

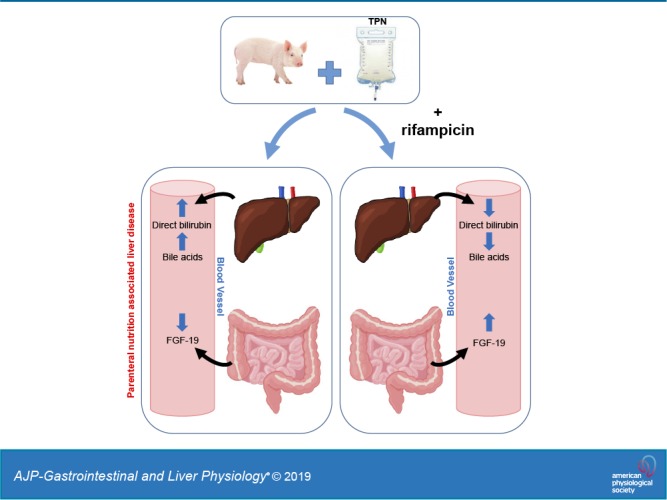
Keywords: bile acid metabolism, cholestasis, parenteral nutrition-associated liver disease, rifampicin, α-tocopherol, vitamin E
Abstract
Infants receiving long-term parenteral nutrition (PN) develop PN-associated liver disease (PNALD). We previously (Ng K et al. JPEN J Parenter Enteral Nutr 40: 656–671, 2016. doi:10.1177/0148607114567900.) showed that PN containing soy-based lipid supplemented with vitamin E (α-tocopherol) prevents the development of PNALD. We hypothesize that this occurs via vitamin E activation of pregnane X receptor (PXR)-mediated pathways involved in bile acid metabolism. Neonatal piglets received PN for 14 days containing Intralipid (IL; soy-based lipid emulsion), IL supplemented with 12.6 mg·kg−1·day−1 vitamin E (VITE), or IL with 10 mg·kg−1·day−1 Rifadin IV (RIF), a PXR agonist. Pigs treated with IL and VITE, but not RIF, developed cholestasis and hyperbilirubinemia, markers of liver disease. The hepatic PXR target genes CYP3A29 and UGT1A6 increased during RIF treatment. RIF also modestly increased metabolism of chenodeoxycholic acid to the more hydrophilic bile acid hyocholic acid. Serum fibroblast growth factor (FGF)-19, a key regulator in suppressing hepatic bile acid synthesis, significantly increased in the RIF group. We conclude rifampicin modified markers of PNALD development by increased metabolism of bile acids and potentially suppressed bile acid synthesis. Vitamin E was ineffective at high lipid doses in preventing PNALD.
NEW & NOTEWORTHY Intravenous vitamin E and rifampicin were administered to neonatal piglets receiving parenteral nutrition to determine their efficacy in reducing the progression of parenteral nutrition-associated liver disease (PNALD). Rifampicin increased serum FGF-19 concentrations and synthesis of the bile acid hyocholic acid which led to a reduction of PNALD parameters at 2 wk of administration. This result has potential clinical implications for the use of rifampicin as a safe and inexpensive treatment for short-term development of PNALD.
INTRODUCTION
The use of parenteral nutrition (PN) to maintain growth and nutrient status of infants with intractable enteral feeding was first initiated 50 years ago (53, 54). The use of soy-oil (SO) based lipid emulsions short-term in PN is very effective in reducing the morbidity and mortality of infants that are intractable to enteral feeding. However, long-term use of SO in infants results in cholestasis, hepatic steatosis, increased markers of liver injury, and elevated serum direct bilirubin, which is collectively referred to as parenteral nutrition-associated liver disease (PNALD). If PNALD is not resolved, there is an increased risk of end-stage liver disease and liver transplant in this population (8). Use of alternative lipid emulsions containing fish oil (FO) or mixed-oil (MO) preparations containing SO, FO, medium chain triglycerides (MCT), and olive oil have proven beneficial in resolving (17, 27, 34) or reducing (11, 40) the development of PNALD. The underlying mechanisms that mediate the beneficial effect of both FO and MO preparations compared with SO is not clear. Plant-based lipids, such as SO, contain phytosterols that have been implicated as a cause of hepatic inflammation (13, 14) and suppress activation of genes involved in bile acid homeostasis (7, 50) when administered parenterally. A confounder to this explanation is that both MO and SO lipid emulsions contain phytosterols, yet MO administration can resolve the development of PNALD and infants on SO develop PNALD. For this reason, we examined vitamin E (α-tocopherol), which is enriched in MO and FO lipid emulsions, but in low concentration in SO lipid emulsions. In our previous report, we found the addition of vitamin E to SO lipid emulsions was protective against PNALD development in preterm piglets (35).
Vitamin E comprises eight different forms, α-, β-, γ-, δ- tocopherols and tocotrienols that differ in biological activities. The α-tocopherol form is the most biologically active, although all forms of vitamin E have potent antioxidant activity that protects against lipid (47) and phytosterol oxidation (45). Unlike other fat-soluble vitamins, vitamin E (α-tocopherol) does not readily accumulate and is metabolized via an initial ω-oxidation by the cytochrome P-450 (CYP) gene CYP4F2 (2, 3, 43, 44). However, some reports suggest CYP3A4, the major xenobiotic metabolism enzyme in humans, may also metabolize vitamin E (1, 24, 37). Many compounds that are metabolized through xenobiotic pathways selfregulate by activating CYP3A4 (42). In vitro studies using cellular reporter assays have shown that γ-tocotrienol (26) and α-tocopherol (35) may directly activate drug metabolism genes. The in vivo data are more complicated with earlier reports in rats and mice showing activation of drug metabolism pathways (32, 49). More recent data in humanized mice suggest there may be a species-dependent effect and vitamin E does not activate drug metabolism in humans (24). However, there is no consensus mechanism for vitamin E actions in drug metabolism. We have previously observed that bile acids are cleared more rapidly in piglets administered supplemental vitamin E during TPN without changes in the canonical bile acid metabolism pathways, and we postulate that this is mediated by activation of a drug metabolism pathway (35).
Homeostatic regulation of bile acids is controlled by the nuclear hormone receptor farnesoid X teceptor (FXR) (10). Bile acids function as ligands for FXR, which activates the transcription of genes involved in both bile acid uptake and clearance at the apical and basolateral membranes in hepatocytes and the ileal enterocytes. In addition, FXR can suppress the synthesis of bile acids in the liver through upregulation of the gut enterokine fibroblast growth factor-19 (FGF-19) (22, 25). During PN, it is hypothesized that bile acids accumulate from disruption of the FXR signaling pathway (7). A secondary clearance mechanism for bile acids exist once there is considerable accumulation through the activation of another nuclear hormone receptor, the pregnane X receptor (PXR) (9). PXR is a key regulator of genes involved in the drug metabolism pathway. The primary target of PXR is CYP3A29 in pigs (CYP3A4 ortholog in humans) for phase I of detoxification (19, 39). The CYP3A29 enzyme can convert the primary bile acid chenodeoxycholic acid (CDCA) to hyocholic acid (HCA) by the addition of the 6-α hydroxyl group. PXR also upregulates several phase II conjugation enzymes including sulfonate (sulfoureatransferases; SULT), UDP glucuronosyltransferase (UGT), and glutathione S-transferase (GST). The resulting bile acid metabolites are more hydrophilic than the precursor primary bile acids. The final phase III process in drug metabolism is transport and several genes are targeted by PXR, including multidrug resistance-associated protein (MRP2) and multidrug resistance protein 1 (MDR1). The activation of this pathway is effective in relieving the itching caused by cholestatic pruritus, and likely this is through enhanced clearance of bile acids (12), which makes this an interesting target for treating PNALD.
The aim of this study is to clarify the potential role of vitamin E in PNALD as a means of preventing disease development, and whether this function is through activation of the PXR pathway. Furthermore, we aim to determine the efficacy of direct activation of the PXR pathway to prevent the development of PNALD. We hypothesize activation of PXR by vitamin E will increase the pool size of the hydrophilic bile acid HCA and facilitate HCA clearance to decrease cholestasis, thereby preventing PNALD.
MATERIALS AND METHODS
Animals and nutritional support.
All animal research was conducted under an approved protocol by the Animal Protocol Review Committee of the Baylor College of Medicine and performed according to the Guide for the Care and Use of Laboratory Animals (Department of Health and Human Services publication no. 85–23, revised 1985, Office of Science and Health Reports, NIH, Bethesda, MD). Term-born piglets aged 3–6 days were delivered to the Children’s Nutrition Research Center (Baylor College of Medicine) from a commercial pig farm. On the same day of arrival, piglets were placed under general anesthesia using isoflurane and surgically implanted with a jugular catheter as described previously (35, 50). Piglets were administered total nutritional support parenterally through the implanted jugular catheter consisting of the following per kilogram body weight: 14 g protein, 24 g carbohydrate, and 10 g lipid. The total fluid volume daily was 240 mL/kg. The piglets were on PN for 14 days. PN administration was increased stepwise from 50% of daily needs at day 0 to 100% at day 5 until the completion of the study. Historical controls (n = 7) from current unpublished studies were included to reduce the number of animals used. The controls consisted of term-born piglets from the same sow supplier that received Litter Life, a milk-based piglet formula. The piglets received feeds for 2 wk so they matched the age and maturity of the piglets receiving TPN in this study.
