

Hepatitis B virus (HBV) affects an estimated 250 million chronic carriers worldwide. Though several vaccines exist, they are ineffective for those already infected. HBV persists due to the formation of covalently closed circular DNA (cccDNA)—the viral minichromosome—in the nucleus of hepatocytes. Current nucleoside analogs and interferon therapies rarely clear cccDNA, requiring lifelong treatment.
KEYWORDS: capsid, hepatitis B virus, antiviral, cccDNA
ABSTRACT
Hepatitis B virus (HBV) affects an estimated 250 million chronic carriers worldwide. Though several vaccines exist, they are ineffective for those already infected. HBV persists due to the formation of covalently closed circular DNA (cccDNA)—the viral minichromosome—in the nucleus of hepatocytes. Current nucleoside analogs and interferon therapies rarely clear cccDNA, requiring lifelong treatment. Our group identified GLP-26, a novel glyoxamide derivative that alters HBV nucleocapsid assembly and prevents viral DNA replication. GLP-26 exhibited single-digit nanomolar anti-HBV activity, inhibition of HBV e antigen (HBeAg) secretion, and reduced cccDNA amplification, in addition to showing a promising preclinical profile. Strikingly, long term combination treatment with entecavir in a humanized mouse model induced a decrease in viral loads and viral antigens that was sustained for up to 12 weeks after treatment cessation.
INTRODUCTION
Despite the availability of effective vaccines, epidemiologic data estimate that approximately 250 million people are chronically infected with hepatitis B virus (HBV) (more than the carriers of HIV and hepatitis C virus [HCV] combined) and are at high risk for development of hepatitis, cirrhosis, and hepatocellular carcinoma (1). Current anti-HBV treatment options include pegylated interferon alpha2a (pegIFN) and/or nucleoside analogs that require lifetime use to suppress the virus. Two key events in the HBV replication cycle involve first the generation of a covalently closed circular (cccDNA) transcriptional template, either from input viral DNA or newly replicated capsid-associated DNA, and second, reverse transcription of the viral pregenomic (pg) RNA to form HBV DNA genomes that are encapsidated into de novo viral particles (2, 3). HBV persists in long-lived hepatocytes due to the establishment and maintenance of cccDNA in the nucleus of host cells (4), where it is not targeted by current therapies and serves as a viral reservoir (5). Since hepatocytes have a long half-life, elimination of cccDNA by hepatocyte turnover can be considered a means of viral clearance only if the cccDNA is disrupted or silenced while replication of new HBV is stopped. HBV rebounds after cessation of treatment with currently approved nucleoside analog inhibitors. To address this issue, novel antiviral agents are now being investigated, including entry inhibitors, hepatitis B surface antigen (HBsAg) inhibitors, and capsid assembly modulators (CAM) (6).
HBV capsid assembly plays an essential role in many steps of the viral replication cycle (7). Notably, the HBV capsid is responsible for trafficking relaxed circular DNA (rcDNA) to the nucleus, thereby establishing and maintaining cccDNA levels as a “refill” mechanism. Further, the HBV capsid protein is found in the nucleus of hepatocytes and interacts with host factors responsible for transcriptional regulation (8). Therefore, it is hypothesized that targeting disruption of the nucleocapsid could impact cccDNA stability and potentially lead to eradication of HBV (9). Based on the promise of sustained antiviral activity, several CAM have been studied, such as GLS4 (1) (phase II clinical trial) (10), RG-7907 (Roche, phase I), AT-130 (2) (11), DVR-23 (12), NVR 3 to 778 (3) (13, 14) (Novira/JnJ, phase IIa), AB-423 (15) and AB-506 (16) (Arbutus), and JNJ-379 (phase IIa) (17) and ABI H0731 (18) (Assembly Bioscience, Phase 1a) (Fig. S1 in the supplemental material). Structurally these compounds are heteroaryldihydro-pyrimidines (HAPs), phenylpropenamides (PP), or sulfamoylbenzamides (SBA). Here, we report the discovery and the preclinical characterization of GLP-26, a novel CAM with a unique glyoxamidopyrrolo backbone, obtained through chemical optimization of early SBA derivatives identified by our team (19).
GLP-26 (Fig. 1) is an HBV capsid assembly modulator (CAM) displaying substantial effects at low nanomolar ranges on both HBV DNA replication and HBV e antigen (HBeAg) secretion, with greater than 1 log reduction of cccDNA amplification along with a promising preclinical profile. Most interestingly, sustained decreases in HBeAg and HBV surface antigen (HBsAg) levels were observed at up to 3 months after drug cessation in an HBV-infected humanized mouse model treated with GLP-26 in combination with entecavir.
FIG 1.
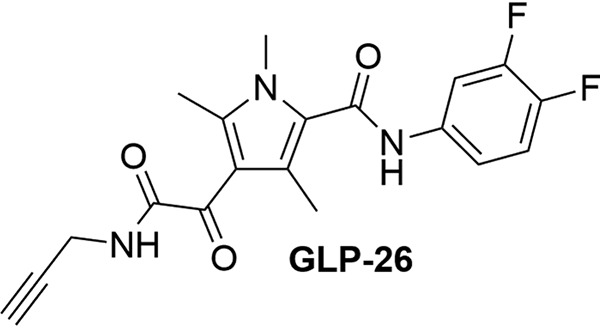
Structure of GLP-26.
RESULTS
GLP-26 is a nontoxic inhibitor of HBV DNA, HBeAg, and cccDNA production in vitro.
