

Abstract
Downregulated expression of K+ channels and decreased K+ currents in pulmonary artery smooth muscle cells (PASMC) have been implicated in the development of sustained pulmonary vasoconstriction and vascular remodeling in patients with idiopathic pulmonary arterial hypertension (IPAH). However, it is unclear exactly how K+ channels are downregulated in IPAH-PASMC. MicroRNAs (miRNAs) are small non-coding RNAs that are capable of posttranscriptionally regulating gene expression by binding to the 3′-untranslated regions of their targeted mRNAs. Here, we report that specific miRNAs are responsible for the decreased K+ channel expression and function in IPAH-PASMC. We identified 3 miRNAs (miR-29b, miR-138, and miR-222) that were highly expressed in IPAH-PASMC in comparison to normal PASMC (>2.5-fold difference). Selectively upregulated miRNAs are correlated with the decreased expression and attenuated activity of K+ channels. Overexpression of miR-29b, miR-138, or miR-222 in normal PASMC significantly decreased whole cell K+ currents and downregulated voltage-gated K+ channel 1.5 (KV1.5/KCNA5) in normal PASMC. Inhibition of miR-29b in IPAH-PASMC completely recovered K+ channel function and KV1.5 expression, while miR-138 and miR-222 had a partial or no effect. Luciferase assays further revealed that KV1.5 is a direct target of miR-29b. Additionally, overexpression of miR-29b in normal PASMC decreased large-conductance Ca2+-activated K+ (BKCa) channel currents and downregulated BKCa channel β1 subunit (BKCaβ1 or KCNMB1) expression, while inhibition of miR-29b in IPAH-PASMC increased BKCa channel activity and BKCaβ1 levels. These data indicate upregulated miR-29b contributes at least partially to the attenuated function and expression of KV and BKCa channels in PASMC from patients with IPAH.
Keywords: KCNA5, KCNMB1, microRNA, posttranscriptional regulation, potassium channels
INTRODUCTION
Sustained pulmonary vasoconstriction is an important early cause of elevated pulmonary vascular resistance in patients with idiopathic pulmonary arterial hypertension (IPAH) (25, 61). Pulmonary arterial tone and vasoconstriction are controlled by the resting membrane potential (Em) in pulmonary artery smooth muscle cells (PASMC) (30, 49, 73). A change in Em plays a key role in excitation-contraction coupling in PASMC by regulating the cytosolic-free Ca2+ concentration ([Ca2+]cyt). Membrane depolarization leads to an increase in [Ca2+]cyt by opening voltage-dependent Ca2+ channels (VDCC) in PASMC (31, 32, 69). Increased [Ca2+]cyt not only causes PASMC contraction and pulmonary vasoconstriction but also stimulates PASMC proliferation and migration, which are the major contributors to concentric pulmonary arterial wall remodeling (10, 15).
K+ channel activity in PASMC contributes significantly to Em regulation (2, 41). Decreased K+ currents by downregulating channel expression (leading to a decreased number of K+ channels in the plasma membrane) and/or inhibiting channel activity would depolarize PASMC, open VDCC, and increase Ca2+ influx. Downregulated expression of K+ channels and decreased K+ currents in PASMC have been implicated in the development and progression of pulmonary hypertension; however, the underlying mechanisms are still unknown.
To date, more than eight different K+ channel families have been identified in the pulmonary vasculature; voltage-gated (KV) and Ca2+-activated K+ (KCa) channels appear to participate in the regulation of Em in PASMC. Each KV channel comprises four pore-forming α subunits and four regulatory cytoplasmic β subunits that modulate channel activity by inactivating α subunits (47). The diversity of mammalian KV channels is derived from genetic diversity, with over 40 genes encoding KV channel α subunits from 12 subtypes (KV1–12) (18, 42, 74). There are five families of Ca2+-activated K+ channels (KCa1–KCa5), including the large-conductance KCa channels [MaxiK or large-conductance Ca2+-activated K+ (BKCa)] (60). The BKCa channel consists of four α subunits (BKCaα) that create a pore in the membrane associated with four ancillary β (BKCaβ1–4) subunits. BKCaβ2 (KCNMB2), BKCaβ3 (KCNMB3), and BKCaβ4 (KCNMB4) subunits are predominantly expressed in endocrine tissue, testis, and brain tissue, respectively; however, BKCaβ1 (KCNMB1) appears to be solely expressed in smooth muscle tissue (5, 12). The function of BKCa channels in PASMC is finely tuned by its regulatory β subunits via enhancing α subunit sensitivity to intracellular Ca2+ and voltage (13). The presence of BKCa channels has been established in human and animal PASMC (3, 9, 20, 26). Downregulated KV channel expression (e.g., KV1.5/KCNA5) and decreased whole cell KV currents in PASMC have been established in pulmonary hypertension (PH), including IPAH (8, 63, 75), whereas the role of BKCa channels in PH is not well established (1, 4, 19, 45).
MicroRNAs (miRNAs) are small non-coding regulatory RNAs that posttranscriptionally regulate gene expression by binding to the 3′-untranslated region (UTR) of their targeted mRNA, thereby preventing translation and/or decreasing stability of target mRNAs. miRNAs have been implicated in the development and progression of pulmonary hypertension by using lung specimen and isolated PASMC from patients with IPAH as well as genetically engineered mice, including miR-204, miR-21, miR-130/301, and the miR-17/92 cluster (11, 28, 46, 56, 76). It has been found that certain miRNAs target multiple K+ channels, including KV7.5/KCNQ5 (34), KV4.2/KCND2 (38), KCa2.3/KCNN3 (36), and KIR2.1/KCNJ2 (40) channels in vascular smooth muscle cells and cardiac myocytes. The aim of this study is to investigate the role of miRNAs in the regulation of KV channels and BKCa channels in PASMC from patients with IPAH.
MATERIALS AND METHODS
Cell culture.
The approval for using human cells was granted by the University Institutional Review Board. PASMC from six healthy (normal) subjects without pulmonary hypertension and six patients with IPAH were provided by the Pulmonary Hypertension Breakthrough Initiative (PHBI). Normal and IPAH-PASMC were cultured in 5% fetal bovine serum (FBS) smooth muscle growth media (LifeLine) and incubated in a humidified 5% CO2 atmosphere at 37°C. Pulmonary arterial endothelial cells (PAEC) isolated from 4 normal subjects and 4 IPAH patients (PHBI) were cultured in 2% FBS growth media (LifeLine) and incubated in a humidified 5% CO2 atmosphere at 37°C. Human PASMC and PAEC were isolated at PHBI facility as previously described (27, 67). PASMC authentication was carried out with fluorescence-activated cell sorting (FACS) and immunocytochemistry (ICC) with smooth muscle actin-α (SMA), smooth muscle 22-α (SM22α), and smooth muscle myosin heavy chain (SMMHC). The percentage of positive cells that exhibit SMA, SM22α, and SMMHC signal was over 95%. PAEC authentication was carried out with FACS and ICC using CD31 (FACS), von Willebrand factor (vWF), and vascular endothelial cadherin (VE-cadherin) (ICC). The percentage of positive cells that exhibit CD31, vWF, and VE-cadherin signal was over 93% in cultures. The cells were acquired at passages 2–3. Passages 4–6 were used for the experiments. The demographic information of human PASMC and PAEC is listed in Table 1.
Table 1.
Demographic information of human subjects from whom PASMC and PAEC were isolated for molecular biological and electrophysiological experiments
| Subjects | Sex | Age, yr | Race | Type of Cells |
|---|---|---|---|---|
| Healthy subject-1 | Female | 36 | White | PASMC |
| Healthy subject-2 | Female | 33 | White | PASMC |
| Healthy subject-3 | Male | 34 | White | PASMC |
| Healthy subject-4 | Female | 34 | Asian | PASMC |
| Healthy subject-5 | Male | 46 | Asian | PASMC |
| Healthy subject-6 | Female | 56 | White | PASMC |
| IPAH patient-1 | Female | 56 | White | PASMC |
| IPAH patient-2 | Female | 41 | White | PASMC |
| IPAH patient-3 | Male | 27 | White | PASMC |
| IPAH patient-4 | Male | 56 | White | PASMC |
| IPAH patient-5 | Female | 33 | White | PASMC |
| IPAH patient-6 | Male | 51 | White | PASMC |
| Healthy subject-1 | Female | 49 | White | PAEC |
| Healthy subject-2 | Male | 51 | White | PAEC |
| Healthy subject-3 | Female | 55 | White | PAEC |
| Healthy subject-4 | Male | 49 | White | PAEC |
| IPAH patient-1 | Male | 51 | White | PAEC |
| IPAH patient-2 | Male | 16 | White | PAEC |
| IPAH patient-3 | Female | 16 | White | PAEC |
| IPAH patient-4 | Female | 22 | White | PAEC |
IPAH, idiopathic pulmonary arterial hypertension; PAEC, pulmonary arterial endothelial cells; PASMC, pulmonary artery smooth muscle cells.
miRNA PCR array.
To determine the miRNA expression profile in PASMC, RNA was isolated from normal, and IPAH-PASMC and miRNA expression was explored using the Human Cancer Pathway Finder miRNA PCR Array from SABiosciences. The lung vasculopathy in IPAH patients was recently demonstrated to be similar to pathological chances in cancer, and miRNA dysregulation in affected lung vascular tissues from IPAH patients shares similarities with miRNA profile in cancer tissues (17, 52). The miRNA PCR array consisted of a 96-well plate, which contained distinct primers for a total of 89 specific miRNAs plus 4 housekeeping small nuclear RNAs for normalization. The miRNAs were reverse transcribed to generate cDNA using miScript Reverse Transcription Kit (Qiagen) and then added to each well of the miRNA PCR array containing distinct miRNA primers. miRNA expression levels were determined by using the CFX96 real-time PCR machine (Bio-Rad Laboratories). The results were analyzed via the ΔΔCt method, and miRNA expression was normalized to the four small housekeeping RNAs U6, SNORD44, SNORD47, and SNORD48 (Qiagen). Small housekeeping RNA U6 displayed the least variability between samples, making it a strong candidate as control. miRNA expression normalized to U6 in normal PASMC was defined as onefold, and miRNAs with a fold change of equal to or greater than 2.0 in IPAH-PASMC are shown in the heatmap.
In silico analysis.
We performed in silico analysis using the standard online software TargetScanHuman v7.2 (http://www.targetscan.org/) to predict the potential target mRNAs encoding KV and BKCa channels for the specific miRNAs. The assigned context score to each 3′-UTR mRNA was used as a tool to select targets. The context score is the sum of the contributions of site-type, 3′ pairing, local AU, and position criteria. For each of these criteria, a more negative score indicates a more favorable site.
miRNA RT-PCR analysis.
The total RNAs were isolated from normal and IPAH-PASMC using a phenol/guanidine-based QIAzol reagent (Qiagen). To generate cDNA for sensitive and specific miRNA detection, the miScript II RT Kit (Qiagen) was used according to the manufacturer’s protocol. Real-time PCR was then performed by using the miScript SYBR Green PCR Kit (Qiagen) on the CFX384 real-time PCR machine (Bio-Rad Laboratories). miRNA specific primers (Qiagen) for human miR-29b, miR-138, and miR-222 were used to amplify miRNAs. The results were analyzed via the ΔΔCt method and miRNA expression was normalized to the small housekeeping RNA U6 since U6 displayed the least variability between samples. U6 is thus a good normalizing control compared with SNORD44, SNORD47, and SNORD48. To implement the ΔΔCt method, we compared our results with an average of the “healthy” cells. miRNA level was first normalized to the U6 level in all healthy subjects and the mean was calculated; then, the ratio of miRNA to U6 from each IPAH patient was normalized to the mean ratio from normal subjects. The mean ratio of miRNA expression level to U6 in normal PASMC was defined as onefold.
Patch-clamp experiments.
