

Repurposing of currently approved medications is an attractive option for the development of novel treatment strategies against physiological and infectious diseases. The antidiabetic sulfonylurea glyburide has demonstrated off-target capacity to inhibit activation of the NLRP3 inflammasome in a variety of disease models, including vaginal candidiasis, caused primarily by the fungal pathogen Candida albicans.
Keywords: Candida albicans, sulfonylureas, vaginitis, inflammasome, immunopathogenesis, vulvovaginal, antidiabetic, repurposing, Candidalysin
ABSTRACT
Repurposing of currently approved medications is an attractive option for the development of novel treatment strategies against physiological and infectious diseases. The antidiabetic sulfonylurea glyburide has demonstrated off-target capacity to inhibit activation of the NLRP3 inflammasome in a variety of disease models, including vaginal candidiasis, caused primarily by the fungal pathogen Candida albicans. Therefore, we sought to determine which of the currently approved sulfonylurea drugs prevent the release of interleukin 1β (IL-1β), a major inflammasome effector, during C. albicans challenge of the human macrophage-like THP1 cell line. Findings revealed that the second-generation antidiabetics (glyburide, glisoxepide, gliquidone, and glimepiride), which exhibit greater antidiabetic efficacy than prior iterations, demonstrated anti-inflammatory effects with various degrees of potency as determined by calculation of 50% inhibitory concentrations (IC50s). These same compounds were also effective in reducing IL-1β release during noninfectious inflammasome activation (e.g., induced by lipopolysaccharide [LPS] plus ATP), suggesting that their anti-inflammatory activity is not specific to C. albicans challenge. Moreover, treatment with sulfonylurea drugs did not impact C. albicans growth and filamentation or THP1 viability. Finally, the use of ECE1 and Candidalysin deletion mutants, along with isogenic NLRP3−/− cells, demonstrated that both Candidalysin and NLRP3 are required for IL-1β secretion, further confirming that sulfonylureas suppress inflammasome signaling. Moreover, challenge of THP1 cells with synthetic Candidalysin peptide demonstrated that this toxin is sufficient to activate the inflammasome. Treatment with the experimental inflammasome inhibitor MCC950 led to similar blockade of IL-1β release, suggesting that Candidalysin-mediated inflammasome activation can be inhibited independently of potassium efflux. Together, these results demonstrate that the second-generation antidiabetic sulfonylureas retain anti-inflammatory activity and may be considered for repurposing against immunopathological diseases, including vaginal candidiasis.
INTRODUCTION
Vulvovaginal candidiasis (VVC) is a common mucosal infection in immunocompetent women, overwhelmingly caused by the opportunistic fungus Candida albicans (1). VVC is the most prevalent human candidal infection, affecting ∼75% of the female population at least once in their lifetime (2). Moreover, 5 to 8% of all women suffer from recurrent infections (RVVC), defined as >3 episodes per year, often necessitating continuous antifungal therapy (3). Although not lethal, VVC poses major quality-of-life issues for women worldwide and is associated with rising health care costs (>$1.8 billion in the United States alone) (4). Fulminant infection typically results in symptoms such as itching, burning, pain, and redness of the vaginal mucosa, often accompanied by vaginal discharge. A seminal study by Fidel et al. using women volunteers intravaginally challenged with C. albicans established VVC as an immunopathology (5). While a majority of volunteers were asymptomatically colonized, those complaining of the aforementioned symptoms demonstrated comparatively high numbers of neutrophils recovered from vaginal lavage fluid. Similar to human infection, the robust mouse model of VVC demonstrates marked neutrophil recruitment within 2 to 3 days following vaginal infection with C. albicans, establishing this model as an excellent system to study immunopathogenesis from the perspective of both host and fungus (6, 7).
Using this model, our laboratory, along with others, has made significant progress in understanding the sequence of the events that contribute to the immunopathology. As C. albicans is polymorphic, it can switch between yeast and hyphal forms—a key virulence attribute for this fungal opportunist (8). Hyphal forms not only allow for mature biofilm formation and tissue invasion, they also represent a genetic reprogramming of the cell that is associated with production of adhesins and secreted factors (e.g., protease and lipases). The use of hypha-defective mutants revealed that loss of filamentation significantly impaired the development of immunopathology, despite high colonization levels in vivo (6). Moreover, recent work from our laboratory has shown that one such secreted factor that is highly encoded during the yeast-to-hypha transition, Candidalysin (a peptide toxin and product of the ECE1 gene), is crucial for driving neutrophil recruitment, inflammatory cytokine production (e.g., interleukin-1β [IL-1β]), and damage at the vaginal mucosa (9). Unsurprisingly, as it is a similar epithelial infection, the capacity to form hyphae and secrete Candidalysin is also required for similar responses during oropharyngeal candidiasis (10).
Given that fungus-mediated damage via hypha formation and Candidalysin secretion is required for the immunopathological response, it was reasonable to conclude that activation of the danger-sensing NLRP3 inflammasome complex may be involved as an early step in the innate immune response and subsequent secretion of IL-1β (11). Signaling via the inflammasome occurs via two broad steps: priming and activation. Priming results from ligation of pattern recognition receptors (e.g., Toll-like receptors) on the cell surface, which leads to upregulation of pro-IL-1β and pro-IL-18, major effectors of the inflammasome response. However, these precursor forms are immature and are unable to be secreted from the cell. Priming also leads to upregulation of other associated inflammasome components, such as NLRP3 and caspase 1. Recognition of exogenous (e.g., toxin or uric acid crystals) or endogenous (e.g., ATP or K+ efflux) effectors induces conformational changes in NLRP3 that further activate caspase 1. Mature caspase 1 is then capable of cleaving pro-IL-1β and pro-IL-18 into their secreted forms, in which they can act in both autocrine and paracrine manners to elicit inflammation, ultimately leading to neutrophil recruitment.
Using an in vitro model system of THP1 cell infection, our laboratory demonstrated that NLRP3 is essential for eliciting robust IL-1β secretion during C. albicans challenge (12). Others have recently demonstrated that Candidalysin is sufficient for activating the NLRP3 inflammasome in murine bone marrow-derived macrophages (BMDM) (13, 14). Moreover, using an unbiased transcriptomic approach, our laboratory revealed that inflammasome-related genes were highly upregulated in vivo during murine vaginal candidiasis and that genetic ablation of NLRP3 significantly attenuated neutrophil recruitment and IL-1β secretion in the vaginal lumen (15).
