

Abstract
Today, bio-medical efforts are entering the subcellular level, which is witnessed with the fast-developing fields of nanomedicine, nanodiagnostics and nanotherapy in conjunction with the implementation of nanoparticles for disease prevention, diagnosis, therapy and follow-up. Nanoparticles or nanocontainers offer advantages including high sensitivity, lower toxicity and improved safety—characteristics that are especially valued in the oncology field. Cancer cells develop and proliferate in complex microenvironments leading to heterogeneous diseases, often with a fatal outcome for the patient. Although antibody-based therapy is widely used in the clinical care of patients with solid tumours, its efficiency definitely needs improvement. Limitations of antibodies result mainly from their big size and poor penetration in solid tissues. Nanobodies are a novel and unique class of antigen-binding fragments, derived from naturally occurring heavy-chain-only antibodies present in the serum of camelids. Their superior properties such as small size, high stability, strong antigen-binding affinity, water solubility and natural origin make them suitable for development into next-generation biodrugs. Less than 30 years after the discovery of functional heavy-chain-only antibodies, the nanobody derivatives are already extensively used by the biotechnology research community. Moreover, a number of nanobodies are under clinical investigation for a wide spectrum of human diseases including inflammation, breast cancer, brain tumours, lung diseases and infectious diseases. Recently, caplacizumab, a bivalent nanobody, received approval from the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) for treatment of patients with thrombotic thrombocytopenic purpura.
Key Points
| Antibodies, major macromolecules used for targeted therapy, led to significant improvement in clinical care and quality of life of cancer patients. |
| However, antibody limitations in terms of size, incomplete tumour penetration and possible immunogenicity led to the development of a new generation of petite drugs and medicines. |
| Biological (nano)drugs, including nanobodies, offer new possibilities for treatment of not only cancer, but also a variety of human diseases on a subcellular level that will revolutionize the (bio)medical fields, as confirmed by the EMA and FDA approval of caplacizumab. |
Introduction
Antibodies for Cancer Therapy
Cancer is considered a cluster of diseases with different molecular changes, including gene mutations and amplifications, copy number alterations, changes in tumour suppressor and DNA repair genes, and epigenetic modifications [1, 2]. Development of a successful tumour therapy is challenging due to low specificity of the drug and toxic effect on adjacent non-tumour cells. An active targeting therapy relies on the specific delivery of an active drug to the target using different possible affinity reagents such as those mediated by a lectin-carbohydrate, ligand-receptor or antibody-antigen recognition [3–5]. Obviously, for maximal effect, the specific receptor targeted by the affinity reagent should be overexpressed at the surface of the diseased cells. Thus, active targeting refers to site-specific ligand-mediated accumulation of drugs into the diseased site due to an increased expression of a specific biomarker for that malignancy [6].
Immunoglobulins (Ig) or antibodies are soluble glycoproteins playing an essential role as the natural therapeutic compound in vertebrates [7]. Five different classes of antibodies (IgG, IgM, IgA, IgD and IgE) are elicited by the immune system as a response to non-self molecules (antigens), except in autoimmune disease conditions, with the purpose of their neutralization or elimination. The complex structure of antibodies is highly conserved among mammals. Antibodies consist of two identical heavy and two identical light chains connected by interchain disulphide bonds and non-covalent interactions as shown in Fig. 1a [8]. The antigen-binding site of antibodies comprises three loops of variable sequence and length within the variable heavy (VH) domain and three variable loops within the variable light (VL) domain [8]. Due to their specificity and affinity for their cognate antigen, antibodies play an important role in biomedical research and they are a preferred vehicle for the specific delivery of a therapeutic drug to its target.
Fig. 1.

Graphical representation of different antibody structures. a Classical antibodies consist of two identical heavy (variable—VH and constant—CH1/2/3 domains) and two identical light (variable—VL and constant—CL domain) chains connected with disulfide bonds. The antigen-binding region (variable fragment—Fv) consists of VH and VL connected with a linker peptide or stabilized with a disulfide bond in the cases of single-chain variable fragment (scFv) and disulfide-stabilized variable fragment (dsFv), respectively. b Camelid heavy-chain antibodies consist of two identical heavy chains only (variable—VHH and constant—CH2/3 domains). The antigen-binding region consists of a single variable domain VHH or nanobody
Antibodies were the first macromolecules used for targeted delivery [9]. Monoclonal antibodies have been employed for several decades in clinical practice for treatment of malignancies [10]. These include anti-carcinoembryonic antigen (CEA) antibody arcitumomab, anti-prostate-specific membrane antigen (PSMA) antibody capromab, anti-cluster of differentiation 20 (CD20) antibody rituximab, anti-human epidermal growth factor receptor 2 (HER2) antibody trastuzumab, anti-vascular endothelial growth factor (VEGF) antibody bevacizumab, anti-cluster of differentiation 52 (CD52) antibody alemtuzumab and three anti-epidermal growth factor receptor (EGFR) antibodies cetuximab, panitumumab and matuzumab [11–13], which are extensively reviewed elsewhere [14, 15]. These monoclonal antibodies are used for systemic tumour treatment and aim to block or neutralize growth factors or their receptors [16]. Antibodies are used for targeting cancer cells using one of the following mechanisms: killing by immune cells (opsonization); modification of biological processes like apoptosis; or delivery of cytotoxic agents like chemotherapeutics [17]. However, an issue with the use of murine antibodies in clinical care is that they elicit an immunogenic response in patients, that is, the generation of human anti-mouse antibodies (HAMA) targeting the murine idiotopes on the administered antibodies, which leads to their neutralization [18]. Different methods have been implemented to reduce immunogenicity, one of which is the construction of chimeric antibodies—monoclonal antibodies comprising human heavy and light chain constant regions and murine variable domains. The introduction of chimeric antibodies led to a decrease of immunogenicity from 50–75% in case of HAMA to 30% in case of human anti-chimeric antibodies (HACA) [18]. A further reduction of the immunogenicity could be obtained by grafting the antigen-binding loops of the VH and VL into a human IgG antibody. Another complication of using antibodies originates from their relatively large size, with dimensions of 14.2 nm × 8.2 nm × 3.8 nm, and corresponding large molecular mass of 150 kDa, which limits their penetration into tumour tissue. In vivo studies suggest that only 0.001–0.01% of the injected antibodies accumulate per gram of solid tumour [19, 20]. The poor penetration can also be a result of the ‘binding site barrier’ effect, first postulated by Weinstein [21, 22], in which antibodies with high affinity towards their antigens bind strongly to the antigen they first encounter. It was previously reasoned that antibodies with the highest affinity towards their antigens would lead to the best tumour targeting affect, as their slow koff rates would ensure prolonged in vitro retention of the antibody on the tumour cells. However, Adams et al. showed that high affinity limits tumour penetration and intratumoural diffusion [23] (i.e., antibodies are trapped at the tumour periphery). Other factors that contribute to uneven and heterogeneous distribution of the antibodies are antigen density, vascularization, capillary permeability, tissue structure and composition, extracellular matrix components, interstitial pressure and degree of necrosis [17, 22]. In the case of cancer therapy, the ‘binding site barrier’ phenomenon leads to incomplete tumour penetration and therefore suboptimal therapeutic efficiency. Consequently, to improve the effect of antibody therapy, antibodies with moderate affinity towards their antigens are considered more advantageous than high affinity antibodies; the latter can be used as appropriate vehicles for delivery of different agents (e.g., toxins and chemotherapeutics) [23]. As a result of the suboptimal antibody concentrations, many patients also develop resistance to the antibody-based therapies, which leads to additional treatment failure [24].
Another weakness of antibodies is their fragility, which allows only intravenous or subcutaneous administration. Moreover, due to the complex hetero-tetrameric structure and posttranslational modifications, therapeutic monoclonal antibodies are mainly expressed in mammalian cells, which results in high expenses during their large-scale production [25]. As a result, so far in tumour management, benefit from the employment of monoclonal antibodies has been proven for only a small number of patients [26].
Passive targeting is usually preferred for the treatment of solid malignancies. Passive targeting employs the increased vascular permeability and retention effect, as well as the poor lymphatic drainage to achieve drug accumulation into the tumour microenvironment [3, 27, 28]. Increased vascular permeability allows for small molecules to pass freely into the tumour interstitium, while the lack of intact lymphatic drainage limits their removal, leading to an effective accumulation of the drugs into the tumour [9, 29, 30]. This results in up to 100 times higher drug concentration into the tumour, which leads to greater tumour cytotoxicity [31, 32]. The ability of a therapeutic drug to penetrate the tumour is not dependent solely on the physicochemical properties of the drug, but it is also influenced by the biology of the tumour, the microvasculature and the interstitial pressure [33]. Once the therapeutic antibody is administered, it can either enter the tumour from the vasculature (extravasation) or from the surrounding tissue (surface uptake) [34]. Besides this, antibody targeting involves diffusion and binding into the tumour interstitium, plasma clearance and internalization as well as catabolism into tumour cells [35]. As described by the Stokes–Einstein equation (, where kB is Boltzmann’s constant, T is the absolute temperature, η is the dynamic viscosity and r is the radius of the spherical particle) for diffusion of spherical particles through a liquid, the diffusion rate is inversely proportional to the molecular radius [17]. In the case of antibodies, the diffusion coefficient varies between 5 and 50 µm2/s [34]. Hence, passive targeting is a non-controllable process since not all drug molecules diffuse at an equal rate. Problems that arise from passive targeting mechanisms are unequal permeability of blood vessels throughout the tumour, which leads to uneven drug distribution and appearance of multidrug resistance (MDR). Transporter proteins that are overexpressed on the surface of cancer cells expel drugs from cells leading to development of MDR that ultimately results in drug resistance and treatment failure [30]. It is suggested that for effective therapy, nanoparticles ought to be in the size range between 5 and 200 nm. This will allow them to pass through the pores between endothelial cells, which vary in size from 50 to 200 nm [9, 36]. Nanoparticles < 5 nm are not recommended for therapeutic use as they might be quickly eliminated from circulation through renal clearance, while those > 200 nm will be captured by the liver and spleen reticuloendothelial system [36]. The best performing nanoparticles are those with a diameter of < 100 nm and with a hydrophilic surface [5, 10].
Although the introduction of antibodies into clinical care initially showed great success, their limitations in size, poor penetration, long serum half-life, strong background signal, heterogeneous distribution and immunogenicity strongly suggests that further progress could be achieved in this field. As the trend in targeted drug delivery moves towards the use of smaller and highly specific molecules, switching from classical antibodies to camelid nanobodies can help in circumventing some of the existing complications in patient care. In this review, we briefly outline the beneficial properties of camelid nanobodies in comparison with human immunoglobulins in treating cancer patients. We evaluate the potential role of nanobodies for in vivo imaging and therapy. We also illustrate the development of nanobodies as biodrugs for treatment of breast cancer, lung diseases and brain tumours. Finally, we present the prospective use of nanobodies as next-generation therapeutic biodrugs by describing caplacizumab, the first EMA- and FDA-approved therapeutic nanobody construct acting against the rare blood disorder thrombotic thrombocytopenic purpura.