Whole cell and single-channel K+ currents were recorded with patch-clamp technique using an Axopatch-1D amplifier and a DigiData 1322 interface (Molecular Devices). The borosilicate patch pipettes (2–3 MΩ) were fabricated on a P-97 electrode puller (Sutter Instrument, Novato, CA) and polished with a Narishige MF-63 microforge for the current recording. The command voltage pulse protocols and the data acquisition were performed with pCLAMP 8.1 software. The currents, filtered at 1–2 kHz and digitized at 2–4 kHz, were elicited by depolarizing the cells from a holding potential of −80 mV to a series of test potentials (ranging from −100 to +100 mV, for 500 ms) in increments of 20 mV (every 15 s). The extracellular (bath) solution for recording KV currents [IK(V)] had an ionic composition of 140 mM NaCl, 4.7 mM KCl, 3 mM MgCl2, 10 mM glucose, 10 mM HEPES, and 1 mM EGTA (at pH 7.4), while the intracellular (pipette) solution contained 140 mM KCl, 4 mM MgCl2, 10 mM HEPES, 10 mM EGTA, and 5 mM Na2ATP (at pH 7.2). To record whole cell KCa currents (IK(Ca)), the bath (extracellular) solution contained 140 mM NaCl, 4.7 mM KCl, 1.8 mM CaCl2, 1.2 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH was adjusted to 7.4 by NaOH). The pipette (intracellular) solution contained 140 mM KCl, 4.4 mM CaCl2, 4 mM MgCl2, 5 mM Na2ATP, 10 mM HEPES, and 5 mM EGTA (pH was adjusted to 7.2 by KOH); the pCa of the pipette solution under this condition was calculated to be ~6.0. The digital subtraction of the currents recorded in PASMC during application of iberiotoxin (IbTX, 100 nM) or Penitrem A (100 nM) from the currents recorded in PASMC before the application of blocker were defined as IbTX-sensitive or Penitrem A-sensitive BKCa currents. The single-channel BKCa currents [iK(Ca)] in the cell-attached membrane patch were measured as we previously reported (50). We held the attached membrane patches at −70 mV before applying various test potentials. Open probability (Popen) and amplitude of the currents were measured with Fetchan and PStat software (Axon Instruments). High-K+ pipette (extracellular) solution was used in recording iK(Ca) so that the K+ equilibrium potential (EK) for the attached membrane was close to 0 mV (i.e., the [K+] in the pipette is close to the [K+] in the cytosol). The bath (extracellular) solution for measuring iK(Ca) in the cell-attached membrane included 141 mM NaCl, 1.8 mM CaCl2, 4.7 mM KCl, 1.2 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH = 7.4). The pipette (extracellular) solution included 125 mM KCl, 10 mM NaCl, 4 mM MgCl2, 5 mM ATP, and 10 mM HEPES (pH = 7.2). Results are presented as a function of the command potential (Ecomm), which is the inverse of the applied (holding or test) potential. Because of a negative resting Em (approximately −40 mV in cultured human PASMC), the actual transmembrane potential across the patched membrane is equal to the difference between the Ecomm and resting Em.
Gene (mRNA) expression RT-PCR analysis.
The total RNAs were isolated from human cells using QIAzol reagent (Qiagen). Real-time RT-PCR was performed as previously described (59). Human oligonucleotide primers (Integrated DNA Technologies) that were used in this study are listed as follows: KCNA1, CCCAGCAAGACGGACTTCTTCAAA (forward)/TCTGGTTTCCTTCCTGCTCAGCTA (reverse); KCNA2, GAACCGCCCTAGCTTTGATG (forward)/TCTCCCAGCTCATAAAACCGA (reverse); KCNA5, TTCTACCACCGGGAAACGGA (forward)/CTTCGGGCACTGTCTGCATT (reverse); KCNMB1, CTGTACCACACGGAGGACACT (forward)/GTAGAGGCGCTGGAATAGGAC (reverse); GAPDH, GCACCGTCAAGGCTGAGAAC (forward)/ATGGTGGTGAAGACGCCAGT (reverse). GAPDH, a housekeeping gene, was used to normalize the expression of the gene or transcript of interest.
Western blot analysis.
Cells were washed with PBS and resuspended into 1 × RIPA buffer (Millipore), which contained protease inhibitor cocktail tablets (Roche). Cells were incubated for 20 min on ice, followed by a 15-min centrifugation at 12,000 revolutions/min. The supernatant was collected and protein concentration was determined using a Nano-Drop spectrophotometer (Thermo Scientific). Samples were applied on 10% SDS-PAGE, and proteins were transferred onto nitrocellulose membranes. The membranes were blocked in 5% nonfat milk and incubated overnight at 4°C with primary antibodies for KCNA1 (cat. no. sc-11184), KCNA2 (cat. no. sc-292447), KCNA5 (cat. no. sc-25681), and KCNMB1 (cat. no. sc-377023) followed by 1-h incubation with secondary antibodies (either anti-rabbit, Cell Signaling, cat. no. 7074S, or anti-mouse, Cell Signaling, cat. no. 7076S). All samples were reprobed for β-actin (cat. no. sc-47778) as a loading control. Blots were developed using the SuperSignal West Pico Chemiluminescent Substrate (Pierce Biotechnology). To compare the levels of target proteins in PASMC between normal subjects and IPAH patients, we first normalized the level of target protein to β-actin level in each normal subject and calculated the mean ratio from all normal subjects as control. Then, the ratio of target protein to β-actin from each IPAH patient was normalized to the mean ratio from normal subjects. The mean ratio of target protein level to β-actin level in normal PASMC was defined as onefold.
Transfection experiments.
For the miRNA transfection, normal PASMC were transfected with 10 nM human miScript miRNA mimics for miR-29b, miR138, and miR-222 (Qiagen), while IPAH cells were transfected with 100 nM miR-29b, miR138, and miR-222 inhibitors (Qiagen) using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) according to the manufacturer’s protocol. AllStars Control siRNA (Qiagen, cat. no. 1027280) and miScript Inhibitor Negative Control (Qiagen, cat. no. 1027271) were utilized as a negative control (NC) for miRNA mimic and miRNA inhibitor transfection experiments, respectively (35, 39, 53, 65). The transfection was performed in Opti-MEM Reduced Serum Medium (Gibco) for 4–6 h. Then, the Opti-MEM was replaced with 5% FBS medium. After 48–72 h, the cells were collected for RNA or protein isolation and were used for electrophysiological recordings. The transfection efficiency was confirmed by real-time RT-PCR. For the gene transfection, human embryonic kidney (HEK) 293 cells were transiently transfected with 2 μg of KCNA5 (OriGene, plasmid no. RG219793) using X-tremeGENE 9 DNA Transfection Reagent (Roche) according to the manufacturer’s instructions. Experiments were performed 24–48 h after transfection.
Antibody validation.
All antibodies used in this study have undergone validation in the course of this study. Representative full-length blots and details of antibody validation are presented in Supplemental Fig. S1 (available at https://doi.org/10.6084/m9.figshare.9636893).
Luciferase assays.
gBlocks gene fragments containing the 3′-UTR of the human KCNA5, including the putative miR-29b binding site as well as enzyme restriction sites for PmeI and XbaI, were purchased from Integrated DNA Technologies. Cloning was performed according to the manufacturer’s protocol. The gBlocks and the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega) were digested with PmeI and XbaI, followed by ligation and transformation. To mutate the miR-29b seed region, a site-directed mutagenesis kit (Invitrogen) with specific primers (Integrated DNA Technologies) was used according to the manufacturer’s protocol. Vector constructs (wild type, mutant-1, and mutant-2) were confirmed by DNA sequencing. Wild type, mutant-1, and mutant-2 3′-UTR vectors as well as an empty vector for a control were used in luciferase assays. To prepare for the luciferase assay, HEK293 cells were transfected in 6-well plates with 10 nM miR-29b mimic (Qiagen), 2 μg luciferase vector, and 40 ng Renilla luciferase vector (pRL-TK) using Lipofectamine RNAiMAX (Invitrogen). Forty-eight hours after transfection, luciferase assays were performed using the Dual-Luciferase Reporter Assay System (Promega), according to the manufacturer’s protocol. Cells were lysed using the passive lysis buffer. Twenty microliters of the resulting cell lysate was added to a white 96-well plate, which was then placed in a GloMax 96 Microplate Luminometer with Dual Injectors (Promega). The firefly luminescence from each experimental condition was normalized to the luminescence from empty control vectors.
Statistical analysis.
The data are expressed as means ± SE. The differences between groups were analyzed for statistical significance with Student’s t test (paired or unpaired as applicable) or ANOVA and post hoc tests (Student-Newman-Keuls) where appropriate. The differences were considered to be statistically significant when P < 0.05.
RESULTS
To discover novel miRNAs that might be involved in the development and progression of pulmonary hypertension, we first conducted an miRNA PCR array experiment to determine and compare miRNA expression levels in PASMC between normal subjects and patients with IPAH. To determine which miRNAs are potentially involved in downregulating K+ channel expression and decreasing whole cell K+ currents in PASMC, we focused on the miRNAs that were selectively upregulated in IPAH-PASMC compared with normal cells. Then, we performed a series of experiments to determine whether the upregulated miRNAs affected expression and activity of various K+ channels in PASMC from IPAH patients.
Decreased activity of KV channels is associated with selectively upregulated miRNAs in IPAH-PASMC.
We first compared whole cell KV currents in PASMC isolated from healthy control subjects and patients with IPAH. The amplitude (Fig. 1Aa and Ab) and current density (Fig. 1Ac) of whole cell KV currents, elicited by depolarizing the cells from a holding potential of −70 mV to a series of test potentials ranging from −90 mV to +90 mV, were significantly (P < 0.01) lower in IPAH-PASMC than in normal (Nor) cells. To determine whether the dysfunction of K+ channels or the decreased current density of KV channels in IPAH-PASMC was due to miRNA-mediated posttranscriptional regulation, we then performed a miRNA PCR microarray experiment to investigate the human miRNA expression profile in normal PASMC and IPAH-PASMC. Among 89 miRNAs screened, 5 miRNAs were significantly (twofold or greater) downregulated (blue) and 6 miRNAs were significantly (twofold or greater) upregulated (red) in IPAH-PASMC compared with normal PASMC (Fig. 1B). The real-time PCR experiments confirmed 3.3-fold, 3.3-fold, and 2.9-fold upregulation for miR-29b, miR-138, and miR-222, respectively, in IPAH-PASMC (Fig. 1C). We were unable to detect any significant differences in miR-15a, miR-29a, and miR-let-7e levels between normal and IPAH cells using the real-time PCR method. In addition to the upregulated miRNAs, miRNA PCR microarray showed there are 5 miRNAs (miR-18a, miR-183, miR-181b, miR-378, and miR-126) that are downregulated in IPAH-PASMC compared with normal PASMC. Therefore, miR-29b, miR-138, and miR-222 were selected for the downstream experiments to investigate whether they were involved in (or required for) the regulation of activity and/or expression of K+ channels in PASMC isolated from patients with IPAH.
Fig. 1.

Upregulated microRNAs (miRNAs) correlate with reduced voltage-gated K+ (KV) currents in pulmonary artery smooth muscle cells (PASMC) from patients with idiopathic pulmonary arterial hypertension (IPAH). A: representative records (a) showing whole cell KV currents elicited by depolarization from a holding potential of −70 mV to a series of test potentials ranging from −80 to +80 mV (in 20-mV increments) in PASMC isolated from normal subjects (Nor) and IPAH patients (IPAH). Summarized data (mean ± SE) showing the averaged current amplitude at +80 mV in Nor and IPAH PASMC (b). The current density-voltage (I-V) relationship curves (c) of whole cell KV currents measured from normal (n = 25 cells from 4–5 different cell lines) and IPAH (n = 25 cells from 4–5 different cell lines) PASMC. I-V curves of normal and IPAH PASMC are significantly different (P < 0.01). B: a heatmap showing miRNAs that are upregulated (red) and downregulated (blue) in normal (n = 2 different cell lines) and IPAH (n = 2 different cell lines) PASMC. Six miRNAs (with greater than twofold upregulation) highlighted in red were further investigated by using real-time RT-PCR analysis. C: real-time RT-PCR analysis of miR-29b, miR-138, and miR-222 expression (normalized to U6) in PASMC from normal subjects (Nor, n = 6 different cell lines) and IPAH patients (IPAH, n = 6 different cell lines). The expression of miR-15a, miR-29a, and miR-let-7e in normal and IPAH PASMC is not significantly different (data not shown). Data are expressed as median (solid line)/mean (dashed line) ± confidence interval or as mean ± SE from 3–5 independent experiments. **P < 0.01, ***P < 0.001 vs. Nor. Statistical analysis was performed using unpaired Student’s t test.
Downregulation of specific K+ channels in PASMC from patients with IPAH.
