

Abstract
Oxygen tension in the kidney is mostly determined by O2 consumption (Qo2), which is, in turn, closely linked to tubular Na+ reabsorption. The objective of the present study was to develop a model of mitochondrial function in the proximal tubule (PT) cells of the rat renal cortex to gain more insight into the coupling between Qo2, ATP formation (GATP), ATP hydrolysis (QATP), and Na+ transport in the PT. The present model correctly predicts in vitro and in vivo measurements of Qo2, GATP, and ATP and Pi concentrations in PT cells. Our simulations suggest that O2 levels are not rate limiting in the proximal convoluted tubule, absent large metabolic perturbations. The model predicts that the rate of ATP hydrolysis and cytoplasmic pH each substantially regulate the GATP-to-Qo2 ratio, a key determinant of the number of Na+ moles actively reabsorbed per mole of O2 consumed. An isolated increase in QATP or in cytoplasmic pH raises the GATP-to-Qo2 ratio. Thus, variations in Na+ reabsorption and pH along the PT may, per se, generate axial heterogeneities in the efficiency of mitochondrial metabolism and Na+ transport. Our results also indicate that the GATP-to-Qo2 ratio is strongly impacted not only by H+ leak permeability, which reflects mitochondrial uncoupling, but also by K+ leak pathways. Simulations suggest that the negative impact of increased uncoupling in the diabetic kidney on mitochondrial metabolic efficiency is partly counterbalanced by increased rates of Na+ transport and ATP consumption. This model provides a framework to investigate the role of mitochondrial dysfunction in acute and chronic renal diseases.
Keywords: kidney cortex, mathematical model, mitochondria, proton motive force
INTRODUCTION
The renal tubules reabsorb >99% of the glomerular filtrate, in part via active transport processes that necessitate energy. The transcellular concentration gradients that drive tubular transport are primarily established by Na+-K+-ATPase pumps that use ATP hydrolysis as a source of energy, which, in turn, requires a sustained rate of ATP generation, mostly by aerobic processes. The kidneys thus have the second highest O2 consumption rate (Qo2) after the heart (14, 68). The greatest density of mitochondria is found in the proximal tubule (PT) and thick ascending limb (8, 44).
In contrast to most other organs, metabolic control of blood flow is poor in the kidney. To stabilize glomerular filtration, renal blood flow and, therefore, the O2 supply are kept within tight limits and do not increase in proportion with Qo2 (70). The inability of the kidney to regulate perfusion independently, coupled to its high metabolic demand, contributes to the kidney’s particular susceptibility to hypoxia. It also implies that intrarenal Po2 is predominantly determined by Qo2 (70). Since chronic hypoxia is now firmly recognized as a unifying pathway that drives the progression of kidney disease (13, 30, 38, 65, 66), understanding the factors that govern renal Qo2 is essential.
Understanding the determinants of Qo2 in the kidney requires, in turn, linking explicitly tubular Na+ transport and Qo2. We have previously built detailed cell-based models of water and solute transport along the nephron, accounting for the specific apical, basolateral, and paracellular transport pathways in each segment, and applied these models to predict segmental Qo2 (55–59). We have also developed organ-scale models that describe the interactions among renal tubules and the vasculature and predict medullary oxygenation (18, 36, 37). However, none of these models included O2 metabolism and instead postulated a fixed Na+ transport-to-Qo2 ratio. We developed the present model of mitochondrial function to couple Na+-driven ATP hydrolysis to Qo2 in the PT.
The PT oxidizes mostly fatty acids and some intermediates of the tricarboxylic acid (TCA) cycle that are reabsorbed from the lumen (3). Under normal conditions, the PT reabsorbs the full load of glucose but only metabolizes <1% of that load (63). The PT also has a significant capacity for gluconeogenesis and may account for 45% of glucose production during prolonged starvation (69, 86). In the model described below, the concentration of pyruvate is specified without explicitly considering the substrates that generated it.
As shown in Fig. 1, pyruvate enters the mitochondria, where it is converted to acetyl CoA, which then enters the TCA cycle. The cycle is formed by a series of eight redox, dehydration, hydration, and decarboxylation reactions that transfer electrons stripped from metabolic substrates to NADH and FADH2. During the process of oxidative phosphorylation, these electrons are sequentially transferred to electron carriers in the respiratory chain, the last step of which consists of the reduction of O2 to H2O. The energy released during electron transfer is used by respiratory complexes I, III, and IV to pump protons from the inner matrix into the intermembrane space, thereby creating an electrochemical potential gradient known as the proton motive force (pmf). The reentry of protons into the matrix across F0F1-ATP synthase drives ADP phosphorylation, i.e., the formation of ATP, which dissipates the pmf. However, some protons reenter the matrix across H+ leak pathways: this shunt also dissipates the pmf and reduces the coupling between oxidative phosphorylation and ATP formation, a phenomenon hence referred to as mitochondrial uncoupling. The newly formed ATP is exchanged for ADP across the inner mitochondrial membrane via adenine nucleotide translocase (ANT).
Fig. 1.

Schematic representation of the transport and metabolic processes included in the model: the tricarboxylic acid cycle, oxidative phosphorylation, chemical reactions, and metabolite transport between the cytoplasm and mitochondria. AK, adenylate kinase; ANT, adenine nucleotide translocase; CI, complex I; CIII, complex III; CIV, complex IV; CoQ, coenzyme Q; Cyt c(ox), cytochrome c (Cyt c) oxidase; Cyt c(red), cytochrome c reductase; HKE, H+/K+ exchange; NDK, nucleoside diphosphokinase; PiHt, Pi-H+ transporter.
In the present study, we examined the impact of ATP hydrolysis, Po2, pH, and electrolyte concentration on the mitochondrial processes described above to better understand the coupling between Qo2, ATP formation, ATP hydrolysis, and Na+ transport in the PT.
MODEL DESCRIPTION
We adapted to the proximal convoluted tubule the model of mitochondria metabolism and electrophysiology developed by Wu and colleagues for cardiac and skeletal muscle (96). The model represents the TCA cycle, oxidative phosphorylation, and metabolite transport between the cytosol and mitochondria and is based on mass and charge conservation equations that consider detailed reaction kinetics and transmembrane fluxes. We adjusted the model’s parameter values to represent mitochondria in the cortical PT of the rat kidney.
Differential Equations
The full set of model equations can be found in Wu et al. (96). Given our focus on the production and consumption of ATP in the PT, the equations that govern the concentration of ATP in the inner mitochondrial matrix (subscript x), intermembrane space (subscript i), and cytoplasm (subscript c) are recapitulated below:
| (1) |
| (2) |
| (3) |
where Wx, Wi, and Wc are the fractional volume occupied by water in the inner matrix, intermembrane space, and cytoplasm, respectively; Vmito and Vcyto are the proportion of cell volume occupied by the mitochondria and cytoplasm, respectively; GATP is the rate of ATP formation by F0F1-ATP synthase; Qndk is the nucleoside diphosphokinase reaction rate; JANT is the flux of ATP across ANT; JATPt is the flux of ATP across the outer mitochondrial membrane; QAKi and QAKc are the mitochondrial and cytoplasmic adenylate kinase reaction rates, respectively; QCK is the creatine kinase reaction rate; and QATP is the ATP consumption rate in the cytosol.
ATP formation is driven by the pmf, which is a function of the electric potential (Ψ) and proton concentration gradients across the inner membrane:
| (4) |
where R is the ideal gas constant, F is the Faraday constant, and T is the absolute temperature. We adopt here the bioenergetics convention (11), such that ΔΨ, ΔpH, and pmf all have positive signs under typical conditions. Given the permeability properties of the outer mitochondrial membrane, pHi is assumed to equal pHc (96). The model equation for pHx is based on conservation of mass and takes into account the generation and consumption of H+ via chemical reactions as well as H+ binding and unbinding. Note that the model considers the binding of H+, K+, and Mg2+ to multiple reactants, including ATP, GTP, GDP, and isocitrate (96). As an example, four ionic forms of ATP are considered: ATP4−, HATP3−, KATP3−, and MgATP2−.
In the original model of Wu et al. (96), hereafter referred to as the Wu et al. model, the flux of protons via leak pathways (JH,leak) was computed based on the Goldman equation. To account for the observed non-Ohmic, quasi-exponential dependence of proton leak on ΔΨ (76), the corresponding flux in the present study is calculated as in later models (5, 47):
| (5) |
where PH,leak is H+ leak permeability. We also include herein a K+ leak pathway (not considered in the Wu et al. model) to represent the diffusion of K+ from the intermembrane space into the inner matrix (40). K+ leak flux (JK,leak) is computed as follows:
| (6) |
K+ leak permeability (PK,leak) is taken as PH,leak × 10−6 in the base case. Note that K+ entry is counterbalanced by K+ exit via H+/K+ exchangers (Fig. 1).
Model Parameters
Structural/volume parameters for renal cortex mitochondria are shown in Table 1. The model assumes fixed cytoplasmic K+, H+, and Mg2+ concentrations. These are, respectively, taken as 110 mM (26), 0.40 mM (49), and 10−7.2 M (71, 97), as measured in PT cells. The concentration of O2 in the matrix is also fixed and set to 67 μM (i.e., Po2 = 50.0 mmHg) under basal conditions. The initial values of the cytoplasmic concentrations of Pi ([Pi]c), citrate, α-ketoglutarate, fumarate, malate, aspartate, pyruvate, glutamate, glucose, and glucose-6-phosphate are taken from Lemieux et al. (61), assuming a wet weight fraction of 0.80 (6). The initial values of the cytoplasmic concentration of succinate and phosphate creatine are taken from Horn et al. (48) and Toma et al. (90). The total concentration of adenosine phosphate species (ATP + ADP + AMP) is set to 2.75 mM (23, 61). Other initial values are those of the original model (96).
