

Abstract
Stacking bonds formed between two blunt‐ended DNA double helices can be used to reversibly stabilize higher‐order complexes that are assembled from rigid DNA components. Typically, at low cation concentrations, stacking bonds break and thus higher‐order complexes disassemble. Herein, we present a site‐specific photochemical mechanism for the reversible covalent stabilization of stacking bonds in DNA assemblies. To this end, we modified one blunt end with the 3‐cyanovinylcarbazole (cnvK) moiety and positioned a thymine residue (T) at the other blunt end. In the bound state, the two blunt‐ended helices are stacked together, resulting in a co‐localization of cnvK and T. Such a configuration induces the formation of a covalent bond across the stacking contact upon irradiation with 365 nm light. This bond can also be cleaved upon irradiation with 310 nm light, allowing repeated formation and cleavage of the same covalent bond on the timescale of seconds. Our system will expand the range of conditions under which stacking‐bond‐stabilized objects may be utilized.
Keywords: 3-cyanovinylcarbazole, covalent stabilization, DNA nanotechnology, DNA origami, photo-crosslinking
Stack and bond: A site‐specific photochemical mechanism for the reversible covalent stabilization of stacking bonds in DNA assemblies is presented.
The sequence‐programmable self‐assembly of DNA single‐strands enables the bottom‐up fabrication of arbitrarily shaped, rigid, three‐dimensional DNA components.1 These components can be used as building blocks for the self‐assembly of yet larger objects that can reach molecular weights up to the gigadalton‐scale with dimensions comparable to those of natural viruses.2 Such higher‐order complexes can be stabilized through non‐covalent base pair stacking bonds between helical blunt ends.2a, 3 Depending on the number and type of blunt‐end stacking contacts used in the system, forming bound states may require concentrations of cations much beyond those found, for example, in physiological fluids. The strong dependence on cation concentration therefore limits the applicability of the higher‐order objects formed through shape‐complementary stacking bond schemes. Herein, we solve the stability problem and expand the range of conditions under which stacking‐bond‐stabilized objects may be utilized. To this end, we present a photochemical strategy for site‐specifically introducing and breaking covalent bonds across stacking contacts within higher‐order DNA complexes.
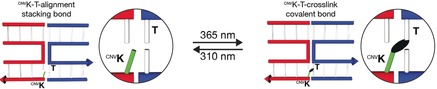
In 2008, Yoshimura and Fujimoto reported that a nucleobase modified with 3‐cyanovinylcarbazole (cnvK) may be covalently linked to an adjacent pyrimidine base on the complementary strand.4 Both the cnvK and the pyrimidine base were located within a continuous DNA double helix and crosslinking proceeds through a [2+2]‐cycloaddition reaction induced by UVA irradiation5 (Figure 1 a). Our mechanism builds on these findings, but in our case, the covalent bond will be formed between the blunt ends of two separate DNA double helices when they form a stacking bond (Figure 1 b). To this end, we replaced a terminal base with the cnvK moiety in one blunt end and positioned a thymine residue at the other corresponding blunt end. To test the possibility for photo‐crosslinking and photo‐cleavage of covalent bonds across stacking contacts, we used our previously described DNA origami switch object2a, 6 (Figure 1 b). The object is built from two rigid bundles of DNA double helices that represent the two arms of the switch. The arms are connected in the middle by a single Holliday junction that acts as the pivot point. The closed state is stabilized by up to 32 stacking interactions between blunt‐ended double helical protrusions on one arm and recessions on the other arm. At low ionic strength, switch particles assume open x‐shaped conformations, while at high ionic strength switch particles adopt the closed brick‐shaped conformation.
Figure 1.
