG132 in the presence or absence of 7.5 µM sunitinib for different times, and TP53 protein levels were measured by western blot analysis (left panel). The half-life of TP53 was detected. The TP53 expression levels from biological triplicates were normalized with GAPDH and quantified with Quantity One. The data represent the average of 3 independent experiments (right panel). (d) Immunocytochemistry for LC3 and nuclear staining with DAPI in HCT116 cells treated with 7.5 µM sunitinib for 24 h. Scale bar: 60 μm. (e) Representative western blots show LC3 and SQSTM1 expression in the same cells as in (a). (f) Representative western blots show the effect of knockdowns of ATG5 and ATG7 on the sunitinib-induced degradation of TP53 in HCT116 cells. (g) Representative western blots show TP53 expression in untreated and sunitinib-treated WT, atg5−/− and atg7−/− MEFs. SUNI, sunitinib; CHX, cycloheximide; SE, short exposure time; LE, long exposure time. All experiments were performed in triplicate. For western blots, the data from 1 of 3 similar experiments are shown.
To explore the mechanism of the sunitinib-mediated decrease in TP53 protein levels, we first examined the effect of sunitinib on TP53 transcription levels using quantitative real-time PCR analysis [12]. Sunitinib treatment failed to decrease TP53 transcription levels (Figure S4). Next, we analyzed the effect of sunitinib on TP53 protein translation by treating the cells with the protein biosynthesis inhibitor cycloheximide (CHX) [13]. Sunitinib treatment decreased the half-life of TP53 protein (Figure S5), confirming that the reduction in TP53 protein levels is due to increased degradation.
Intracellular TP53 levels are primarily controlled by proteasome-dependent degradation [14], prompting us to examine the role of the proteasome in sunitinib-induced TP53 degradation. We inhibited proteasomal proteolytic activity using MG132, a specific proteasome inhibitor [15]. Consistent with the essential role of the proteasome in TP53 degradation, MG132 increased TP53 expression levels and prolonged its half-life. However, the addition of MG132 failed to inhibit sunitinib-induced TP53 degradation (Figure S6). Most importantly, sunitinib shortened the half-life of TP53 when the proteasome pathway was inhibited (Figure 1(c)). Collectively, these studies clearly demonstrated that sunitinib-induced TP53 degradation could occur independently of the proteasome.
Given that sunitinib activates autophagy (Figure 1(d,e)), and autophagy is the other primary system responsible for protein degradation in addition to the proteasome [16], it is possible that sunitinib-induced TP53 degradation is mediated by autophagy. As expected, 3-methyladenine (3-MA) and chloroquine (CQ), inhibitors of autophagy and lysosome function, respectively, efficiently prevented the TP53 degradation induced by sunitinib (Figure S7). Considering that ATG5 and ATG7 are essential for autophagosome formation and are required for autophagy [17], we used siRNA to knock down ATG5 and ATG7. We found that silencing ATG5 or ATG7 could block sunitinib-induced autophagy and TP53 degradation (Figure 1(f)). In addition, we examined atg5−/− or atg7−/− MEFs to further analyze the autophagic degradation of TP53. The knockout of Atg5 or Atg7 also significantly inhibited sunitinib-induced TP53 degradation (Figure 1(g)). These observations depart from the generally accepted notion that TP53 protein levels are mainly controlled by the proteasome. Here, we provide the first evidence that autophagy might be an alternative mechanism for WT TP53 degradation.
Nuclear export is essential for sunitinib-induced TP53 autophagic degradation, which does not rely on the nuclear export signals of TP53
The current literature indicates that protein degradation by autophagy mainly occurs in the cytoplasm [18]. We further aimed to detect whether sunitinib affected the nuclear export of TP53, given that TP53 exhibits a typical strong nuclear distribution in most cell types [19]. We used immunocytochemistry to detect the subcellular localization of TP53 after sunitinib treatment, and as shown in Figure 2(a) and S8A, most of the nuclear TP53 had been shifted to the cytoplasm. To confirm the effect of sunitinib on TP53 nuclear export, a subcellular fractionation analysis was performed on the cell lysates. After sunitinib treatment, TP53 levels decreased in the nuclear fraction but increased in the cytosolic fraction (Figure 2(b)). Based on these signature features, we then determined whether TP53 nuclear export was critical for the sunitinib-induced autophagic degradation of TP53. Leptomycin B (LMB), a specific inhibitor of the nuclear export receptor XPO1/CRM1, is able to increase TP53 via stabilizing TP53 in the nucleus and preventing TP53 degradation by inhibiting the activity of MDM2 [20]. As expected, LMB could inhibit sunitinib-induced degradation of TP53 (Figure 2(c) and S8B), suggesting that the nucleus-to-cytoplasm distribution of TP53 is crucial for its autophagic degradation.
Figure 2.
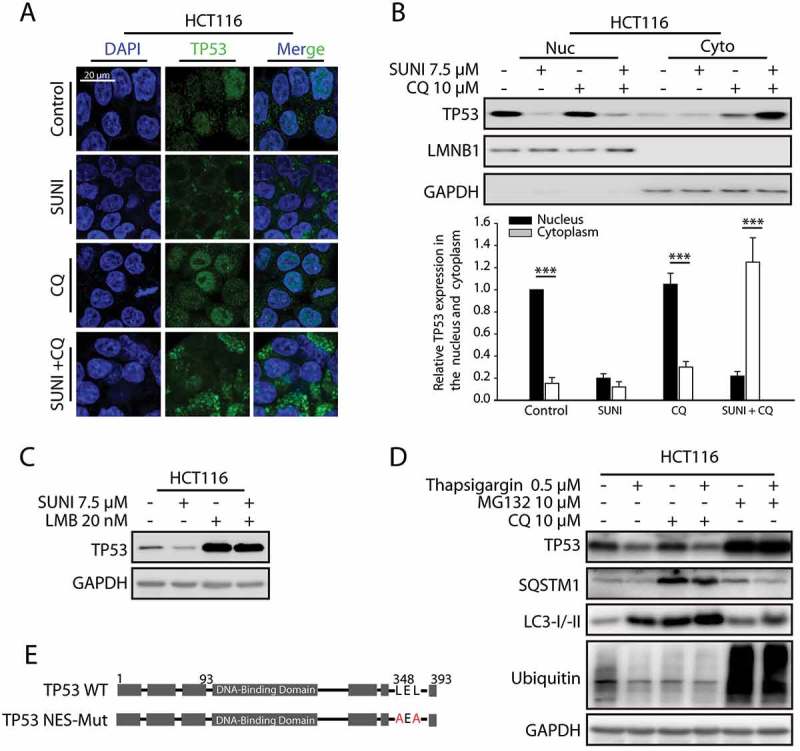
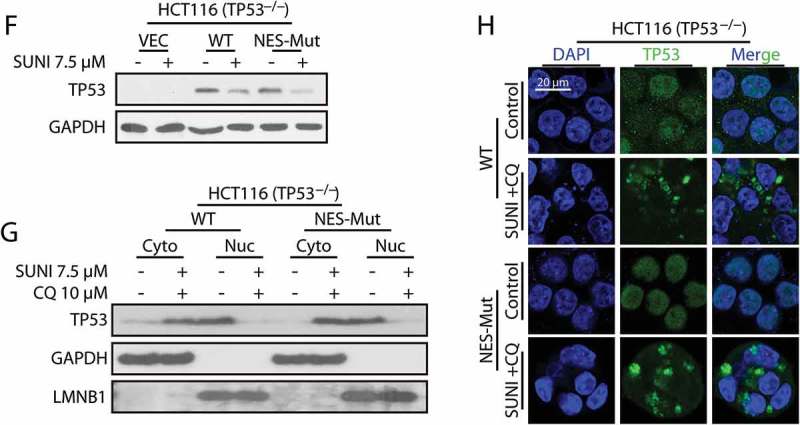
Nuclear export is essential for the induced autophagic degradation of TP53, which does not rely on TP53 nuclear export signals. (a) Immunocytochemistry for TP53 and nuclear staining by DAPI in HCT116 cells. The cells were treated with 7.5 µM sunitinib for 24 h in the absence or presence of 10 µM CQ added 2 h before the sunitinib addition. (b) Representative western blots show the cytoplasmic and nuclear distribution of TP53 in cells similar to those in (a) (upper panel). TP53 expression levels in the nuclear or cytoplasmic fractions of triplicate samples were normalized using LMNB1 (lamin B1) or GAPDH expression levels and quantified with Quantity One. The data represent the average of 3 independent experiments. Each value represents a ratio of the level of TP53 in the nucleus of control groups (lower panel). (***P = 0.009, nucleus versus cytoplasm in control groups; ***P = 0.0008, nucleus versus cytoplasm in CQ groups; ***P = 0.0006, nucleus versus cytoplasm in sunitinib plus CQ groups.) (c) Representative western blots show the effect of the inhibition of nucleus-to-cytoplasm transport by 20 nM LMB on the sunitinib-induced degradation of TP53 in HCT116 cells. Cells were treated with 20 nM LMB or 7.5 µM sunitinib alone or in combination for 24 h. (d) Representative western blots show the effect of TP53 cytoplasmic transport on the degradation of TP53 in HCT116 cells following thapsigargin treatment. Cells were treated with 0.5 µM thapsigargin for 24 h in the absence or presence of 10 µM CQ for 2 h before the addition of thapsigargin or in the absence or presence of 10 µM MG132 for 8 h before the experiments ended. (e) A schematic representation of the nuclear export signal (NES) mutation of TP53 used in this study. (f) Representative western blots show the effect of the NES mutation on the sunitinib-induced degradation of TP53 in TP53−/− cells in which the vector, WT or NES-mutated TP53 was re-expressed. The cells were treated with 7.5 µM sunitinib for 24 h. (g) Representative western blots show the effect of the NES mutation on the sunitinib-induced nuclear export of TP53 in TP53−/− cells in which WT or NES-mutated TP53 was re-expressed. Cells were treated with 7.5 µM sunitinib for 24 h. (h) Immunocytochemistry for TP53 and nuclear staining by DAPI in cells similar to that in (g). Scale bar: 20 μm. SUNI, sunitinib; LMB, leptomycin B; CQ, chloroquine. All experiments were performed in triplicate. For western blots, data from 1 of 3 experiments are shown.