This study was designed to examine the change in PNALD development with two interventional treatments, so all animals received TPN. The piglets were randomly assigned into three treatment groups: 1) Control (IL), 2) vitamin E supplemented (VITE), and 3) rifampicin supplemented (RIF; Rifadin IV, Sanofi US, Bridgewater, NJ). The IL group received the standard PN as outlined above with no additional supplementation. The α-tocopherol concentration in IL according to the manufacturer’s data sheets is 37 ± 1.3 mg/L. The VITE group received PN with the lipid emulsion component supplemented with d-α-tocopherol (Sparkhawk Laboratories, Lanexa, KS). The d-α-tocopherol concentration was selected to match the concentration of d-α-tocopherol equivalents present in Omegaven (31). The d-α-tocopherol was added to IL heated to 50°C under constant stirring for 15 min as described in detail previously (35). The final concentration of d-α-tocopherol in the supplemented IL was 251 mg d-α-tocopherol/L. The RIF group received 10 mg·kg−1·day−1 Rifadin IV starting on day 1 to day 14 via infusion through the jugular catheter. Rifadin IV was given as a single bolus infused over a 10-min period. The concentration of Rifadin IV was based on concentrations of rifampicin determined to be safe and effective for treatment of children with cholestatic pruritus (12, 55).
The piglets were weighed every other day to adjust infusion rates to maintain the targeted nutrient administration. On day 13 of the study, piglets were administered a constant infusion of [2H4]CDCA (Cambridge Isotope Laboratories, Tewksbury, MA) for 12 h to assess the conversion of CDCA to HCA in all treatment groups. The piglets received an initial priming dose of 23 μg/kg [2H4]CDCA and subsequent constant infusion of 23 μg·kg-1·h -1[2H4]CDCA at a concentration of 23 μg/0.2 mL. Blood samples were collected every 2 h from 0 to 12 h of the infusion. From this measurement, the % fractional synthetic rate (% FSR) of hyocholic acid was calculated using established equations (15).
On day 14, piglets received TPN infusions up to the moment of tissue collection. Piglets were removed from the infusion apparatus and administered a cocktail of pentobarbital and phenytoin through the jugular catheter for euthanasia. Samples were collected immediately following euthanasia.
Blood and tissue analysis.
Serum samples collected from piglets on the final day of the study were analyzed for multiple serum chemistry parameters as done in our previous studies (35, 50). Analysis was performed by the Center for Comparative Medicine (Baylor College of Medicine) on a Cobas Integra 400 plus analyzer (Roche Diagnostics, Rotkreuz, Switzerland). Liver triglyceride was determined using 350 mg of frozen tissue as described in detail previously. Incubation was overnight at 55°C in 1,000 μL of ethanolic KOH (2:1 ethanol:30% KOH). The supernatant was collected after 5 min centrifugation and 300 μL of H2O:ethanol (1:1) was added. From the mixture, 200 μL was removed and mixed with 200 μL 1 M MgCl2. The sample was left to sit on ice for 10 min. After a 5-min centrifugation, the supernatant was collected. The saponified lipid sample was assayed using a commercially available kit (Infinity Triglycerides, Thermo Fisher Scientific) to determine triglyceride concentration.
Phytosterol and oxysterol analysis.
Sample preparation and analysis of plasma samples by liquid chromatography tandem mass spectrometry (LC-MS/MS) for the phytosterols sitosterol, stigmasterol, and campesterol concentrations were performed as described previously (30). The serum sample preparation and analysis of oxysterols 7α-hydroxy (OH)-campesterol, 7α-OH-sitosterol, 7β-OH-campesterol, 7β-OH-sitosterol, 7-keto-campesterol, 7-keto-sitosterol, 7α-OH-cholesterol, and 27α-OH-cholesterol by gas-liquid chromatography-mass spectroscopy (GC-MS) were performed according to the protocol described in detail previously (21).
Vitamin E analysis.
Vitamin E (α-tocopherol) was analyzed in both plasma and liver by high performance liquid chromatography (HPLC), as described in detail previously (18). A minimum of 200 mg of liver was homogenized in ethanol. Samples were run through a 4.0 × 125-mm Perkin-Elmer HS-5-Silica column (D-7770, Perkin-Elmer, Uberlingen, Germany). Merck (D-6100, Damstadt, Germany) external standards were used for identification and quantification of α-tocopherol and γ-tocopherol.
Gene and protein expression analysis.
Gene expression was performed as detailed previously with minor modifications (35). Real time PCR gene expression analysis was performed on the cDNA using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific) and primers for our target genes (available upon request) (35). Fold-change was calculated using the ΔΔCt method.
Western blot analysis was performed on protein isolated from 50 mg of frozen tissue as described previously (35). Total protein per lane was 30 μg, which was incubated with antibodies for our target proteins: phase I drug metabolism hydroxylase anti-CYP3A4 (Millipore Sigma, St. Louis, MO), phase II drug metabolism transferase anti-UGT1A1 (Abcam, Cambridge, UK), and housekeeping protein anti-β-actin (Santa Cruz Biotechnology, Dallas, TX). The membranes were then incubated in the appropriate secondary horseradish peroxidase-conjugated antibodies (Santa Cruz Biotechnology). The membrane was visualized on a CCD camera using the ChemiDoc XRS system (Bio-Rad).
FGF-19 plasma protein expression was determined using a porcine FGF-19 enzyme-linked immunosorbent assay (ELISA; RayBiotech Life, Norcross, GA) according to the manufacturer’s instructions. This kit was developed using a recombinant porcine FGF-19 protein generated in our laboratory and validated using purified protein and extract from cells overexpressing the recombinant FGF-19 plasmid. The plasma samples were from the final blood collection that had been snap frozen and stored at −80°C until use for the ELISA.
Bile acid analysis.
Total bile acids were assayed in the plasma, liver tissue, gall bladder contents (bile), urine, and cecal content. All samples were quantified using a commercially available kit (BQ kits). The liver bile acid pool was calculated from the micromolar content of the total liver divided by the weight of the piglet in kilograms. The gall bladder bile acid content was determined by calculating the total bile weight collected from each piglet divided by the weight of the piglet in kilograms.
Various conjugated and free bile acid enrichments and concentrations in plasma and tissues were measured by LC-MS/MS (Q-Exactive Orbitrap, ThermoScientific). The samples were prepared by mixing with an appropriate amount of an internal standard (d9-CDCA or d9-Glyco-CDCA to determine free and conjugated bile acids concentration by isotope dilution). Sample extraction and analysis were performed as detailed previously (35).
Serum antioxidant analysis.
Commercially available kits for free radical marker and hydrogen peroxide scavenger superoxide dismutase (SOD; Abcam) and two markers of lipid peroxidation, malondialdehyde (MDA, Abcam), and 8-iso-prostaglandin F2α (PGF-2α; Enzo Life Sciences, Farmingham, NY), were used according to the manufacturer’s protocol. Serum samples were from the final day tissue collection that had been snap frozen and stored at −80°C until being thawed on the day of assay use.
Statistics.
Statistical analyses were performed using SigmaPlot 11.0 software (Systat Software, San Jose, CA). The historical controls were not included in any statistical analysis and are supplied as reference data only. Differences among the three groups were first analyzed using one-way ANOVA, and post hoc analysis was performed using Tukey's test as described in the figure legends. For results that were not of normal distribution, the nonparametric Kruskal-Wallis ANOVA on ranks followed by pairwise comparisons using Dunn’s test was used to determine significance. Two-way repeated measures ANOVA was used to calculate significance for % FSR to determine a time-dependent effect on bile acid synthesis. P values <0.05 were considered significant. Results are presented as means ± SE.
RESULTS
A total of 24 piglets (n = 8/group) were used for the study. Of these, 19 completed the study, and the numbers in each group in the final analysis were IL (n = 7), VITE (n = 5), and RIF (n = 7). The excluded piglets were removed for having sustained elevated body temperatures, without a determined cause. In Table 1, we present that the final body weight and the body weight gain from day 0 to day 14 did not differ among groups. The body and tissue weights did not differ among groups.
Table 1.
Body and tissue weights
| IL | VITE | RIF | CON | |
|---|---|---|---|---|
| Body weight | ||||
| Day 0, g | 1,915 ± 87.0 | 2,0535 ± 168 | 1,879 ±5 102 | (905–1,763) |
| Day 14, g | 4,010 ± 155 | 4,175 ± 314 | 4,153 ± 211 | (2,064–3,688) |
| Growth rate, g·kg−1·day−1 | 53.3 ± 2.80 | 48.7 ± 1.51 | 54.5 ± 2.75 | (45.6–55.8) |
| Tissue weight, g/kg | ||||
| Liver | 65.3 ± 4.19 | 65.4 ± 3.68 | 55.0 ± 2.21 | (30–34) |
| Spleen | 7.02 ± 1.46 | 10.61 ± 1.48 | 7.15 ± 1.07 | (2.6–3.8) |
| Jejunum | 8.39 ± 0.24 | 8.95 ± 0.67 | 9.98 ± 0.51 | (17.0–20.8) |
| Ileum | 10.8 ± 0.57 | 8.83 ± 0.35 | 10. ± 0.44 | (24.9–29.2) |
| Brain | 10.4 ± 0.29 | 10.0 ± 0.67 | 10.4 ± 0.37 | (11.6–16.2) |
| Heart | 5.65 ± 0.98 | 6.60 ± 0.22 | 5.38 ± 0.94 | (6.3–7.3) |
| Kidney | 9.21 ± 0.32 | 10.38 ± 0.90 | 10.14 ± 0.67 | (6.5–8.4) |
Values are means ± SE. IL, Intralipid; RIF, Rifadin IV; VITE, vitamin E.