In silico analysis revealed that miR-29b, miR-138, and miR-222 could target several KV (i.e., KV1.1/KCNA1, KV1.2/KCNA2, KV1.3/KCNA3, KV1.5/KCNA5, KV1.6/KCNA6, KV1.7/KCNA7, KVβ1.1/KCNAB1, KVβ1.3/KCNAB3) and BKCa (BKCaβ1/KCNMB1, BKCaβ3/KCNMB3, BKCaβ4/KCNMB4) channels. It has been established that KV1.1/KCNA1, KV1.2/KCNA2, KV1.5/KCNA5, and BKCaβ1/KCNMB1 are involved in the development of pulmonary hypertension (4, 9, 75). In this study, we aimed to determine whether miR-29b, miR-138, and miR-222 are responsible for the regulation of these channels in PASMC isolated from patients with IPAH. To test this possibility, we first compared the expression level of certain K+ channels (KV1.1, KV1.2, KV1.5, and BKCaβ1) in PASMC from normal subjects and patients with IPAH. Real-time RT-PCR experiments showed that the mRNA levels of KV1.2 (KCNA2), KV1.5 (KCNA5), and BKCaβ1 (KCNMB1) were downregulated in PASMC isolated from patients with IPAH (Fig. 2A). However, there were no significant differences in KV1.1 (KCNA1) mRNA expression between normal and IPAH-PASMC. Utilizing Western blot technique, we confirmed these results. The protein levels of KCNA2 (KV1.2), KCNA5 (KV1.5), and KCNMB1 (BKCaβ1) were significantly reduced in IPAH-PASMC compared with control cells, while the protein level of KCNA1 did not change (Fig. 2, B and C). These data indicate that mRNA expression of certain K+ channels is significantly downregulated in PASMC from IPAH patients. Downregulated K+ channel expression is potentially responsible for the decreased amplitude and current density of whole cell K+ currents due to changes in K+ channel pore-forming subunits (i.e., KCNA2 and KCNA5) or from changes in K+ channel regulatory subunits (i.e., KCNMB1) in IPAH patients.
Fig. 2.

Downregulation of selective K+ channels in pulmonary artery smooth muscle cells (PASMC) isolated from patients with idiopathic pulmonary arterial hypertension (IPAH). A: real-time RT-PCR analysis of KCNA1, KCNA2, KCNA5, and KCNMB1 (normalized to GAPDH) in PASMC isolated from normal subjects (Nor, n = 6 different cell lines) and IPAH patients (IPAH, n = 6 different cell lines). *P < 0.05, ***P < 0.001 vs. Nor. B: representative Western blot images showing the protein expression level of KCNA1, KCNA2, KCNA5, and β-actin (a) as well as KCNMB1 and β-actin (b) in normal and IPAH-PASMC. C: summarized data showing protein level of KCNA1, KCNA2, KCNA5, and KCNMB1 (normalized to β-actin) in normal subjects (Nor, n = 6 different cell lines) and IPAH patients (IPAH, n = 6 different cell lines). *P < 0.05, **P < 0.01 vs. Nor. D: representative Western blot images (a) showing the protein expression level of KCNA2, KCNA5, KCNMB1, and β-actin and summarized data (b) in pulmonary arterial endothelial cells (PAEC) isolated from normal subjects (Nor, n = 4 different cell lines) and IPAH patients (IPAH, n = 4 different cell lines). *P < 0.05 vs. Nor. E: real-time RT-PCR analysis of microRNA (miR)-29b, miR-138, and miR-222 expression (normalized to U6) in PAEC from normal subjects (Nor, n = 4 different cell lines) and IPAH patients (IPAH, n = 4 different cell lines). Data are expressed as median/mean ± confidence interval from 3–5 independent experiments. **P < 0.01, ***P < 0.001 vs. Nor. Statistical analysis was performed using unpaired Student’s t test.
Moreover, we compared levels of K+ channels in pulmonary arterial endothelial cells (PAEC) between healthy subjects and IPAH patients to identify whether KCNA2, KCNA5, and KCNMB1 were decreased in endothelial cells. As shown in Fig. 2Da, we were able to detect KCNA2, KCNA5, and KCNMB1 channels in human PAEC but did not reveal significant changes in protein expression of KCNA2 and KCNA5 between normal and IPAH PAEC. KCNMB1 level was slightly increased in IPAH cells compared with controls (Fig. 2Db). We did not measure KCNA1 protein levels in normal and IPAH PAEC since there were no differences in KCNA1 expression between normal and IPAH-PASMC. These results demonstrate that the downregulation of KCNA2, KCNA5, and KCNMB1 are unique to PASMC in IPAH patients.
To further demonstrate that miR-29b, miR-138, and miR-222 specifically regulate K+ channels in PASMC, we also quantified the miRNAs in normal and IPAH PAEC. Real-time PCR experiments showed that the miR-138 level was significantly decreased, whereas the miR-222 level was markedly increased in PAEC isolated from IPAH patients compared with controls (Fig. 2E). We did not reveal any changes in miR-29b expression between normal and IPAH cells. These findings led us to speculate that unchanged K+ channel expression is related to miR-29b but not miR138 and miR-222 levels in IPAH PAEC.
The miR-29b, miR-138, and miR-222 mimics decrease the activity and downregulate the expression of KV channels in normal human PASMC.
This set of experiments was designed to investigate whether miR-29b, miR-138, and miR-222 regulate the expression and/or activity of K+ channels in human PASMC. As shown in Supplemental Table S1, in silico analysis revealed that miR-29b, miR-138, and miR-222 target KCNA2, KCNA5, and KCNMB1 in human PASMC (Supplemental Material is available at https://doi.org/10.6084/m9.figshare.9636893). First, we aimed to determine whether the overexpression of miR-29b, miR-138, or miR-222 (which were detected at higher levels in IPAH-PASMC) in normal PASMC decreased K+ channel expression and/or activity. Normal PASMC transfected with miR-29b, miR-138, and miR-222 mimics had a dramatic increase of miR-29b, miR-138, and miR-222 compared with nontransfected (Mock) cells and cells transfected with AllStars Control siRNA (NC mimic) (Fig. 3A). There was no significant difference between mock and NC mimic groups. It is important to note that we should measure the level of functional (RNA induced silencing complex-associated) miRNA in a manner such that we can avoid detecting miRNA mimic trapped in nonfunctional locations. Because of the limitations of real-time RT-PCR technique, however, we were only able to detect the total, but not the functional, miRNA level (Fig. 3A). Then, we compared KV channel activity in normal PASMC transfected with NC mimic or miRNA mimics (miR-29b/miR-138/miR-222). The amplitude and current density of whole cell KV currents were significantly decreased by miR-29b (P < 0.001), miR-138 (P < 0.001), or miR-222 (P < 0.001) mimics (Fig. 3B); the miR-29b-induced inhibition (~80%) seemed to be greater than the miR-138/miR-222-mediated inhibition (at 60%–70% compared with NC mimic-transfected cells) (Fig. 3Bb). We then aimed to investigate which KV channels were responsible for the miRNA-mediated decrease in KV currents in normal PASMC. As shown earlier in this study (see Fig. 2), we found that both mRNA and protein expression levels of KCNA2 and KCNA5 were significantly downregulated in IPAH-PASMC compared with cells from healthy subjects. In the next set of experiments, we examined whether expression of KCNA2 and KCNA5 channels were regulated by miR-29b, miR-138, and miR-222 in normal PASMC. As shown in Fig. 3C, all three miRNAs (miR-29b/miR-138/miR-222) remarkably decreased the mRNA expression level of KCNA5 but not the mRNA expression of KCNA2 in normal PASMC. We were able to confirm the real-time PCR results in the Western blot experiments. However, we could not detect any changes in KCNA2 protein expression between the NC mimic-transfected cells and the miRNA-transfected cells, while the KCNA5 level was dramatically reduced in PASMC overexpressed miR-29b, miR-138, or miR-222 (Fig. 3D). These results indicate that the upregulation of miR-29b, miR-138, or miR-222 (observed in IPAH-PASMC) reduces whole cell KV currents by, at least partially, downregulating KCNA5 channels in normal PASMC.
Fig. 3.

MicroRNA (miR)-29b, miR-138, and miR-222 reduce voltage-gated K+ (KV) currents and downregulate KCNA5 expression in normal human pulmonary artery smooth muscle cells (PASMC). A: real-time RT-PCR analysis of miR-29b, miR-138, and miR-222 expression (normalized to U6) in nontransfected PASMC (mock) and PASMC transfected with AllStars Control siRNA [negative control (NC) mimic], miR-29b, miR-138, or miR-222 mimic (n = 3 different cell lines). Data are expressed as mean ± SE from 3–4 independent experiments. **P < 0.01, ***P < 0.001 vs. Mock; ###P < 0.001 vs. NC mimic. B: representative whole cell KV currents (a) elicited by depolarizing the cells from a holding potential of −70 mV to a series of test potentials ranging from −80 to +80 mV (in 20-mV increments) and summarized current-voltage (I-V) relationship curves (b) in normal human PASMC transfected with NC, miR-29b, miR-138, or miR-222 mimic. Summarized data showing that the current density of K+ currents induced by the test potentials of +40 mV to +80 mV in PASMC transfected with miR-29b, miR-138, or miR-222 mimic (n = 25 cells from 4–5 different cell lines) is significantly different from the NC mimic group (P < 0.001). C: real-time RT-PCR analysis (mean ± SE) of KCNA2 and KCNA5 (normalized to GAPDH) in normal PASMC transfected with NC, miR-29b, miR-138, or miR-222 mimic (n = 5 different cell lines). **P < 0.01, ***P < 0.001 vs. NC mimic. D: representative Western blot images (a) and summarized data (b) showing the protein expression levels of KCNA2, KCNA5, and β-actin in normal PASMC transfected with NC, miR-29b, miR-138, or miR-222 mimic (n = 5 different cell lines). Data are expressed as mean ± SE or mean/median ± confidence interval (b) from at least 3 independent experiments. *P < 0.05, **P < 0.01 vs. NC mimic. Statistical analysis was performed using one-way ANOVA and post hoc test (Student-Newman-Keuls).
Inhibition of miR-29b rescues the activity and expression of KV channels in PASMC that were isolated from IPAH patients.
To investigate whether the inhibition of selected miRNAs (miR-29b, miR-138, or miR-222) in IPAH-PASMC results in recovering of KV channel activity and/or expression, we transfected IPAH-PASMC with the miRNA inhibitors (or antagomiRs). As expected, transfection of the specific miRNA inhibitors, antagomiR-29b, antagomiR-138, or antagomiR-222 in IPAH-PASMC significantly decreased the levels of miR-29b, miR-138, and miR-222, respectively (Fig. 4A), whereas transfection of the miScript Inhibitor Negative Control (NC Inhibitor) had no effect on the miRNA levels (Fig. 4A) in comparison to the nontransfected cells (Mock). We then compared KV channel activity in IPAH-PASMC transfected with the NC inhibitor or the miRNA inhibitors. As shown in Fig. 4Ba, antagomiR-29b completely rescued whole cell KV currents in IPAH-PASMC to the normal level, while antagomiR-138 partially restored whole cell KV currents. The amplitude of the whole cell KV currents was significantly (P < 0.01) higher in IPAH-PASMC transfected with antagomiR-29b or antagomiR-138 than in NC inhibitor-transfected IPAH-PASMC (Fig. 4Bb). Interestingly, inhibition of miR-222 with antagomiR-222 had no effect on whole cell KV currents in IPAH-PASMC (Fig. 4B).
Fig. 4.

Inhibition of microRNA (miR)-29b restores voltage-gated K+ (KV) channel activity and KCNA5 expression in idiopathic pulmonary arterial hypertension (IPAH)-pulmonary artery smooth muscle cells (PASMC). A: real-time RT-PCR analysis (means ± SE) of miR-29b, miR-138, and miR-222 expression levels (normalized to U6) in nontransfected IPAH-PASMC (mock) and IPAH-PASMC transfected with miScript Inhibitor Negative Control (NC inhibitor), antagomiR-29b, antagomiR-138, or antagomiR-222 (n = 3 different cell lines). Data are expressed as mean ± SE from 3–4 independent experiments. *P < 0.05, **P < 0.01 vs. Mock; #P < 0.05, ##P < 0.01 vs. NC inhibitor. B: representative whole cell KV currents (a) elicited by depolarizing the cells from a holding potential of −70 mV to a series of test potentials ranging from −60 mV to +80 mV (in 20-mV increments) and summarized data (means ± SE) showing current-voltage (I-V) relationship curves (b) of the KV currents in IPAH-PASMC transfected with NC inhibitor, antagomiR-29b, antagomiR-138, or antagomiR-222. Summarized data (b) showing that the current density of K+ currents induced by the test potentials of +40 mV to +80 mV in IPAH-PASMC transfected with antagomiR-29b and antagomiR-138 (n = 25 cells from 4–5 different cell lines) is significantly different from the NC inhibitor group (P < 0.01). C: real-time RT-PCR analysis of KCNA5 (normalized to GAPDH) in IPAH-PASMC transfected with NC inhibitor, antagomiR-29b, antagomiR-138, or antagomiR-222 (n = 5 different cell lines). D: representative Western blot images (a) and summarized data (b) showing the protein expression level of KCNA5 and β-actin in IPAH-PASMC transfected with NC inhibitor, antagomiR-29b, antagomiR-138, or antagomiR-222 (n = 5 different cell lines). Data are expressed as mean ± SE or mean/median ± confidence interval (b) from at least 3 independent experiments. ***P < 0.001 vs. NC inhibitor. Statistical analysis was performed using one-way ANOVA and post hoc test (Student-Newman-Keuls). miRNA, microRNA.