The in vitro anti-HBV activity of GLP-26 was determined by measuring secreted HBV DNA from HepAD38 cells and from infected primary human hepatocytes (PHH). GLP-26 displayed potent antiviral activity, with an EC50 of 0.003 μM and 0.04 μM in HepAD38 cells (Table 1) and PHH (Table 2), respectively. GLP-26 did not show toxicity at up to 100 μM in human hepatoma cell lines (HepG2) nor in a panel of other relevant cell types (Table S1 in the supplemental material), yielding a wide selectivity index (SI) (HepG2 cells >33,333). It is noteworthy that GLP-26 was 25 to 120× more potent in these assays than GLS4, a CAM currently evaluated in clinical trials. In addition, GLP-26 did not show signs of mitochondrial toxicity at concentrations of up to 50 μM and no increases in lactic acid production (measured as % of lactic acid to % of nuclear DNA) was observed at concentrations of up to 25 μM, which is well above the EC50/90 antiviral values (Table S2). As a correlate to cccDNA levels, GLP-26 was evaluated for inhibition of HBeAg production in HepAD38 cells (20). GLP-26 effectively inhibited HBeAg secretion with an EC50 of 0.003 μM, which was over 50 times more potent than GLS4 (EC50 of 0.16 μM, not shown). As expected, the nucleoside analog lamivudine (3TC) had a minimal effect on the inhibition of HBeAg secretion (Fig. 2A). The same system was used to investigate the effects of agents on cccDNA using reverse transcriptase quantitative PCR (RT-qPCR). GLP-26 and GLS4 exhibited equally potent inhibition of HBV cccDNA amplification, with a >1 log reduction relative to the untreated mock control (Fig. 2B).
TABLE 1.
Anti-HBV activity of GLP-26 in HepAD38 cells
| Druga | Anti-HBV activityb
(μM) |
|
|---|---|---|
| EC50 | EC90 | |
| GLP-26 | 0.003 ± 0.002 | 0.014 ± 0.002 |
| GLS4 | 0.08 ± 0.02 | 0.28 ± 0.06 |
| 3TC | 0.41 ± 0.36 | 1.65 ± 0.92 |
| ETV | 0.0006 ± 0.0003 | 0.011 ± 0.002 |
| TDF | 0.005 ± 0.0004 | 0.070 ± 0.010 |
3TC, lamivudine; ETV, entecavir; TDF, tenofovir disoproxil fumarate.
All values represent the average of at least two independent experiments and samples were performed in duplicate ± SD.
TABLE 2.
Anti-HBV activity of GLP-26 in primary human hepatocytes (PHH)
| Druga | Anti-HBV activityb at EC50 (μM) | Toxicity at CC50 (μM) |
|---|---|---|
| GLP-26 | 0.04 ± 0.01 | >10 |
| GLS4 | 4.34 ± 1.62 | >10 |
| TDF | 0.27 ± 0.23 | >10 |
TDF: Tenofovir disoproxil fumarate.
All values represent the average of at least two independent experiments and samples were performed in duplicate ± SD.
FIG 2.

Decrease of cccDNA markers in HepAD38 cells by GLP-26. (A) Percent inhibition of HBeAg secretion was visualized by ELISA. Cells with or without drugs were incubated in the absence of tetracycline (Tet) for 7 days, followed by the addition of Tet to the culture of both untreated and drug-treated cells for another 7 days (from day 7 to day 14). Tet ON, cells cultured in the presence of tetracycline only for 14 days. Inhibition (%) of HBeAg secretion was determined relative to untreated Tet off/on control. (B) The levels of HBV DNA and cccDNA were quantified by qPCR and log viral reduction was determined relative to the untreated mock Tet off control. All values represent the average of at least two independent experiments and samples were performed in duplicate ± standard deviation (SD).
GLP-26 stabilizes HBV capsid particles and induces their accumulation in the cytoplasm.
Direct binding of GLP-26 to HBV capsid protein was evaluated using a fluorescent thermal shift assay by measuring changes in the thermal stability of capsids upon complexation with GLP-26 (21, 22). The HBV capsid protein fragment of 1 to 149 amino acids (aa) (Cp149) was expressed and isolated as dimers as previously described, and capsid particles were formed from Cp149 dimers by decreasing pH and increasing salt concentration. GLP-26 reproducibly increased the melting temperature of HBV Cp149 capsids (Tm = 87 ± 0.3°C) to a greater extent than did GLS4 (Tm = 85 ± 0.3°C) (Fig. 3A). Fitting the titration of GLP-26 to HBV Cp149 capsids provided a Kd (dissociation constant) of 0.7 ± 1.5 μM, which was ∼60-fold lower than GLS4 (Kd = 41 ± 13 μM), indicating that GLP-26 binds to and stabilizes HBV capsids.
FIG 3.

Effects of GLP-26 binding to HBV capsids. (A) Thermal shift fluorescence assay thermograms for vehicle (dotted line) and GLS4-treated (dashed line) and GLP-26-treated (solid line) HBV Cp149 capsids. (B to D) Confocal immunofluorescence microscopy images showing HBV core protein (green) distribution in HepAD38 hepatocytes after 24 h for vehicle (in DMSO) (B), GLP-26 (1μM) (C), and GLS4 (1 μM) (D). Nuclei are DAPI stained (blue).
The effect of GLP-26 on the cellular localization of capsids was determined by confocal microscopy in HepAD38 cells (Fig. S3). In the absence of either drug (Fig. 3B), 50% of the cells contained HBV core proteins corresponding to HBV capsids in both the nucleus and the cytoplasm. In contrast, treatment with GLP-26 for 24 h emptied the nucleus and led to an accumulation of capsid particles exclusively in the cytoplasm (Fig. 3C), while treatment with GLS4 caused capsids to form large aggregates spread in the cell (Fig. 3D), as previously reported for this type of HAP derivatives (23–26).
GLP-26 induces the formation of firm HBV capsid particles.
The effects of GLP-26 on capsid assembly were observed using negative-stain transmission electron microscopy (TEM). To determine the effects on the assembly process, Cp149 dimers were first incubated with the drug and then followed by addition of salt to initiate assembly. In the absence of either drug, HBV capsids formed regular icosahedrons with a diameter of approximately 40 nm (Fig. 4A and B). Addition of GLP-26 to Cp149 dimers followed by assembly generated clusters of small and misshapen particles. In contrast, large, aberrant capsid morphologies were induced by GLS4 (Fig. S4). To determine the effects postassembly, images of preformed HBV capsid particles treated with compounds were collected. Addition of GLS4 to preformed capsids resulted in larger, broken assemblies, similar in appearance to cracked egg shells (Fig. S4). Unlike GLS4, fewer particles were observed upon addition of GLP-26, and those that remained exhibited smaller and firmer morphologies more like “hard boiled eggs” (Fig. 4E and F).
FIG 4.
