

Abstract
Factors secreted from tumors/tumor cells are hypothesized to cause skeletal muscle wasting in cancer patients. We examined whether cancer cells secrete factors to promote atrophy by evaluating the effects of conditioned media (CM) from murine lung cancer cells and primary cultures of human lung tumor cells on cultured myotubes. We evaluated murine Lewis lung carcinoma (LLC) and KRASG12D cells, and primary cell lines derived from tumor biopsies from patients with lung cancer (hTCM; n = 6). In all experiments, serum content was matched across treatment groups. We hypothesized that CM from murine and human tumor cells would reduce myotube myosin content, decrease mitochondrial content, and increase mitochondrial reactive oxygen species (ROS) production. Treatment of myotubes differentiated for 7 days with CM from LLC and KRASG12D cells did not alter any of these variables. Effects of murine tumor cell CM were observed when myotubes differentiated for 4 days were treated with tumor cell CM and compared with undiluted differentiation media. However, these effects were not apparent if tumor cell CM treatments were compared with control cell CM or dilution controls. Finally, CM from human lung tumor primary cell lines did not modify myosin content or mitochondrial content or ROS production compared with either undiluted differentiated media, control cell CM, or dilution controls. Our results do not support the hypothesis that factors released from cultured lung cancer/tumor cells promote myotube wasting or mitochondrial abnormalities, but we cannot dismiss the possibility that these cells could secrete such factors in vivo within the native tumor microenvironment.
Keywords: conditioned media, human, murine, myotubes, tumor
INTRODUCTION
Cancer cachexia is a syndrome characterized by unintentional loss of fat and skeletal muscle tissue (19) that affects ~50% of patients (18, 62). Patients with solid tumors are most likely to experience muscle atrophy, particularly in advanced stages (44). Muscle wasting increases treatment side effects (15, 24, 57), decreases response to chemotherapy (31, 35, 55), and increases mortality (17). Despite these deleterious consequences, the causes of cancer cachexia remain unclear.
How tumors that are anatomically distal to skeletal muscle cause skeletal muscle atrophy is unknown. One long-standing theory holds that factors released from tumor cells promote muscle wasting through direct effects on muscle. Data supporting this theory comes primarily from preclinical models (2, 39, 49). Although these studies have provided a wealth of mechanistic data, their translatability to human cancer remains questionable. In animal models, tumors grow rapidly and comprise a large fraction of body mass (7). For instance, in the C26 xenoplant model, the onset of cachexia occurs when the tumor reaches ~5% of body mass (58), which occurs 2–4 wk after inoculation (10, 32). Extrapolated to humans, the size of this tumor would equate to an ~8-lb (3.6 kg) tumor in men and an ~6 lb (2.7 kg)-tumor in women. Because of the large tumor size, the pathoetiology of cachexia in these preclinical models may skew disproportionately toward tumor-derived factors. In contrast, human tumors develop relatively slower and comprise <<1% of body mass.
A common model used to test for effects of tumor-derived factors on skeletal muscle involves treating cultured muscle cells with conditioned media (CM) from cancer/tumor cells (11, 22, 54, 60, 61). CM experiments provide a focused model to examine the direct effects of tumor/cancer cell-derived factors on skeletal muscle. Data from these approaches using various cell lines support the notion that tumor-/cancer cell-derived factors promote skeletal muscle atrophy (40, 65, 66, 71). In the present study, we sought to use this model to examine the effects of lung cancer cell-derived factors on cultured murine myotubes. We chose lung cancer because patients experience a high prevalence of cachexia (6) and because lung cancer is a common model used for preclinical cancer cachexia studies. We utilized CM from two murine lung tumor cell lines: the Lewis lung carcinoma (LLC) and the KRASG12D cell lines. The former potently induces cachexia in mice (50, 71) and is used widely. The latter contains the most frequently occurring mutation in human non-small-cell lung tumors (64) and provides a near genetically identical cell line for control studies. Additionally, we report, for the first time to our knowledge, the effects of CM from primary cell lines derived from clinical biopsies of human lung tumors. We tested the effects of CM from these cells on differentiated C2C12 murine myotubes. Myotubes were differentiated for 7 days before treatment with CM, a time when they develop anatomical and physiological attributes similar to in vivo skeletal muscle and demonstrate atrophy in response to myotoxic chemotherapeutics, such as anthracyclines (25, 26). On the basis of prior work in the field using these and other cell lines, we hypothesized that CM from murine and human tumor cells would reduce myotube myosin content, decrease mitochondrial content, and increase mitochondrial reactive oxygen species (ROS) production.
METHODS
Cell culture.
C2C12 myoblasts (CRL1772; ATCC, Manassas, VA) were cultured in low glucose (1 g/L), Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Gibco Thermo Fisher Scientific, Waltham, MA) and antibiotics (50 U/mL penicillin and 50 μg/mL streptomycin). Cells were plated (2 × 104 cells/cm2) on Matrigel (60 μg/cm2; Corning, Bedford, MA) and switched to differentiation medium (DM) of low serum (1% heat-inactivated FBS), high glucose (4.5 g/L) DMEM when they reached 90–100% confluence to induce differentiation, as described (34). Each experiment contained at least three technical replicates, and each experiment was independently replicated two to three times.
LLC (ATCC CRL-1642) cells, a murine lung cancer cell line that promotes cachexia in mice (48), were cultured, as described (22). Briefly, 2 × 105 LLC cells were plated on 10-cm-diameter culture dishes in DMEM supplemented with 10% FBS, 50 U/mL penicillin, and 50 μg/mL streptomycin and grown for 48 h to a final density of 0.8–11 × 106 cells per culture dish. NL20 (ATCC CRL-2503) cells were used as a control cell line for LLC cells, as they are nontumorigenic and have been used as controls for LLC CM treatment of C2C12 myotubes previously (72). NL20 cells were grown as described (22). Briefly, 2 × 105 NL20 cells were plated into 10-cm-diameter culture dishes in DMEM supplemented with 10% FBS, 50 U/mL penicillin, and 50 μg/mL streptomycin and grown for 48 h to a final density of 0.8–11 × 106 cells per dish. For both LLC and NL20 cells, CM from this 48-h growth period was collected, centrifuged to remove cell debris, and stored at −80°C until use.
Primary mouse tracheal epithelial cells (MTECs) were isolated from 14-wk-old male and female LSL-KRASG12D (The Jackson Laboratory, no. 008179), as described (1). The LSL-KRASG12D (30) mice were generated by crossing CCSP-rtTA (The Jackson Laboratory, CCSP-rtTA no. 006222) and TetO7-Cre (The Jackson Laboratory no. 006224) bitransgenic mice, to create CCSP-rtTA/TetO-Cre/LSL-KRASG12D mice. Cells were cultured on collagen-coated plates in DMEM-F-12 medium containing 10 μg/mL cholera toxin (Sigma), 2 mg/mL insulin (Roche), 2.5 mg/mL transferrin (Sigma), 12.5 mg/mL bovine pituitary extract (Invitrogen), 10 μg/mL epithelial growth factor (Calbiochem), 50 μM dexamethasone (Sigma), U/50 μg/mL penicillin-streptomycin (Gibco), 4.5 mM l-glutamine (Invitrogen), and 1 mL Primocin (Invitrogen), as described (1). MTECs were treated with either a Cre recombinant adenovirus to activate the oncogenic KRAS mutation (KrasG12D) or an empty vector (KrasWT), grown until confluent in a T75 flask, washed, and cultured in serum-free medium for 2 days and then the medium was collected, centrifuged to remove cell debris, and stored at −80°C until use. KrasWT cells served as a control cell line. KrasG12D cells were validated with PCR, along with the observation that KrasWT cells do not survive passaging, whereas KrasG12D cells do.
To obtain human tumor cell CM, lung tumor biopsies from patients were taken with endobronchial ultrasound-guided transbronchial needle aspiration (EBUS-TBNA) sampling or percutaneous biopsy and were used to establish primary cultures, as described (45), with modifications. Briefly, tissue obtained from the biopsy was treated with elastase and DNAse and then cultured in RPMI 1640 supplemented with l-glutamine and HEPES on collagen-coated dishes. Primary broncheal/tracheal endothelial cells (HBEC; ATCC no. PCS-300-010) (2.5 × 105 cells/cm2) were used as a nontumorigenic, control cell line. They were cultured as adherent monolayers in minimum essential medium (MEM; Invitrogen) supplemented with 9% FBS (Invitrogen), 2 mM l-glutamine, 100 U penicillin and 100 μg/mL streptomycin (Sigma-Aldrich). For both primary tumor and HBEC cells, CM was collected following 24-h incubation in RPMI 1640 + 9% FBS, centrifuged to remove cell debris, and stored at −80°C until use.
Preparation of CM and dilution controls.
