

Abstract
Epithelial Na+ channels (ENaCs) are members of a family of cation channels that function as sensors of the extracellular environment. ENaCs are activated by specific proteases in the biosynthetic pathway and at the cell surface and remove embedded inhibitory tracts, which allows channels to transition to higher open-probability states. Resolved structures of ENaC and an acid-sensing ion channel revealed highly organized extracellular regions. Within the periphery of ENaC subunits are unique domains formed by antiparallel β-strands containing the inhibitory tracts and protease cleavage sites. ENaCs are inhibited by Na+ binding to specific extracellular site(s), which promotes channel transition to a lower open-probability state. Specific inositol phospholipids and channel modification by Cys-palmitoylation enhance channel open probability. How these regulatory factors interact in a concerted manner to influence channel open probability is an important question that has not been resolved. These various factors are reviewed, and the impact of specific factors on human disorders is discussed.
Keywords: ASIC, ENaC, gating, palmitoylation, phosphatidylinositol, protease, sodium
INTRODUCTION
Epithelial Na+ channels (ENaCs) were initially identified as a key pathway for Na+ transit across the apical membrane of high-resistance, Na+-transporting epithelia (57, 89). These channels were subsequently found to be expressed in a variety of epithelial and nonepithelial tissues (89, 172). In the kidney, ENaCs are expressed in the aldosterone-sensitive distal nephron (ASDN) and have a key role in fine-tuning Na+ absorption from the ultrafiltrate, serving as a final pathway for Na+ absorption in the nephron, where key hormones involved in the regulation of extracellular fluid volume and blood pressure exert their effects.
In addition to its role in transepithelial Na+ transport, electrogenic Na+ absorption via ENaC is coupled to K+ secretion in the ASDN, which is mediated by several apical K+ secretory channels. These include the inwardly rectifying K+ channel Kir1.1 [or renal outer medullary K+ channel (ROMK)] and the large-conductance Ca2+-activated K+ (BK) channel (36). Early inhibitors of ENaC, amiloride and triamterene, were developed as K+-sparing diuretics (57, 88). In the distal nephron, Na+ is absorbed with Cl− via the thiazide-sensitive Na+-Cl− cotransporter (NCC) in the distal convoluted tubule, or Na+ is absorbed via ENaC in exchange for K+ or in parallel with Cl− in later segments of the nephron. Recent studies have highlighted the role of plasma K+ concentration ([K+]) in regulation of NCC, as well as ENaC and K+ secretory channels (60, 160). When plasma [K+] is low, NCC is activated and ENaC is suppressed, favoring Na+ and Cl− absorption. When plasma [K+] is high, NCC is suppressed and ENaC is activated, favoring Na+ and K+ secretion.
Monogenetic mutations of ENaC in individuals with inherited forms of hypertension associated with hypokalemia (Liddle syndrome) or hypotension associated with hyperkalemia (pseudohypoaldosteronism type 1) underscore the importance of ENaC in controlling blood pressure as well as blood K+ (62, 143). Mutations associated with Liddle syndrome disrupt a Pro-Tyr (PY) motif in the COOH terminus of the β- or γ-subunit of ENaC, which impairs interaction of the channel with the ubiquitin ligase Nedd4-2, resulting in an increase in expression of channels at the plasma membrane as well as an increase in channel open probability (1, 49, 90).
It is likely that a subtle dysregulation of ENaC contributes to essential hypertension. A few specific ENaC variants have been associated with hypertension in specific populations, although they have not been associated with a gain of function when examined in heterologous expression systems (7, 16, 120, 151, 155). A gain-of-function ENaC variant was recently described in siblings with a Liddle syndrome phenotype (142). This variant is in the extracellular region of the α-subunit and is one of a growing number of variants in the extracellular regions of ENaC subunits that exhibit a gain-of-function phenotype (38, 136, 137). Whether other gain-of-function ENaC variants are associated with hypertension remains to be shown.
In addition to its role in the kidney in the regulated reabsorption of filtered Na+ and facilitation of K+ secretion, ENaCs are expressed at other sites that influence blood pressure. For example, the channel is expressed in lingual epithelia, where it has a role in salt taste (37, 96), and in the distal colon, where it absorbs ingested Na+ (30, 57, 89, 180). ENaCs are expressed in endothelia and vascular smooth muscle (14, 76, 91, 118, 154, 172). Its role at these sites in regulating vascular tone and blood pressure is still being explored. ENaCs are also expressed in antigen-presenting cells, where increases in extracellular Na+ activate an ENaC-dependent signaling cascade, resulting in release of proinflammatory cytokines that increase blood pressure (19, 118, 166).
ENaCs are also expressed in airway and alveolar epithelium, where they have important roles in regulating airway and alveolar surface liquid volumes (72, 106, 158). For example, enhanced ENaC activity in the airway may lead to reduced airway surface liquid volume, impaired mucociliary clearance, and inflammation (102). ENaC activity appears to be enhanced in cystic fibrosis (CF) and has been suggested to contribute to CF pathogenesis (45, 69). However, the role of ENaC in CF pathogenesis has not been settled (40, 74), and ENaC inhibitors have not proved to be beneficial in the treatment of CF (113, 144).
ENaC STRUCTURE
Studies following the initial cloning of ENaC and other members of the ENaC/degenerin family revealed a similar subunit structure: short cytoplasmic NH2 and COOH termini and two membrane-spanning domains connected by a large extracellular loop (30, 31, 150). The resolved structure of an acid-sensing ion channel (ASIC), a member of the ENaC/degenerin family, provided important insights regarding the structural organization of ASICs and other members of the ENaC/degenerin family (15, 75, 177). These features were confirmed with the recently resolved structure of ENaC (121). ENaCs are heterotrimers composed of structurally related subunits, referred to as α, β, and γ, whereas ASICs are homo- or heterooligomeric trimers. A δ-subunit is expressed in specific tissues, substituting for the α-subunit to form δβγ, with properties that differ from αβγ-subunit channels (58). δ-Subunits are not expressed in rodents (58, 62). Resolved structures revealed highly organized extracellular regions composed of discrete domains resembling a hand holding a ball (Fig. 1). Two of these domains, referred to as the palm and β-ball, are formed by β-strands and are in close proximity to the lipid bilayer. More peripheral domains, referred to as the thumb, finger, and knuckle, are formed primarily by α-helices.
Fig. 1.
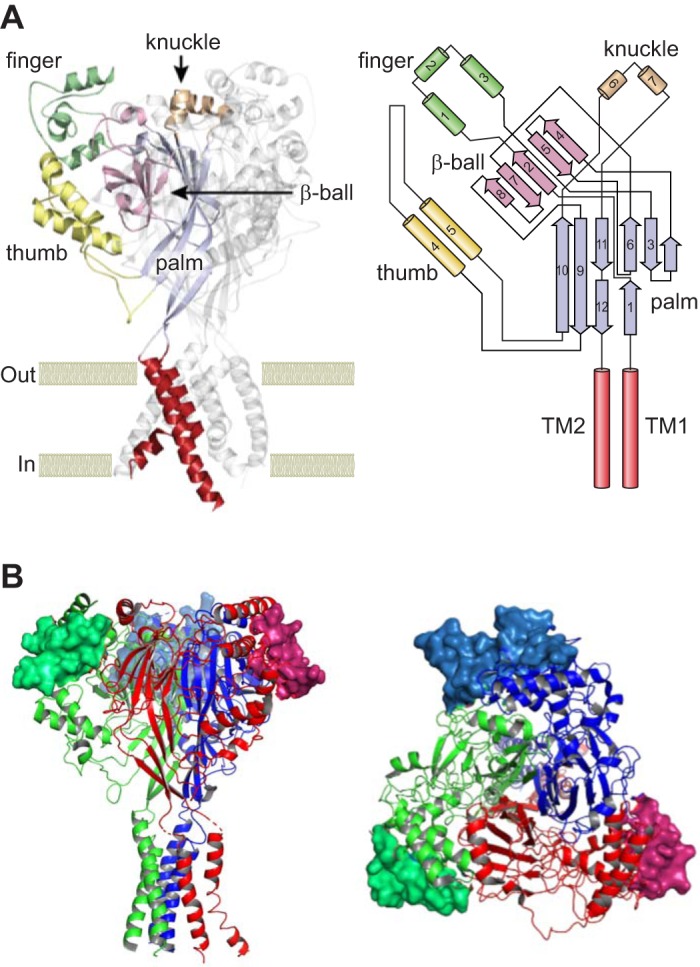
Acid-sensing ion channel (ASIC) type 1 and epithelial Na+ channel (ENaC) structures. A, left: ribbon illustration of the ASIC1 homotrimer, highlighting the extracellular and transmembrane regions of 1 subunit. Discrete domains within the extracellular region include the proximal palm and β-ball formed by β-strands and the peripheral α-helical thumb, finger, and knuckle domains. Transmembrane helices are indicated in red. A, right: organization of an ASIC1 subunit. Contiguous peripheral domains with α-helices (cylinders) arise from noncontiguous proximal domains with β-strands (arrows). TM, transmembrane. B: ribbon illustration of a human ENaC heterotrimer. Residues in the gating relief of inhibition by proteolysis (GRIP) domains that are unique to ENaC and contain the α- and γ-subunit-embedded inhibitory tracts are shown as surface rendering. α-Subunit (red), β-subunit (blue), and γ-subunit (green) are shown. Side (left) and top (right) views are shown. ASIC1 and ENaC structures are based on Protein Data Bank codes 4NYK and 6BQN (75, 121). [Top right and top left were modified from Kashlan and Kleyman (82), with permission.]
ENaC REGULATION
Most members of the ENaC/degenerin family are silent at baseline and activated by factors in the extracellular environment, including specific ions, peptides, or mechanical forces. On the other hand, ENaC is constitutively active, and its open probability is modified by extracellular factors, including ions, proteases, and mechanical forces (82, 83, 89). There is increasing evidence that these factors interact at specific sites within the extracellular regions of ENaC/degenerin family members, resulting in structural transitions that alter the conformation of the channel gate within the transmembrane pore and, in turn, changes in channel open probability. Within the extracellular region, the least-conserved domains are those containing α-helices, particularly the finger domain (78, 82). We and others have speculated that these poorly conserved regions have key roles in conferring specificity with regard to the factors that regulate distinct members of the ENaC/degenerin family. Perhaps this is best highlighted by the selective activation of ENaC by proteases that target unique regions in the extracellular domains of the α- and γ-subunits (78, 82, 121) (see below).
Functional ENaC expression is largely regulated by altering the number of channels at the plasma membrane and/or by altering open probability. As mentioned above, Liddle syndrome mutations affect both channel density at the cell surface and open probability. Aldosterone increases Na+ transport by increasing transcription and translation of specific ENaC subunits and by stabilizing channels at the plasma membrane. Frindt and Palmer (50) showed that this is indeed true in isolated rat tubules, but the increase in subunit density accounts for <25% of the increase in transtubular Na+ current, implying that the remaining 75% is likely due to an increase in single-channel open probability. Single-channel recordings in an amphibian principal cell culture model showed that acute application of aldosterone dramatically increases single-channel open probability (84). Aldosterone also enhances expression of proteolytically processed channels at the plasma membrane (see below). There have been extensive reviews on the regulation of ENaC surface density (25, 46, 124, 153). The remainder of our review examines specific factors that regulate ENaC open probability, focusing on αβγ-subunit channels. Several of these factors regulate ENaC open probability by interacting at sites within the extracellular regions of ENaC (see below).
Regulation by Proteases
The observation that Na+ transport across toad urinary bladder is reduced by the serine protease inhibitor aprotinin (122) provided the first hint that ENaCs are regulated by proteases. Vallet, Rossier, and colleagues (165) subsequently showed that ENaC is activated by the protease trypsin. This group identified prostasin as a channel-activating serine protease (165). They and others went on to identify a series of serine proteases and metalloproteases that can activate ENaC (63, 167). While it was initially unclear whether ENaC itself was the target of proteases, Masilamani, Knepper, and colleagues provided the first clue that an ENaC subunit (γ) was cleaved (105). Using ENaC subunits with NH2- and COOH-terminal epitope tags, Hughey, Kleyman, and co-workers showed that the α- and γ-subunits of ENaC were cleaved at defined sites within these subunits by furin, a trans-Golgi resident member of the proprotein family of serine proteases (70, 71). Introduction of mutations at key cleavage sites prevented both ENaC proteolytic processing and channel activation. The use of cell lines that lacked furin expression and selective furin inhibitors provided further evidence regarding the role of furin in cleaving and activating ENaC.
Studies using heterologous expression systems have clearly shown that proteases have a role in activating ENaC. Evidence from studies with in vivo systems is not as clear. These studies have focused on administration of serine protease inhibitors to rodents or have used mice where selected proteases have been knocked out. For example, administration of aprotinin, a nonselective serine protease inhibitor, to mice led to a natriuresis (21) and is consistent with protease-dependent activation of ENaC in vivo. Prostasin knockout mice exhibit abnormal skin development and early mortality (94) and, therefore, are not a useful model to study the role of prostasin in regulating ENaC in specific tissues. Selective knockout of prostasin in alveolae was associated with reduced fluid clearance (129), and a colonic knockout led to a moderate reduction in colonic potential difference (103). However, a kidney tubule-specific prostasin knockout has not yet been described. While kallikrein activates ENaC in heterologous expression systems, kallikrein knockout mice exhibited enhanced Na+ absorption in cortical collecting ducts (48). The increased Na+ transport may reflect an electroneutral process, as transepithelial voltage was unchanged. One concern is that multiple proteases might activate ENaC in specific settings and in specific cell types. Simply blocking one protease might not be sufficient to reduce ENaC activity in vivo (89).
A related question is whether proteolysis of ENaC subunits correlates with its activation in vivo. Frindt, Palmer, and co-workers recently found a discrepancy between the appearance of cleaved ENaC subunits (early response) and the increase in functional ENaC expression (late response) when rats were fed a low-salt diet (52). While cleaved subunits appeared early after initiation of the low-salt diet, the increase in channel activity was delayed by days. Inasmuch as a variety of factors influence ENaC open probability and/or channel trafficking, this observation is not surprising. Some of the factors that regulate ENaC open probability, in addition to proteolysis, are discussed below.
How do proteases activate the channel? A large body of evidence suggests that proteases activate ENaC by removing embedded inhibitory tracts in the α- and γ-subunits, transitioning channels to higher open-probability states (for reviews see Refs. 82, 87, 89, 141). The α-subunit is cleaved twice by furin. Channels lacking cleavage sites have a very low open probability and have been referred to as near-silent channels (29, 70, 146). Cleavage at both α-subunit furin sites is required to activate the channel, releasing a 26-residue embedded inhibitory tract and transitioning channels to a moderate-activity state (35, 146). Channels with a mutant α-subunit lacking this 26-residue inhibitory tract, as well as the α-subunit furin sites, are functional, although the α-subunit is not cleaved, demonstrating that release of the inhibitory tract, rather than cleavage per se, activates the channel (35). A peptide corresponding to the 26-residue tract inhibits ENaC, and within this 26-residue tract is a key 8-residue (LPHPLQRL) tract that retains inhibitory activity (34).
This paradigm for ENaC activation by proteases is also relevant to the γ-subunit. However, furin cleaves the γ-subunit only once (70). A growing number of proteases cleave the γ-subunit distal to the furin site, releasing an embedded inhibitory tract of >40 residues and transitioning channels to a high open-probability state (22). These proteases include prostasin, transmembrane serine protease 4, matriptase, cathepsins B and S, neutrophil and pancreatic elastase, kallikrein, meprin, urokinase, plasmin, and specific bacterial proteases (2, 5, 22, 26–28, 55, 56, 61, 63, 77, 126–128, 156, 159, 165, 167). Several proteases, including prostasin and kallikrein, appear to target a polybasic RKRK sequence distal to the furin site. In combination with furin, these proteases release a 43-residue tract (22, 126, 128). Within this 43-residue tract is an 11-residue (RFLNLIPLLVF) tract that retains inhibitory activity (125). Other proteases, such as plasmin and neutrophil and pancreatic elastase, cleave the γ-subunit at sites that are just distal to the RKRK tract (2, 127). Channels with a mutant γ-subunit lacking the 43-residue inhibitory tract as well as γ-subunit furin site have a high open probability, although the mutant γ-subunit is not cleaved (22).
Both the α- and γ-subunits have embedded inhibitory tracts that, when released, result in channel activation. Using a combination of wild-type and mutant ENaC subunits where the inhibitory tracts were either retained within a subunit or deleted, Carattino, Hughey, and Kleyman showed that the loss of the γ-subunit inhibitory tract has a dominant role in channel activation (32). They suggested that proteolytic processing and associated channel activation constitute a stepwise process (89) (Fig. 2). Noncleaved channels, in general, have a low open probability. Channels that have been processed by furin, where the α-subunit has lost its inhibitory tract and the γ-subunit has been cleaved once, have, in general, an intermediate open probability. Channels that have been processed by furin and a second protease, where the α- and γ-subunits have lost their inhibitory tracts, have, in general, a high open probability. An important caveat is that other important factors, in addition to proteases, influence channel open probability. Several key factors are discussed below. How these factors interact to modulate channel open probability requires further examination.
Fig. 2.

Epithelial Na+ channel (ENaC) activation by proteases. ENaC subunits assemble in the endoplasmic reticulum. As channels transit through the biosynthetic pathway, the α- and γ-subunits are processed by furin, releasing the α-subunit inhibitory tract. This favors transition of channels from a low to an intermediate open probability (Po). The γ-subunit is cleaved once by furin. Cleavage of this subunit by a second protease results in release of its inhibitory tract and favors transition of channels to a high open probability. There are other important factors that influence channel open probability. [Modified from Ray et al. (139), with permission from Elsevier.]
Several studies have reported COOH-terminal α- or γ-subunit cleavage fragments smaller than fragments generated by proteolytic processing associated with release of inhibitory tracts and channel activation. As discussed above, subunit cleavage is not necessary for channel activation (55, 119, 140, 179). We are not aware of studies demonstrating that ENaC subunit proteolysis resulting in smaller COOH-terminal α- or γ-subunit cleavage fragments modifies channel activity or is associated with the release of an inhibitory tract.
The α- and γ-subunit inhibitory tracts are present in the peripheral finger domains of these subunits and represent inserts that are not present in other members of the ENaC/degenerin family. For example, the α-subunit inhibitory tract is within a unique 72-residue insert that is not present in ASIC1 (75, 78). While the resolved structure of ASIC1 provided important insights into the organization of the extracellular regions of members of this ion channel family, it was unable to inform structural details regarding the inhibitory tracts in ENaC. Kashlan, Kleyman, and co-workers assessed the effects of an α-subunit inhibitory peptide on a large number of α-subunit mutants and, in combination with cross-linking studies, suggested that the inhibitory tract resides at a thumb-and-finger domain interface (78, 79, 81, 82). These investigators hypothesize that this interface is dynamic and that the inhibitory tract reduces channel open probability by stabilizing the interface (79). Subsequent work by Balchak, Thompson, Kashlan, and co-workers suggested that the γ-subunit inhibitory tract reduces channel activity by a similar mechanism (17). Noreng, Baconguis, and co-workers recently resolved the structure of the ENaC αβγ-subunit by cryoelectron microscopy (121). The structure confirmed that the inhibitory tract, which they referred to as the “gating relief of inhibition by proteolysis” (or GRIP) domain, is located at a thumb-and-finger domain interface. The domain is formed by antiparallel β-strands with accessible protease cleavage sites (Fig. 1).
ENaC subunit proteolysis has been observed in states of extracellular volume depletion and decreased effective arterial volume, such as heart failure, and with aldosterone administration in volume-replete states (50, 51, 183). As furin cleaves the α-subunit twice, α-subunit fragments with a size consistent with furin processing likely reflect release of the α-subunit inhibitory tract. However, an increase in γ-subunit cleavage does not necessarily reflect release of the γ-subunit inhibitory tract. Several lines of evidence, including use of an antibody against the γ-subunit inhibitory tract, suggest that this inhibitory tract is released from ENaCs expressed in kidneys in the settings of volume depletion or aldosterone administration (33, 156, 163, 179).
ENaC also appears to be activated by urinary proteases in nephrotic syndrome (21, 127, 138, 139, 156). In this setting, damaged glomeruli allow plasminogen to be filtered, and tubular urokinase converts this filtered plasminogen to activate protease plasmin. Plasmin can directly cleave the γ-subunit, facilitating the release of its inhibitory tract and activating the channel (127, 156). Plasmin may also influence ENaC activity by interacting with other proteases, such as prostasin (157). Other urinary proteases may also have a role in activating ENaC in nephrotic syndrome (77, 92). By cleaving the γ-subunit and activating ENaC, filtered proteases such as plasmin may contribute to urinary Na+ retention in nephrotic syndrome. Urinary plasminogen and plasmin have been found in a growing number of disease processes associated with glomerular proteinuria (8, 10, 23, 24, 138). The role of ENaC-mediated Na+ absorption in humans with proteinuria is still unclear. Few trials or case reports have examined the efficacy of amiloride in humans with proteinuria, and results have been inconsistent, with hyperkalemia as a complicating factor (9, 68, 123, 164, 176).
Regulation by Extracellular Na+
ENaC is a Na+- and Li+-selective channel. In addition to transporting Na+, the channel is inhibited by extracellular Na+, a process that has been termed Na+ self-inhibition and was first described more than 40 years ago (53). A growing body of evidence suggests that extracellular Na+ interacts at site(s) within the extracellular regions of ENaC subunits, driving allosteric changes that are transmitted to the channel gate to reduce channel open probability (20, 80, 81, 146). We have suggested that this regulatory response allows ENaCs in the ASDN to alter rates of Na+ influx in response to changes in urinary Na+ concentration (89).
The Na+ self-inhibition response is tightly linked to the extent of ENaC processing by proteases and is an excellent example of the interplay between different factors that regulate ENaC gating (20, 41, 146). Noncleaved channels exhibit a markedly enhanced Na+ self-inhibition response, reflecting the fact that the channels are largely closed when extracellular Na+ concentration is high (>100 mM). However, in the presence of low extracellular Na+ concentration, the channels are active, with an intermediate open probability reflecting relief from the inhibitory effect of Na+ (146). Channels that have been processed by furin exhibit a less-robust Na+ self-inhibition response and have an intermediate open probability in the presence of a high Na+ concentration, while channels processed by furin and a second protease, such as prostasin, have lost the Na+ self-inhibition response and have a notably high open probability in the presence of a high Na+ concentration (20, 22, 41, 146).
A large number of sites in the different domains of the extracellular regions of the α- and γ-subunits where amino acid substitutions alter the Na+ self-inhibition response and channel activity have been identified (38, 39, 42, 43, 81, 100, 145–149, 175). These mutations are at sites that may directly participate in Na+ binding or affect allosteric transitions that occur following Na+ binding. It has been challenging to differentiate site(s) involved in Na+ binding from other sites that affect Na+ self-inhibition. In native channels, this inhibitory response has a clear cation preference of Na+ > Li+ >> K+ (20, 80). Kashlan, Kleyman, and co-workers identified sites on an extracellular loop connecting the β6- and β7-strands and in the vicinity of an α-subunit acidic cleft, where mutations affected the cation selectivity of the Na+ self-inhibition response, suggesting that residues in this region serve as an effector-binding site for Na+ (80).