While treatment with azole drugs remains largely effective in resolving VVC, new strategies to more timely arrest immunopathological symptoms are needed (16). Given the clear role of the inflammasome in partially driving the immunopathogenesis of VVC, blockade of inflammasome activation may serve as a rational drug target (15). Previous studies using BMDM and the murine model of vaginitis have established that glyburide, a second-generation sulfonylurea drug used clinically to treat type 2 diabetes mellitus (second-generation sulfonylureas exhibit greater antidiabetic efficacy than prior iterations), is capable of inhibiting inflammasome activation (13, 15). Therefore, the objective of the current study was to determine whether other currently approved sulfonylurea drugs retain similar capacity to inhibit inflammasome activation and identify those which may be considered for repurposing as novel treatment agents against VVC.
RESULTS
Using differentiated THP1 cells, initial screening of 10 clinically marketed sulfonylurea compounds including both first-generation (acetohexamide, chlorapropamide, gliclazide, tolazamide, and tolbutamide) and second-generation (glimepiride, gliquidone, glisoxepide, and glyburide) antidiabetics was undertaken (17). We also utilized the glyburide analog 16673-34-0 and known highly potent experimental inflammasome inhibitor MCC950 (18). Both C. albicans-challenged and unchallenged THP1 macrophages were treated with each compound at 250 μM (Fig. 1A). Such a high dosage was initially selected to eliminate any compounds that did not exhibit anti-inflammasome activity. Unsurprisingly, MCC950 and glyburide demonstrated significant inhibition of IL-1β secretion compared to the results for the vehicle control. Interestingly, other second-generation drugs, including gliquidone, glimepiride, and glisoxepide, also showed significant IL-1β inhibitory activity. Importantly, treatment of unchallenged THP1 macrophages with equivalent dosing of the sulfonylureas demonstrated that none of the compounds led to secretion of IL-1β in the absence of C. albicans challenge, suggesting that sulfonylureas alone do not elicit inflammasome activity (Fig. 1A).
FIG 1.

Second-generation antidiabetic sulfonylureas demonstrate the capacity to inhibit IL-1β release without affecting cell viability. (A) THP1 cells were treated with 250 μM of each sulfonylurea or vehicle alone (0.5% DMSO) for 1 h, followed by challenge with C. albicans using an MOI of 2:1 for 4 h. Cell-free supernatants were collected and assessed for IL-1β by ELISA. Experiments were conducted in technical quadruplicate, and the results are reported as the mean concentrations + SD from independent experiments (n = 3). (B) THP1 cells were treated with 250 μM of each sulfonylurea for 1 h, followed by 20 ng LPS for 3.5 h and then 5 mM ATP for 30 min. Cell-free supernatants were collected and assessed for IL-1β by ELISA. Experiments were conducted in technical quadruplicate, and the results ae reported as the mean concentrations + SD from independent experiments (n = 3). (C) THP1 cells were treated with 250 μM of each sulfonylurea or 1% SDS for 5 h as described above, supernatant removed, XTT reagent added for 2 h, and color change measured at 450 nm. Percent viability values were calculated by comparing the results for samples to the results for the vehicle-treated control. Data are cumulative from technical quadruplicates and reported as the mean values + SD from independent experiments (n = 3). *, P < 0.05.
In order to eliminate the possibility that the sulfonylureas identified above had an unanticipated effect on C. albicans and confirm that the results were truly due to NLRP3 inhibition, we carried out a complementary assay using lipopolysaccharide (LPS) to prime cells, followed by stimulation with ATP (LPS+ATP challenge), an established robust inflammasome activation signal (19). With the exception of glimepiride, these results revealed that the same sulfonylureas identified by the results shown in Fig. 1A also significantly inhibited IL-1β release during LPS+ATP challenge (Fig. 1B). It is conceivable that treatment of THP1 cells with high doses of sulfonylureas may impart cellular toxicity, which could indirectly impact IL-1β secretion. However, use of the XTT [2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide salt] viability assay demonstrated that even at this high dose, the sulfonylurea-treated cells showed viability equal to that of the vehicle control-treated cells (Fig. 1C). Cells treated with medium supplemented with 1% sodium dodecyl sulfate (SDS) displayed significant toxicity. Collectively, these results suggested that second-generation sulfonylureas retain the capacity to inhibit inflammasome activation.
Further investigation was carried out using only the compounds that had exhibited significant inhibition in either of the challenge assays. While agreement between the C. albicans and LPS+ATP assays strongly suggested that sulfonylureas do not impact Candida biology directly, we wanted to rule out this possibility. Therefore, we undertook standard growth assays in the presence (250 μM) or absence of lead sulfonylureas. C. albicans treated with all five compounds showed growth similar to that of the vehicle-treated control (Fig. 2A). Because filamentation is required for activation of the inflammasome, we wanted to determine whether sulfonylureas may impact the capacity of C. albicans to undergo the yeast-to-hypha switch (20). Growth in RPMI medium supplemented with vehicle or sulfonylureas (250 μM) did not impact hypha formation (Fig. 2B). Therefore, these results demonstrate that sulfonylureas retain their anti-inflammatory effect independent of obvious effectors of C. albicans virulence.
FIG 2.