From the above results, it was concluded that autophagy activation and nucleus-to-cytoplasm translocation of TP53 were 2 requirements for the degradation of TP53 by autophagy. We suspected that other stresses which can both induce TP53 cytoplasmic translocation and autophagy, such as ER stress, may cause the degradation of TP53 by autophagy [21,22]. The ER stress inducer thapsigargin (TG) activates autophagosome formation with LC3 conversion from the LC3-I to LC3-II form via the EIF2AK3/PERK-EIF2S1/eIF2α and ERN1/IRE1-MAPK/JNK pathways, as well as nucleus-to-cytoplasm translocation of TP53 [22,23]. Here we found that inhibiting autophagy by CQ failed to block TG induced-TP53 degradation (Figure S9 and Figure 2(d)). Our findings provided the first evidence that autophagy might be an alternative mechanism for WT TP53 degradation, and sunitinib-induced TP53 nuclear export was different from the canonical mechanism.
To further understand the relationship between nucleus-cytoplasm trafficking and the sunitinib-induced autophagic degradation of TP53, we generated TP53-NES-mutant cells by transfecting a TP53-NES-Mut plasmid into HCT116 TP53−/− cells. Surprisingly, the impaired NES of TP53 had little influence on sunitinib-induced TP53 cytoplasmic aggregation and degradation, indicating that sunitinib influenced the nucleus-to-cytoplasm shift of TP53 independent of its NESs (Figure 2(f-h)). Thus, these results suggest that other precise mechanisms might participate in sunitinib-induced TP53 nuclear export.
Figure 2.
(Continued).
Nuclear export of HMGB1 is essential for sunitinib-induced autophagic degradation of TP53
Based on the above findings, we sought other explanations for the sunitinib-evoked nucleus-to-cytoplasm shift of TP53. Because LMB is a powerful export blocker of CRM1 with respect to Rev-like NES proteins, such as TP53 [24], we first determined whether additional factors in export pathways that are CRM1 dependent could affect the nuclear export of TP53. HMGB1, as a TP53-interacting protein, is proposed to be a highly conversed nuclear protein that forms a complex with nuclear TP53 and is a nucleus-cytoplasmic shuttling protein [25]. To confirm whether HMGB1 is involved in the sunitinib-induced degradation of TP53 by autophagy, we targeted the exon of HMGB1 in HCT116 cells using the CRISPR/Cas9 system to generate HMGB1−/− cells. HMGB1 deletion almost completely prevented the degradation of TP53 induced by sunitinib (Figure 3(a)). Notably, the re-expression of HMGB1 in HMGB1−/− cells reactivated this degradation (Figure 3(b,d)), indicating a critical role for HMGB1 in sunitinib-induced TP53 degradation. In addition, the nucleus-to-cytoplasm shift of TP53 caused by sunitinib was completely inhibited in HMGB1−/− cells, whereas sunitinib-induced TP53 nuclear export was reactivated when HMGB1 was re-expressed in HMGB1−/− cells (Figure 3(e)). These data suggest that HMGB1 might participate in the sunitinib-induced transport of TP53 from the nucleus to the cytoplasm and its subsequent degradation by autophagy.
Figure 3.
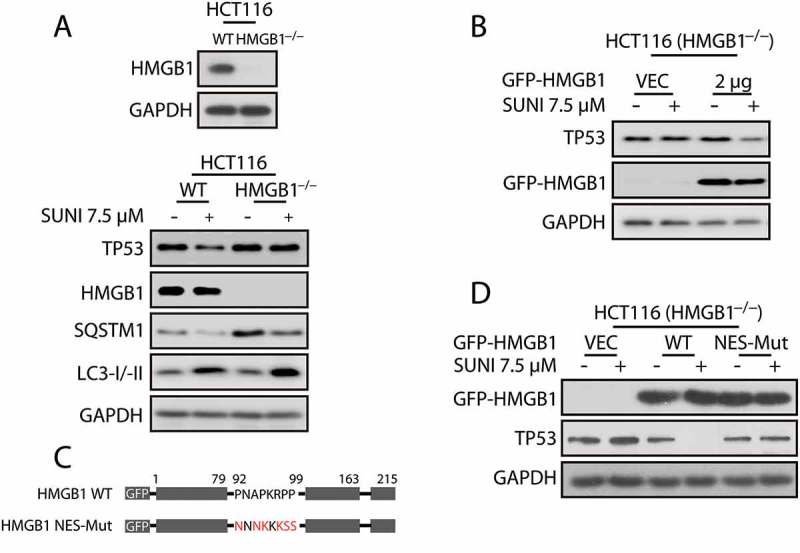
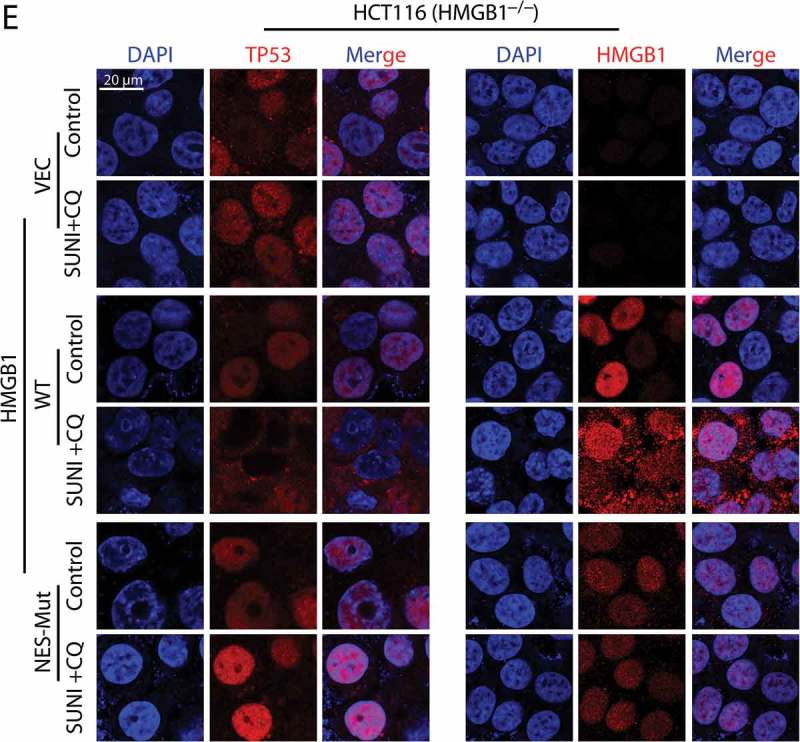
Nuclear export of HMGB1 is crucial for sunitinib-induced TP53 nuclear export and autophagic degradation. (a) Representative western blots show the effect of HMGB1 knockout on the sunitinib-induced degradation of TP53. Cells were treated with 7.5 µM sunitinib for 24 h. (b) Representative western blots show the sunitinib-induced degradation of TP53 in HMGB1−/− cells in which WT HMGB1 was re-expressed. Cells were treated with sunitinib for 24 h. (c) A schematic representation of the NES mutation of HMGB1 was used in this study. (d) Representative western blots show the effect of the NES mutation on the sunitinib-induced degradation of TP53 in HMGB1−/− cells in which vector, WT or NES-mutated HMGB1 were re-expressed. The cells were treated with 7.5 µM sunitinib for 24 h. (e) Immunocytochemistry for TP53, HMGB1 and nuclei (DAPI) in cells similar to that in (d). Scale bar: 20 μm. SUNI, sunitinib; CQ, chloroquine. All experiments were performed in triplicate. For western blots, data from 1 of 3 experiments are shown.
Figure 3.
(Continued).