Next, we investigated whether inhibition of miR-29b, miR-138, or miR-222 resulted in increased KCNA5 expression in IPAH-PASMC. Out of the three miRNA inhibitors, only antagomiR-29b was able to significantly upregulate both mRNA and protein level of KCNA5 channels (Fig. 4, C and D). Inhibition of miR-138 or miR-222 by transfecting antagomiR-138 or antagomiR-222 had no effect on KCNA5 protein expression levels in IPAH-PASMC (Fig. 4, C and D). We did not quantify KCNA2 protein levels since KCNA2 mRNA was not changed in miRNA-transfected cells (see Fig. 3, C and D). These data demonstrate that only miR-29b inhibition is sufficient to recover or restore the whole cell KV currents by increasing KCNA5 protein expression in PASMC isolated from patients with IPAH.
To further characterize the recovered or restored currents in antagomiR-29b-transfected IPAH-PASMC, we conducted pharmacological experiments using diphenyl phosphine oxide-1 (DPO-1), a specific KV1.5/KCNA5 channel blocker (21, 23). To confirm that DPO-1 specifically blocks KV1.5/KCNA5 currents, we used HEK293 cells transiently transfected with the KCNA5 gene (Fig. 5A) to determine whether DPO-1 decreases whole cell KCNA5 currents. Extracellular application of DPO-1 (0.3 µM) to the KCNA5-transfected cells significantly and reversibly decreased the amplitude and current density of whole cell KCNA5 currents (Fig. 5B). These results indicate that DPO-1 is a potent inhibitor of KCNA5 channels. Then, we examined the effect of DPO-1 on the recovered K+ currents in IPAH-PASMC transfected with antagomiR-29b. As shown in Fig. 5C, antagomiR-29b transfection resulted in an increase in KCNA5 protein expression level in IPAH-PASMC (see also Fig. 4, C and D). Extracellular application of 0.3 μM DPO-1 significantly and reversibly decreased the whole cell KV currents in IPAH-PASMC transfected with antagomiR-29b (Fig. 5D). The amplitude and current density of KV currents, elicited by a test potential of +80 mV from a holding potential of −70 mV, in antagomiR-29b-transfected IPAH-PASMC were reduced from 343.1 ± 49.8 pA and 22.9 ± 3.3 pA/pF to 128.6 ± 36.4 pA and 6.4 ± 1.8 pA/pF (P < 0.001) before (Control) and during (DPO-1) extracellular application of 0.3 μM DPO-1 (Fig. 5D, b and c). The DPO-1-mediated inhibitory effect on whole cell KV currents was also reversible in some cells. These data led us to conclude that increased miR-29b and miR-29b-mediated KCNA5 downregulation are involved in decreasing whole cell KV currents in IPAH-PASMC.
Fig. 5.

Whole cell K+ currents in idiopathic pulmonary arterial hypertension (IPAH)-pulmonary artery smooth muscle cells (PASMC) transfected with microRNA (miR)-29b inhibitor are sensitive to the specific voltage-gated K+ channel 1.5 (KV1.5) channel blocker, diphenyl phosphine oxide-1 (DPO-1). A: Western blot analysis of KCNA5 and β-actin in control human embryonic kidney (HEK) cells (Control) and cells transfected with empty vector (Vector) or KCNA5. B: representative whole cell K+ currents (a), elicited by depolarization from a holding potential of −70 mV to a series of test potentials ranging from −80 mV to +80 mV (in 20-mV increments) in a KCNA5-transfected HEK cell before (Control), during (DPO-1), and after (Washout) extracellular application of 0.3 µM DPO-1. Summarized data (means ± SE) showing the amplitude of the KCNA5 currents at +80 mV (n = 10–15 cells from 3 different cell lines) (b) and the current-voltage (I-V) curves (n = 10–15 cells from 3 different cell lines) (c) in KCNA5-transfected HEK cells before (Control), during (DPO-1), and after (Washout) extracellular application of DPO-1. ***P < 0.001 vs. Control and Washout. The I-V curve of the KCNA5 currents in KCNA5-transfected cells superfused with DPO-1 is significantly different (P < 0.001) from the curves in the cells before (Control) and after (Washout) DPO-1 application. C: Western blot analysis of KCNA5 and β-actin in nontransfected control IPAH-PASMC (Mock) and IPAH-PASMC transfected with negative control (NC) inhibitor or antagomiR-29b. D: representative currents (a) elicited by depolarization from a holding potential of −70 mV to a series of test potentials ranging from −80 mV to +80 mV (in 20-mV increments) in an antagomir-29b-transfected IPAH-PASMC before (Control), during (DPO-1), and after (Washout) extracellular application of 0.3 µM DPO-1. Summarized data (means ± SE) showing the amplitude of the outward K+ currents at +80 mV (n = 10–15 cells from 3 different cell lines) (b) and the current-voltage (I-V) curves (n = 10–15 cells from 3 different cell lines) (c) in antagomir-29b-transfected IPAH-PASMC before (Control), during (DPO-1), and after (Washout) extracellular application of DPO-1. Data are expressed as mean ± SE from at least 3 independent experiments. ***P < 0.001 vs. Control and Washout. The I-V curve of the outward K+ currents in antagomir-29b-transfected IPAH-PASMC superfused with DPO-1 is significantly different (P < 0.001) from the curves in the cells before (Control) and after (Washout) DPO-1 application. Statistical analysis was performed using one-way ANOVA and post hoc test (Student-Newman-Keuls).
miR-29b directly targets 3′-UTR of human KCNA5.
There is a potential complementary binding site for miR-29b in the 3′-UTR of human KCNA5 mRNA (Fig. 6A). To determine if miR-29b directly binds KCNA5, we performed luciferase assays in which wild-type 3′-UTR of KCNA5 was cloned into a pmirGLO vector. To further confirm direct binding of miR-29b to the putative seed sequence in the 3′-UTR of KCNA5, we additionally designed 2 different luciferase constructs containing specific point mutations within the seed sequence (Fig. 6B). The firefly luminescence was significantly decreased in HEK293 cells transfected with wild-type KCNA5 3′-UTR vector and miR-29b mimic compared with cells transfected with wild-type KCNA5 3′-UTR vector and NC mimic (Fig. 6C). This decrease indicates that miR-29b binds to the 3′-UTR of KCNA5. As expected, the transfections with mutated plasmids (mutant-1 and mutant-2) did not significantly alter the luciferase activity between the NC inhibitor-transfected cells and the miR-29b-transfected cells (Fig. 6C). These results demonstrate that miR-29b directly binds to the 3′-UTR of KCNA5. Therefore, a selective miR-29b inhibitor would be an ideal therapeutic agent that may be able to rescue the attenuated K+ channel function and the decreased KCNA5 expression in PASMC from patients with IPAH.
Fig. 6.
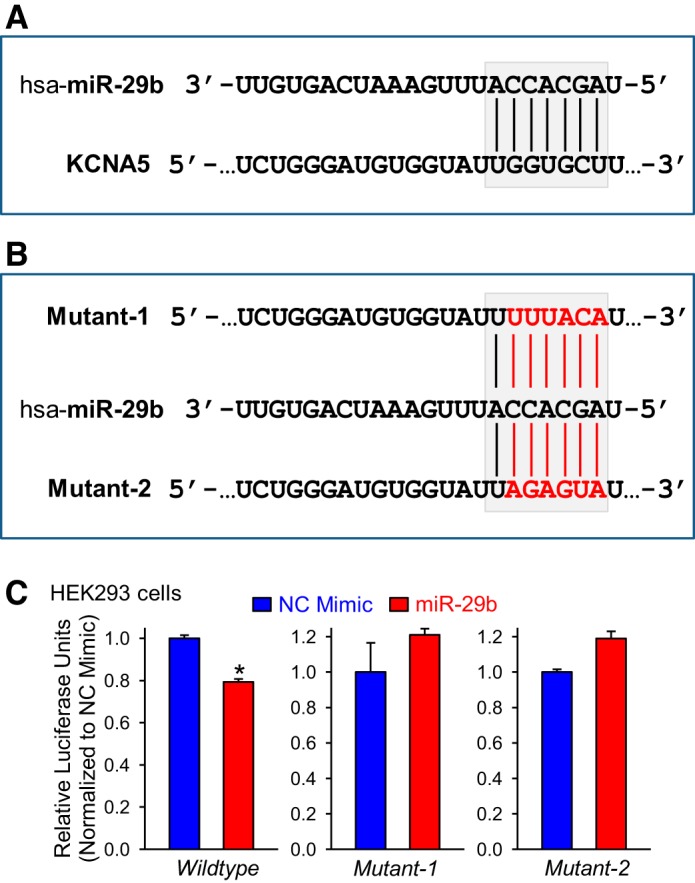
MicroRNA (miR)-29b directly binds to the 3′-untranslated region (UTR) of KCNA5. A: the potential binding site of miR-29b on the 3′-UTR of human KCNA5. B: the sequences mutated in miR-29b binding site within the 3′-UTR of KCNA5. C: the summarized data showing luciferase activity in human embryonic kidney (HEK) 293 cells cotransfected with negative control (NC) or miR-29b mimic and pRL-TK vector containing the 3′-UTR region of KCNA5 (wild type or mutant) and the luciferase gene (n = 4). Data are expressed as mean ± SE from at least 3 independent experiments. *P < 0.05 vs. NC mimic. Statistical analysis was performed using unpaired Student’s t test.
Activity of BKCa channels is attenuated in PASMC isolated from IPAH patients.
Earlier in this study, we showed that KCNMB1 mRNA and protein level is lower in IPAH-PASMC than normal cells (see Fig. 2, Bb and C). In silico analysis revealed that miR-29b, miR-138, and miR-222 could also target the BKCaβ1 channel encoded by the KCNMB1 gene. Therefore, we aimed to measure the activity of BKCa channels in normal and IPAH-PASMC. In these experiments, whole cell BKCa currents were measured under conditions in which the intracellular [Ca2+] (in the pipette solution) was maintained at a high level (1 μM by using appropriate concentrations of EGTA and CaCl2), and extracellular [Ca2+] (in the bath solution) was kept at 1.8 mM. Extracellular application of 100 nM iberiotoxin (IbTX), a selective blocker of BKCa channels, significantly reduced the whole cell BKCa currents in both normal and IPAH-PASMC (Fig. 7Aa). The subtraction of the currents recorded during the application of IbTX from the currents recorded before application of IbTX revealed IbTX-sensitive BKCa currents (Fig. 7Ab).
Fig. 7.