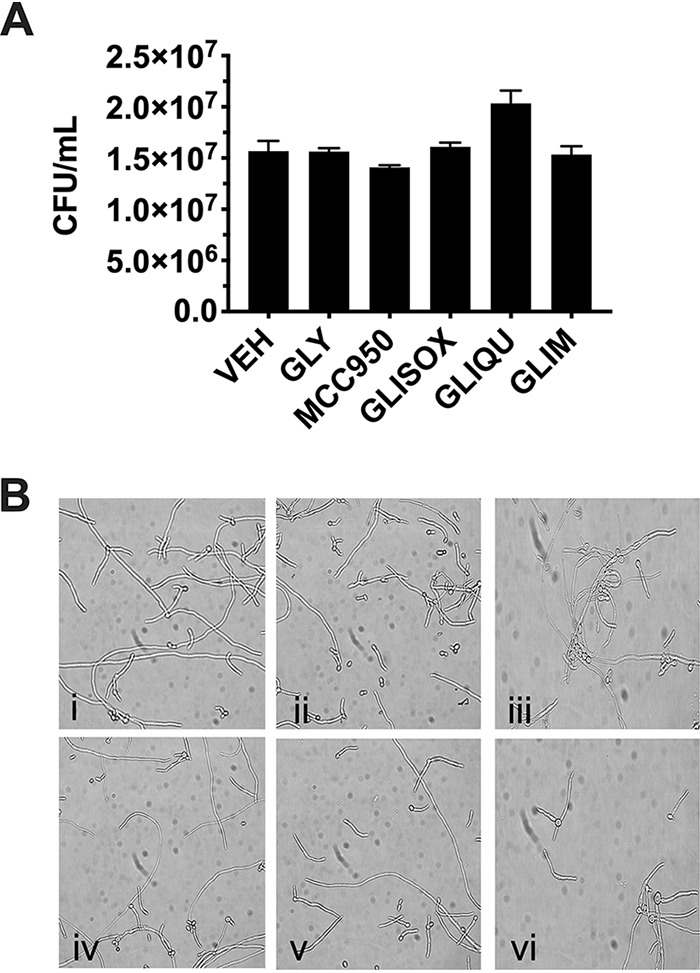
Sulfonylureas do not inhibit C. albicans growth or filamentation. (A) C. albicans was cultured in YNB growth medium with sulfonylureas (250 μM) or vehicle (0.5% DMSO) for 24 h with shaking at 30°C. Quantitative CFU counts were reported after serial dilution onto agar plates and overnight incubation. Data are reported as the mean fungal burdens + SD from independent experiments (n = 3). GLY, glyburide; GLIM, glimepiride; GLIQU, gliquidone; GLISOX, glisoxepide; VEH, vehicle. (B) Standardized C. albicans suspensions were diluted to 2.5 × 106 cells/ml in RPMI 1640 medium containing sulfonylureas (250 μM) or vehicle (0.5% DMSO). Cultures were incubated at 37°C with shaking, and on the following day, images were captured by standard light microscopy. Images represent the following compound treatments from 5 random fields of view of independent experiments (n = 3): (i) glyburide, (ii) glimepiride, (iii) gliquidone, (iv) glisoxepide, (v) MCC950, and (vi) vehicle.
We next wished to determine the relative potency of each lead sulfonylurea by calculating 50% inhibitory concentrations (IC50s) using both the C. albicans and the LPS+ATP challenge experimental assay (Fig. 3). The IC50s were remarkably similar in both assays, in the low to middle micromolar range, with the exception of glimepiride (also demonstrating discrepancy in prior LPS+ATP assays). Unfortunately, none of the other lead antidiabetic sulfonylureas showed as high a potency as glyburide (∼6.4 to 12.3 μM). The established inhibitor MCC950 had a 1,000-fold-increased potency over that of glyburide. Notable differences in the structure of this molecule (as discussed below) compared to the structures of the antidiabetics likely contribute to its greater efficacy.
FIG 3.

IC50s for sulfonylureas demonstrating IL-1β inhibition. IC50s were established by assessing IL-1β inhibition via ELISA using serial dilutions of sulfonylureas and converting values for optical density at 450 nm to percent inhibition values by comparing them to the values for the vehicle-treated control for both C. albicans (red font) and LPS+ATP (blue font) challenge. IC50 curves were constructed by plotting log concentrations of the inhibitor versus percent inhibition values using a four-parameter variable slope and best-fit model. IC50s ± SD are reported. n.c., not calculated—represents potential IC50s that exceed 250 μM, the highest dose used in these assays. Results are the mean values from independent experiments (n = 3). Structures were imported using ChemDraw. Braces group first and second generations of antidiabetic sulfonylureas.
It has been established that Candidalysin (product of the ECE1 gene) is sufficient and/or necessary to drive inflammation in murine BMDM cells and the mouse model of VVC, but it has yet to be shown that this same virulence determinant is responsible for NLRP3-dependent IL-1β secretion in THP1 cells (9, 13, 14). Therefore, wild-type (WT) and NLRP3−/− THP1 cells were challenged with WT C. albicans or an ECE1 deletion mutant (ece1Δ/Δ), Candidalysin deletion mutant (ece1Δ/Δ + ECE1Δ184–279), or ECE1 revertant (ece1Δ/Δ + ECE1). The results clearly demonstrated that C. albicans-mediated secretion of IL-1β in THP1 cells requires Candidalysin and is predominantly governed by NLRP3 (Fig. 4A). Finally, we wanted to determine whether Candidalysin was sufficient for activation of the NLRP3 inflammasome and, if so, whether it could be inhibited via sulfonylurea treatment. Treatment of cells with 20 μM synthetic Candidalysin peptide alone for 30 min led to a very modest increase in IL-1β secretion (Fig. 4B). However, when cells were prestimulated with LPS, large amounts of IL-1β were elicited following Candidalysin treatment over the same time period. These activation kinetics strongly support an inflammasome model of IL-1β release. Treatment of THP1 cells prior to priming with sulfonylurea drugs used at their respective IC50s established for C. albicans challenge led to approximately 50% inhibition of IL-1β release compared to the results for the vehicle-treated control when challenged with Candidalysin (Fig. 4B). Overall, these data suggest that currently approved antidiabetics display moderate levels of anti-inflammatory activity via inhibition of the NLRP3 inflammasome.
FIG 4.
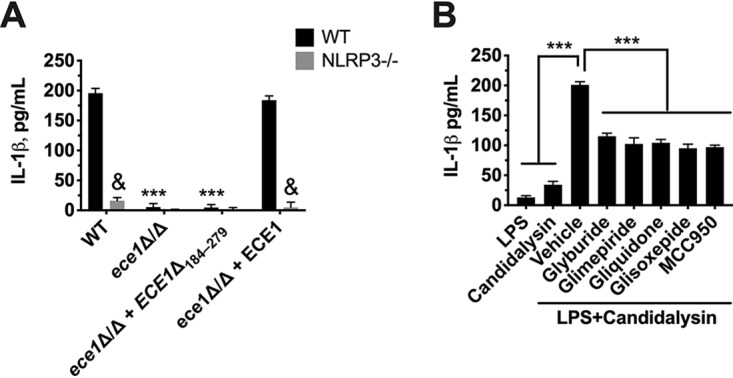
Candidalysin is necessary and sufficient for activating the NLRP3 inflammasome and can be inhibited by second-generation sulfonylureas. (A) WT and NLRP3−/− THP1 cells were challenged with WT (BWP17 + cIP30), ECE1 deletion mutant (ece1Δ/Δ), Candidalysin deletion mutant (ece1Δ/Δ + ECE1Δ184–279), or ECE1 revertant (ece1Δ/Δ + ECE1) C. albicans strains for 4 h using an MOI of 2:1. IL-1β in culture supernatants was measured by ELISA, and the results are reported as mean concentrations + SD. Data were derived from technical quadruplicates of independent experiments (n = 3). (B) WT THP1 cells were treated with LPS alone (20 ng) or Candidalysin alone (20 μM) in the presence of vehicle (0.5% DMSO). Similarly, THP1 cells were treated with vehicle or sulfonylureas (respective IC50s from the experiments whose results are shown in Fig. 3) for 1 h, followed by priming with LPS (20 ng for 3.5 h) and then inflammasome activation by ATP (5 mM for 30 min). Culture supernatants were assessed for IL-1β by ELISA, and data are reported as mean concentrations + SD. Data were calculated from the results for quadruplicate technical replicates and averaged from independent experiments (n = 3). &, P < 0.001; ***, P < 0.001.