There are an increasing number of nonsynonymous single-nucleotide variants (nsSNVs, or missense mutations) in the genes encoding the three (α, β, and γ) ENaC subunits expressed in the ASDN that are reported in publicly available databases. We and others have identified rare human ENaC nsSNVs that result in either a gain or a loss of function that was associated with changes in the Na+ self-inhibition response. For example, both αW593R and γL511Q are gain-of-function variants that showed an increase in open probability and a loss of Na+ self-inhibition (38, 136) (Fig. 3). A recent report described siblings with a Liddle syndrome phenotype and gain-of-function mutation in the α-subunit (C479R) that appeared to reflect an increase in channel open probability (142). It was previously reported that an alanine substitution at the equivalent site in the mouse α-subunit exhibited a reduced Na+ self-inhibition response (147). Further studies are needed to answer the following questions. Do humans with ENaC gain-of-function variants that have a loss of the Na+ self-inhibition response have an increased risk of hypertension? Do humans with ENaC loss-of-function variants that have an enhanced the Na+ self-inhibition response have a reduced risk of hypertension.
Fig. 3.
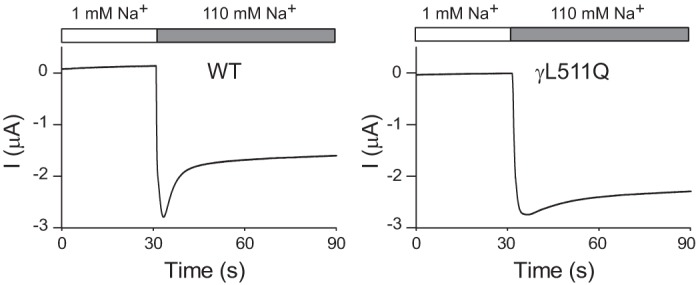
Epithelial Na+ channel (ENaC) Na+ self-inhibition response. Current traces illustrate the Na+ self-inhibition response assessed in Xenopus oocytes expressing wild-type (WT) human ENaC (left) or the γL511Q mutant (right). Oocytes were initially bathed in a solution containing 1 mM Na and 109 mM N-methyl-d-glucamine. After a rapid transition to a 110 mM Na bath, a rapid increase in inward Na+ current (I; downward deflection) was followed by a slow reduction in current, reflecting Na+ self-inhibition. The Na+ self-inhibition response in oocytes expressing the γL511Q mutant was markedly dampened. [Modified from Chen et al. (38), with permission.]
Regulation by Lipids
ENaC activity requires inositol lipid phosphates.
It has been known for a number of years that ENaC could be activated by application of phosphatidylinositol 4,5-bisphosphate (PIP2) to the cytosolic surface of channels in excised, inside-out patches (99, 130, 132, 133, 162, 178). Ma, Saxena, and Warnock found that the regulation of ENaC by PIP2 and phosphatidylinositol 3,4,5-trisphosphate (PIP3) did not involve a change in surface expression of ENaC, nor did it involve ENaC trafficking (99); i.e., it was a result of a change in open probability. The implication of these electrophysiological experiments is that PIP2 must interact with one or more of the ENaC subunits. In fact, an anti-PIP2 antibody coimmunoprecipitates the β- and γ-subunits, but not the α-subunit (178). Other anionic lipids also seemed capable of activating ENaC, but to a lesser extent than PIP2 (99). Subsequently, two binding sites for PIP2 were identified at the NH2 terminus of ENaC β- and γ-subunits (98), and one binding site for PIP3 was identified at the NH2 terminus of the ENaC γ-subunit (65). The same work also showed that all the early aldosterone-induced increase in Na+ transport was due to increases in inositol lipid phosphate binding. Other investigators described additional PIP2 and PIP3 binding sites to sites immediately following second-transmembrane domains (131–133, 152), but the NH2-terminal sites appear to be the critical sites for PIP2 regulation of ENaC.
Several other methods have been used to measure PIP2 binding to ENaC. “PIP strips” are nitrocellulose membranes prespotted with different lipids (3). When the strips are exposed to GST fusion proteins with different β- and γ-subunit domains, the fusion proteins bind to specific lipids. This method avoids the problem of the binding of lipid to nonspecific proteins associated with the antibody or beads in coimmunoprecipitations. It also avoids the problem of vagaries in transfection or expression of constructs regulating lipid production. PIP strip overlay binding assays showed that the NH2 terminus of β-subunits binds PIP2 and PIP3. In excised patches of apical membranes from Na+-transporting epithelial cells, both lipids activate ENaC (99, 178). Presumably, the β-subunit preferentially binds to PIP2 and PIP3, while the α- and γ-subunits do not because of differences within the NH2-terminal domains of the three subunits. The β- and γ-subunits also bind strongly to phosphatidic acid, a degradation product of inositol phospholipids that was recently shown to inhibit ENaC activity (182). These results suggest that hydrophobicity and anionic-character membrane phospholipids are critical for binding and that the inositol phosphate head group is necessary for ENaC activation. Investigators have hypothesized that ENaC activity is regulated by interaction of the positive charges within the NH2 terminus of ENaC with anionic phospholipids of the inner leaflet of the plasma membrane (65, 97, 133, 182).
PIP2 and PIP3 are localized in distinct nanoscale regions within the plasma membrane of cultured epithelial cells. Wang and Richards (171) used anti-phospholipid antibodies directly conjugated with Alexa Fluor 647 to show that PIP2 and PIP3 cluster in membrane domains of ~60 and ~130 nm, respectively. ENaC appears to be in lipid domains enriched in inositol lipid phosphates. Specifically, these are domains that contain high concentrations of inositol phospholipids and the enzymes that produce the lipids (95, 107, 161). Other investigators, using differential detergent solubility, also showed that some fraction of ENaC is present in PIP2-rich domains (3, 4, 66, 67). They concluded that ENaC was inserted into the membrane from the Golgi already in fully formed PIP2-rich domains but that the presence of ENaC in these domains was not necessary for function. However, while their evidence for the presence of ENaC in lipid domains is convincing, the conclusion that ENaC can function just as well in non-PIP2-rich as in PIP2-rich domain regions may not be correct. They concluded that if it were necessary for ENaC to be in these domains, then disrupting the domains should rapidly reduce ENaC function. Indeed, when they applied a domain-disrupting agent, methyl-β-cyclodextrin, (MβCD), Na+ current did decrease, but at a rate that they concluded was more consistent with disrupting the membrane insertion of rafts (with their embedded ENaC) than requiring the presence of ENaC in lipid domains for ENaC activity.
The problem with applying either MβCD or cholesterol to the apical surface of the cells is that lipids, including cholesterol in the outer membrane leaflet, are more ordered and, therefore, less accessible than in the less-ordered inner leaflet (73). Therefore, removal of cholesterol from the outer leaflet by MβCD may require high concentrations and may act more slowly, while removing cholesterol from the inner leaflet should be faster and occur at lower concentrations. In fact, in excised membrane patches, MβCD applied to the cytosolic surface of the membrane at a concentration 1,000 times lower than had been applied to the luminal surface (10–50 μM, instead of 10–50 mM) reduced ENaC channel activity to nearly zero in <5 min (181). The idea that disrupting the lipid domains eliminates all channel activity implies that only functional channels are in these domains.
MARCKS acts to maintain PIP2 in lipid domains and to promote ENaC-PIP2 interaction.
PIP2 is necessary to open ENaC. However, there is a conceptual problem with a simple model of ENaC and PIP2 associating by simple lateral diffusion in the membrane. ENaC is a relatively rare protein (only a few functional channels per μm2 in the apical membrane). PIP2 is also a rare molecule, constituting <1 in 1,000 membrane lipid molecules (73). Since PIP2 association is necessary for ENaC activity, it is possible to estimate the likelihood of random diffusional interaction between ENaC and PIP2 if we consider that PIP2 follows a random walk to reach ENaC. PIP2 moves by filling vacancies in the inner leaflet phospholipid structure; i.e., each step in the random walk is the cross-sectional size of a single PIP2 molecule, ~0.55 nm2 (73). The diffusion constant of PIP2 measured by fluorescence correlation spectroscopy is ~4 × 10−12 m2/s (101). For a typical principal cell, ENaC density will be ~7 channels/cm2. With these values, the mean time between collision of PIP2 and ENaC would be 6.3 × 102 s, or approximately once in 10 min. In fact, in principal cells, ENaC channels open every 1 or 2 s in a typical patch (18). The implication of these observations is that there must be a mechanism by which the local concentration of PIP2 could be increased close enough to ENaC to account for the apparently anonymously high opening rate. This could be accomplished by a protein that is associated with apical membrane lipid domains and is capable of binding and sequestering PIP2 with an affinity that would also allow PIP2 to also bind and activate ENaC. One such protein is MARCKS (myristoylated alanine-rich C-kinase substrate) or its closely related isoform MLP-1 (MARCKS-like protein 1). Both MARCKS and MLP-1 consist of a myristoylated NH2-terminal domain and an effector domain containing a large number of positively charged, basic amino acids. The positive charge within this domain electrostatically binds anionic lipids (e.g., PIP2) and causes MARCKS and MLP-1 to associate with PIP2-rich lipid domains (Fig. 4) (13, 54, 108, 110, 169, 170). The basic effector domain also contains protein kinase C (PKC) phosphorylation sites, cytoskeletal binding sites, and Ca2+/calmodulin-binding sites (64). MARCKS and MLP-1 reversibly associate with the inner leaflet of PIP2-rich lipid domains through hydrophobic and electrostatic interactions of their myristoyl group and basic effector domain, respectively (86). MARCKS and MLP-1 cross-link to actin, but the binding is regulated by PKC phosphorylation and calmodulin binding. As the name MARCKS implies, PKC phosphorylates three of the serine residues within the basic effector domain of MARCKS or MLP-1, adding anionic charge to the otherwise-positive effector domain. This change in overall charge disrupts the interaction with membrane PIP2 and leads to the translocation of MARCKS or MLP-1 from the membrane to the cytoplasm. Besides MARCKS and MLP-1, other members of the MARCKS family of proteins (growth-associated protein 43 and cardioactive peptide 23) also sequester PIPs (93), but only MARCKS or MLP-1 is present in Na+-transporting epithelial tissue (47, 173, 174). MARCKS has a predicted molecular mass of ~32 kDa but migrates slowly and close to 75 kDa on SDS-PAGE, probably due to its rod-shaped structure, unusual concentration of positive charge, and ability to bind SDS molecules. On SDS-PAGE, 26-kDa MLP-1 runs at 52 kDa.
Fig. 4.

Schematic diagram of myristoylated alanine-rich C-kinase substrate (MARCKS) and MARCKS-like protein 1 (MLP-1). The effector domain has multiple sites for phosphatidylinositol 4,5-bisphosphate (PIP2) interaction (note positive-charged residues) and sites for phosphorylation (note serine residues). MARCKS and MLP-1 function as a reversible source of PIP2 at the membrane. MLP-1 is the predominant MARCKS isoform in the mouse kidney. The ability of MARCKS and MLP-1 to function as a PIP2-sequestering protein at the membrane is dependent on hydrophobic interactions between the myristoylation domain and the membrane, and electrostatic forces between the effector domain and anionic lipids in the membrane.
ENaC is associated with MARCKS.
To be most effective, MARCKS needs to associate with PIP2-rich lipid domains, which it does through its myristoyl modification and association with PIP2, as we and others have shown (3, 11–13, 44, 59, 85, 110, 117, 168, 169), but MARCKS should also interact directly with ENaC. In fact, it is possible to coimmunoprecipitate the β-subunit of ENaC with MARCKS antibody (178).
MARCKS/MLP-1 association with PIP2-rich lipid domains is regulated.
Besides acting as a reversible source of PIP2 at the cytosolic surface of the apical membrane, MARCKS and MLP-1 can be regulated by controlling their level of association with the apical membrane, since after translocation from the apical membrane to the cytosol, MARCKS and MLP-1 associate with PIP2 (85, 86, 108, 109). Thus, MARCKS-mediated delivery of PIP2 to ENaC with consequent ENaC activation can be regulated by controlling MARCKS translocation. This is significant, since it shows an entirely new mechanism for altering ENaC activity. PKC-mediated phosphorylation reduces MARCKS association with the membrane and, thereby, diminishes the ability of MARCKS to present PIP2 to ENaC, with a concomitant reduction in ENaC activity; therefore, a reduction in PKC activity should increase ENaC activity by increasing open probability. The open probability of ENaC is higher in isolated, split-open tubules from PKCα knockout than wild-type mice. This increase in ENaC activity is associated with a substantial increase in the blood pressure of knockout mice (18), reinforcing the idea that MARCKS and MLP-1 are relevant physiologically.
Ca2+ might also neutralize the electrostatic interactions between MARCKS effector domain and PIP2 and promote phosphorylation and translocation (Fig. 5). Ca2+ can also activate calmodulin, which is known to promote MARCKS translocation (11).
Fig. 5.

Interaction of myristoylated alanine-rich C-kinase substrate (MARCKS) with the plasma membrane. Dephosphorylated MARCKS binds to the plasma membrane and cross-links to actin. Phosphorylated MARCKS detaches from the plasma membrane and disrupts actin filaments. PIP2, phosphatidylinositol 4,5-bisphosphate.
A model for MARCKS/MLP-1 regulation of ENaC and Na+ transport.
PIP2-rich membrane domains are formed in the Golgi in association with another chaperone protein, MAL/VIP17 (6, 104, 111, 112, 134, 135), and transit to the plasma membrane. To be open, ENaC must associate with an inositol lipid phosphate (e.g., PIP2). This interaction is facilitated by localizing ENaC in a PIP2-rich, cholesterol-containing apical membrane domain. Both PIP2 and ENaC can be stabilized in these domains by several chaperone proteins, including MARCKS or its closely related isoform MLP-1, both of which associate with the inner lipid leaflet. Under appropriate conditions (e.g., PKC activation), MARCKS/MLP-1 is phosphorylated and can move from association with the membrane lipid domain into the cytosol and then no longer stabilizes the PIP2-ENaC complex, allowing PIP2 to be hydrolyzed and ENaC to leave the specialized lipid domains. In the absence of PIP2 association, ENaC can be ubiquitinated and internalized.
ENaC is activated by Cys-palmitoylation.
Cys-palmitoylation is a reversible attachment of palmitate to cytoplasmic cysteine residues on proteins. Posttranslational modification by palmitoylation is another mechanism by which lipid molecules activate ENaC (114–116). The β- and γ-subunits of ENaC are palmitoylated at specific cytoplasmic cysteine residues (βCys43, βCys557, γCys33, and γCys41). Mutating these cysteine residues prevented palmitoylation and dramatically reduced channel open probability, while surface expression and proteolytic processing of channel subunits were unaffected (114, 115). Preventing γ-subunit palmitoylation had a more dominant role in inhibiting ENaC than preventing β-subunit palmitoylation (115). Five of the 23 known palmitoyltransferases (referred to as DHHCs) activate ENaC when coexpressed in Xenopus oocytes (116). The functional roles of palmitoyltransferases in regulating ENaC in vivo are unclear. The specific palmitoyltransferases that regulate ENaCs in principal cells in the kidney in vivo or in other cells are unknown. As multiple palmitoyltransferases could have a role in regulation of ENaC in a specific cell, knocking out a single DHHC may not be sufficient to prevent or reduce ENaC palmitoylation. Furthermore, specific DHHCs could modify other proteins that influence ENaC.
How do subunit palmitoylation and subunit interactions with PIP2 lead to an increase in channel open probability? We have proposed that palmitoylation or PIP2 binding facilitates interactions between cytoplasmic domains and the plasma membrane, resulting in conformational changes that are transmitted to the transmembrane domains and channel gating (89, 114, 115). However, the conformational changes associated with ENaC subunit palmitoylation or subunit interactions with PIP2 have not been defined. As the resolved structure of ENaC did not include cytoplasmic domains and constructs with truncated cytoplasmic domains were used to generate these structures, the detailed structural information needed to address these questions is lacking.
SUMMARY
A variety of extracellular and intracellular factors regulate ENaC open probability. Our overview of several important ENaC regulators provides insights into the complexity of this process. The vast majority of studies have examined these regulatory factors in isolation. How they work in concert to modulate ENaC gating is still unclear, as is an understanding of conformational changes that ultimately impinge on the channel’s gate. Our review has focused on the regulation of αβγ-subunit channels, with studies largely performed using heterologous expression systems and established cell lines. How these factors influence channels with differing subunit compositions and how they affect ENaCs expressed in different tissues are questions that continue to be addressed.
GRANTS
This work was supported by National Institutes of Health Grants HL-147818, DK-038470, and DK-079307 (to T. R. Kleyman) and DK-110409 (to D. C. Eaton).
DISCLOSURES
T. R. Kleyman receives an honorarium from Wiley, Inc., as Editor-in-Chief of Physiological Reports. D. C. Eaton has no conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
T.R.K. and D.C.E. drafted manuscript; T.R.K. and D.C.E. edited and revised manuscript; T.R.K. and D.C.E. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Drs. Shaohu Sheng, Ossama Kashlan, and Evan Ray for generating figures for this review.
REFERENCES
- 1.Abriel H, Loffing J, Rebhun JF, Pratt JH, Schild L, Horisberger JD, Rotin D, Staub O. Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle’s syndrome. J Clin Invest 103: 667–673, 1999. doi: 10.1172/JCI5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adebamiro A, Cheng Y, Rao US, Danahay H, Bridges RJ. A segment of γ-ENaC mediates elastase activation of Na+ transport. J Gen Physiol 130: 611–629, 2007. doi: 10.1085/jgp.200709781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alli AA, Bao HF, Alli AA, Aldrugh Y, Song JZ, Ma HP, Yu L, Al-Khalili O, Eaton DC. Phosphatidylinositol phosphate-dependent regulation of Xenopus ENaC by MARCKS protein. Am J Physiol Renal Physiol 303: F800–F811, 2012. doi: 10.1152/ajprenal.00703.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alli AA, Bao HF, Liu BC, Yu L, Aldrugh S, Montgomery DS, Ma HP, Eaton DC. Calmodulin and CaMKII modulate ENaC activity by regulating the association of MARCKS and the cytoskeleton with the apical membrane. Am J Physiol Renal Physiol 309: F456–F463, 2015. doi: 10.1152/ajprenal.00631.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alli AA, Song JZ, Al-Khalili O, Bao HF, Ma HP, Alli AA, Eaton DC. Cathepsin B is secreted apically from Xenopus 2F3 cells and cleaves the epithelial sodium channel (ENaC) to increase its activity. J Biol Chem 287: 30073–30083, 2012. doi: 10.1074/jbc.M111.338574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alonso MA, Millán J. The role of lipid rafts in signalling and membrane trafficking in T lymphocytes. J Cell Sci 114: 3957–3965, 2001. [DOI] [PubMed] [Google Scholar]
- 7.Ambrosius WT, Bloem LJ, Zhou L, Rebhun JF, Snyder PM, Wagner MA, Guo C, Pratt JH. Genetic variants in the epithelial sodium channel in relation to aldosterone and potassium excretion and risk for hypertension. Hypertension 34: 631–637, 1999. doi: 10.1161/01.HYP.34.4.631. [DOI] [PubMed] [Google Scholar]
- 8.Andersen H, Friis UG, Hansen PB, Svenningsen P, Henriksen JE, Jensen BL. Diabetic nephropathy is associated with increased urine excretion of proteases plasmin, prostasin and urokinase and activation of amiloride-sensitive current in collecting duct cells. Nephrol Dial Transplant 30: 781–789, 2015. doi: 10.1093/ndt/gfu402. [DOI] [PubMed] [Google Scholar]
- 9.Andersen H, Hansen PB, Bistrup C, Nielsen F, Henriksen JE, Jensen BL. Significant natriuretic and antihypertensive action of the epithelial sodium channel blocker amiloride in diabetic patients with and without nephropathy. J Hypertens 34: 1621–1629, 2016. doi: 10.1097/HJH.0000000000000967. [DOI] [PubMed] [Google Scholar]
- 10.Andersen RF, Buhl KB, Jensen BL, Svenningsen P, Friis UG, Jespersen B, Rittig S. Remission of nephrotic syndrome diminishes urinary plasmin content and abolishes activation of ENaC. Pediatr Nephrol 28: 1227–1234, 2013. doi: 10.1007/s00467-013-2439-2. [DOI] [PubMed] [Google Scholar]
- 11.Arbuzova A, Murray D, McLaughlin S. MARCKS, membranes, and calmodulin: kinetics of their interaction. Biochim Biophys Acta 1376: 369–379, 1998. doi: 10.1016/S0304-4157(98)00011-2. [DOI] [PubMed] [Google Scholar]
- 12.Arbuzova A, Wang J, Murray D, Jacob J, Cafiso DS, McLaughlin S. Kinetics of interaction of the myristoylated alanine-rich C kinase substrate, membranes, and calmodulin. J Biol Chem 272: 27167–27177, 1997. doi: 10.1074/jbc.272.43.27167. [DOI] [PubMed] [Google Scholar]
- 13.Arbuzova A, Wang L, Wang J, Hangyás-Mihályné G, Murray D, Honig B, McLaughlin S. Membrane binding of peptides containing both basic and aromatic residues. Experimental studies with peptides corresponding to the scaffolding region of caveolin and the effector region of MARCKS. Biochemistry 39: 10330–10339, 2000. doi: 10.1021/bi001039j. [DOI] [PubMed] [Google Scholar]
- 14.Ashley Z, Mugloo S, McDonald FJ, Fronius M. Epithelial Na+ channel differentially contributes to shear stress-mediated vascular responsiveness in carotid and mesenteric arteries from mice. Am J Physiol Heart Circ Physiol 314: H1022–H1032, 2018. doi: 10.1152/ajpheart.00506.2017. [DOI] [PubMed] [Google Scholar]
- 15.Baconguis I, Bohlen CJ, Goehring A, Julius D, Gouaux E. X-ray structure of acid-sensing ion channel 1-snake toxin complex reveals open state of a Na+-selective channel. Cell 156: 717–729, 2014. doi: 10.1016/j.cell.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker EH, Dong YB, Sagnella GA, Rothwell M, Onipinla AK, Markandu ND, Cappuccio FP, Cook DG, Persu A, Corvol P, Jeunemaitre X, Carter ND, MacGregor GA. Association of hypertension with T594M mutation in β-subunit of epithelial sodium channels in black people resident in London. Lancet 351: 1388–1392, 1998. doi: 10.1016/S0140-6736(97)07306-6. [DOI] [PubMed] [Google Scholar]