Next, we focused on the precise mechanism by which HMGB1-mediated the TP53 nucleus-to-cytoplasm shift and autophagic degradation. Previous studies demonstrated that the nuclear export of HMGB1 is NES-dependent [26]; thus, we hypothesized that the nucleus-to-cytoplasm shift of HMGB1 may contribute to the TP53 nuclear export and autophagic degradation that occurred during sunitinib treatment. Vector, WT or NES-mutant HMGB1 plasmids were transfected into HMGB1−/− cells. As expected, the impaired nucleus-to-cytoplasm transport of HMGB1 completely inhibited the cytoplasmic aggregation and autophagic degradation of TP53, whereas the re-expression of WT HMGB1 reactivated these events (Figure 3(c-e)). These results indicate that HMGB1 mediates the sunitinib-induced autophagic degradation of TP53 by shifting nuclear TP53 to the cytoplasm.
Sunitinib induces the nuclear export of TP53 by HMGB1 for degradation by autophagy
To define the role of sunitinib in the HMGB1-mediated nuclear export of TP53 and the subsequent degradation of TP53 by autophagy, we used immunoprecipitation to analyze the influence of sunitinib on the interaction between TP53 and HMGB1. As shown in Figure 4(a,b), the enhanced interaction of TP53 and HMGB1 by sunitinib was detected only when the cells were co-treated with CQ to prevent TP53 from degradation by autophagy, suggesting that sunitinib induced HMGB1-associated autophagic degradation of TP53 by promoting the interaction of these 2 proteins. In parallel, we analyzed the direct effect of sunitinib on the nuclear export of HMGB1 in WT TP53 or TP53−/− cells using immunocytochemistry and subcellular fractionation analysis. Sunitinib directly caused transport of nuclear HMGB1 to the cytoplasm, regardless of the status of TP53 (Figure 4(c-e)). From these results we concluded that HMGB1 nuclear export is required for the transport of TP53 from the nucleus to the cytoplasm and for its subsequent degradation by autophagy. Next, we tested whether HMGB1 translocation caused the autophagic degradation of TP53 in general conditions. LPS treatment (the typical stress for HMGB1 translocation) had little effect on the degradation of TP53 (Figure 4(f) and S10), suggesting that sunitinib-induced HMGB1 translocation may be different from the canonical HMGB1 translocation. Moreover, sunitinib still induced autophagy in HMGB1−/− cells, where the autophagic degradation of TP53 was inhibited (Figure 3(a)). These data demonstrate that sunitinib-induced HMGB1 nuclear export is a prerequisite for the nuclear export and autophagic degradation of TP53.
Figure 4.
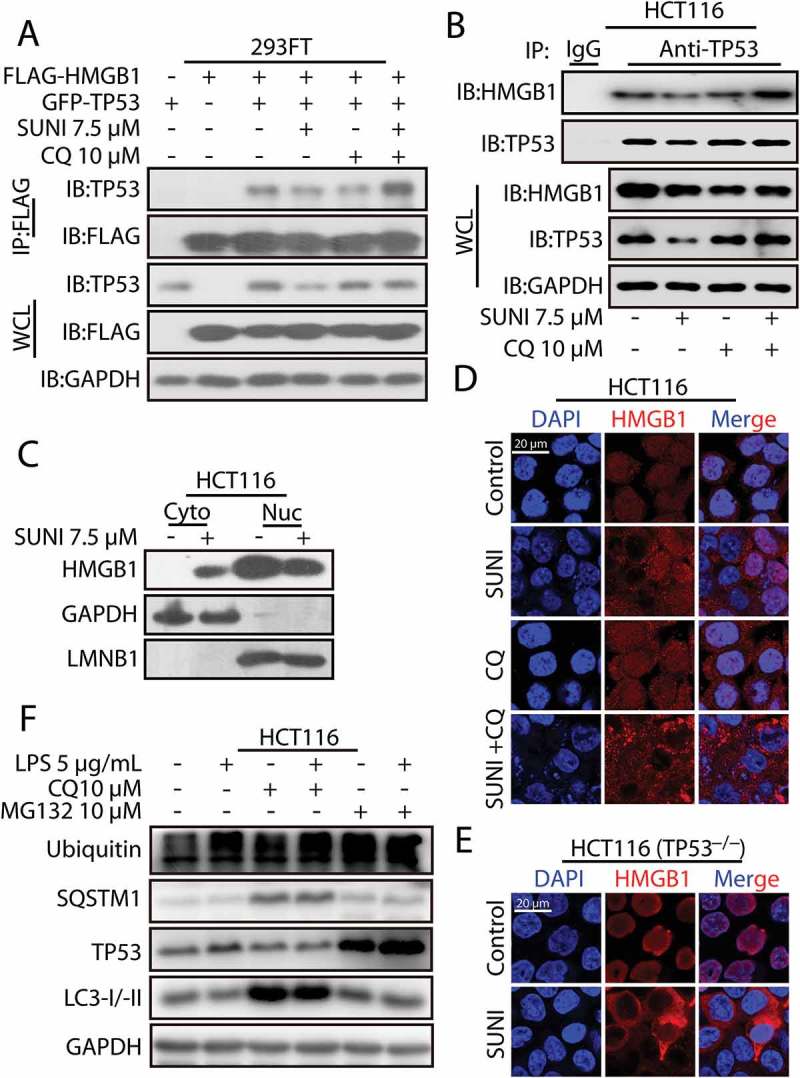
Sunitinib promotes the interaction of TP53 and HMGB1 and the nucleus-to-cytoplasm transport of HMGB1. (a) 293FT cells were transfected with plasmids encoding FLAG-HMGB1 and GFP-TP53 and were then treated with 7.5 µM sunitinib for 24 h in the absence or presence of 10 µM CQ for 2 h prior to the addition of sunitinib. The cell lysates were subjected to immunoprecipitation using an antibody against FLAG, and the resulting immunoprecipitate was analyzed by TP53 and FLAG immunoblots. (b) The cell lysates from the treated HCT116 cells were subjected to an immunoprecipitation using an antibody against TP53, followed by TP53 and HMGB1 western blots. Cells were treated with 10 µM CQ for 2 h before the addition of 7.5 µM sunitinib for 24 h. (c) Representative western blots show the cytoplasmic (Cyto) and nuclear (Nuc) distribution of HMGB1 in untreated or sunitinib-treated HCT116 cells for 24 h. (d) Immunocytochemistry for HMGB1 and nuclear staining in untreated or sunitinib-treated HCT116 cells. Cells were treated with 7.5 µM sunitinib for 24 h in the absence or presence of CQ for 2 h prior to the addition of sunitinib. Scale bar: 20 μm. (e) Immunocytochemistry for HMGB1 and nuclear staining in untreated or sunitinib-treated TP53−/− HCT116 cells. Cells were treated with 7.5 µM sunitinib for 24 h. Scale bar: 20 µm. (f) Representative western blots show the effect of HMGB1 cytoplasmic transport following LPS treatment on the degradation of TP53 in HCT116 cells. Cells were treated with LPS for 24 h in the absence or presence of 10 µM CQ for 2 h before the addition of LPS or in the absence or presence of 10 µM MG132 for 8 h before the end of the experiments. SUNI, sunitinib; CQ, chloroquine. All experiments were performed in triplicate. For western blots, 1 of 3 similar experiments is shown.
The autophagy cargo protein SQSTM1 directly interacts with TP53 for its autophagic degradation
The degradation of proteins by autophagy is specifically mediated by direct interaction with autophagy cargo receptors [27]. To determine which autophagy cargo receptor is involved in the sunitinib-induced degradation of TP53 by autophagy, we used an siRNA pool targeting the known autophagy cargo receptors (Figure S16). The siRNA-mediated depletion of SQSTM1 severely inhibited sunitinib-induced autophagic degradation of TP53, implying that SQSTM1 might be the specific cargo receptor promoting TP53 degradation (Figure 5(a)). SQSTM1 recognizes protein aggregates and delivers them to the lysosome for degradation [28]. Sunitinib promoted the formation of large TP53 puncta that colocalized with dots of SQSTM1 in the cytoplasm, supporting a possible role of SQSTM1 in sunitinib-mediated TP53 degradation (Figure 5(b)).
Figure 5.