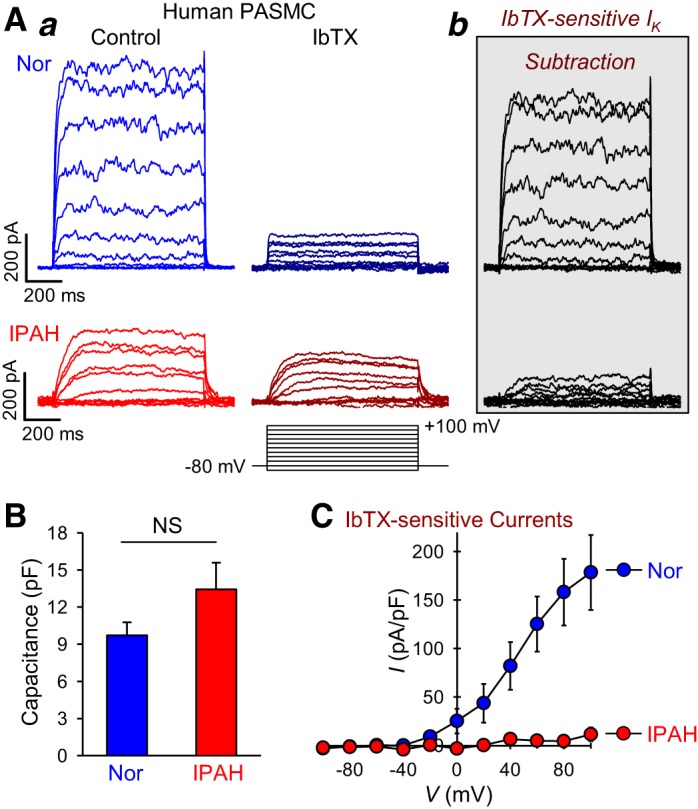
Large-conductance Ca2+-activated K+ (BKCa) channel currents are decreased in pulmonary artery smooth muscle cells (PASMC) from patients with idiopathic pulmonary arterial hypertension (IPAH). A: representative whole cell K+ currents (a) elicited by depolarizing cells from a holding potential of −70 mV to a series of test potentials ranging from −100 mV to +100 mV (in 20-mV increments, for 500 ms every 15 s) in PASMC from normal subjects (Nor) and PASMC from IPAH patients (IPAH) before (Control) and during [iberotoxin (IbTX)] extracellular application of 100 nM IbTX. IbTX-sensitive components of the currents (b) were obtained by subtracting the currents recorded during application of IbTX from the currents recorded under control condition. B: bar graphs summarizing the capacitance of PASMC isolated from normal subjects and IPAH patients (means ± SE, n = 6 cells, NS). C: summarized data (means ± SE) showing the current-voltage (I-V) relationship curves of the IbTX-sensitive K+ currents in normal PASMC and IPAH-PASMC (n = 25 cells from 4–5 different cell lines, P < 0.05 vs. Normal). Data are expressed as mean ± SE from at least 3 independent experiments. Statistical analysis was performed using unpaired Student’s t test.
In normal PASMC, the outward K+ currents elicited by depolarizing the cells from a holding potential of −80 mV to a series of test potentials ranging from −90 to +100 mV were mostly inhibited by 100 nM IbTX. On the other hand, the amplitude of whole cell outward K+ currents (including KV and BKCa currents) in IPAH-PASMC was smaller than that in normal PASMC (Fig. 7A), and the sensitivity of the whole cell outward K+ currents to IbTX was also much less in IPAH-PASMC than in normal PASMC (Fig. 7A). It is important to note that the IbTX-insensitive currents, which are mainly generated by K+ efflux through KV channels (IK(V)), are very low in normal PASMC, indicating that high intracellular [Ca2+] reduces IK(V) (55). There was no significant difference in cell capacitance between normal and IPAH-PASMC (Fig. 7B). The amplitude and current density of the IbTX-sensitive BKCa currents in IPAH-PASMC were significantly (P < 0.05) lower than in normal PASMC (Fig. 7C). These results indicate that the activity of BKCa channels is dramatically reduced in PASMC isolated from IPAH patients.
miR-29b regulates BKCa channel activity and KCNMB1 expression.
This set of experiments was designed to test whether the selected upregulated miRNAs (miR-29b/miR-138/miR-222) contributed to the attenuated BKCa channel activity and/or expression in PASMC from IPAH patients. At first, we compared KCNMB1 expression in normal PASMC transfected with miR-29b, miR-138, or miR-222 mimics. Western blot experiments showed that the protein level of KCNMB1 was decreased by miR-29b, miR-138, and miR-222 in normal PASMC (Fig. 8A). We were also able to confirm our results on channel activities in the single-channel cell-attached mode of patch-clamp experiments. The amplitude of single-channel current at holding potential of +40 mV indicates that this is the large-conductance BKCa channel current (Fig. 8Ba). The open probability (Popen) of the channels was significantly decreased in cells transfected with miR-29b mimic compared with NC mimic-transfected cells (Fig. 8B, a and b). Overexpression of miR-138 also had an inhibitory effect on BKCa channel activity in normal PASMC, but the Popen in miR-138-transfected cells was only partially decreased in comparison to cells transfected with NC mimic (Fig. 8C). We did not measure BKCa channel activity in miR-222-transfected cells because we showed earlier that miR-222 had no effect on the whole cell K+ currents in IPAH-PASMC (see Fig. 4B).
Fig. 8.
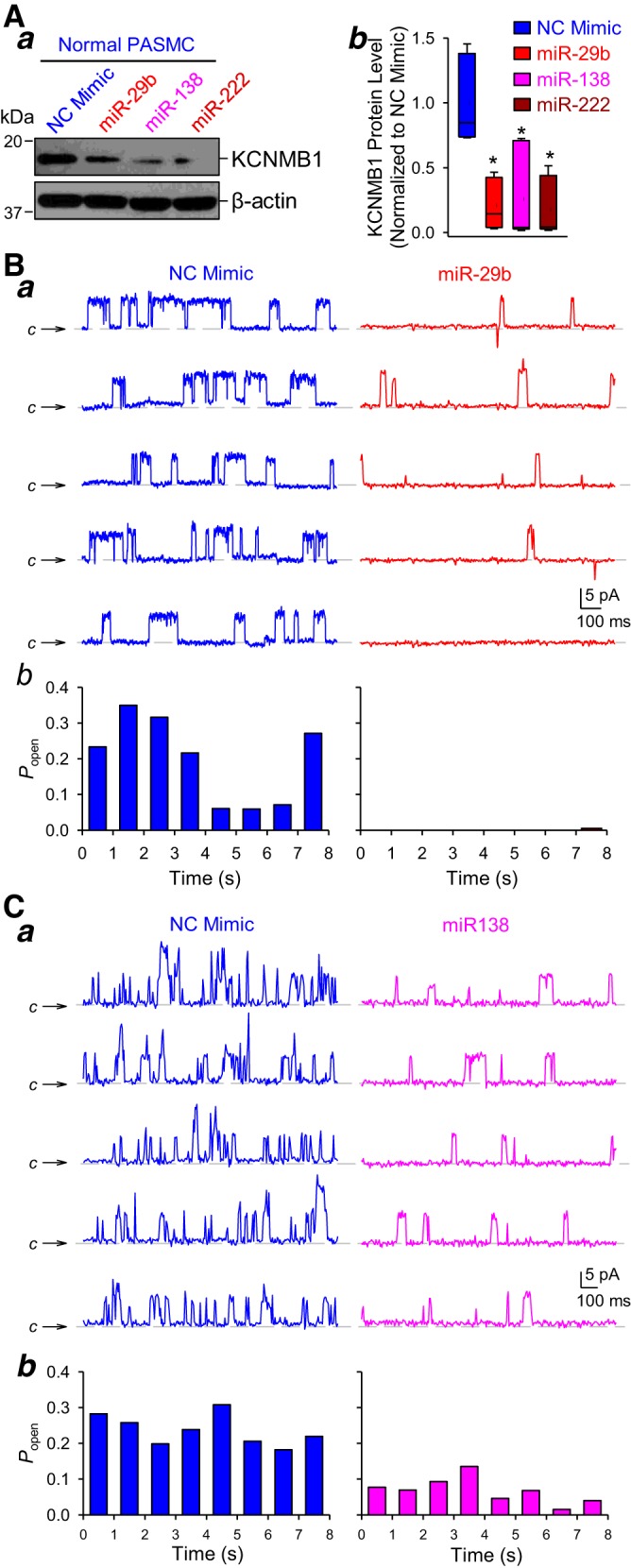
MicroRNA (miR)-29b, miR-138, and miR-222 downregulate KCNMB1 and reduce large-conductance Ca2+-activated K+ (BKCa) currents in normal pulmonary artery smooth muscle cells (PASMC). A: representative Western blot images (a) and summarized data (means ± SE) (b) showing the protein expression level of the BKCa channel β1-subunit (KCNMB1) and β-actin in normal PASMC transfected with AllStars Control siRNA [negative control (NC) mimic], miR-29b, miR-138, or miR-222 mimic (n = 5 different cell lines). Data are expressed as mean/median ± confidence interval from at least three independent experiments. *P < 0.05 vs. NC. B: representative large-conductance single-channel K+ currents in cell-attached membrane patches held at +40 mV (a) in normal PASMC transfected with NC mimic or miR-29b. The symbol C and horizontal broken lines denote the current level when the channel is closed. Summarized data (means ± SE) (b) showing the steady-state open probability (Popen) of the currents recorded in normal PASMC transfected with NC or miR-29b mimic. C: representative single-channel BKCa current recordings in cell-attached membrane patches held at +40 mV (a) in normal PASMC transfected with NC and miR138 mimic. The symbol C and horizontal broken lines denote the current level when the channel is closed. Bar graph (b) summarizing the steady-state Popen of the single-channel currents in normal PASMC transfected with NC or miR-138 mimic. Statistical analysis was performed using one-way ANOVA and post hoc test (Student-Newman-Keuls) or unpaired Student’s t test.
Then, we aimed to determine whether antagomiR-29b, antagomiR-138, and antagomiR-222 recovered the expression of BKCa channels in PASMC isolated from patients with IPAH. As shown in Fig. 9A, only antagomiR-29b significantly restored KCNMB1 protein expression in IPAH-PASMC, whereas antagomiR-138 and antagomiR-222 had little effect on the protein level of KCNMB1 in IPAH-PASMC. These data suggest that miR-29b had the greatest ability to regulate BKCa channel expression and KCa currents in human PASMC; therefore, we excluded miR-138 and miR-222 from subsequent experiments. To further confirm miR-29b-mediated regulation of BKCa channels, we recorded the whole cell K+ current in the presence of a specific blocker of KCa channels. Extracellular application of Penitrem A (100 nM), a selective blocker of BKCa channels, was able to reduce the whole cell K+ currents in normal PASMC transfected with NC mimic but not in cells transfected with miR-29b mimic (Fig. 9Ba). In NC mimic-transfected cells, the outward K+ currents were significantly reduced by the BKCa channel blocker, Penitrem A. On the other hand, the whole cell outward K+ currents in miR-29b-transfected cells were smaller than that in NC mimic-transfected PASMC and insensitive to Penitrem A. The subtraction of the currents recorded during the application of Penitrem A from the currents recorded before application of the blocker revealed Penitrem A-sensitive BKCa currents (Fig. 9Bb). The amplitude of the Penitrem A-sensitive BKCa currents in PASMC transfected with miR-29b was significantly (P < 0.05) lower than that in NC mimic-transfected cells (Fig. 9Bc). These data indicate that miR-29b, in addition to its inhibitory effect on KV channels, also decreases BKCa channel activity by, at least in part, reducing KCNMB1 mRNA and protein level in PASMC.
Fig. 9.

Inhibition of miR-29b restores KCNMB1 expression level and whole cell large-conductance Ca2+-activated K+ (BKCa) currents in pulmonary artery smooth muscle cells (PASMC) from patients with idiopathic pulmonary arterial hypertension (IPAH). A: representative Western blot images (a) and summarized data (mean ± SE or mean/median ± confidence interval) (b) showing the protein expression level of KCNMB1 and β-actin in IPAH-PASMC transfected with miScript Inhibitor Negative Control (NC inhibitor), antagomiR-29b, antagomiR-138, or antagomiR-222 (n = 5 different cell lines). *P < 0.05 vs. NC inhibitor. B: representative whole cell K+ currents, elicited by depolarization from a holding potential of −70 mV to a series of test potentials ranging from −80 mV to +80 mV (in 20-mV increments) in normal PASMC transfected with AllStars Control siRNA (NC mimic) or miR-29b mimic before (Control) and during extracellular application of 100 nM penitrem A (Penitrem A). Penitrem A-sensitive component of the currents (b) was obtained by subtracting the currents recorded during application of Penitrem A from the currents recorded under control condition. Summarized data (means ± SE) (c) showing the I-V curves of the Penitrem A-sensitive K+ currents in NC mimic- and miR-29b-transfected PASMC (n = 25 cells from 4 to 5 different cell lines, P < 0.05 vs. NC mimic). C: representative whole cell K+ currents, elicited by depolarizing an antagomir-29b-transfected IPAH-PASMC from a holding potential of −70 mV to a series of test potentials ranging from −80 mV to +80 mV (in 20-mV increments) before (Control), during (Penitrem A) and after (Washout) extracellular application of 100 nM penitrem A. Summarized data (means ± SE (b) showing the amplitude of K+ currents at +80 mV and the I-V curves of the currents (c) in IPAH-PASMC transfected with antagomiR-29b before (Control), during (Penitrem A) and after (Washout) extracellular application of Penitrem A (n = 10–15 cells from 3 different cell lines, ***P < 0.001 vs. Control). D: representative single-channel current recorded in cell-attached membrane patches held at +40 mV (a) in IPAH-PASMC transfected with NC inhibitor (top in red) and antagomir-29b (bottom in blue). Steady-state Popen in IPAH-PASMC transfected with NC inhibitor or antagomiR-29b (b). The symbolC and horizontal broken lines denote the current level when the channel is closed. Statistical analysis was performed using one-way ANOVA and post hoc test (Student-Newman-Keuls) or unpaired Student’s t test.