Table 1.
Model parameters for proximal convoluted tubule mitochondria
| Parameter | Value | Reference(s) |
|---|---|---|
| Fractional mitochondrial volume, L mitochondria/L cell | 0.25 | 15, 27, 46, 53 |
| Fractional cytoplasmic volume, L cytoplasm/L cell | 0.72 | Based on Ref. 72 |
| Cytoplasm water fraction, L water/L cytoplasm | 0.77 | 41, 60 |
| Mitochondria water fraction, L water/L mitochondria | 0.659 | 60 |
| Protein density of mitochondria, mg protein/L mitochondria | 3.274 × 105 | Based on Refs. 83, 94 |
| Total matrix concentration of NADH, mol/L matrix water | 0.824 × 10−3 | 19 |
| Total matrix concentration of coenzyme Q, mol/L matrix water | 2.15 × 10−3 | 7 |
| Total intermembrane concentration of cytochrome c, mol/L intermembrane water | 1.96 × 10−3 | 7 |
| Kidney-to-heart complex I activity ratio | 0.75 | 20, 98 |
| Kidney-to-heart complex III activity ratio | 0.50 | 7 |
| Kidney-to-heart complex IV activity ratio | 0.25 | 7 |
The model assumes that the total concentrations of NADH, coenzyme Q (CoQ), and cytochrome c (Cyt c) remain fixed. The total concentration of CoQ and Cyt c in renal cortex mitochondria is estimated based on kidney-to-heart content ratios (7), taking into account differences in mitochondrial protein density. Similarly, the activity of complexes I, III, and IV in PT mitochondria is estimated based on kidney-to-heart activity ratios (7, 20, 98). The hexokinase reaction rate is set to zero, given the very low hexokinase activity in the PT (82, 93). Other parameter values (including the adenylate kinase and creatine kinase reaction rate constants) are those of the original model. Kidney-specific parameter values are shown in Table 1.
ATP Consumption Rate
The baseline rate of ATP consumption in the proximal convoluted tubule is estimated as 9.57 pmol ATP·s−1·tubule−1 based on our published model of transport along the PT (58). The length and inner radius of the proximal convoluted tubule are taken as 0.97 and 12.5 × 10−4 cm, respectively; assuming that the cytoplasmic water volume equals 10−3 cm3/cm2 epithelium (95), the volume of water in the cytoplasm is 7.62 × 10−6 cm3/tubule, and the ATP consumption rate is equivalent to 1.26 × 10−3 mol ATP·s−1·L cytoplasm−1.
RESULTS
Isolated Mitochondria
The model was validated by comparing its predictions with our laboratory’s recent measurements of mitochondrial function in the renal cortex of Sprague-Dawley rats: the simulations mimicked the experimental protocol and bath conditions previously described in Schiffer et al. (79). In addition to the parameter choices described above (which were based on experimental data), PH,leak (Eq. 5) was set to 345 mol·s−1·M−1·L mitochondrial volume−1 to match measured leak respiration rates.
State 2 respiration, which is determined in the presence of glutamate and malate without ADP, characterizes Qo2 in the absence of ATP formation; under these “idle conditions,” the protons pumped into the intermembrane space reenter the matrix via H+ leak pathways that consume the pmf. On the subsequent addition of ADP, during state 3 respiration, F0F1-ATP synthase becomes the dominant pathway for proton reentry. Thus, the state 3-to-state 2 Qo2 ratio (Jo2,s3/Jo2,s2), also known as the respiratory control ratio (RCR), is a common measure of mitochondrial coupling. P/O is defined as the amount of ATP produced by F0F1-ATP synthase per oxygen atom reduced by the respiratory chain. The leak respiration is usually measured in the presence of oligomycin, which inhibits the F0 unit of ATP synthase and prevents ATP formation (51). As shown in Table 2, model predictions agree well with measured values of the state 3 respiration rate, RCR, leak respiration, and P/O (79).
Table 2.
Model predictions for isolated mitochondria
| Experimental Values (79) | Model Predictions | |
|---|---|---|
| State 3 respiration, nmol O2·mg−1·s−1 | 4.08 ± 0.60 | 4.09 |
| Respiratory control ratio | 8.5 ± 0.9 | 9.6 |
| Leak respiration, nmol O2·mg−1·s−1 | 0.37 ± 0.06 | 0.46 |
| P/O at half-maximum respiration | 1.8 ± 0.1 | 1.92 |
Respiration fluxes are per milligram of mitochondrial protein. P/O, amount of ATP produced by F0F1-ATP synthase per oxygen atom reduced by the respiratory chain.
The most uncertain parameters in this study are the leak permeabilities. To assess the sensitivity of model predictions to PH,leak, we varied its value by a factor ranging from 0.1 to 10 and computed the corresponding respiration rates under the experimental conditions of Schiffer et al. (79). As shown in Fig. 2, increasing PH,leak raises Jo2,s2 more than Jo2,s3, thereby lowering RCR. Indeed, as more protons reenter the matrix via H+ leak pathways, they dissipate a greater fraction of the pmf generated by substrate oxidation and reduce the driving force for ATP formation (i.e., the difference between Jo2,s3 and Jo2,s3). A twofold increase in PH,leak from its baseline value is predicted to reduce RCR from 9.6 to 6.9, if PK,leak is set to its baseline value.
Fig. 2.

Predicted effect of proton leak permeability (PH,leak) on state 2 and state 3 O2 consumption (A) and the respiratory control ratio (RCR; B) in isolated mitochondria. Results are shown for three different values of K+ leak permeability: 0, baseline (), and baseline × 10.
We also examined the impact of PK,leak. By itself, increasing PK,leak stimulates the cycling of K+ across the inner mitochondrial membrane; the resulting increase in H+/K+ exchange, in turn, stimulates the cycling of H+ and raises the respiration rate in both states 2 and 3 (Fig. 2A). Conversely, decreasing PK,leak reduces H+/K+ exchange, thereby diminishing H+ reentry via pathways other than F0F1-ATP synthase, lowering Jo2,s2 more than Jo2,s3, and elevating RCR (Fig. 2B).
Mitochondria In Vivo
Base case.
We then performed simulations of renal mitochondria function in vivo; the cytosolic concentration of pyruvate was set to 0.15 mM. Mitochondrial O2 concentration, which is also fixed in the model, was set to 67 μM (50 mmHg) in the base case and pHc to 7.20. Based on our previously published model of rat PT function (58), the baseline rate of ATP consumption in the cytoplasm (QATP) was set to 1.26 mmol·s−1·(L·cytoplasm)−1 [or 0.904 mmol·s−1·(L·cell)−1], as described above. Base-case results at steady state are shown in Table 3.
Table 3.
Model predictions for mitochondria in vivo
| Qo2*, nmol O2·mg−1·s−1 | GATP*, nmol ATP·mg−1·s−1 | P/O | pmf, mV | [ATP]c, mM | [ATP]c/[ADP]c | |
|---|---|---|---|---|---|---|
| Base case | ||||||
| Po2 = 50 mmHg, pHc = 7.20 | 2.49 | 10.05 | 2.02 | 172.76 | 2.51 | 10.94 |
| Variations in Po2 | ||||||
| Po2 = 60 mmHg | 2.49 | 10.05 | 2.02 | 173.12 | 2.51 | 11.10 |
| Po2 = 20 mmHg | 2.48 | 10.06 | 2.03 | 169.75 | 2.47 | 9.47 |
| Po2 = 10 mmHg | 2.46 | 10.07 | 2.05 | 164.93 | 2.35 | 6.44 |
| Variations in QATP | ||||||
| QATP × 0.75 | 1.93 | 7.52 | 1.95 | 173.52 | 2.54 | 12.84 |
| QATP × 1.25 | 3.05 | 12.59 | 2.06 | 172.02 | 2.43 | 8.24 |
| QATP × 1.50 | 3.61 | 15.12 | 2.09 | 171.14 | 2.18 | 4.43 |
| Variations in pHc | ||||||
| pHc = 7.40 | 2.47 | 10.06 | 2.04 | 175.54 | 2.40 | 7.48 |
| pHc = 7.00 | 2.52 | 10.04 | 1.99 | 169.06 | 2.57 | 14.49 |
| pHc = 6.80 | 2.57 | 10.02 | 1.95 | 164.55 | 2.58 | 16.06 |
| Variations in [K+]c | ||||||
| [K+]c = 60 mM | 2.42 | 10.08 | 2.08 | 172.84 | 2.52 | 11.92 |
| [K+]c = 140 mM | 2.53 | 10.04 | 1.99 | 172.70 | 2.50 | 10.48 |
| Variations in [Mg2+]c | ||||||
| [Mg2+]c = 0.2 mM | 2.49 | 10.05 | 2.02 | 172.71 | 2.45 | 8.66 |
| [Mg2+]c = 0.8 mM | 2.49 | 10.05 | 2.02 | 172.89 | 2.57 | 14.74 |
| Variations in H+ and K+ leak permeability | ||||||
| PH,leak = 0, PK,leak = 0 | 2.25 | 10.15 | 2.25 | 173.82 | 2.52 | 11.39 |
| PH,leak = 0, PK,leak × 1 | 2.40 | 10.09 | 2.10 | 173.17 | 2.51 | 11.12 |
| PH,leak = 0, PK,leak × 10 | 3.52 | 9.64 | 1.37 | 168.69 | 2.45 | 8.78 |
| PH,leak × 10, PK,leak = 0 | 3.08 | 9.82 | 1.59 | 169.96 | 2.47 | 9.58 |
| PH,leak × 10, PK,leak × 1 | 3.21 | 9.77 | 1.52 | 169.50 | 2.47 | 9.33 |
| PH,leak × 10, PK,leak × 10 | 4.17 | 9.38 | 1.13 | 165.87 | 2.38 | 7.05 |
[ADP]c, cytoplasmic ADP concentration; [ATP]c, cytoplasmic ATP concentration; [ATP]c/[ADP]c, ratio of [ATP]c to [ADP]c; GATP, ATP generation; [K+]c, cytoplasmic K+ concentration; [Mg2+]c, cytoplasmic Mg2+ concentration; pHc, cytoplasmic pH; PH,leak, H+ leak permeability; PK,leak, K+ leak permeability; pmf, proton motive force; P/O, amount of ATP produced by F0F1-ATP synthase per oxygen atom reduced by the respiratory chain; QATP, rate of ATP hydrolysis in the cytoplasm; Qo2, O2 consumption.