- 17.Balchak DM, Thompson RN, Kashlan OB. The epithelial Na+ channel γ subunit autoinhibitory tract suppresses channel activity by binding the γ subunit’s finger-thumb domain interface. J Biol Chem 293: 16217–16225, 2018. doi: 10.1074/jbc.RA118.004362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bao HF, Thai TL, Yue Q, Ma HP, Eaton AF, Cai H, Klein JD, Sands JM, Eaton DC. ENaC activity is increased in isolated, split-open cortical collecting ducts from protein kinase Cα knockout mice. Am J Physiol Renal Physiol 306: F309–F320, 2014. doi: 10.1152/ajprenal.00519.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, Mernaugh RL, Itani HA, Loperena R, Chen W, Dikalov S, Titze JM, Knollmann BC, Harrison DG, Kirabo A. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep 21: 1009–1020, 2017. doi: 10.1016/j.celrep.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bize V, Horisberger JD. Sodium self-inhibition of human epithelial sodium channel: selectivity and affinity of the extracellular sodium sensing site. Am J Physiol Renal Physiol 293: F1137–F1146, 2007. doi: 10.1152/ajprenal.00100.2007. [DOI] [PubMed] [Google Scholar]
- 21.Bohnert BN, Menacher M, Janessa A, Wörn M, Schork A, Daiminger S, Kalbacher H, Häring HU, Daniel C, Amann K, Sure F, Bertog M, Haerteis S, Korbmacher C, Artunc F. Aprotinin prevents proteolytic epithelial sodium channel (ENaC) activation and volume retention in nephrotic syndrome. Kidney Int 93: 159–172, 2018. doi: 10.1016/j.kint.2017.07.023. [DOI] [PubMed] [Google Scholar]
- 22.Bruns JB, Carattino MD, Sheng S, Maarouf AB, Weisz OA, Pilewski JM, Hughey RP, Kleyman TR. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the γ-subunit. J Biol Chem 282: 6153–6160, 2007. doi: 10.1074/jbc.M610636200. [DOI] [PubMed] [Google Scholar]
- 23.Buhl KB, Friis UG, Svenningsen P, Gulaveerasingam A, Ovesen P, Frederiksen-Møller B, Jespersen B, Bistrup C, Jensen BL. Urinary plasmin activates collecting duct ENaC current in preeclampsia. Hypertension 60: 1346–1351, 2012. doi: 10.1161/HYPERTENSIONAHA.112.198879. [DOI] [PubMed] [Google Scholar]
- 24.Buhl KB, Oxlund CS, Friis UG, Svenningsen P, Bistrup C, Jacobsen IA, Jensen BL. Plasmin in urine from patients with type 2 diabetes and treatment-resistant hypertension activates ENaC in vitro. J Hypertens 32: 1672–1677, 2014. doi: 10.1097/HJH.0000000000000216. [DOI] [PubMed] [Google Scholar]
- 25.Butterworth MB. Regulation of the epithelial sodium channel (ENaC) by membrane trafficking. Biochim Biophys Acta 1802: 1166–1177, 2010. doi: 10.1016/j.bbadis.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butterworth MB, Zhang L, Heidrich EM, Myerburg MM, Thibodeau PH. Activation of the epithelial sodium channel (ENaC) by the alkaline protease from Pseudomonas aeruginosa. J Biol Chem 287: 32556–32565, 2012. doi: 10.1074/jbc.M112.369520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butterworth MB, Zhang L, Liu X, Shanks RM, Thibodeau PH. Modulation of the epithelial sodium channel (ENaC) by bacterial metalloproteases and protease inhibitors. PLoS One 9: e100313, 2014. doi: 10.1371/journal.pone.0100313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caldwell RA, Boucher RC, Stutts MJ. Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am J Physiol Lung Cell Mol Physiol 288: L813–L819, 2005. doi: 10.1152/ajplung.00435.2004. [DOI] [PubMed] [Google Scholar]
- 29.Caldwell RA, Boucher RC, Stutts MJ. Serine protease activation of near-silent epithelial Na+ channels. Am J Physiol Cell Physiol 286: C190–C194, 2004. doi: 10.1152/ajpcell.00342.2003. [DOI] [PubMed] [Google Scholar]
- 30.Canessa CM, Merillat AM, Rossier BC. Membrane topology of the epithelial sodium channel in intact cells. Am J Physiol Cell Physiol 267: C1682–C1690, 1994. doi: 10.1152/ajpcell.1994.267.6.C1682. [DOI] [PubMed] [Google Scholar]
- 31.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367: 463–467, 1994. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 32.Carattino MD, Hughey RP, Kleyman TR. Proteolytic processing of the epithelial sodium channel γ-subunit has a dominant role in channel activation. J Biol Chem 283: 25290–25295, 2008. doi: 10.1074/jbc.M803931200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carattino MD, Mueller GM, Palmer LG, Frindt G, Rued AC, Hughey RP, Kleyman TR. Prostasin interacts with the epithelial Na+ channel and facilitates cleavage of the γ-subunit by a second protease. Am J Physiol Renal Physiol 307: F1080–F1087, 2014. doi: 10.1152/ajprenal.00157.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carattino MD, Passero CJ, Steren CA, Maarouf AB, Pilewski JM, Myerburg MM, Hughey RP, Kleyman TR. Defining an inhibitory domain in the α-subunit of the epithelial sodium channel. Am J Physiol Renal Physiol 294: F47–F52, 2008. doi: 10.1152/ajprenal.00399.2007. [DOI] [PubMed] [Google Scholar]
- 35.Carattino MD, Sheng S, Bruns JB, Pilewski JM, Hughey RP, Kleyman TR. The epithelial Na+ channel is inhibited by a peptide derived from proteolytic processing of its α-subunit. J Biol Chem 281: 18901–18907, 2006. doi: 10.1074/jbc.M604109200. [DOI] [PubMed] [Google Scholar]
- 36.Carrisoza-Gaytan R, Carattino MD, Kleyman TR, Satlin LM. An unexpected journey: conceptual evolution of mechanoregulated potassium transport in the distal nephron. Am J Physiol Cell Physiol 310: C243–C259, 2016. doi: 10.1152/ajpcell.00328.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chandrashekar J, Kuhn C, Oka Y, Yarmolinsky DA, Hummler E, Ryba NJ, Zuker CS. The cells and peripheral representation of sodium taste in mice. Nature 464: 297–301, 2010. doi: 10.1038/nature08783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen J, Kleyman TR, Sheng S. Gain-of-function variant of the human epithelial sodium channel. Am J Physiol Renal Physiol 304: F207–F213, 2013. doi: 10.1152/ajprenal.00563.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen J, Ray EC, Yates ME, Buck TM, Brodsky JL, Kinlough CL, Winarski KL, Hughey RP, Kleyman TR, Sheng S. Functional roles of clusters of hydrophobic and polar residues in the epithelial Na+ channel knuckle domain. J Biol Chem 290: 25140–25150, 2015. doi: 10.1074/jbc.M115.665398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen JH, Stoltz DA, Karp PH, Ernst SE, Pezzulo AA, Moninger TO, Rector MV, Reznikov LR, Launspach JL, Chaloner K, Zabner J, Welsh MJ. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell 143: 911–923, 2010. doi: 10.1016/j.cell.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chraïbi A, Horisberger JD. Na self inhibition of human epithelial Na channel: temperature dependence and effect of extracellular proteases. J Gen Physiol 120: 133–145, 2002. doi: 10.1085/jgp.20028612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collier DM, Snyder PM. Extracellular chloride regulates the epithelial sodium channel. J Biol Chem 284: 29320–29325, 2009. doi: 10.1074/jbc.M109.046771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collier DM, Snyder PM. Identification of epithelial Na+ channel (ENaC) intersubunit Cl− inhibitory residues suggests a trimeric αγβ channel architecture. J Biol Chem 286: 6027–6032, 2011. doi: 10.1074/jbc.M110.198127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Denisov G, Wanaski S, Luan P, Glaser M, McLaughlin S. Binding of basic peptides to membranes produces lateral domains enriched in the acidic lipids phosphatidylserine and phosphatidylinositol 4,5-bisphosphate: an electrostatic model and experimental results. Biophys J 74: 731–744, 1998. doi: 10.1016/S0006-3495(98)73998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Donaldson SH, Boucher RC. Sodium channels and cystic fibrosis. Chest 132: 1631–1636, 2007. doi: 10.1378/chest.07-0288. [DOI] [PubMed] [Google Scholar]
- 46.Eaton DC, Malik B, Bao HF, Yu L, Jain L. Regulation of epithelial sodium channel trafficking by ubiquitination. Proc Am Thorac Soc 7: 54–64, 2010. doi: 10.1513/pats.200909-096JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El Amri M, Fitzgerald U, Schlosser G. MARCKS and MARCKS-like proteins in development and regeneration. J Biomed Sci 25: 43, 2018. doi: 10.1186/s12929-018-0445-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.El Moghrabi S, Houillier P, Picard N, Sohet F, Wootla B, Bloch-Faure M, Leviel F, Cheval L, Frische S, Meneton P, Eladari D, Chambrey R. Tissue kallikrein permits early renal adaptation to potassium load. Proc Natl Acad Sci USA 107: 13526–13531, 2010. doi: 10.1073/pnas.0913070107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Firsov D, Schild L, Gautschi I, Mérillat AM, Schneeberger E, Rossier BC. Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: a quantitative approach. Proc Natl Acad Sci USA 93: 15370–15375, 1996. doi: 10.1073/pnas.93.26.15370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frindt G, Palmer LG. Acute effects of aldosterone on the epithelial Na channel in rat kidney. Am J Physiol Renal Physiol 308: F572–F578, 2015. doi: 10.1152/ajprenal.00585.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frindt G, Palmer LG. Surface expression of sodium channels and transporters in rat kidney: effects of dietary sodium. Am J Physiol Renal Physiol 297: F1249–F1255, 2009. doi: 10.1152/ajprenal.00401.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frindt G, Yang L, Bamberg K, Palmer LG. Na restriction activates epithelial Na channels in rat kidney through two mechanisms and decreases distal Na delivery. J Physiol 596: 3585–3602, 2018. doi: 10.1113/JP275988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fuchs W, Larsen EH, Lindemann B. Current-voltage curve of sodium channels and concentration dependence of sodium permeability in frog skin. J Physiol 267: 137–166, 1977. doi: 10.1113/jphysiol.1977.sp011805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gambhir A, Hangyás-Mihályné G, Zaitseva I, Cafiso DS, Wang J, Murray D, Pentyala SN, Smith SO, McLaughlin S. Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys J 86: 2188–2207, 2004. doi: 10.1016/S0006-3495(04)74278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.García-Caballero A, Dang Y, He H, Stutts MJ. ENaC proteolytic regulation by channel-activating protease 2. J Gen Physiol 132: 521–535, 2008. doi: 10.1085/jgp.200810030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garcia-Caballero A, Ishmael SS, Dang Y, Gillie D, Bond JS, Milgram SL, Stutts MJ. Activation of the epithelial sodium channel by the metalloprotease meprin β subunit. Channels (Austin) 5: 14–22, 2011. doi: 10.4161/chan.5.1.13759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev 77: 359–396, 1997. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 58.Giraldez T, Rojas P, Jou J, Flores C, Alvarez de la Rosa D. The epithelial sodium channel δ-subunit: new notes for an old song. Am J Physiol Renal Physiol 303: F328–F338, 2012. doi: 10.1152/ajprenal.00116.2012. [DOI] [PubMed] [Google Scholar]
- 59.Glaser M, Wanaski S, Buser CA, Boguslavsky V, Rashidzada W, Morris A, Rebecchi M, Scarlata SF, Runnels LW, Prestwich GD, Chen J, Aderem A, Ahn J, McLaughlin S. Myristoylated alanine-rich C kinase substrate (MARCKS) produces reversible inhibition of phospholipase C by sequestering phosphatidylinositol 4,5-bisphosphate in lateral domains. J Biol Chem 271: 26187–26193, 1996. doi: 10.1074/jbc.271.42.26187. [DOI] [PubMed] [Google Scholar]