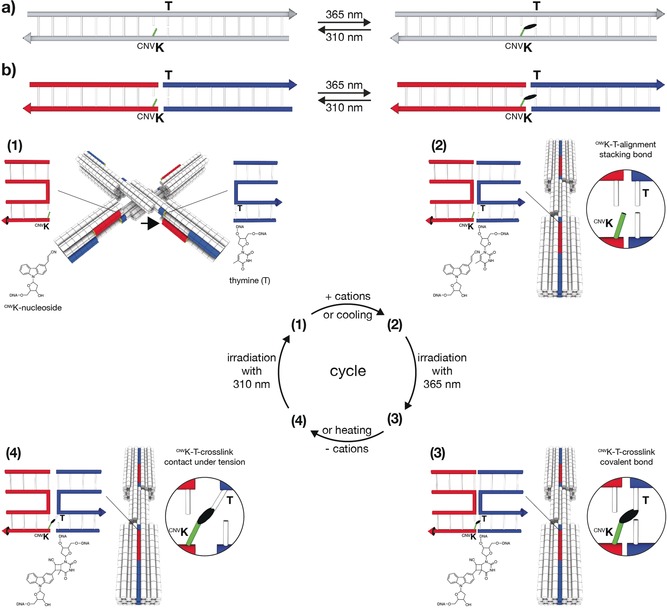
Schematic of photo‐crosslinking and photo‐cleavage of cnvK‐modified nucleosides across stacking contacts. a) Covalent photo‐crosslinking within a continuous DNA double helix. The cyanovinyl group of the cnvK‐modified nucleobase (light green) can be covalently linked to the thymine residue (T) on the “‐1” position (from 5′ to 3′) of the complementary strand. The black ellipsoid represents the covalent bond. b) Covalent photo‐crosslinking within the context of a stacking contact in DNA assemblies. The stacking contact consists of double helical protrusions (red) and double helical recessions (blue) that form discontinuous DNA double helices in the bound state. (1)–(4): Scheme of photo‐crosslinking and photo‐cleavage using the DNA origami switch object. (1) Open conformation of the switch under low ionic strength. The cnvK moiety and T are separated in space. The black arrow indicates the position of the second cnvK‐modification (Figure S1). (2) Closed conformation of the switch under high ionic strength. The cnvK moiety and T are co‐localized in space. (3) Formation of the covalent bond between the cnvK and T upon 365 nm irradiation. (4) The stacking contact is under tension when decreasing the cation concentration. Cleavage of the cnvK−T covalent bond can be triggered upon 310 nm irradiation.
To implement our mechanism, we introduced two cnvK moieties at stacking contacts next to the pivot point of the switch object (Figure S1). At low ionic strength conditions switch particles sample open states (Figure 1 b, (1)). In these states, the cnvK moiety and the thymine residue are far apart from each other, and we expect that the covalent bond cannot be formed across the stacking contact. Increasing the concentration of cations in solution shifts the conformational equilibrium of the switch towards the closed state. In this state, we expect that the cnvK moiety and the thymine residue will be properly positioned (Figure 1 b, (2)) to enable the formation of a covalent bond upon 365 nm light irradiation (Figure 1 b, (3)). Decreasing the concentration of cations in solution after irradiation will drive the two arms of the switch again away from each other towards the open state. However, the object will remain in the closed state due to the newly created covalent bond across the stacking contact (Figure 1 b, (4)). The conformational transition to the open state may then be remotely triggered through irradiation with light of shorter wavelength (e.g., 310 nm), which will break the covalent bond between the cnvK moiety and the thymine residue.