Results are given per milligram mitochondrial protein.
The predicted rate of ATP generation by F0F1-ATP synthase, 10.05 nmol·s−1·mg protein−1, is equivalent to 0.823 mmol·s−1·(L·cell)−1, which means that ATP synthase provides 91.0% of the ATP that is hydrolyzed in the cytoplasm. Some ATP is also generated by the conversion of phosphocreatine to creatine (catalyzed by creatine kinase) and by the conversion of ADP into AMP and ATP (catalyzed by adenylate kinase).
The cytoplasmic concentration of ATP ([ATP]c) at steady state is computed as 2.5 mM, in good agreement with measured values (1.5–2.8 mM) in rat renal tissue (23, 61). The predicted [Pi]c (1.7 mM) also falls within the published range of values (1–10 mM) (1, 23, 32, 61), and the predicted ATP-to-ADP cytoplasmic concentration ratio ([ATP]c/[ADP]c), 10.9, is comparable to that measured (~9) in mouse PTs (99). Freeman and colleagues (31) reported a P/O of 1.7–2.0 for the whole kidney in vivo (32) and 2.5 in the perfused kidney, and the model value (2.0) lies within these measured values. The predicted pmf (173 mV) is also within the typical 170- to 200-mV range (25).
Renal cortical Po2 is not limiting at the basal ATP consumption rate.
Renal tissue Po2 varies between 50 and 60 mmHg (67–80 μM) in the renal cortex and drops to 10–20 mmHg (13–27 μM) in the renal medulla (13, 62, 88). To determine whether O2 may be rate limiting in this range, we computed [ATP]c/[ADP]c as a function of mitochondrial Po2 for different values of QATP. The dependence of [ATP]c/[ADP]c on Po2 and QATP, as shown in Fig. 3, is qualitatively similar to that observed by Balaban et al. (4) in suspensions of rabbit cortical tubules: a decrease in QATP raises [ATP]c/[ADP]c, whereas low Po2 reduces [ATP]c/[ADP]c.
Fig. 3.
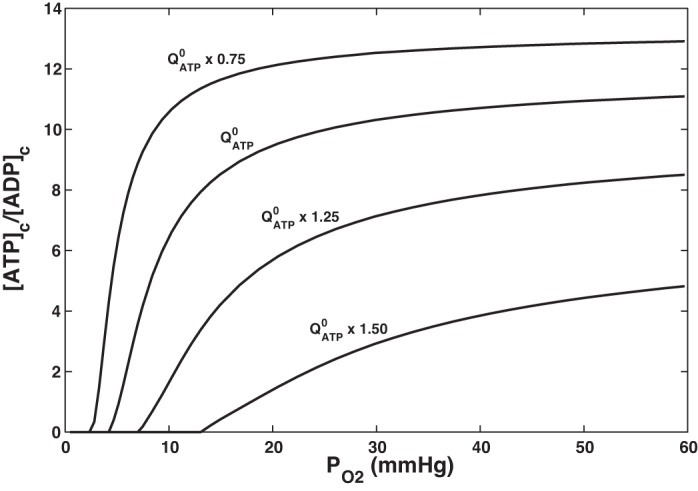
Predicted effect of Po2 on the ATP-to-ADP cytoplasmic concentration ratio ([ATP]c/[ADP]c) for different rates of ATP hydrolysis (QATP). denotes the baseline value of QATP. Cytoplasmic pH is set to 7.20.
At high Po2 and low QATP values, [ATP]c/[ADP]c exhibits a weak dependence on Po2. At baseline QATP, the predicted [ATP]c/[ADP]c remains > 10 if Po2 remains >25 mmHg. Below that threshold, the concentration ratio begins to fall rapidly with decreasing Po2 and is close to zero at Po2 ~5 mmHg. Furthermore, at baseline QATP, the predicted Qo2 and GATP change very little over the physiological Po2 range (10–60 mmHg). Toward the lower end of that interval, Qo2 and GATP are maintained to the detriment of [ATP]c, but the latter is predicted to remain >2 mM (Table 3).
We should note that QATP may not be independent of [ATP]c at lower ATP levels. The affinity of Na+-K+-ATPase for ATP is between 0.3 and 0.8 mM in most cells, and a value of 0.4 mM was measured in the rabbit PT (87). Hence, fixing the rate of ATP hydrolysis independently of [ATP]c is a reasonable assumption, unless the latter falls below millimolar levels.
Altogether, these results suggest that renal cortex O2 levels are not rate limiting when QATP remains close to its base-case value in the proximal convoluted tubule.
Raising QATP increases P/O.
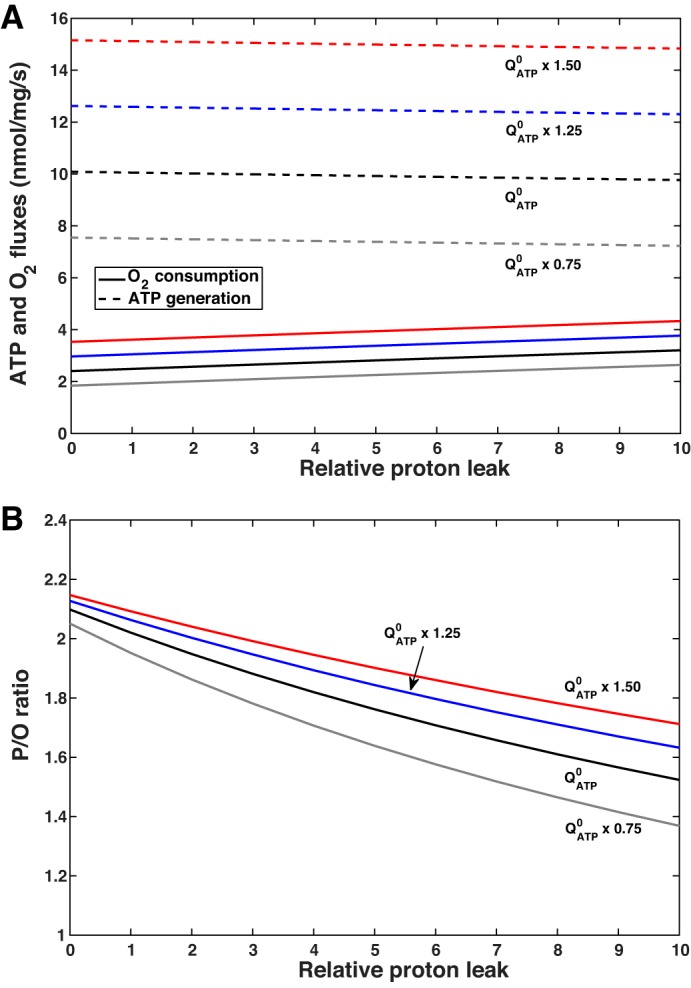
The rate of Na+ reabsorption, which governs the rate of ATP hydrolysis, may vary substantially in the PT, depending on many hemodynamic, hormonal, and neurogenic factors. We then examined the impact of ATP consumption on Qo2 and GATP at a fixed Po2 (50 mmHg). Raising QATP reduces [ATP]c/[ADP]c, which increases the driving force across ANT, in turn stimulating the production of ATP by F0F1-ATP synthase and Qo2 (Fig. 4A). When QATP was raised from 75% to 150% of its base-case value (at pHc = 7.20), the computed Qo2 and GATP doubled, and P/O increased from 1.95 to 2.09 (Table 3).
Fig. 4.

Predicted effect of ATP hydrolysis (QATP) on ATP formation by F0F1-ATP synthase and O2 consumption (A), P/O (amount of ATP produced by F0F1-ATP synthase per oxygen atom reduced by the respiratory chain; B), and the proton motive force (pmf; C) for different values of cytosolic pH (pHc): 7.4, 7.2, 7.0 and 6.8. Po2 is fixed at 50 mmHg.