DISCUSSION
As novel drug discovery faces challenges of high development costs and lengthy regulatory approval processes, repurposing of established medications as new therapeutic options has become attractive in recent years (21). Given the relatively low toxicity profiles of approved drugs, the window from discovery to application can theoretically be shortened considerably. One such class of compounds, the sulfonylureas originally designed to treat type 2 diabetes, has gained considerable attention as repurposed agents to treat a variety of infectious and immunological and physiological complications (17, 22). Among these are inflammatory disorders (at least partially driven by NLRP3), including melioidosis, bronchopulmonary dysplasia, allergic asthma, cystitis, endotoxemia, subarachnoid hemorrhage, and autoimmune encephalomyelitis (19, 23–30).
Given the multifactorial causes of and cellular mechanisms contributing to these diseases, protection mediated by a single molecular structure is impressive. But precisely how does glyburide work to inhibit inflammation across a spectrum of disorders? Glyburide and other hypoglycemic sulfonylurea drugs exert their antidiabetic effects by inhibiting the canonical Sur1-Kir6.2 receptor, an ATP-sensitive potassium channel (KATP) found in β cells of the pancreas (17). Blockade of Sur1 depolarizes the cell membrane, resulting in inhibition of potassium efflux, ultimately leading to calcium influx and triggering the release of insulin to signal glucose metabolism. Exactly how glyburide impairs NLRP3 inflammasome signaling is still somewhat enigmatic. As potassium efflux is a known trigger for inflammasome activation via the P2X7 receptor, it was likely assumed that glyburide (and now potentially other sulfonylureas) reduce IL-1β signaling simply by blocking potassium efflux (31). That said, genetic deletion of KATP subunits in macrophages does not abrogate inflammasome inhibition by glyburide (19). Furthermore, the antidiabetic sulfonylurea drug glipizide inhibits Sur1 but fails to block inflammasome activation (19). Therefore, an ion efflux inhibitory model likely does not explain the anti-inflammatory mechanism in its entirety. Recent work using the highly potent nonantidiabetic inflammasome inhibitor MCC950 has demonstrated that this compound interferes with further-upstream processes of inflammasome activity, by forcing NLRP3 into a closed conformational state via interference with ATP hydrolysis (32). It is currently unclear whether the antidiabetic sulfonylureas may exert anti-inflammasome effects via a similar mechanism.
The peptide toxin Candidalysin has recently been shown to be necessary and sufficient for NLRP3 inflammasome activation of both human and murine LPS-primed primary macrophages in vitro, as challenge with synthetic Candidalysin promoted IL-1β release and the use of ECE1 or Candidalysin deletion mutants abrogated this response (13, 14). Candidalysin intercalates into the membrane of a variety of cell types, including epithelial, endothelial, and hematopoietic cells, presumably resulting in ion efflux (10, 33, 34). Logically, this could serve as an activation signal for the NLRP3 inflammasome, as has been extensively described (31). A recent study by Kasper et al. demonstrated that treatment of murine BMDM with glyburide (also referred to as glibenclamide) was capable of very modestly inhibiting IL-1β release following Candidalysin challenge (13). The authors concluded this activity was due to blockade of potassium efflux (although changes in K+ levels were not explicitly monitored). However, given that the sulfonylurea glipizide (and others used in this study) is also capable of inhibiting potassium efflux and yet is incapable of blocking inflammasome activation, it is possible that glyburide does not exert its effects against Candidalysin exclusively via this proposed mechanism (19). In fact, our results with MCC950 somewhat challenge this paradigm, as this compound does not exhibit affinity for the KATP receptor, nor does it block potassium efflux, and yet it still robustly inhibits IL-1β with nanomolar potency.
To further emphasize the complexity of signaling events, Candidalysin permeabilizes the cellular membrane, including that of the TR146 oral epithelial cell line (10). However, presumably calcium influx (and not potassium efflux) contributes to immune activation in these cells, as treatment with the calcium chelator BAPTA-AM [1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)] does, but glyburide does not, impact IL-1β release (33). This could be due to lack of sufficient inflammasome expression in this cell line and mobilization of innate immune responses via mitogen-activated protein kinase signaling (35). In any case, results from this study confirm that THP1 cells are highly dependent on NLRP3 to elicit IL-1β in response to C. albicans or Candidalysin peptide. Moving forward, it will be important to determine the capacity for sulfonylurea drugs to block inflammasome-mediated inflammation in a variety of cell lines and types, including vaginal epithelial cells, if repurposing or future design is sought for efficacy at disparate mucosal sites.
It is striking that nearly all second-generation sulfonylureas (exhibiting greater antidiabetic efficacy than prior iterations) tested in this study displayed activity against both C. albicans- and LPS+ATP-mediated inflammasome activation, whereas first-generation compounds seemingly had no effect. This begs the question of whether structural compound modifications designed to enhance potency and extend half-life also contribute to increased anti-inflammasome function. Although not entirely elucidated, the bulkier para position substitutions of the second-generation compounds relative to the small para position groups of the first-generation compounds appear to play an important role in anti-inflammasome function (Fig. 3). However, the notable absence of activity with the sulfonamide glyburide analog 16673-34-40 indicates that the substituted sulfonylurea group in the antidiabetics also plays a role in their anti-inflammasome function. Interestingly, a prior study showed that anti-inflammasome activity was retained with a different glyburide truncation, substituting a sulfonyl chloride for the sulfonylurea moiety (19). Loss of either the benzamido or sulfonyl group partially reduced the capacity of glyburide to inhibit IL-1β release from BMDM; loss of both groups totally abolished inhibitory activity. Although MCC950 retains the sulfonylurea group relative to the structures of the antidiabetics, the indacene ring adjacent to the urea, compared to the alicyclic portion of the antidiabetics, is essential for its nearly 1,000-fold increase in anti-inflammatory potency. In support of this, work by Hill et al. has demonstrated that replacement of the alicyclic groups of the antidiabetic sulfonylureas with the indacene ring of MCC950 led to increased anti-inflammasome activity while also retaining antidiabetic function (36). Clearly, additional structure-activity studies will be needed to fully elucidate the structural basis for anti-inflammasome activity of this class of compounds.