We performed experiments to test for the possibility of forming covalent bonds across stacking contacts and to elucidate the kinetics of photo‐crosslinking. We incubated switch particles at the high cation concentrations (30 mm MgCl2) that stabilize the closed state and exposed samples to 365 nm light. Subsequently, we performed gel electrophoresis experiments on the irradiated samples at the low cation concentrations that induce the transition to the open state. As can be seen by evaluating the band intensities in the gel image shown in Figure 2 a (left), the fraction of high‐mobility, closed particles increases with increasing irradiation time, even though all samples are exposed to object‐opening conditions. Therefore, we conclude that the high‐mobility particles were covalently crosslinked and thus trapped in the closed state. The fraction of crosslinked particles reaches saturation after approximately 10 s of exposure to 365 nm light (Figure 2 b, left). The yield of crosslinked particles not only depends on the irradiation time but also increases with increasing MgCl2 concentrations when comparing fixed irradiation times (Figure S3). The crosslinking reaction may be considered as a first‐order rate equation. For a constant illumination intensity and constant cation concentration, we expect the concentration of crosslinked particles to increase exponentially with illumination time, which is what we observe (Figure 2 b; left, solid line). However, in this model, the yield of crosslinked particles should always go to 100 % for a sufficiently long illumination time. Instead, we observe that the maximal attainable yield is around 75 %. We attribute the incomplete crosslinking to structurally defective switch particles that cannot adopt the closed state on the one hand and to particles missing the cnvK modifications owing to synthesis defects on the other hand. The first factor could be addressed by purifying particles capable of forming the closed state as we previously described.2a The second factor could be addressed by introducing more cnvK moieties. In support of this, we note that reducing the number of cnvK modifications per switch particle from two to one resulted in a reduced maximal attainable crosslinking yield (Figure S4). Direct imaging with negative‐stain transmission electron microscopy (nsTEM) provided complementary evidence for successful photo‐induced crosslinking. As expected, non‐irradiated switch particles dissolved in low ionic strength buffer adopted the open x‐shaped conformation (Figure 2 c, left). In contrast, switch particles that we previously irradiated at high ionic strength conditions and then dissolved in low ionic strength buffer to induce opening appear as closed brick‐shaped particles (Figure 2 c, right), which thus shows successful crosslinking.
Figure 2.
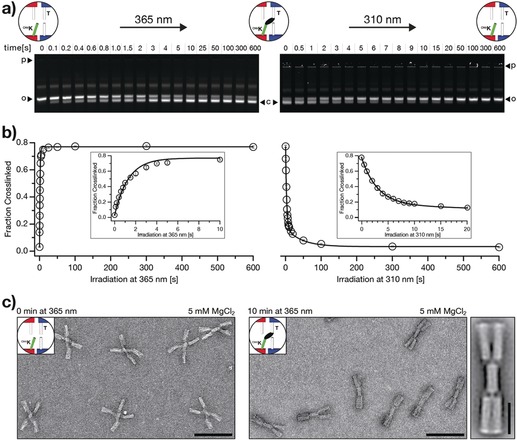
Kinetic characterization of photo‐crosslinking and photo‐cleavage using the DNA origami switch object. a) Fluorescent images of 1.5 % agarose gels laser‐scanned in the Cy5‐channel (635 nm). Time series of photo‐crosslinking at 365 nm irradiation (left) and photo‐cleavage at 310 nm irradiation (right). p=pocket, o=open (non‐crosslinked) switch particles, c=closed (crosslinked) switch particles. The images of the gels are globally auto‐leveled. b) Evaluation of the gel data shown in (a). We performed the experiment in triplicate (Figure S2); error bars show the standard deviation. The fraction of crosslinked particles is computed as described in the Supporting Information (Materials and Methods, 4. UV‐irradiation). We fitted the photo‐crosslinking data by using a single exponential with rate constant k c and the photo‐cleavage data by using a double exponential with rate constants k s1 and k s2 (solid lines). Values of k c=0.7 s−1, k s1=0.02 s−1, and k s2=0.3 s−1 were obtained, respectively. c) Negative‐stain TEM data of non‐irradiated (non‐crosslinked) switch particles (left) and irradiated (crosslinked) switch particles (right) at object‐opening conditions, scale bars=100 nm. See Figure S6 for field‐of‐view images. 2D‐particle micrograph of the cnvK‐crosslinked switch object (rightmost), scale bar=25 nm.