Nanobodies, the Smaller Variant of Antibodies
The occurrence of functional heavy-chain-only antibodies in the serum of camelids (dromedaries, camels, llamas, alpacas, guanacos and vicuñas) was discovered serendipitously nearly 30 years ago [37]. As the name suggests, heavy-chain-only antibodies lack light chains and also the first constant CH1 domain within the heavy chain is absent (Fig. 1b), which leads to their size reduction. Heavy-chain-only antibodies have a molecular mass of 95 kDa, while their variable antigen-binding domains (VHH) have a prolate shape with dimensions of 4 nm × 2.5 nm × 3 nm (Fig. 2) and are usually 12–14 kDa [38–41]. This variable domain of these heavy-chain antibodies comprises full antigen-binding potential, strong affinity to its cognate antigen and so it is considered to be the smallest naturally occurring, intact antigen binding fragment [42]. Their petite size in the low nanometre size range inspired Ablynx to introduce the name ‘nanobodies’. Nanobodies are extensively used for research purposes in academia, but are also identified and produced by numerous commercial companies, including Ablynx (now Sanofi), VHsquared, Chromotek, Camel-IDS, Hybrigenics and many others [10].
Fig. 2.
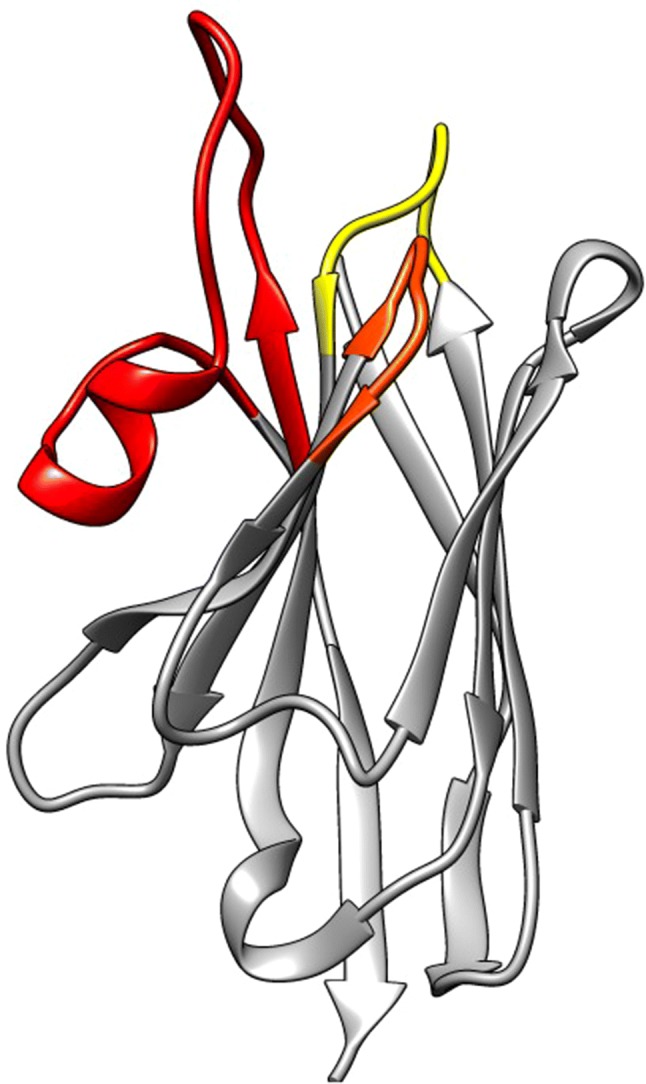
Ribbon representation of a nanobody (pdb 1JTT). The framework regions are in grey, the hypervariable H1, H2 and H3 antigen binding loops are in yellow, orange and red, respectively
Nanobodies have the same structural architecture as VH domains of human immunoglobulins: four conserved sequence regions (framework regions—FR1/2/3/4) are surrounding three hypervariable antigen-binding loops (complementarity determining regions—CDR1/2/3) [42–44]. The paratope of nanobodies comprises three CDRs, and also, regularly, framework residues are involved in the antigen interaction. Nevertheless, CDR3 is the main contributor for antigen recognition and specificity, whereas CDR1 and CDR2 assist in the binding strength [45–47]. The CDR3 of nanobodies is on average 18 amino acids long, which is substantially longer than the average 12- or 14-amino acid-long CDR3 loop of VH domains from mouse or human antibodies, respectively [20, 44]. A notable exception to this extended nanobody CDR3 loop are the llama nanobodies, where a fraction has a significantly shorter CDR3 consisting of only 4–8 amino acids [42]. The longer length of the majority of the CDR3 suggests a greater structural flexibility in the antigen-free form, which supposedly will be fixed in one single conformation upon antigen binding and thus creates an entropic penalty for binding. The presence of an additional interloop disulphide bond between either CDR1 (in camels) or CDR2 (in llamas) and CDR3 reduces this entropic penalty and at the same time increases the conformational stability of nanobodies [48]. Furthermore, a hallmark difference between a human VH and a camelid VHH is noted within the framework 2 region where four highly conserved and hydrophobic amino acids (V42, G49, L50 and W52 in VH) are substituted in nanobodies by smaller and/or more hydrophilic amino acids (F42 or Y42, E49, R50 and G52) (amino acid numbering is according to the international ImMunoGeneTics (IMGT) information system). This increased frequency of occurrence of polar and charged amino acids at the solvent-exposed site of the autonomous VHH explains their enhanced solubility in polar solvents (i.e. water) [42]. The extended CDR3 loop of nanobodies has the capacity to form a finger-like structure or a convex paratope (Fig. 2) that penetrates into the small cavities on the surface of the antigen or interacts with antigenic concave surfaces, respectively. Therefore, the paratope of nanobodies are perfectly shaped to recognize specifically the catalytic site enzymes and so many nanobodies act as modulators of the enzymatic activity [20, 45, 49, 50]. In sharp contrast, the paratopes of classical immunoglobulins form cavities, grooves or flat surfaces that are associated with small chemical groups (haptens), peptides or epitopes on large proteins, respectively. Hence, since nanobodies and classical antibodies prefer differently shaped epitopes, they will normally not compete for binding on the same antigen. This is an extremely useful property if one wants to quantify antigens of infectious agents in the presence of host serum. Moreover, nanobodies can be selected or developed to distinguish between different protein isoforms, a very important property to develop a highly specific diagnostic test. In addition, the utility of nanobodies as probes in immunocytochemistry, immunohistochemistry and immunoblotting has been demonstrated [51, 52]. However, as nanobodies recognize mostly conformational epitopes, their application as probes in western blot is less successful [53].
Moreover, nanobodies in general have unexpected physical properties: prolonged shelf life at + 4 °C and at − 20 °C, tolerance to increased temperature (60–80 °C, several weeks at 37 °C), resistance to proteolytic degradation, exposure to non-physiological pH (pH range 3.0–9.0), elevated pressure (500–750 MPa) and chemical denaturants (2–3 M guanidinium chloride, 6–8 M urea), all of which barely harm their antigen-binding capacity [54]. The nanobody robustness is mainly attributed to its efficient refolding capacity after chemical or thermal denaturation [55], although this reversible refolding upon thermal denaturation was recently questioned [56]. The monomeric structure of nanobodies and the lack of post-translational modifications allow for their expression in microbial systems including Escherichia coli, Saccharomyces cerevisiae and Pichia pastoris, which reduces the manufacturing costs [20, 57]. Nanobodies can be produced in milligram quantities per litre of culture in shake flasks, which means low-cost manufacturing and availability of sufficient amounts of purified nanobodies with consistent properties [58].
The sequence identity between nanobodies and the VH of human immunoglobulins of family III is above 80% [59]. In their extensive analysis, Klarenbeek et al. assessed the homology between the camelid germline IgV gene repertoire and their human counterparts and found 95% sequence identity of the camelid IGHV family 3 with its human FR counterpart [60]. This means that nanobodies will have a low immunogenic profile and are thus suitable for human administration [61]. Still, the sequence of a therapeutic nanobody can always be ‘humanized’ if wanted [62]. However, additional engineering might be required after the humanization process to restore the original affinity of the nanobody for its target. A number of nanobodies have been identified as lead compounds and reached advanced preclinical stages, while several of them are currently being tested in clinical trials as presented in Table 1 (data obtained from https://clinicaltrials.gov) [63–65]. Diagnosing cancer in early stages requires the use of imaging agents that are able to penetrate tumour tissues and bind to their targets with high specificity while the excess, unbound agent is rapidly removed from the body. The size of nanobodies makes them suitable agents for in vivo imaging [66–68]. Their short half-life in the bloodstream assures a high tumour to background ratio shortly after administration, which is a desired property of imaging agents [10, 38, 40, 69]. High antigen specificity is crucial for reducing side effects and minimizing the chance of obtaining false positives [54]. However, for molecular imaging, nanobodies have to be labelled with a diagnostic radioisotope, which can be gamma-emitting nuclides for single photon emission computed tomography (SPECT) or positron-emitting nuclides for positron emission tomography (PET) [70]. PET isotopes 68Ga and 18F are most appropriate for human use due to the short half-lives of 68 and 110 min, respectively. One such example is the use of an anti-HER2 nanobody for detection of HER2 expression in breast cancer using PET–computer tomography (PET/CT) and for clinical PET/CT and molecular imaging (iPET) [68, 71]. The authors examined safety, biodistribution, dosimetry and tumour targeting potential of 68Ga-HER2 nanobody in breast cancer patients and observed a complete absence of adverse effects. Therefore, they concluded that the procedure is safe to use in human patients. They noticed only a low background signal, rapid renal clearance of the tracer (60–90 min post-injection) and highest uptake in the metastatic lesions, kidneys, liver and the intestines. Early and late half-lives of the 68Ga-NOTA anti-HER2 nanobody were calculated to be 2.9 and 25.5 min, respectively, while at 1-h post-injection only 10% of the tracer was monitored. The 68Ga-NOTA anti-HER2 nanobody is currently being prepared for a phase II clinical trial (ClinicalTrials.gov NCT03331601) evaluating its potential to detect brain metastasis in breast cancer patients; the trial is expected to be completed in 2021. Although the biodistribution of nanobodies is found to be antigen-specific, a potential problem in using nanobodies as in vivo imaging probes is their accumulation in the kidneys, which is a consequence of their renal elimination [66]. Accumulation in the kidneys might limit the use of nanobodies as detection probes to screen organs located in the vicinity of kidneys, such as the pancreas [54].
Table 1.