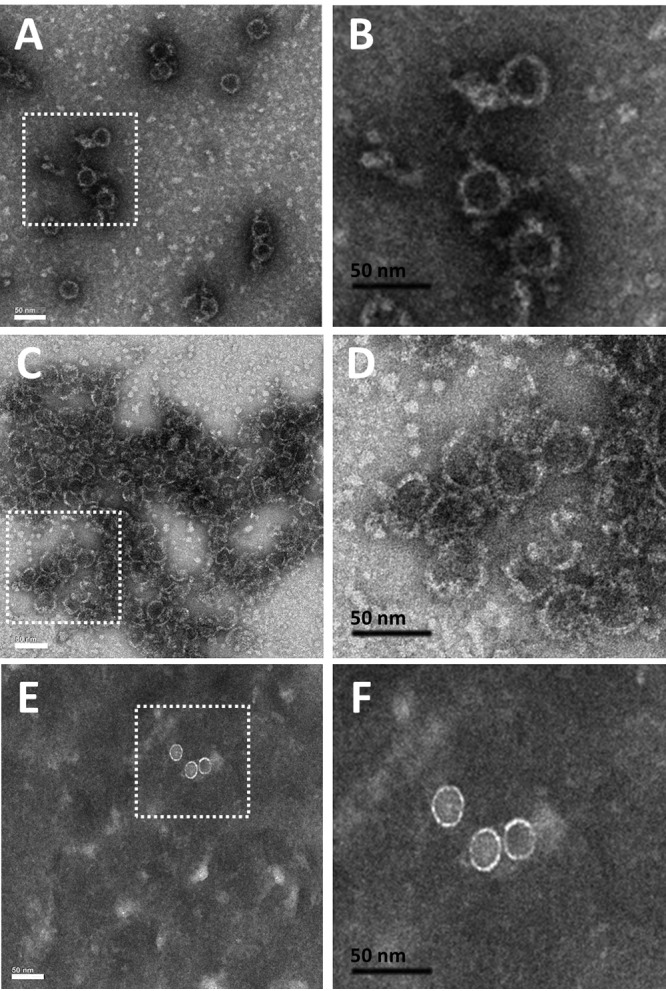
Effects of GLP-26 on the HBV Cp149 capsid morphology determined by negative-stain transmission electron microscopy. HBV Cp149 capsid particles were treated with vehicle (A and B) or GLP-26 (25 μM) prior to assembly initiation (C and D). For postassembly assessment, preformed HBV-Cp149 capsid particles were treated with GLP-26 (25 μM) (E and F). Black bars represent 50 nm scale.
GLP-26 demonstrated synergistic antiviral activity with nucleoside analogs in vitro.
Anticipating that HBV CAMs such as GLP-26 will be administered in combination with existing direct-acting agents, we evaluated its interaction with entecavir (ETV), a potent nucleoside analog inhibitor of HBV replication. For the median-effect analysis, the drugs were combined at a 5:1 ratio (GLP-26 + ETV) based on their EC50 values. These two agents resulted in a combination index (CI) of 0.6 (Table S3), indicating that GLP-26 interacted synergistically with ETV in the HepAD38 system.
GLP-26 has a favorable in vitro and in vivo pharmacokinetic profile.
In preparation for in vivo applications, the stability of GLP-26 was evaluated. GLP-26 demonstrated favorable stability profiles in plasma with half-lives of >24 h in human, mouse, and dog, and a t½ of ∼8.5 h in rat plasma samples (Table S4). Liver microsome stability was also satisfactory, with a t½ of 71 min and 7.6 h in mouse and human, respectively (Table S6).
The pharmacokinetic characteristics of GLP-26 were evaluated in CD-1 mice, where it displayed favorable oral bioavailability (Fig. S5) with an observed area under the concentration-time curve from 0 to 7 h (AUC0–7hobs) of 1,587 and 1,306 ng · h/ml by oral (p.o.) and intravenous (i.v.) routes, respectively. The t½ was much longer from p.o. administration (>6 h) than from i.v. (1.5 h), providing prolonged concentrations well above the in vitro EC50. Due to the favorable oral absorption profile and long half-life, we decided to deliver GLP-26 in vivo using oral dosing.
GLP-26 decreases HBV DNA, HBsAg, and HBeAg levels in HBV-infected humanized mice.
To evaluate GLP-26 activity in vivo, HBV-infected BRGS-uPA mice with chimeric humanized livers (HUHEP mice) (27, 28) were treated with either GLP-26 alone (60 mg/kg/day) or in combination with ETV (0.3 mg/kg/day) by oral administration (Fig. 5A to D). At the start of treatment, all mice had serum human albumin (hAlb) levels above 100 μg/ml and serum HBV DNA levels above 106 copies/ml. Over a period of 10 weeks, the untreated cohort showed increased levels of HBV DNA (>1 log10) and HBsAg (0.5 to 2 log10) with no significant changes in HBeAg expression. Treatment with GLP-26 alone led to decreases in viral loads (1 to 3 log10), HBsAg (0.3 to 2 log10), and HBeAg (0.3 to 1 log10). In comparison, mice treated with ETV alone had a 2.7 to 3.3 log10 decrease in HBV DNA (one mouse had levels below limits of detection); however, the ETV-treated group showed minimal decreases in HBsAg and HBeAg (0.7 log10 and 0.2 log10, respectively).
FIG 5.

Sustained antiviral activity of GLP-26 in HBV-infected humanized mice by oral administration. Effect of entecavir (ETV) (0.3 mg/kg/day), GLP-26 (60 mg/kg/day) or the combination of ETV/ GLP-26 at the same doses on HBV DNA (A), HBsAg (B), HBeAg (C), and human albumin (D) in HBV-infected HUHEP mice. Treatment period is indicated in the gray shaded area followed by a rebound period. The lower limit of detection is shown as a thin dotted horizontal line in A and B. The thick dotted horizontal line shows the reference point of one for evaluating fold changes. Each full line represents the longitudinal results from an individual HBV-infected HUHEP mouse either untreated (black line), ETV-treated (green line), GLP-26-treated (blue line), or ETV/GLP-26-treated (red line).
GLP-26 and ETV act synergistically to reduce viral DNA and antigens with long-term sustained activity posttreatment in HBV-infected humanized mice.