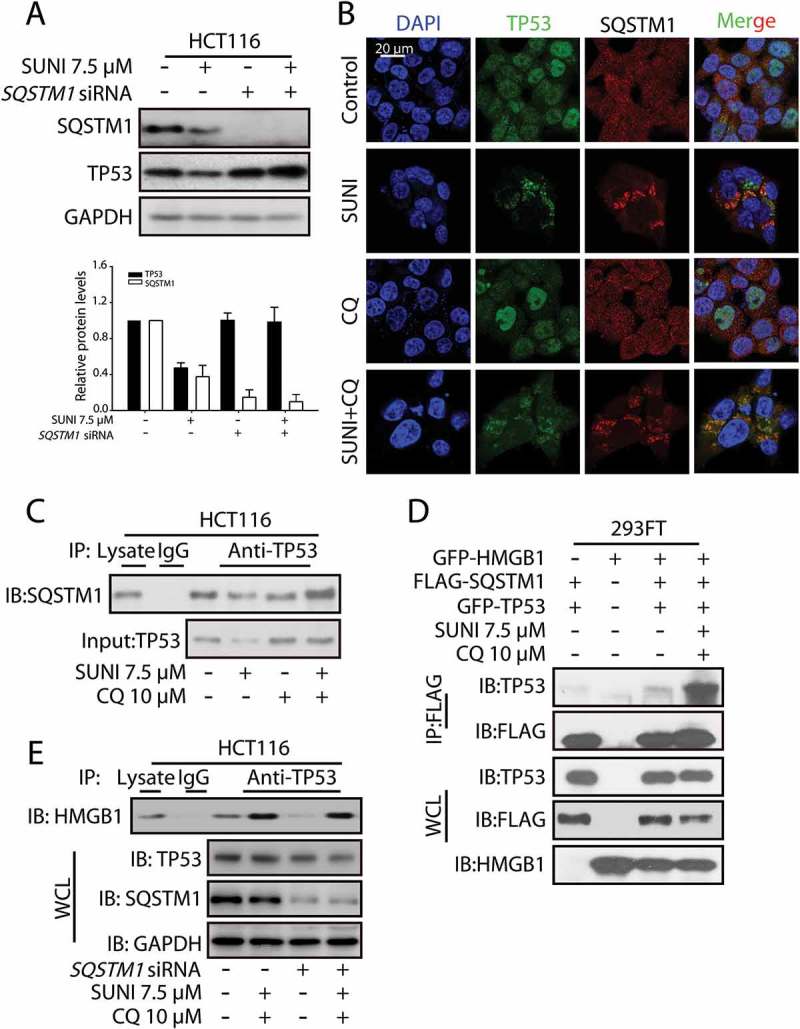
Sunitinib induces TP53 autophagic degradation in an SQSTM1-dependent manner. (a) Representative western blots show the effect of the knockdown of SQSTM1 on the sunitinib-induced degradation of TP53. Cells were treated with 7.5 µM sunitinib for 24 h in the absence or presence of 10 µM CQ for 2 h before the addition of sunitinib. The TP53 or SQSTM1 expression levels from biological triplicates were normalized with GAPDH and quantified with Quantity One. The data represent the average of 3 independent experiments. Each value represents a ratio of the maximal level of TP53 or SQSTM1. (b) The colocalization of TP53 with SQSTM1 in untreated or sunitinib-treated HCT116 cells. The cells were stained for TP53 (green), SQSTM1 (red) and nuclei (blue). Scale bar: 20 µm. (c) HCT116 cells were treated with sunitinib for 24 h in the absence or presence of CQ for 2 h prior to the addition of sunitinib. The cell lysates were subjected to immunoprecipitation using an antibody against TP53, followed by western blots to detect TP53 and SQSTM1 expression. (d) 293FT cells were transfected with GFP-HMGB1, FLAG-SQSTM1 and GFP-TP53 and were then treated with sunitinib and CQ for 24 h. The cell lysates were subjected to immunoprecipitation using an antibody against FLAG, followed by western blots to detect TP53, FLAG and HMGB1 expression. (e) HCT116 cells and SQSTM1 knockdown HCT116 cells were treated with sunitinib plus CQ. The cell lysates were subjected to immunoprecipitation using an antibody against TP53, followed by western blots to detect HMGB1, TP53 and SQSTM1 expression. SUNI, sunitinib; CQ, chloroquine; WCL, whole cell lysates. All experiments were performed in triplicate. For western blots, data from 1 of 3 experiments are shown.
To further detect the effect of sunitinib on the interaction between SQSTM1 and TP53, an immunoprecipitation analysis was performed. Sunitinib enhanced the interaction between TP53 and SQSTM1 in HCT116 cells (Figure 5(c)). In addition, consistent with the endogenous protein immunoprecipitation experiment, sunitinib increased the interaction between SQSTM1 and TP53 in 293FT cells expressing GFP-HMGB1, FLAG-SQSTM1 and GFP-TP53 (Figure 5(d)). To test the effect of SQSTM1 on the interaction of TP53 and HMGB1, and the nuclear export of TP53, we generated SQSTM1 knockdown cells. Sunitinib plus CQ still enhanced the interaction between TP53 and HMGB1 as well as induced the nuclear export of TP53 in SQSTM1 knockdown HCT116 cells (Figure 5(e) and S11). Collectively, these data indicate that sunitinib treatment causes HMGB1 to trigger the traffic of nuclear TP53 to the cytoplasm, and once in the cytoplasm, TP53 interacts with SQSTM1 for its degradation by autophagy.
Modulation of HMGB1 enhances the anti-cancer activity of sunitinib by upregulating TP53 function
From the above findings, we concluded that HMGB1 played a vital role in TP53 protein turnover. Thus, next we investigated the potential tumor-suppressive effect of HMGB1 on sunitinib-induced anti-cancer activity. As shown in Figure 6(a,b), the shRNA-mediated knockdown of HMGB1 attenuated the sunitinib-induced TP53 degradation and enhanced the efficacy of sunitinib in HCT116 cells. Of note, we generated TP53 knockdown cells in TP53 WT HCT116 cells to determine the effect of HMGB1 silencing on sunitinib-induced cell death in WT TP53 or knockdown cells. HMGB1 knockdown significantly enhanced the sunitinib-induced cell death in WT TP53 cells, whereas it had little effect on TP53 knockdown cells (Figure 6(c,d)), suggesting that HMGB1 knockdown contributed to sunitinib-induced cell death by blocking TP53 degradation. In addition, the sunitinib-induced anti-cancer activity was enhanced in HMGB1 knockdown cancer cells, and this effect resulted from rescuing the pro-apoptotic function of TP53 in vivo (Figure 6(e-g)). This finding strongly suggests that HMGB1 might also function as a negative regulator in sunitinib-treated cancer cells, which is reminiscent of a previous finding that low HMGB1 expression strongly correlates with enhanced relapse-free survival in human cancer patients [29]. Moreover, HMGB1 inhibition might be a reliable strategy for improving the outcome of sunitinib.
Figure 6.
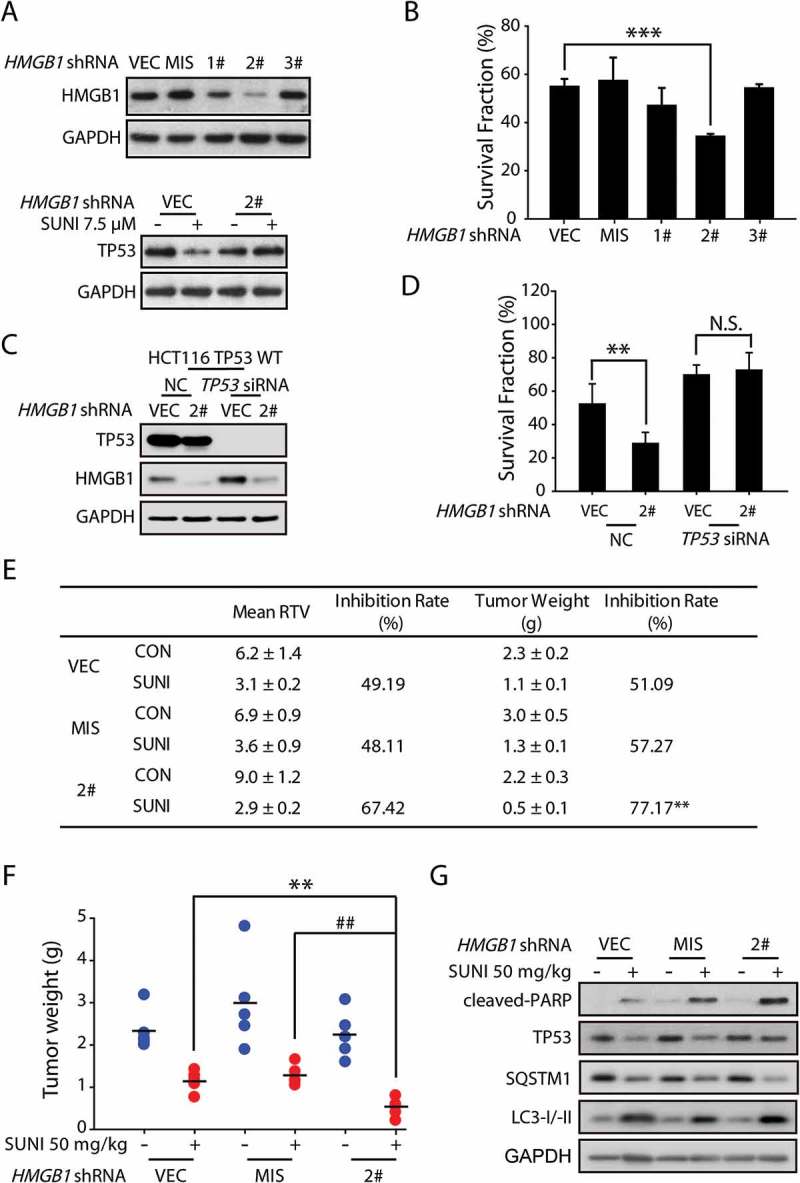
HMGB1 inhibition increases the anti-cancer activity of sunitinib by rescuing TP53 expression. (a) Representative western blots show the effect of HMGB1 knockdown on sunitinib-induced degradation of TP53 in HCT116 cells. The cells were treated with sunitinib for 24 h. (b) Results from the SRB staining assay show the effect of HMGB1 knockdown on the anti-cancer activity of sunitinib in HCT116 cells. The data are representative of 3 independent experiments. The data are expressed as the mean ± SD and P-values were determined using an unpaired two-tailed Student’s t-test (***P = 0.0002, HMGB1 shRNA2# versus vector). (c) Representative western blots show the efficiency of the HMGB1 and TP53 knockdown in HCT116 TP53 WT cells. (d) Results from the SRB staining assay show the effect of the HMGB1 and TP53 knockdown on the anti-cancer activity of sunitinib in HCT116 TP53 WT cells. The data are expressed as the mean ± SD and the P-values were determined using an unpaired two-tailed Student’s t-test (**P = 0.002, sunitinib versus control; N.S. = 0.26, HMGB1 shRNA versus vector). (e-g) Nude mice transplanted with HCT116 cells transfected with the vector (VEC), mismatched (MIS) or stable knockdown for HMGB1 were randomly divided into 2 groups (n = 5/group) and were treated with either the vehicle or sunitinib (50 mg/kg once a day). (e) The mean RTV values, inhibition rate of RTV, mean tumor weight and inhibition rate of tumor weights are shown. The mean RTV and mean tumor weight are expressed as the mean ± SEM. A one-way ANOVA followed by a post hoc Tukey’s test was used to identify significant differences (**P = 0.004, HMGB1 shRNA2# versus vector). (f) The dots represent the tumor weights of each of the tumors in 6 groups and the lines represent the mean values from each group. The data are expressed as the mean ± SEM. A one-way ANOVA followed by a post hoc Tukey’s test was used to identify significant differences (**P = 0.004, HMGB1 shRNA2# versus vector; ##P = 0.001, HMGB1 shRNA2# versus mismatched shRNA). (g) Representative western blots show the expression levels of cleaved-PARP and TP53 in each tumor group. SUNI, sunitinib. Three independent in vitro experiments were performed. For western blots, the data from 1 of 3 experiments are shown.