Nanobodies that have entered in clinical trials
| Nanobody drug | Disease | Target | Clinical trial | Phase of clinical development | References |
|---|---|---|---|---|---|
|
Caplacizumab ALX-0681 ALX-0081 |
Thrombotic thrombocytopenic purpura | Ultra large von Willebrand factor |
I III II I III II |
[98, 139–143] | |
|
Ozoralizumab ATN-103 |
Rheumatoid arthritis | TNF |
I/II I/II II I |
[144] | |
|
Vobarilizumab ALX-0061 |
Rheumatoid arthritis | IL6 |
II II II I I/II |
[145] | |
|
Vobarilizumab ALX-0061 |
Systemic lupus erythematosus | IL6 | NCT02437890 | II | |
| ALX-0171 | Lower respiratory tract infection | RSV |
II I/II I I I II II |
[114, 146] | |
|
ALX-0761 M1095 MSB0010841 |
Psoriasis | IL17A, IL17F and IL17A/F |
II I |
[147] | |
| Bispecific nanobody-derived CAR-T cells | Refractory/ relapsed B-cell lymphoma | CD19/CD20 |
I I |
[105, 148] | |
| α-ADAMTS-5 nanobody M6495 | Osteoarthritis | ADAMTS-5 |
I I |
[149, 150] | |
| VHH batch 203027 | Diarrhoea | Rotavirus | NCT01259765 | II | [151] |
| [131I]-SGMIB anti-HER2 VHH1 | Breast cancer | HER2 | NCT02683083 | I | |
| 68-GaNOTA-anti-HER2 VHH1 | Brain metastasis of breast carcinoma | HER2 | NCT03331601 | II | |
| ALX-0651 | Healthy volunteers | CXCR4 | NCT01374503 | I | |
| PF-05230905 | Healthy volunteers | NCT01284036 | I |
Phase I: dosing, toxicity and excretion in healthy subjects; Phase II: safety and efficacy in large patient cohorts; Phase III: multi-centred, randomized, placebo-controlled trials; Phase IV: post-marketing studies
ADAMTS-5 α-A disintegrin and metalloproteinase with thrombospondin motifs-5, CAR chimeric antigen receptor, CD cluster of differentiation, CXCR4 chemokine receptor type 4, HER2 human epidermal growth factor receptor 2, IL interleukin, RSV respiratory syncytial virus, TNF tumour necrosis factor
Although the small size of a monomeric nanobody is beneficial in many cases, it can be a disadvantage for therapy as nanobodies will be rapidly cleared through the kidneys since their molecular mass is well below the 50–60 kDa renal threshold for glomerular filtration. Therefore, if the antigen is not circulating in the patient’s blood, only a marginal fraction of the administered nanobody will reach its cognate target. Thus, in order to obtain a high target load in vivo, a monomeric nanobody should be frequently administered. Alternatively, multimeric nanobody constructs or nanobodies fused to serum albumin (either directly or via an albumin-binding nanobody) can be produced for increasing their half-life in blood [40, 72]. In one study, the authors fused a bivalent α-EGFR nanobody to a nanobody binding to human serum albumin and managed to increase the serum half-life of this α-EGFR-α-EGFR-α-Alb nanobody to 2–3 days [26].
Nanoparticles in Nanomedicine
Important developments in the clinical care of cancer patients including genomic profiling, immunotherapy and targeted therapy, emerged in the last decades [73]. One such revolutionary approach is the use of nanoparticles (metallic, inorganic, organic and biological) for medical purposes.
Nanomedicine is a rising interdisciplinary field that is based on the use of nanoparticles in health and medicine with the objective to prevent, to diagnose and to treat disease at a molecular level [29, 74, 75]. In general, nanoparticles are small molecules with sizes ranging from 1 nm to 1 µm; for biological purposes, however, the size usually varies between 10 and 800 nm [76]. The aim of nanomedicine is development of novel imaging and therapeutic agents with enhanced efficacy, improved safety and lower toxicity [27, 77]. Besides nanomedicine, the fields of nanodiagnosis and nanotherapy are also rapidly developing. Nanodiagnosis stands for the planned use of devices with at least one dimension in the nanometre scale for detecting events that occur at a molecular level [78]. Nanodiagnosis will provide early disease monitoring with high sensitivity. For performing a successful diagnosis, nanoparticles need to meet several criteria: rapid extravasation from the blood vessels, fast penetration into the desired tissue, and eventually the unbound agent should quickly re-enter the blood vessels and be eliminated by the body, preferably via the kidneys, to generate a high signal-to-noise ratio in targeted tissues [29]. Until now, several nanoparticles have been developed for use in diagnostics with the most common being gold nanoparticles, dendrimers and quantum dots [10, 76]. In addition, nanoparticles can be combined with existing imaging technologies to obtain a more precise diagnosis.
The term nanotherapy describes the use of nanoparticles for drug delivery and therapy. The failure of current tumour therapy often results from a lack of effective drug delivery, toxicity issues, acquired drug resistance and poor drug solubility in aqueous solutions [28, 79]. Implementation of nanoparticles in tumour therapy can significantly improve the clinical outcome due to their beneficial properties such as small size, water solubility, improved stability, increased bioavailability and better pharmacogenetic features [80, 81].
The use of nanoparticles in oncology (i.e., nano-oncology) includes diagnostics as well as therapy [81, 82]. Thus, nano-oncology is a hybrid field between biochemistry, engineering technology and medicine with the aim of advancing tumour detection, diagnosis and therapy [83]. For successful implementation in clinical care, nanoparticles should be biocompatible, biodegradable, non-toxic, stable after administration and easily produced at large scale with controllable physical and chemical properties [84]. Different nanoparticles like liposomes, gold nanoparticles, iron-oxide nanocrystals and quantum dots are being manufactured for use in nano-oncology [3, 6, 10, 76]. However, our current knowledge of nanoparticle pharmacokinetics, biodistribution, metabolism and clearance is limited due to the small number of such particles being tested in clinical trials [83]. Additionally, potential toxicity issues for humans and the environment of the employment of inorganic nanoparticles are still to be determined [27, 85–87]. Once administered, nanoparticles will have the opportunity to interact with different biological environments (like cytoplasm and extracellular matrix), multiple cells, organs and tissues before they reach their specific target [27]. It is anticipated that nanoparticles that contain heavy metals can accumulate in vital organs such as the liver and brain, and can lead to tissue-specific toxicity [86]. Moreover, metallic (iron oxide and silver) nanoparticles can generate reactive oxygen species and lead to genotoxicity, while gold nanoparticles present risks for humans due to their affinity towards DNA and a low clearance rate. The properties of nanoparticles including their benefits and pitfalls have to be thoroughly examined and understood before they reach the patient. Nevertheless, the conjugation of nanobodies to nanoparticles could become an interesting combination, especially for nanomedicine and nano-oncology. Nanobodies are easily tailored for a directional conjugation to nanoparticles, which is expected to lead to an improved active targeting of the decorated nanoparticles to diseased tissues. However, the nanoparticle, or nanocontainer that can be loaded with drugs, will increase the size of the nanobodies, so that extravasation and tissue penetration will be less favourable.
Nanobodies as Therapeutics
Because nanobodies combine the beneficial properties of small molecules and monoclonal antibodies, they are an attractive agent for development of novel therapeutic strategies. Their small size makes them useful for targeting antigens residing in tissues that are weakly vascularized and poorly accessible. Nanobodies show better extravasation and tissue penetration than classical monoclonal antibodies, which is obviously crucial for therapeutic applications [39, 61]. The growth of solid tumours is dependent on the formation of new blood vessels (i.e. angiogenesis) and many nanobodies have been generated to interfere with this vascularization [88]. In one study, a nanobody against VEGFR2 was identified, which showed high antigen binding affinity (KD = 5.4 nM) and inhibitory potential of capillary tube formation in vitro [89].
Blood and lymphatic vasculature differ significantly between tumours and healthy tissue. While normal blood vessels are compact and continuous, tumour blood vessels are leaky, containing pores that can reach over 100 nm in size. This leads to a phenomenon that is referred to as the “enhanced permeability and retention effect” (EPR) [80]. So far, potential targets for nanobody-based therapies are extracellular targets such as ligand receptors or transmembrane proteins with differential expression in the target cells of interest. To this end, nanobodies against transmembrane growth factor receptors EGFR1 (HER1), EGFR2 (HER2), VEGFR2, c-Met and chemokine receptor type 7 (CXCR7) have been developed [61, 89–92]. These receptors have been linked to different malignancies. For example, VEGFR has been found expressed in different cancers including brain, lung, breast and colon cancer; c-Met is implicated in colon, breast and ovarian cancers and haematological malignancies; and the overexpression of CXCR7 is correlated with breast and lung cancers [61, 90]. Nanobodies against extracellular targets like HGF and chemokines have also been developed [26].
In addition, nanobodies can be used as drug carriers (nanocarriers) and other delivery systems for targeted therapy. This way, systemic toxicity will be avoided, hydrophilic drugs can be solubilized into lipid bilayers or micelles, and large amounts can be administered at once [53]. For such purposes, nanocarriers should be between 5 and 200 nm in size with hydrophilic surface [5, 36]. Examples of such cargoes with enhanced specificity for tumour cells expressing EGFR are nanobody-liposomes [93], nanobody-micelles [94] and nanobody-albumin nanoparticles [95].
For cancer therapy, nanobodies can be used either as antagonists, to prevent ligand binding and causing conformational changes that lead to activation of signalling cascades, or as allosteric inhibitors to modulate the enzymatic activity of their target proteins [61]. It is notable that anti-ligand nanobodies can be effective only if one single ligand is involved in the induction of the signalling cascade of the receptor [53].
Nanobodies show thermal resistance and stability against harsh conditions, chaotropic agents and pH extremes, so the route of administration can be intravenous, oral, intraperitoneal or intratumor. In each case, the nanobodies should be formatted in line with the stringent conditions that they will meet, like extreme pH and proteases after oral administration, and serum stability after an intravenous injection. Proper formulation is essential to ensure stability and drug release at the desired site, and to avoid unwanted side effects. In general, an extreme isoelectric point (pI) of the nanobody makes it less suitable for in vivo use. Monomeric nanobodies are rapidly cleared from the bloodstream via the kidneys and if such nanobodies are conjugated with toxic substances, their accumulation in the kidneys might lead to unwanted renal toxicity [66].
Despite the weaknesses of nanobodies for therapeutic applications (short serum half-life and rapid renal clearance), their superior properties (natural origin, small size, low immunogenicity, strong antigen-binding affinity, stability and water solubility) allowed for a number of them to enter clinical trials [64, 68, 96–98]. Three major points have to be addressed for drug candidates to enter clinical trials:
does the drug reach the target organ in sufficient amounts to give the desired effect?
is the specific binding of the drug and the target enough for the biological activity?
does the compound exert the functional modulation of the target? [99].
The clinical trial database https://clinicaltrials.gov was searched for the keywords ‘nanobody’, ‘nanobodies’, ‘VHH’ and ‘Ablynx’ (access date 19 April 2019) and yielded the results presented in Table 1. Nanobodies used in clinical trials are of different formats such as bivalent monospecific, bivalent bispecific, bivalent bispecific albumin-conjugated, trivalent bispecific and bispecific chimeric antigen receptor (CAR) T cell.