Based on our in vitro results showing a synergistic effect between capsid assembly modulator GLP-26 and nucleoside analog ETV, a similar combination was evaluated in vivo. HBV-infected mice were treated concomitantly with both agents for 10 weeks. In the six mice treated with the combination of ETV (0.3 mg/kg/day) and GLP-26 (60 mg/kg/day), viral loads strongly decreased, with a mean of −4 log10 HBV DNA, and half the mice had undetectable viremia at the end of treatment (Fig. 5A). Furthermore, all the mice had significantly decreased viral antigen loads, with means of −1.8 log10 and −1 log10 for HBsAg and HBeAg, respectively (Fig. 5B and C).
As half the mice in the ETV/GLP-26 group had viral loads below the limit of detection after 10 weeks, viral kinetics were monitored for 11 to 12 weeks after cessation of treatment. During the rebound phase, viral loads returned rapidly in HBV-infected HUHEP mice that had received ETV alone, consistent with previous results (29). However, in the ETV/GLP-26 combination treatment group, of the three mice that had undetectable viremia, two remained aviremic for several weeks. HBV DNA was undetectable in one mouse for 5 weeks and the other for 11 weeks after cessation of treatment (Fig. 5A). Interestingly, even if HBeAg levels remained stable or slightly increased in most mice during the rebound phase, reduction of HBeAg up to −2 log10 was observed in two mice (Fig. 5C). HBsAg levels decreased substantially even after treatment cessation with the exception of two mice. One mouse showed undetectable levels of HBsAg (lower limit of detection = 0.1 IU/ml) despite being weakly viremic (Fig. 5B).
DISCUSSION
Current treatments for chronic HBV are limited to nucleoside polymerase inhibitors and/or pegylated interferon (pegIFN). These strategies rarely achieve a functional cure and thus development of novel therapeutic agents interfering with other essential steps of the viral replication cycle are needed. GLP-26 is a novel nontoxic CAM that displays low nanomolar activity against HBV in both HepAD38 cells and primary human hepatocytes (PHH). GLP-26 is highly specific for HBV and did not show activity against a panel of other viruses, including Dengue virus, West Nile virus, Chikungunya virus, Zika virus, and HIV-1 up to 30 μM (Table S6). Unlike heteroaryldihydropyrimidine derivatives (HAP) such as GLS4, GLP-26 binds to the capsid and induces formation of tight, intact particles. These biochemical outcomes are similar to those observed for the alternate class of CAMs that includes the propenamide (AT-130) and sulfamoylbenzamide (SBA, such as AB-423 or NVR-3-778) derivatives.
GLP-26 decreases cccDNA in vitro and decreases HBeAg (a biomarker of cccDNA) levels in vitro and in vivo. HBV maintains cccDNA levels by recycling mature relaxed circular DNA (rcDNA) to the nucleus (30). This process relies on the proper biophysical properties of the HBV capsid protein for rcDNA formation and nuclear transport. Since GLP-26 affects capsid assembly and possibly transport into the nucleus, the agent likely disrupts cccDNA maintenance within this cycle, leading to an overall reduction in cccDNA levels.
GLP-26 in combination with ETV potently decreased HBsAg and HBeAg levels in a humanized mouse model of infection, both during and, more importantly, after treatment. Previous studies have shown decreased HBV viral antigens during combination treatments (31), yet continued antiviral effects on these markers after treatment have not been yet reported for other CAMs from the same class, such as AB-423 (15) and JNJ-632 (32). The mechanisms resulting in the observed sustained response could arise from the very potent antiviral activity of GLP-26 (10 to 100 times more potent than AB-423 and JNJ-632) combined with a prolonged exposure from oral administration, leading to sustained efficacious levels of GLP-26. Since HBsAg seroconversion is more likely with low levels of HBsAg (33), these observations suggest there is potential for seroconversion when these biomarkers are decreased with GLP-26 treatment. In addition, the model used in this study did not reconstitute the mice with humanized immune systems, and we anticipate improved activity and seroconversion in immunocompetent animal models or humans upon treatment with GLP-26.
Overall, GLP-26 displayed favorable metabolic stability with high oral bioavailability. No adverse effects were observed after oral administration for up to 10 weeks, highlighting the relative safety of this compound. Optimization of the treatment period, oral dosing, and drug combinations (with ETV, pegIFN, etc.) will be essential to deliver a more pronounced and lasting antiviral effect in animal models and eventually in humans.
In conclusion, we identified GLP-26 as a highly potent and promising HBV CAM. Direct effects of GLP-26 on HBV capsid assembly were established using electron microscopy, confocal microscopy, and thermal shift assays. GLP-26 inhibited HBV DNA, HBeAg, and cccDNA amplification, and did not display any toxicity in vitro. Oral bioavailability in mice and stability in both plasma and liver microsomes strengthen an already excellent preclinical profile. Combination treatment of GLP-26 with ETV in a humanized mouse model of HBV infection delivered sustained antiviral response for up to 12 weeks after treatment cessation, offering the hope that similar effects can be reached in humans with this novel CAM.
MATERIALS AND METHODS
Synthesis of compounds.
Synthesis and characterization of GLP-26 is described in detail in the supplemental material. GLS4 was prepared according to the chemistry and methods previously described (34). Both compounds had a purity of >95% as determined by 1H, 13C, 19F nuclear magnetic resonance (NMR) and high-pressure liquid chromatography (HPLC) analysis. Entecavir (ETV), lamivudine (3TC), and tenofovir disoproxil fumarate (TDF) were purchased from commercial vendors and confirmed at >95% purity using standard analytical methods such as mass spectrometry and NMR.
Cytotoxicity assays.
In vitro cytotoxicity was determined using the CellTiter 96 nonradioactive cell proliferation colorimetric assay (MTT assay; Promega) in primary human peripheral blood mononuclear cells (PBMC) and in human T lymphoblast (CEM) and human hepatocellular carcinoma (HepG2) cell lines. Toxicity levels were measured as the concentration of test compound that inhibited cell proliferation by 50% (50% cytotoxic concentration, CC50).
HBV assay in HepAD38 cells.