Interestingly, we found that glimepiride inhibited C. albicans activation of the inflammasome but had no effect when cells were challenged with LPS and ATP. These data suggest that glimepiride affects the fungus itself. It is possible that this drug may somehow modify the fungal cell, making pathogen-associated molecular patterns less optimal or accessible for priming the inflammasome. It is also possible that glimepiride disrupts Candidalysin-mediated damage. However, Candidalysin is strongly linked to hyphal growth, and this process was not disrupted by glimepiride. That said, reduced expression, impaired secretion, or altered intercalation of Candidalysin into the host membrane may all potentially contribute to reduced damage responses. Future work using gene-specific reporters and toxin assays will help elucidate this mechanism.
Obviously, inhibiting inflammation via inflammasome blockade may not be beneficial in the case of disseminated disease, where immune responses are required for containment of microbial pathogens. For example, the use of inflammasome component knockout mice and inflammasome inhibitors (e.g., MCC950) during viral-bacterial coinfection led to reduced inflammation but ultimately resulted in higher bacterial burdens than in WT or untreated mice (37). Additionally, NLRP3 and inflammasome signaling components are required for robust protection against oropharyngeal and systemic candidiasis (20, 38, 39). While inflammasome inhibition may require a delicate balance for optimal efficacy, we feel that such challenges can be leveraged for treatment of certain mucosal infections, such as vaginitis. Targeted and genome-wide association studies have uncovered that NLRP3 is more highly activated in VVC-susceptible patients, and thus, presumably, higher levels of this target exist for inhibition (40). Moreover, vaginitis is a localized, nonlethal infection that rarely (if ever) disseminates from the vaginal lumen, and its symptoms are largely antagonized by the associated inflammatory response. Therefore, inflammasome inhibitors can be delivered locally as topical gels or creams that can exert their action specifically at the mucosal surface to avoid systemic complications. Aside from blocking inflammation, coadministration of effective antifungal drugs would also be required, thus limiting the potential for worsening fungal load in the face of localized immunosuppression.
Collectively, our data contribute to a growing body of literature regarding repurposing of sulfonylurea drugs as potential anti-inflammatory agents and identify both glyburide and MCC950 as potent inhibitors of NLRP3-dependent inflammasome activation by C. albicans and Candidalysin in human macrophage-like cells.
MATERIALS AND METHODS
Growth of microorganisms.
C. albicans reference isolate SC5314, strain BWP17 + cIP30, and ece1Δ/Δ, ece1Δ/Δ + ECE1Δ184–279, and ece1Δ/Δ + ECE1 mutants were maintained in glycerol stocks stored at −80°C (10, 41). A small amount of each stock was spread onto yeast extract-peptone-dextrose (YPD) agar and incubated at 30°C for 48 h to obtain isolated colonies. A single colony was transferred to 1.5 ml of YPD and incubated at 30°C with shaking at 200 rpm for 16 h prior to use.
Growth of cell lines.
WT (THP1-null) and NLRP3−/− (THP1-defNLRP3) THP1 cells (Invivogen) were cultured according to the manufacturer’s protocol in RPMI 1640 medium containing 25 mM HEPES supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin-streptomycin, and 100 μg/ml normocin as described previously (12). THP1 cells were counted on a Countess II FL instrument (Life Technologies) and frozen as aliquots of ∼5 × 106 cells in liquid nitrogen. Upon recovery from cryopreservation, THP1 cells were incubated for 3 days at 37°C, 5% CO2, and 90% humidity in a T25 flask in complete culture medium (RPMI 1640 medium containing 10% heat-inactivated FBS and 100 U/ml penicillin-streptomycin). After 3 days, THP1 cells were counted on the Countess II, assessed for high viability by exclusionary trypan blue staining, and diluted to 5.56 × 105 cells/ml in complete culture medium, and 180-μl aliquots were seeded at a final density of 1 × 105 cells/well in 96-well cell culture-treated polystyrene plates. Phorbol 12-myristate 13-acetate (PMA) (InvivoGen) was added at a final concentration of 100 nM to differentiate cells to a macrophage phenotype, and the cells were incubated at 37°C with 5% CO2 for 24 h.
Preparation of C. albicans.
One-milliliter aliquots of overnight cultures of C. albicans were washed three times in phosphate-buffered saline (PBS) by centrifugation at 8,000 rpm. After resuspending in an equivalent volume of complete cell culture medium (RPMI 1640 medium containing 10% heat-inactivated FBS and 100 U/ml penicillin-streptomycin), cells were diluted 1:100 in sterile water and counted on a hemocytometer.
Preparation of sulfonylurea stocks.
Both approved (glyburide, gliclazide, glimepiride, tolbutamide, tolazamide, glisoxepide, gliquidone, and acetohexamide [Sigma-Aldrich] and chlorpropamide [VWR]) and experimental (MCC950 [Cayman Chemical] and 16673-34-0 [Sigma-Aldrich]) sulfonylurea compounds were purchased from commercial suppliers. Using sterilized instruments, sulfonylureas were prepared as 50 mM working stocks in 100% dimethyl sulfoxide (DMSO). These stocks were further diluted 1:200 in 10 ml of phenol red-free RPMI 1640 medium containing 25 mM HEPES to obtain 250 μM working stocks for initial screening experiments.
Inflammasome activation by C. albicans and inhibition by the sulfonylureas.