Piglets were administered SO that contained different amounts of vitamin E in the form of d-α-tocopherol. In Fig. 1, A and B, the piglets that received VITE had significantly higher levels of serum (P < 0.001) and liver (P < 0.001) α-tocopherol compared with IL and RIF. The primary form of vitamin E in Intralipid is γ-tocopherol (35). We found the serum γ-tocopherol levels (Fig. 1, C and D) were significantly lower in the VITE compared with IL (P = 0.025). Similarly, the liver γ-tocopherol levels were significantly lower in the VITE (P = 0.049) and RIF (P = 0.049) compared with IL. Given what appears to be a treatment-dependent effect on the metabolism of hepatic γ-tocopherol, we examined the expression of the primary gene associated with metabolism of all forms of vitamin E, Cyp4f2 (Fig. 1E), and the α-tocopherol transport (α-tocopherol transport protein; TTP) (Fig. 1F), and found that there was no difference in expression among the groups.
Fig. 1.
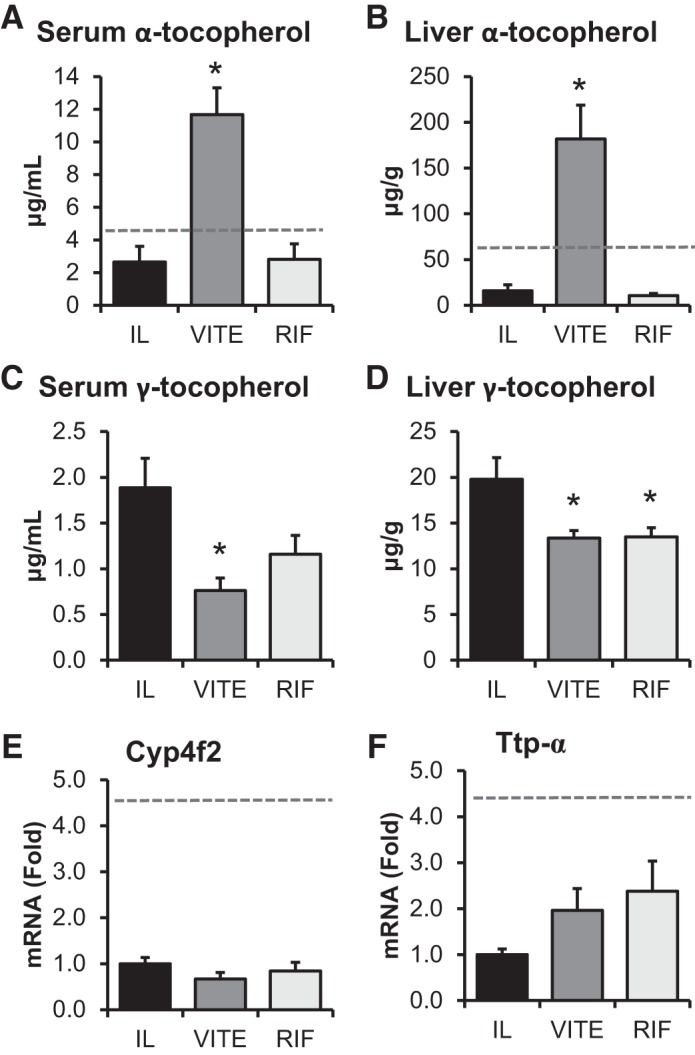
Enrichment of soy oil (SO) lipid emulsion with α-tocopherol increases α-tocopherol and decreases γ-tocopherol in serum and liver. Term piglets were administered lipid emulsions for 14 days containing standard SO emulsion (IL), α-tocopherol-supplemented SO (VITE), or SO supplemented with daily injections of 10 mg/L Rifadin IV (RIF). Samples were collected on day 14 and measurements for α-tocopherol in the serum (A) and liver (B) and γ-tocopherol in the serum (C) and liver (D). Real-time qPCR was performed on liver samples to quantify Cyp4f1 expression (E) and Ttp-α (F). Enteral control piglet historical mean values, when available, are represented by the dashed line. Values are expressed as means ± SE. One-way ANOVA; n = 5–7 piglets/group. *P < 0.05 vs. IL.
The primary biological activity of α-tocopherol is as an antioxidant. We examined a marker of global free radical antioxidant and hydrogen peroxide scavenger serum super oxide dismutase (SOD) and two markers of lipid peroxidation: malondialdehyde (MDA) and 8-iso-prostaglandin F2α (PGE-F2α) (Table 2). The VITE group did not differ from either IL or RIF.
Table 2.
Oxidant stress markers
| IL | VITE | RIF | P Value | |
|---|---|---|---|---|
| Serum SOD, % inhibition | 36.9 ± 3.1 | 38.1 ± 3.4 | 40.3 ± 2.9 | 0.90 |
| Serum MDA, pmol | 21.1 ± 2.9 | 26.5 ± 4.1 | 43.8 ± 18.5 | 0.36 |
| Serum 8-iso-PGE, pg/mL | 515 ± 38.2 | 493 ± 48.6 | 539 ± 22.8 | 0.69 |
Values are means ± SE. IL, Intralipid; VITE, vitamin E; RIF, Rifadin IV; SOD, superoxide dismutase; MDA, malondialdehyde; PGE, prostaglandin.
In our previous study, addition of vitamin E reduced serum phytosterol concentration in the supplemented groups (35). We examined this effect in the current study and found that serum concentrations of phytosterols (Table 3) did not differ among any of the groups; however, the serum total phytosterols were all markedly higher than our previously reported serum total phytosterols in SO of (347 ± 26.3 vs. 28.45 ± 3.40 μM, respectively) (35). We also measured oxidized phytosterol concentration. Overall, the levels of oxidized phytosterols were much lower than phytosterols (nanomolar vs. micromolar, respectively).
Table 3.
Plasma phytosterols
| IL | VITE | RIF | |
|---|---|---|---|
| Plasma phytosterol, μM | |||
| β-Sitosterol | 236 ± 17.8 | 206 ± 12.5 | 274 ± 26.5 |
| Stigmasterol | 34.0 ± 2.56 | 30.0 ± 2.58 | 40.1 ± 3.52 |
| Campesterol | 77. ± 6.16 | 68.3 ± 4.62 | 95.9 ± 9.91 |
| Total phytosterol | 347 ± 26.3 | 304 ± 20.0 | 410 ± 40.0 |
| Plasma oxidized phytosterol, nM | |||
| 7α-OH campesterol | 3.07 ± 0.26 | 2.07 ± 0.27 | 3.47 ± 0.22 |
| 7α-OH sitosterol | 3.45 ± 0.29 | 2.53 ± 0.30 | 3.69 ± 0.28 |
| 7β-OH campesterol | 1.9 ± 0.22 | 1.35 ± 0.21 | 1.93 ± 0.17 |
| 7β-OH sitosterol | 2.95 ± 0.31 | 1.9 ± 0.31 | 2.79 ± 0.21 |
| 7-Keto campesterol | 7.11 ± 0.63 | 5.63 ± 0.83 | 5.46 ± 0.57 |
| 7-Keto sitosterol | 33.1 ± 3.26 | 27.2 ± 2.22 | 33.2 ± 2.34 |
| Total oxidized phytosterol | 51. ± 4.63 | 40.8 ± 3.63 | 50.5 ± 2.69 |
Values are means ± SE. IL, Intralipid; RIF, Rifadin IV; VITE, vitamin E.
Administering TPN containing SO to piglets for 14 days results in elevated serum direct bilirubin levels, similar to what is observed in infants (23, 35, 50). Pigs fed enterally for 14 days in our previous research had low direct bilirubin (0.016 ± 0.004 mg/dL) (50). Our current results agree with PN increasing direct bilirubin levels (Fig. 2A). The direct bilirubin levels in IL and VITE piglets were 2.9 ± 0.3 and 3.0 ± 0.5 mg/dL, respectively, which were significantly higher than direct bilirubin levels in the RIF group (1.4 ± 0.2 mg/dL; P = 0.016 vs. VITE; P = 0.011 vs. IL). Overall, direct bilirubin levels were two to five times higher than in our previous studies (23, 35, 50). The increased direct bilirubin levels did not lead to increased liver injury, as ALT levels (Fig. 2C) were no different among groups and within the normal range for enteral piglets (50). We did not observe any change among groups for serum or liver lipid markers (Fig. 2, D–H).
Fig. 2.

Rifampicin treatment exhibits improved serum chemistry profile. Laboratory chemistry values for term piglets administered lipid emulsions for 14 days containing standard soy oil (SO) emulsion (IL), α-tocopherol supplemented SO (VITE), or SO supplemented with daily injections of 10 mg/L Rifadin IV (RIF) are presented. Serum chemistry marker of cholestasis direct bilirubin (A), cholangiocyte injury marker γ-glutamyl transferase (GGT) (B), liver injury marker alanine amino transferase (ALT) (C), serum lipid markers cholesterol (D), 7α-OH-cholesterol (E), 27α-OH-cholesterol (F), triglycerides (G), and liver triglycerides (H) were analyzed. Enteral control piglet historical mean values, when available, are represented by the dashed line. Values are expressed as means ± SE. *P < 0.05 vs. IL; #P < 0.05 vs. VITE. One-way ANOVA; n = 5–7 piglets per group.