One volume of tumor or control cell CM was mixed with three volumes of serum-free DMEM to treat myotubes. In all cases, serum content was standardized so that tumor and control cell CM and untreated/undiluted DM treatments had the same final serum concentration (2.5% for LLC and NL20 cells, 1% for KrasG12D and KrasWT cells, and 2.25% for human tumor and HBEC cells). We also included a dilution control to our experiments, which consisted of DM diluted 1:3 with Hank’s balanced salt solution (HBSS; 1 g/L glucose). As with other treatments/controls, dilution controls contained the same serum concentration as the respective treatments listed above. Thus, we had four treatment groups in each experiment: untreated/undiluted controls (i.e., no CM added and no dilution), dilution control (1:3 dilution of HBSS/DM), nontumorigenic cell CM control (1:3 dilution of CM/DM), and cancer/tumor cell CM (1:3 dilution of CM/DM). In all conditions, media were changed daily during the 3-day treatment period.
Patients.
Patients (76 ± 7 yr; Table 1 for details) with known or suspected lung cancer were recruited from the University of Vermont Medical Center Lung Multidisciplinary Clinic. Written informed consent was obtained from all volunteers before their participation, and protocols were approved by the Committees on Human Research at the University of Vermont. Tumor cells were obtained when patients underwent either clinically indicated bronchoscopy or percutaneous biopsy. Following on-site cytopathological diagnosis of cancer, one additional fine needle aspiration was performed to obtain cells.
Table 1.
Physical and disease characteristics in cancer patients
| Means ± SE | |
|---|---|
| n | 6 |
| Age, yr | 69 ± 11 |
| Sex (M/F) | 2/4 |
| BMI, kg/m2 | 26.9 ± 7 |
| Patients reporting weight loss n, kg | 1 (6.4) |
| Cancer stage (n for I/II/III/IV) | 2/1/1/2 |
| Diagnosis (n) | |
| NSCLC | 5 |
| SCLC | 1 |
| Histology (n) | |
| Adenocarcinoma | 4 |
| Basaloid squamous cell | 2 |
| Neuroendocrine | 1 |
Data represent number of patients (n) or means ± SE. NSCLS, non-small-cell lung cancer; SCLC, small-cell lung cancer.
Protein expression.
Protein expression was measured by Western blot. Myotubes were washed with phosphate-buffered saline, lysed [50 mM Tris, 150 mM NaCl, 10% (vol/vol) glycerol, 0.5% IGEPAL CA-630, 1 mM EDTA containing protease inhibitor cocktail (1:100, cat. no. P8340, Sigma) and phosphatase inhibitor cocktail 3 (1:100, cat. no. P0044, Sigma)], incubated on ice for 30 min, and then centrifuged at 14,000 g at 4°C for 10 min. Lysate protein contents were measured (Bio-Rad DC Protein Assay, Hercules, CA) and diluted in sample prep buffer (62.5 mM Tris·HCl, pH 6.8, 10% glycerol, 1% SDS 0.005% bromophenol blue, 5% 2-mercaptoethanol). Proteins were separated by SDS-PAGE (Bio-Rad), transferred to polyvinylidene difluride membranes, and blocked with TBST buffer (150 mM NaCl, 0.05% Tween-20, and 20 mM Tris·HCl, pH 7.4) containing 5% nonfatty milk or BSA. After blocking, membranes were incubated overnight with myosin primary antibody (1:20,000, Sigma M4276, RRID AB_477190) and GAPDH (1:15,000, Bio-Rad, RRID AB_1720065). Membranes were washed for 30 min in TBST and then incubated for 1 h at room temperature in 5% milk-TBST containing anti-mouse IgG-peroxidase (1:15,000, Sigma A2304, RRID AB_257993) secondary antibody with Clarity Western ECL Substrate (Bio-Rad).
Myotube mitochondrial content and reactive oxygen species production.
Mitochondrial content and reactive oxygen species (ROS) production were measured with fluorometric dyes, as described (25). Briefly, C2C12 myotubes grown in black-walled 96-well plates were loaded with fluorescent dyes to assess mitochondrial content (1 μM MitoTracker Green FM; 490/516 nm) and ROS production (1 μM MitoSOX Red; 510/580 nm; both Molecular Probes, Eugene, OR) 15 min before measurement. Fluorescence was measured for 15 min in the basal condition on a microplate reader (BioTek, Winooski, VT), and the MitoSox signal was expressed relative to MitoTracker signal to control ROS production for mitochondrial content.
Myotube size.
Myotube size was assessed from myotube diameter measurements. Digital images of myotube cultures were acquired at ×4 magnification. Average diameters (5 diameters per tube) of n = 10–25 myotubes per field from n = 2 random fields per well on n = 6 replicate wells per group were measured using ImageJ software (National Institutes of Health, Frederick, MD) by an assessor blinded to treatment status.
Myotube oxygen consumption rate.
The oxygen consumption rate (OCR) of C2C12 myotubes was measured using a Seahorse XF96 analyzer (Seahorse Biosciences, North Billerica, MA). Briefly, 1 × 104 C2C12 cells per well were seeded in a Seahorse XFe96- or XF24-well cell culture microplate, as described above and differentiated for 7 days. At day 7 (d7), Myotubes were treated with tumor CM or control cell CM or were diluted with HBSS, as detailed above, for 3 days. Thereafter, culture medium was removed; cells were washed twice with HBSS, and media were replaced with Seahorse assay medium supplemented with (10 mM sodium pyruvate and 10 mM glucose, pH 7.4). This medium was used as a diluent for compounds injected into the culture dishes during measurements. Plates were equilibrated for 1 h at 37°C with no CO2 before they were transferred into the XFe96 or XF24 analyzer. After the basal OCR was measured, the analyzer sequentially injected the following compounds (final concentration): oligomycin (4 μM), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP; 4 μM), and rotenone-antimycin A (4 μM each) (all from Sigma), with each well measured in triplicate.
Statistics.
Analysis of variance (ANOVA) was used to compare treatments using GraphPad Prism 8.2.0 (San Diego, CA). A P value of <0.05 was used as the threshold for statistical significance. All data are presented as means ± SE unless otherwise specified.
RESULTS
Characteristics of C2C12 myotube cultures.
C2C12 myotubes were differentiated for 7 days before treatment with CM. Under our culture conditions, there was rapid growth and accumulation of myosin between d3 and d7, followed by a relative plateauing of myotube myosin content and diameter (26). At d7, myotubes expressed myofilament proteins, including myosin, actin, and α-actinin, and organized these proteins into myofilaments (26). Myofilaments and components of the excitation-contraction coupling system were functional at this time, as electrical field stimulation caused intracellular Ca2+ cycling and contraction, which could be prevented by pharmacological inhibition of excitation contraction coupling with the sodium channel blocker tetrodotoxin or inhibitors of myosin-actin ATPase (26). Thus, at this stage of differentiation, myotubes exhibited numerous structural and functional hallmark characteristics of in vivo muscle.
LLC and Kras CM.
We began by testing the effects of LLC CM, as this cell line is widely used to initiate tumors that promote cachexia in mouse xenoplant models (4, 8, 11). In C2C12 myotubes differentiated for 7 days, we found no differences in myosin content between untreated myotubes (i.e., nothing added to DM; labeled CTRL in figure) and LLC CM, HBSS dilution control (DIL CTRL) and NL20 control cell CM (Fig. 1A). To verify that myosin content measures reflected changes in myotube size, we compared diameters of 7d myotubes in LLC CM and untreated myotube groups. As observed with myosin content measures, there were no differences between these two groups (17.6 ± 0.3 vs. 18.3 ± 0.4 µm; n = 252 and 311 myotubes, respectively). In contrast to LLC, we found an effect of KrasG12D CM on myotube myosin content (Fig. 1B). However, it paradoxically increased myosin content compared with all control conditions (P = 0.002). Here again, we evaluated myotube diameters in KrasG12D and untreated control groups to validate myosin content measurements. In agreement with myosin measurements, we found an increase in the diameter of the KrasG12D compared with untreated controls (16.7 ± 0.4 vs. 14.8 ± 0.4 µm, P < 0.01; n = 276 and 281 myotubes, respectively).
Fig. 1.