Next, we tested for the possibility of breaking the covalent bonds again and evaluated the kinetics of photo‐ cleavage (Figure 2 a, right). We incubated previously crosslinked switch particles at low, object‐opening cation concentrations (5 mm MgCl2) and exposed the samples to 310 nm light irradiation. Gel electrophoresis under object‐opening, low‐ionic strength conditions (5 mm MgCl2) reveals that the fraction of low‐mobility, open particles increases with increasing irradiation time (Figure 2 a, right). Hence, the covalent bonds across stacking contacts can also be broken again. The photo‐cleavage reaction goes to completion after about 100 s of exposure to 310 nm light (Figure 2 b, right). Moreover, we observe that the yield of photo‐cleavage increases with decreasing MgCl2 concentrations (Figure S5). This finding suggests that a destabilizing force is needed to drive the cnvK moiety and the thymine residue away from each other. In support of this, Yoshimura and Fujimoto previously achieved photo‐cleavage of cnvK bonds within continuous double helices only in the presence of denaturing urea.4 The time‐dependence of the yield of photo‐cleavage (Figure 2 b, right) may be understood by considering that the closed species can contain particles having two or one intact cnvK bonds, which cannot be discriminated by their electrophoretic mobilities. Breaking the bond in the species having only one bond should be a simple single‐exponential decay reaction. In contrast, the conversion of the species with two bonds to the open state with zero bonds proceeds via an obligate intermediate state with one bond that is hidden to the observer resulting in overall slower decay kinetics. Our data obtained from gel electrophoresis can be described favorably with the superposition of two single exponentials having a fast and a slow time constant (Figure 2 b; right, solid line).
We also tested whether multiple iterations through the photo‐crosslinking and photo‐cleavage cycle put forward in Figure 1 can be experimentally realized. For each cycle, we irradiated the sample for 5 min with 365 nm light for crosslinking, exchanged the buffer, and irradiated the sample for 5 min with 310 nm light to break the covalent bonds again. We followed one cycle in detail by directly imaging switch particles with nsTEM (Figure 3 a) and monitored nine successive cycles through gel electrophoresis mobility analysis (Figure 3 b). Evaluation of the gel data reveals that the fraction of crosslinked switch particles gradually decreases from cycle to cycle (Figure 3 c), which we attribute to photochemical side reactions that lead to non‐functional cnvK modifications. However, this effect can be mitigated by reducing the exposure time to 310 nm light (Figure S9).
Figure 3.
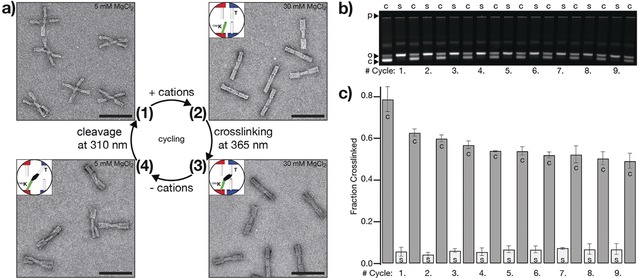
Reversibility and cycling. a) Negative‐stain TEM data of one cycle of photo‐crosslinking and photo‐cleavage that is illustrated in Figure 1, scale bars=100 nm. See Figure S7 for field‐of‐view images. b) Fluorescent image of a 1.5 % agarose gel laser‐scanned in the Cy5‐channel (635 nm). Samples of nine successive cycles of photo‐crosslinking and photo‐cleavage are loaded on the gel. c=crosslinking with 365 nm light for 5 min, s=cleavage with 310 nm light for 5 min (top labels). p=pocket, o=open (non‐crosslinked) switch particles, c=closed (crosslinked) switch particles (left labels). The image of the gel is globally auto‐leveled. c) Evaluation of the gel data shown in (b). We performed the experiment in duplicate (Figure S8); error bars show the standard deviation. c=crosslinking at 365 nm, s=cleavage at 310 nm.