Sunitinib promotes mutant TP53 degradation based on the interaction domain of TP53 with HMGB1
To elucidate which part of TP53 is responsible for its interaction with HMGB1, we screened the effect of sunitinib on TP53 degradation in several cell lines with different TP53 mutations. Interestingly, sunitinib only decreased the expression of TP53 proteins with a mutation that occurred before amino acid 272, implying that the 272–393 section of amino acids may be the interaction domain (Figure 7(a,b)). Next, we used TP53−/− HCT116 cells transfected with different GFP-tagged TP53-deletion constructs to detect the effect of sunitinib on mutant TP53 degradation. Interestingly, sunitinib only decreased TP53 expression in cells that expressed the deletion mutant Δ353-393, whereas the deletion mutants Δ313-393 or Δ273-393 were not affected (Figure 7(c)). In addition, we used 293FT cells transfected with different GFP-tagged TP53-deletion constructs to determine the exact section of TP53 that is responsible for its interaction with HMGB1 during sunitinib treatment. Expression of TP53 deletion mutant Δ353-393 or TP53 WT still resulted in its interaction with HMGB1 (Figure 7(d)), whereas the deletion mutants Δ313-393 and Δ273-393 lost this effect, indicating that the 313–352 domain of TP53 is required for its interaction with HMGB1 during sunitinib treatment.
Figure 7.
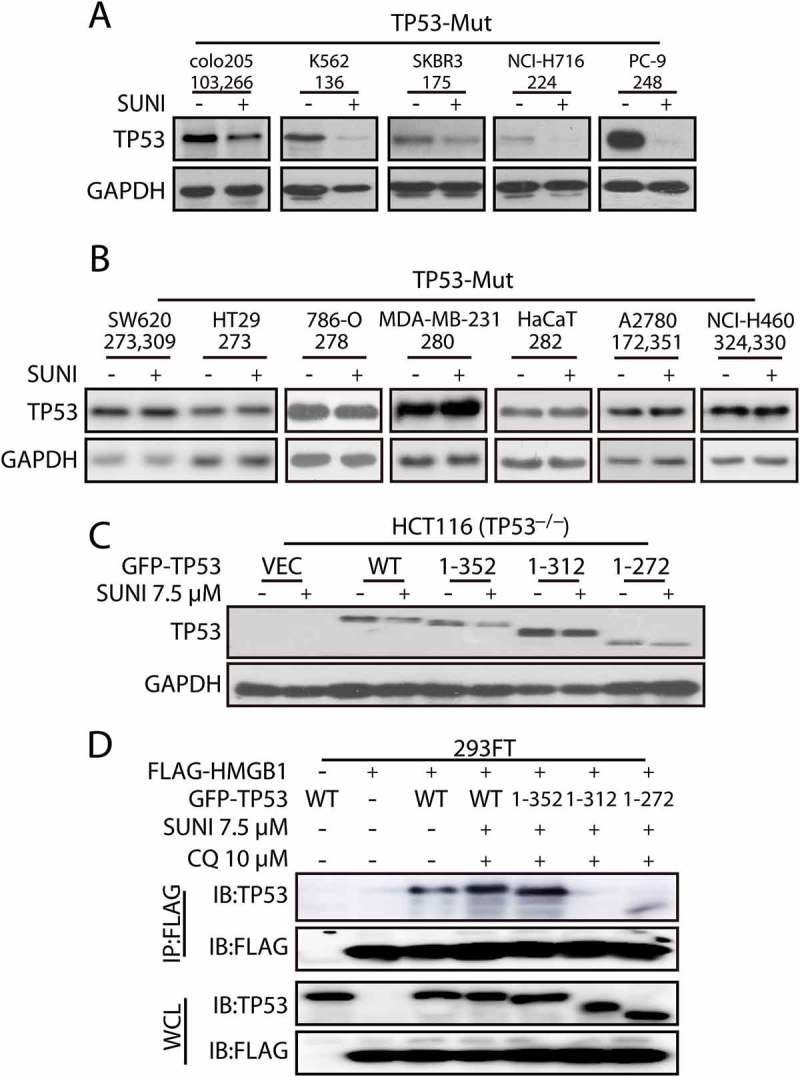
Sunitinib induces TP53 degradation in cells with different TP53 mutations. (a-b) Representative western blots show the expression of TP53 in cells with different TP53 mutations treated with sunitinib. The cells were treated with sunitinib for 24 h. Representative western blots show the effects of sunitinib on cells with different TP53 mutations. (c) HCT116 TP53−/− cells were transfected with plasmids encoding truncated GFP-TP53 mutants and were then exposed to 7.5 µM sunitinib for 24 h. (d) Mapping of the TP53 domains responsible for binding to HMGB1. 293FT cells were co-transfected with plasmids expressing FLAG-HMGB1 and the truncated GFP-TP53 mutants as indicated, and the cell lysates were immunoprecipitated with an anti-FLAG antibody, followed by immunoblots to detect TP53 and FLAG expression. The cells were treated with 7.5 μM sunitinib and 10 μM CQ for 24 h. WCL, whole cell lysates; SUNI, sunitinib. For western blots, data from 1 of 3 experiments are shown.
Discussion
Resistance mechanisms such as autophagy could be the reason sunitinib produces a low response rate in many cancers, and in responsive cancers, the duration of response can often be short [30]. Understanding the underlying mechanisms of sunitinib resistance is critical to improving its outcomes. Here, we first showed that the autophagic degradation of TP53 was closely related to the resistance to sunitinib. Mechanistically, sunitinib induced the binding of TP53 to its specific nucleus-localized target, HMGB1, leading to the nuclear export of TP53, dependent on the NES of HMGB1. Reciprocally, the HMGB1-transported cytoplasmic TP53 directly interacted with the autophagy cargo receptor SQSTM1, forming aggregates for degradation by autophagy. HMGB1 inhibition might be a reliable strategy for overcoming the resistance of sunitinib. We also found that sunitinib induced the degradation of TP53 protein expression levels, except for those with a mutation in the 313 to 352 amino acid section, which is based on its interaction domain with HMGB1. Therefore, the status and mutation sites of TP53 can be used as a response marker for the application of sunitinib.
The regulation of TP53 stability has always been a study focus. The intracellular levels of TP53 are regulated through a proteasome-dependent pathway. Here, we found that sunitinib still activated the autophagic degradation of TP53 even when the proteasome-pathway was inhibited (Figure 1(c) and S6), indicating that the proteasome-pathway is not involved in this type of TP53 degradation. In fact, whether autophagy-lysosome degradation is involved in the regulation of TP53 turnover is widely debated [31–34]. Mutant TP53 is degraded through chaperone-mediated autophagy (CMA) by a small-molecule agent, and autophagy is inhibited in this process. Glucose restriction in cancer cells induces MDM2-mediated autophagic degradation of mutant TP53. Our data reveal, for the first time, that the autophagy-lysosome pathway plays a role in the degradation of WT TP53.