- 60.Hadchouel J, Ellison DH, Gamba G. Regulation of renal electrolyte transport by WNK and SPAK-OSR1 kinases. Annu Rev Physiol 78: 367–389, 2016. doi: 10.1146/annurev-physiol-021115-105431. [DOI] [PubMed] [Google Scholar]
- 61.Haerteis S, Krappitz M, Bertog M, Krappitz A, Baraznenok V, Henderson I, Lindström E, Murphy JE, Bunnett NW, Korbmacher C. Proteolytic activation of the epithelial sodium channel (ENaC) by the cysteine protease cathepsin-S. Pflugers Arch 464: 353–365, 2012. doi: 10.1007/s00424-012-1138-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hanukoglu I, Hanukoglu A. Epithelial sodium channel (ENaC) family: phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 579: 95–132, 2016. doi: 10.1016/j.gene.2015.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harris M, Firsov D, Vuagniaux G, Stutts MJ, Rossier BC. A novel neutrophil elastase inhibitor prevents elastase activation and surface cleavage of the epithelial sodium channel expressed in Xenopus laevis oocytes. J Biol Chem 282: 58–64, 2007. doi: 10.1074/jbc.M605125200. [DOI] [PubMed] [Google Scholar]
- 64.Hartwig JH, Thelen M, Rosen A, Janmey PA, Nairn AC, Aderem A. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium-calmodulin. Nature 356: 618–622, 1992. doi: 10.1038/356618a0. [DOI] [PubMed] [Google Scholar]
- 65.Helms MN, Liu L, Liang YY, Al-Khalili O, Vandewalle A, Saxena S, Eaton DC, Ma HP. Phosphatidylinositol 3,4,5-trisphosphate mediates aldosterone stimulation of epithelial sodium channel (ENaC) and interacts with γ-ENaC. J Biol Chem 280: 40885–40891, 2005. doi: 10.1074/jbc.M509646200. [DOI] [PubMed] [Google Scholar]
- 66.Hill WG, An B, Johnson JP. Endogenously expressed epithelial sodium channel is present in lipid rafts in A6 cells. J Biol Chem 277: 33541–33544, 2002. doi: 10.1074/jbc.C200309200. [DOI] [PubMed] [Google Scholar]
- 67.Hill WG, Butterworth MB, Wang H, Edinger RS, Lebowitz J, Peters KW, Frizzell RA, Johnson JP. The epithelial sodium channel (ENaC) traffics to apical membrane in lipid rafts in mouse cortical collecting duct cells. J Biol Chem 282: 37402–37411, 2007. doi: 10.1074/jbc.M704084200. [DOI] [PubMed] [Google Scholar]
- 68.Hinrichs GR, Mortensen LA, Jensen BL, Bistrup C. Amiloride resolves resistant edema and hypertension in a patient with nephrotic syndrome; a case report. Physiol Rep 6: e13743, 2018. doi: 10.14814/phy2.13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hobbs CA, Da Tan C, Tarran R. Does epithelial sodium channel hyperactivity contribute to cystic fibrosis lung disease? J Physiol 591: 4377–4387, 2013. doi: 10.1113/jphysiol.2012.240861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD, Johnson JP, Stockand JD, Kleyman TR. Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem 279: 18111–18114, 2004. doi: 10.1074/jbc.C400080200. [DOI] [PubMed] [Google Scholar]
- 71.Hughey RP, Mueller GM, Bruns JB, Kinlough CL, Poland PA, Harkleroad KL, Carattino MD, Kleyman TR. Maturation of the epithelial Na+ channel involves proteolytic processing of the α- and γ-subunits. J Biol Chem 278: 37073–37082, 2003. doi: 10.1074/jbc.M307003200. [DOI] [PubMed] [Google Scholar]
- 72.Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A, Boucher R, Rossier BC. Early death due to defective neonatal lung liquid clearance in α-ENaC-deficient mice. Nat Genet 12: 325–328, 1996. doi: 10.1038/ng0396-325. [DOI] [PubMed] [Google Scholar]
- 73.Ingólfsson HI, Melo MN, van Eerden FJ, Arnarez C, Lopez CA, Wassenaar TA, Periole X, de Vries AH, Tieleman DP, Marrink SJ. Lipid organization of the plasma membrane. J Am Chem Soc 136: 14554–14559, 2014. doi: 10.1021/ja507832e. [DOI] [PubMed] [Google Scholar]
- 74.Itani OA, Chen JH, Karp PH, Ernst S, Keshavjee S, Parekh K, Klesney-Tait J, Zabner J, Welsh MJ. Human cystic fibrosis airway epithelia have reduced Cl− conductance but not increased Na+ conductance. Proc Natl Acad Sci USA 108: 10260–10265, 2011. doi: 10.1073/pnas.1106695108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature 449: 316–323, 2007. doi: 10.1038/nature06163. [DOI] [PubMed] [Google Scholar]
- 76.Jernigan NL, Drummond HA. Vascular ENaC proteins are required for renal myogenic constriction. Am J Physiol Renal Physiol 289: F891–F901, 2005. doi: 10.1152/ajprenal.00019.2005. [DOI] [PubMed] [Google Scholar]
- 77.Ji HL, Zhao R, Komissarov AA, Chang Y, Liu Y, Matthay MA. Proteolytic regulation of epithelial sodium channels by urokinase plasminogen activator: cutting edge and cleavage sites. J Biol Chem 290: 5241–5255, 2015. doi: 10.1074/jbc.M114.623496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kashlan OB, Adelman JL, Okumura S, Blobner BM, Zuzek Z, Hughey RP, Kleyman TR, Grabe M. Constraint-based, homology model of the extracellular domain of the epithelial Na+ channel α-subunit reveals a mechanism of channel activation by proteases. J Biol Chem 286: 649–660, 2011. doi: 10.1074/jbc.M110.167098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kashlan OB, Blobner BM, Zuzek Z, Carattino MD, Kleyman TR. Inhibitory tract traps the epithelial Na+ channel in a low activity conformation. J Biol Chem 287: 20720–20726, 2012. doi: 10.1074/jbc.M112.358218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kashlan OB, Blobner BM, Zuzek Z, Tolino M, Kleyman TR. Na+ inhibits the epithelial Na+ channel by binding to a site in an extracellular acidic cleft. J Biol Chem 290: 568–576, 2015. doi: 10.1074/jbc.M114.606152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kashlan OB, Boyd CR, Argyropoulos C, Okumura S, Hughey RP, Grabe M, Kleyman TR. Allosteric inhibition of the epithelial Na+ channel through peptide binding at peripheral finger and thumb domains. J Biol Chem 285: 35216–35223, 2010. doi: 10.1074/jbc.M110.167064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kashlan OB, Kleyman TR. ENaC structure and function in the wake of a resolved structure of a family member. Am J Physiol Renal Physiol 301: F684–F696, 2011. doi: 10.1152/ajprenal.00259.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev 82: 735–767, 2002. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- 84.Kemendy AE, Kleyman TR, Eaton DC. Aldosterone alters the open probability of amiloride-blockable sodium channels in A6 epithelia. Am J Physiol Cell Physiol 263: C825–C837, 1992. doi: 10.1152/ajpcell.1992.263.4.C825. [DOI] [PubMed] [Google Scholar]
- 85.Kim J, Blackshear PJ, Johnson JD, McLaughlin S. Phosphorylation reverses the membrane association of peptides that correspond to the basic domains of MARCKS and neuromodulin. Biophys J 67: 227–237, 1994. doi: 10.1016/S0006-3495(94)80473-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim J, Shishido T, Jiang X, Aderem A, McLaughlin S. Phosphorylation, high ionic strength, and calmodulin reverse the binding of MARCKS to phospholipid vesicles. J Biol Chem 269: 28214–28219, 1994. [PubMed] [Google Scholar]
- 87.Kleyman TR, Carattino MD, Hughey RP. ENaC at the cutting edge: regulation of epithelial sodium channels by proteases. J Biol Chem 284: 20447–20451, 2009. doi: 10.1074/jbc.R800083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kleyman TR, Cragoe EJ Jr. The mechanism of action of amiloride. Semin Nephrol 8: 242–248, 1988. [PubMed] [Google Scholar]
- 89.Kleyman TR, Kashlan OB, Hughey RP. Epithelial Na+ channel regulation by extracellular and intracellular factors. Annu Rev Physiol 80: 263–281, 2018. doi: 10.1146/annurev-physiol-021317-121143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Knight KK, Olson DR, Zhou R, Snyder PM. Liddle’s syndrome mutations increase Na+ transport through dual effects on epithelial Na+ channel surface expression and proteolytic cleavage. Proc Natl Acad Sci USA 103: 2805–2808, 2006. doi: 10.1073/pnas.0511184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kusche-Vihrog K, Jeggle P, Oberleithner H. The role of ENaC in vascular endothelium. Pflugers Arch 466: 851–859, 2014. doi: 10.1007/s00424-013-1356-3. [DOI] [PubMed] [Google Scholar]
- 92.Larionov A, Dahlke E, Kunke M, Zanon Rodriguez L, Schiessl IM, Magnin JL, Kern U, Alli AA, Mollet G, Schilling O, Castrop H, Theilig F. Cathepsin B increases ENaC activity leading to hypertension early in nephrotic syndrome. J Cell Mol Med 23: 6543–6553, 2019. doi: 10.1111/jcmm.14387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Laux T, Fukami K, Thelen M, Golub T, Frey D, Caroni P. GAP43, MARCKS, and CAP23 modulate PI4,5P2 at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J Cell Biol 149: 1455–1472, 2000. doi: 10.1083/jcb.149.7.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Leyvraz C, Charles RP, Rubera I, Guitard M, Rotman S, Breiden B, Sandhoff K, Hummler E. The epidermal barrier function is dependent on the serine protease CAP1/Prss8. J Cell Biol 170: 487–496, 2005. doi: 10.1083/jcb.200501038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li X, Leu S, Cheong A, Zhang H, Baibakov B, Shih C, Birnbaum MJ, Donowitz M. Akt2, phosphatidylinositol 3-kinase, and PTEN are in lipid rafts of intestinal cells: role in absorption and differentiation. Gastroenterology 126: 122–135, 2004. doi: 10.1053/j.gastro.2003.10.061. [DOI] [PubMed] [Google Scholar]
- 96.Lin W, Finger TE, Rossier BC, Kinnamon SC. Epithelial Na+ channel subunits in rat taste cells: localization and regulation by aldosterone. J Comp Neurol 405: 406–420, 1999. doi: 10.1002/(SICI)1096-9861(19990315)405:3<406:AID-CNE10>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 97.Ma HP, Chou CF, Wei SP, Eaton DC. Regulation of the epithelial sodium channel by phosphatidylinositides: experiments, implications, and speculations. Pflugers Arch 455: 169–180, 2007. doi: 10.1007/s00424-007-0294-3. [DOI] [PubMed] [Google Scholar]
- 98.Ma HP, Eaton DC. Acute regulation of epithelial sodium channel by anionic phospholipids. J Am Soc Nephrol 16: 3182–3187, 2005. doi: 10.1681/ASN.2005040434. [DOI] [PubMed] [Google Scholar]
- 99.Ma HP, Saxena S, Warnock DG. Anionic phospholipids regulate native and expressed epithelial sodium channel (ENaC). J Biol Chem 277: 7641–7644, 2002. doi: 10.1074/jbc.C100737200. [DOI] [PubMed] [Google Scholar]