Similar to serum bilirubin levels, bile acids accumulate in the plasma during parenteral nutrition. The RIF group accumulated significantly fewer bile acids in the plasma (17.1 ± 2.5 μM) compared with IL (37.8 ± 6.5 μM; P = 0.015) (Fig. 3A). There was no difference between the IL and VITE groups. We have observed typical plasma bile acids in enterally fed piglets to be 3.1 ± 1.0 μM, so the RIF treatment did not completely return bile acids to normal concentrations (50). For there to be fewer bile acids in the plasma, bile could accumulate in the liver or be excreted. The liver bile acid pool was not different among any groups (Fig. 3B). The standard flux of bile acids that would progress from the liver to the gall bladder and then into the intestine was significantly lower in the RIF bile acid pool compared with VITE (P = 0.004) (Fig. 3C). A similar pattern in bile volume was observed with significantly less in the RIF group compared with VITE (P = 0.003) (Fig. 3D). Surprisingly, the VITE group had the largest pool of bile acids in the gall bladder. The VITE group had a much greater fluid content in the gall bladder, which was likely a large contributor to the overall larger pool size. Despite the larger gall bladder pool in VITE, the RIF group trended toward higher bile acid content in the cecum, with 2.2 times greater than the IL treatment compared with VITE’s 1.6 times greater bile acid content compared with IL (Fig. 3E). There can be increased urinary clearance of xenobiotics in the urine when PXR is activated by rifampicin (52). We did not see any increase in urinary bile acids by rifampicin, nor any difference in urinary bile acid excretion among any groups (Fig. 3F).
Fig. 3.
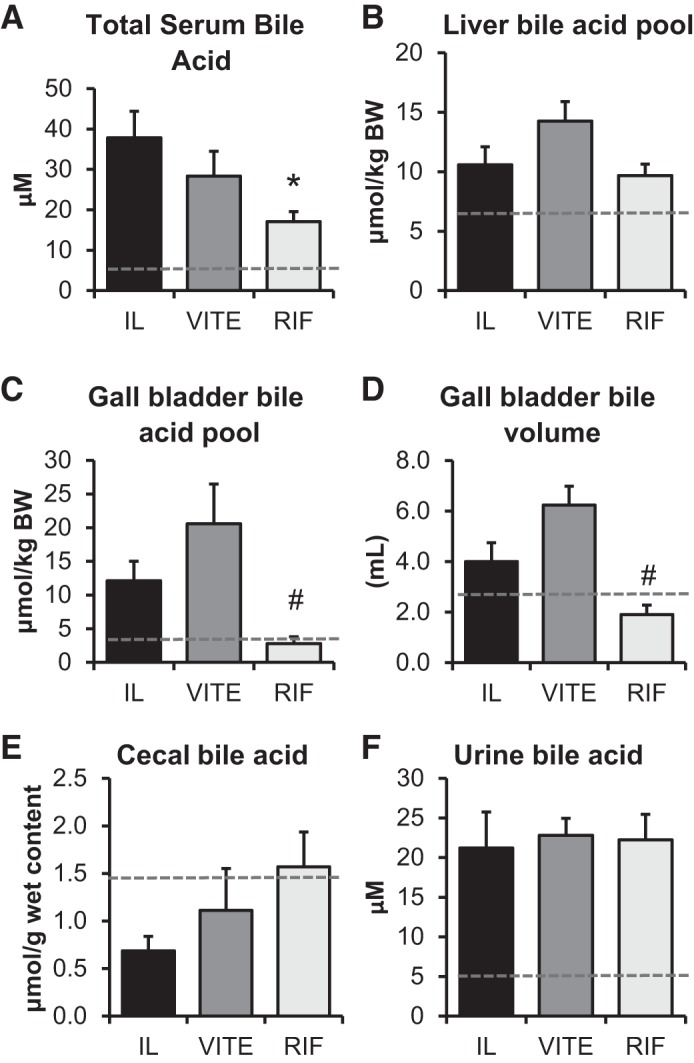
Rifampicin treatment increases the clearance of bile acids from the serum to the gall bladder. Term piglets were administered lipid emulsions for 14 days containing standard soy oil (SO) emulsion (IL), α-tocopherol-supplemented SO (VITE), or SO supplemented with daily injections of 10 mg·kg−1·day−1 Rifadin IV (RIF). Total bile acid was determined in the 3 major compartments for bile acid circulation: serum (A), liver (B), and gall bladder (C). The total volume of bile content in the gall bladder (D) is also shown. The relative concentration of bile acids was determined in the cecum (E). The bile acid concentration of bile collected from urine in the bladder is shown (F). Enteral control piglet historical mean values, when available, are represented by the dashed line. Values are expressed as means ± SE. One-way ANOVA; n = 4–7 piglets/group. *P < 0.05 vs. IL. #P < 0.05 vs. VITE.
Clearance of bile acid from the plasma in the RIF treatment group could be due to enhanced facilitated clearance of bile acids. We used the potent PXR agonist rifampicin to activate the drug-metabolizing pathway to determine whether vitamin E targets this pathway and whether PXR activation can facilitate clearance of bile acids to prevent PNALD. The phase I PXR target Cyp3a29 significantly increased compared with IL (P = 0.001) and VITE (P = 0.015) at the mRNA level (Fig. 4A) and the protein level for IL (P = 0.006) and VITE (P < 0.001) (Fig. 4C) in RIF piglets. The phase II conjugation enzyme UGT1A6 was not different at the mRNA level (Fig. 4B), but protein expression was significantly increased in the RIF treatment group (P < 0.001) (Fig. 4D). There were no observed differences between the IL and VITE treatment groups in either phase I or phase II target genes.
Fig. 4.
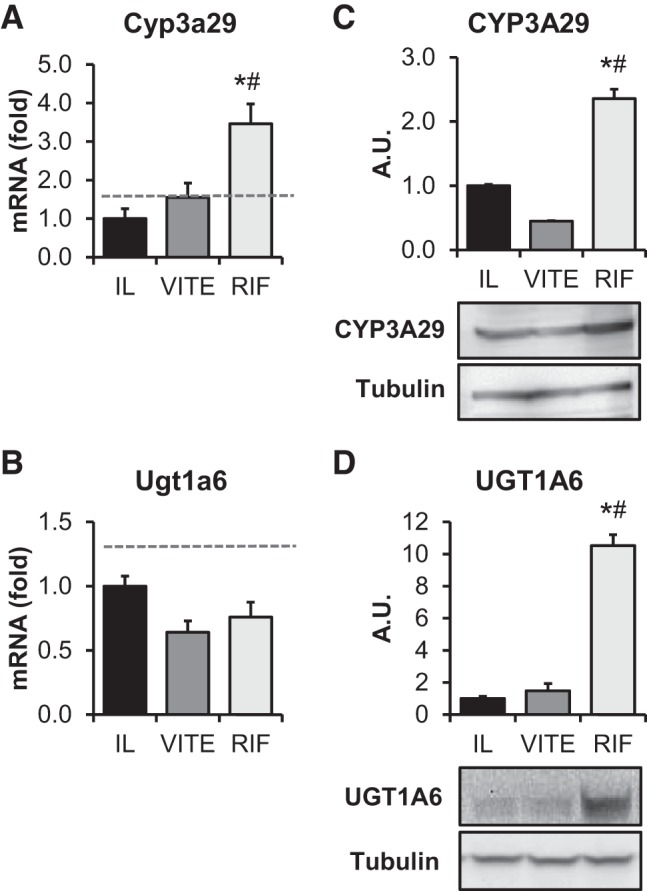
Rifampicin, but not α-tocopherol, activates downstream targets of the hepatic pregnane X receptor (PXR) pathway. Term piglets were administered lipid emulsions for 14 days containing standard soy oil (SO) emulsion (IL), α-tocopherol-supplemented SO (VITE), or SO supplemented with daily injections of 10 mg·kg−1·day−1 Rifadin IV (RIF). Quantitative PCR analysis was done of hepatic PXR downstream targets Cyp3a29 (A) and Ugt1a6 (B) normalized to β-actin. Western blot analysis of hepatic PXR downstream targets Cyp3a29 (C) and Ugt1a6 (D) is also shown. Tubulin protein values were used as a loading control. Blot images are pooled representative samples of each treatment. Graphs were quantified using n = 5–7 piglets/group. Enteral control piglet historical mean values, when available, are represented by the dashed line. Values are expressed as means ± SE. One-way ANOVA. *P < 0.05 vs. IL. #P < 0.05 vs. VITE.
The activation of the PXR pathway by rifampicin should facilitate clearance of bile acids. CYP3A29 can convert CDCA to HCA via addition of a 6α-hydroxy to CDCA (Fig. 5A). We used stably labeled [2H4]CDCA infusion to look at the fractional synthetic rate (FSR) of [2H4]HCA and observed a significant twofold increase over 12 h in FSR in the RIF group (P = 0.015) compared with the IL group (Fig. 5B). VITE and IL treatment did not differ. Bile acid conjugation to glycine and taurine occurs in the liver following synthesis. Because of limited sample availability, we did not quantify the [2H4]CDCA conjugates; so, instead, we looked at the ratio of HCA to CDCA and their conjugates in the plasma to achieve a secondary measure for CDCA conversion to HCA. There is between 70 and 150 times more glycine-conjugated bile acids compared with unconjugated bile acids (Table 4). The conjugation rate to glycine does not differ among groups. The total amount of taurine-conjugated bile acids was 16-fold lower than the total glycine bile acids, and there was no difference among groups in their rates of conjugation. HCA concentrations were up to ninefold higher than CDCA in the piglets receiving IL treatment. Since the total concentration of HCA, hyodeoxycholic acid, and CDCA were lower in the RIF group than IL and VITE in the unconjugated and glycine-conjugated groups, we also analyzed the treatments by the ratio of HCA to CDCA to observe whether there was a shift in HCA production by RIF. For the unconjugated and conjugated bile acids, we did not observe a statistical difference in the ratio of CDCA to HCA (Fig. 5, C–E). There was a nonsignificant increase in glycine-HCA (4%) in the RIF group compared with the IL group (Fig. 5D). We observed a similar increase in taurine-HCA (7%) in the RIF group compared with the IL group (Fig. 5E).
Fig. 5.

Conversion of [2H4]CDCA to [2H4]HCA and serum bile acid profiles of chenodeoxycholic acid (CDCA), hyocholic acid (HCA), and hyodeoxycholic acid (HDCA). Term piglets were administered lipid emulsions for 14 days containing standard soy oil (SO) emulsion (IL), α-tocopherol-supplemented SO (VITE), or SO supplemented with daily injections of 10 mg·kg−1·day−1 Rifadin IV (RIF). A: schematic of Cyp3a29 enzymatic conversion of labeled CDCA to HCA. B: calculated fractional synthetic rate of [2H4]HCA from continuous infusion of [2H4]CDCA at a rate of 23 μg·kg−1·day−1 for 12 h. The relative percent serum concentration of CDCA to HCA at day 14 for unconjugated (C), glycine-conjugated (D), and taurine-conjugated (E) bile acids are also shown. Values are expressed as means ± SE; n = 4–7 piglets/group. %FSR significance calculated with repeated measures 2-way ANOVA. *P < 0.05 vs. IL.
Table 4.
Plasma CDCA, HCA, HDCA, and conjugates
| IL | VITE | RIF | |
|---|---|---|---|
| Unconjugated bile acids, μM | |||
| CDCA | 0.14 ± 0.05 | 0.11 ± 0.02 | 0.09 ± 0.02 |
| HCA | 1.30 ± 0.72 | 1.26 ± 0.30 | 0.48 ± 0.16 |
| HDCA | 0.22 ± 0.04 | 0.16 ± 0.03 | 0.11 ± 0.02 |
| Total bile acid | 1.67 ± 0.77 | 1.52 ± 0.35 | 0.61 ± 0.20 |
| Glycine-conjugated bile acids, μM | |||
| CDCA | 9.19 ± 5.59 | 7.15 ± 5.46 | 3.73 ± 2.16 |
| HCA | 96.4 ± 35.4 | 114 ± 40.9 | 82.5 ± 16.1 |
| HDCA | 11.0 ± 4.53 | 6.92 ± 3.49 | 4.97 ± 1.96 |
| Total bile acid | 116 ± 39.9 | 128 ± 48.4 | 93.5 ± 20.8 |
| Taurine-conjugated bile acids, μM | |||
| CDCA | 2.25 ± 1.05 | 2.97 ± 2.50 | 2.74 ± 2.15 |
| HCA | 4.26 ± 2.30 | 6.15 ± 4.00 | 3.02 ± 1.31 |
| HDCA | 0.98 ± 0.36 | 1.55 ± 1.15 | 1.37 ± 0.77 |
| Total bile acid | 6.57 ± 3.03 | 9.44 ± 6.97 | 5.81 ± 3.34 |
Values are means ± SE. IL, Intralipid; RIF, Rifadin IV; VITE, vitamin E; CDCA, chenodeoxycholic acid; HCA, hyocholic acid; HDCA, hyodeoxycholic acid.
We examined expression of a panel of bile acid transport- and synthesis-associated genes but did not observe any differences in expression among IL, VITE, or RIF with transport genes Bsep, Ntcp, or Ost-α (Fig. 6A). Ost-α decreased 50% in the RIF group compared with IL, although based on the piglet numbers this failed to reach significance. The rate-limiting enzyme for bile acid synthesis, Cyp7a1, the alternative bile acid acidic pathway gene Cyp27a1, and the pig-specific HCA synthesis gene Cyp4a21 were all unchanged by treatment groups (Fig. 6B). We also examined the expression of Cyp8b1, which is expressed in fetal piglets and decreases following birth (29), but by the end of the study expression was undetectable in our piglets. Other transporters for phospholipids, bilirubin, and bile acids were also unchanged in the liver (Fig. 6C). Other potential targets for liver injury were also examined, including cytokine expression of Il-1β, Il-6, and Tnf-α, which did not differ among groups (data not shown). Consistent with our urinary bile acid findings, bile acid transporters (Asbt, Ost-α, Mrp2) in the kidneys did not differ among treatments (Fig. 6D).
Fig. 6.

Expression of hepatic and kidney genes involved in bile acid synthesis and transport. Term piglets were administered lipid emulsions for 14 days containing standard soy oil (SO) emulsion (IL), α-tocopherol-supplemented SO (VITE), or SO supplemented with daily injections of 10 mg·kg−1·day−1 Rifadin IV (RIF). Quantitative PCR analysis of hepatic genes associated with bile acid transport (A), bile acid synthesis (B), and phospholipid and xenobiotic clearance (C) was performed. Kidney genes associated with bile acid transport (D) are also shown. All gene expression was normalized to β-actin. Enteral control piglet historical mean values, when available, are represented by the dashed line. Values are expressed as means ± SE. One-way ANOVA; n = 5–7 piglets/group.
There was a significant increase in serum FGF-19 levels in the RIF group compared with both IL (P < 0.001) and VITE (P = 0.003) (Fig. 7). We examined the transcript levels of Fgf-19 in both the liver and intestine to determine a source of the plasma FGF-19; however, both tissues’ Fgf-19 mRNA levels were unchanged among IL, VITE, or RIF. The gall bladder is another large source of FGF-19 (56); however, we did not have tissue collected from this organ to determine whether this was a source of our observed circulating FGF-19.
Fig. 7.

Rifampicin treatment increases expression of fibroblast growth factor (FGF)-19. Term piglets were administered lipid emulsions for 14 days containing standard soy oil (SO) emulsion (IL), α-tocopherol-supplemented SO (VITE), or SO supplemented with daily injections of 10 mg·kg−1·day−1 Rifadin IV (RIF). Quantitative PCR analysis of hepatic Fgf-19 (A) and distal ileum FGF-19 (B) is shown. Gene expression was normalized to β-actin. Serum FGF-19 concentration from day 14 samples of FGF-19 (C) are also shown. Enteral control piglet historical mean values, when available, are represented by the dashed line. Values are expressed as means ± SE. One-way ANOVA; n = 5–7 piglets/group. *P < 0.05 vs. IL.
DISCUSSION
The administration of parenteral nutrition that has a lipid emulsion comprised primarily of SO leads to the development of PNALD. In our previous study, we showed that supplementation of SO with vitamin E in the form of natural α-tocopherol prevented the development of PNALD in preterm piglets (35). This observation led us to hypothesize that vitamin E is protective against the development of PNALD and may do so through enhanced clearance of bile acids by activation of the PXR drug metabolism pathway. A more recent publication by Muto et al. (33), however, found that administration of SO supplemented with vitamin E to term piglets failed to prevent the development of PNALD. The two studies differed by the age (preterm vs. term) of the piglets and the overall dose of lipid emulsion (5 vs. 10 g·kg−1·day−1). Given the disparity between the study results, we conducted a follow-up study in term piglets supplemented with α-tocopherol to determine whether this vitamin E supplementation is effective in preventing PNALD. We also tested whether vitamin E achieves this through facilitated clearance of bile acids by activation of PXR, or if direct activation of PXR is equally protective. We failed to see any benefit from vitamin E supplementation to prevent PNALD, nor did it activate PXR-mediated drug metabolism. However, the direct activation of PXR-mediated drug metabolism by rifampicin significantly reduced serum bilirubin and bile acids.
The first aim of our study was to determine the effectiveness of vitamin E supplementation in the prevention of PNALD and whether this functions through facilitated clearance of bile acids by activation of drug metabolism pathways. Contrary to our hypothesis, the addition of vitamin E was ineffective in preventing elevations in direct bilirubin or conjugated bile acids. We speculate that the primary reason supplemental vitamin E did not prevent PNALD is the increased lipid dose used in the current study. High parenteral lipid doses in humans cause fat overload syndrome, which causes hematological abnormalities as well as jaundice, hepatosplenomegaly, and respiratory distress (6, 16, 20, 51). We cannot confirm any alterations in hematological parameters in our piglets, but we did observe increased tissue weights, jaundice, and respiratory distress. Furthermore, in our previous study, direct bilirubin concentration with Intralipid administration for 2 wk only reached 0.39 ± 0.09 mg/dL (SE) (50). The high lipid dose of 10 g·kg−1·day−1 increased the direct bilirubin by about fourfold, so the severity of disease onset was remarkably increased with a doubling of intravenous lipid load. The implication of these findings is that the benefits of α-tocopherol supplementation may be limited to mild or moderate disease onset rather than the more severe case we observed.
As expected, supplementing α-tocopherol to the lipid emulsion increased the serum and hepatic levels of α-tocopherol. The administration of rifampicin did not change the concentration of α-tocopherol, suggesting that α-tocopherol is not a substrate for Cyp3a29 modification. The pathway for α-tocopherol hepatic clearance is not entirely understood, but the primary rate-limiting enzyme for α-tocopherol metabolism is thought to be Cyp4f2, which facilitates an initial ω-hydroxylation (36, 43). In the α-tocopherol-treated piglets, Cyp4f2 expression was unchanged, which is in agreement with both rat (32) and mouse (49) models. However, γ-tocopherol concentrations decreased in the liver and serum of α-tocopherol- and rifampicin-treated piglets. Abe et al. (1) observed a similar isoform-dependent effect by treating rats with an inhibitor of CYP enzyme function, ketoconazole, which caused an increase in γ-tocopherol and no change in α-tocopherol concentrations. Tocopherol transport protein-α was unchanged in our groups, so is likely not a contributor to the observed difference in γ-tocopherol concentrations. These results confirm there is an isoform-dependent effect on vitamin E metabolism that works through CYP genes, but α-tocopherol is not the targeted isoform. We did not assess the accumulation of α-tocopherol in other tissues, so we cannot be certain that nonhepatic α-tocopherol accumulation occurred. Overall, hepatic levels of vitamin E did not affect hepatic function. The biological effect of α-tocopherol was in agreement with results from Muto et al. (33), as there was no benefit to antioxidant defense in the plasma of piglets that were administered supplemental α-tocopherol. Typically, there is a high level of oxidative stress associated with premature birth (38). The piglets used in the current study and by Muto and colleagues (33) were term born, which may account for the lack of change in antioxidant markers. A caveat to this is we did not assess any hepatic markers of oxidation, so we cannot rule out that there are tissue-specific oxidant stress differences among treatments.
We additionally tested whether α-tocopherol activates drug metabolism through PXR. The role of α-tocopherol as a ligand for PXR is unclear. Our results support the conclusion that α-tocopherol is not a ligand for PXR, as we did not see any increase in vivo in PXR target genes Cyp3a29 nor Ugt1a6 in piglets receiving supplemental α-tocopherol. In mouse studies, other transport proteins in the liver are upregulated including multidrug resistance 1 (MDR1), multidrug resistance protein 2 (MRP2), and multidrug resistance protein 3 (MRP3) (32, 48). However, none of the genes examined increased following α-tocopherol supplementation. We did see a large increase in gall bladder fluid volume in the α-tocopherol-treated piglets. This result would suggest that either there is an increase in transport into the gall bladder or there is strong inhibition of gall bladder clearance. To our knowledge, there is no literature to suggest that α-tocopherol inhibits gall bladder emptying, and we do not consider this the likely cause. Therefore, there may be additional genes facilitating this increased gall bladder volume, or this may be through enhanced transport activity or protein expression, which could not be assessed given limited working antibodies in the piglet for these target genes.
The second aim of our study was to determine whether direct activation of the PXR pathway prevented the development of PNALD. Treatment with rifampicin was effective in reducing the primary markers of PNALD, serum direct bilirubin, and plasma bile acids. We did not see any change in lipid metabolism among groups, as cholesterol and serum and hepatic triglycerides were similar. This result suggests that the main benefit of rifampicin likely occurred through modification of bile acid metabolism and transport, not through improved metabolism of hepatic lipids. Rifampicin activated the downstream targets of PXR, Cyp3a29 and Ugt1a6. In humans, rifampicin increases the ratio of urinary excreted HCA and glucuronidated bile acids (52). HCA forms by the 6α-hydroxlyation of CDCA via Cyp3a29. The conversion of CDCA to HCA doubled at 12 h in the rifampicin-treated piglets. Despite this, we did not see much of a change in the in total HCA pool in the piglets. Piglets do not synthesize cholic acid due to a lack of Cyp8b1 expression; rather, they produce large quantities of HCA through Cyp4a21 (28). This large basal production of HCA could have masked our ability to see significantly increased synthesis of HCA driven by Cyp3a29, as we did observe some modest increase in conjugated bile acids. In the serum, we observed an overall decrease in serum bile acids with no alteration in the rate of amidation following rifampicin treatment, which is consistent with clearance of bile acids through facilitated transport via PXR activation (45). We did not analyze the rate of glucuronidation of bile acids, but we would expect there is an increased rate given the elevated UGT1A6 protein expression. We did not see a change in total urinary output from the treatment of RIF, which would suggest the majority of bile acid clearance in still occurring through enterohepatic circulation. To support this, there was an increase in cecal bile acid content in the RIF-treated group. This result would suggest that despite the lack of change in transporter expression, there may be some movement of bile acids through enterohepatic circulation in the RIF group. Furthermore, this could account for the lower bile volume and bile acid pool in the gall bladder. The RIF group may be moving bile acids into the intestine, and the VITE group may have reduced bile circulation into the common bile duct, which is common in most instances of TPN (33). We did not access hepatic transporter activity or gall bladder emptying, which may be two important components to the benefit observed by rifampicin treatment. Rifampicin may also alter bile acids by modifying the microbiome, given it is an antibiotic. Multiple studies observed differences in bile acid composition and effect on metabolism from antibiotic administration (5, 41, 46). All groups did receive some intravenous antibiotics as postoperative care, which may have altered some bacteria in all groups. We did not assess changes to the microbiome so cannot rule out that this did not contribute to some of the observed effects in the RIF group.
An important finding with rifampicin treatment was the strong induction of circulating FGF-19. The canonical pathway for FGF-19 signaling is through activation of FXR-mediated transcription of FGF-19 in the enterocyte, which is secreted into the blood to activate hepatic surface receptor heterodimers FGFR4/β-Klotho. FGF-19 binding suppresses Cyp7a1 and decreases the synthesis of bile acids. Activation of this pathway is consistent with the reduced bile acid concentration in the serum of the rifampicin-treated piglets. The reduced gall bladder bile acid pool in these piglets also supports the suppression of bile acid synthesis. However, the concentration of 7α-OH-cholesterol, the product of CYP7A1 activity, was unchanged. There was also no change in the product of CYP27A1 activity, 27α-OH-cholesterol, which is surprising given the high concentrations of FGF-19. Wistuba et al. (54a) were the first group to observe PXR-mediated induction of FGF-19. However, what is unique to our study is that we observed this induction independent of enteral feeding. We postulate that the source of the circulating FGF-19 was the intestine, yet transcript levels were not different in either the ileum or hepatocytes. Other sources of FGF-19 are possible, with the gall bladder also producing large quantities of FGF-19 (56). We cannot determine the activity of the FXR-regulated transporters or their protein concentration given the limitations of available antibodies in piglets, so it is possible that there are FXR-mediated events occurring in the rifampicin-treated group. However, we speculate that the reduced bile acid and bilirubin concentrations are primarily through PXR-mediated FGF-19 regulation.
A current hypothesis for the development of PNALD in infants involves the suppression of FXR-mediated bile acid signaling by phytosterols present in the soy-based lipid emulsion. ATP-binding cassette transporter G5/G8 (ABCG5/8) clears phytosterols from circulation. Presumably, there are a few other pathways for the clearance of phytosterols, as mutations in ABCG5/8 result in the development of the disease β-sitosterolemia, which is characterized by ultraphysiological elevations of all phytosterols in the blood. To our knowledge, this is the first study to examine whether direct activation of PXR drug metabolism pathways can facilitate clearance of phytosterols. Phytosterols cleared faster would potentially reduce the inhibition of FXR signaling; however, we did not see any difference in their clearance. In fact, the concentration of phytosterols in the RIF trended toward higher concentrations than in the other treatment groups. We also examined several forms of phytosterol oxidation products, which are formed in vivo, yet the mechanism is not clear (46); however, they too were unchanged by rifampicin treatment. The supplementation of α-tocopherol did not change the production of oxidized phytosterols either, which is consistent with a recent human study that examined the effects of several antioxidant sources, including vitamin E, on oxidized phytosterol synthesis in vivo (4).
In conclusion, we have found that treatment with rifampicin, a PXR agonist, reduces serum bile acid concentration caused by a high lipid dose during TPN. In contrast, the addition of vitamin E did not reduce any markers of PNALD development. Given some of the variables in our study, we still cannot exclude vitamin E as having any benefit in the prevention of PNALD in all instances of disease development. We have established that at high lipid doses and in term-born piglets, the supplementation of α-tocopherol is not beneficial. However, in conditions of prematurity or lower lipid dose, our previous data support a beneficial effect (35). Additional study is warranted to clarify the mechanism for the protective effect of vitamin E. Vitamin E was not an effective agonist for activation of PXR. The use of rifampicin to activate PXR may have promising clinical benefits for the treatment of PNALD, although more studies on the long-term benefit to reducing PNALD progression and risk of hepatic injury are needed. Furthermore, the activation of FGF-19 by rifampicin and the function it may have in reducing the progression of PNALD warrant additional research.