The HBV assay was performed in HepAD38 cells as previously described (35). Briefly, HepAD38 cells were seeded onto 96-well plates in medium containing tetracycline (Tet) and incubated for 2 days at 37°C in a humidified 5% CO2 atmosphere. On day two, medium with Tet was removed and cells were washed with 1× phosphate-buffered saline (PBS). Antiviral drugs were prepared in medium without Tet and added in duplicate at various concentrations. After a total of 7 days incubation, total DNA was extracted using DNeasy 96 tissue kit (Qiagen), and HBV DNA was amplified by real-time PCR (20). Antiviral activity was measured by determining the average threshold cycle for the HBV DNA amplification with the compounds (alone or in combination), which was subtracted from the average cycle of the untreated-tetracycline control (ΔCT). Drugs were first tested individually for effective concentration, which inhibited 50% and 90% of HBV DNA replication (EC50 and EC90) using the CalcuSyn software program (Biosoft, Ferguson, MO, USA).
Evaluation of HBeAg secretion.
The effect of GLP-26 on the levels of cccDNA amplification was assessed using the HepAD38 cells to measure HBeAg as a cccDNA-dependent marker (36). In this system, HBV replication is controlled with tetracycline, where its presence in the medium blocks pregenomic (pg) RNA synthesis, whereas its absence allows synthesis of pgRNA and HBV DNA replication to occur. In addition, when cells are retreated with Tet, new cccDNA formation is restored and HBeAg production can be measured as a reporter for levels of intracellular cccDNA. HepAD38 cells were incubated with or without test compounds for 7 days in medium without Tet, and another 7 days in medium with Tet, in which cccDNA formation and virus production rely exclusively on restored cccDNA and not on the transgene. Supernatants were harvested at day 14, clarified by centrifugation at 2,550 × g for 5 min, and stored at −70°C until use. Levels of HBeAg secreted in the culture medium were measured by using an HBeAg enzyme-linked immunosorbent assay (ELISA) kit (BioChain Institute Inc. Hayward, CA) according to the manufacturer’s protocol. The concentration of compound that reduced levels of secreted HBeAg by 50% (EC50) was determined by linear regression.
Evaluation of intracellular HBV DNA and cccDNA levels.
DNA was extracted from HepAD38 cells for cccDNA detection. On day 14 of the experiments, DNA was purified from cells using a commercially available kit (plasmid miniprep kit; Qiagen) or a modified Hirt extraction (37). All samples were treated with plasmid-safe ATP-dependent DNase (PSAD) (Epicentre, Lucigen Corporation, Middleton, WI) at 37°C for 18 h, followed by incubation at 70°C for 30 min to inactivate PSAD. For HBV cccDNA amplification, we used TaqMan primers as previously shown (38) to specifically amplify cccDNA (forward primer 5′- ACTCTTGGACTCBCAGCAATG-3′; reverse primer 5′-CTTTATACGGGTCAATGTCCA-3′; and probe 5′-FAM-CTTTTTCACCTCTGCCTAATCATCTCWTGTTCA-TAMRA-3′) using the LightCycler 480 instrument (Roche).
Anti-HBV evaluation in primary human hepatocytes.
Primary human hepatocytes (PHH) were seeded on collagen-coated 48-well plates at 1.4 × 105 cells/well in InVitroGro cryoplateable (CP) hepatocyte medium (BioIVT) for 3 h, and then replenished with maintenance hepatocyte incubation (HI) medium (InVitroGro HI plus Torpedo antibiotic mix; BioIVT). After 24 h, cells were incubated with HBV inoculum at multiplicity of infection (MOI) of 1,000 genome equivalents per cell in maintenance HI medium containing 4% polyethylene glycol (PEG)-8000. The HBV inoculum was removed 24 h postinfection, and the cultures were maintained in HI medium for 4 days. Infected PHH cells were then incubated with the indicated concentrations of test compounds for 7 days, when the medium with or without compound was replenished. After a total of 10 days, supernatants were harvested and HBV DNA production was quantified by qRT-PCR using the following HBV-specific primers: HBV-AD38-qF1 (5′-CCGTCT GTG CCT TCT CAT CTG-3′), HBV-AD38-qR1 (5′-AGT CCA AGA GTY CTC TTATRY AAG ACC TT-3′), and HBV-AD38-qP1 (5′-FAM-CCG TGT GCA/ZEN/CTT CGCTTC ACC TCT GC-3′ BHQ1).
Electron microscopy.
Samples of HBV Cp149 dimers and capsids were prepared as previously described (19). Samples were fixed onto a charged carbon grid and stained by uranyl acetate contrast agent for 15 min. EM images were collected using a JEOL JEM-1400 electron microscope operating at 120 kV at 25,000× to 35,000× magnification (Emory University, Robert P. Apkarian electron microscopy core facility).
Thermal shift fluorescence assay.
Samples were prepared containing 2 μM HBV Cp149 capsids and various concentrations of compounds (1 to 80 μM) with <1% dimethyl sulfoxide (DMSO) to a final volume of 40 μl. Sypro orange was added to each well at 2 μl of a 1:50 dilution. Each measurement was made in triplicate across two samples. Sypro orange fluorescence was monitored continuously as temperature scanned from 45 to 95°C at a rate of 1°C/min on a Light Cycler 480 (Roche).
Confocal microscopy.
Experiments were performed on HepAD38 cells. Cells were maintained in Dulbecco modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, France) and 1% antibiotics (penicillin/streptomycin; Gibco, France) at 37°C in 5% CO2. HepAD38 cells were seeded for 6 h, washed, and incubated in DMEM. Next, 1% vol/vol DMSO or 100 μM GLP-26 (in 1% vol/vol DMSO) were added to this medium. After 24 h of treatment, the medium was refreshed and cells were incubated for an additional 24 h. HepAD38 cells were fixed with 4% paraformaldehyde/PBS, permeabilized with 0.2% triton/PBS, and blocked for 45 min with 0.4% bovine serum albumin (BSA). Cells were then incubated with a human anti-HBc antibody (39) and, after successive washing in PBS, with Alexa fluor 488 goat anti-human (Thermo Fisher Scientific). The last wash contained 4′,6-diamidino-2-phenylindole (DAPI) and cells were kept in PBS at 4°C until observation. Fluorescence confocal images were taken using a confocal microscope LEICA SP8 gSTED equipped with 63× PL APO 1.40 CS2 oil, a laser diode at 405 nm for DAPI, and an argon laser at 488 nm for Alexa 488.