Meanwhile, emerging evidence suggests that WT TP53 protein controls the canonical metastasis pathways to block cell adhesion, motility and invasion and that the loss of WT TP53 greatly enhances the risk of the development of cancer malignancies [35,36]. The underlying mechanism is largely unknown, and the targeting of TP53 for degradation by autophagy in both cancer cells and non-cancerous cells might provide a possible explanation for the issues encountered when using sunitinib. Of note, WT TP53 is strongly upregulated in response to a variety of stresses, and normally acts to suppress cell growth or promote apoptosis in cells of the lung, liver and heart [37–39]. The removal of overactivated WT TP53 by sunitinib-induced degradation by autophagy might serve as the basis for a possible cure for diseases in these organs. This hypothesis is strongly supported by the observations that the morphological alterations and fibrotic content seen following bleomycin challenge are significantly reduced by sunitinib treatment. Sunitinib prevents idiopathic pulmonary fibrosis by decreasing bleomycin-induced WT TP53 expression. In addition, the mouse model results are certainly encouraging, showing that the death rate of bleomycin-induced idiopathic pulmonary fibrosis decreased from 5 out of 6 to 3 out of 6 with sunitinib treatment (data not shown).
Mutations in the TP53 gene abrogate its normal functions, leading to genomic instability and loss of growth control. Mutant TP53 correlates with advanced tumor grade, progression, therapy and survival. Promoting the degradation of mutant TP53 can combat these fatal cancers. However, strategies that are based on known proteasome-dependent degradation only stabilize the expression of TP53, as in the case of nutlins (which inhibit the interaction between MDM2 and TP53). To date, very little is known regarding mutant TP53 turnover through the autophagy pathway. Our study not only provides a clue for the precise administration of sunitinib but also elucidates a novel mutant TP53 degradation pathway.
Regarding basic research, we discovered that TP53 is a selective autophagy substrate that is degraded upon sunitinib treatment. In addition, the nucleus-to-cytoplasm transfer of TP53 is essential for its autophagic degradation during sunitinib treatment, a process that is independent of its NES but is dependent on its specific, nucleus-localized target, HMGB1. Hence, we conclude that HMGB1 defines a conserved pathway of degradation under autophagy-activated circumstances that negatively regulates the function of TP53 by mediating its turnover in both WT and mutant TP53 cells treated with sunitinib (Figure 3 and S2). Since sunitinib also failed to reduce TP53 expression levels in cells with TP53 mutation in the 273 to 312 amino acid section (Figure 7), we speculated although this domain of TP53 is not the binding sites for HMGB1, it might influence the interaction with HMGB1. HMGB1 plays a potential context-dependent role in the regulation of autophagy and participates in the autophagy process at several levels [40]. Here, sunitinib still activated autophagy in HMGB1−/− cells (Figure 3(a)), and silencing BECN1/beclin 1, a co-factor of HMGB1-mediated autophagy, had little effect on sunitinib-induced TP53 degradation and translocation (Figure S14). These results indicate that the role of HMGB1 in the sunitinib-induced autophagic degradation of TP53 is distinct from its canonical function in the regulation of autophagy. Our findings highlight the importance of further identifying the mechanism that exists in sunitinib-induced autophagy.
It is also worthwhile to mention that based on our results of the anti-cancer activity of sunitinib in vitro and in vivo (Figure 6), HMGB1 inhibition might be a reliable strategy for overcoming the resistance of sunitinib. In the last 10 years several relatively small molecules derived either from natural sources or from chemical synthesis were explored for their ability to inhibit HMGB1 pathological activity. Among these small-molecule inhibitors of HMGB1, the most selective one is the natural steroidal pigment tanshinone IIA, which protects mice against lethal endotoxemia by selectively blocking endotoxin-induced HMGB1 cytoplasmic translocation [41]. We found that tanshinone IIA could significantly enhance the anti-cancer activity of sunitinib as well as block sunitinib-induced TP53 degradation (Figure S15). Because tanshinone IIA has already been used in China in the treatment of cardiovascular disorders, it can be rapidly translated into the clinic as a therapeutic strategy.
In conclusion, the data presented in this study advance our understanding of the autophagy response within tumor tissues and reveal key roles played by the HMGB1-mediated autophagic degradation of TP53 in the resistance to sunitinib as a cancer therapy. These findings represent a proof of concept that the suppression of HMGB1 enhances the anti-cancer activity of sunitinib. More importantly, this study identifies a new pathway controlling TP53 protein turnover, adds valuable information on the nucleus-to-cytoplasm transport pattern of TP53 and indicates a novel function of HMGB1 that involves the transportation of nuclear components to the cytoplasm.
Materials and methods
Cells culture and treatment
HCT116, colo205, K562, RCC4, Lovo, HepG2, SMMC-7721, NCI-H716, SKBR3, HL-7702, 3T-6, HT29, SW620, 786-O, MDA-MB-231, HaCaT, A2780, PC-9, NCI-H460 and 293FT cells were obtained from the Institute of Biochemistry and Cell Biology (Shanghai, China). HCT116 TP53−/− cells (a TP53-null immortalized cell line) was provided by Linhua Meng (Shanghai Institute of Materia Medica, Chinese Academy of Science). WT, atg5−/− and atg7−/− mouse embryonic fibroblasts were provided by Wei Liu (Zhejiang University). Six human colon cancer cell lines, HCT116, colo205, Lovo, HT29, NCI-H716 and SW620, 2 human liver cancer cell lines, HepG2 and SMMC-7721, 2 human renal cell carcinoma cells, RCC4 and 786-O, 1 human ovarian cancer cell line, A2780, 2 lung cancer cell lines, PC-9 and NCI-H460, 2 human breast cancer cell lines, SKBR3 and MDA-MB-231, 1 human leukemia cell line, K562, 1 human hepatocyte cell line, HL-7702, and 1 mouse fibroblast cell line, 3T-6, were maintained in DMEM (Gibco, 10569010) supplemented with 10% fetal bovine serum (HyClone, SV30160.03), 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco, 10378016) in a humid atmosphere of 5% CO2 and 95% air at 37°C. 293FT cells and WT, atg5−/− and atg7−/− mouse embryonic fibroblasts were cultured in DMEM. Reagents used were as follows: Sunitinib (Selleck Chemicals, S7781), cycloheximide (CHX; MedChemExpress, HY-12320), leptomycin B (LMB; Santa Cruz Biotechnology, sc-358688), chloroquine (CQ; Sigma-Aldrich, C6628), 3-methyladenine (3-MA; Sigma-Aldrich, M9281), MG132 (Sigma-Aldrich, M8699), thapsigargin (TG; Sigma-Aldrich, T9033), and LPS (Sigma-Aldrich, L2630). In selected samples, 35 μM CHX, 20 nM LMB, 0.5 μM TG, 5 μg/mL LPS, 5 μM tanshinone IIA (Sigma-Aldrich, 51704) and 1 μM MG132 were used. In some samples, 10 μM CQ was added 2 h before sunitinib treatment and 10 μM MG132 was added 8 h before cell collection.
Isolation of cancer cells from colon cancer patients
Anonymized tumor tissues from men aged 44–85 who underwent surgery were collected with their informed consent, according to the procedures approved by the Ethics Committee at Hangzhou First People’s Hospital (REC reference no. 2015/98–01). Primary, short-term, endothelial colon cancer cell cultures were established from the tumor tissues of colon cancer patients. Tumor samples were subjected to mechanical and enzymatic dissociation. The cancer cells were cultured in serum-free medium supplemented with 20 ng/mL EGF (PeproTech, AF-100–15) and 10 ng/mL FGF2 (PeproTech, 100-18B). After enzymatic dissociation, the cells were plated onto collagen-coated dishes in DMEM medium containing 10% FBS, to obtain primary tumor cell cultures.
Plasmid constructs and transfection
Plasmids used in this study were as follows: pCMV6-XL5-TP53 (Origene, SC119832), pCMV6-Entry-Myc-DDK-SQSTM1 (Origene, RC203214) and pCMV6-AC-GFP-HMGB1 (Origene, RG205918). HMGB1 cDNA was inserted into the pcDNA3.0 plasmid (a gift from Dr. Ronggui Hu) with FLAG tag. pCMV6-XL5-TP53 was GFP-tagged at the amino terminus. The pCMV6-XL5-TP53-NES-Mut and the pCMV6-AC-GFP-HMGB1-NES-Mut were generated by site-directed mutagenesis and multi-site mutagenesis using the Hieff MutTM Site-Directed Mutagenesis kit (Yeasen, 11003ES10) and Hieff MutTM Multi Site-Directed Mutagenesis Kit (Yeasen, 11004ES10). The cells were transfected with the plasmids using Lipofectamine 2000 (Invitrogen, 11668019) according to the manufacturer’s instructions.
Transfection of shRNA
HMGB1 shRNAs (TR316576) were purchased from Origene. The cells were transfected with plasmids using Lipofectamine 2000 (Invitrogen, 11668019) according to the manufacturer’s instructions [42].
Quantitative real-time PCR
Total RNA was extracted according to the manufacturer’s protocol for TRIzol Reagent (Invitrogen, 15596026). cDNA was prepared using a cDNA reverse transcription kit (Transgene Biotech, AT311-03). Quantitative RT-PCR was performed by using iTaq Universal SYBR Green Supermix (Bio-Rad, 1725125) in a CFX96TM Real-Time System (Bio-Rad, Hercules, CA, USA). The reactions were performed in triplicate. The primer sequences were as follows:
TP53 forward, 5´- GAGGCCTTGGAACTCAAGGATG-3´;
TP53 reverse, 5´- TCAGTCTGAGTCAGGCCCTTC-3´;
GAPDH forward, 5´-TGATGACATCAAGAAGGTGGTGAAG-3´;
and GAPDH reverse, 5´-TCCTTGGAGGCCATGTGGGCCAT-3´.
Transfection of siRNA oligonucleotides
siRNA oligonucleotides were transfected at a final concentration of 12 nM using Oligofectamine (Invitrogen, 12252011). The siRNAs targeting
SQSTM1 (5ʹ-GCAUUGAAGUUGAUAUCGAdTdT-3ʹ),
ATG5 (5ʹ-CAUCUGAGCUACCCGGAUAUU-3ʹ),
ATG7 (5ʹ-CGUGGAAUUGAUGGUAUUUdTdT-3ʹ),
BECN1 (5ʹ-CAAGUUCAUGCUGACGAAUdTdT-3ʹ),
STBD1 (5ʹ-AAUGGACAUUUGAUUUCUAdTdT-3ʹ),
CALCOCO2/NDP52 (5ʹ-UUCAGUUGAAGCAGCUCUGUCUCCC-3ʹ),
CBL/c-Cbl (5ʹ-GGGAAGGCUUCUAUUUGUUdTdT-3ʹ),
NBR1 (5ʹ-GGAGUGGAUUUACCAGUUAUU-3ʹ),
TOLLIP (5ʹ-GCUGGAAUAAGGUCAUCCAdTdT-3ʹ)
and the siRNA Negative Control (5ʹ-UUCUCCGAACGUGUCACGUdTdT-3ʹ) were purchased from GenePharma (Shanghai, China) [43].
Generation of HMGB1-knockout cell lines with the CRISPR/Cas9 system
The backbone plasmids Lenti-sgRNA-EGFP and Lenti-CAS9-puro were obtained from GeneChem CO., Ltd (Shanghai, China). To construct the double nicking HMGB1-sgRNA-guided CRISPR/Cas9 plasmids, a pair of oligos (sgRNA1: TTTCTAAGAAGTGCTCAGAG and sgRNA2: GGAGATCCTAAGAAGCCGAG) were designed and subcloned into a Cas9 backbone. Cells were first infected with lenti-cas9 and then selected using puromycin. The stable sub-lines were then infected with lenti-sgRNA to specifically knock out the target genes [44].
Western blot
Protein lysates (30–50 μg per sample) were separated on 8, 10 or 12% SDS-polyacrylamide gels and transferred to PVDF membranes (Merck Millipore, IPVH00010). Incubations with primary and secondary antibodies and signal detection followed the manufacturer’s protocol using Western Lightning Plus-ECL (PerkinElmer, NEL105001EA). Quantitative image analysis was performed by using Quantity One software. The following antibodies were used: anti-GAPDH (Santa Cruz Biotechnology, sc-25778), anti-TP53 (Santa Cruz Biotechnology, sc-47698, sc-6243), anti-HMGB1 (Santa Cruz Biotechnology, sc-26351), anti-SQSTM1 (Santa Cruz Biotechnology, sc-48402), anti-LC3B (Cell Signaling Technology, 2775s), anti-SQSTM1 (Cell Signaling Technology, 5114s), anti-HMGB1 (Cell Signaling Technology, 6893s), anti-ATG7 (Cell Signaling Technology, 2631s), anti-ATG5 (Cell Signaling Technology, 8540s), anti-cleaved-PARP (Cell Signaling Technology, 5625s), and HRP-labeled secondary antibodies (MultiSciences Biotech, GAR007, GAM007, and RAG007). The signal intensity was quantified using Quantity One® software and was shown to be within the linear range of detection.
Immunoprecipitation
Protein lysates were prepared in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40 (Beyotime, ST366) with protease inhibitor cocktail (Cell Signaling Technology, 5871) and were used for an overnight immunoprecipitation (500 μg per sample) at 4°C with 10 μL of the following antibodies: anti-FLAG (GenScript, L00425); anti-TP53 (Santa Cruz Biotechnology, sc-47698); anti-HMGB1 (Santa Cruz Biotechnology, sc-26351) and anti-SQSTM1 (Santa Cruz Biotechnology, sc-48402). A total of 20 μL of protein A/G Plus-agarose (Santa Cruz Biotechnology, sc-2003) was included in the incubation with the anti-TP53, anti-HMGB1 and anti-SQSTM1 antibodies. The resulting immunoprecipitates were analyzed using western blot assays.
Immunocytochemistry
Cells grown on poly-D-Lysine-coated cover slips were washed with phosphate-buffered saline (PBS; Gibco, 10010023) and fixed with 4% paraformaldehyde (Sigma-Aldrich, P6148) in PBS for 15 min at room temperature. They were then permeabilized with 0.2% Triton X-100 (BioFROXX, 1139ML100) in PBS for 15 min, blocked with nonfat dried milk in Tris-buffered saline (20 mM Tris-HCl, 500 mM NaCl, pH 7.4) with Tween-20 (Aladdin, T104863) (TTBS) for 1 h and incubated with primary antibodies at room temperature for 2 h or 4°C overnight. After washing with PBS, the cells were incubated with Alexa Fluor 488 or 568-conjugated secondary antibodies (Thermo Fisher Scientific, A21202, A10042, A10037, A11057) for 2 h, and then stained with DAPI (Dojindo, D212) for 5 min and mounted for fluorescence microscopy. The following primary antibodies were used: anti-TP53 (Santa Cruz Biotechnology, sc-47698), anti-HMGB1 (Santa Cruz Biotechnology, sc-26351), anti-SQSTM1 (Santa Cruz Biotechnology, sc-48402) and anti-LC3B (Cell Signaling Technology, 2775s).
Nuclear/cytoplasmic fractionation
Nuclear/cytoplasmic fractionation was performed according to Abcam’s subcellular and nuclear fractionation protocols. The nuclei were isolated by centrifugation and the supernatant containing the cytosolic fraction was collected. Equal volumes of the nuclear and cytoplasmic lysates were assayed by immunoblotting [45].
Cell survival assay
Cell survival rate was assessed using a sulforhodamine B (SRB; Sigma-Aldrich, S1402) colorimetric assay as previously described [46]. The absorbance at 575 nm was measured using a multiscan spectrum (Thermo Fisher Scientific, Marietta, OH, USA) until the absorbance values remained unchanged. Assays were performed in 3 independent experiments.
Animal xenograft model and in vivo activity measurements
HCT116 xenograft model was established by implanting tumor cells subcutaneously into nude mice. When the tumors reached a mean size of 90 mm3, the mice were randomly divided into 6 groups and treated as indicated in the experiment. Tumor volumes were measured every 4 days and calculated as (length × width2)/2. The individual relative tumor volume (RTV) and tumor growth inhibition T/C were calculated as previously reported [47]. The individual tumor weights were measured after 20 days of administration. All mice were bred according to Institutional Animal Care and Use Committee (IACUC) protocols and the Animal Research Committee at Zhejiang University approved all animal studies.
Statistical analyses
In vitro data are expressed as the mean ± standard deviation (SD), and in vivo data are expressed as the mean ± standard error of the mean (SEM). Statistical comparisons between 2 groups were performed using Student’s t-test; multiple group comparisons were made using a one-way analysis of variance (ANOVA). Once the significance of the group differences (P < 0.05) was established, Tukey’s post hoc tests were used for pairwise comparisons. Otherwise, the Kruskal-Wallis test was used to assess the significance of the differences among several groups. If the group differences were significant (P < 0.05), Dunn’s test was used for post hoc comparisons between pairs of groups. The data were analyzed using SPSS 20.0 for Windows. The statistical test and its associated P-values are indicated in the legends of each figure.
Funding Statement
This work was supported by National Natural Science Foundation for Distinguished Young Scholar of China (No.81625024), National Natural Science Foundation of China (Nos. 81673457 and 81473288) and Public Projects of Zhejiang Province (No.2016C31005).