In summary, we have presented a mechanism that allows reversible formation and cleavage of covalent bonds between separate blunt‐ended DNA double helices when they form a stacking bond. To this end, the terminal base of one blunt‐ended helix of the stacking contact is modified with the cnvK moiety and a thymine residue is positioned at the other corresponding blunt‐end. The structural context provided by the DNA origami object enables positioning of the two blunt‐ends of the separate DNA double helices into a base pair stacking configuration. As a result, the cnvK moiety and the thymine residue are co‐localized in such a manner that 365 nm light irradiation can induce the efficient formation of a covalent bond across the stacking contact. Hence, the DNA origami object acts as a templating device for the photochemical reaction. Moreover, the system that we implemented allows the repeated formation and cleavage of the same covalent bond in multiple cycles. We also found that a destabilizing force is necessary to efficiently cleave the thus formed bond through irradiation with 310 nm light. In our experiments, we generated this force by increasing the electrostatic repulsion between the two blunt‐end‐carrying interfaces of the switch object, thereby pulling the cnvK moiety and its thymine binding partner apart.
The strategy described herein for the reversible covalent stabilization of stacking contacts in DNA assemblies has direct practical utility. Our mechanism opens access to the preparation of pure and dense solutions of stacking‐bond stabilized higher‐order DNA complexes, as needed for potential in vitro and in vivo applications, crystallization assays, or cryo‐EM imaging. With cnvK‐crosslinking across stacking contacts, higher‐order complexes may be covalently linked in their fully assembled states. Once covalently stabilized, higher‐order assemblies can be subjected to purification and enrichment procedures, which typically require low ionic strength buffers to obtain satisfying recovery yields. Under these conditions, higher‐order assemblies would normally break down into their individual building blocks unless further stabilized. Furthermore, cnvK‐crosslinking of stacking contacts may be also used for photo‐caging of mechanically interlocked DNA assemblies such as rotaxanes7 and other dynamic mechanisms in which movable parts need to be encapsulated.8 Taken together, our mechanism opens up new possibilities for sample handling and photo‐caging applications in the context of structural and dynamic DNA nanotechnology.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Florian Praetorius and Fernanda Pereira for discussions. This work was supported by a European Research Council Consolidator Grant to H.D. (GA no. 724261) and the Deutsche Forschungsgemeinschaft through grants provided within the Gottfried‐Wilhelm‐Leibniz Program, the SFB863 TPA9, the Excellence Cluster CIPSM, and the Technische Universität München (TUM) Institute for Advanced Study.
T. Gerling, H. Dietz, Angew. Chem. Int. Ed. 2019, 58, 2680.
Contributor Information
Dr. Thomas Gerling, Email: thomas.gerling@tum.de, https://www.dietzlab.org.
Prof. Dr. Hendrik Dietz, Email: dietz@tum.de.
References
- 1.