For activating T cells in vitro, anti-‘cluster of differentiation 3’ (CD3) antibodies are used. Still, therapeutic anti-CD3 antibodies induce systemic inflammation and adverse effects. This led to the development of bispecific T-cell engagers (BiTE) that are smaller than antibodies (11 nm in length and 55 kDa in size) and are composed of two single-chain variable fragments (scFv) with unique antigen specificity connected by a short amino acid linker [100]. What distinguishes BiTE from other scFv is that one receptor usually targets the CD3 subunit of the T-cell receptor, which allows for targeting T cells, while the other receptor targets a specific tumour antigen. In this regard, smaller sized BiTEs can be generated by substituting one or both the scFv with nanobodies. An example is the Her2 × CD3 bispecific antibody using anti-CD3 clone UTCH1 scFv and anti-Her2 nanobody, which showed strong anti-tumour effect in vitro and inhibits tumour growth in vivo [101, 102]. A similar approach was used by Mølgaard et al. with the generation of the bispecific light T-cell engager (LiTE) consisting of anti-CD3 UTCH1 scFv and anti-EGFR nanobody [103]. The model shows high tissue penetration, but requires continuous injection for therapeutic purposes. As an alternative, chimeric antigen receptor (CAR) T cells expressing non major histocompatibility complex (MHC) antigen receptors can be used. CAR T cells with nanobodies as the antigen-recognizing agent against MUC1 and EGFR have already been described [104, 105].
A different approach is the use of nanobodies in photodynamic therapy (PDT)—a minimally invasive and non-toxic treatment that induces cell death by activating a photosensitizer with light exposure [106]. The use of antibodies (both classical and heavy-chain) in PDT is termed ‘photoimmunotherapy’. Van de Broek et al. developed a method for conjugation of gold nanoparticles with anti-HER2 nanobodies to target HER2-expressing carcinoma cells [6]. The authors proved specific interaction of the carcinoma cells with the anti-HER2 conjugated gold nanoparticles, while binding was not observed for the controls.
In addition to these, nanobodies conjugated to various nanoparticles (liposomes, micelles and albumin nanoparticles) have also been explored [53]. A multivalent nanobody-liposome system targeting EGFR-overexpressing tumour cells was described by Oliveira et al. [93]. In this study, the authors show that liposomes conjugated to anti-EGFR ectodomain (EGa1) associate to a greater extent with human tumour cells than liposomes without the conjugated nanobody. Later, an improved liposome-nanobody system loaded with an anti-IGF-1R kinase inhibitor (AG538) was introduced [107]. This AG538-loaded liposome-nanobody system showed good growth inhibition of head and neck cancer cells in vitro. Similar systems were developed by conjugating polymeric micelles to anti-EGFR (EGa1) nanobodies [94, 108]. The results show that anti-EGFR nanobody-conjugated polymeric micelles bind more effectively to EGFR-expressing cells compared with the unconjugated micelles. In addition, the authors stated that unspecific binding of the micelle-nanobody conjugate to EGFR-negative cells was not observed. Lastly, development of anti-EGFR nanobody-albumin nanoparticles (NANAPs) has also been reported [95]. The study by Altintas et al. showed successful internalization of the NANAPs and inhibition of 14C squamous head and neck cancer cell proliferation, while no such effect was observed for non-targeted formulations [95]. Overall, these studies present the potential that nanobodies hold for the development of targeted therapies either alone or in combination with different molecular structures. However, further studies are needed to determine the efficacy and safety of these combined therapeutic systems.
Nanobodies Against Breast Cancer
Breast cancer is a heterogeneous disease with different morphologies and molecular profiles. The human epidermal growth factor receptor (HER) consists of four receptor tyrosine kinases (HER1/2/3/4), which play important roles in extracellular transmission of signals that lead to cancer cell growth, survival and differentiation [109]. Activation of these receptors leads to signalling cascades that promote cell proliferation, angiogenesis, invasion and metastasis. HER2 is a transmembrane protein that is found overexpressed in breast cancers, non-small-cell lung cancers, gastric cancer, colon cancers and ovarian cancers [110]. Although the humanized IgG1 monoclonal antibody trastuzumab is used to treat breast cancer patients, the therapy is most effective for patients with HER2-positive malignancies. The patients with low or heterogeneous HER2 expression benefit only marginally from the therapy [110]. A nanobody directed against HER2 that can be internalized by breast carcinoma cells expressing the receptor was developed [110]. The authors observed different tumour targeting properties of their nanobody, depending on the strategy that was used for labelling ([131I]IB-Mal-D-GEEEK vs Iodogen). A higher ratio of tumour to healthy tissue signal was obtained when labelling with [131I]IB-Mal-D-GEEEK [110]. However, the issue with kidney toxicity still remains to be solved.
Another receptor molecule that is frequently overexpressed and dysregulated in breast cancer is VEGFR2 [53]. Behdani et al. selected an anti-VEGFR2 nanobody that binds to cell surface VEGFR2, and proved that it efficiently inhibits capillary tube formation in vitro [89]. Further characterization of the nanobody and evaluation of its potential in vivo is still to be published.
Nanobodies Against Lung Diseases
For the treatment of lung diseases, drugs can be either administered by systemic injection (intravenously or subcutaneously) or by inhalation [111]. Inhaled drug delivery offers a number of advantages like a direct drug delivery at high concentrations, a fast onset of the drug action, a smaller dose needed to achieve a therapeutic effect and a lower systemic exposure that decreases the appearance of side effects. Administration by inhalation of anti-IL13 Fab fragment on murine models for asthma and a chimeric anti-EGFR monoclonal antibody (cetuximab) for lung cancer is being tested [112, 113]. If delivered through the lungs either as an aerosol or dry powder, it is important for the drugs to be able to maintain their structural integrity and bioactivity [111]. In this regard, the high stability of nanobodies, their resistance to harsh conditions, robustness and solubility are favourable properties for development of alternative administration routes like pulmonary delivery. A 42-kDa trivalent nanobody (ALX-0171) targeting the surface glycoprotein fusion (F) protein of human respiratory syncytial virus (RSV) is currently in clinical development for pulmonary administration in the form of an aerosol [114]. ALX-0171 drug inhalation allows for rapid drug delivery to the site of infection, which will shorten disease time. A phase I clinical trial confirmed that ALX-0171 was well received in healthy adult volunteers and in subjects with hyper-reactive airways [111]. However, Sanofi, which acquired Ablynx in mid-2018, has decided to discontinue the clinical tests of the inhaler-administered ALX-0171 for treatment of RSV-infected children.
Another example for the use of nanobodies in the development of lung-targeting drugs is the targeting of pulmonary surfactant protein A (SPA) [115]. Due to the exclusive expression of SPA on alveolar epithelial type II cells, it can serve as a molecule for specific targeting of lung diseases. In one study, the authors selected two nanobodies, Nb6 and Nb17, that showed high specificity for SPA [115]. To evaluate the lung-targeting ability of Nb17, the authors performed in vivo imaging using murine models and showed fast accumulation of Nb17 in the lungs, and complete clearance from the blood circulation within 6 h after injection. In addition, at least in mice studies, a minimal retention of Nb17 in liver and spleen was detected.
Nanobody Applications for Central Nervous System Pathologies
Nanobodies Passing the Blood–Brain Barrier
The human brain is an immunologically unique and privileged organ as it is protected by the semi-permeable physical blood–brain barrier (BBB) [116]. Although the main role of the BBB is regulation of homeostasis and protection of the brain from damaging agents, for therapeutic applications it becomes an obstacle as it prevents the passage of high molecular weight drugs [117]. Different mechanisms, including physical methods (disruption with osmotic shock, magnetic gradient and ultrasound) [118], use of cell-penetrating peptides [119] as well as receptor-mediated transcytosis (e.g., transferrin receptor, insulin receptor and low-density lipoprotein receptor-related protein) [120, 121] have been explored for increasing BBB permeability. Among these strategies are the so-called ‘Trojan horses’ that are ligands or antibodies targeting receptors of the BBB resulting in the transfer of various molecules into the brain either by non-specific charge-mediated adsorptive endocytosis or by energy-dependant receptor-specific endocytosis/transcytosis [122]. One of the most interesting technologies is the transport pathway through the transferrin receptors on the surface of microvascular endothelial cells [123]. The transferrin receptor is highly expressed at the BBB and mediates iron delivery to the brain through binding and trafficking of transferrin protein [124]. Hence, different antibodies against the transferrin receptor have been developed [125, 126]; antibody release into the in vitro model of the brain parenchyma can be improved by binding to the transferrin receptor at acidic pH and decreasing antibody affinity to the receptor [127]. In addition to antibodies, nanobodies against the transferrin receptor have also been raised [128]. Furthermore, two nanobodies able to cross the BBB in vitro, FC5 and FC44, were described previously [129, 130]. Abulrob et al. examined the mechanism by which FC5 transmigrates through the BBB in vitro and in vivo [131]. The results of their study show that transcytosis of FC5 through the BBB is independent of charge, but undergoes actin- and PI3 kinase-dependent transcytosis through clathrin-coated endocytic vesicles and depends on the recognition of specific oligosaccharide epitopes on the surface of human brain cerebromicrovascular endothelial cells. The authors also show that FC5 migration through the BBB does not involve interactions with the transferrin receptor [131]. In a different study, Haqqani et al. compared single domain FC5, monovalent and bivalent FC5 fused to the N-terminus of human Fc (Mono-FC5Fc and Bi-FC5Fc, respectively) and demonstrated that both Mono-FC5Fc and Bi-FC5Fc exhibit better BBB transcytosis than the single domain FC5 [132]. Although FC5 shows binding potential towards brain endothelium when compared with endothelium from lung, umbilical cord and even astrocytes, its specificity is not restricted to humans, but also binds to mouse and rat cerebral endothelial cells [131]. The above findings predict a good potential for the use of FC5 as a transport mechanism through the BBB, albeit further optimization will be necessary.
As administered nanobodies distribute widely and rapidly through the human body, they give new opportunities for molecular diagnosis of pathologies of the central nervous system. However, accessing the brain for therapeutic purposes is a challenge due to the existence of the BBB [67]. Without active transport, this biological barrier is only permeable for lipophilic molecules with a size below 400 Da [91]. One of the reasons for the limited ability of nanobodies to cross the BBB is thought to be their short half-life in the bloodstream [133]. However, monomeric nanobodies have been reported to be able to cross the BBB freely, even if their molecular mass (15 kDa) is well above the natural size barrier [129, 134, 135]. Nevertheless, this presumed passive BBB passage of nanobodies failed to deliver therapeutic amounts into the brain [136]. It is well established that proteins can cross the BBB by adsorptive-mediated endocytosis (AME). The AME process is triggered by electrostatic interactions between cationic proteins with basic pI and anionic charges present on the target cell surface [134]. The authors of one study reported that nanobodies with basic pI targeted against glial fibrillary acidic protein (GFAP) will spontaneously cross the BBB, and/or will be able to penetrate cells to bind its intracellular target in vivo [134]. This suggests that nanobodies with high pI could also be used as intrabodies. A limitation of this approach was the relatively large amount of nanobody (2 mg) that was injected into the murine model to obtain the desired effect [134].
We thus conclude that nanobodies are superior to currently used antibodies for transport through the BBB because they show better penetration and more homogeneous distribution. However, in the case of systemic administration, their small size is considered a limitation due to rapid clearance from the body.