These results can also be analyzed based on the proton circuit. Increasing QATP is predicted to lower the pmf (Fig. 4C), because it stimulates ATP formation and enhances proton reentry through F0F1-ATP synthase; the relative contribution of proton leak pathways to H+ reentry then diminishes. The drop in pmf, in turn, stimulates the electron transport chain and Qo2, since it lowers the energy required to pump protons across the inner mitochondrial membrane (11). As H+ leak pathways consume a smaller fraction of the pmf, GATP rises proportionally more than Qo2 and their ratio (i.e., P/O) increases.
P/O decreases with decreasing cytoplasmic pH.
Cytoplasmic pH (pHc) decreases along the PT. In rat tubules, pHc decreased from 7.14 in early proximal convolutions to 6.87 in late proximal convolutions under free-flow conditions and from 7.26 to 7.15 when tubules were perfused with 15 mM luminal (71). In the next set of simulations, we varied pHc between 7.4 and 6.8.
Lowering pHc (at a given QATP) is predicted to decrease the mitochondrial membrane potential and pmf (Fig. 4C), in accordance with the results of Wu et al. (96). This reduces the driving force across F0F1-ATP synthase, stimulates H+ pumping along the electron transport chain, and raises Qo2 (Fig. 4A). As a result, P/O is predicted to decrease with decreasing pHc (Fig. 4B). At baseline QATP, the computed P/O is 2.04 at pHc = 7.4 and 1.95 at pHc = 6.8.
Effects of cytosolic potassium, magnesium, and phosphate.
In all simulations, the cytosolic and intermembrane concentrations of K+ are taken to be equal and fixed, and so are those of Mg2+. As noted above, the model accounts for the binding of K+, Mg+, and H+ to pertinent reactants, including ATP. Thus, varying [K+]c (between 60 and 140 mM) and [Mg2+]c (between 0.2 and 0.8 mM) is predicted to affect the kinetic (time-dependent) behavior of the system, but has a small impact on the steady-state values of Qo2 and GATP (Table 3), as further described below.
Note first that the intracellular concentration and activity of K+ are significantly lower in PT cells than in myocytes (21, 26, 52, 73), and our baseline [K+]c value (110 mM) may be in the upper range (16, 26, 50). Decreasing [K+]c from 110 to 60 mM reduces H+/K+ exchange across the inner mitochondrial membrane (i.e., H+ reentry into the matrix) and raises the pmf. While this stimulates the activity of F0F1-ATP synthase, it also increases the electrochemical gradient against which the respiratory complexes have to pump H+, which acts to reduce Qo2 and raise P/O (Table 3).
A twofold increase or decrease in [Mg2+]c has a negligible effect on GATP, Qo2, and P/O; it has a more noticeable effect on [ATP]c/[ADP]c (Table 3) since the largest fraction of the Mg2+ pool in the cytoplasm and mitochondria is bound to ATP (77). Similarly, if the rate of ATP hydrolysis is fixed, reducing [Pi]c has a small impact on ATP production and Qo2, but it shifts the [ATP]c/[ADP]c toward lower values, in both the cytoplasm and mitochondria (results not shown).
Impact of H+ and K+ leak pathways.
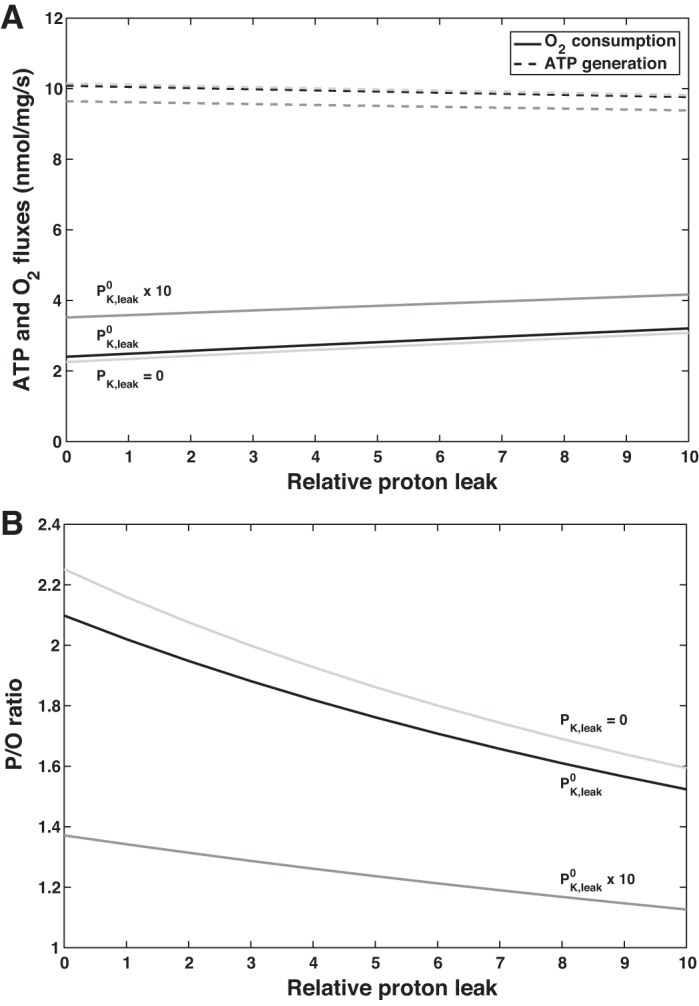
The main determinant of P/O is the conductance of H+ leak pathways, which shunt protons across the inner mitochondrial membrane, bypassing F0F1-ATP synthase. As discussed below, mitochondrial uncoupling is thought to help maintain homeostasis and reduce oxidative damage, but it may be harmful in the kidney as it elevates Qo2 and promotes hypoxia (80).
In the absence of H+ leak pathways, P/O is computed as 2.10. As expected, augmenting the leak of protons into the inner matrix decreases ΔpH and the pmf, thereby raising Qo2 while reducing ATP formation and P/O (Fig. 5). If PH,leak is increased 10-fold relative to the base case, Qo2 is predicted to rise by 29%, and ATP formation to decrease by 3%, yielding a P/O of 1.52. Note that if the rate of ATP hydrolysis is augmented at a given PH,leak value, ATP formation increases more than Qo2 (see above), and P/O is higher (Fig. 5).
Fig. 5.
Predicted effect of proton leak permeability on ATP formation by F0F1-ATP synthase and O2 consumption (A) and P/O (amount of ATP produced by F0F1-ATP synthase per oxygen atom reduced by the respiratory chain; B) for different values of ATP hydrolysis (QATP). K+ leak permeability is kept constant. denotes the baseline value of QATP.
Similarly, if K+ leak permeability is augmented at a given PH,leak value, K+ cycling across the inner membrane is enhanced, and more protons enter the inner matrix via H+/K+ exchangers, which lowers the pmf and reduces ATP formation. Conversely, Qo2 increases slightly, since less energy is then required to pump protons across the electron transport chain complexes (Fig. 6A). Thus, P/O decreases with increasing PK,leak, all else being constant (Fig. 6B).
Fig. 6.
Predicted effect of proton leak permeability on ATP formation by F0F1-ATP synthase and O2 consumption (A) and P/O (amount of ATP produced by F0F1-ATP synthase per oxygen atom reduced by the respiratory chain; B) for different values of K+ leak permeability (PK,leak). ATP hydrolysis (QATP) is maintained equal to the baseline value of QATP (). denotes the baseline value of PK,leak.
Uncoupling in diabetic PTs.
The expression of uncoupling protein (UCP)2 is enhanced in the diabetic kidney, and leak respiration is augmented (34, 67). A 50% increase in leak respiration in isolated mitochondria, as measured in diabetic rats (67), can be simulated by multiplying PH,leak by a factor of 2.5. Per se, a 2.5-fold increase in PH,leak is predicted to lower P/O by 5% (from 2.02 to 1.91) in mitochondria in vivo. However, other diabetes-related changes may conversely elevate P/O. Uncoupling contributes to increased Qo2 and intrarenal hypoxia in the diabetic kidney. A 15-mmHg decrease in cortical Po2, as previously reported in Friederich-Persson et al. (34), is predicted to very slightly raise P/O (see Table 3). In addition, increased Na+ reabsorption in the diabetic PT raises the rate of ATP hydrolysis, which by itself also elevates P/O (see above). Specifically, a 15% increase in QATP, reflecting the measured 15% increase in 86Rb uptake by PTs in diabetic rats (2), is predicted to raise P/O from 1.91 to 1.95. As discussed below, the present model can serve to elucidate the contribution of individual factors to altered mitochondrial metabolism in diabetes.
DISCUSSION
The goal of the present study was to develop a model of mitochondrial function in proximal convoluted tubule cells to gain more insight into the coupling between Qo2, ATP formation, ATP hydrolysis, and Na+ transport in that segment. We considered the proximal convoluted tubule in isolation herein, without taking into account cross-talk with other segments and possible compensating mechanisms. For this purpose, we extended the model developed by Wu et al. (96) for cardiac tissue and skeletal muscle mitochondria. The structural parameters and concentration values of the present model were adjusted based on PT-specific measurements (Table 1). We also modified the equation that characterizes the proton leak flux (Eq. 5) and added K+ leak conductance (Eq. 6). With these changes, the model correctly predicted both in vitro and in vivo measurements of Qo2, RCR, and P/O as well as ATP and Pi levels in PT cells (Table 2).