To further characterize the outward K+ currents recovered by transfecting the miR-29b inhibitor, we measured whole cell BKCa currents before, during, and after application of Penitrem A in IPAH-PASMC transfected with antagomiR-29b (Fig. 9C). The extracellular application of 100 nM Penitrem A significantly and reversibly reduced the whole cell outward K+ currents in antagomiR-29b-transfected IPAH-PASMC (Fig. 9C). The sensitivity of antagomiR-29b-recovered or restored whole cell K+ currents to Penitrem A indicates that BKCa currents are part of the K+ currents in IPAH-PASMC restored by antagomir-29b (Fig. 9C). These data also imply that increased miR-29b in IPAH-PASMC is involved in decreasing whole cell K+ currents by downregulating KV channels (see Fig. 5D) and BKCa channels (see Fig. 9C) in PASMC.
Next, we performed single-channel patch-clamp experiments to further confirm that antagomiR-29b elevated BKCa currents in IPAH-PASMC. miR-29b inhibitor or antagomir-29b significantly increased the channel activity and Popen in IPAH-PASMC (Fig. 9D). The amplitude (~10 pA) of a single-channel current at a holding potential of +40 mV for the cell-attached membrane patch indicates that the antagomiR-29b-recovered K+ currents in IPAH-PASMC (Fig. 9D) are likely the large-conductance (~200 pS) BKCa channel currents. Based on these results, we conclude that miR-29b inhibition is sufficient to restore BKCa channel activity in PASMC isolated from patients with IPAH.
DISCUSSION
In this study, we demonstrate that miR-29b is specifically upregulated and negatively exerts posttranscriptional regulation onto K+ (KV and BKCa) channels in PASMC isolated from IPAH patients in comparison to PASMC isolated from normal subjects. Additionally, we showed that inhibitors or antagomiRs specifically targeting miR-29b could be novel potential therapeutic agents for patients with IPAH. Our results indicate that 1) upregulated miR-29b, miR-138, and miR-222 are associated with reduced K+ channel activity and downregulated KV1.2 (KCNA2), KV1.5 (KCNA5), and BKCaβ1 (KCNMB1) channel subunits in PASMC from IPAH patients; 2) miR-29b decreases KV channel activity in IPAH-PASMC by directly targeting KCNA5; and 3) miR-29b also contributes to attenuated BKCa channel activity and BKCaβ1 expression in IPAH-PASMC. The miR-29b-mediated posttranscriptional suppression of the KV channel pore-forming α subunit, KV1.5/KCNA5, and the large-conductance KCa channel regulatory β subunit, BKCaβ1/KCNMB1, in PASMC is thus one of the important mechanisms responsible for attenuated whole cell K+ currents in PASMC from patients and animals with pulmonary hypertension. The decreased function and expression of KV (e.g., KV1.5/KCNA5) and BKCa (e.g., BKCaβ1/KCNMB1) channel subunits by miR-29b in PASMC would contribute to causing pulmonary vasoconstriction (by membrane depolarization-mediated increase in [Ca2+]cyt) and pulmonary vascular remodeling (by enhancing PASMC proliferation and inhibiting apoptosis).
We previously identified the expression of many different K+ channels in normal human PASMC (26). We and other investigators showed that mRNA and protein expression of KV channels (e.g., KV1.2/KCNA2, KV1.5/KCNA5) was downregulated, and amplitude and current density of KV currents were reduced in PASMC isolated from patients with IPAH compared with PASMC isolated from normal subjects (or patients without pulmonary hypertension) and in PASMC isolated from animals with experimental pulmonary hypertension (e.g., hypoxia-induced pulmonary hypertension) compared with the normoxic controls (8, 22, 24, 44, 63, 64, 75). Our current findings are consistent with the previous results: the whole cell KV currents are reduced and KV1.2/KCNA2 and KV1.5/KCNA5 channels are downregulated in IPAH-PASMC in comparison to normal cells. However, we could not detect any significant changes in KV1.1 (KCNA1) mRNA and protein levels between normal and IPAH-PASMC. Given that KV1.5/KCNA5 channel is an important K+ channel involved in regulating the membrane potential and inducing membrane repolarization and hyperpolarization in PASMC (48), the downregulated KV1.5/KCNA5 expression is responsible, at least partially, for the decreased amplitude and current density of whole cell IK(V) and the membrane depolarization-associated increases in [Ca2+]cyt in PASMC from IPAH patients under normoxia.
When we used intracellular pipette solution containing high [Ca2+]cyt to record BKCa currents in human PASMC, we found that the IbTX-insensitive currents or the remaining IbTX-insensitive KV currents in PASMC superfused with a high concentration of IbTX (100 nM) were very small in normal PASMC (see Fig. 7A). These results are consistent with early studies showing that intracellular Ca2+ may inhibit KV channels in PASMC (51, 55). Post et al. (51) first reported in 1992 that acute hypoxia inhibited KV channels in PASMC by an increase in [Ca2+]cyt; the initial increase in [Ca2+]cyt was likely due to hypoxia-mediated Ca2+ release (55) from IP3-sensitive intracellular Ca2+ stores and/or Ca2+ influx through voltage-independent cation channels (57, 66). The resulting membrane depolarization induced by Ca2+-mediated KV channel inhibition further results in Ca2+ influx through voltage-dependent Ca2+ channels, causing pulmonary vasoconstriction.
Unlike KV channels, the expression and function of BKCa channels are not well understood in pulmonary hypertension (1, 4, 19, 45). In this study, we revealed that BKCa channel activity is attenuated and KCNMB1 expression is reduced in PASMC from IPAH patients in comparison to cells from healthy subjects. Our findings are consistent with the observations in animal models of pulmonary hypertension (7, 71, 77). BKCa channel activator NS1619 was able to reduce right ventricular systolic pressure, a surrogate of pulmonary arterial systolic pressure, in rats with monocrotaline-induced pulmonary hypertension (54). KCNMB1 is a regulatory subunit that modulates the electrophysiological property of the pore-forming subunit (e.g., KCNMA1) of the large-conductance Ca2+-activated and voltage-dependent K+ channels (BKCa channels) (13). Coassembly of the β subunit (KCNMB1) with the pore-forming α subunit (KCNMA1) significantly affects the biophysical (e.g., voltage- and Ca2+-sensitivity) and pharmacological (e.g., toxin selectivity and efficacy) properties of the BKCa channel (42). The modifications of Ca2+ sensitivity (which is regulated by β subunit/KCNMB1) are mainly responsible for the decreased activity of BKCa channels in PASMC isolated from rats with hypoxia-induced pulmonary hypertension (9), while kcnmb1−/− mice develop worse pulmonary hypertension under hypoxic conditions (4). Interestingly, kcnmb1−/− mice are also hypertensive owing to the inability of BKCa channels to be activated by Ca2+ sparks and the compensated pressure-induced vasoconstriction (13). Additionally, whole cell BKCa current density and Ca2+ sensitivity were also lower in vascular smooth muscle cells from patients with systemic hypertension due to KCNMB1 downregulation (72). The striking parallels to systemic hypertension suggest that impaired BKCa channel function may be an important contributor to sustained vasoconstriction in pulmonary hypertension.
In contrast to our results, another group revealed the upregulation of KCNMB1 mRNA and protein in lung tissue from donors and IPAH patients (45). Although KCNMB1 appeared to be restricted mainly to smooth muscle tissue, we at first detected its expression in human pulmonary arterial endothelial cells (PAEC). Using Western blot experiments, we demonstrated that KCNMB1 is decreased in PASMC but increased in PAEC from IPAH patients. We previously reported that the whole-lung tissue is mainly composed of lung vascular endothelial cells (62); therefore, the divergent changes in KCNMB1 expression in PASMC and PAEC could explain, at least partially, the different data on KCNMB1 level in PASMC and lung tissue-isolated patients with IPAH or animals with experimental pulmonary hypertension.
Various miRNAs have been implicated in the development and progression of pulmonary hypertension (11, 28, 46, 56, 76); however, miRNA-mediated regulation of KV and BKCa channels is not well understood. Recently, Mondejar-Parreño et al. (43) revealed that KCNA5 is inhibited by the upregulated miR-1 in a hypoxia/SU5416-induced pulmonary hypertension model in rats. In contrast, Machado’s group reported that hypoxia attenuated miR-1 in in vitro and in vivo experiments (58). Additional studies are needed to further investigate these differing findings. Interestingly, it is reported that miR-1 is downregulated in PASMC isolated from IPAH patients (58), suggesting that downregulation of KCNA5 is not directly due to miR-1 in IPAH-PASMC. Our data showed for the first time that miR-29b directly inhibits KCNA5 and decreases KCNMB1, resulting in reduced whole cell K+ currents in IPAH-PASMC. The upregulation of miR-138 and miR-222 were also previously established in pulmonary hypertension (6, 29, 33, 37, 68), but in this study, we demonstrated that miR-29b has the most potential to regulate K+ channel function and expression in PASMC from patients with IPAH. Although miR-138 and miR-222 attenuate K+ channel activity and downregulate KCNA5 and KCNMB1, inhibition of miR-138 and miR-222 are not sufficient to restore K+ channel activity and expression in IPAH-PASMC. It is reasonable to speculate that miR-29b could be a potential interesting target in developing a novel therapeutic approach for different forms of pulmonary hypertension.
In human cancer, miR-29b is known to be a tumor suppressor; however, it may also act as a tumor promoter under certain conditions (70). Data about miR-29b in pulmonary hypertension is limited but suggest that miR-29b is involved in the development and progression of disease. In contrast, a recent study found that hypoxia decreases miR-29b in vivo and in vitro (16). Chen et al. showed that miR-29b suppresses proliferation and promotes apoptosis by targeting CCND2 and Mcl-1. Similar to cancer research miR-29b could have a different effect in hypoxia-induced pulmonary hypertension and idiopathic PAH. It is also unclear whether and how miR-29b affects PASMC contraction, proliferation, migration, and/or apoptosis in normal subjects and IPAH patients, this line of investigation will be pursued in future studies.
In conclusion, we identified three miRNAs (miR-29b, miR-138, miR-222) in this study that are upregulated in IPAH-PASMC compared with normal PASMC in addition to many miRNAs that have been implicated in the development and progression of pulmonary hypertension. Our results from this study indicate that downregulated KCNA5, a pore-forming α subunit for forming KV channels, and KCNMB1, a regulatory β subunit for BKCa channels, and the reduced whole cell KV and BKCa currents in IPAH-PASMC may be due partially to the upregulation of selected miRNAs (e.g., miR-29b, miR-138, miR-222). Overexpression of these miRNAs in normal PASMC makes normal cells phenotypically resemble IPAH-PASMC (e.g., with reduced whole cell K+ currents and downregulated KCNA5/KCNMB1), whereas inhibition of miR-29b only in IPAH-PASMC is sufficient to rescue the decreased K+ currents and KCNA5/KCNMB1 expression. AntagomiR-29b-recovered whole cell K+ currents in IPAH-PASMC contain both Penitrem A-insensitive KV currents and Penitrem A-sensitive BKCa currents. These observations indicate that upregulated miR-29b in IPAH-PASMC is sufficient to decrease both KV currents (by downregulating KCNA5) and BKCa currents (by downregulating KCNMB1). Significantly reduced whole cell K+ currents (e.g., by miR-29b, miR-138, miR-222) would subsequently cause membrane depolarization, enhance Ca2+ influx through VDCC, increase [Ca2+]cyt and eventually cause pulmonary vasoconstriction and induce pulmonary vascular medial hypertrophy (by stimulating PASMC proliferation and migration). Furthermore, decreased K+ currents or K+ efflux through KV and BKCa channels in IPAH-PASMC would also inhibit PASMC apoptosis by attenuating apoptotic volume decrease and inhibiting cytoplasmic caspase activity and further contribute to pulmonary vascular wall thickening (14). The impaired BKCa channel function or inhibited Ca2+- and voltage-mediated BKCa channel activation would also contribute to increasing myogenic vascular tone and further enhance sustained pulmonary vasoconstriction. These data imply that the specific inhibition of miR-29b in PASMC (by the use of chemically modified oligonucleotide antisense inhibitors or antagomirs) may be an important strategy to develop novel and therapeutically effective approaches to treat patients with IPAH.
GRANTS
This work was supported, in part, by grants from the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL125208, HL135807, and HL126609).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.B. and J.X.-J.Y. conceived and designed research; A.B., R.J.A., T.Z., J.F.E.V., N.M.P., A.Y., H.Y., and B.A.Q. performed experiments; A.B., R.J.A., T.Z., J.F.E.V., N.M.P., A.Y., H.Y., B.A.Q., S.R., F.B., A.H., R.R.V., A.M., and J.X.-J.Y. analyzed data; A.B., R.R.V., P.A.T., A.M., and J.X.-J.Y. interpreted results of experiments; A.B., R.J.A., T.Z., J.F.E.V., N.M.P., A.Y., H.Y., S.R., F.B., A.H., R.R.V., and J.X.-J.Y. prepared figures; A.B., N.M.P., and J.X.-J.Y. drafted manuscript; A.B., R.J.A., M.B., L.W., K.S.R., R.R.V., P.A.T., A.M., and J.X.-J.Y. edited and revised manuscript; A.B., R.J.A., T.Z., J.F.E.V., N.M.P., A.Y., H.Y., B.A.Q., M.B., L.W., K.S.R., S.R., F.B., A.H., R.R.V., P.A.T., A.M., and J.X.-J.Y. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the CMREF-PHBI (Cardiovascular Medical Research and Education Fund/Pulmonary Hypertension Breakthrough Initiative) Research Network for providing human lung cells for this study.
REFERENCES
- 1.Ahn YT, Kim YM, Adams E, Lyu SC, Alvira CM, Cornfield DN. Hypoxia-inducible factor-1α regulates KCNMB1 expression in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 302: L352–L359, 2012. doi: 10.1152/ajplung.00302.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Archer SL, Souil E, Dinh-Xuan AT, Schremmer B, Mercier JC, El Yaagoubi A, Nguyen-Huu L, Reeve HL, Hampl V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest 101: 2319–2330, 1998. doi: 10.1172/JCI333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barman SA, Zhu S, White RE. Hypoxia modulates cyclic AMP activation of BkCa channels in rat pulmonary arterial smooth muscle. Hai 183: 353–361, 2005. doi: 10.1007/s00408-005-2547-2. [DOI] [PubMed] [Google Scholar]
- 4.Barnes EA, Lee L, Barnes SL, Brenner R, Alvira CM, Cornfield DN. β1-Subunit of the calcium-sensitive potassium channel modulates the pulmonary vascular smooth muscle cell response to hypoxia. Am J Physiol Lung Cell Mol Physiol 315: L265–L275, 2018. doi: 10.1152/ajplung.00060.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Behrens R, Nolting A, Reimann F, Schwarz M, Waldschütz R, Pongs O. hKCNMB3 and hKCNMB4, cloning and characterization of two members of the large-conductance calcium-activated potassium channel beta subunit family. FEBS Lett 474: 99–106, 2000. doi: 10.1016/S0014-5793(00)01584-2. [DOI] [PubMed] [Google Scholar]
- 6.Bienertova-Vasku J, Novak J, Vasku A. MicroRNAs in pulmonary arterial hypertension: pathogenesis, diagnosis and treatment. J Am Soc Hypertens 9: 221–234, 2015. doi: 10.1016/j.jash.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 7.Bonnet S, Dumas-de-La-Roque E, Bégueret H, Marthan R, Fayon M, Dos Santos P, Savineau JP, Baulieu EE. Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc Natl Acad Sci USA 100: 9488–9493, 2003. doi: 10.1073/pnas.1633724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 113: 2630–2641, 2006. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 9.Bonnet S, Savineau JP, Barillot W, Dubuis E, Vandier C, Bonnet P. Role of Ca2+-sensitive K+ channels in the remission phase of pulmonary hypertension in chronic obstructive pulmonary diseases. Cardiovasc Res 60: 326–336, 2003. doi: 10.1016/S0008-6363(03)00527-3. [DOI] [PubMed] [Google Scholar]
- 10.Boucherat O, Chabot S, Antigny F, Perros F, Provencher S, Bonnet S. Potassium channels in pulmonary arterial hypertension. Eur Respir J 46: 1167–1177, 2015. doi: 10.1183/13993003.00798-2015. [DOI] [PubMed] [Google Scholar]
- 11.Boucherat O, Potus F, Bonnet S. microRNA and pulmonary hypertension. Adv Exp Med Biol 888: 237–252, 2015. doi: 10.1007/978-3-319-22671-2_12. [DOI] [PubMed] [Google Scholar]
- 12.Brenner R, Jegla TJ, Wickenden A, Liu Y, Aldrich RW. Cloning and functional characterization of novel large conductance calcium-activated potassium channel beta subunits, hKCNMB3 and hKCNMB4. J Biol Chem 275: 6453–6461, 2000. doi: 10.1074/jbc.275.9.6453. [DOI] [PubMed] [Google Scholar]
- 13.Brenner R, Peréz GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature 407: 870–876, 2000. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- 14.Brevnova EE, Platoshyn O, Zhang S, Yuan JX. Overexpression of human KCNA5 increases IK V and enhances apoptosis. Am J Physiol Cell Physiol 287: C715–C722, 2004. doi: 10.1152/ajpcell.00050.2004. [DOI] [PubMed] [Google Scholar]
- 15.Burg ED, Remillard CV, Yuan JX. Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: pharmacotherapeutic implications. Br J Pharmacol 153, Suppl 1: S99–S111, 2008. doi: 10.1038/sj.bjp.0707635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen J, Li Y, Li Y, Xie L, Wang J, Zhang Y, Xiao T. Effect of miR-29b on the proliferation and apoptosis of pulmonary artery smooth muscle cells by targeting Mcl-1 and CCND2. BioMed Res Int 2018: 1–10, 2018. doi: 10.1155/2018/6051407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Courboulin A, Ranchoux B, Cohen-Kaminsky S, Perros F, Bonnet S. MicroRNA networks in pulmonary arterial hypertension: share mechanisms with cancer? Curr Opin Oncol 28: 72–82, 2016. doi: 10.1097/CCO.0000000000000253. [DOI] [PubMed] [Google Scholar]
- 18.Cox RH. Molecular determinants of voltage-gated potassium currents in vascular smooth muscle. Cell Biochem Biophys 42: 167–195, 2005. doi: 10.1385/CBB:42:2:167. [DOI] [PubMed] [Google Scholar]
- 19.Detweiler ND, Song L, McClenahan SJ, Versluis RJ, Kharade SV, Kurten RC, Rhee SW, Rusch NJ. BK channels in rat and human pulmonary smooth muscle cells are BKα-β1 functional complexes lacking the oxygen-sensitive stress axis regulated exon insert. Pulm Circ 6: 563–575, 2016. doi: 10.1086/688838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dopico AM, Bukiya AN, Jaggar JH. Calcium- and voltage-gated BK channels in vascular smooth muscle. Pflugers Arch 470: 1271–1289, 2018. doi: 10.1007/s00424-018-2151-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du YM, Zhang XX, Tu DN, Zhao N, Liu YJ, Xiao H, Sanguinetti MC, Zou A, Liao YH. Molecular determinants of Kv1.5 channel block by diphenyl phosphine oxide-1. J Mol Cell Cardiol 48: 1111–1120, 2010. doi: 10.1016/j.yjmcc.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 22.Fan Z, Liu B, Zhang S, Liu H, Li Y, Wang D, Liu Y, Li J, Wang N, Liu Y, Zhang B. YM155, a selective survivin inhibitor, reverses chronic hypoxic pulmonary hypertension in rats via upregulating voltage-gated potassium channels. Clin Exp Hypertens 37: 381–387, 2015. doi: 10.3109/10641963.2014.987390. [DOI] [PubMed] [Google Scholar]
- 23.Fancher IS, Butcher JT, Brooks SD, Rottgen TS, Skaff PR, Frisbee JC, Dick GM. Diphenyl phosphine oxide-1-sensitive K+ channels contribute to the vascular tone and reactivity of resistance arteries from brain and skeletal muscle. Microcirculation 22: 315–325, 2015. doi: 10.1111/micc.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fike CD, Kaplowitz MR, Zhang Y, Madden JA. Voltage-gated K+ channels at an early stage of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 291: L1169–L1176, 2006. doi: 10.1152/ajplung.00117.2006. [DOI] [PubMed] [Google Scholar]
- 25.Firth AL, Mandel J, Yuan JX. Idiopathic pulmonary arterial hypertension. Dis Model Mech 3: 268–273, 2010. doi: 10.1242/dmm.003616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Firth AL, Remillard CV, Platoshyn O, Fantozzi I, Ko EA, Yuan JX. Functional ion channels in human pulmonary artery smooth muscle cells: voltage-dependent cation channels. Pulm Circ 1: 48–71, 2011. doi: 10.4103/2045-8932.78103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goncharov DA, Kudryashova TV, Ziai H, Ihida-Stansbury K, DeLisser H, Krymskaya VP, Tuder RM, Kawut SM, Goncharova EA. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 129: 864–874, 2014. doi: 10.1161/CIRCULATIONAHA.113.004581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta S, Li L. Modulation of miRNAs in pulmonary hypertension. Int J Hypertens 2015: 1–10, 2015. doi: 10.1155/2015/169069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong Z, Chen KH, DasGupta A, Potus F, Dunham-Snary K, Bonnet S, Tian L, Fu J, Breuils-Bonnet S, Provencher S, Wu D, Mewburn J, Ormiston ML, Archer SL. MicroRNA-138 and microRNA-25 down-regulate mitochondrial calcium uniporter, causing the pulmonary arterial hypertension cancer phenotype. Am J Respir Crit Care Med 195: 515–529, 2017. doi: 10.1164/rccm.201604-0814OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang JB, Liu YL, Sun PW, Lv XD, Bo K, Fan XM. Novel strategy for treatment of pulmonary arterial hypertension: enhancement of apoptosis. Hai 188: 179–189, 2010. doi: 10.1007/s00408-010-9233-8. [DOI] [PubMed] [Google Scholar]
- 31.Jernigan NL, Resta TC. Calcium homeostasis and sensitization in pulmonary arterial smooth muscle. Microcirculation 21: 259–271, 2014. doi: 10.1111/micc.12096. [DOI] [PubMed] [Google Scholar]
- 32.Kuhr FK, Smith KA, Song MY, Levitan I, Yuan JX. New mechanisms of pulmonary arterial hypertension: role of Ca2+ signaling. Am J Physiol Heart Circ Physiol 302: H1546–H1562, 2012. doi: 10.1152/ajpheart.00944.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S, Ran Y, Zhang D, Chen J, Li S, Zhu D. MicroRNA-138 plays a role in hypoxic pulmonary vascular remodelling by targeting Mst1. Biochem J 452: 281–291, 2013. doi: 10.1042/BJ20120680. [DOI] [PubMed] [Google Scholar]
- 34.Li SS, Ran YJ, Zhang DD, Li SZ, Zhu D. MicroRNA-190 regulates hypoxic pulmonary vasoconstriction by targeting a voltage-gated K+ channel in arterial smooth muscle cells. J Cell Biochem 115: 1196–1205, 2014. doi: 10.1002/jcb.24771. [DOI] [PubMed] [Google Scholar]
- 35.Ling L, Kokoza VA, Zhang C, Aksoy E, Raikhel AS. MicroRNA-277 targets insulin-like peptides 7 and 8 to control lipid metabolism and reproduction in Aedes aegypti mosquitoes. Proc Natl Acad Sci USA 114: E8017–E8024, 2017. doi: 10.1073/pnas.1710970114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ling TY, Wang XL, Chai Q, Lu T, Stulak JM, Joyce LD, Daly RC, Greason KL, Wu LQ, Shen WK, Cha YM, Lee HC. Regulation of cardiac CACNB2 by microRNA-499: potential role in atrial fibrillation. BBA Clin 7: 78–84, 2017. doi: 10.1016/j.bbacli.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu JJ, Zhang H, Xing F, Tang B, Wu SL, Xuan L, Kang PF, Xu Q, Wang HJ, Zhang NR, Wang XJ. MicroRNA-138 promotes proliferation and suppresses mitochondrial depolarization in human pulmonary artery smooth muscle cells through targeting TASK-1. Mol Med Rep 17: 3021–3027, 2018. doi: 10.3892/mmr.2017.8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu X, Zhang Y, Du W, Liang H, He H, Zhang L, Pan Z, Li X, Xu C, Zhou Y, Wang L, Qian M, Liu T, Yin H, Lu Y, Yang B, Shan H. MiR-223-3p as a novel microRNA regulator of expression of voltage-gated K+ channel Kv4.2 in acute myocardial infarction. Cell Physiol Biochem 39: 102–114, 2016. doi: 10.1159/000445609. [DOI] [PubMed] [Google Scholar]