Following overnight incubation of THP1 cells, spent culture medium was replaced with 180 μl sulfonylurea working stocks or vehicle (0.5% DMSO) prepared in phenol red-free RPMI medium. Cells were returned to a 37°C CO2 incubator for 1 h prior to challenge. Overnight cultures of C. albicans were prepared as described above and adjusted to 1 × 107 cells/ml, and 20-μl amounts of the suspension were added to wells containing THP1 cells, generating a 2:1 multiplicity of infection (MOI). Mock-infected controls using medium alone supplemented with sulfonylureas or vehicle were also included. Cells were returned to a 37°C CO2 incubator for 4 h to allow for sufficient inflammasome activation. Cells were then gently centrifuged at 200 × g for 2 min to settle the cells/fungi, and 100 μl of culture supernatant was transferred to a polystyrene plate containing 100 μl of prediluted 1× enzyme-linked immunosorbent assay (ELISA)/enzyme-linked immunosorbent spot (ELISPOT) assay buffer (eBioscience) and stored at −20°C. Culture supernatants were assessed for IL-1β using the human Ready-Set-Go! ELISA kit (eBioScience). ELISA optical density values from mock-infected controls were subtracted from those of C. albicans-challenged samples. Experiments were conducted in technical replicates (n = 4) and repeated independently in triplicate. Data are reported as mean values plus standard deviations (SD).
Inflammasome activation by LPS+ATP and inhibition by the sulfonylureas.
Inflammasome activation and sulfonylurea-mediated inhibition were also assessed using a setup identical to the one described above, except that C. albicans was omitted in the challenge step and replaced by 1 μg/ml lipopolysaccharide (Escherichia coli 0111:B4; InvivoGen) prepared in phenol red-free RPMI medium and incubated for 3.5 h, followed by the addition of 5 mM ATP (InvivoGen) 30 min prior to the endpoint. IL-1β ELISA optical density values from mock-activated controls (no ATP) were subtracted from those of LPS+ATP-challenged samples. Experiments were conducted in technical replicates (n = 4) and repeated independently in triplicate. Data are reported as mean values plus SD.
Calculation of IC50s for IL-1β inhibition by the sulfonylureas.
Assays to determine IC50s for sulfonylurea-mediated inhibition of IL-1β release for both C. albicans and LPS+ATP challenge were conducted exactly as described above except that serial dilutions (1:10 for MCC950 and 1:2 for the rest) of sulfonylurea working stocks were prepared in RPMI 1640 containing 25 mM HEPES and 0.5% DMSO. All values were in comparison to the value for the vehicle-treated control and calculated as a percentage of maximum IL-1β release. Concentrations were log transformed and plotted in GraphPad Prism 7.0. IC50s were obtained by using a four-parameter variable slope and best-fit values. Experiments were conducted in technical replicates (n = 4) and repeated independently in triplicate. Data are reported as mean values plus SD.
Hyphal growth assay.
C. albicans cultures were adjusted to 1 × 106 cells/ml in RPMI 1640 medium containing 25 mM HEPES and vehicle alone (0.5% DMSO) or 250 μM each select sulfonylurea. One-milliliter aliquots were placed in 15-ml snap cap tubes and incubated in a 37°C incubator with shaking (200 rpm). Aliquots were removed at 4 and 24 h postinoculation, wet mounts prepared, and images captured by standard light microscopy and a digital camera.
Quantitative growth assay.
C. albicans cultures were adjusted to 1 × 106 cells/ml in 1× complete yeast nitrogen base (YNB) medium containing vehicle alone (0.5% DMSO) or 250 μM each select sulfonylurea. One-milliliter aliquots were placed in 15-ml snap cap tubes and incubated in a 30°C incubator with shaking (200 rpm) for 16 h. Aliquots were removed, serially diluted 1:10 in sterile distilled water, and plated onto YPD agar plates using the drop plate method. After sufficient drying, plates were inverted and placed in a 37°C incubator for 16 h. The following day, fungal burdens were determined by enumeration of resultant CFU. Experiments were repeated in biological triplicate, and results expressed as the mean values plus SD.
THP1 viability assay.
The XTT [2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide salt] reduction assay was used to determine the metabolic activity of THP1 cells after treatment with 250 μM each sulfonylurea (42, 43). Cells were cultivated and treated with sulfonylurea working stocks or vehicle alone (0.5% dimethyl sulfoxide [DMSO]) prepared in phenol red-free medium as described above. An additional treatment group containing vehicle plus 1% SDS was also included as a control for cell death. After treatment with sulfonylureas for 5 h, medium was removed, cells gently washed in sterile PBS, and 200 μl of XTT working reagent (0.5 mg/ml XTT and 1 μM menadione) added for 2 h. Conversion of the XTT substrate to a soluble colored formazan product correlates with cell viability. Plates were gently centrifuged at 200 × g for 2 min to settle the cells, 100 μl of XTT reagent transferred to a fresh polystyrene plate, and the resulting absorbance read at 490 nm. The toxicity of each sulfonylurea or SDS was expressed as the percentage relative to the value for the vehicle-only control. Experiments were conducted using technical replicates (n = 4) and performed in biological triplicates. Data are represented as the mean values plus SD.
Candidalysin challenge of THP1 cells.
THP1 cells were grown and treated with sulfonylureas at their respective IC50s or with vehicle exactly as described for LPS+ATP challenge experiments above. However, instead of the addition of ATP 30 min prior to the experimental endpoint, the Candidalysin peptide (SIIGIIMGILGNIPQVIQIIMSIVKAFKGNK; Peptide Synthetics, UK) was added to each well at a final concentration of 20 μM (13). In some instances, non-LPS-primed cells or no-Candidalysin controls were also included. The IL-1β responses elicited were assessed in culture supernatants by ELISA. Data are expressed as the mean values plus SD.
Statistics and image construction.
All experiments were performed in biological triplicates. ELISA, XTT, and CFU data were compared using one-way analysis of variance (ANOVA) and Dunnet’s post test. Differences were considered significant at a P value of <0.05. All statistical analyses were performed and graphs were composed with GraphPad Prism. Images of hyphal growth were arranged in PowerPoint. Adobe Photoshop version 5.0 was used to process final images for publication quality.
ACKNOWLEDGMENTS
We thank Duncan Wilson (University of Aberdeen) for constructing and providing WT (BWP17 + cIP30) and ece1Δ/Δ, ece1Δ/Δ + ECE1, and ece1Δ/Δ + ECE1Δ184–279 strains.
The present work was funded by the National Institutes of Health, National Institute of Allergy and Infectious Diseases, under grants number R21AI127942 (B.M.P.) and R01AI134796 (B.M.P.).