Nanobodies Against Brain Tumours
The application of nanobodies for targeting tumours of the central nervous system is also explored. Grade IV gliomas, glioblastomas, are brain tumours that are among the deadliest human malignancies. Amplification of the transmembrane glycoprotein EGFR is common in glioblastomas. EGFR gene amplifications and activating mutations are observed in up to 70% of glioblastomas and play an important role in gliomagenesis [72]. α-EGFR nanobodies that inhibit EGF-induced signalling and cell proliferation in vitro have already been developed [137]. A combination of a nanobody targeted against EGFR and a pro-apoptotic EGFR-specific nanobody and their immune-conjugates against tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) was used as a targeted treatment for glioblastoma [72]. Nanobody conjugates were secreted from neural stem cells on site with sustained release and showed sufficient tumour uptake level in glioblastoma. The combination caused decreased glioblastoma growth and invasiveness both in vitro and in vivo.
Caplacizumab for Thrombotic Thrombocytopenic Purpura
The first EMA- and FDA-approved 28-kDa nanobody is the bivalent nanobody caplacizumab (ALX-0681) for treatment of thrombotic thrombocytopenic purpura (TTP) [98]. TTP is a blood disorder where the activity of von Willebrand factor (vWf)-cleaving protease ADAMTS-13 is inhibited (enzyme activity < 10%), which leads to platelet consumption in vWf-platelet aggregates and microvascular thrombosis [98]. In approximately 20% of the cases diagnosed with TTP, the disease has a fatal outcome. The current treatment consists of daily plasma exchange to restore ADAMTS-13 and remove vWf autoantibodies, as well as immunosuppressive therapy to supress anti-vWf antibodies [98]. Relapses are commonly encountered after treatment and are a permanent risk as they can occur within 30 days after the last plasma exchange or even 10–20 years later. Almost 15 years ago, a nanobody that recognizes the active state of vWf was developed [138]. The nanobody, named caplacizumab, allows the detection of vWf in plasma of patients with ADAMTS-13 deficiency [64]. There are two types of caplacizumab, ALX-0681 and ALX-0081, for subcutaneous and intravenous administration, respectively. The bivalent nanobody shows an inhibitory effect on platelet aggregation in blood, a good safety profile and efficiency. The phase III HERCULES double-blinded, randomized, placebo-controlled trial proved that, in combination with current therapy, caplacizumab treatment with subcutaneous administration aids in faster resolution of the disease, fewer TTP-related deaths and less disease recurrence [98]. The trial was multi-centred and took place in 92 laboratories worldwide. During the trial, patients that received caplacizumab showed faster platelet count normalization than the ones receiving placebo. In addition, lower incidence of TTP recurrence and TTP-related deaths were observed in the caplacizumab-treated patients compared with those receiving placebo [98].
Expert Opinion
Since the discovery of naturally occurring heavy-chain antibodies in sera of camelids and the development of technologies to clone and identify their antigen-binding fragments, known as nanobodies, the number and range of possible applications with nanobodies seem to have exploded. Supported by intrinsic beneficial biochemical and biophysical properties, nanobodies are a robust targeting entity that is easily assembled or incorporated into more complex, pluripotent constructs. Even in the absence of an intrinsic therapeutic effect, their conjugation with chemotherapeutic agents generates highly promising targeted drug delivery compounds. Although small monomeric nanobodies are often superior over classical antibodies for therapeutic applications, they still have their own drawbacks such as a fast renal clearance preventing a high load at the diseased tissue and inducing kidney toxicity. However, tools and strategies are available to engineer the nanobodies into next-generation constructs of higher efficacy and with fewer side effects.
It can be tricky to compare the performance of classical antibodies with nanobodies for therapeutic applications. Although their size, complexity, glycosylation, presence or absence of Fc, epitope-specificity and manufacturing are widely different, they both have an IgG origin and share a high specificity for their cognate target, which they bind with high affinity as well. Consequently, the bivalent antibodies might perform better due to longer blood circulation time, avidity effects, the capacity to cross-link or dimerize their antigens and in triggering an Fc-mediated immune response. In contrast, monomeric nanobodies have a much faster extravasation and an improved tumour penetration and better targeting of cryptic epitopes but result in lower loads at lesions due to their very fast blood clearance via the kidneys. Although nanobodies are easily dimerized or multimerized and could be equipped with a serum albumin binding nanobody to obtain avidity effects and to improve their blood retention, such engineering will inevitably reduce their diffusion advantages. Finally, if wanted, nanobodies can also easily be equipped with the Fc regions (hinge, CH2 and CH3 domain, most likely of human IgG1) to reconstitute a humanized heavy-chain-only antibody format to include the Fc-mediated effector functions, and increase the blood retention time as well.
Compliance with Ethical Standards
Funding
I.J. acknowledges financing from the Interreg EC Project 2014-2020, Ref. No. 146, Acronym: TRANS-GLIOMA; the Research Programme Grant P1-0390; and the Z3-1869 project from the Slovenian Research Agency (ARRS). The funders had no role in manuscript design, writing or decision to publish.
Conflict of interest
Ivana Jovčevska and Serge Muyldermans declare they have no conflict of interest.
References
- 1.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer—a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6(2):107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 2.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 3.Jabir NR, Anwar K, Firoz CK, Oves M, Kamal MA, Tabrez S. An overview on the current status of cancer nanomedicines. Curr Med Res Opin. 2018;34(5):911–921. doi: 10.1080/03007995.2017.1421528. [DOI] [PubMed] [Google Scholar]
- 4.Lammers T, Hennink WE, Storm G. Tumour-targeted nanomedicines: principles and practice. Br J Cancer. 2008;99(3):392–397. doi: 10.1038/sj.bjc.6604483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang MD, Shin DM, Simons JW, Nie S. Nanotechnology for targeted cancer therapy. Expert Rev Anticancer Ther. 2007;7(6):833–837. doi: 10.1586/14737140.7.6.833. [DOI] [PubMed] [Google Scholar]
- 6.Van de Broek B, Devoogdt N, D’Hollander A, Gijs HL, Jans K, Lagae L, et al. Specific cell targeting with nanobody conjugated branched gold nanoparticles for photothermal therapy. ACS Nano. 2011;5(6):4319–4328. doi: 10.1021/nn1023363. [DOI] [PubMed] [Google Scholar]
- 7.Tillib SV. “Camel nanoantibody” is an efficient tool for research, diagnostics and therapy. Mol Biol (Mosk) 2011;45(1):77–85. doi: 10.1134/S0026893311010134. [DOI] [PubMed] [Google Scholar]
- 8.Padlan EA. Anatomy of the antibody molecule. Mol Immunol. 1994;31(3):169–217. doi: 10.1016/0161-5890(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 9.Alexis F, Rhee JW, Richie JP, Radovic-Moreno AF, Langer R, Farokhzad OC. New frontiers in nanotechnology for cancer treatment. Urol Oncol. 2008;26(1):74–85. doi: 10.1016/j.urolonc.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 10.Hu Y, Liu C, Muyldermans S. Nanobody-based delivery systems for diagnosis and targeted tumor therapy. Front Immunol. 2017;8:1442. doi: 10.3389/fimmu.2017.01442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sunada H, Magun BE, Mendelsohn J, MacLeod CL. Monoclonal antibody against epidermal growth factor receptor is internalized without stimulating receptor phosphorylation. Proc Natl Acad Sci USA. 1986;83(11):3825–3829. doi: 10.1073/pnas.83.11.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7(4):301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 13.Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20(3):719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 14.Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10(5):317–327. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23(9):1147–1157. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 16.Scott AM, Allison JP, Wolchok JD. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012;12:14. [PMC free article] [PubMed] [Google Scholar]
- 17.Beckman RA, Weiner LM, Davis HM. Antibody constructs in cancer therapy: protein engineering strategies to improve exposure in solid tumors. Cancer. 2007;109(2):170–179. doi: 10.1002/cncr.22402. [DOI] [PubMed] [Google Scholar]
- 18.Majidi J, Barar J, Baradaran B, Abdolalizadeh J, Omidi Y. Target therapy of cancer: implementation of monoclonal antibodies and nanobodies. Hum Antibodies. 2009;18(3):81–100. doi: 10.3233/HAB-2009-0204. [DOI] [PubMed] [Google Scholar]
- 19.Epenetos AA, Snook D, Durbin H, Johnson PM, Taylor-Papadimitriou J. Limitations of radiolabeled monoclonal antibodies for localization of human neoplasms. Cancer Res. 1986;46(6):3183–3191. [PubMed] [Google Scholar]
- 20.Kolkman JA, Law DA. Nanobodies—from llamas to therapeutic proteins. Drug Discov Today Technol. 2010;7(2):e95–e146. doi: 10.1016/j.ddtec.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 21.Weinstein JN, van Osdol W. Early intervention in cancer using monoclonal antibodies and other biological ligands: micropharmacology and the “binding site barrier”. Cancer Res. 1992;52(9 Suppl):2747s–2751s. [PubMed] [Google Scholar]
- 22.Juweid M, Neumann R, Paik C, Perez-Bacete MJ, Sato J, van Osdol W, et al. Micropharmacology of monoclonal antibodies in solid tumors: direct experimental evidence for a binding site barrier. Cancer Res. 1992;52(19):5144–5153. [PubMed] [Google Scholar]
- 23.Adams GP, Schier R, McCall AM, Simmons HH, Horak EM, Alpaugh RK, et al. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001;61(12):4750–4755. [PubMed] [Google Scholar]
- 24.Cruz E, Kayser V. Monoclonal antibody therapy of solid tumors: clinical limitations and novel strategies to enhance treatment efficacy. Biologics. 2019;13:33–51. doi: 10.2147/BTT.S166310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farajpour Z, Rahbarizadeh F, Kazemi B, Ahmadvand D. A nanobody directed to a functional epitope on VEGF, as a novel strategy for cancer treatment. Biochem Biophys Res Commun. 2014;446(1):132–136. doi: 10.1016/j.bbrc.2014.02.069. [DOI] [PubMed] [Google Scholar]
- 26.Tijink BM, Laeremans T, Budde M, Stigter-van Walsum M, Dreier T, de Haard HJ, et al. Improved tumor targeting of anti-epidermal growth factor receptor nanobodies through albumin binding: taking advantage of modular nanobody technology. Mol Cancer Ther. 2008;7(8):2288–2297. doi: 10.1158/1535-7163.MCT-07-2384. [DOI] [PubMed] [Google Scholar]
- 27.Ventola CL. Progress in nanomedicine: approved and investigational nanodrugs. Pharm Ther. 2017;42(12):742–755. [PMC free article] [PubMed] [Google Scholar]
- 28.Miller MA, Arlauckas S, Weissleder R. Prediction of anti-cancer nanotherapy efficacy by imaging. Nanotheranostics. 2017;1(3):296–312. doi: 10.7150/ntno.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rizzo LY, Theek B, Storm G, Kiessling F, Lammers T. Recent progress in nanomedicine: therapeutic, diagnostic and theranostic applications. Curr Opin Biotechnol. 2013;24(6):1159–1166. doi: 10.1016/j.copbio.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 31.Maeda H, Matsumura Y. Tumoritropic and lymphotropic principles of macromolecular drugs. Crit Rev Ther Drug Carrier Syst. 1989;6(3):193–210. [PubMed] [Google Scholar]
- 32.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392. [PubMed] [Google Scholar]
- 33.Zhan W, Gedroyc W, Xu XY. The effect of tumour size on drug transport and uptake in 3-D tumour models reconstructed from magnetic resonance images. PLoS One. 2017;12(2):e0172276. doi: 10.1371/journal.pone.0172276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thurber GM, Dane Wittrup K. A mechanistic compartmental model for total antibody uptake in tumors. J Theor Biol. 2012;314:57–68. doi: 10.1016/j.jtbi.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thurber GM, Schmidt MM, Wittrup KD. Factors determining antibody distribution in tumors. Trends Pharmacol Sci. 2008;29(2):57–61. doi: 10.1016/j.tips.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Huo M, Chen Y, Shi J. Tumor microenvironment-enabled nanotherapy. Adv Healthc Mater. 2018;7(8):e1701156. doi: 10.1002/adhm.201701156. [DOI] [PubMed] [Google Scholar]
- 37.Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, et al. Naturally occurring antibodies devoid of light chains. Nature. 1993;363(6428):446–448. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- 38.Huang L, Muyldermans S, Saerens D. Nanobodies(R): proficient tools in diagnostics. Expert Rev Mol Diagn. 2010;10(6):777–785. doi: 10.1586/erm.10.62. [DOI] [PubMed] [Google Scholar]
- 39.Chakravarty R, Goel S, Cai W. Nanobody: the “magic bullet” for molecular imaging? Theranostics. 2014;4(4):386–398. doi: 10.7150/thno.8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Audenhove I, Gettemans J. Nanobodies as versatile tools to understand, diagnose, visualize and treat cancer. EBioMedicine. 2016;8:40–48. doi: 10.1016/j.ebiom.2016.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bannas P, Hambach J, Koch-Nolte F. Nanobodies and nanobody-based human heavy chain antibodies as antitumor therapeutics. Front Immunol. 2017;8:1603. doi: 10.3389/fimmu.2017.01603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82:775–797. doi: 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- 43.Ingram JR, Schmidt FI, Ploegh HL. Exploiting nanobodies’ singular traits. Annu Rev Immunol. 2018;36:695–715. doi: 10.1146/annurev-immunol-042617-053327. [DOI] [PubMed] [Google Scholar]
- 44.Muyldermans S, Baral TN, Retamozzo VC, De Baetselier P, De Genst E, Kinne J, et al. Camelid immunoglobulins and nanobody technology. Vet Immunol Immunopathol. 2009;128(1–3):178–183. doi: 10.1016/j.vetimm.2008.10.299. [DOI] [PubMed] [Google Scholar]
- 45.Zavrtanik U, Lukan J, Loris R, Lah J, Hadzi S. Structural basis of epitope recognition by heavy-chain camelid antibodies. J Mol Biol. 2018;430(21):4369–4386. doi: 10.1016/j.jmb.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 46.Mitchell LS, Colwell LJ. Analysis of nanobody paratopes reveals greater diversity than classical antibodies. Protein Eng Des Sel. 2018;31(7–8):267–275. doi: 10.1093/protein/gzy017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitchell LS, Colwell LJ. Comparative analysis of nanobody sequence and structure data. Proteins. 2018;86(7):697–706. doi: 10.1002/prot.25497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Govaert J, Pellis M, Deschacht N, Vincke C, Conrath K, Muyldermans S, et al. Dual beneficial effect of interloop disulfide bond for single domain antibody fragments. J Biol Chem. 2012;287(3):1970–1979. doi: 10.1074/jbc.M111.242818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Konning D, Zielonka S, Grzeschik J, Empting M, Valldorf B, Krah S, et al. Camelid and shark single domain antibodies: structural features and therapeutic potential. Curr Opin Struct Biol. 2017;45:10–16. doi: 10.1016/j.sbi.2016.10.019. [DOI] [PubMed] [Google Scholar]
- 50.Lauwereys M, Arbabi Ghahroudi M, Desmyter A, Kinne J, Holzer W, De Genst E, et al. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. 1998;17(13):3512–3520. doi: 10.1093/emboj/17.13.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beghein E, Gettemans J. Nanobody Technology: a versatile toolkit for microscopic imaging, protein–protein interaction analysis, and protein function exploration. Front Immunol. 2017;8:771. doi: 10.3389/fimmu.2017.00771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Koningsbruggen S, de Haard H, de Kievit P, Dirks RW, van Remoortere A, Groot AJ, et al. Llama-derived phage display antibodies in the dissection of the human disease oculopharyngeal muscular dystrophy. J Immunol Methods. 2003;279(1–2):149–161. doi: 10.1016/S0022-1759(03)00232-1. [DOI] [PubMed] [Google Scholar]
- 53.Oliveira S, Heukers R, Sornkom J, Kok RJ, van Bergen PM. Targeting tumors with nanobodies for cancer imaging and therapy. J Controll Release. 2013;172(3):607–617. doi: 10.1016/j.jconrel.2013.08.298. [DOI] [PubMed] [Google Scholar]
- 54.De Vos J, Devoogdt N, Lahoutte T, Muyldermans S. Camelid single-domain antibody-fragment engineering for (pre)clinical in vivo molecular imaging applications: adjusting the bullet to its target. Expert Opin Biol Ther. 2013;13(8):1149–1160. doi: 10.1517/14712598.2013.800478. [DOI] [PubMed] [Google Scholar]
- 55.Harmsen MM, De Haard HJ. Properties, production, and applications of camelid single-domain antibody fragments. Appl Microbiol Biotechnol. 2007;77(1):13–22. doi: 10.1007/s00253-007-1142-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kunz P, Zinner K, Mucke N, Bartoschik T, Muyldermans S, Hoheisel JD. The structural basis of nanobody unfolding reversibility and thermoresistance. Sci Rep. 2018;8(1):7934. doi: 10.1038/s41598-018-26338-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arbabi-Ghahroudi M, Tanha J, MacKenzie R. Prokaryotic expression of antibodies. Cancer Metastasis Rev. 2005;24(4):501–519. doi: 10.1007/s10555-005-6193-1. [DOI] [PubMed] [Google Scholar]
- 58.Dmitriev OY, Lutsenko S, Muyldermans S. Nanobodies as probes for protein dynamics in vitro and in cells. J Biol Chem. 2016;291(8):3767–3775. doi: 10.1074/jbc.R115.679811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muyldermans S, Cambillau C, Wyns L. Recognition of antigens by single-domain antibody fragments: the superfluous luxury of paired domains. Trends Biochem Sci. 2001;26(4):230–235. doi: 10.1016/S0968-0004(01)01790-X. [DOI] [PubMed] [Google Scholar]
- 60.Klarenbeek A, El Mazouari K, Desmyter A, Blanchetot C, Hultberg A, de Jonge N, et al. Camelid Ig V genes reveal significant human homology not seen in therapeutic target genes, providing for a powerful therapeutic antibody platform. Mabs-Austin. 2015;7(4):693–706. doi: 10.1080/19420862.2015.1046648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kijanka M, Dorresteijn B, Oliveira S, van Bergen en Henegouwen PM. Nanobody-based cancer therapy of solid tumors. Nanomedicine (Lond). 2015;10(1):161–174. doi: 10.2217/nnm.14.178. [DOI] [PubMed] [Google Scholar]
- 62.Vincke C, Loris R, Saerens D, Martinez-Rodriguez S, Muyldermans S, Conrath K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem. 2009;284(5):3273–3284. doi: 10.1074/jbc.M806889200. [DOI] [PubMed] [Google Scholar]
- 63.Romao E, Morales-Yanez F, Hu Y, Crauwels M, De Pauw P, Hassanzadeh GG, et al. Identification of useful nanobodies by phage display of immune single domain libraries derived from camelid heavy chain antibodies. Curr Pharm Des. 2016;22(43):6500–6518. doi: 10.2174/1381612822666160923114417. [DOI] [PubMed] [Google Scholar]
- 64.Fernandes JC. Therapeutic application of antibody fragments in autoimmune diseases: current state and prospects. Drug Discov Today. 2018;23(12):1996–2002. doi: 10.1016/j.drudis.2018.06.003. [DOI] [PubMed] [Google Scholar]
- 65.ClinicalTrials.gov. NIH U.S. National Library of Medicine. 2019. https://clinicaltrials.gov/ct2/results?cond=&term=ablynx&cntry=&state=&city=&dist=. https://clinicaltrials.gov/ct2/results?cond=&term=nanobody&cntry=&state=&city=&dist=. https://clinicaltrials.gov/ct2/results?cond=&term=nanobodies&cntry=&state=&city=&dist=. https://clinicaltrials.gov/ct2/results?cond=&term=vhh&cntry=&state=&city=&dist. Accessed 19 Apr 2019.
- 66.Schoonooghe S, Laoui D, Van Ginderachter JA, Devoogdt N, Lahoutte T, De Baetselier P, et al. Novel applications of nanobodies for in vivo bio-imaging of inflamed tissues in inflammatory diseases and cancer. Immunobiology. 2012;217(12):1266–1272. doi: 10.1016/j.imbio.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 67.Devoogdt N, Xavier C, Hernot S, Vaneycken I, D’Huyvetter M, De Vos J, et al. Molecular imaging using nanobodies: a case study. Methods Mol Biol. 2012;911:559–567. doi: 10.1007/978-1-61779-968-6_35. [DOI] [PubMed] [Google Scholar]
- 68.Keyaerts M, Xavier C, Heemskerk J, Devoogdt N, Everaert H, Ackaert C, et al. Phase I study of 68 Ga-HER2-nanobody for PET/CT assessment of HER2 expression in breast carcinoma. J Nucl Med. 2016;57(1):27–33. doi: 10.2967/jnumed.115.162024. [DOI] [PubMed] [Google Scholar]
- 69.Hassanzadeh-Ghassabeh G, Devoogdt N, De Pauw P, Vincke C, Muyldermans S. Nanobodies and their potential applications. Nanomedicine (Lond). 2013;8(6):1013–1026. doi: 10.2217/nnm.13.86. [DOI] [PubMed] [Google Scholar]
- 70.Debie P, Devoogdt N, Hernot S. Targeted nanobody-based molecular tracers for nuclear imaging and image-guided surgery. Antibodies. 2019;8(1):12. doi: 10.3390/antib8010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xavier C, Vaneycken I, D’Huyvetter M, Heemskerk J, Keyaerts M, Vincke C, et al. Synthesis, preclinical validation, dosimetry, and toxicity of 68 Ga-NOTA-anti-HER2 Nanobodies for iPET imaging of HER2 receptor expression in cancer. J Nucl Med. 2013;54(5):776–784. doi: 10.2967/jnumed.112.111021. [DOI] [PubMed] [Google Scholar]
- 72.van de Water JA, Bagci-Onder T, Agarwal AS, Wakimoto H, Roovers RC, Zhu Y, et al. Therapeutic stem cells expressing variants of EGFR-specific nanobodies have antitumor effects. Proc Natl Acad Sci USA. 2012;109(41):16642–16647. doi: 10.1073/pnas.1202832109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Quader S, Kataoka K. Nanomaterial-enabled cancer therapy. Mol Ther J Am Soc Gene Ther. 2017;25(7):1501–1513. doi: 10.1016/j.ymthe.2017.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jain KK. Nanomedicine: application of nanobiotechnology in medical practice. Med Princ Pract. 2008;17(2):89–101. doi: 10.1159/000112961. [DOI] [PubMed] [Google Scholar]
- 75.Uchegbu IF, Siew A. Nanomedicines and nanodiagnostics come of age. J Pharm Sci. 2013;102(2):305–310. doi: 10.1002/jps.23377. [DOI] [PubMed] [Google Scholar]
- 76.Jain KK. Advances in the field of nanooncology. BMC Med. 2010;8:83. doi: 10.1186/1741-7015-8-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR. Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm Res. 2016;33(10):2373–2387. doi: 10.1007/s11095-016-1958-5. [DOI] [PubMed] [Google Scholar]
- 78.Jain KK. Nanodiagnostics: application of nanotechnology in molecular diagnostics. Expert Rev Mol Diagn. 2003;3(2):153–161. doi: 10.1586/14737159.3.2.153. [DOI] [PubMed] [Google Scholar]
- 79.Fernandes C, Suares D, Yergeri MC. Tumor microenvironment targeted nanotherapy. Front Pharmacol. 2018;9:1230. doi: 10.3389/fphar.2018.01230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aslan B, Ozpolat B, Sood AK, Lopez-Berestein G. Nanotechnology in cancer therapy. J Drug Target. 2013;21(10):904–913. doi: 10.3109/1061186X.2013.837469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jain KK. Recent advances in nanooncology. Technol Cancer Res Treat. 2008;7(1):1–13. doi: 10.1177/153303460800700101. [DOI] [PubMed] [Google Scholar]
- 82.Jain KK. Nanobiotechnology and personalized medicine. Prog Mol Biol Transl Sci. 2011;104:325–354. doi: 10.1016/B978-0-12-416020-0.00008-5. [DOI] [PubMed] [Google Scholar]
- 83.Wang M, Thanou M. Targeting nanoparticles to cancer. Pharmacol Res. 2010;62(2):90–99. doi: 10.1016/j.phrs.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 84.Hua S, de Matos MBC, Metselaar JM, Storm G. Current trends and challenges in the clinical translation of nanoparticulate nanomedicines: pathways for translational development and commercialization. Front Pharmacol. 2018;9:790. doi: 10.3389/fphar.2018.00790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sanna V, Pala N, Sechi M. Targeted therapy using nanotechnology: focus on cancer. Int J Nanomed. 2014;9:467–483. doi: 10.2147/IJN.S36654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim BY, Rutka JT, Chan WC. Nanomedicine. N Engl J Med. 2010;363(25):2434–2443. doi: 10.1056/NEJMra0912273. [DOI] [PubMed] [Google Scholar]
- 87.Yoon JW, Jiang W, Rutka JT, Huang Y, Kim BYS. Perspectives of nanotechnology in the management of gliomas. Prog Neurol Surg. 2018;32:196–210. doi: 10.1159/000469691. [DOI] [PubMed] [Google Scholar]
- 88.Arezumand R, Alibakhshi A, Ranjbari J, Ramazani A, Muyldermans S. Nanobodies as novel agents for targeting angiogenesis in solid cancers. Front Immunol. 2017;8:1746. doi: 10.3389/fimmu.2017.01746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Behdani M, Zeinali S, Khanahmad H, Karimipour M, Asadzadeh N, Azadmanesh K, et al. Generation and characterization of a functional nanobody against the vascular endothelial growth factor receptor-2; angiogenesis cell receptor. Mol Immunol. 2012;50(1–2):35–41. doi: 10.1016/j.molimm.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 90.Vosjan MJ, Vercammen J, Kolkman JA, Stigter-van Walsum M, Revets H, van Dongen GA. Nanobodies targeting the hepatocyte growth factor: potential new drugs for molecular cancer therapy. Mol Cancer Ther. 2012;11(4):1017–1025. doi: 10.1158/1535-7163.MCT-11-0891. [DOI] [PubMed] [Google Scholar]
- 91.Rissiek B, Koch-Nolte F, Magnus T. Nanobodies as modulators of inflammation: potential applications for acute brain injury. Front Cell Neurosci. 2014;8:344. doi: 10.3389/fncel.2014.00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ebrahimizadeh W, Mousavi Gargari SL, Javidan Z, Rajabibazl M. Production of novel VHH nanobody inhibiting angiogenesis by targeting binding site of VEGF. Appl Biochem Biotechnol. 2015;176(7):1985–1995. doi: 10.1007/s12010-015-1695-y. [DOI] [PubMed] [Google Scholar]
- 93.Oliveira S, Schiffelers RM, van der Veeken J, van der Meel R, Vongpromek R, van Bergen En Henegouwen PM, et al. Downregulation of EGFR by a novel multivalent nanobody-liposome platform. J Controll Release. 2010;145(2):165–175. doi: 10.1016/j.jconrel.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 94.Talelli M, Rijcken CJ, Oliveira S, van der Meel R, van Bergen En Henegouwen PM, Lammers T, et al. Nanobody-shell functionalized thermosensitive core-crosslinked polymeric micelles for active drug targeting. J Controll Release. 2011;151(2):183–192. doi: 10.1016/j.jconrel.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 95.Altintas I, Heukers R, van der Meel R, Lacombe M, Amidi M, van Bergen En Henegouwen PM, et al. Nanobody-albumin nanoparticles (NANAPs) for the delivery of a multikinase inhibitor 17864 to EGFR overexpressing tumor cells. J Controll Release. 2013;165(2):110–118. doi: 10.1016/j.jconrel.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 96.Mima T, Nishimoto N. Clinical value of blocking IL-6 receptor. Curr Opin Rheumatol. 2009;21(3):224–230. doi: 10.1097/BOR.0b013e3283295fec. [DOI] [PubMed] [Google Scholar]
- 97.Roovers RC, Vosjan MJ, Laeremans T, el Khoulati R, de Bruin RC, Ferguson KM, et al. A biparatopic anti-EGFR nanobody efficiently inhibits solid tumour growth. Int J Cancer. 2011;129(8):2013–2024. doi: 10.1002/ijc.26145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Scully M, Cataland SR, Peyvandi F, Coppo P, Knobl P, Kremer Hovinga JA, et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2019;380(4):335–346. doi: 10.1056/NEJMoa1806311. [DOI] [PubMed] [Google Scholar]
- 99.Xu X, Vugmeyster Y. Challenges and opportunities in absorption, distribution, metabolism, and excretion studies of therapeutic biologics. AAPS J. 2012;14(4):781–791. doi: 10.1208/s12248-012-9388-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290–296. doi: 10.1038/icb.2014.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xing J, Lin L, Li J, Liu J, Zhou C, Pan H, et al. BiHC, a T-cell-engaging bispecific recombinant antibody, has potent cytotoxic activity against Her2 tumor cells. Transl Oncol. 2017;10(5):780–785. doi: 10.1016/j.tranon.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lin L, Li L, Zhou C, Li J, Liu J, Shu R, et al. A HER2 bispecific antibody can be efficiently expressed in Escherichia coli with potent cytotoxicity. Oncol Lett. 2018;16(1):1259–1266. doi: 10.3892/ol.2018.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Molgaard K, Harwood SL, Compte M, Merino N, Bonet J, Alvarez-Cienfuegos A, et al. Bispecific light T-cell engagers for gene-based immunotherapy of epidermal growth factor receptor (EGFR)-positive malignancies. Cancer Immunol Immunother CII. 2018;67(8):1251–1260. doi: 10.1007/s00262-018-2181-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Iri-Sofla FJ, Rahbarizadeh F, Ahmadvand D, Rasaee MJ. Nanobody-based chimeric receptor gene integration in Jurkat cells mediated by phiC31 integrase. Exp Cell Res. 2011;317(18):2630–2641. doi: 10.1016/j.yexcr.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 105.Albert S, Arndt C, Feldmann A, Bergmann R, Bachmann D, Koristka S, et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunology. 2017;6(4):e1287246. doi: 10.1080/2162402X.2017.1287246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Steeland S, Vandenbroucke RE, Libert C. Nanobodies as therapeutics: big opportunities for small antibodies. Drug Discov Today. 2016;21(7):1076–1113. doi: 10.1016/j.drudis.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 107.van der Meel R, Oliveira S, Altintas I, Haselberg R, van der Veeken J, Roovers RC, et al. Tumor-targeted Nanobullets: Anti-EGFR nanobody-liposomes loaded with anti-IGF-1R kinase inhibitor for cancer treatment. J Controll Release. 2012;159(2):281–289. doi: 10.1016/j.jconrel.2011.12.027. [DOI] [PubMed] [Google Scholar]
- 108.Talelli M, Iman M, Varkouhi AK, Rijcken CJ, Schiffelers RM, Etrych T, et al. Core-crosslinked polymeric micelles with controlled release of covalently entrapped doxorubicin. Biomaterials. 2010;31(30):7797–7804. doi: 10.1016/j.biomaterials.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 109.Moulder S, Hortobagyi GN. Advances in the treatment of breast cancer. Clin Pharmacol Ther. 2008;83(1):26–36. doi: 10.1038/sj.clpt.6100449. [DOI] [PubMed] [Google Scholar]
- 110.Pruszynski M, Koumarianou E, Vaidyanathan G, Revets H, Devoogdt N, Lahoutte T, et al. Targeting breast carcinoma with radioiodinated anti-HER2 nanobody. Nucl Med Biol. 2013;40(1):52–59. doi: 10.1016/j.nucmedbio.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Van Heeke G, Allosery K, De Brabandere V, De Smedt T, Detalle L, de Fougerolles A. Nanobodies(R) as inhaled biotherapeutics for lung diseases. Pharmacol Ther. 2017;169:47–56. doi: 10.1016/j.pharmthera.2016.06.012. [DOI] [PubMed] [Google Scholar]
- 112.Hacha J, Tomlinson K, Maertens L, Paulissen G, Rocks N, Foidart JM, et al. Nebulized anti-IL-13 monoclonal antibody Fab’ fragment reduces allergen-induced asthma. Am J Respir Cell Mol Biol. 2012;47(5):709–717. doi: 10.1165/rcmb.2012-0031OC. [DOI] [PubMed] [Google Scholar]
- 113.Maillet A, Guilleminault L, Lemarie E, Lerondel S, Azzopardi N, Montharu J, et al. The airways, a novel route for delivering monoclonal antibodies to treat lung tumors. Pharm Res. 2011;28(9):2147–2156. doi: 10.1007/s11095-011-0442-5. [DOI] [PubMed] [Google Scholar]
- 114.Detalle L, Stohr T, Palomo C, Piedra PA, Gilbert BE, Mas V, et al. Generation and characterization of ALX-0171, a potent novel therapeutic nanobody for the treatment of respiratory syncytial virus infection. Antimicrob Agents Chemother. 2016;60(1):6–13. doi: 10.1128/AAC.01802-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang SM, He X, Li N, Yu F, Hu Y, Wang LS, et al. A novel nanobody specific for respiratory surfactant protein A has potential for lung targeting. Int J Nanomed. 2015;10:2857–2869. doi: 10.2147/IJN.S77268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Louveau A, Harris TH, Kipnis J. Revisiting the mechanisms of CNS immune privilege. Trends Immunol. 2015;36(10):569–577. doi: 10.1016/j.it.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shilo M, Motiei M, Hana P, Popovtzer R. Transport of nanoparticles through the blood–brain barrier for imaging and therapeutic applications. Nanoscale. 2014;6(4):2146–2152. doi: 10.1039/c3nr04878k. [DOI] [PubMed] [Google Scholar]
- 118.Zhang TT, Li W, Meng G, Wang P, Liao W. Strategies for transporting nanoparticles across the blood–brain barrier. Biomater Sci. 2016;4(2):219–229. doi: 10.1039/c5bm00383k. [DOI] [PubMed] [Google Scholar]
- 119.Eugenin EA, Clements JE, Zink MC, Berman JW. Human immunodeficiency virus infection of human astrocytes disrupts blood–brain barrier integrity by a gap junction-dependent mechanism. J Neurosci. 2011;31(26):9456–9465. doi: 10.1523/JNEUROSCI.1460-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cena V, Jativa P. Nanoparticle crossing of blood–brain barrier: a road to new therapeutic approaches to central nervous system diseases. Nanomedicine (Lond). 2018;13(13):1513–1516. doi: 10.2217/nnm-2018-0139. [DOI] [PubMed] [Google Scholar]
- 121.Saraiva C, Praca C, Ferreira R, Santos T, Ferreira L, Bernardino L. Nanoparticle-mediated brain drug delivery: overcoming blood–brain barrier to treat neurodegenerative diseases. J Controll Release. 2016;235:34–47. doi: 10.1016/j.jconrel.2016.05.044. [DOI] [PubMed] [Google Scholar]
- 122.Stanimirovic DB, Sandhu JK, Costain WJ. Emerging technologies for delivery of biotherapeutics and gene therapy across the blood–brain barrier. BioDrugs. 2018;32(6):547–559. doi: 10.1007/s40259-018-0309-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Paterson J, Webster CI. Exploiting transferrin receptor for delivering drugs across the blood–brain barrier. Drug Discov Today Technol. 2016;20:49–52. doi: 10.1016/j.ddtec.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 124.Lajoie JM, Shusta EV. Targeting receptor-mediated transport for delivery of biologics across the blood–brain barrier. Annu Rev Pharmacol Toxicol. 2015;55:613–631. doi: 10.1146/annurev-pharmtox-010814-124852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Niewoehner J, Bohrmann B, Collin L, Urich E, Sade H, Maier P, et al. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron. 2014;81(1):49–60. doi: 10.1016/j.neuron.2013.10.061. [DOI] [PubMed] [Google Scholar]
- 126.Pardridge WM, Buciak JL, Friden PM. Selective transport of an anti-transferrin receptor antibody through the blood–brain barrier in vivo. J Pharmacol Exp Ther. 1991;259(1):66–70. [PubMed] [Google Scholar]
- 127.Sade H, Baumgartner C, Hugenmatter A, Moessner E, Freskgard PO, Niewoehner J. A human blood–brain barrier transcytosis assay reveals antibody transcytosis influenced by pH-dependent receptor binding. PLoS One. 2014;9(4):e96340. doi: 10.1371/journal.pone.0096340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Farrington GK, Caram-Salas N, Haqqani AS, Brunette E, Eldredge J, Pepinsky B, et al. A novel platform for engineering blood–brain barrier-crossing bispecific biologics. FASEB J. 2014;28(11):4764–4778. doi: 10.1096/fj.14-253369. [DOI] [PubMed] [Google Scholar]
- 129.Muruganandam A, Tanha J, Narang S, Stanimirovic D. Selection of phage-displayed llama single-domain antibodies that transmigrate across human blood-brain barrier endothelium. FASEB J. 2002;16(2):240–242. doi: 10.1096/fj.01-0343fje. [DOI] [PubMed] [Google Scholar]
- 130.Tanha J, Muruganandam A, Stanimirovic D. Phage display technology for identifying specific antigens on brain endothelial cells. Methods Mol Med. 2003;89:435–449. doi: 10.1385/1-59259-419-0:435. [DOI] [PubMed] [Google Scholar]
- 131.Abulrob A, Sprong H, Van Bergen en Henegouwen P, Stanimirovic D. The blood–brain barrier transmigrating single domain antibody: mechanisms of transport and antigenic epitopes in human brain endothelial cells. J Neurochem. 2005;95(4):1201–1214. doi: 10.1111/j.1471-4159.2005.03463.x. [DOI] [PubMed] [Google Scholar]
- 132.Haqqani AS, Delaney CE, Brunette E, Baumann E, Farrington GK, Sisk W, et al. Endosomal trafficking regulates receptor-mediated transcytosis of antibodies across the blood–brain barrier. J Cereb Blood Flow Metab. 2018;38(4):727–740. doi: 10.1177/0271678X17740031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Suffredini G, East JE, Levy LM. New applications of nanotechnology for neuroimaging. AJNR Am J Neuroradiol. 2014;35(7):1246–1253. doi: 10.3174/ajnr.A3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li T, Bourgeois JP, Celli S, Glacial F, Le Sourd AM, Mecheri S, et al. Cell-penetrating anti-GFAP VHH and corresponding fluorescent fusion protein VHH-GFP spontaneously cross the blood–brain barrier and specifically recognize astrocytes: application to brain imaging. FASEB J. 2012;26(10):3969–3979. doi: 10.1096/fj.11-201384. [DOI] [PubMed] [Google Scholar]
- 135.Rutgers KS, Nabuurs RJ, van den Berg SA, Schenk GJ, Rotman M, Verrips CT, et al. Transmigration of beta amyloid specific heavy chain antibody fragments across the in vitro blood–brain barrier. Neuroscience. 2011;190:37–42. doi: 10.1016/j.neuroscience.2011.05.076. [DOI] [PubMed] [Google Scholar]
- 136.Caljon G, Caveliers V, Lahoutte T, Stijlemans B, Ghassabeh GH, Van Den Abbeele J, et al. Using microdialysis to analyse the passage of monovalent nanobodies through the blood–brain barrier. Br J Pharm. 2012;165(7):2341–2353. doi: 10.1111/j.1476-5381.2011.01723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Roovers RC, Laeremans T, Huang L, De Taeye S, Verkleij AJ, Revets H, et al. Efficient inhibition of EGFR signaling and of tumour growth by antagonistic anti-EFGR nanobodies. Cancer Immunol Immunother CII. 2007;56(3):303–317. doi: 10.1007/s00262-006-0180-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hulstein JJ, de Groot PG, Silence K, Veyradier A, Fijnheer R, Lenting PJ. A novel nanobody that detects the gain-of-function phenotype of von Willebrand factor in ADAMTS13 deficiency and von Willebrand disease type 2B. Blood. 2005;106(9):3035–3042. doi: 10.1182/blood-2005-03-1153. [DOI] [PubMed] [Google Scholar]
- 139.Bhoopalan SV, Hankins J, George J, Ryder A, Onder AM, Puri L. Use of caplacizumab in a child with refractory thrombotic thrombocytopenic purpura. Pediatr Blood Cancer. 2019;66(7):e27737. doi: 10.1002/pbc.27737. [DOI] [PubMed] [Google Scholar]
- 140.Kaczmarek V, Holle J, Astudillo R, Kempf C, Bufler P, Muller D. Caplacizumab for relapsing thrombotic thrombocytopenic purpura. Pediatr Nephrol. 2019 doi: 10.1007/s00467-019-04281-z. [DOI] [PubMed] [Google Scholar]
- 141.Peyvandi F, Callewaert F. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2016;374(25):2497–2498. doi: 10.1056/NEJMc1603180. [DOI] [PubMed] [Google Scholar]
- 142.Peyvandi F, Scully M, Kremer Hovinga JA, Cataland S, Knobl P, Wu H, et al. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2016;374(6):511–522. doi: 10.1056/NEJMoa1505533. [DOI] [PubMed] [Google Scholar]
- 143.Peyvandi F, Scully M, Kremer Hovinga JA, Knobl P, Cataland S, De Beuf K, et al. Caplacizumab reduces the frequency of major thromboembolic events, exacerbations and death in patients with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost JTH. 2017;15(7):1448–1452. doi: 10.1111/jth.13716. [DOI] [PubMed] [Google Scholar]
- 144.Kratz F, Elsadek B. Clinical impact of serum proteins on drug delivery. J Controll Release. 2012;161(2):429–445. doi: 10.1016/j.jconrel.2011.11.028. [DOI] [PubMed] [Google Scholar]
- 145.Van Roy M, Ververken C, Beirnaert E, Hoefman S, Kolkman J, Vierboom M, et al. The preclinical pharmacology of the high affinity anti-IL-6R Nanobody(R) ALX-0061 supports its clinical development in rheumatoid arthritis. Arthr Res Ther. 2015;17:135. doi: 10.1186/s13075-015-0651-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Palomo C, Mas V, Detalle L, Depla E, Cano O, Vazquez M, et al. Trivalency of a nanobody specific for the human respiratory syncytial virus fusion glycoprotein drastically enhances virus neutralization and impacts escape mutant selection. Antimicrob Agents Chemother. 2016;60(11):6498–6509. doi: 10.1128/AAC.00842-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Svecova D, Lubell MW, Casset-Semanaz F, Mackenzie H, Grenningloh R, Krueger JG. A randomized, double-blind, placebo-controlled phase 1 study of multiple ascending doses of subcutaneous M1095, an anti-interleukin 17A/F nanobody, in moderate-to-severe psoriasis. J Am Acad Dermatol. 2019;81(1):196–203. doi: 10.1016/j.jaad.2019.03.056. [DOI] [PubMed] [Google Scholar]
- 148.De Munter S, Ingels J, Goetgeluk G, Bonte S, Pille M, Weening K, et al. Nanobody based dual specific CARs. Int J Mol Sci. 2018;19(2):1. doi: 10.3390/ijms19020403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pereira J, Ottevaere I, Serruys B, Dejonckheere E, Bay-Jensen AC, Siebuhr AS, et al. Pharmacokinetic and pharmacodynamic modelling of the novel anti-ADAMTS-5 nanobody M6495 using the neo-epitope ARGS as a biomarker. Osteoarthr Cartil. 2018;26:S176. doi: 10.1016/j.joca.2018.02.381. [DOI] [Google Scholar]
- 150.Siebuhr A, Bay-Jensen AC, Thudium CT, Karsdal MA, Serruys B, Werkmann D, et al. The anti-ADAMTS-5 nanobody® M6495, protects against cartilage breakdown in cartilage and synovial joint tissue explant models. Osteoarthr Cartil. 2018;26:S187. doi: 10.1016/j.joca.2018.02.402. [DOI] [Google Scholar]
- 151.Sarker SA, Jakel M, Sultana S, Alam NH, Bardhan PK, Chisti MJ, et al. Anti-rotavirus protein reduces stool output in infants with diarrhea: a randomized placebo-controlled trial. Gastroenterology. 2013;145(4):740-8 e8. doi: 10.1053/j.gastro.2013.06.053. [DOI] [PubMed] [Google Scholar]