Drug combinations study.
Drug interactions were analyzed using CalcuSyn (Biosoft, Ferguson, MO, USA) computer software. For the median-effect analysis, the drugs were combined at a 5:1 ratio of GLP-26 to entecavir (ETV) based on their EC50 values. Five to six concentrations of each single drug, or in combination, were performed in at least two independent experiments (40).
Stability of GLP-26 in mouse, rat, dog, and human plasma.
One milliliter of mouse, rat, dog, or human plasma containing 5 mM MgCl2 was used for the stability assay. Propantheline bromide at 10 μM was used as a positive control. The reaction was started by adding 10 μl of 1 mM stock solution of GLP-26 to give a final concentration of 10 μM and incubating at 37°C. At selected times of 0, 0.25, 0.5, 1, 2, 4, 6, and 24 h, 100 μl aliquots were taken and the reaction was stopped by mixing with 400 μl of ice-cold acetonitrile. The samples were centrifuged and 100 μl of the supernatant was mixed with 100 μl of liquid chromatography-mass spectrometry (LC-MS) mobile phase modifier and then subjected to LC-MS/MS analysis.
Stability in mouse and human liver microsomes.
The reaction mixture was prepared in a total volume of 1.5 ml containing 5 mM MgCl2, 100 mM potassium phosphate buffer (pH 7.4), 1 mg/ml mouse or human liver microsomes, and 1 μM compound. The reaction was initiated by adding 1 mM NADPH to the mixture and incubating at 37°C. At selected times of 0, 5, 15, 30, 45, 60, and 90 min, 200 μl aliquots were taken and the reaction stopped by mixing with 200 μl of 70% ice-cold methanol. The samples were centrifuged and the supernatant were subjected to LC-MS/MS analysis. Propranolol at 10 μM was used as a positive control.
Pharmacokinetic studies.
GLP-26 (3 mg/ml) in PBS containing 20% DMSO and 20% PEG-400 was given by intravenous (i.v.) injection (15 mg/kg) and orally (p.o.) (30 mg/kg) to female CD-1 mice. At the given time points of 0.5 h, 2 h, 4 h, and 7 h, blood samples were collected using heparinized capillaries. Samples were centrifuged at 15,000 × g for 10 min. Subsequently, blood plasma was collected and frozen at –80°C until analysis using LC-MS/MS (ACQUITY UPLC BEH C18 Column, 130 Å, 1.7 μm, 3 mm × 30 mm and Turbo-Ionspray Interface in the negative ion-mode on AB Sciex 5500Qtrap).
Generation of HUHEP mice, HBV infection, and treatment.
BALB/c Rag2−/−IL-2Rγc−/−NOD.sirpa uPAtg/tg (BRGS-uPA) mice were intrasplenically injected with 7 × 105 freshly thawed human hepatocytes (BD Biosciences, Corning) to generate HUHEP mice as previously described (28). Liver chimerism of HUHEP mice was evaluated with a species-specific human albumin (hAlb) ELISA (Bethyl Laboratories) on plasma samples as previously described (28). HUHEP mice with ≥100 μg/ml hAlb were intraperitoneally infected with 1 × 107 HBV genome equivalents as previously described (27). HBV-infected mice with >106 HBV DNA copies/ml were treated per os with either ETV (0.3 mg/kg/day) (Baraclude, BMS), GLP-26 (60 mg/kg/day, dissolved in PEG-400) (Sigma), or the combination of ETV/GLP-26 at the same doses, delivered in MediDrop sucralose (clear H2O) continuously for 10 weeks. For the rebound phase, mice were returned to regular drinking water. Animals were housed in isolators under pathogen-free conditions with humane care.
Virological measurements in HUHEP mice.
HBV DNA was extracted from plasma and quantified by qPCR as previously described (27). HBeAg was quantified with an ELISA chemiluminescent immunoassay kit (Autobio, China) and HBsAg was quantified with the MONOLISA HBsAg Ultra kit (Bio-Rad) following the manufacturer’s protocols.
Ethics approval.
IACUC approval was obtained prior to initiation of these mouse studies. Experiments were approved by an institutional ethical committee at the Institut Pasteur (Paris, France) and validated by the French Ministry of Education and Research (MENESR number 02162.02).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by NIH grants 1-R01-AI-132833 (to R.F.S.) and 5P30-AI-50409 (CFAR) (to R.F.S.), by Agence Nationale de Recherches sur le Sida et les Hépatites Virales (ANRS) grants number 2016-16180 and number 2016-16365, European Commission Seventh Framework Program PATHCo (HEALTH-F3-2012-305578), the Institut Pasteur, and Institut national de la santé et de la recherche médicale (INSERM) (to H.S.M.), and by ANRS and INSERM (to H.D.R.).
We gratefully acknowledge the Center for Translational Science and the Animalerie Centrale of the Institut Pasteur for productive collaboration.