GRANTS
This work was supported in part by federal funds from the USDA, Agricultural Research Service under Cooperative Agreement No. 58-6250-6-001, the American Society for Parenteral and Enteral Nutrition and the Texas Medical Center Digestive Diseases Center [National Institutes of Health (NIH) Grant P30 DK-56338], and NIH Grant DK-094616 (to D. Burrin). G. Guthrie was supported by a training fellowship from NIH Grant T32-DK07664.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
DISCLOSURES
D. G. Burrin received lipid emulsions donated from Fresenius Kabi for the study. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
G.G. and D.G.B. conceived and designed research; G.G. and B.S. performed experiments; G.G., B.S., S.C., C.L., and J.P. analyzed data; G.G., B.S., S.C., C.L., J.P., and D.G.B. interpreted results of experiments; G.G. prepared figures; G.G. drafted manuscript; G.G., B.S., S.C., C.L., J.P., and D.G.B. edited and revised manuscript; G.G., B.S., S.C., C.L., J.P., and D.G.B. approved final version of manuscript.
REFERENCES
- 1.Abe C, Uchida T, Ohta M, Ichikawa T, Yamashita K, Ikeda S. Cytochrome P450-dependent metabolism of vitamin E isoforms is a critical determinant of their tissue concentrations in rats. Lipids 42: 637–645, 2007. doi: 10.1007/s11745-007-3064-2. [DOI] [PubMed] [Google Scholar]
- 2.Bardowell SA, Ding X, Parker RS. Disruption of P450-mediated vitamin E hydroxylase activities alters vitamin E status in tocopherol supplemented mice and reveals extra-hepatic vitamin E metabolism. J Lipid Res 53: 2667–2676, 2012. doi: 10.1194/jlr.M030734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bardowell SA, Duan F, Manor D, Swanson JE, Parker RS. Disruption of mouse cytochrome p450 4f14 (Cyp4f14 gene) causes severe perturbations in vitamin E metabolism. J Biol Chem 287: 26077–26086, 2012. doi: 10.1074/jbc.M112.373597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baumgartner S, Mensink RP, Haenen GR, Bast A, Binder CJ, Bekers O, Husche C, Lütjohann D, Plat J. The effects of vitamin E or lipoic acid supplementation on oxyphytosterols in subjects with elevated oxidative stress: a randomized trial. Sci Rep 7: 15288, 2017. doi: 10.1038/s41598-017-15615-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, Littmann E, van den Brink MR, Jenq RR, Taur Y, Sander C, Cross JR, Toussaint NC, Xavier JB, Pamer EG. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517: 205–208, 2015. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell AN, Freedman MH, Pencharz PB, Zlotkin SH. Bleeding disorder from the “fat overload” syndrome. JPEN J Parenter Enteral Nutr 8: 447–449, 1984. doi: 10.1177/0148607184008004447. [DOI] [PubMed] [Google Scholar]
- 7.Carter BA, Taylor OA, Prendergast DR, Zimmerman TL, Von Furstenberg R, Moore DD, Karpen SJ. Stigmasterol, a soy lipid-derived phytosterol, is an antagonist of the bile acid nuclear receptor FXR. Pediatr Res 62: 301–306, 2007. doi: 10.1203/PDR.0b013e3181256492. [DOI] [PubMed] [Google Scholar]
- 8.Chan S, McCowen KC, Bistrian BR, Thibault A, Keane-Ellison M, Forse RA, Babineau T, Burke P. Incidence, prognosis, and etiology of end-stage liver disease in patients receiving home total parenteral nutrition. Surgery 126: 28–34, 1999. doi: 10.1067/msy.1999.98925. [DOI] [PubMed] [Google Scholar]
- 9.Chen J, Zhao KN, Chen C. The role of CYP3A4 in the biotransformation of bile acids and therapeutic implication for cholestasis. Ann Transl Med 2: 7, 2014. doi: 10.3978/j.issn.2305-5839.2013.03.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang JY. Bile acids: regulation of synthesis. J Lipid Res 50: 1955–1966, 2009. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deshpande G, Simmer K, Deshmukh M, Mori TA, Croft KD, Kristensen J. Fish oil (SMOFlipid) and olive oil lipid (Clinoleic) in very preterm neonates. J Pediatr Gastroenterol Nutr 58: 177–182, 2014. [DOI] [PubMed] [Google Scholar]
- 12.El-Karaksy H, Mansour S, El-Sayed R, El-Raziky M, El-Koofy N, Taha G. Safety and efficacy of rifampicin in children with cholestatic pruritus. Indian J Pediatr 74: 279–281, 2007. doi: 10.1007/s12098-007-0044-8. [DOI] [PubMed] [Google Scholar]
- 13.El Kasmi KC, Anderson AL, Devereaux MW, Vue PM, Zhang W, Setchell KD, Karpen SJ, Sokol RJ. Phytosterols promote liver injury and Kupffer cell activation in parenteral nutrition-associated liver disease. Sci Transl Med 5: 206ra137, 2013. doi: 10.1126/scitranslmed.3006898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El Kasmi KC, Vue PM, Anderson AL, Devereaux MW, Ghosh S, Balasubramaniyan N, Fillon SA, Dahrenmoeller C, Allawzi A, Woods C, McKenna S, Wright CJ, Johnson L, D’Alessandro A, Reisz JA, Nozik-Grayck E, Suchy FJ, Sokol RJ. Macrophage-derived IL-1β/NF-κB signaling mediates parenteral nutrition-associated cholestasis. Nat Commun 9: 1393, 2018. doi: 10.1038/s41467-018-03764-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foster DM, Barrett PH, Toffolo G, Beltz WF, Cobelli C. Estimating the fractional synthetic rate of plasma apolipoproteins and lipids from stable isotope data. J Lipid Res 34: 2193–2205, 1993. [PubMed] [Google Scholar]
- 16.Goulet O, Girot R, Maier-Redelsperger M, Bougle D, Virelizier JL, Ricour C. Hematologic disorders following prolonged use of intravenous fat emulsions in children. JPEN J Parenter Enteral Nutr 10: 284–288, 1986. doi: 10.1177/0148607186010003284. [DOI] [PubMed] [Google Scholar]
- 17.Gura KM, Duggan CP, Collier SB, Jennings RW, Folkman J, Bistrian BR, Puder M. Reversal of parenteral nutrition-associated liver disease in two infants with short bowel syndrome using parenteral fish oil: implications for future management. Pediatrics 118: e197–e201, 2006. doi: 10.1542/peds.2005-2662. [DOI] [PubMed] [Google Scholar]
- 18.Guthrie G, Kulkarni M, Vlaardingerbroek H, Stoll B, Ng K, Martin C, Belmont J, Hadsell D, Heird W, Newgard CB, Olutoye O, van Goudoever J, Lauridsen C, He X, Schuchman EH, Burrin D. Multi-omic profiles of hepatic metabolism in TPN-fed preterm pigs administered new generation lipid emulsions. J Lipid Res 57: 1696–1711, 2016. doi: 10.1194/jlr.M069526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He Y, Zhou X, Li X, Jin X, Wang X, Pan X, Bi D. Relationship between CYP3A29 and pregnane X receptor in landrace pigs: pig CYP3A29 has a similar mechanism of regulation to human CYP3A4. Comp Biochem Physiol C Toxicol Pharmacol 214: 9–16, 2018. doi: 10.1016/j.cbpc.2018.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Heyman MB, Storch S, Ament ME. The fat overload syndrome. Report of a case and literature review. Am J Dis Child 135: 628–630, 1981. doi: 10.1001/archpedi.1981.02130310034012. [DOI] [PubMed] [Google Scholar]
- 21.Husche C, Weingärtner O, Pettersson H, Vanmierlo T, Böhm M, Laufs U, Lütjohann D. Validation of an isotope dilution gas chromatography-mass spectrometry method for analysis of 7-oxygenated campesterol and sitosterol in human serum. Chem Phys Lipids 164: 425–431, 2011. doi: 10.1016/j.chemphyslip.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 22.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, Gerard RD, Repa JJ, Mangelsdorf DJ, Kliewer SA. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2: 217–225, 2005. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 23.Jain AK, Stoll B, Burrin DG, Holst JJ, Moore DD. Enteral bile acid treatment improves parenteral nutrition-related liver disease and intestinal mucosal atrophy in neonatal pigs. Am J Physiol Gastrointest Liver Physiol 302: G218–G224, 2012. doi: 10.1152/ajpgi.00280.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson CH, Bonzo JA, Cheng J, Krausz KW, Kang DW, Luecke H, Idle JR, Gonzalez FJ. Cytochrome P450 regulation by α-tocopherol in Pxr-null and PXR-humanized mice. Drug Metab Dispos 41: 406–413, 2013. doi: 10.1124/dmd.112.048009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, Kliewer SA, Gonzalez FJ. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res 48: 2664–2672, 2007. doi: 10.1194/jlr.M700330-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Landes N, Pfluger P, Kluth D, Birringer M, Rühl R, Böl GF, Glatt H, Brigelius-Flohé R. Vitamin E activates gene expression via the pregnane X receptor. Biochem Pharmacol 65: 269–273, 2003. doi: 10.1016/S0006-2952(02)01520-4. [DOI] [PubMed] [Google Scholar]