- 100.Maarouf AB, Sheng N, Chen J, Winarski KL, Okumura S, Carattino MD, Boyd CR, Kleyman TR, Sheng S. Novel determinants of epithelial sodium channel gating within extracellular thumb domains. J Biol Chem 284: 7756–7765, 2009. doi: 10.1074/jbc.M807060200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Macháň R, Hof M. Lipid diffusion in planar membranes investigated by fluorescence correlation spectroscopy. Biochim Biophys Acta 1798: 1377–1391, 2010. doi: 10.1016/j.bbamem.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 102.Mall M, Grubb BR, Harkema JR, O’Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med 10: 487–493, 2004. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- 103.Malsure S, Wang Q, Charles RP, Sergi C, Perrier R, Christensen BM, Maillard M, Rossier BC, Hummler E. Colon-specific deletion of epithelial sodium channel causes sodium loss and aldosterone resistance. J Am Soc Nephrol 25: 1453–1464, 2014. doi: 10.1681/ASN.2013090936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martín-Belmonte F, Puertollano R, Millán J, Alonso MA. The MAL proteolipid is necessary for the overall apical delivery of membrane proteins in the polarized epithelial Madin-Darby canine kidney and Fischer rat thyroid cell lines. Mol Biol Cell 11: 2033–2045, 2000. doi: 10.1091/mbc.11.6.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC α, β, and γ subunit proteins in rat kidney. J Clin Invest 104: R19–R23, 1999. doi: 10.1172/JCI7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Matalon S, Bartoszewski R, Collawn JF. Role of epithelial sodium channels in the regulation of lung fluid homeostasis. Am J Physiol Lung Cell Mol Physiol 309: L1229–L1238, 2015. doi: 10.1152/ajplung.00319.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Matsuura D, Taguchi K, Yagisawa H, Maekawa S. Lipid components in the detergent-resistant membrane microdomain (DRM) obtained from the synaptic plasma membrane of rat brain. Neurosci Lett 423: 158–161, 2007. doi: 10.1016/j.neulet.2007.05.068. [DOI] [PubMed] [Google Scholar]
- 108.McLaughlin S, Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438: 605–611, 2005. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- 109.McLaughlin S, Aderem A. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem Sci 20: 272–276, 1995. doi: 10.1016/S0968-0004(00)89042-8. [DOI] [PubMed] [Google Scholar]
- 110.McLaughlin S, Wang J, Gambhir A, Murray D. PIP2 and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct 31: 151–175, 2002. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 111.Millán J, Alonso MA. MAL, a novel integral membrane protein of human T lymphocytes, associates with glycosylphosphatidylinositol-anchored proteins and Src-like tyrosine kinases. Eur J Immunol 28: 3675–3684, 1998. doi: 10.1002/(SICI)1521-4141(199811)28:11<3675:AID-IMMU3675>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 112.Millán J, Puertollano R, Fan L, Alonso MA. Caveolin and MAL, two protein components of internal detergent-insoluble membranes, are in distinct lipid microenvironments in MDCK cells. Biochem Biophys Res Commun 233: 707–712, 1997. doi: 10.1006/bbrc.1997.6530. [DOI] [PubMed] [Google Scholar]
- 113.Moore PJ, Tarran R. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis lung disease. Expert Opin Ther Targets 22: 687–701, 2018. doi: 10.1080/14728222.2018.1501361. [DOI] [PubMed] [Google Scholar]
- 114.Mueller GM, Maarouf AB, Kinlough CL, Sheng N, Kashlan OB, Okumura S, Luthy S, Kleyman TR, Hughey RP. Cys palmitoylation of the β subunit modulates gating of the epithelial sodium channel. J Biol Chem 285: 30453–30462, 2010. doi: 10.1074/jbc.M110.151845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mukherjee A, Mueller GM, Kinlough CL, Sheng N, Wang Z, Mustafa SA, Kashlan OB, Kleyman TR, Hughey RP. Cysteine palmitoylation of the γ subunit has a dominant role in modulating activity of the epithelial sodium channel. J Biol Chem 289: 14351–14359, 2014. doi: 10.1074/jbc.M113.526020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mukherjee A, Wang Z, Kinlough CL, Poland PA, Marciszyn AL, Montalbetti N, Carattino MD, Butterworth MB, Kleyman TR, Hughey RP. Specific palmitoyltransferases associate with and activate the epithelial sodium channel. J Biol Chem 292: 4152–4163, 2017. doi: 10.1074/jbc.M117.776146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Murray D, Ben-Tal N, Honig B, McLaughlin S. Electrostatic interaction of myristoylated proteins with membranes: simple physics, complicated biology. Structure 5: 985–989, 1997. doi: 10.1016/S0969-2126(97)00251-7. [DOI] [PubMed] [Google Scholar]
- 118.Mutchler SM, Kleyman TR. New insights regarding epithelial Na+ channel regulation and its role in the kidney, immune system and vasculature. Curr Opin Nephrol Hypertens 28: 113–119, 2019. doi: 10.1097/MNH.0000000000000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nielsen MR, Frederiksen-Møller B, Zachar R, Jørgensen JS, Hansen MR, Ydegaard R, Svenningsen P, Buhl K, Jensen BL. Urine exosomes from healthy and hypertensive pregnancies display elevated level of α-subunit and cleaved α- and γ-subunits of the epithelial sodium channel—ENaC. Pflugers Arch 469: 1107–1119, 2017. doi: 10.1007/s00424-017-1977-z. [DOI] [PubMed] [Google Scholar]
- 120.Nkeh B, Samani NJ, Badenhorst D, Libhaber E, Sareli P, Norton GR, Woodiwiss AJ. T594M variant of the epithelial sodium channel β-subunit gene and hypertension in individuals of African ancestry in South Africa. Am J Hypertens 16: 847–852, 2003. doi: 10.1016/S0895-7061(03)01016-1. [DOI] [PubMed] [Google Scholar]
- 121.Noreng S, Bharadwaj A, Posert R, Yoshioka C, Baconguis I. Structure of the human epithelial sodium channel by cryo-electron microscopy. eLife 7: e39340, 2018. doi: 10.7554/eLife.39340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Orce GG, Castillo GA, Margolius HS. Inhibition of short-circuit current in toad urinary bladder by inhibitors of glandular kallikrein. Am J Physiol Renal Fluid Electrolyte Physiol 239: F459–F465, 1980. doi: 10.1152/ajprenal.1980.239.5.F459. [DOI] [PubMed] [Google Scholar]
- 123.Oxlund CS, Buhl KB, Jacobsen IA, Hansen MR, Gram J, Henriksen JE, Schousboe K, Tarnow L, Jensen BL. Amiloride lowers blood pressure and attenuates urine plasminogen activation in patients with treatment-resistant hypertension. J Am Soc Hypertens 8: 872–881, 2014. [Erratum in J Am Soc Hypertens 9: 76, 2015.] doi: 10.1016/j.jash.2014.09.019. [DOI] [PubMed] [Google Scholar]
- 124.Palmer LG, Patel A, Frindt G. Regulation and dysregulation of epithelial Na+ channels. Clin Exp Nephrol 16: 35–43, 2012. doi: 10.1007/s10157-011-0496-z. [DOI] [PubMed] [Google Scholar]
- 125.Passero CJ, Carattino MD, Kashlan OB, Myerburg MM, Hughey RP, Kleyman TR. Defining an inhibitory domain in the γ-subunit of the epithelial sodium channel. Am J Physiol Renal Physiol 299: F854–F861, 2010. doi: 10.1152/ajprenal.00316.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Passero CJ, Mueller GM, Myerburg MM, Carattino MD, Hughey RP, Kleyman TR. TMPRSS4-dependent activation of the epithelial sodium channel requires cleavage of the γ-subunit distal to the furin cleavage site. Am J Physiol Renal Physiol 302: F1–F8, 2012. doi: 10.1152/ajprenal.00330.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Passero CJ, Mueller GM, Rondon-Berrios H, Tofovic SP, Hughey RP, Kleyman TR. Plasmin activates epithelial Na+ channels by cleaving the γ-subunit. J Biol Chem 283: 36586–36591, 2008. doi: 10.1074/jbc.M805676200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Patel AB, Chao J, Palmer LG. Tissue kallikrein activation of the epithelial Na channel. Am J Physiol Renal Physiol 303: F540–F550, 2012. doi: 10.1152/ajprenal.00133.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Planès C, Randrianarison NH, Charles RP, Frateschi S, Cluzeaud F, Vuagniaux G, Soler P, Clerici C, Rossier BC, Hummler E. ENaC-mediated alveolar fluid clearance and lung fluid balance depend on the channel-activating protease 1. EMBO Mol Med 2: 26–37, 2010. doi: 10.1002/emmm.200900050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pochynyuk O, Bugaj V, Stockand JD. Physiologic regulation of the epithelial sodium channel by phosphatidylinositides. Curr Opin Nephrol Hypertens 17: 533–540, 2008. doi: 10.1097/MNH.0b013e328308fff3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pochynyuk O, Tong Q, Medina J, Vandewalle A, Staruschenko A, Bugaj V, Stockand JD. Molecular determinants of PI4,5P2 and PI3,4,5P3 regulation of the epithelial Na+ channel. J Gen Physiol 130: 399–413, 2007. doi: 10.1085/jgp.200709800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pochynyuk O, Tong Q, Staruschenko A, Ma HP, Stockand JD. Regulation of the epithelial Na+ channel (ENaC) by phosphatidylinositides. Am J Physiol Renal Physiol 290: F949–F957, 2006. doi: 10.1152/ajprenal.00386.2005. [DOI] [PubMed] [Google Scholar]
- 133.Pochynyuk O, Tong Q, Staruschenko A, Stockand JD. Binding and direct activation of the epithelial Na+ channel (ENaC) by phosphatidylinositides. J Physiol 580: 365–372, 2007. doi: 10.1113/jphysiol.2006.127449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Puertollano R, Alonso MA. MAL, an integral element of the apical sorting machinery, is an itinerant protein that cycles between the trans-Golgi network and the plasma membrane. Mol Biol Cell 10: 3435–3447, 1999. doi: 10.1091/mbc.10.10.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Puertollano R, Alonso MA. A short peptide motif at the carboxyl terminus is required for incorporation of the integral membrane MAL protein to glycolipid-enriched membranes. J Biol Chem 273: 12740–12745, 1998. doi: 10.1074/jbc.273.21.12740. [DOI] [PubMed] [Google Scholar]
- 136.Rauh R, Diakov A, Tzschoppe A, Korbmacher J, Azad AK, Cuppens H, Cassiman JJ, Dötsch J, Sticht H, Korbmacher C. A mutation of the epithelial sodium channel associated with atypical cystic fibrosis increases channel open probability and reduces Na+ self inhibition. J Physiol 588: 1211–1225, 2010. doi: 10.1113/jphysiol.2009.180224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ray EC, Chen J, Kelly TN, He J, Hamm LL, Gu D, Shimmin LC, Hixson JE, Rao DC, Sheng S, Kleyman TR. Human epithelial Na+ channel missense variants identified in the GenSalt study alter channel activity. Am J Physiol Renal Physiol 311: F908–F914, 2016. doi: 10.1152/ajprenal.00426.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ray EC, Miller RG, Demko JE, Costacou T, Kinlough CL, Demko CL, Unruh ML, Orchard TJ, Kleyman TR. Urinary plasmin(ogen) as a prognostic factor for hypertension. Kidney Int Rep 3: 1434–1442, 2018. doi: 10.1016/j.ekir.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ray EC, Rondon-Berrios H, Boyd CR, Kleyman TR. Sodium retention and volume expansion in nephrotic syndrome: implications for hypertension. Adv Chronic Kidney Dis 22: 179–184, 2015. doi: 10.1053/j.ackd.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Reihill JA, Walker B, Hamilton RA, Ferguson TE, Elborn JS, Stutts MJ, Harvey BJ, Saint-Criq V, Hendrick SM, Martin SL. Inhibition of protease-epithelial sodium channel signaling improves mucociliary function in cystic fibrosis airways. Am J Respir Crit Care Med 194: 701–710, 2016. doi: 10.1164/rccm.201511-2216OC. [DOI] [PubMed] [Google Scholar]
- 141.Rossier BC, Stutts MJ. Activation of the epithelial sodium channel (ENaC) by serine proteases. Annu Rev Physiol 71: 361–379, 2009. doi: 10.1146/annurev.physiol.010908.163108. [DOI] [PubMed] [Google Scholar]