RCR is often used to assess mitochondrial function in vitro, because it encompasses three key metabolic processes, namely substrate oxidation, ATP formation, and proton leak (11). However, many factors affect RCR, including the nature of the exogenous substrate, and the concentrations of mitochondria and ATP (89). In isolated mitochondria from the rat renal cortex, RCR varies from ~4 to 9 (28, 64, 75, 78). We compared our theoretical results with the recently published in vitro measurements of Schiffer et al. (79) because experimental conditions in other studies were not fully characterized, and/or measurements related to succinate-based respiration, whereas the Wu et al. model does not account for succinate dehydrogenase (complex II).
The model predicts pmf values on the lower end of the typical 170–200 mV range (25), owing to a small contribution of the pH gradient (Eq. 4). Whereas the predicted ΔΨ is comparable to typical values (150–180 mV; see Ref. 11), the predicted ΔpH (~0.1 pH units, or 6 mV) is lower than most reported values (~0.5 pH units, or 30 mV), including indirect measurements in renal cortical tubules (1, 84). There are several possible explanations for these discrepancies. First, the mitochondrial model is based in part on the study of Bose et al. (9), who suggested that the low ΔpH values (~0.05 mV) they measured reflect the complex incubation ionic medium and higher temperature (37°C) they used. Second, accurate ΔpH measurements are difficult, owing to substantial heterogeneity within both the cytosol and mitochondria. Rieger et al. (74a) observed significant lateral ΔpH variations in HeLa cells, where the local pH at F0F1 dimers was 0.3 units less acidic than that at complex IV. Aw and Jones (1) ascribed inconsistencies in their data to a transcellular pH gradient within the cytosol of PT cells. Finally, theoretical analysis by Dzbek and Korzeniewski (25) suggests that the magnitude of ΔpH is strongly determined by K+ leak conductance. This conductance, which represents the K+ diffusion pathway that contributes to the cycling of K+ across the inner membrane (40), was not considered in the original Wu et al. model. In the present study, we assumed a small K+ leak conductance in PT mitochondria, as this pathway remains to be properly characterized. Moreover, the inner mitochondrial membrane expresses a variety of K+ channels, such as the ATP-regulated (mitoKATP) and Ca2+-activated (mitoBKCa) K+ channels that also regulate mitochondrial respiration (54). Experimental investigations of K+ transport pathways in renal mitochondria could shed more light on the specific impact of K+ on respiration in the kidney.
We should also note that Po2 is fixed in our computations. This assumption is appropriate for the proximal convoluted tubule, which is surrounded by a dense network of peritubular capillaries that maintain Po2 in the renal cortex at 50–60 mmHg. The simulations shown in Table 3 indicate that Qo2 and ATP generation vary imperceptibly when Po2 varies within the 50- to 60-mmHg range, if QATP is kept constant. Taken together, our results suggest that O2 levels are not rate limiting in the proximal convoluted tubule absent large metabolic (or pathological) perturbations.
Po2 may drop to 10–20 mmHg in the renal medulla (88), and our recent investigation suggests that the mitochondria of medullary cells are able to adapt to their hypoxic environment because of their higher efficiency: their RCR, P/O, and O2 affinity are higher than those of cortical mitochondria (81). Whether these adjustments result from greater abundance of TCA cycle components and/or electron transport chain complexes, as suggested by findings in porcine renal cells (91), remains to be ascertained.
The metabolic efficiency of a nephron segment is usually assessed by measuring the number of Na+ moles reabsorbed (TNa) per O2 mole consumed, often designated as the TNa-to-Qo2 ratio (TNa/Qo2). Three key factors determine TNa/Qo2: 1) the relative contributions of transcellular and paracellular Na+ transport, 2) the basal energy requirements of the cells, and 3) mitochondrial P/O. First, transcellular Na+ reabsorption is driven by basolateral Na+-K+-ATPase, which couples the hydrolysis of 1 ATP to the transport of 3 Na+ and 2 K+, but a significant fraction of luminal Na+ is passively reabsorbed across the paracellular route: ~30–50% of total TNa in the PT (42, 43, 58, 74). Hence, the ratio of TNa to the rate of ATP hydrolysis by Na+-K+-ATPase may differ substantially from 3. Second, not all of the ATP that is generated by oxidative phosphorylation is subsequently hydrolyzed by Na+-K+-ATPase. ATP serves to fuel other biochemical processes (including gluconeogenesis and H+-ATPase-mediated H+ secretion in the PT), and it is also generated or consumed in reactions such as the interconversion of ADP into ATP and AMP in the mitochondria and cytosol and the exchange of terminal phosphate between GTP and ATP in the mitochondria, both of which are considered in this study. Finally, some ATP is generated in the cytosol, and ATP generation in the mitochondria is not perfectly coupled to Qo2, owing to H+ leak pathways. The reentry of H+ into the inner matrix by pathways other than F0F1-ATP synthase dissipates part of the pmf generated by substrate oxidation and diminishes the driving force for ATP synthesis.
The coupling between Qo2 and ATP generation is encapsulated by P/O: the larger the H+ leak conductance, the smaller the P/O. In the base case, P/O is computed as 2.02. Assuming that all ATP generated by F0F1-ATP synthase is hydrolyzed by the Na+-K+ pump to actively transport Na+ and that Na+ transport is 40% passive and 60% active in the PT (see above), extrapolation of this P/O value yields a TNa/Qo2 of (1 + 0.40/0.60)(3)(2.02)(2) = 20.2, which is consistent with the reported range of 15–26 in the rat kidney (29).
Our simulations of mitochondrial function in vivo suggest that, if the levels of TCA cycle substrates and enzymes are maintained constant, P/O is strongly determined by QATP (Fig. 4B). An isolated increase in QATP raises P/O because it stimulates mitochondrial formation and export of ATP, thereby augmenting the fraction of the pmf that is consumed by ATP synthesis, relative to that consumed by H+ leak pathways. These results suggest that stimulating the activity of Na+-K+-ATPase may by itself increase the number of Na+ moles actively reabsorbed per O2 mole consumed (/Qo2), provided that Po2 levels are not limiting. /Qo2 ratio has been assumed until now to be independent of the Na+ transport rate (58, 59). Overall Na+ transport efficiency (TNa/Qo2) depends not only on basolateral Na+-K+-ATPase activity but also on Na+ fluxes across apical transporters as well as transepithelial concentration and electric potential gradients. These driving forces vary substantially along the PT, thereby modulating local TNa/Qo2. Our results suggest that -induced variations in mitochondrial metabolism efficiency also contribute to axial heterogeneities in Na+ transport efficiency.
Our results also suggest that pH has a significant impact on Na+ transport efficiency in the PT. Pastoriza-Munoz et al. (71) reported a decrease in pHc from the early to the late PT. Our simulations indicate that, at a fixed QATP, lowering pHc reduces the pmf; this, in turn, decreases ATP formation and P/O (Fig. 4B). In PT cells, pHc is linked to the activity of the apical Na+/H+ exchanger (NHE3), which is itself coupled to the activity of basolateral Na+-K+-ATPase and thus to QATP; in other words, the different factors (e.g., QATP and pHc) that regulate P/O and TNa/Qo2 in vivo are interwoven.
Since proton leak pathways partially uncouple Qo2 and ATP synthesis, they substantially increase metabolic expenditures; yet they must serve broadly beneficial purposes given their ubiquity (22). The electron transport chain generates reactive oxygen species (ROS), such as superoxide and hydrogen peroxide, as a byproduct of oxidative metabolism. According to the “uncoupling to survive” hypothesis, H+ leaks act to minimize the formation of ROS by lowering the pmf (10). The present model does not include the formation of ROS, but it does predict a significant impact of H+ leak fluxes on the pmf (Table 3).
Mitochondrial H+ leaks occur via basal and inducible pathways. Basal H+ conductance is mostly catalyzed by ANT, independently of its ATP/ADP exchange activity (12). The inducible H+ conductance is catalyzed by UCPs; UCP2 is the only isoform detected in the rat kidney, where its expression is confined to the PT and medullary thick ascending limb (35). Diabetes stimulates UCP2-mediated uncoupling in the rat kidney; interestingly, blockade of diabetes-induced UCP2 regulation activates mitochondrial uncoupling via ANT (33). However, the H+ transport properties of these pathways remain poorly understood, and the present model does not distinguish between constitutive and inducible proton leak conductances.
Mitochondrial uncoupling is thought to help maintain homeostasis and reduce oxidative damage, but, in pathological conditions where uncoupling is enhanced in the kidney, it may be harmful since it elevates Qo2 and promotes hypoxia (80). Our laboratory recently demonstrated that leak respiration and Qo2 are significantly elevated, and Po2 conversely decreased, in diabetic mice and that deletion of UCP2 lowers Qo2 and restores intrarenal O2 to normal levels (34). Baines and Ho (2) reported that /Qo2 decreases from 10.8 in nondiabetic PTs to 8.8 in diabetic tubules, an 18% reduction that they postulated stems from increased fatty acid metabolism. Simulations using the present model indicate that, by itself, a diabetes-induced 50% increase in leak respiration, as measured in diabetic rats (67), would lower P/O (a proxy for /Qo2) by 5%, but enhanced ATP consumption would attenuate this decrease. Diabetes is accompanied by hyperfiltration, hypertrophy (particularly of the PT) (92), increased flux of carbohydrate substrates into the mitochondria, and altered levels of TCA cycle enzymes (85); a decrease in mitochondrial protein content has also been observed in the diabetic mouse kidney (24). The present model may serve to assess the impact of each of these changes, in isolation and together, on mitochondrial function, when quantification of diabetes-induced changes in enzymatic and substrate content in PT cells becomes available.