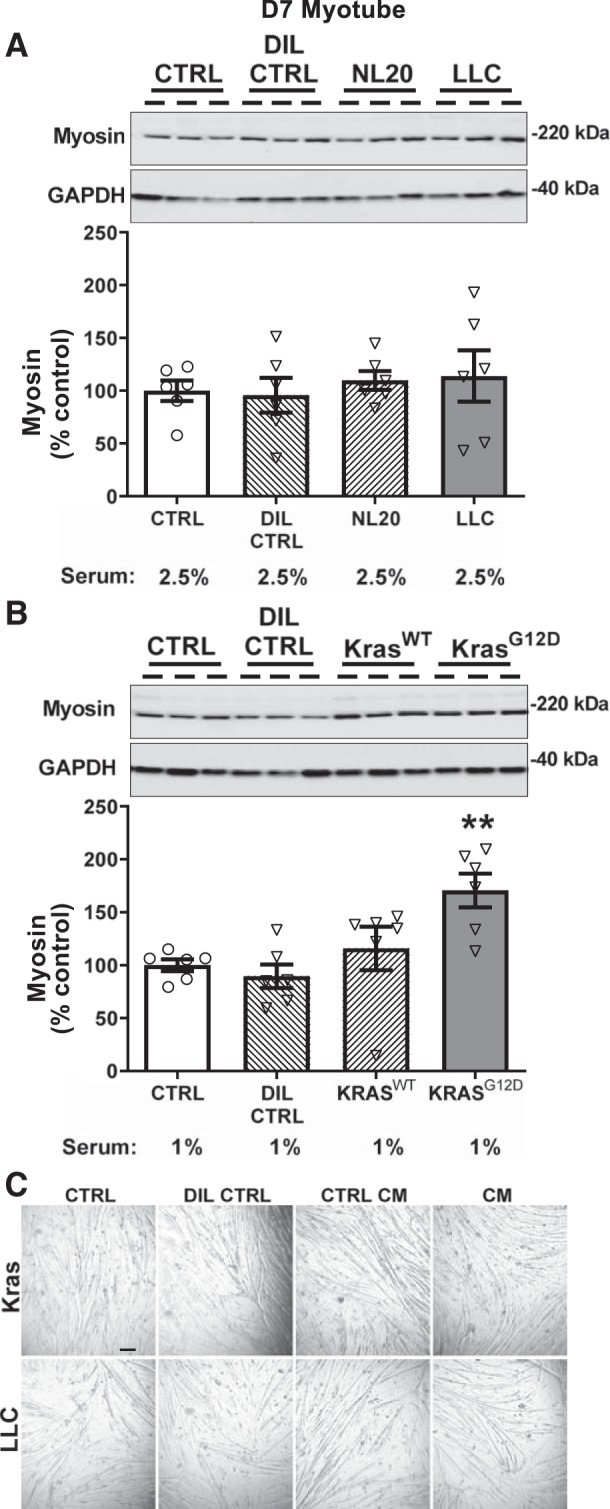
Effects of 3 days of murine Lewis lung carcinoma (LLC) cells (A, gray bar) or (KrasG12D; B, gray bar) conditioned medium (CM; 1:3) treatment on myosin content in day 7 (d7) differentiated myotubes compared with untreated/undiluted (CTRL; open bars), dilution (DIL CTRL; 1:3; hatched bar), and control cell CM (NL20 and KrasWT cells; 1:3; hatched bars) controls. A: no effect of LLC CM was found compared with CTRL, NL20 CM, or DIL CTRL (n = 6/bar for all conditions). B: KrasG12D CM increased myotube myosin content compared with CTRL, and both KrasWT and DIL CTRL (n = 6/bar for all conditions). ANOVA was used to test for group effects. If a group effect was found, post hoc contrasts were used to test for location of differences between groups. C: representative wide-field images of myotubes at day 10 post-differentiation following 3 days of treatment with CTRL, DIL CTRL, or control/cancer cell CM (scale bar, 100 µm). Representative gel images for myosin are shown at the top of A and B for a subset of replicates with corresponding GAPDH bands as a loading control. Serum content was similar in all treatments and is listed below bar labels. All data are means ± SE, with individual data points shown with each bar. **P < 0.01, greater than CTRL, DIL CTRL, and KrasWT.
Timing of CM application.
Because the results above disagreed with numerous reports in the literature and our initial hypotheses, we sought to evaluate the cause of these divergent findings. One notable difference between our protocol and the majority of studies in the field is that we differentiated myotubes for 7 days before CM application, whereas most prior studies had treated myotubes 4 or 5 days post-differentiation. To examine whether the timing of treatment explained our inability to observe effects of CM, we examined the effects of CM treatment of myotubes starting at d4 post-differentiation. Under these conditions, we observed a 32% reduction in myosin content (P < 0.05) in response to LLC CM compared with untreated control (Fig. 2A). Importantly, however, a similar reduction in myosin content was noted with CM from the nontumorigenic NL20 cell line (33%, P < 0.05), and a trend for lower myosin content was observed in the DIL CTRL group (P = 0.08) compared with untreated controls (Fig. 2A). Treating myotubes with Kras CM starting at d4 post-differentiation, we found a 36% reduction in myosin content with KrasG12D CM (P < 0.05). Similar to the data with LLC CM, however, we also observed a reduction of 38% in myosin content with CM from KrasWT cells (P < 0.05) and a 39% reduction with the DIL CTRL (P < 0.05) compared with untreated controls (Fig. 2B). Collectively, and considered in the context of growth curves for C2C12 myotubes in our laboratory (26), the effects of murine lung cancer CM observed on d4 myotubes appear to be due to growth inhibition.
Fig. 2.
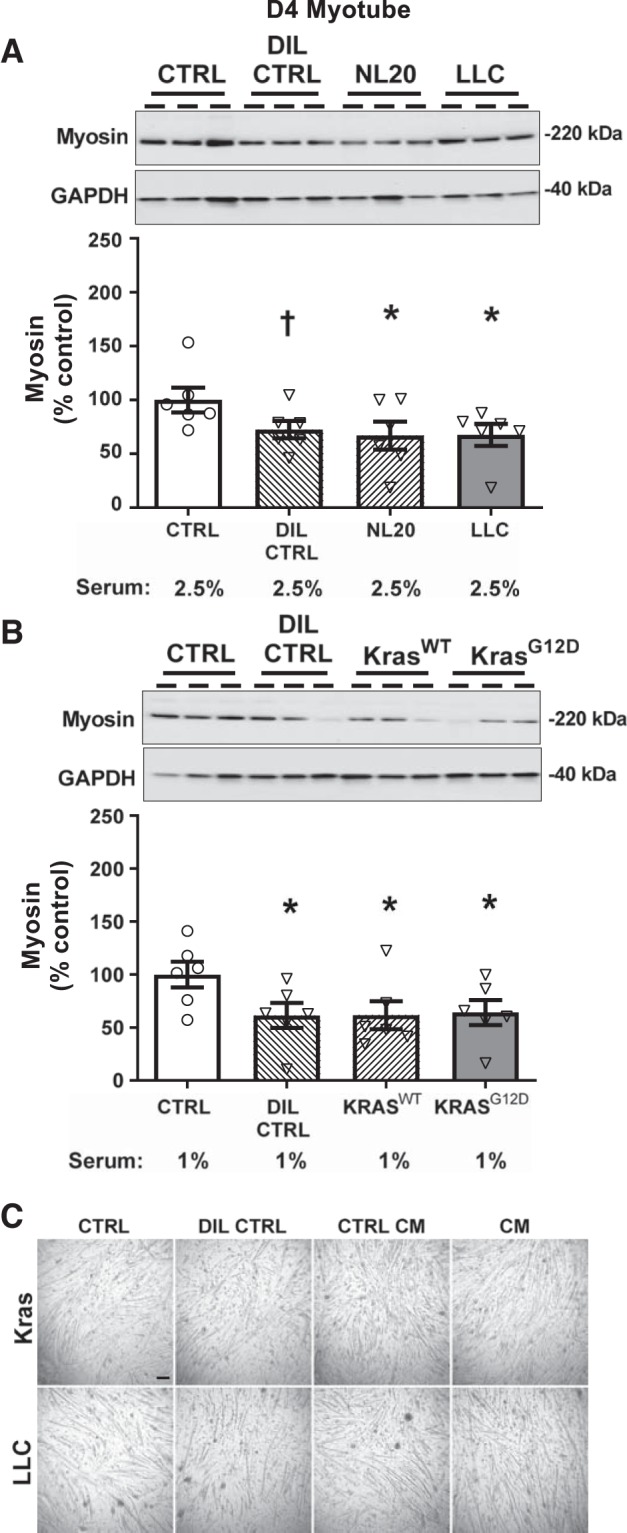
Effects of 3 days of murine LLC (A; gray bar) or KrasG12D (B; gray bar) conditioned medium (CM; 1:3) treatment on myosin content in day 4 (d4) differentiated myotubes compared with untreated/undiluted (CTRL; open bars), dilution (DIL CTRL; 1:3; hatched bars), and control cell CM (NL20 and KrasWT cells; 1:3; hatched bars) controls. A: LLC and NL20 CM reduced, and DIL CTRL tended to reduce, myotube myosin content compared with CTRL (n = 6/bar for all conditions). B: KrasG12D and KrasWT CM and DIL CTRL reduced myotube myosin content compared with CTRL (n = 6/bar for all conditions). ANOVA was used to test for group effects. If a group effect was found, post hoc contrasts were used to test for location of differences between groups. C: representative wide-field images of myotubes at day 4 post-differentiation following 3 days of treatment with CTRL, DIL CTRL, or control/cancer cell CM (scale bar, 100 µm). Representative gel images for myosin are shown at the top of A and B for a subset of replicates with corresponding GAPDH bands as a loading control. Serum content was similar in all treatments and is listed below bar labels All data are means ± SE, with individual data points shown with each bar. *P < 0.05, lower than CTRL; †P < 0.10, lower than CTRL.
Mitochondrial content, ROS production, and mitochondrial respiration with murine CM.
Recent reports have suggested a role for oxidant stress in the atrophic effect of tumor-related factors (3, 16, 20, 33, 62). Thus, we examined the effects of CM on mitochondrial content and ROS production. LLC treatment of myotubes 7 days post-differentiation did not alter mitochondrial ROS production (Fig. 3A), nor did treatment with any of the control conditions. Mitochondrial content was also unchanged with DIL CTRL, NL20, or LLC CM treatment (Fig. 3B). With Kras CM, using d7 post-differentiation myotubes, there were no changes in ROS production compared with untreated controls (Fig. 3C), but both KrasG12D (P < 0.01) and KrasWT (P < 0.01) CM increased mitochondrial content (Fig. 3D).
Fig. 3.

Effects of 3 days of murine LLC (A and B; gray bar) or KrasG12D (C and D; gray bar) conditioned medium (CM; 1:3) treatment on mitochondrial reactive oxygen species (ROS) production and mitochondrial content in day 7 (d7) differentiated myotubes compared with untreated/undiluted (CTRL; open bars), dilution (DIL CTRL; 1:3; hatched bar), and control cell CM (NL20 and KrasWT cells; 1:3; hatched bars) controls. No differences were found among LLC, NL20 CM, DIL CTRL, or CTRL conditions for mitochondrial ROS (A) or content (B); n = 12/bar for all conditions. C: no differences were found among KrasG12D, KrasWT CM, DIL CTRL, or CTRL for mitochondrial ROS; n = 12/bar for all conditions. D: both KrasG12D and KrasWT CM increased mitochondrial content compared with DIL CTRL and CTRL conditions; n = 12/bar for all conditions. Serum content was similar across conditions within each experiment, as noted in Figs. 1 and 2. ANOVA was used to test for group effects. If a group effect was found, post hoc contrasts were used to test for location of differences between groups. All data are means ± SE, with individual data points shown with each bar. **P < 0.01, greater than CTRL and DIL CTRL.
As with myotube myosin content, we also examined the effects of LLC and Kras CM on d4 myotubes. With LLC CM., no changes in ROS were found (Fig. 4A). However, mitochondrial content was elevated with NL20 CM (P < 0.05; Fig. 4B). Kras CM increased ROS production in d4 myotubes with both KrasG12D (P < 0.01) and KrasWT (P < 0.01) CM (Fig. 4C) but not with DIL CTRL. Mitochondrial content was also increased by KrasG12D (P < 0.01) and KrasWT (P < 0.01) CM (Fig. 4D) relative to untreated controls.
Fig. 4.

Effects of 3 days of murine LLC (A and B; gray bar) or KrasG12D (C and D; gray bar) conditioned medium (CM; 1:3) treatment on mitochondrial reactive oxygen species (ROS) production and mitochondrial content in day 4 (d4) differentiated myotubes compared with untreated/undiluted (CTRL; open bars), dilution (DIL CTRL; 1:3; hatched bar), and control cell CM (NL20 and KrasWT cells; 1:3; hatched bars) controls. No differences were found among LLC, NL20 CM, DIL CTRL, and CTRL conditions for mitochondrial ROS; n = 12/bar for all conditions. B: NL20 CM increased mitochondrial content compared with LLC, DIL CTRL, or CTRL conditions; n = 12/bar for all conditions. Both KrasG12D and KrasWT CM increased mitochondrial ROS production (C) and content (D) compared with DIL CTRL or CTRL conditions; n = 12/bar. Serum content was similar across conditions within each experiment, as noted in Figs. 1 and 2. ANOVA was used to test for group effects. If a group effect was found, post hoc contrasts were used to test for location of differences between groups. All data are means ± SE, with individual data points shown with each bar. *P < 0.05, greater than CTRL, **P < 0.01, greater than CTRL and DIL CTRL.
To clarify whether there was any effect of murine tumor CM to impair mitochondrial function of d7 differentiated myotubes, we measured myotube respiration 3 days following CM or control conditions. We chose d7, rather than d4, myotubes because this was when dilution effects (i.e., lower myosin content) were not apparent. For the LLC-treated group relative to untreated controls, we found no effect on basal respiration (LLC, 105 ± 12%; NL20, 105 ± 2%; DIL CTRL, 99 ± 21%), maximal respiration (LLC, 104 ± 15%; NL20, 110 ± 2%; DIL CTRL, 96 ± 24%), calculated ATP production (i.e., basal respiration minus proton leak and nonmitochondrial oxygen consumption; LLC, 102 ± 3%; NL20, 92 ± 5%; DIL CTRL, 101 ± 7%) or calculated spare respiratory capacity (maximal respiration minus basal respiration; LLC, 104 ± 15%; NL20, 112 ± 3%; DIL CTRL, 98 ± 24%). For Kras CM, we similarly found no effect on basal respiration (KrasWT, 125 ± 22%; KrasG12D, 117 ± 10%; DIL CTRL, 115 ± 17%), maximal respiration (KrasWT, 105 ± 21%; KrasG12D, 98 ± 7%; DIL CTRL, 93 ± 12%), calculated ATP production (KrasWT, 122 ± 20%; KrasG12D, 123 ± 10%; DIL CTRL, 117 ± 13%) or calculated spare respiratory capacity (KrasWT, 94 ± 22%; KrasG12D, 86 ± 5%; DIL CTRL, 81 ± 9%). Moreover, none of the other measured or calculated variables available via this analysis differed between CM treatments and controls (data not shown). These results further support the lack of effects of CM on mitochondrial function that could contribute to lower myosin content and atrophy through energetic stress signaling pathways.
Myotube myosin content with human CM.
Informed by the results of LLC and Kras studies, we decided to test CM from patient lung cancer biopsies on d7 myotubes, since we reasoned that effects of CM observed on d4 myotubes were due to growth inhibition. We treated d7 myotubes with patient tumor CM from six patients (hTCM), control cell CM (HBEC), DIL CTRL, and untreated controls (CTRL) for 3 days. Individual patient data are represented in figures by different symbols of different color to demonstrate variation in myosin response in different patient tumor cell CM. As with murine tumor CM experiments, serum content was constant among all groups. We found no effect of patient lung tumor cell CM on myosin content compared with untreated controls nor any effect of DIL CTRL or HBEC CM (Fig. 5A). Similarly, we found no effect of patient lung tumor cell CM, HBEC CM, or DIL CTRL compared with untreated controls to alter mitochondrial ROS production (Fig. 5B). However, we did observe an effect of hTCM (+19%, P < 0.05) and HBEC CM (+16%, P < 0.05) to increase mitochondrial content compared with untreated controls, but no effect of DIL CTRL (Fig. 5C).
Fig. 5.
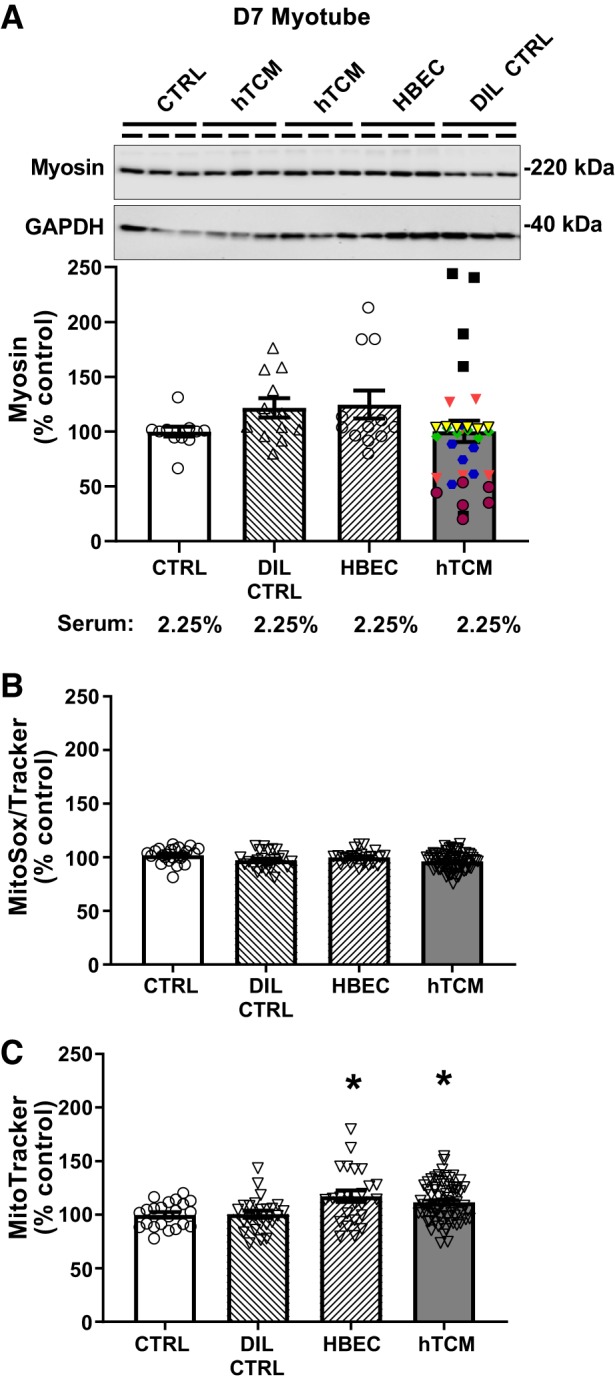
Effects of 3 days of treatment with primary human tumor cell conditioned medium (hTCM; 1:3; gray bars; n = 6 patients) on myosin protein content (A) and mitochondrial reactive oxygen species (ROS) production (B) and content (C) in day 7 (d7) differentiated myotubes compared with untreated/undiluted (CTRL), dilution (DIL CTRL; hatched bars), and control primary human bronchial epithelial cell CM (HBEC; 1:3 dilution; hatched bars) controls. No differences were found between treatments for myotube myosin content (n = 12/bar for CTRL, DIL CTRL, HBEC, and n = 36/bar for hTCM; A) or mitochondrial ROS production; (n = 24/bar for CTRL, DIL CTRL, HBEC and n = 72/bar for hTCM; B), with data for individual patients for myosin data shown using different symbols of different color. C: both hTCM and HBEC increased mitochondrial content compared with DIL CTRL and CTRL (n = 24/bar for CTRL, DIL CTRL, HBEC and n = 72/bar for hTCM). Representative gel images for myosin are shown at the top of A for a subset of replicates with corresponding GAPDH bands as a loading control. Serum content was similar across conditions within each experiment. ANOVA was used to test for group effects. If a group effect was found, post hoc contrasts were used to test for the location of differences between groups. All data are means ± SE, with individual data points shown with each bar. *P < 0.05, greater than CTRL and DIL CTRL.
DISCUSSION
Conventional wisdom holds that tumors release soluble factors that act on distal skeletal muscle tissue to cause atrophy and other metabolic derangements in cancer patients. We evaluated this proposition by testing the effects of murine and human lung cancer/tumor cell CM on C2C12 myotube cultures, a common model to test for cancer/tumor cell-derived atrophic factors (11, 37, 38, 40, 46, 53, 70). Our studies included controls to account for dilution of media and factors released into media by growing nontumorigenic/cachexia-inducing cells and evaluated the effects of timing of CM application relative to the start of differentiation. Additionally, we matched serum content across all treatment and control conditions. When we examined the effects of murine lung cancer cell CM 7 days post-differentiation, when myotubes express structural and functional phenotypes common to in vivo skeletal muscle (26), we found no atrophic effects of CM on myotubes. Murine lung cancer cell CM reduced myosin content and caused mitochondrial adaptations compared with untreated controls when applied to myotubes differentiated for 4 days, a time when most studies in the literature had studied the effects of tumor/cancer cell CM. However, similar atrophic effects were observed with control cell CM and dilution controls. To our knowledge, this is the first report of the effects of CM from primary cultures of human lung tumor cells on cultured myotubes. Similar to murine cancer cell CM, however, we found no evidence for an effect of human tumor CM on myotubes differentiated for 7 days. Our results do not support the long-standing hypothesis that factors released from tumor/cancer cells contribute to skeletal muscle atrophy and mitochondrial abnormalities.
Cultured muscle cells are a unique and valuable system that permits examination of muscle in isolation, with the ability to manipulate the extracellular environment. Despite these strengths, studies in cultured muscle cells are often criticized because of the artificial nature of this system. While a myotube is arguably not a perfect model of a muscle fiber, how well this system models in vivo muscle may vary depending on its stage of differentiation. Our results underscore this caveat, as treatment of myotubes with either murine or human cancer/tumor cell CM starting at 7 days post-differentiation had no effect on either myotube myosin content or mitochondrial content/ROS production compared with untreated controls. In fact, in the case of the KRASG12D cells, CM paradoxically enhanced myotube myosin and mitochondrial content compared with untreated controls. In contrast to results on d7 differentiated myotubes, CM had effects on cells treated at 4 days post-differentiation, causing myosin depletion and some mitochondrial maladaptations. The failure to observe effects at 7 days post-differentiation is unlikely related to the cell lines used, as LLC is well proven to cause cachexia when transplanted into mice (12, 36, 47, 67) and when LLC CM has been applied to cultured myotubes (22, 38, 40, 41, 69). The effects of KRASG12D CM are less well described, but data from our laboratories (unpublished data) and others show that KRASG12D mice experience muscle atrophy (42). Alternatively, one could argue that myotubes at this later developmental stage are recalcitrant to atrophic stimuli. However, we found that application of various chemotherapeutics provokes atrophy at this time (25), in agreement with studies by other laboratories on myotubes studied earlier following differentiation (23).
Our data from experiments treating d4 differentiated myotubes with LLC and KRASG12D CM argue that a dilution effect caused by the addition of the CM to standard DM impairs myotube growth. Numerous studies have shown that C2C12 cells are still increasing in size and myosin content from day 4 to day 7 post-differentiation (13, 14, 27, 43, 56, 59, 63), which we similarly observed (26). Medium dilution with CM may simply starve cells of nutrients required for normal growth. A recent study by Jackman et al. (28) supports this interpretation. In this study, 4-day post-differentiation myotubes were treated for 3 days with cancer cell CM from two different cell lines and were compared with untreated DM, similar to the experiment conducted in the current study. Time course data showed that the effect of CM was not to induce atrophy but to impair the growth of myotubes. Our HBSS dilution control further supports the effects of the CM dilution on myotube growth. Thus, compared with control cells maintained in undiluted DM, the effects of tumor cell CM on d4 differentiated myotubes primarily reflects growth inhibition.
If medium dilution impairs myotube growth, a more valid experimental control would be a cell type similar to the tumor cell line, but one that does not release factors into the medium that cause muscle wasting. Because the identity of these secreted factors is unknown, the selection of appropriate control cell lines is difficult. We used CM from a similar lung epithelial cell type compared with tumor cell lines. In the control for LLC CM, we used the nontumorigenic NL20 cells (22, 51, 52, 73), which are used widely in the field as a control for LLC CM (68, 70, 71). This is not an ideal cell line because of species differences, but it is nontumorigenic in animals, grows well under similar medium conditions as LLC cells, and has been used as a control for LLC CM in past studies (48, 72).
Concern about the appropriateness of the control cell line is lessened in the Kras CM experiments, where we used a Cre-recombinant adenovirus to activate the oncogenic KRAS mutation (KrasG12D) or treatment of cells with an empty vector as a control (KrasWT). Thus, with the exception of Cre expression and transgene activation, the KrasWT cells are identical to KrasG12D cells. CM from KrasWT controls, therefore, should reflect the effects of nutrient depletion, secreted factors, and/or metabolic byproducts typical of growing cells, only differing from KrasG12D CM for those secreted factors that contribute to cachexia. As with the LLC CM experiments, data from these controls largely mirrored that of the HBSS dilution controls and KrasG12D CM, further supporting the conclusion that most of the effects of tumor cell CM that we observed in d4 differentiated myotubes is related to a dilution effect when comparisons are made to cells maintained in untreated (i.e., undiluted) control medium.
Animal models generally contain large tumors or a heavy tumor burden before an atrophic phenotype is observed. In contrast, tumors of atrophic lung cancer patients are relatively smaller. Thus, if human tumor cells secrete factors that act on muscle to cause atrophy, these factors are either secreted in greater amounts than in murine models and/or they are more effective at inducing muscle wasting. Our results from the CM of cells obtained from tumors of lung cancer patients do not support either conclusion. When we compared human tumor CM (hTCM) with control cell CM (HBEC), dilution controls or untreated controls, no change in myosin content or ROS production was measured. Moreover, we observed a paradoxical increase in mitochondrial content with both hTCM and control cell CM. One major caveat to these experiments is that most of our patients did not report weight loss. This may have been due to the fact that tumor biopsies were obtained at the time of diagnosis and one-half of the samples were from early-stage patients. The presentation of cachexia is more apparent in patients later in the disease (21) and/or further into the course treatment. Nonetheless, in the one patient that did report weight loss, no effect of their tumor CM on myotube myosin content or mitochondrial variables was noted. We also observed considerable variability in the response of myotubes to hTCM compared with murine tumor cell CM. This is likely explained, in part, by the outbred nature of the human population. In light of this variability, subject-specific control cell CM (e.g., airway epithelial primary cell cultures) may be required as within-subject controls to test for effects of tumor-specific atrophic factors. On balance, our data do not support the hypothesis that factors secreted from primary cultures of human lung tumor cells act directly on skeletal muscle cells to provoke atrophy and mitochondrial maladaptations. We are careful to note, however, that further studies that compare the response to CM from tumor cells derived from later stage, cachectic, and noncachectic patients are needed to elucidate whether tumor-secreted factors cause myotube atrophy associated with clinical cancer cachexia in humans (21).
Several caveats to our studies deserve discussion. First, we examined the direct effects of CM on skeletal muscle. Muscles contain an array of other cell types (immune cells, satellite cells, fibroblasts, etc.) that could respond to tumor-derived factors to provoke atrophy. For instance, tumor CM may mediate its effects through host inflammatory response to the presence of tumor(s) either systemically or locally within the muscle (5, 9). Our experiments did not test these potential host-derived factors, such as cytokines, chemokines, and growth factors. Second, we used the common 1:3 dilution of DM with CM to treat the cultured myotubes. The amount of secreted factors in this dilution, and the fact that we exposed myotubes to a single bolus of the CM (medium changed daily), may have been insufficient to promote skeletal muscle myotube atrophy. To the first point, considering the relatively small size of most human tumors relative to the systemic circulation and interstitial fluid volume, such a dilution would seem far in excess of what skeletal muscles are exposed to in vivo. To this latter point, recent studies have argued that coculture of tumor cells and muscle myotubes is required to observe a true atrophic effect of tumor-secreted factors to cause atrophy (28). While this may be the case, as we have demonstrated with our various control conditions, appropriate control cell lines will be needed to ensure that myotube atrophy is not provoked by medium substrate depletion and/or accumulation of metabolic waste products. Third, differences in experimental conditions and materials across studies, such as the specific strain of C2C12 and cancer cell lines, culture conditions, and methods for the preparation of CM could explain variation in findings. To this last point, in some prior studies it is difficult to discern the final serum concentration of CM because of insufficient detail in descriptions of experimental methods. The serum content of CM is an important factor that could influence myotube size/myosin content. In our studies, we controlled for serum content across all CM and control conditions. Fourth, we stored CM frozen before application to myotubes. This could have affected the stability of cachectic factors sensitive to cycles of freeze-thaw. However, only one cycle of freeze-thaw was performed, and prior work has shown that frozen CM promotes myotube wasting/growth inhibition when treated at 4 days post-differentiation (28), as we similarly observed in the current study. Finally, our model is uncoupled from the complex clinical condition present in cancer patients, such as pulmonary insufficiency, chemotherapeutics, and muscle disuse. The effects of these clinical factors may cause skeletal muscle to be more responsive to tumor-related factors that incite muscle wasting.
In summary, although the common experimental model of using 4-day differentiated myotubes for treatment with tumor/cancer cell CM yielded lower myotube myosin content compared with untreated controls, these decreases were not apparent in 7-day differentiated myotubes or when compared with controls for medium dilution or other secreted factors. These results suggest that CM effects were related to medium dilution and growth inhibition at a time when myotubes were undergoing rapid growth (26), rather than atrophy, a finding in accord with recent work by other laboratories (28). Our results serve as a cautionary note to using the myotube culture model for these types of CM experiments. More specifically, our data suggest careful consideration of the timing of treatment and appropriate controls. These considerations, particularly the issue of timing of treatment, may not apply only to tumor/cancer cell CM experiments but to any atrophic or hypertrophic stimuli. Finally, to our knowledge, we provide the first report of the effects of human tumor primary cell CM on cultured myotubes. Similar to results from murine tumor cell CM, we found no effect of human tumor cell CM on myotube myosin content or mitochondrial content or ROS production. Collectively, our results do not support the long-standing hypothesis that tumor-secreted factors act directly on skeletal muscle to cause atrophy and metabolic dysfunction. Our results should not be construed, however, as a refutation of this hypothesis, as these cells could secrete such factors in vivo in the native tumor microenvironment and/or require other host-related factors to mediate their effects on muscle.
GRANTS
This study was funded by National Institutes of Health Grants R01 AR-065826, R21 CA-191532, S10 OD-017969, and P30 RR-032135. B. A. Guigni was funded by a Department of Defense SMART Scholarship ID 2016-85335.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.A.G., J.A.C., and M.J.T. conceived and designed research; B.A.G., J.v.d.V., and C.M.K. performed experiments; B.A.G. analyzed data; B.A.G., J.A.C., and M.J.T. interpreted results of experiments; B.A.G. prepared figures; B.A.G., J.v.d.V., and M.J.T. drafted manuscript; B.A.G., J.v.d.V., C.M.K., J.A.C., and M.J.T. edited and revised manuscript; B.A.G., J.v.d.V., C.M.K., J.A.C., and M.J.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the patients who generously volunteered for these studies. We thank Cheryl van de Wetering and Bradley Anair for their technical support.
REFERENCES
- 1.Alcorn JF, Guala AS, van der Velden J, McElhinney B, Irvin CG, Davis RJ, Janssen-Heininger YM. Jun N-terminal kinase 1 regulates epithelial-to-mesenchymal transition induced by TGF-beta1. J Cell Sci 121: 1036–1045, 2008. doi: 10.1242/jcs.019455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ando K, Takahashi F, Kato M, Kaneko N, Doi T, Ohe Y, Koizumi F, Nishio K, Takahashi K. Tocilizumab, a proposed therapy for the cachexia of Interleukin6-expressing lung cancer. PLoS One 9: e102436, 2014. doi: 10.1371/journal.pone.0102436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antunes D, Padrão AI, Maciel E, Santinha D, Oliveira P, Vitorino R, Moreira-Gonçalves D, Colaço B, Pires MJ, Nunes C, Santos LL, Amado F, Duarte JA, Domingues MR, Ferreira R. Molecular insights into mitochondrial dysfunction in cancer-related muscle wasting. Biochim Biophys Acta 1841: 896–905, 2014. doi: 10.1016/j.bbalip.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Au ED, Desai AP, Koniaris LG, Zimmers TA. The MEK-inhibitor selumetinib attenuates tumor growth and reduces IL-6 expression but does not protect against muscle wasting in lewis lung cancer cachexia. Front Physiol 7: 682, 2017. doi: 10.3389/fphys.2016.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer-associated cachexia. Nat Rev Dis Primers 4: 17105, 2018. doi: 10.1038/nrdp.2017.105. [DOI] [PubMed] [Google Scholar]
- 6.Baracos VE, Reiman T, Mourtzakis M, Gioulbasanis I, Antoun S. Body composition in patients with non-small cell lung cancer: a contemporary view of cancer cachexia with the use of computed tomography image analysis. Am J Clin Nutr 91: 1133S–1137S, 2010. doi: 10.3945/ajcn.2010.28608C. [DOI] [PubMed] [Google Scholar]
- 7.Bennani-Baiti N, Walsh D. Animal models of the cancer anorexia-cachexia syndrome. Support Care Cancer 19: 1451–1463, 2011. doi: 10.1007/s00520-010-0972-0. [DOI] [PubMed] [Google Scholar]
- 8.Blackwell TA, Cervenka I, Khatri B, Brown JL, Rosa-Caldwell ME, Lee DE, Perry RA Jr, Brown LA, Haynie WS, Wiggs MP, Bottje WG, Washington TA, Kong BC, Ruas JL, Greene NP. Transcriptomic analysis of the development of skeletal muscle atrophy in cancer-cachexia in tumor-bearing mice. Physiol Genomics 50: 1071–1082, 2018. doi: 10.1152/physiolgenomics.00061.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bohnert KR, McMillan JD, Kumar A. Emerging roles of ER stress and unfolded protein response pathways in skeletal muscle health and disease. J Cell Physiol 233: 67–78, 2018. doi: 10.1002/jcp.25852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonetto A, Rupert JE, Barreto R, Zimmers TA. The colon-26 carcinoma tumor-bearing mouse as a model for the study of cancer cachexia. J Vis Exp (117): 2016. doi: 10.3791/54893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown JL, Lee DE, Rosa-Caldwell ME, Brown LA, Perry RA, Haynie WS, Huseman K, Sataranatarajan K, Van Remmen H, Washington TA, Wiggs MP, Greene NP. Protein imbalance in the development of skeletal muscle wasting in tumour-bearing mice. J Cachexia Sarcopenia Muscle 9: 987–1002, 2018. doi: 10.1002/jcsm.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown JL, Rosa-Caldwell ME, Lee DE, Blackwell TA, Brown LA, Perry RA, Haynie WS, Hardee JP, Carson JA, Wiggs MP, Washington TA, Greene NP. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J Cachexia Sarcopenia Muscle 8: 926–938, 2017. doi: 10.1002/jcsm.12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaturvedi V, Dye DE, Kinnear BF, van Kuppevelt TH, Grounds MD, Coombe DR. Interactions between skeletal muscle myoblasts and their extracellular matrix revealed by a serum free culture system. PLoS One 10: e0127675, 2015. doi: 10.1371/journal.pone.0127675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng CS, El-Abd Y, Bui K, Hyun YE, Hughes RH, Kraus WE, Truskey GA. Conditions that promote primary human skeletal myoblast culture and muscle differentiation in vitro. Am J Physiol Cell Physiol 306: C385–C395, 2014. doi: 10.1152/ajpcell.00179.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi Y, Oh DY, Kim TY, Lee KH, Han SW, Im SA, Kim TY, Bang YJ. Skeletal muscle depletion predicts the prognosis of patients with advanced pancreatic cancer undergoing palliative chemotherapy, independent of body mass index. PLoS One 10: e0139749, 2015. doi: 10.1371/journal.pone.0139749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Constantinou C, Fontes de Oliveira CC, Mintzopoulos D, Busquets S, He J, Kesarwani M, Mindrinos M, Rahme LG, Argilés JM, Tzika AA. Nuclear magnetic resonance in conjunction with functional genomics suggests mitochondrial dysfunction in a murine model of cancer cachexia. Int J Mol Med 27: 15–24, 2011. doi: 10.3892/ijmm.2010.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dewys WD, Begg C, Lavin PT, Band PR, Bennett JM, Bertino JR, Cohen MH, Douglass HO Jr, Engstrom PF, Ezdinli EZ, Horton J, Johnson GJ, Moertel CG, Oken MM, Perlia C, Rosenbaum C, Silverstein MN, Skeel RT, Sponzo RW, Tormey DC; Eastern Cooperative Oncology Group . Prognostic effect of weight loss prior to chemotherapy in cancer patients. Am J Med 69: 491–497, 1980. doi: 10.1016/S0149-2918(05)80001-3. [DOI] [PubMed] [Google Scholar]
- 18.Fearon KC. Cancer cachexia and fat-muscle physiology. N Engl J Med 365: 565–567, 2011. doi: 10.1056/NEJMcibr1106880. [DOI] [PubMed] [Google Scholar]
- 19.Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 16: 153–166, 2012. doi: 10.1016/j.cmet.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 20.Fontes-Oliveira CC, Busquets S, Toledo M, Penna F, Paz Aylwin M, Sirisi S, Silva AP, Orpí M, García A, Sette A, Inês Genovese M, Olivan M, López-Soriano FJ, Argilés JM. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: altered energetic efficiency? Biochim Biophys Acta 1830: 2770–2778, 2013. doi: 10.1016/j.bbagen.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 21.Gannavarapu BS, Lau SKM, Carter K, Cannon NA, Gao A, Ahn C, Meyer JJ, Sher DJ, Jatoi A, Infante R, Iyengar P. Prevalence and survival impact of pretreatment cancer-associated weight loss: a tool for guiding early palliative care. J Oncol Pract 14: e238–e250, 2018. doi: 10.1200/JOP.2017.025221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao S, Carson JA. Lewis lung carcinoma regulation of mechanical stretch-induced protein synthesis in cultured myotubes. Am J Physiol Cell Physiol 310: C66–C79, 2016. doi: 10.1152/ajpcell.00052.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilliam LA, Moylan JS, Patterson EW, Smith JD, Wilson AS, Rabbani Z, Reid MB. Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. Am J Physiol Cell Physiol 302: C195–C202, 2012. doi: 10.1152/ajpcell.00217.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Go SI, Park MJ, Song HN, Kang MH, Park HJ, Jeon KN, Kim SH, Kim MJ, Kang JH, Lee GW. Sarcopenia and inflammation are independent predictors of survival in male patients newly diagnosed with small cell lung cancer. Support Care Cancer 24: 2075–2084, 2016. doi: 10.1007/s00520-015-2997-x. [DOI] [PubMed] [Google Scholar]
- 25.Guigni BA, Callahan DM, Tourville TW, Miller MS, Fiske B, Voigt T, Korwin-Mihavics B, Anathy V, Dittus K, Toth MJ. Skeletal muscle atrophy and dysfunction in breast cancer patients: role for chemotherapy-derived oxidant stress. Am J Physiol Cell Physiol 315: C744–C756, 2018. doi: 10.1152/ajpcell.00002.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guigni BA, Fix DK, Bivona JJ III, Palmer BM, Carson JA, Toth MJ. Electrical stimulation prevents doxorubicin-induced atrophy and mitochondrial loss in cultured myotubes. Am J Physiol Cell Physiol 317: C1213–C1228, 2019. doi: 10.1152/ajpcell.00148.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Issa ME, Muruganandan S, Ernst MC, Parlee SD, Zabel BA, Butcher EC, Sinal CJ, Goralski KB. Chemokine-like receptor 1 regulates skeletal muscle cell myogenesis. Am J Physiol Cell Physiol 302: C1621–C1631, 2012. doi: 10.1152/ajpcell.00187.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jackman RW, Floro J, Yoshimine R, Zitin B, Eiampikul M, El-Jack K, Seto DN, Kandarian SC. Continuous release of tumor-derived factors improves the modeling of cachexia in muscle cell culture. Front Physiol 8: 738, 2017. [Erratum in: Front Physiol 10: 394, 2019.] doi: 10.3389/fphys.2017.00738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15: 3243–3248, 2001. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johns N, Stephens NA, Fearon KCH. Muscle wasting in cancer. Int J Biochem Cell Biol 45: 2215–2229, 2013. doi: 10.1016/j.biocel.2013.05.032. [DOI] [PubMed] [Google Scholar]
- 32.Jourdain M, Melly S, Summermatter S, Hatakeyama S. Mouse models of cancer-induced cachexia: hind limb muscle mass and evoked force as readouts. Biochem Biophys Res Commun 503: 2415–2420, 2018. doi: 10.1016/j.bbrc.2018.06.170. [DOI] [PubMed] [Google Scholar]
- 33.Julienne CM, Dumas JF, Goupille C, Pinault M, Berri C, Collin A, Tesseraud S, Couet C, Servais S. Cancer cachexia is associated with a decrease in skeletal muscle mitochondrial oxidative capacities without alteration of ATP production efficiency. J Cachexia Sarcopenia Muscle 3: 265–275, 2012. doi: 10.1007/s13539-012-0071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langen RC, Schols AM, Kelders MC, Wouters EF, Janssen-Heininger YM. Enhanced myogenic differentiation by extracellular matrix is regulated at the early stages of myogenesis. In Vitro Cell Dev Biol Anim 39: 163–169, 2003. doi: 10.1007/s11626-003-0011-2. [DOI] [PubMed] [Google Scholar]
- 35.Lanic H, Kraut-Tauzia J, Modzelewski R, Clatot F, Mareschal S, Picquenot JM, Stamatoullas A, Leprêtre S, Tilly H, Jardin F. Sarcopenia is an independent prognostic factor in elderly patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Leuk Lymphoma 55: 817–823, 2014. doi: 10.3109/10428194.2013.816421. [DOI] [PubMed] [Google Scholar]
- 36.Lee DE, Brown JL, Rosa-Caldwell ME, Blackwell TA, Perry RA Jr, Brown LA, Khatri B, Seo D, Bottje WG, Washington TA, Wiggs MP, Kong BW, Greene NP. Cancer cachexia-induced muscle atrophy: evidence for alterations in microRNAs important for muscle size. Physiol Genomics 49: 253–260, 2017. doi: 10.1152/physiolgenomics.00006.2017. [DOI] [PubMed] [Google Scholar]
- 37.Lee H, Lee SJ, Bae GU, Baek NI, Ryu JH. Canadine from Corydalis turtschaninovii stimulates myoblast differentiation and protects against myotube atrophy. Int J Mol Sci 18: 2748, 2017. doi: 10.3390/ijms18122748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee HW, Baker E, Lee KM, Persinger AM, Hawkins W, Puppa M. Effects of low-dose leucine supplementation on gastrocnemius muscle mitochondrial content and protein turnover in tumor-bearing mice. Appl Physiol Nutr Metab 44: 997–1004, 2019. doi: 10.1139/apnm-2018-0765. [DOI] [PubMed] [Google Scholar]
- 39.Llovera M, García-Martínez C, López-Soriano J, Agell N, López-Soriano FJ, Garcia I, Argilés JM. Protein turnover in skeletal muscle of tumour-bearing transgenic mice overexpressing the soluble TNF receptor-1. Cancer Lett 130: 19–27, 1998. doi: 10.1016/S0304-3835(98)00137-2. [DOI] [PubMed] [Google Scholar]
- 40.McLean JB, Moylan JS, Andrade FH. Mitochondria dysfunction in lung cancer-induced muscle wasting in C2C12 myotubes. Front Physiol 5: 503, 2014. doi: 10.3389/fphys.2014.00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McLean JB, Moylan JS, Horrell EM, Andrade FH. Proteomic analysis of media from lung cancer cells reveals role of 14-3-3 proteins in cachexia. Front Physiol 6: 136, 2015. doi: 10.3389/fphys.2015.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller A, McLeod L, Alhayyani S, Szczepny A, Watkins DN, Chen W, Enriori P, Ferlin W, Ruwanpura S, Jenkins BJ. Blockade of the IL-6 trans-signalling/STAT3 axis suppresses cachexia in Kras-induced lung adenocarcinoma. Oncogene 36: 3059–3066, 2017. doi: 10.1038/onc.2016.437. [DOI] [PubMed] [Google Scholar]