- 142.Salih M, Gautschi I, van Bemmelen MX, Di Benedetto M, Brooks AS, Lugtenberg D, Schild L, Hoorn EJ. A missense mutation in the extracellular domain of αENaC causes Liddle syndrome. J Am Soc Nephrol 28: 3291–3299, 2017. doi: 10.1681/ASN.2016111163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schild L, Lu Y, Gautschi I, Schneeberger E, Lifton RP, Rossier BC. Identification of a PY motif in the epithelial Na channel subunits as a target sequence for mutations causing channel activation found in Liddle syndrome. EMBO J 15: 2381–2387, 1996. doi: 10.1002/j.1460-2075.1996.tb00594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shei RJ, Peabody JE, Kaza N, Rowe SM. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis. Curr Opin Pharmacol 43: 152–165, 2018. doi: 10.1016/j.coph.2018.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sheng S, Bruns JB, Kleyman TR. Extracellular histidine residues crucial for Na+ self-inhibition of epithelial Na+ channels. J Biol Chem 279: 9743–9749, 2004. doi: 10.1074/jbc.M311952200. [DOI] [PubMed] [Google Scholar]
- 146.Sheng S, Carattino MD, Bruns JB, Hughey RP, Kleyman TR. Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am J Physiol Renal Physiol 290: F1488–F1496, 2006. doi: 10.1152/ajprenal.00439.2005. [DOI] [PubMed] [Google Scholar]
- 147.Sheng S, Maarouf AB, Bruns JB, Hughey RP, Kleyman TR. Functional role of extracellular loop cysteine residues of the epithelial Na+ channel in Na+ self-inhibition. J Biol Chem 282: 20180–20190, 2007. doi: 10.1074/jbc.M611761200. [DOI] [PubMed] [Google Scholar]
- 148.Shi S, Carattino MD, Kleyman TR. Role of the wrist domain in the response of the epithelial sodium channel to external stimuli. J Biol Chem 287: 44027–44035, 2012. doi: 10.1074/jbc.M112.421743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Shi S, Ghosh DD, Okumura S, Carattino MD, Kashlan OB, Sheng S, Kleyman TR. Base of the thumb domain modulates epithelial sodium channel gating. J Biol Chem 286: 14753–14761, 2011. doi: 10.1074/jbc.M110.191734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Snyder PM, McDonald FJ, Stokes JB, Welsh MJ. Membrane topology of the amiloride-sensitive epithelial sodium channel. J Biol Chem 269: 24379–24383, 1994. [PubMed] [Google Scholar]
- 151.Soundararajan R, Pearce D, Hughey RP, Kleyman TR. Role of epithelial sodium channels and their regulators in hypertension. J Biol Chem 285: 30363–30369, 2010. doi: 10.1074/jbc.R110.155341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Staruschenko A, Pochynyuk O, Vandewalle A, Bugaj V, Stockand JD. Acute regulation of the epithelial Na+ channel by phosphatidylinositide 3-OH kinase signaling in native collecting duct principal cells. J Am Soc Nephrol 18: 1652–1661, 2007. doi: 10.1681/ASN.2007010020. [DOI] [PubMed] [Google Scholar]
- 153.Staub O, Abriel H, Plant P, Ishikawa T, Kanelis V, Saleki R, Horisberger JD, Schild L, Rotin D. Regulation of the epithelial Na+ channel by Nedd4 and ubiquitination. Kidney Int 57: 809–815, 2000. doi: 10.1046/j.1523-1755.2000.00919.x. [DOI] [PubMed] [Google Scholar]
- 154.Sternak M, Bar A, Adamski MG, Mohaissen T, Marczyk B, Kieronska A, Stojak M, Kus K, Tarjus A, Jaisser F, Chlopicki S. The deletion of endothelial sodium channel α (αENaC) impairs endothelium-dependent vasodilation and endothelial barrier integrity in endotoxemia in vivo. Front Pharmacol 9: 178, 2018. doi: 10.3389/fphar.2018.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sugiyama T, Kato N, Ishinaga Y, Yamori Y, Yazaki Y. Evaluation of selected polymorphisms of the Mendelian hypertensive disease genes in the Japanese population. Hypertens Res 24: 515–521, 2001. doi: 10.1291/hypres.24.515. [DOI] [PubMed] [Google Scholar]
- 156.Svenningsen P, Bistrup C, Friis UG, Bertog M, Haerteis S, Krueger B, Stubbe J, Jensen ON, Thiesson HC, Uhrenholt TR, Jespersen B, Jensen BL, Korbmacher C, Skøtt O. Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol 20: 299–310, 2009. doi: 10.1681/ASN.2008040364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Svenningsen P, Uhrenholt TR, Palarasah Y, Skjødt K, Jensen BL, Skøtt O. Prostasin-dependent activation of epithelial Na+ channels by low plasmin concentrations. Am J Physiol Regul Integr Comp Physiol 297: R1733–R1741, 2009. doi: 10.1152/ajpregu.00321.2009. [DOI] [PubMed] [Google Scholar]
- 158.Talbot CL, Bosworth DG, Briley EL, Fenstermacher DA, Boucher RC, Gabriel SE, Barker PM. Quantitation and localization of ENaC subunit expression in fetal, newborn, and adult mouse lung. Am J Respir Cell Mol Biol 20: 398–406, 1999. doi: 10.1165/ajrcmb.20.3.3283. [DOI] [PubMed] [Google Scholar]
- 159.Tan CD, Hobbs C, Sameni M, Sloane BF, Stutts MJ, Tarran R. Cathepsin B contributes to Na+ hyperabsorption in cystic fibrosis airway epithelial cultures. J Physiol 592: 5251–5268, 2014. doi: 10.1113/jphysiol.2013.267286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int 89: 127–134, 2016. doi: 10.1038/ki.2015.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tong J, Nguyen L, Vidal A, Simon SA, Skene JH, McIntosh TJ. Role of GAP-43 in sequestering phosphatidylinositol 4,5-bisphosphate to raft bilayers. Biophys J 94: 125–133, 2008. doi: 10.1529/biophysj.107.110536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tong Q, Gamper N, Medina JL, Shapiro MS, Stockand JD. Direct activation of the epithelial Na+ channel by phosphatidylinositol 3,4,5-trisphosphate and phosphatidylinositol 3,4-bisphosphate produced by phosphoinositide 3-OH kinase. J Biol Chem 279: 22654–22663, 2004. doi: 10.1074/jbc.M401004200. [DOI] [PubMed] [Google Scholar]
- 163.Uchimura K, Kakizoe Y, Onoue T, Hayata M, Morinaga J, Yamazoe R, Ueda M, Mizumoto T, Adachi M, Miyoshi T, Shiraishi N, Sakai Y, Tomita K, Kitamura K. In vivo contribution of serine proteases to the proteolytic activation of γENaC in aldosterone-infused rats. Am J Physiol Renal Physiol 303: F939–F943, 2012. doi: 10.1152/ajprenal.00705.2011. [DOI] [PubMed] [Google Scholar]
- 164.Unruh ML, Pankratz VS, Demko JE, Ray EC, Hughey RP, Kleyman TR. Trial of amiloride in type 2 diabetes with proteinuria. Kidney Int Rep 2: 893–904, 2017. doi: 10.1016/j.ekir.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, Rossier BC. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature 389: 607–610, 1997. doi: 10.1038/39329. [DOI] [PubMed] [Google Scholar]
- 166.Van Beusecum JP, Barbaro NR, McDowell Z, Aden LA, Xiao L, Pandey AK, Itani HA, Himmel LE, Harrison DG, Kirabo A. High salt activates CD11c+ antigen-presenting cells via SGK (serum glucocorticoid kinase) 1 to promote renal inflammation and salt-sensitive hypertension. Hypertension 74: 555–563, 2019. doi: 10.1161/HYPERTENSIONAHA.119.12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Vuagniaux G, Vallet V, Jaeger NF, Hummler E, Rossier BC. Synergistic activation of ENaC by three membrane-bound channel-activating serine proteases (mCAP1, mCAP2, and mCAP3) and serum- and glucocorticoid-regulated kinase (Sgk1) in Xenopus oocytes. J Gen Physiol 120: 191–201, 2002. doi: 10.1085/jgp.20028598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wang J, Arbuzova A, Hangyás-Mihályné G, McLaughlin S. The effector domain of myristoylated alanine-rich C kinase substrate binds strongly to phosphatidylinositol 4,5-bisphosphate. J Biol Chem 276: 5012–5019, 2001. doi: 10.1074/jbc.M008355200. [DOI] [PubMed] [Google Scholar]
- 169.Wang J, Gambhir A, Hangyás-Mihályné G, Murray D, Golebiewska U, McLaughlin S. Lateral sequestration of phosphatidylinositol 4,5-bisphosphate by the basic effector domain of myristoylated alanine-rich C kinase substrate is due to nonspecific electrostatic interactions. J Biol Chem 277: 34401–34412, 2002. doi: 10.1074/jbc.M203954200. [DOI] [PubMed] [Google Scholar]
- 170.Wang J, Gambhir A, McLaughlin S, Murray D. A computational model for the electrostatic sequestration of PI4,5P2 by membrane-adsorbed basic peptides. Biophys J 86: 1969–1986, 2004. doi: 10.1016/S0006-3495(04)74260-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wang J, Richards DA. Segregation of PIP2 and PIP3 into distinct nanoscale regions within the plasma membrane. Biol Open 1: 857–862, 2012. doi: 10.1242/bio.20122071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Warnock DG, Kusche-Vihrog K, Tarjus A, Sheng S, Oberleithner H, Kleyman TR, Jaisser F. Blood pressure and amiloride-sensitive sodium channels in vascular and renal cells. Nat Rev Nephrol 10: 146–157, 2014. doi: 10.1038/nrneph.2013.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Widmer F, Caroni P. Identification, localization, and primary structure of CAP-23, a particle-bound cytosolic protein of early development. J Cell Biol 111: 3035–3047, 1990. doi: 10.1083/jcb.111.6.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Widmer F, Caroni P. Phosphorylation-site mutagenesis of the growth-associated protein GAP-43 modulates its effects on cell spreading and morphology. J Cell Biol 120: 503–512, 1993. doi: 10.1083/jcb.120.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Winarski KL, Sheng N, Chen J, Kleyman TR, Sheng S. Extracellular allosteric regulatory subdomain within the γ-subunit of the epithelial Na+ channel. J Biol Chem 285: 26088–26096, 2010. doi: 10.1074/jbc.M110.149963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yamaguchi E, Yoshikawa K, Nakaya I, Kato K, Miyasato Y, Nakagawa T, Kakizoe Y, Mukoyama M, Soma J. Liddle’s-like syndrome associated with nephrotic syndrome secondary to membranous nephropathy: the first case report. BMC Nephrol 19: 122, 2018. doi: 10.1186/s12882-018-0916-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yoder N, Yoshioka C, Gouaux E. Gating mechanisms of acid-sensing ion channels. Nature 555: 397–401, 2018. doi: 10.1038/nature25782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yue G, Malik B, Yue G, Eaton DC. Phosphatidylinositol 4,5-bisphosphate (PIP2) stimulates epithelial sodium channel activity in A6 cells. J Biol Chem 277: 11965–11969, 2002. doi: 10.1074/jbc.M108951200. [DOI] [PubMed] [Google Scholar]
- 179.Zachar RM, Skjødt K, Marcussen N, Walter S, Toft A, Nielsen MR, Jensen BL, Svenningsen P. The epithelial sodium channel γ-subunit is processed proteolytically in human kidney. J Am Soc Nephrol 26: 95–106, 2015. doi: 10.1681/ASN.2013111173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zeiske W, Wills NK, Van Driessche W. Na+ channels and amiloride-induced noise in the mammalian colon epithelium. Biochim Biophys Acta 688: 201–210, 1982. doi: 10.1016/0005-2736(82)90595-8. [DOI] [PubMed] [Google Scholar]
- 181.Zhai YJ, Liu BC, Wei SP, Chou CF, Wu MM, Song BL, Linck VA, Zou L, Zhang S, Li XQ, Zhang ZR, Ma HP. Depletion of cholesterol reduces ENaC activity by decreasing phosphatidylinositol-4,5-bisphosphate in microvilli. Cell Physiol Biochem 47: 1051–1059, 2018. doi: 10.1159/000490170. [DOI] [PubMed] [Google Scholar]
- 182.Zhang ZR, Chou CF, Wang J, Liang YY, Ma HP. Anionic phospholipids differentially regulate the epithelial sodium channel (ENaC) by interacting with α-, β-, and γ-ENaC subunits. Pflugers Arch 459: 377–387, 2010. doi: 10.1007/s00424-009-0733-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zheng H, Liu X, Rao US, Patel KP. Increased renal ENaC subunits and sodium retention in rats with chronic heart failure. Am J Physiol Renal Physiol 300: F641–F649, 2011. doi: 10.1152/ajprenal.00254.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]