In conclusion, our model of mitochondrial function in proximal convoluted tubule cells predicts that the rate of ATP hydrolysis and pHc are essential determinants of mitochondria metabolic efficiency, as assessed by P/O. Since Na+ reabsorption and pH vary along the PT, these results suggest that axial heterogeneities in Na+ transport efficiency stem in part from axial variations in mitochondrial metabolism. Our results also suggest that P/O may be significantly modulated by mitochondria K+ leak pathways, which remain to be characterized in the PT. Incorporating this model into cellular models of water and solute transport along the nephron and accounting for ROS formation will allow us to gain more insight into the role of mitochondrial dysfunction in acute and chronic renal diseases (17, 39, 45).
GRANTS
This work was supported by the Canada 150 Research Chair program and by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-106102 (to A. T. Layton).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.E. and A.T.L. conceived and designed research; A.E. performed experiments; A.E. analyzed data; A.E., F.P., and A.T.L. interpreted results of experiments; A.E. prepared figures; A.E. drafted manuscript; A.E., F.P., and A.T.L. edited and revised manuscript; A.E., F.P., and A.T.L. approved final version of manuscript.
REFERENCES
- 1.Aw TY, Jones DP. Heterogeneity of pH in the aqueous cytoplasm of renal proximal tubule cells. FASEB J 3: 52–58, 1989. doi: 10.1096/fasebj.3.1.2910737. [DOI] [PubMed] [Google Scholar]
- 2.Baines A, Ho P. Glucose stimulates O2 consumption, NOS, and Na/H exchange in diabetic rat proximal tubules. Am J Physiol Renal Physiol 283: F286–F293, 2002. doi: 10.1152/ajprenal.00330.2001. [DOI] [PubMed] [Google Scholar]
- 3.Balaban RS, Mandel LJ. Metabolic substrate utilization by rabbit proximal tubule. An NADH fluorescence study. Am J Physiol Renal Physiol 254: F407–F416, 1988. doi: 10.1152/ajprenal.1988.254.3.F407. [DOI] [PubMed] [Google Scholar]
- 4.Balaban RS, Mandel LJ, Soltoff SP, Storey JM. Coupling of active ion transport and aerobic respiratory rate in isolated renal tubules. Proc Natl Acad Sci USA 77: 447–451, 1980. doi: 10.1073/pnas.77.1.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bazil JN, Beard DA, Vinnakota KC. Catalytic coupling of oxidative phosphorylation, ATP demand, and reactive oxygen species generation. Biophys J 110: 962–971, 2016. doi: 10.1016/j.bpj.2015.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beck FB, Dörge A, Mason J, Rick R, Thurau K. Element concentrations of renal and hepatic cells under potassium depletion. Kidney Int 22: 250–256, 1982. doi: 10.1038/ki.1982.162. [DOI] [PubMed] [Google Scholar]
- 7.Benard G, Faustin B, Passerieux E, Galinier A, Rocher C, Bellance N, Delage JP, Casteilla L, Letellier T, Rossignol R. Physiological diversity of mitochondrial oxidative phosphorylation. Am J Physiol Cell Physiol 291: C1172–C1182, 2006. doi: 10.1152/ajpcell.00195.2006. [DOI] [PubMed] [Google Scholar]
- 8.Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol 13: 629–646, 2017. doi: 10.1038/nrneph.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bose S, French S, Evans FJ, Joubert F, Balaban RS. Metabolic network control of oxidative phosphorylation: multiple roles of inorganic phosphate. J Biol Chem 278: 39155–39165, 2003. doi: 10.1074/jbc.M306409200. [DOI] [PubMed] [Google Scholar]
- 10.Brand MD. Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp Gerontol 35: 811–820, 2000. doi: 10.1016/S0531-5565(00)00135-2. [DOI] [PubMed] [Google Scholar]
- 11.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J 435: 297–312, 2011. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, Brookes PS, Cornwall EJ. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem J 392: 353–362, 2005. doi: 10.1042/BJ20050890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brezis M, Rosen S. Hypoxia of the renal medulla−its implications for disease. N Engl J Med 332: 647–655, 1995. doi: 10.1056/NEJM199503093321006. [DOI] [PubMed] [Google Scholar]
- 14.Brezis M, Rosen S, Silva P, Epstein FH. Renal ischemia: a new perspective. Kidney Int 26: 375–383, 1984. doi: 10.1038/ki.1984.185. [DOI] [PubMed] [Google Scholar]
- 15.Calvo-Rubio M, Burón MI, López-Lluch G, Navas P, de Cabo R, Ramsey JJ, Villalba JM, González-Reyes JA. Dietary fat composition influences glomerular and proximal convoluted tubule cell structure and autophagic processes in kidneys from calorie-restricted mice. Aging Cell 15: 477–487, 2016. doi: 10.1111/acel.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cemerikić D, Wilcox CS, Giebisch G. Intracellular potential and K+ activity in rat kidney proximal tubular cells in acidosis and K+ depletion. J Membr Biol 69: 159–165, 1982. doi: 10.1007/BF01872275. [DOI] [PubMed] [Google Scholar]
- 17.Che R, Yuan Y, Huang S, Zhang A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am J Physiol Renal Physiol 306: F367–F378, 2014. doi: 10.1152/ajprenal.00571.2013. [DOI] [PubMed] [Google Scholar]
- 18.Chen J, Edwards A, Layton AT. Effects of pH and medullary blood flow on oxygen transport and sodium reabsorption in the rat outer medulla. Am J Physiol Renal Physiol 298: F1369–F1383, 2010. doi: 10.1152/ajprenal.00572.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coughlan MT, Nguyen TV, Penfold SA, Higgins GC, Thallas-Bonke V, Tan SM, Van Bergen NJ, Sourris KC, Harcourt BE, Thorburn DR, Trounce IA, Cooper ME, Forbes JM. Mapping time-course mitochondrial adaptations in the kidney in experimental diabetes. Clin Sci (Lond) 130: 711–720, 2016. doi: 10.1042/CS20150838. [DOI] [PubMed] [Google Scholar]
- 20.Daley E, Wilkie D, Loesch A, Hargreaves IP, Kendall DA, Pilkington GJ, Bates TE. Chlorimipramine: a novel anticancer agent with a mitochondrial target. Biochem Biophys Res Commun 328: 623–632, 2005. doi: 10.1016/j.bbrc.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 21.Désilets M, Baumgarten CM. K+, Na+, and Cl− activities in ventricular myocytes isolated from rabbit heart. Am J Physiol Cell Physiol 251: C197–C208, 1986. doi: 10.1152/ajpcell.1986.251.2.C197. [DOI] [PubMed] [Google Scholar]
- 22.Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 26: 192–205, 2011. doi: 10.1152/physiol.00046.2010. [DOI] [PubMed] [Google Scholar]
- 23.Dowd T, Barac-Nieto M, Gupta RK, Spitzer A. 31P nuclear magnetic resonance and saturation transfer studies of the isolated perfused rat kidney. Ren Physiol Biochem 12: 161–170, 1989. [DOI] [PubMed] [Google Scholar]
- 24.Dugan LL, You Y-H, Ali SS, Diamond-Stanic M, Miyamoto S, DeCleves A-E, Andreyev A, Quach T, Ly S, Shekhtman G, Nguyen W, Chepetan A, Le TP, Wang L, Xu M, Paik KP, Fogo A, Viollet B, Murphy A, Brosius F, Naviaux RK, Sharma K. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest 123: 4888–4899, 2013. doi: 10.1172/JCI66218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dzbek J, Korzeniewski B. Control over the contribution of the mitochondrial membrane potential(ΔΨ) and proton gradient(ΔpH) to the protonmotive force (Δp). J Biol Chem 283: 33232–33239, 2008. doi: 10.1074/jbc.M802404200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edelman A, Curci S, Samarzija I, Frömter E. Determination of intracellular K+ activity in rat kidney proximal tubular cells. Pflugers Arch 378: 37–45, 1978. doi: 10.1007/BF00581956. [DOI] [PubMed] [Google Scholar]
- 27.Else PL, Hulbert AJ. An allometric comparison of the mitochondria of mammalian and reptilian tissues: the implications for the evolution of endothermy. J Comp Physiol B 156: 3–11, 1985. doi: 10.1007/BF00692920. [DOI] [PubMed] [Google Scholar]
- 28.Elzinga LW, Mela-Riker LM, Widener LL, Bennett WM. Renal cortical mitochondrial integrity in experimental cyclosporine nephrotoxicity. Transplantation 48: 102–106, 1989. doi: 10.1097/00007890-198907000-00024. [DOI] [PubMed] [Google Scholar]
- 29.Evans RG, Harrop GK, Ngo JP, Ow CPC, O’Connor PM. Basal renal O2 consumption and the efficiency of O2 utilization for Na+ reabsorption. Am J Physiol Renal Physiol 306: F551–F560, 2014. doi: 10.1152/ajprenal.00473.2013. [DOI] [PubMed] [Google Scholar]
- 30.Fine LG, Norman JT. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: from hypothesis to novel therapeutics. Kidney Int 74: 867–872, 2008. doi: 10.1038/ki.2008.350. [DOI] [PubMed] [Google Scholar]