- 43.Murphy SM, Kiely M, Jakeman PM, Kiely PA, Carson BP. Optimization of an in vitro bioassay to monitor growth and formation of myotubes in real time. Biosci Rep 36: e00330, 2016. doi: 10.1042/BSR20160036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naito T, Okayama T, Aoyama T, Ohashi T, Masuda Y, Kimura M, Shiozaki H, Murakami H, Kenmotsu H, Taira T, Ono A, Wakuda K, Imai H, Oyakawa T, Ishii T, Omori S, Nakashima K, Endo M, Omae K, Mori K, Yamamoto N, Tanuma A, Takahashi T. Unfavorable impact of cancer cachexia on activity of daily living and need for inpatient care in elderly patients with advanced non-small-cell lung cancer in Japan: a prospective longitudinal observational study. BMC Cancer 17: 800, 2017. doi: 10.1186/s12885-017-3795-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, Katayama R, Costa C, Ross KN, Moran T, Howe E, Fulton LE, Mulvey HE, Bernardo LA, Mohamoud F, Miyoshi N, VanderLaan PA, Costa DB, Jänne PA, Borger DR, Ramaswamy S, Shioda T, Iafrate AJ, Getz G, Rudin CM, Mino-Kenudson M, Engelman JA. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun 6: 6377, 2015. doi: 10.1038/ncomms7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oraldi M, Maggiora M, Paiuzzi E, Canuto RA, Muzio G. CLA reduces inflammatory mediators from A427 human lung cancer cells and A427 conditioned medium promotes differentiation of C2C12 murine muscle cells. Lipids 48: 29–38, 2013. doi: 10.1007/s11745-012-3734-6. [DOI] [PubMed] [Google Scholar]
- 47.Paul PK, Gupta SK, Bhatnagar S, Panguluri SK, Darnay BG, Choi Y, Kumar A. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol 191: 1395–1411, 2010. doi: 10.1083/jcb.201006098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Penna F, Busquets S, Argilés JM. Experimental cancer cachexia: Evolving strategies for getting closer to the human scenario. Semin Cell Dev Biol 54: 20–27, 2016. doi: 10.1016/j.semcdb.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 49.Puppa MJ, White JP, Velázquez KT, Baltgalvis KA, Sato S, Baynes JW, Carson JA. The effect of exercise on IL-6-induced cachexia in the Apc (Min/+) mouse. J Cachexia Sarcopenia Muscle 3: 117–137, 2012. doi: 10.1007/s13539-011-0047-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Puyol M, Martín A, Dubus P, Mulero F, Pizcueta P, Khan G, Guerra C, Santamaría D, Barbacid M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 18: 63–73, 2010. doi: 10.1016/j.ccr.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 51.Schiller JH, Bittner G. Loss of the tumorigenic phenotype with in vitro, but not in vivo, passaging of a novel series of human bronchial epithelial cell lines: possible role of an alpha 5/beta 1-integrin-fibronectin interaction. Cancer Res 55: 6215–6221, 1995. [PubMed] [Google Scholar]
- 52.Schiller JH, Kao C, Bittner G, Harris C, Oberley TD, Meisner LF. Establishment and characterization of a SV40 T-antigen immortalized human bronchial epithelial cell line. In Vitro Cell Dev Biol 28: 461–464, 1992. doi: 10.1007/BF02634125. [DOI] [PubMed] [Google Scholar]
- 53.Seo E, Kang H, Lim OK, Jun HS. Supplementation with IL-6 and muscle cell culture conditioned media enhances myogenic differentiation of adipose tissue-derived stem cells through STAT3 activation. Int J Mol Sci 19: 1557, 2018. doi: 10.3390/ijms19061557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seto DN, Kandarian SC, Jackman RW. A key role for leukemia inhibitory factor in C26 cancer cachexia. J Biol Chem 290: 19976–19986, 2015. doi: 10.1074/jbc.M115.638411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shachar SS, Deal AM, Weinberg M, Williams GR, Nyrop KA, Popuri K, Choi SK, Muss HB. Body composition as a predictor of toxicity in patients receiving anthracycline and taxane-based chemotherapy for early-stage breast cancer. Clin Cancer Res 23: 3537–3543, 2017. doi: 10.1158/1078-0432.CCR-16-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shu L, Houghton PJ. The mTORC2 complex regulates terminal differentiation of C2C12 myoblasts. Mol Cell Biol 29: 4691–4700, 2009. doi: 10.1128/MCB.00764-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sjøblom B, Grønberg BH, Wentzel-Larsen T, Baracos VE, Hjermstad MJ, Aass N, Bremnes RM, Fløtten Ø, Bye A, Jordhøy M. Skeletal muscle radiodensity is prognostic for survival in patients with advanced non-small cell lung cancer. Clin Nutr 35: 1386–1393, 2016. doi: 10.1016/j.clnu.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 58.Strassmann G, Fong M, Kenney JS, Jacob CO. Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J Clin Invest 89: 1681–1684, 1992. doi: 10.1172/JCI115767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stuelsatz P, Pouzoulet F, Lamarre Y, Dargelos E, Poussard S, Leibovitch S, Cottin P, Veschambre P. Down-regulation of MyoD by calpain 3 promotes generation of reserve cells in C2C12 myoblasts. J Biol Chem 285: 12670–12683, 2010. doi: 10.1074/jbc.M109.063966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun R, Zhang S, Hu W, Lu X, Lou N, Yang Z, Chen S, Zhang X, Yang H. Valproic acid attenuates skeletal muscle wasting by inhibiting C/EBPβ-regulated atrogin1 expression in cancer cachexia. Am J Physiol Cell Physiol 311: C101–C115, 2016. doi: 10.1152/ajpcell.00344.2015. [DOI] [PubMed] [Google Scholar]
- 61.Sustova H, De Feudis M, Reano S, Alves Teixeira M, Valle I, Zaggia I, Agosti E, Prodam F, Filigheddu N. Opposing effects of 25-hydroxy- and 1α,25-dihydroxy-vitamin D3 on pro-cachectic cytokine-and cancer conditioned medium-induced atrophy in C2C12 myotubes. Acta Physiol (Oxf) 226: e13269, 2019. doi: 10.1111/apha.13269. [DOI] [PubMed] [Google Scholar]
- 62.Tan BH, Fearon KC. Cachexia: prevalence and impact in medicine. Curr Opin Clin Nutr Metab Care 11: 400–407, 2008. doi: 10.1097/MCO.0b013e328300ecc1. [DOI] [PubMed] [Google Scholar]
- 63.Tang J, He A, Yan H, Jia G, Liu G, Chen X, Cai J, Tian G, Shang H, Zhao H. Damage to the myogenic differentiation of C2C12 cells by heat stress is associated with up-regulation of several selenoproteins. Sci Rep 8: 10601, 2018. doi: 10.1038/s41598-018-29012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tímár J. The clinical relevance of KRAS gene mutation in non-small-cell lung cancer. Curr Opin Oncol 26: 138–144, 2014. doi: 10.1097/CCO.0000000000000051. [DOI] [PubMed] [Google Scholar]
- 65.Tisdale MJ. Are tumoral factors responsible for host tissue wasting in cancer cachexia? Future Oncol 6: 503–513, 2010. doi: 10.2217/fon.10.20. [DOI] [PubMed] [Google Scholar]
- 66.Yano CL, Ventrucci G, Field WN, Tisdale MJ, Gomes-Marcondes MC. Metabolic and morphological alterations induced by proteolysis-inducing factor from Walker tumour-bearing rats in C2C12 myotubes. BMC Cancer 8: 24, 2008. doi: 10.1186/1471-2407-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yoshimura T, Saitoh K, Sun L, Wang Y, Taniyama S, Yamaguchi K, Uchida T, Ohkubo T, Higashitani A, Nikawa T, Tachibana K, Hirasaka K. Morin suppresses cachexia-induced muscle wasting by binding to ribosomal protein S10 in carcinoma cells. Biochem Biophys Res Commun 506: 773–779, 2018. doi: 10.1016/j.bbrc.2018.10.184. [DOI] [PubMed] [Google Scholar]
- 68.Zhang G, Jin B, Li YP. C/EBPβ mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J 30: 4323–4335, 2011. doi: 10.1038/emboj.2011.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang G, Li YP. p38β MAPK upregulates atrogin1/MAFbx by specific phosphorylation of C/EBPβ. Skelet Muscle 2: 20, 2012. doi: 10.1186/2044-5040-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang G, Lin RK, Kwon YT, Li YP. Signaling mechanism of tumor cell-induced up-regulation of E3 ubiquitin ligase UBR2. FASEB J 27: 2893–2901, 2013. doi: 10.1096/fj.12-222711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang G, Liu Z, Ding H, Miao H, Garcia JM, Li YP. Toll-like receptor 4 mediates Lewis lung carcinoma-induced muscle wasting via coordinate activation of protein degradation pathways. Sci Rep 7: 2273, 2017. doi: 10.1038/s41598-017-02347-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang G, Liu Z, Ding H, Zhou Y, Doan HA, Sin KWT, Zhu ZJ, Flores R, Wen Y, Gong X, Liu Q, Li YP. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat Commun 8: 589, 2017. doi: 10.1038/s41467-017-00726-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou D, Xie M, He B, Gao Y, Yu Q, He B, Chen Q. Microarray data re-annotation reveals specific lncRNAs and their potential functions in non-small cell lung cancer subtypes. Mol Med Rep 16: 5129–5136, 2017. doi: 10.3892/mmr.2017.7244. [DOI] [PMC free article] [PubMed] [Google Scholar]