B.M.P. and D.J.L. designed the study. D.J.L. collected the data. B.M.P., K.E.H., and D.J.L. performed the data analysis and interpreted study results. B.M.P., K.E.H., and D.J.L. wrote the paper. All authors gave approval of the final version to be submitted.
REFERENCES
- 1.Sobel JD. 2007. Vulvovaginal candidosis. Lancet 369:1961–1971. doi: 10.1016/S0140-6736(07)60917-9. [DOI] [PubMed] [Google Scholar]
- 2.Yano J, Sobel JD, Nyirjesy P, Sobel R, Williams VL, Yu Q, Noverr MC, Fidel PL Jr. 2019. Current patient perspectives of vulvovaginal candidiasis: incidence, symptoms, management and post-treatment outcomes. BMC Womens Health 19:48. doi: 10.1186/s12905-019-0748-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sobel JD. 1997. Vaginitis. N Engl J Med 337:1896–1903. doi: 10.1056/NEJM199712253372607. [DOI] [PubMed] [Google Scholar]
- 4.Foxman B, Barlow R, D’Arcy H, Gillespie B, Sobel JD. 2000. Candida vaginitis: self-reported incidence and associated costs. Sex Transm Dis 27:230–235. doi: 10.1097/00007435-200004000-00009. [DOI] [PubMed] [Google Scholar]
- 5.Fidel PL Jr, Barousse M, Espinosa T, Ficarra M, Sturtevant J, Martin DH, Quayle AJ, Dunlap K. 2004. An intravaginal live Candida challenge in humans leads to new hypotheses for the immunopathogenesis of vulvovaginal candidiasis. Infect Immun 72:2939–2946. doi: 10.1128/iai.72.5.2939-2946.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peters BM, Palmer GE, Nash AK, Lilly EA, Fidel PL Jr, Noverr MC. 2014. Fungal morphogenetic pathways are required for the hallmark inflammatory response during Candida albicans vaginitis. Infect Immun 82:532–543. doi: 10.1128/IAI.01417-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yano J, Kolls JK, Happel KI, Wormley F, Wozniak KL, Fidel PL Jr. 2012. The acute neutrophil response mediated by S100 alarmins during vaginal Candida infections is independent of the Th17-pathway. PLoS One 7:e46311. doi: 10.1371/journal.pone.0046311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters BM, Yano J, Noverr MC, Fidel PL Jr. 2014. Candida vaginitis: when opportunism knocks, the host responds. PLoS Pathog 10:e1003965. doi: 10.1371/journal.ppat.1003965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richardson JP, Willems HME, Moyes DL, Shoaie S, Barker KS, Tan SL, Palmer GE, Hube B, Naglik JR, Peters BM. 2018. Candidalysin drives epithelial signaling, neutrophil recruitment, and immunopathology at the vaginal mucosa. Infect Immun 86:e00645-17. doi: 10.1128/IAI.00645-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moyes DL, Wilson D, Richardson JP, Mogavero S, Tang SX, Wernecke J, Hofs S, Gratacap RL, Robbins J, Runglall M, Murciano C, Blagojevic M, Thavaraj S, Forster TM, Hebecker B, Kasper L, Vizcay G, Iancu SI, Kichik N, Hader A, Kurzai O, Luo T, Kruger T, Kniemeyer O, Cota E, Bader O, Wheeler RT, Gutsmann T, Hube B, Naglik JR. 2016. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 532:64–68. doi: 10.1038/nature17625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vande Walle L, Lamkanfi M. 2011. Inflammasomes: caspase-1-activating platforms with critical roles in host defense. Front Microbiol 2:3. doi: 10.3389/fmicb.2011.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willems HME, Lowes DJ, Barker KS, Palmer GE, Peters BM. 2018. Comparative analysis of the capacity of the Candida species to elicit vaginal immunopathology. Infect Immun 86:e00527-18. doi: 10.1128/IAI.00527-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kasper L, König A, Koenig P-A, Gresnigt MS, Westman J, Drummond RA, Lionakis MS, Groß O, Ruland J, Naglik JR, Hube B. 2018. The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nat Commun 9:4260. doi: 10.1038/s41467-018-06607-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogiers O, Frising UC, Kucharikova S, Jabra-Rizk MA, van Loo G, Van Dijck P, Wullaert A. 2019. Candidalysin crucially contributes to Nlrp3 inflammasome activation by Candida albicans hyphae. mBio 10:e02221-18. doi: 10.1128/mBio.02221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruno VM, Shetty AC, Yano J, Fidel PL Jr, Noverr MC, Peters BM. 2015. Transcriptomic analysis of vulvovaginal candidiasis identifies a role for the NLRP3 inflammasome. mBio 6:e00182-15. doi: 10.1128/mBio.00182-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carr PL, Felsenstein D, Friedman RH. 1998. Evaluation and management of vaginitis. J Gen Intern Med 13:335–346. doi: 10.1046/j.1525-1497.1998.00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thule PM, Umpierrez G. 2014. Sulfonylureas: a new look at old therapy. Curr Diab Rep 14:473. doi: 10.1007/s11892-014-0473-5. [DOI] [PubMed] [Google Scholar]
- 18.Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Núñez G, Latz E, Kastner DL, Mills KHG, Masters SL, Schroder K, Cooper MA, O’Neill LAJ. 2015. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21:248–255. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM, Dixit VM. 2009. Glyburide inhibits the cryopyrin/Nalp3 inflammasome. J Cell Biol 187:61–70. doi: 10.1083/jcb.200903124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joly S, Ma N, Sadler JJ, Soll DR, Cassel SL, Sutterwala FS. 2009. Cutting edge: Candida albicans hyphae formation triggers activation of the Nlrp3 inflammasome. J Immunol 183:3578–3581. doi: 10.4049/jimmunol.0901323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parvathaneni V, Kulkarni NS, Muth A, Gupta V. 22 June 2019. Drug repurposing: a promising tool to accelerate the drug discovery process. Drug Discov Today 24:2076–2085. doi: 10.1016/j.drudis.2019.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salazar JJ, Ennis WJ, Koh TJ. 2015. Diabetes medications: impact on inflammation and wound healing. J Diabetes Complications 30:746–752. doi: 10.1016/j.jdiacomp.