- 1a. Benson E., Mohammed A., Gardell J., Masich S., Czeizler E., Orponen P., Hogberg B., Nature 2015, 523, 441–444; [DOI] [PubMed] [Google Scholar]
- 1b. Dietz H., Douglas S. M., Shih W. M., Science 2009, 325, 725–730; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Douglas S. M., Dietz H., Liedl T., Hogberg B., Graf F., Shih W. M., Nature 2009, 459, 414–418; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Rothemund P. W., Nature 2006, 440, 297–302; [DOI] [PubMed] [Google Scholar]
- 1e. Sobczak J. P., Martin T. G., Gerling T., Dietz H., Science 2012, 338, 1458–1461; [DOI] [PubMed] [Google Scholar]
- 1f. Veneziano R., Ratanalert S., Zhang K., Zhang F., Yan H., Chiu W., Bathe M., Science 2016, 352, 1534; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1g. Wei B., Dai M., Yin P., Nature 2012, 485, 623–626; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1h. Han D., Pal S., Nangreave J., Deng Z., Liu Y., Yan H., Science 2011, 332, 342–346; [DOI] [PubMed] [Google Scholar]
- 1i. Zhang F., Jiang S., Wu S., Li Y., Mao C., Liu Y., Yan H., Nat. Nanotechnol. 2015, 10, 779–784; [DOI] [PubMed] [Google Scholar]
- 1j. Seeman N. C., J. Theor. Biol. 1982, 99, 237–247. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Gerling T., Wagenbauer K. F., Neuner A. M., Dietz H., Science 2015, 347, 1446–1452; [DOI] [PubMed] [Google Scholar]
- 2b. Iinuma R., Ke Y., Jungmann R., Schlichthaerle T., Woehrstein J. B., Yin P., Science 2014, 344, 65–69; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Ong L. L., Hanikel N., Yaghi O. K., Grun C., Strauss M. T., Bron P., Lai-Kee-Him J., Schueder F., Wang B., Wang P., Kishi J. Y., Myhrvold C., Zhu A., Jungmann R., Bellot G., Ke Y., Yin P., Nature 2017, 552, 72; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Wagenbauer K. F., Sigl C., Dietz H., Nature 2017, 552, 78–83; [DOI] [PubMed] [Google Scholar]
- 2e. Hong F., Zhang F., Liu Y., Yan H., Chem. Rev. 2017, 117, 12584–12640. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Kilchherr F., Wachauf C., Pelz B., Rief M., Zacharias M., Dietz H., Science 2016, 353, aaf5508; [DOI] [PubMed] [Google Scholar]
- 3b. Woo S., Rothemund P. W., Nat. Chem. 2011, 3, 620–627; [DOI] [PubMed] [Google Scholar]
- 3c. Aghebat Rafat A., Pirzer T., Scheible M. B., Kostina A., Simmel F. C., Angew. Chem. Int. Ed. 2014, 53, 7665–7668; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7797–7801; [Google Scholar]
- 3d. Wang R., Kuzuya A., Liu W., Seeman N. C., Chem. Commun. 2010, 46, 4905–4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yoshimura Y., Fujimoto K., Org. Lett. 2008, 10, 3227–3230. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Fujimoto K., Yamada A., Yoshimura Y., Tsukaguchi T., Sakamoto T., J. Am. Chem. Soc. 2013, 135, 16161–16167; [DOI] [PubMed] [Google Scholar]
- 5b. He X., Sha R., Zhuo R., Mi Y., Chaikin P. M., Seeman N. C., Nat. Mater. 2017, 16, 993–997; [DOI] [PubMed] [Google Scholar]
- 5c. Rajendran A., Endo M., Katsuda Y., Hidaka K., Sugiyama H., J. Am. Chem. Soc. 2011, 133, 14488–14491; [DOI] [PubMed] [Google Scholar]
- 5d. Tagawa M., Shohda K.-i., Fujimoto K., Suyama A., Soft Matter 2011, 7, 10931–10934. [Google Scholar]
- 6.
- 6a. Bruetzel L. K., Gerling T., Sedlak S. M., Walker P. U., Zheng W., Dietz H., Lipfert J., Nano Lett. 2016, 16, 4871–4879; [DOI] [PubMed] [Google Scholar]
- 6b. Bruetzel L. K., Walker P. U., Gerling T., Dietz H., Lipfert J., Nano Lett. 2018, 18, 2672–2676. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. List J., Falgenhauer E., Kopperger E., Pardatscher G., Simmel F. C., Nat. Commun. 2016, 7, 12414; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Powell J. T., Akhuetie-Oni B. O., Zhang Z., Lin C., Angew. Chem. Int. Ed. 2016, 55, 11412–11416; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11584–11588. [Google Scholar]
- 8.
- 8a. Ketterer P., Willner E. M., Dietz H., Sci. Adv. 2016, 2, e1501209; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Marras A. E., Zhou L., Su H.-J., Castro C. E., Proc. Natl. Acad. Sci. USA 2015, 112, 713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary