
Fig. 2.
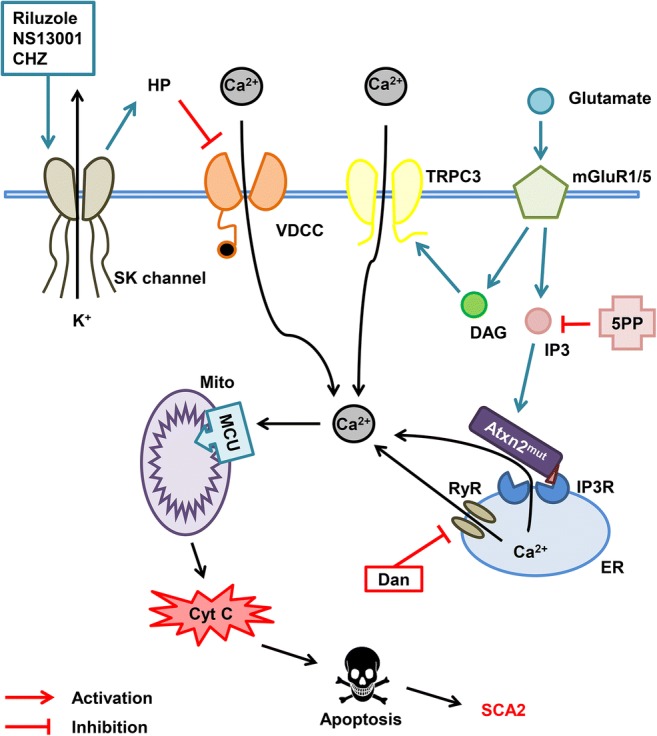
Calcium hypothesis of SCA2 pathogenesis. Molecules of glutamate released into the synaptic cleft activate metabotropic glutamate receptor (mGluR) promoting the release of inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG) molecules into the cytoplasm and further activation of the IP3 receptor (IP3R) on the endoplasmic reticulum (ER) membrane leading to the calcium entry from the ER to the cytoplasm. This process is called IP3-induced calcium release (IICR). DAG directly activates transient receptor potential channel 3 (TRPC3) and more calcium ions pass to the cytoplasm from extracellular space. It was shown that mutant ataxin-2 protein (Atxn2mut) with pathological polyglutamine expansion, but not wild-type Atxn2, associates with IP3R and increases its sensitivity to IP3. Hyperactivation of IP3R results in the abnormal calcium signaling in cerebellar Purkinje cells. Overloaded calcium (Ca2+) ions are pumped into the mitochondria (Mito) through the mitochondrial calcium uniporter (MCU) leading to the mitochondrial swelling, followed later by the rupture of outer mitochondrial membrane and further release of pro-apoptotic factors like cytochrome C (Cyt C) into the cytoplasm thus initiating apoptosis. Dramatically increased IICR can be suppressed by adeno-associated virus-mediated expression of the IP3-5-phosphatase enzyme (5PP) which converts IP3 into nonactive form IP2. Another way to reduce calcium release from ER may be the inhibition of ryanodine receptors (RyRs) with dantrolene (Dan). Small-conductance calcium-activated potassium channels (SK channels) are required for proper PC pacemaker activity. SK channel activation with riluzole, NS13001, and chlorzoxazone (CHZ) increases hyperpolarization (HP) of the PC membrane thus inhibiting the voltage-dependent calcium channels (VDCC) leading to the decrease of calcium influx from extracellular space