- 31.Freeman D, Bartlett S, Radda G, Ross B. Energetics of sodium transport in the kidney: saturation transfer 31P-NMR. Biochim Biophys Acta 762: 325–336, 1983. doi: 10.1016/0167-4889(83)90087-3. [DOI] [PubMed] [Google Scholar]
- 32.Freeman DM, Chan L, Yahaya H, Holloway P, Ross BD. Magnetic resonance spectroscopy for the determination of renal metabolic rate in vivo. Kidney Int 30: 35–42, 1986. doi: 10.1038/ki.1986.147. [DOI] [PubMed] [Google Scholar]
- 33.Friederich-Persson M, Aslam S, Nordquist L, Welch WJ, Wilcox CS, Palm F. Acute knockdown of uncoupling protein-2 increases uncoupling via the adenine nucleotide transporter and decreases oxidative stress in diabetic kidneys. PLoS One 7: e39635, 2012. doi: 10.1371/journal.pone.0039635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friederich-Persson M, Persson P, Hansell P, Palm F. Deletion of uncoupling protein-2 reduces renal mitochondrial leak respiration, intrarenal hypoxia and proteinuria in a mouse model of type 1 diabetes. Acta Physiol (Oxf) 223: e13058, 2018. doi: 10.1111/apha.13058. [DOI] [PubMed] [Google Scholar]
- 35.Friederich M, Nordquist L, Olerud J, Johansson M, Hansell P, Palm F. Identification and distribution of uncoupling protein isoforms in the normal and diabetic rat kidney. Adv Exp Med Biol 645: 205–212, 2009. doi: 10.1007/978-0-387-85998-9_32. [DOI] [PubMed] [Google Scholar]
- 36.Fry BC, Edwards A, Layton AT. Impact of nitric-oxide-mediated vasodilation and oxidative stress on renal medullary oxygenation: a modeling study. Am J Physiol Renal Physiol 310: F237–F247, 2016. doi: 10.1152/ajprenal.00334.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fry BC, Edwards A, Sgouralis I, Layton AT. Impact of renal medullary three-dimensional architecture on oxygen transport. Am J Physiol Renal Physiol 307: F263–F272, 2014. doi: 10.1152/ajprenal.00149.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fu Q, Colgan SP, Shelley CS. Hypoxia: the force that drives chronic kidney disease. Clin Med Res 14: 15–39, 2016. doi: 10.3121/cmr.2015.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galvan DL, Green NH, Danesh FR. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int 92: 1051–1057, 2017. doi: 10.1016/j.kint.2017.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garlid KD, Paucek P. Mitochondrial potassium transport: the K+ cycle. Biochim Biophys Acta 1606: 23–41, 2003. doi: 10.1016/S0005-2728(03)00108-7. [DOI] [PubMed] [Google Scholar]
- 41.Gershon ND, Porter KR, Trus BL. The cytoplasmic matrix: its volume and surface area and the diffusion of molecules through it. Proc Natl Acad Sci USA 82: 5030–5034, 1985. doi: 10.1073/pnas.82.15.5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Green R, Bishop JH, Giebisch G. Ionic requirements of proximal tubular sodium transport. III. Selective luminal anion substitution. Am J Physiol Renal Physiol 236: F268–F277, 1979. doi: 10.1152/ajprenal.1979.236.3.F268. [DOI] [PubMed] [Google Scholar]
- 43.Green R, Moriarty RJ, Giebisch G. Ionic requirements of proximal tubular fluid reabsorption flow dependence of fluid transport. Kidney Int 20: 580–587, 1981. doi: 10.1038/ki.1981.180. [DOI] [PubMed] [Google Scholar]
- 44.Guder WG, Ross BD. Enzyme distribution along the nephron. Kidney Int 26: 101–111, 1984. doi: 10.1038/ki.1984.143. [DOI] [PubMed] [Google Scholar]
- 45.Hall AM, Unwin RJ. The not so ‘mighty chondrion’: emergence of renal diseases due to mitochondrial dysfunction. Nephron Physiol 105: 1–10, 2007. doi: 10.1159/000096860. [DOI] [PubMed] [Google Scholar]
- 46.He X, Liu Y, Usa K, Tian Z, Cowley AWJ Jr, Liang M. Ultrastructure of mitochondria and the endoplasmic reticulum in renal tubules of Dahl salt-sensitive rats. Am J Physiol Renal Physiol 306: F1190–F1197, 2014. doi: 10.1152/ajprenal.00073.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heiske M, Letellier T, Klipp E. Comprehensive mathematical model of oxidative phosphorylation valid for physiological and pathological conditions. FEBS J 284: 2802–2828, 2017. doi: 10.1111/febs.14151. [DOI] [PubMed] [Google Scholar]
- 48.Horn M, Frantz S, Remkes H, Laser A, Urban B, Mettenleiter A, Schnackerz K, Neubauer S. Effects of chronic dietary creatine feeding on cardiac energy metabolism and on creatine content in heart, skeletal muscle, brain, liver and kidney. J Mol Cell Cardiol 30: 277–284, 1998. doi: 10.1006/jmcc.1997.0590. [DOI] [PubMed] [Google Scholar]
- 49.Ikari A, Kano T, Suketa Y. Magnesium influx enhanced by nitric oxide in hypertensive rat proximal tubule cells. Biochem Biophys Res Commun 294: 710–713, 2002. doi: 10.1016/S0006-291X(02)00542-9. [DOI] [PubMed] [Google Scholar]
- 50.Khuri RN, Agulian SK, Bogharian K. Electrochemical potentials of potassium in proximal renal tubule of rat. Pflugers Arch 346: 319–326, 1974. doi: 10.1007/BF00596187. [DOI] [PubMed] [Google Scholar]
- 51.Lanza IR, Nair KS. Functional assessment of isolated mitochondria in vitro. Methods Enzymol 457: 349–372, 2009. doi: 10.1016/S0076-6879(09)05020-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laprade R, Lapointe JY, Breton S, Duplain M, Cardinal J. Intracellular potassium activity in mammalian proximal tubule: effect of perturbations in transepithelial sodium transport. J Membr Biol 121: 249–259, 1991. doi: 10.1007/BF01951558. [DOI] [PubMed] [Google Scholar]
- 53.Larsson L. The ultrastructure of the developing proximal tubule in the rat kidney. J Ultrastruct Res 51: 119–139, 1975. doi: 10.1016/S0022-5320(75)80013-X. [DOI] [PubMed] [Google Scholar]
- 54.Laskowski M, Augustynek B, Kulawiak B, Koprowski P, Bednarczyk P, Jarmuszkiewicz W, Szewczyk A. What do we not know about mitochondrial potassium channels? Biochim Biophys Acta 1857: 1247–1257, 2016. doi: 10.1016/j.bbabio.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 55.Layton AT, Laghmani K, Vallon V, Edwards A. Solute transport and oxygen consumption along the nephrons: effects of Na+ transport inhibitors. Am J Physiol Renal Physiol 311: F1217–F1229, 2016. doi: 10.1152/ajprenal.00294.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Layton AT, Vallon V. SGLT2 inhibition in a kidney with reduced nephron number: modeling and analysis of solute transport and metabolism. Am J Physiol Renal Physiol 314: F969–F984, 2018. doi: 10.1152/ajprenal.00551.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Layton AT, Vallon V, Edwards A. A computational model for simulating solute transport and oxygen consumption along the nephrons. Am J Physiol Renal Physiol 311: F1378–F1390, 2016. doi: 10.1152/ajprenal.00293.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Layton AT, Vallon V, Edwards A. Modeling oxygen consumption in the proximal tubule: effects of NHE and SGLT2 inhibition. Am J Physiol Renal Physiol 308: F1343–F1357, 2015. doi: 10.1152/ajprenal.00007.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Layton AT, Vallon V, Edwards A. Predicted consequences of diabetes and SGLT inhibition on transport and oxygen consumption along a rat nephron. Am J Physiol Renal Physiol 310: F1269–F1283, 2016. doi: 10.1152/ajprenal.00543.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.LeFurgey A, Spencer AJ, Jacobs WR, Ingram P, Mandel LJ. Elemental microanalysis of organelles in proximal tubules. I. Alterations in transport and metabolism. J Am Soc Nephrol 1: 1305–1320, 1991. [DOI] [PubMed] [Google Scholar]
- 61.Lemieux G, Berkofsky J, Lemieux C. Renal tissue metabolism in the rat during chronic metabolic alkalosis: importance of glycolysis. Can J Physiol Pharmacol 64: 1419–1426, 1986. doi: 10.1139/y86-240. [DOI] [PubMed] [Google Scholar]
- 62.Lübbers DW, Baumgärtl H. Heterogeneities and profiles of oxygen pressure in brain and kidney as examples of the Po2 distribution in the living tissue. Kidney Int 51: 372–380, 1997. doi: 10.1038/ki.1997.49. [DOI] [PubMed] [Google Scholar]
- 63.Mandel LJ. Metabolic substrates, cellular energy production, and the regulation of proximal tubular transport. Annu Rev Physiol 47: 85–101, 1985. doi: 10.1146/annurev.ph.47.030185.000505. [DOI] [PubMed] [Google Scholar]
- 64.McMartin KE, Wallace KB. Calcium oxalate monohydrate, a metabolite of ethylene glycol, is toxic for rat renal mitochondrial function. Toxicol Sci 84: 195–200, 2005. doi: 10.1093/toxsci/kfi062. [DOI] [PubMed] [Google Scholar]