We also thank Pierre-Ivan Raynal and Julien Burlaud-Gaillard from the electron microscopy (EM) facility (IBiSA) of the Tours University (http://microscopies.med.univ-tours.fr) for technical support. We gratefully acknowledge Elizabeth Wright (University of Wisconsin, Madison) and Hong Yi (Emory University, Robert P. Apkarian electronmicroscopy core) for their advice and assistance with the EM studies.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.World Health Organization. 2018. Hepatitis B fact sheet. World Health Organization, Geneva, Switzerland: http://www.who.int/news-room/fact-sheets/detail/hepatitis-b. [Google Scholar]
- 2.Locarnini S, Zoulim F. 2010. Molecular genetics of HBV infection. Antivir Ther 15, Suppl 3:3–14. [DOI] [PubMed] [Google Scholar]
- 3.Schadler S, Hildt E. 2009. HBV life cycle: entry and morphogenesis. Viruses 1:185–209. doi: 10.3390/v1020185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Block TM, Guo H, Guo JT. 2007. Molecular virology of hepatitis B virus for clinicians. Clin Liver Dis 11:685–706. doi: 10.1016/j.cld.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai D, Mills C, Yu W, Yan R, Aldrich CE, Saputelli JR, Mason WS, Xu X, Guo JT, Block TM, Cuconati A, Guo H. 2012. Identification of disubstituted sulfonamide compounds as specific inhibitors of hepatitis B virus covalently closed circular DNA formation. Antimicrob Agents Chemother 56:4277–4288. doi: 10.1128/AAC.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim WR. 2018. Emerging therapies toward a functional cure for hepatitis B virus infection. Gastroenterol Hepatol (N Y) 14:439–442. [PMC free article] [PubMed] [Google Scholar]
- 7.Cole AG. 2016. Modulators of HBV capsid assembly as an approach to treating hepatitis B virus infection. Curr Opin Pharmacol 30:131–137. doi: 10.1016/j.coph.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Diab A, Foca A, Zoulim F, Durantel D, Andrisani O. 2018. The diverse functions of the hepatitis B core/capsid protein (HBc) in the viral life cycle: implications for the development of HBc-targeting antivirals. Antiviral Res 149:211–220. doi: 10.1016/j.antiviral.2017.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boucle S, Bassit L, Ehteshami M, Schinazi RF. 2016. Toward elimination of hepatitis B virus using novel drugs, approaches, and combined modalities. Clin Liver Dis 20:737–749. doi: 10.1016/j.cld.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu G, Liu B, Zhang Y, Li J, Arzumanyan A, Clayton MM, Schinazi RF, Wang Z, Goldmann S, Ren Q, Zhang F, Feitelson MA. 2013. Preclinical characterization of GLS4, an inhibitor of hepatitis B virus core particle assembly. Antimicrob Agents Chemother 57:5344–5354. doi: 10.1128/AAC.01091-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katen SP, Chirapu SR, Finn MG, Zlotnick A. 2010. Trapping of hepatitis B virus capsid assembly intermediates by phenylpropenamide assembly accelerators. ACS Chem Biol 5:1125–1136. doi: 10.1021/cb100275b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campagna MR, Liu F, Mao R, Mills C, Cai D, Guo F, Zhao X, Ye H, Cuconati A, Guo H, Chang J, Xu X, Block TM, Guo JT. 2013. Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. J Virol 87:6931–6942. doi: 10.1128/JVI.00582-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gane EJ, Schwabe C, Walker K, Flores L, Hartman GD, Klumpp K, Liaw S, Brown NA. 2014. Phase 1a safety and pharmacokinetics of NVR 3–778, a potential first-in-class HBV core inhibitor. Hepatology 60:1279A. [Google Scholar]
- 14.Lam AM, Espiritu C, Vogel R, Ren S, Lau V, Kelly M, Kuduk SD, Hartman GD, Flores OA, Klumpp K. 2018. Preclinical characterization of NVR 3-778, a first-in-class capsid assembly modulator against hepatitis B virus. Antimicrob Agents Chemother 63: e01734-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mani N, Cole AG, Phelps JR, Ardzinski A, Cobarrubias KD, Cuconati A, Dorsey BD, Evangelista E, Fan K, Guo F, Guo H, Guo JT, Harasym TO, Kadhim S, Kultgen SG, Lee ACH, Li AHL, Long Q, Majeski SA, Mao R, McClintock KD, Reid SP, Rijnbrand R, Snead NM, Micolochick Steuer HM, Stever K, Tang S, Wang X, Zhao Q, Sofia MJ. 2018. Preclinical profile of AB-423, an inhibitor of hepatitis B virus pregenomic RNA encapsidation. Antimicrob Agents Chemother 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mani N, Li AHL, Ardzinski A, Bailey L, Phelps JR, Burns R, Chiu T, Cole AG, Cuconati A, Dorsey BD, Evangelista E, Gotchev D, Harasym TO, Jarosz A, Kadhim S, Kondratowicz A, Kultgen SG, Kwak K, Lee ACH, Majeski S, Mcclintock K, Pan J, Pasetka C, Rijnbrand R, Shapiro A, Steuer HM, Stever K, Tang S, Teng X, Wong M, Sofia MJ. 2018. Preclinical antiviral drug combination studies utilizing novel orally bioavailable investigational agents for chronic hepatitis B infection: AB-506, a next generation HBV capsid inhibitor, and AB-452, an HBV RNA destabilizer. J Hepatol 68:S17. doi: 10.1016/S0168-8278(18)30252-6. [DOI] [Google Scholar]
- 17.National Institutes of Health. 2017. An efficacy, safety, and pharmacokinetics study of JNJ-56136379 in participants with chronic hepatitis B virus infection. https://clinicaltrials.gov/ct2/show/NCT03361956.