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koh GC, Maude RR, Schreiber MF, Limmathurotsakul D, Wiersinga WJ, Wuthiekanun V, Lee SJ, Mahavanakul W, Chaowagul W, Chierakul W, White NJ, van der Poll T, Day NP, Dougan G, Peacock SJ. 2011. Glyburide is anti-inflammatory and associated with reduced mortality in melioidosis. Clin Infect Dis 52:717–725. doi: 10.1093/cid/ciq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao J, Kapadia VS, Brown LS, Cheong N, Longoria C, Mija D, Ramgopal M, Mirpuri J, McCurnin DC, Savani RC. 2015. The NLRP3 inflammasome is critically involved in the development of bronchopulmonary dysplasia. Nat Commun 6:8977. doi: 10.1038/ncomms9977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cui W, Zhang S, Cai Z, Hu X, Zhang R, Wang Y, Li N, Chen Z, Zhang G. 2015. The antidiabetic agent glibenclamide protects airway hyperresponsiveness and inflammation in mice. Inflammation 38:835–845. doi: 10.1007/s10753-014-9993-z. [DOI] [PubMed] [Google Scholar]
- 26.Hughes FM Jr, Hill HM, Wood CM, Edmondson AT, Dumas A, Foo WC, Oelsen JM, Rac G, Purves JT. 2016. The NLRP3 inflammasome mediates inflammation produced by bladder outlet obstruction. J Urol 195:1598–1605. doi: 10.1016/j.juro.2015.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdallah DM, Nassar NN, Abd-El-Salam RM. 2011. Glibenclamide ameliorates ischemia-reperfusion injury via modulating oxidative stress and inflammatory mediators in the rat hippocampus. Brain Res 1385:257–262. doi: 10.1016/j.brainres.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 28.Huang K, Gu Y, Hu Y, Ji Z, Wang S, Lin Z, Li X, Xie Z, Pan S. 2015. Glibenclamide improves survival and neurologic outcome after cardiac arrest in rats. Crit Care Med 43:e341–e349. doi: 10.1097/CCM.0000000000001093. [DOI] [PubMed] [Google Scholar]
- 29.Schattling B, Steinbach K, Thies E, Kruse M, Menigoz A, Ufer F, Flockerzi V, Bruck W, Pongs O, Vennekens R, Kneussel M, Freichel M, Merkler D, Friese MA. 2012. TRPM4 cation channel mediates axonal and neuronal degeneration in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med 18:1805–1811. doi: 10.1038/nm.3015. [DOI] [PubMed] [Google Scholar]
- 30.Sheth KN, Simard JM, Elm J, Kronenberg G, Kunte H, Kimberly WT. 2016. Human data supporting glyburide in ischemic stroke. Acta Neurochir Suppl 121:13–18. doi: 10.1007/978-3-319-18497-5_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. 2006. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 32.Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, Chitty JL, Fraser JA, Jennings MP, Robertson AAB, Schroder K. 2019. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol 15:556–559. doi: 10.1038/s41589-019-0277-7. [DOI] [PubMed] [Google Scholar]
- 33.Ho J, Yang X, Nikou SA, Kichik N, Donkin A, Ponde NO, Richardson JP, Gratacap RL, Archambault LS, Zwirner CP, Murciano C, Henley-Smith R, Thavaraj S, Tynan CJ, Gaffen SL, Hube B, Wheeler RT, Moyes DL, Naglik JR. 2019. Candidalysin activates innate epithelial immune responses via epidermal growth factor receptor. Nat Commun 10:2297. doi: 10.1038/s41467-019-09915-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swidergall M, Khalaji M, Solis NV, Moyes DL, Drummond RA, Hube B, Lionakis MS, Murdoch C, Filler SG, Naglik JR. 2019. Candidalysin is required for neutrophil recruitment and virulence during systemic Candida albicans infection. J Infect Dis 220:1477–1488. doi: 10.1093/infdis/jiz322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moyes DL, Murciano C, Runglall M, Kohli A, Islam A, Naglik JR. 2012. Activation of MAPK/c-Fos induced responses in oral epithelial cells is specific to Candida albicans and Candida dubliniensis hyphae. Med Microbiol Immunol 201:93–101. doi: 10.1007/s00430-011-0209-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hill JR, Coll RC, Sue N, Reid JC, Dou J, Holley CL, Pelingon R, Dickinson JB, Biden TJ, Schroder K, Cooper MA, Robertson A. 2017. Sulfonylureas as concomitant insulin secretagogues and NLRP3 inflammasome inhibitors. ChemMedChem 12:1449–1457. doi: 10.1002/cmdc.201700270. [DOI] [PubMed] [Google Scholar]
- 37.Robinson KM, Ramanan K, Clay ME, McHugh KJ, Pilewski MJ, Nickolich KL, Corey C, Shiva S, Wang J, Muzumdar R, Alcorn JF. 2018. The inflammasome potentiates influenza/Staphylococcus aureus superinfection in mice. JCI Insight 3:97470. doi: 10.1172/jci.insight.97470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, Fitzgerald KA. 2009. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 5:487–497. doi: 10.1016/j.chom.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van de Veerdonk FL, Joosten LA, Shaw PJ, Smeekens SP, Malireddi RK, van der Meer JW, Kullberg BJ, Netea MG, Kanneganti TD. 2011. The inflammasome drives protective Th1 and Th17 cellular responses in disseminated candidiasis. Eur J Immunol 41:2260–2268. doi: 10.1002/eji.201041226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roselletti E, Perito S, Gabrielli E, Mencacci A, Pericolini E, Sabbatini S, Cassone A, Vecchiarelli A. 2017. NLRP3 inflammasome is a key player in human vulvovaginal disease caused by Candida albicans. Sci Rep 7:17877. doi: 10.1038/s41598-017-17649-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gillum AM, Tsay EY, Kirsch DR. 1984. Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol Gen Genet 198:179–182. doi: 10.1007/bf00328721. [DOI] [PubMed] [Google Scholar]
- 42.Pierce CG, Uppuluri P, Tristan AR, Wormley FL Jr, Mowat E, Ramage G, Lopez-Ribot JL. 2008. A simple and reproducible 96-well plate-based method for the formation of fungal biofilms and its application to antifungal susceptibility testing. Nat Protoc 3:1494–1500. doi: 10.1038/nprot.2008.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roehm NW, Rodgers GH, Hatfield SM, Glasebrook AL. 1991. An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J Immunol Methods 142:257–265. doi: 10.1016/0022-1759(91)90114-u. [DOI] [PubMed] [Google Scholar]