Abbreviations
- 3-MA
3-methyladenine
- ATG5
autophagy related 5
- ATG7
autophagy related 7
- CHX
cycloheximide
- CMA
chaperone-mediated autophagy
- CQ
chloroquine
- NESs
nuclear export signals
- DAPI
4′,6-diamidino-2-phenylindole
- GISTs
gastrointestinal stromal tumors
- HCQ
hydroxychloroquine
- LC3
microtubule-associated protein 1 light chain 3
- LMB
leptomycin B
- MEFs
mouse embryonic fibroblasts
- mRCC
metastatic renal cell carcinoma
- PNET
primitive neurotodermal tumor
- siATG5
siRNA targeting ATG5
- siATG7
siRNA targeting ATG7
- siRNA
small interfering RNA
- siSQSTM1
siRNA targeting SQSTM1
- WT
wild-type
Acknowledgments
We would like to thank Dr Wei Liu (Zhejiang University, China) for providing the WT, atg5−/− and atg7−/− mouse embryonic fibroblasts. We would like to thank Dr Linhua Meng (Shanghai Institute of Materia Medica, Chinese Academy of Science, China) for providing the HCT116 TP53−/− cells. We would like to thank Dr Ronggui Hu and Wei Liu for their helpful advice. We would also like to thank experimenter Wei Yin from the Image Center, Core Facilities, Zhejiang University School of Medicine for the confocal laser scanning microscope images.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Gotink KJ, Broxterman HJ, Honeywell RJ, et al. Acquired tumor cell resistance to sunitinib causes resistance in a HT-29 human colon cancer xenograft mouse model without affecting sunitinib biodistribution or the tumor microvasculature. Oncoscience. 2014;1:844–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chow LQ, Eckhardt SG.. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25:884–896. [DOI] [PubMed] [Google Scholar]
- [3].Qu L, Ding J, Chen C, et al. Exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell. 2016;29:653–668. [DOI] [PubMed] [Google Scholar]
- [4].DeVorkin L, Hattersley M, Kim P, et al. Autophagy inhibition enhances sunitinib efficacy in clear cell ovarian carcinoma. Mol Cancer Res. 2017;15:250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wiedmer T, Blank A, Pantasis S, et al. Autophagy inhibition improves sunitinib efficacy in pancreatic neuroendocrine tumors via a lysosome-dependent mechanism. Mol Cancer Ther. 2017;16:2502–2515. [DOI] [PubMed] [Google Scholar]
- [6].Melnyk N, Xie XQ, Koh DJY, et al. CTEP #8342 autophagy modulation with antiangiogenic therapy: A phase I trial of sunitinib (Su) and hydroxychloroquine (HCQ). J Clin Oncol. 2013;31. [Google Scholar]
- [7].Cabrera S, Maciel M, Herrera I, et al. Essential role for the ATG4B protease and autophagy in bleomycin-induced pulmonary fibrosis. Autophagy. 2015;11:670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bradner JE. Cancer: an essential passenger with p53. Nature. 2015;520:626–627. [DOI] [PubMed] [Google Scholar]
- [9].Allen MA, Andrysik Z, Dengler VL, et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. ELife. 2014;3:e02200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bieging KT, Attardi LD. Cancer: A piece of the p53 puzzle. Nature. 2015;520:37–38. [DOI] [PubMed] [Google Scholar]
- [11].Hessa T, Sharma A, Mariappan M, et al. Protein targeting and degradation are coupled for elimination of mislocalized proteins. Nature. 2011;475:394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang C, Ge Q, Zhang Q, et al. Targeted p53 activation by saRNA suppresses human bladder cancer cells growth and metastasis. J Exp Clin Cancer Res. 2016;35:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lu K, Psakhye I, Jentsch S. A new class of ubiquitin-Atg8 receptors involved in selective autophagy and polyQ protein clearance. Autophagy. 2014;10:2381–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu J, Zhang C, Wang XL, et al. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ. 2014;21:1792–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Coffill CR, Lee AP, Siau JW, et al. The p53-Mdm2 interaction and the E3 ligase activity of Mdm2/Mdm4 are conserved from lampreys to humans. Genes Dev. 2016;30:281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Munoz-Braceras S, Calvo R, Escalante R. TipC and the chorea-acanthocytosis protein VPS13A regulate autophagy in Dictyostelium and human HeLa cells. Autophagy. 2015;11:918–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gomez-Puerto MC, Folkerts H, Wierenga AT, et al. Autophagy proteins ATG5 and ATG7 are essential for the maintenance of human CD34+ hematopoietic stem-progenitor cells. Stem Cells. 2016;34:1651–1663. [DOI] [PubMed] [Google Scholar]
- [18].Peng Y, Puglielli L. Nϵ-lysine acetylation in the lumen of the endoplasmic reticulum: A way to regulate autophagy and maintain protein homeostasis in the secretory pathway. Autophagy. 2016;12:1051–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].An WG, Kanekal M, Simon MC, et al. Stabilization of wild-type p53 by hypoxia-inducible factor 1α. Nature. 1998;392:405–408. [DOI] [PubMed] [Google Scholar]
- [20].Geyer RK, Yu ZK, Maki CG. The MDM2 RING-finger domain is required to promote p53 nuclear export. Nat Cell Biol. 2000;2:569–573. [DOI] [PubMed] [Google Scholar]
- [21].Ding WX, Ni HM, Gao W, et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282:4702–4710. [DOI] [PubMed] [Google Scholar]
- [22].Qu L, Huang S, Baltzis D, et al. Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3beta. Genes Dev. 2004;18:261–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ogata M, Hino SI, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Smart P, Lane EB, Lane DP, et al. Effects on normal fibroblasts and neuroblastoma cells of the activation of the p53 response by the nuclear export inhibitor leptomycin B. Oncogene. 1999;18:7378–7386. [DOI] [PubMed] [Google Scholar]
- [25].Livesey KM, Kang R, Vernon P, et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012;72:1996–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yang L, Xie M, Yang M, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun. 2014;5:4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hua F, Li K, Yu JJ, et al. TRB3 links insulin/IGF to tumour promotion by interacting with p62 and impeding autophagic/proteasomal degradations. Nat Commun. 2015;6:7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. [DOI] [PubMed] [Google Scholar]
- [29].Ladoire S, Enot D, Senovilla L, et al. The presence of LC3B puncta and HMGB1 expression in malignant cells correlate with the immune infiltrate in breast cancer. Autophagy. 2016;12:864–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mross K, Scheulen M, Strumberg D, et al. FOLFIRI and sunitinib as first-line treatment in metastatic colorectal cancer patients with liver metastases–a CESAR phase II study including pharmacokinetic, biomarker, and imaging data. Int J Clin Pharmacol Ther. 2014;52:642–652. [DOI] [PubMed] [Google Scholar]
- [31].Vakifahmetoglu-Norberg H, Kim M, Xia HG, et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013;27:1718–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rodriguez OC, Choudhury S, Kolukula V, et al. Dietary downregulation of mutant p53 levels via glucose restriction: mechanisms and implications for tumor therapy. Cell Cycle. 2012;11:4436–4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Choudhury S, Kolukula VK, Preet A, et al. Dissecting the pathways that destabilize mutant p53 The proteasome or autophagy? Cell Cycle. 2013;12:1022–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Aggarwal M, Saxena R, Sinclair E, et al. Reactivation of mutant p53 by a dietary-related compound phenethyl isothiocyanate inhibits tumor growth. Cell Death Differ. 2016;23:1615–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hashimoto A, Oikawa T, Hashimoto S, et al. P53- and mevalonate pathway-driven malignancies require Arf6 for metastasis and drug resistance. J Cell Biol. 2016;213:81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Powell E, Piwnica-Worms D, Piwnica-Worms H. Contribution of p53 to metastasis. Cancer Discov. 2014;4:405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nakashima N, Kuwano K, Maeyama T, et al. The p53-Mdm2 association in epithelial cells in idiopathic pulmonary fibrosis and non-specific interstitial pneumonia. J Clin Pathol. 2005;58:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kurinna S, Stratton SA, Coban Z, et al. p53 regulates a mitotic transcription program and determines ploidy in normal mouse liver. Hepatology. 2013;57:2004–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Matsumoto S, Sakata Y, Suna S, et al. Circulating p53-responsive microRNAs are predictive indicators of heart failure after acute myocardial infarction. Circ Res. 2013;113:322–326. [DOI] [PubMed] [Google Scholar]
- [40].Sun X, Tang D. HMGB1-dependent and -independent autophagy. Autophagy. 2014;10:1873–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Musumeci D, Roviello GN, Montesarchio D. An overview on HMGB1 inhibitors as potential therapeutic agents in HMGB1-related pathologies. Pharmacol Ther. 2014;141:347–357. [DOI] [PubMed] [Google Scholar]
- [42].Tang D, Kang R, Cheh CW, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29:5299–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hayakawa K, Pham LDD, Katusic ZS, et al. Astrocytic high-mobility group box 1 promotes endothelial progenitor cell-mediated neurovascular remodeling during stroke recovery. Proc Natl Acad Sci U S A. 2012;109:7505–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Awuah SG, Riddell IA, Lippard SJ. Repair shielding of platinum-DNA lesions in testicular germ cell tumors by high-mobility group box protein 4 imparts cisplatin hypersensitivity. Proc Natl Acad Sci U S A. 2017;114:950–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rabadi MM, Kuo MC, Ghaly T, et al. Interaction between uric acid and HMGB1 translocation and release from endothelial cells. Am J Physiology-Renal Physiol. 2012;302:F730–F741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rubinstein LV, Shoemaker RH, Paull KD, et al. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J Natl Cancer Inst. 1990;82:1113–1118. [DOI] [PubMed] [Google Scholar]
- [47].Trouet A, Passioukov A, Van Derpoorten K, et al. Extracellularly tumor-activated prodrugs for the selective chemotherapy of cancer: application to doxorubicin and preliminary in vitro and in vivo studies. Cancer Res. 2001;61:2843–2846. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.