- 18.Yuen MF, Agarwal K, Gane G, Schwabe C, Cheng W, Sievert W, Kim DJ, Ahn SH, Lim Y-S, Visvanathan K, Ruby E, Liaw S, Colonno R, Lopatin U. 2018. Interim safety, tolerability pharmacokinetics, and antiviral activity of ABI-H0731, a novel core protein allosteric modulator, in healthy volunteers, and non-cirrhotic viremic subjects with chronic hepatitis B. J Hepatol 68:S111. doi: 10.1016/S0168-8278(18)30439-2. [DOI] [Google Scholar]
- 19.Sari O, Boucle S, Cox BD, Ozturk T, Russell O, Bassit L, Amblard F, Schinazi RF. 2017. Synthesis of sulfamoylbenzamide derivatives as HBV capsid assembly effector. Eur J Med Chem 138:407–421. doi: 10.1016/j.ejmech.2017.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pas SD, Fries E, De Man RA, Osterhaus AD, Niesters HG. 2000. Development of a quantitative real-time detection assay for hepatitis B virus DNA and comparison with two commercial assays. J Clin Microbiol 38:2897–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lo MC, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, Ellestad G. 2004. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem 332:153–159. doi: 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 22.Tang J, Huber AD, Pineda DL, Boschert KN, Wolf JJ, Kankanala J, Xie J, Sarafianos SG, Wang Z. 2019. 5-Aminothiophene-2, 4-dicarboxamide analogues as hepatitis B virus capsid assembly effectors. Eur J Med Chem 164:179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lahlali T, Berke JM, Vergauwen K, Foca A, Vandyck K, Pauwels F, Zoulim F, Durantel D. 2018. Novel potent capsid assembly modulators regulate multiple steps of the Hepatitis B virus life-cycle. Antimicrob Agents Chemother 62:e00835-18. doi: 10.1128/AAC.00835-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huber AD, Wolf JJ, Liu D, Gres AT, Tang J, Boschert KN, Puray-Chavez MN, Pineda DL, Laughlin TG, Coonrod EM, Yang Q, Ji J, Kirby KA, Wang Z, Sarafianos SG. 2018. The heteroaryldihydropyrimidine Bay 38-7690 induces hepatitis B virus core protein aggregates associated with promyelocytic leukemia nuclear bodies in infected cells. mSphere 3:e00131-18. doi: 10.1128/mSphereDirect.00131-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corcuera A, Stolle K, Hillmer S, Seitz S, Lee J-Y, Bartenschlager R, Birkmann A, Urban A. 2018. Novel non-heteroarylpyrimidine (HAP) capsid assembly modifiers have a different mode of action from HAPs in vitro. Antiviral Res 158:135–142. doi: 10.1016/j.antiviral.2018.07.011. [DOI] [PubMed] [Google Scholar]
- 26.Rat V, Seigneuret F, Burlaud-Gaillard J, Lemoine R, Hourioux C, Zoulim F, Testoni B, Meunier JC, Tauber C, Roingeard P, de Rocquigny H. 2019. BAY 41-4109-mediated aggregation of assembled and misassembled HBV capsids in cells revealed by electron microscopy. Antiviral Res 169:104557. doi: 10.1016/j.antiviral.2019.104557. [DOI] [PubMed] [Google Scholar]
- 27.Dusséaux M, Masse-Ranson G, Darche S, Ahodantin J, Li Y, Fiquet O, Beaumont E, Moreau P, Rivière L, Neuveut C, Soussan P, Roingeard P, Kremsdorf D, Di Santo JP, Strick-Marchand H. 2017. Viral load affects the immune response to HBV in mice with humanized immune system and liver. Gastroenterology 153:1647–1661. doi: 10.1053/j.gastro.2017.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strick-Marchand H, Dusséaux M, Darche S, Huntington ND, Legrand N, Masse-Ranson G, Corcuff E, Ahodantin J, Weijer K, Spits H, Kremsdorf D, Di Santo JP. 2015. A novel mouse model for stable engraftment of a human immune system and human hepatocytes. PLoS One 10:e0119820. doi: 10.1371/journal.pone.0119820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mueller H, Wildum S, Luangsay S, Walther J, Lopez A, Tropberger P, Ottaviani G, Lu W, Parrott NJ, Zhang JD, Schmucki R, Racek T, Hoflack JC, Kueng E, Point F, Zhou X, Steiner G, Lütgehetmann M, Rapp G, Volz T, Dandri M, Yang S, Young JAT, Javanbakht H. 2018. A novel orally available small molecule that inhibits hepatitis B virus expression. J Hepatol 68:412–420. doi: 10.1016/j.jhep.2017.10.014. [DOI] [PubMed] [Google Scholar]
- 30.Zlotnick A, Venkatakrishnan B, Tan Z, Lewellyn E, Turner W, Francis S. 2015. Core protein: a pleiotropic keystone in the HBV lifecycle. Antiviral Res 121:82–93. doi: 10.1016/j.antiviral.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klumpp K, Shimada T, Allweiss L, Volz T, Lütgehetmann M, Hartman G, Flores OA, Lam AM, Dandri M. 2018. Efficacy of NVR 3–778, alone and in combination with pegylated interferon, vs entecavir in uPA/SCID mice with humanized livers and HBV infection. Gastroenterology 154:652–662. doi: 10.1053/j.gastro.2017.10.017. [DOI] [PubMed] [Google Scholar]
- 32.Vandyck K, Rombouts G, Stoops B, Tahri A, Vos A, Verschueren W, Wu Y, Yang J, Hou F, Huang B, Vergauwen K, Dehertogh P, Berke JM, Raboisson P. 2018. Synthesis and evaluation of N-phenyl-3-sulfamoyl-benzamide derivatives as capsid assembly modulators inhibiting hepatitis B virus (HBV). J Med Chem 61:6247–6260. doi: 10.1021/acs.jmedchem.8b00654. [DOI] [PubMed] [Google Scholar]
- 33.Tseng TC, Liu CJ, Su TH, Wang CC, Chen CL, Chen PJ, Chen DS, Kao JH. 2011. Serum hepatitis B surface antigen levels predict surface antigen loss in hepatitis B e antigen seroconverters. Gastroenterology 141:517–525. doi: 10.1053/j.gastro.2011.04.046. [DOI] [PubMed] [Google Scholar]
- 34.Ren Q, Liu X, Luo Z, Li J, Wang C, Goldmann S, Zhang J, Zhang Y. 2017. Discovery of hepatitis B virus capsid assembly inhibitors leading to a heteroaryldihydropyrimidine based clinical candidate (GLS4). Bioorg Med Chem 25:1042–1056. doi: 10.1016/j.bmc.2016.12.017. [DOI] [PubMed] [Google Scholar]
- 35.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 41:1715–1720. doi: 10.1128/AAC.41.8.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou T, Guo H, Guo JT, Cuconati A, Mehta A, Block TM. 2006. Hepatitis B virus e antigen production is dependent upon covalently closed circular (ccc) DNA in HepAD38 cell cultures and may serve as a cccDNA surrogate in antiviral screening assays. Antiviral Res 72:116–124. doi: 10.1016/j.antiviral.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 37.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Y, Sze J, He ML. 2004. HBV cccDNA in patients’ sera as an indicator for HBV reactivation and an early signal of liver damage. World J Gastroenterol 10:82–85. doi: 10.3748/wjg.v10.i1.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roingeard P, Romet-Lemonne JL, Leturcq D, Goudeau A, Essex M. 1990. Hepatitis B virus core antigen (HBc Ag) accumulation in an HBV nonproducer clone of HepG2-transfected cells is associated with cytopathic effect. Virology 179:113–120. doi: 10.1016/0042-6822(90)90280-5. [DOI] [PubMed] [Google Scholar]
- 40.Bassit L, Grier J, Bennett M, Schinazi RF. 2008. Combinations of 2’-C-methylcytidine analogues with interferon-alpha2b and triple combination with ribavirin in the hepatitis C virus replicon system. Antivir Chem Chemother 19:25–31. doi: 10.1177/095632020801900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.