- 27.Le HD, de Meijer VE, Robinson EM, Zurakowski D, Potemkin AK, Arsenault DA, Fallon EM, Malkan A, Bistrian BR, Gura KM, Puder M. Parenteral fish-oil-based lipid emulsion improves fatty acid profiles and lipids in parenteral nutrition-dependent children. Am J Clin Nutr 94: 749–758, 2011. doi: 10.3945/ajcn.110.008557. [DOI] [PubMed] [Google Scholar]
- 28.Lundell K, Wikvall K. Gene structure of pig sterol 12α-hydroxylase (CYP8B1) and expression in fetal liver: comparison with expression of taurochenodeoxycholic acid 6α-hydroxylase (CYP4A21). Biochim Biophys Acta 1634: 86–96, 2003. doi: 10.1016/j.bbalip.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 29.Lundell K, Wikvall K. Species-specific and age-dependent bile acid composition: aspects on CYP8B and CYP4A subfamilies in bile acid biosynthesis. Curr Drug Metab 9: 323–331, 2008. doi: 10.2174/138920008784220574. [DOI] [PubMed] [Google Scholar]
- 30.Mackay DS, Jones PJ, Myrie SB, Plat J, Lütjohann D. Methodological considerations for the harmonization of non-cholesterol sterol bio-analysis. J Chromatogr B Analyt Technol Biomed Life Sci 957: 116–122, 2014. doi: 10.1016/j.jchromb.2014.02.052. [DOI] [PubMed] [Google Scholar]
- 31.Mundi MS, Salonen BR, Bonnes S. Home parenteral nutrition: fat emulsions and potential complications. Nutr Clin Pract 31: 629–641, 2016. doi: 10.1177/0884533616663635. [DOI] [PubMed] [Google Scholar]
- 32.Mustacich DJ, Leonard SW, Devereaux MW, Sokol RJ, Traber MG. α-Tocopherol regulation of hepatic cytochrome P450s and ABC transporters in rats. Free Radic Biol Med 41: 1069–1078, 2006. doi: 10.1016/j.freeradbiomed.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 33.Muto M, Lim D, Soukvilay A, Field C, Wizzard PR, Goruk S, Ball RO, Pencharz PB, Mi S, Curtis J, Wales PW, Turner JM. Supplemental parenteral vitamin E into conventional soybean lipid emulsion does not prevent parenteral nutrition-associated liver disease in full-term neonatal piglets. JPEN J Parenter Enteral Nutr 41: 575–582, 2017. doi: 10.1177/0148607115612030. [DOI] [PubMed] [Google Scholar]
- 34.Nandivada P, Baker MA, Mitchell PD, O’Loughlin AA, Potemkin AK, Anez-Bustillos L, Carlson SJ, Dao DT, Fell GL, Gura KM, Puder M. Predictors of failure of fish-oil therapy for intestinal failure-associated liver disease in children. Am J Clin Nutr 104: 663–670, 2016. doi: 10.3945/ajcn.116.137083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ng K, Stoll B, Chacko S, Saenz de Pipaon M, Lauridsen C, Gray M, Squires EJ, Marini J, Zamora IJ, Olutoye OO, Burrin DG. Vitamin E in new-generation lipid emulsions protects against parenteral nutrition-associated liver disease in parenteral nutrition-fed preterm pigs. JPEN J Parenter Enteral Nutr 40: 656–671, 2016. doi: 10.1177/0148607114567900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohnmacht S, Nava P, West R, Parker R, Atkinson J. Inhibition of oxidative metabolism of tocopherols with ω-N-heterocyclic derivatives of vitamin E. Bioorg Med Chem 16: 7631–7638, 2008. doi: 10.1016/j.bmc.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parker RS, Sontag TJ, Swanson JE. Cytochrome P4503A-dependent metabolism of tocopherols and inhibition by sesamin. Biochem Biophys Res Commun 277: 531–534, 2000. doi: 10.1006/bbrc.2000.3706. [DOI] [PubMed] [Google Scholar]
- 38.Perrone S, Negro S, Tataranno ML, Buonocore G. Oxidative stress and antioxidant strategies in newborns. J Matern Fetal Neonatal Med 23, Suppl 3: 63–65, 2010. doi: 10.3109/14767058.2010.509940. [DOI] [PubMed] [Google Scholar]
- 39.Rasmussen MK. Induction of cytochrome P450 mRNA in porcine primary hepatocytes cultured under serum free conditions: comparison of freshly isolated cells and cryopreserved. Exp Cell Res 360: 218–225, 2017. doi: 10.1016/j.yexcr.2017.09.011. [DOI] [PubMed] [Google Scholar]
- 40.Repa A, Binder C, Thanhaeuser M, Kreissl A, Pablik E, Huber-Dangl M, Berger A, Haiden N. A mixed lipid emulsion for prevention of parenteral nutrition associated cholestasis in extremely low birth weight infants: a randomized clinical trial. J Pediatr 194: 87–93.e1, 2018. doi: 10.1016/j.jpeds.2017.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17: 225–235, 2013. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 42.Shukla SJ, Sakamuru S, Huang R, Moeller TA, Shinn P, Vanleer D, Auld DS, Austin CP, Xia M. Identification of clinically used drugs that activate pregnane X receptors. Drug Metab Dispos 39: 151–159, 2011. doi: 10.1124/dmd.110.035105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sontag TJ, Parker RS. Cytochrome P450 ω-hydroxylase pathway of tocopherol catabolism. Novel mechanism of regulation of vitamin E status. J Biol Chem 277: 25290–25296, 2002. doi: 10.1074/jbc.M201466200. [DOI] [PubMed] [Google Scholar]
- 44.Sontag TJ, Parker RS. Influence of major structural features of tocopherols and tocotrienols on their ω-oxidation by tocopherol-ω-hydroxylase. J Lipid Res 48: 1090–1098, 2007. doi: 10.1194/jlr.M600514-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Tabee E, Azadmard‐Damirchi S, Jägerstad M, Dutta PC. Effects of α‐tocopherol on oxidative stability and phytosterol oxidation during heating in some regular and high‐oleic vegetable oils. J Am Oil Chem Soc 85: 857–867, 2008. doi: 10.1007/s11746-008-1274-2. [DOI] [Google Scholar]
- 46.Theriot CM, Bowman AA, Young VB. Antibiotic-induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. MSphere 1: e00045-15, 2016. doi: 10.1128/mSphere.00045-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Traber MG, Atkinson J. Vitamin E, antioxidant and nothing more. Free Radic Biol Med 43: 4–15, 2007. doi: 10.1016/j.freeradbiomed.2007.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Traber MG, Labut EM, Leonard SW, Lebold KM. α-Tocopherol injections in rats up-regulate hepatic ABC transporters, but not cytochrome P450 enzymes. Free Radic Biol Med 51: 2031–2040, 2011. doi: 10.1016/j.freeradbiomed.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Traber MG, Siddens LK, Leonard SW, Schock B, Gohil K, Krueger SK, Cross CE, Williams DE. α-Tocopherol modulates Cyp3a expression, increases γ-CEHC production, and limits tissue γ-tocopherol accumulation in mice fed high γ-tocopherol diets. Free Radic Biol Med 38: 773–785, 2005. doi: 10.1016/j.freeradbiomed.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 50.Vlaardingerbroek H, Ng K, Stoll B, Benight N, Chacko S, Kluijtmans LA, Kulik W, Squires EJ, Olutoye O, Schady D, Finegold ML, van Goudoever JB, Burrin DG. New generation lipid emulsions prevent PNALD in chronic parenterally fed preterm pigs. J Lipid Res 55: 466–477, 2014. doi: 10.1194/jlr.M044545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wesson DE, Rich RH, Zlotkin SH, Pencharz PB. Fat overload syndrome causing respiratory insufficiency. J Pediatr Surg 19: 777–778, 1984. doi: 10.1016/S0022-3468(84)80367-X. [DOI] [PubMed] [Google Scholar]
- 52.Wietholtz H, Marschall HU, Sjövall J, Matern S. Stimulation of bile acid 6α-hydroxylation by rifampin. J Hepatol 24: 713–718, 1996. doi: 10.1016/S0168-8278(96)80268-6. [DOI] [PubMed] [Google Scholar]
- 53.Wilmore DW, Dudrick SJ. Growth and development of an infant receiving all nutrients exclusively by vein. JAMA 203: 860–864, 1968. doi: 10.1001/jama.1968.03140100042009. [DOI] [PubMed] [Google Scholar]
- 54.Wilmore DW, Groff DB, Bishop HC, Dudrick SJ. Total parenteral nutrition in infants with catastrophic gastrointestinal anomalies. J Pediatr Surg 4: 181–189, 1969. doi: 10.1016/0022-3468(69)90389-3. [DOI] [PubMed] [Google Scholar]
- 54a.Wistuba W, Gnewuch C, Liebisch G, Schmitz G, Langmann T. Lithocholic acid induction of the FGF19 promoter in intestinal cells is mediated by PXR. World J Gastroenterol 13: 4230–4235, 2007. doi: 10.3748/wjg.v13.i31.4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yerushalmi B, Sokol RJ, Narkewicz MR, Smith D, Karrer FM. Use of rifampin for severe pruritus in children with chronic cholestasis. J Pediatr Gastroenterol Nutr 29: 442–447, 1999. doi: 10.1097/00005176-199910000-00013. [DOI] [PubMed] [Google Scholar]
- 56.Zweers SJ, Booij KA, Komuta M, Roskams T, Gouma DJ, Jansen PL, Schaap FG. The human gallbladder secretes fibroblast growth factor 19 into bile: towards defining the role of fibroblast growth factor 19 in the enterobiliary tract. Hepatology 55: 575–583, 2012. doi: 10.1002/hep.24702. [DOI] [PubMed] [Google Scholar]