- 65.Mimura I, Nangaku M. The suffocating kidney: tubulointerstitial hypoxia in end-stage renal disease. Nat Rev Nephrol 6: 667–678, 2010. doi: 10.1038/nrneph.2010.124. [DOI] [PubMed] [Google Scholar]
- 66.Nangaku M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J Am Soc Nephrol 17: 17–25, 2006. doi: 10.1681/ASN.2005070757. [DOI] [PubMed] [Google Scholar]
- 67.Nordquist L, Friederich-Persson M, Fasching A, Liss P, Shoji K, Nangaku M, Hansell P, Palm F. Activation of hypoxia-inducible factors prevents diabetic nephropathy. J Am Soc Nephrol 26: 328–338, 2015. doi: 10.1681/ASN.2013090990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O’Connor PM. Renal oxygen delivery: matching delivery to metabolic demand. Clin Exp Pharmacol Physiol 33: 961–967, 2006. doi: 10.1111/j.1440-1681.2006.04475.x. [DOI] [PubMed] [Google Scholar]
- 69.Owen OE, Felig P, Morgan AP, Wahren J, Cahill GF Jr. Liver and kidney metabolism during prolonged starvation. J Clin Invest 48: 574–583, 1969. doi: 10.1172/JCI106016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Palm F, Nordquist L. Renal tubulointerstitial hypoxia: cause and consequence of kidney dysfunction. Clin Exp Pharmacol Physiol 38: 474–480, 2011. doi: 10.1111/j.1440-1681.2011.05532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pastoriza-Munoz E, Harrington RM, Graber ML. Axial heterogeneity of intracellular pH in rat proximal convoluted tubule. J Clin Invest 80: 207–215, 1987. doi: 10.1172/JCI113049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pfaller W, Rittinger M. Quantitative morphology of the rat kidney. Int J Biochem 12: 17–22, 1980. doi: 10.1016/0020-711X(80)90035-X. [DOI] [PubMed] [Google Scholar]
- 73.Polimeni PI. Extracellular space and ionic distribution in rat ventricle. Am J Physiol 227: 676–683, 1974. doi: 10.1152/ajplegacy.1974.227.3.676. [DOI] [PubMed] [Google Scholar]
- 74.Rector FC., Jr Sodium, bicarbonate, and chloride absorption by the proximal tubule. Am J Physiol Renal Physiol 244: F461–F471, 1983. doi: 10.1152/ajprenal.1983.244.5.F461. [DOI] [PubMed] [Google Scholar]
- 74a.Rieger B, Junge W, Busch KB. Lateral pH gradient between OXPHOS complex IV and F0F1 ATP-synthase in folded mitochondrial membranes. Nat Commun 5: 3103, 2014. doi: 10.1038/ncomms4103. [DOI] [PubMed] [Google Scholar]
- 75.Rivera MI, Jones TW, Lau SS, Monks TJ. Early morphological and biochemical changes during 2-Br-(diglutathion-S-yl)hydroquinone-induced nephrotoxicity. Toxicol Appl Pharmacol 128: 239–250, 1994. doi: 10.1006/taap.1994.1203. [DOI] [PubMed] [Google Scholar]
- 76.Rolfe DFS, Hulbert AJ, Brand MD. Characteristics of mitochondrial proton leak and control of oxidative phosphorylation in the major oxygen-consuming tissues of the rat. Biochim Biophys Acta 1188: 405–416, 1994. doi: 10.1016/0005-2728(94)90062-0. [DOI] [PubMed] [Google Scholar]
- 77.Romani AMP. Cellular magnesium homeostasis. Arch Biochem Biophys 512: 1–23, 2011. doi: 10.1016/j.abb.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rosca MG, Vazquez EJ, Chen Q, Kerner J, Kern TS, Hoppel CL. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes 61: 2074–2083, 2012. doi: 10.2337/db11-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schiffer TA, Christensen M, Gustafsson H, Palm F. The effect of inactin on kidney mitochondrial function and production of reactive oxygen species. PLoS One 13: e0207728, 2018. doi: 10.1371/journal.pone.0207728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schiffer TA, Friederich-Persson M. Mitochondrial reactive oxygen species and kidney hypoxia in the development of diabetic nephropathy. Front Physiol 8: 211, 2017. doi: 10.3389/fphys.2017.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schiffer TA, Gustafsson H, Palm F. Kidney outer medulla mitochondria are more efficient compared with cortex mitochondria as a strategy to sustain ATP production in a suboptimal environment. Am J Physiol Renal Physiol 315: F677–F681, 2018. doi: 10.1152/ajprenal.00207.2018. [DOI] [PubMed] [Google Scholar]
- 82.Schmidt U, Marosvari I, Dubach UC. Renal metabolism of glucose: anatomical sites of hexokinase activity in the rat nephron. FEBS Lett 53: 26–28, 1975. doi: 10.1016/0014-5793(75)80673-9. [DOI] [PubMed] [Google Scholar]
- 83.Schmitt S, Schulz S, Schropp E-M, Eberhagen C, Simmons A, Beisker W, Aichler M, Zischka H. Why to compare absolute numbers of mitochondria. Mitochondrion 19: 113–123, 2014. doi: 10.1016/j.mito.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 84.Schoolwerth AC, Gesek FA, Culpepper RM. Proton compartmentation in rat renal cortical tubules. Am J Physiol Renal Physiol 256: F986–F993, 1989. doi: 10.1152/ajprenal.1989.256.6.F986. [DOI] [PubMed] [Google Scholar]
- 85.Sharma K. Mitochondrial dysfunction in the diabetic kidney. : Mitochondrial Dynamics in Cardiovascular Medicine, edited by Santulli G. Cham, Switzerland: Springer International, 2017, p. 553–562. [DOI] [PubMed] [Google Scholar]
- 86.Singh P, McDonough AA, Thomson SC. Metabolic basis of solute transport. : Brenner and Rector’s The Kidney (10th Ed.), edited by Skorecki K, Chertow GM, Marsden PA, Taal MW, Yu AS. Philadelphia, PA: Elsevier, 2016, p. 122–143. [Google Scholar]
- 87.Soltoff SP, Mandel LJ. Active ion transport in the renal proximal tubule. III. The ATP dependence of the Na pump. J Gen Physiol 84: 643–662, 1984. doi: 10.1085/jgp.84.4.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stillman IE, Brezis M, Heyman SN, Epstein FH, Spokes K, Rosen S. Effects of salt depletion on the kidney: changes in medullary oxygenation and thick ascending limb size. J Am Soc Nephrol 4: 1538–1545, 1994. [DOI] [PubMed] [Google Scholar]
- 89.Tarjan EM, Von Korff RW. Factors affecting the respiratory control ratio of rabbit heart mitochondria. J Biol Chem 242: 318–324, 1967. [PubMed] [Google Scholar]
- 90.Toma I, Kang JJ, Sipos A, Vargas S, Bansal E, Hanner F, Meer E, Peti-Peterdi J. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J Clin Invest 118: 2526–2534, 2008. doi: 10.1172/JCI33293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tuma Z, Kuncova J, Mares J, Matejovic M. Mitochondrial proteomes of porcine kidney cortex and medulla: foundation for translational proteomics. Clin Exp Nephrol 20: 39–49, 2016. doi: 10.1007/s10157-015-1135-x. [DOI] [PubMed] [Google Scholar]
- 92.Vallon V, Komers R. Pathophysiology of the diabetic kidney. Compr Physiol 1: 1175–1232, 2011. doi: 10.1002/cphy.c100049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vandewalle A, Wirthensohn G, Heidrich HG, Guder WG. Distribution of hexokinase and phosphoenolpyruvate carboxykinase along the rabbit nephron. Am J Physiol Renal Physiol 240: F492–F500, 1981. doi: 10.1152/ajprenal.1981.240.6.F492. [DOI] [PubMed] [Google Scholar]
- 94.Walker DG. A survey of dehydrogenases in various epithelial cells in the rat. J Cell Biol 17: 255–277, 1963. doi: 10.1083/jcb.17.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weinstein AM, Weinbaum S, Duan Y, Du Z, Yan Q, Wang T. Flow-dependent transport in a mathematical model of rat proximal tubule. Am J Physiol Renal Physiol 292: F1164–F1181, 2007. doi: 10.1152/ajprenal.00392.2006. [DOI] [PubMed] [Google Scholar]
- 96.Wu F, Yang F, Vinnakota KC, Beard DA. Computer modeling of mitochondrial tricarboxylic acid cycle, oxidative phosphorylation, metabolite transport, and electrophysiology. J Biol Chem 282: 24525–24537, 2007. doi: 10.1074/jbc.M701024200. [DOI] [PubMed] [Google Scholar]
- 97.Yoshitomi K, Frömter E. Cell pH of rat renal proximal tubule in vivo and the conductive nature of peritubular (OH−) exit. Pflugers Arch 402: 300–305, 1984. doi: 10.1007/BF00585513. [DOI] [PubMed] [Google Scholar]
- 98.Yu H-T, Fu X-Y, Liang B, Wang S, Liu J-K, Wang S-R, Feng Z-H. Oxidative damage of mitochondrial respiratory chain in different organs of a rat model of diet-induced obesity. Eur J Nutr 57: 1957–1967, 2018. doi: 10.1007/s00394-017-1477-0. [DOI] [PubMed] [Google Scholar]
- 99.Zager RA. Mitochondrial free radical production induces lipid peroxidation during myohemoglobinuria. Kidney Int 49: 741–751, 1996. doi: 10.1038/ki.1996.103. [DOI] [PubMed] [Google Scholar]