- 39.Liu XS, Fan BY, Pan WL, Li C, Levin AM, Wang X, Zhang RL, Zervos TM, Hu J, Zhang XM, Chopp M, Zhang ZG. Identification of miRNomes associated with adult neurogenesis after stroke using Argonaute 2-based RNA sequencing. RNA Biol 14: 488–499, 2017. doi: 10.1080/15476286.2016.1196320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo X, Pan Z, Shan H, Xiao J, Sun X, Wang N, Lin H, Xiao L, Maguy A, Qi XY, Li Y, Gao X, Dong D, Zhang Y, Bai Y, Ai J, Sun L, Lu H, Luo XY, Wang Z, Lu Y, Yang B, Nattel S. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J Clin Invest 123: 1939–1951, 2013. doi: 10.1172/JCI62185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Makino A, Firth AL, Yuan JX. Endothelial and smooth muscle cell ion channels in pulmonary vasoconstriction and vascular remodeling. Compr Physiol 1: 1555–1602, 2011. doi: 10.1002/cphy.c100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michelakis ED, Reeve HL, Huang JM, Tolarova S, Nelson DP, Weir EK, Archer SL. Potassium channel diversity in vascular smooth muscle cells. Can J Physiol Pharmacol 75: 889–897, 1997. doi: 10.1139/y97-111. [DOI] [PubMed] [Google Scholar]
- 43.Mondejar-Parreño G, Callejo M, Barreira B, Morales-Cano D, Esquivel-Ruiz S, Moreno L, Cogolludo A, Perez-Vizcaino F. miR-1 is increased in pulmonary hypertension and downregulates Kv1.5 channels in rat pulmonary arteries. J Physiol 597: 1185–1197, 2019. doi: 10.1113/JP276054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moudgil R, Michelakis ED, Archer SL. The role of k+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 13: 615–632, 2006. doi: 10.1080/10739680600930222. [DOI] [PubMed] [Google Scholar]
- 45.Nagaraj C, Tang B, Nagy BM, Papp R, Jain PP, Marsh LM, Meredith AL, Ghanim B, Klepetko W, Kwapiszewska G, Weir EK, Olschewski H, Olschewski A. Docosahexaenoic acid causes rapid pulmonary arterial relaxation via KCa channel-mediated hyperpolarisation in pulmonary hypertension. Eur Respir J 48: 1127–1136, 2016. doi: 10.1183/13993003.01814-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Negi V, Chan SY. Discerning functional hierarchies of microRNAs in pulmonary hypertension. JCI Insight 2: e91327, 2017. doi: 10.1172/jci.insight.91327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nieves-Cintrón M, Syed AU, Nystoriak MA, Navedo MF. Regulation of voltage-gated potassium channels in vascular smooth muscle during hypertension and metabolic disorders. Microcirculation 25: e12423, 2018. doi: 10.1111/micc.12423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park WS, Firth AL, Han J, Ko EA. Patho-, physiological roles of voltage-dependent K+ channels in pulmonary arterial smooth muscle cells. J Smooth Muscle Res 46: 89–105, 2010. doi: 10.1540/jsmr.46.89. [DOI] [PubMed] [Google Scholar]
- 49.Platoshyn O, Golovina VA, Bailey CL, Limsuwan A, Krick S, Juhaszova M, Seiden JE, Rubin LJ, Yuan JX. Sustained membrane depolarization and pulmonary artery smooth muscle cell proliferation. Am J Physiol Cell Physiol 279: C1540–C1549, 2000. doi: 10.1152/ajpcell.2000.279.5.C1540. [DOI] [PubMed] [Google Scholar]
- 50.Platoshyn O, Remillard CV, Fantozzi I, Mandegar M, Sison TT, Zhang S, Burg E, Yuan JX. Diversity of voltage-dependent K+ channels in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 287: L226–L238, 2004. doi: 10.1152/ajplung.00438.2003. [DOI] [PubMed] [Google Scholar]
- 51.Post JM, Hume JR, Archer SL, Weir EK. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am J Physiol Cell Physiol 262: C882–C890, 1992. doi: 10.1152/ajpcell.1992.262.4.C882. [DOI] [PubMed] [Google Scholar]
- 52.Pullamsetti SS, Savai R, Seeger W, Goncharova EA. Translational advances in the field of pulmonary hypertension. From cancer biology to new pulmonary arterial hypertension therapeutics. Targeting cell growth and proliferation signaling hubs. Am J Respir Crit Care Med 195: 425–437, 2017. doi: 10.1164/rccm.201606-1226PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qian H, Yang C, Yang Y. MicroRNA-26a inhibits the growth and invasiveness of malignant melanoma and directly targets on MITF gene. Cell Death Discov 3: 17028, 2017. doi: 10.1038/cddiscovery.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Revermann M, Neofitidou S, Kirschning T, Schloss M, Brandes RP, Hofstetter C. Inhalation of the BKCa-opener NS1619 attenuates right ventricular pressure and improves oxygenation in the rat monocrotaline model of pulmonary hypertension. PLoS One 9: e86636, 2014. doi: 10.1371/journal.pone.0086636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salvaterra CG, Goldman WF. Acute hypoxia increases cytosolic calcium in cultured pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 264: L323–L328, 1993. doi: 10.1152/ajplung.1993.264.3.L323. [DOI] [PubMed] [Google Scholar]
- 56.Sessa R, Hata A. Role of microRNAs in lung development and pulmonary diseases. Pulm Circ 3: 315–328, 2013. doi: 10.4103/2045-8932.114758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith KA, Voiriot G, Tang H, Fraidenburg DR, Song S, Yamamura H, Yamamura A, Guo Q, Wan J, Pohl NM, Tauseef M, Bodmer R, Ocorr K, Thistlethwaite PA, Haddad GG, Powell FL, Makino A, Mehta D, Yuan JX. Notch activation of Ca2+ signaling in the development of hypoxic pulmonary vasoconstriction and pulmonary hypertension. Am J Respir Cell Mol Biol 53: 355–367, 2015. doi: 10.1165/rcmb.2014-0235OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sysol JR, Chen J, Singla S, Zhao S, Comhair S, Natarajan V, Machado RF. Micro-RNA-1 is decreased by hypoxia and contributes to the development of pulmonary vascular remodeling via regulation of sphingosine kinase 1. Am J Physiol Lung Cell Mol Physiol 314: L461–L472, 2018. doi: 10.1152/ajplung.00057.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tang H, Babicheva A, McDermott KM, Gu Y, Ayon RJ, Song S, Wang Z, Gupta A, Zhou T, Sun X, Dash S, Wang Z, Balistrieri A, Zheng Q, Cordery AG, Desai AA, Rischard F, Khalpey Z, Wang J, Black SM, Garcia JGN, Makino A, Yuan JX. Endothelial HIF-2α contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol 314: L256–L275, 2018. doi: 10.1152/ajplung.00096.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Toro L, Wallner M, Meera P, Tanaka Y. Maxi-K(Ca), a unique member of the voltage-gated K channel superfamily. News Physiol Sci 13: 112–117, 1998. [DOI] [PubMed] [Google Scholar]
- 61.Tuder RM. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res 367: 643–649, 2017. doi: 10.1007/s00441-016-2539-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Z, Yang K, Zheng Q, Zhang C, Tang H, Babicheva A, Jiang Q, Li M, Chen Y, Carr SG, Wu K, Zhang Q, Balistrieri A, Wang C, Song S, Ayon RJ, Desai AA, Black SM, Garcia JGN, Makino A, Yuan JX, Lu W, Wang J. Divergent changes of p53 in pulmonary arterial endothelial and smooth muscle cells involved in the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 316: L216–L228, 2019. doi: 10.1152/ajplung.00538.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weir EK, Obreztchikova M, Vargese A, Cabrera JA, Peterson DA, Hong Z. Mechanisms of oxygen sensing: a key to therapy of pulmonary hypertension and patent ductus arteriosus. Br J Pharmacol 155: 300–307, 2008. doi: 10.1038/bjp.2008.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whitman EM, Pisarcik S, Luke T, Fallon M, Wang J, Sylvester JT, Semenza GL, Shimoda LA. Endothelin-1 mediates hypoxia-induced inhibition of voltage-gated K+ channel expression in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 294: L309–L318, 2008. doi: 10.1152/ajplung.00091.2007. [DOI] [PubMed] [Google Scholar]
- 65.Xin H, Li Y, Buller B, Katakowski M, Zhang Y, Wang X, Shang X, Zhang ZG, Chopp M. Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells 30: 1556–1564, 2012. doi: 10.1002/stem.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu L, Chen Y, Yang K, Wang Y, Tian L, Zhang J, Wang EW, Sun D, Lu W, Wang J. Chronic hypoxia increases TRPC6 expression and basal intracellular Ca2+ concentration in rat distal pulmonary venous smooth muscle. PLoS One 9: e112007, 2014. doi: 10.1371/journal.pone.0112007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C, Dweik RA, Tuder RM, Stuehr DJ, Erzurum SC. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA 104: 1342–1347, 2007. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu Y, Bei Y, Shen S, Zhang J, Lu Y, Xiao J, Li X. MicroRNA-222 promotes the proliferation of pulmonary arterial smooth muscle cells by targeting P27 and TIMP3. Cell Physiol Biochem 43: 282–292, 2017. doi: 10.1159/000480371. [DOI] [PubMed] [Google Scholar]
- 69.Yamamura A, Ohara N, Tsukamoto K. Inhibition of excessive cell proliferation by calcilytics in idiopathic pulmonary arterial hypertension. PLoS One 10: e0138384, 2015. doi: 10.1371/journal.pone.0138384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yan B, Guo Q, Fu FJ, Wang Z, Yin Z, Wei YB, Yang JR. The role of miR-29b in cancer: regulation, function, and signaling. Onco Targets Ther 8: 539–548, 2015. doi: 10.2147/OTT.S75899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yan J, Chen R, Liu P, Gu Y. Docosahexaenoic acid attenuates hypoxic pulmonary vasoconstriction by activating the large conductance Ca2+-activated K+ currents in pulmonary artery smooth muscle cells. Pulm Pharmacol Ther 28: 9–16, 2014. doi: 10.1016/j.pupt.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 72.Yang Y, Li PY, Cheng J, Mao L, Wen J, Tan XQ, Liu ZF, Zeng XR. Function of BKCa channels is reduced in human vascular smooth muscle cells from Han Chinese patients with hypertension. Hypertension 61: 519–525, 2013. doi: 10.1161/HYPERTENSIONAHA.111.00211. [DOI] [PubMed] [Google Scholar]
- 73.Yeligar SM, Kang BY, Bijli KM, Kleinhenz JM, Murphy TC, Torres G, San Martin A, Sutliff RL, Hart CM. PPARγ regulates mitochondrial structure and function and human pulmonary artery smooth muscle cell proliferation. Am J Respir Cell Mol Biol 58: 648–657, 2018. doi: 10.1165/rcmb.2016-0293OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu FH, Catterall WA. The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE 2004: re15, 2004. doi: 10.1126/stke.2532004re15. [DOI] [PubMed] [Google Scholar]
- 75.Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV Jr, Gaine SP, Orens JB, Rubin LJ. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 98: 1400–1406, 1998. doi: 10.1161/01.CIR.98.14.1400. [DOI] [PubMed] [Google Scholar]
- 76.Zhou G, Chen T, Raj JU. MicroRNAs in pulmonary arterial hypertension. Am J Respir Cell Mol Biol 52: 139–151, 2015. doi: 10.1165/rcmb.2014-0166TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhu S, White RE, Barman SA. Role of phosphodiesterases in modulation of BKCa channels in hypertensive pulmonary arterial smooth muscle. Ther Adv Respir Dis 2: 119–127, 2008. doi: 10.1177/1753465808091327. [DOI] [PMC free article] [PubMed] [Google Scholar]