

Abstract
The plasma membrane is a dynamic environment with a complex composition of lipids, proteins, and cholesterol. Areas enriched in cholesterol and sphingolipids are believed to form lipid rafts, domains of highly ordered lipids. The unique physical properties of these domains have been proposed to influence many cellular processes. Here, we demonstrate that the activation of insulin receptor (IR) and insulin-like growth factor 1 receptor (IGF1R) depends critically on the structures of membrane sterols. IR and IGF1R autophosphorylation in vivo was inhibited by cholesterol depletion, and autophosphorylation was restored by the replacement with exogenous cholesterol. We next screened a variety of sterols for effects on IR activation. The ability of sterols to support IR autophosphorylation was strongly correlated to the propensity of the sterols to form ordered domains. IR autophosphorylation was fully restored by the incorporation of ergosterol, dihydrocholesterol, 7-dehydrocholesterol, lathosterol, desmosterol, and allocholesterol, partially restored by epicholesterol, and not restored by lanosterol, coprostanol, and 4-cholesten-3-one. These data support the hypothesis that the ability to form ordered domains is sufficient for a sterol to support ligand-induced activation of IR and IGF1R in intact mammalian cells.
Keywords: Receptor tyrosine kinase, Cholesterol, Autophosphorylation
1. Introduction
The lipid environment of biological plasma membranes may exist in multiple different states with various properties: liquid-disordered, liquid-ordered [1], and perhaps the solid-like gel state [2]. The solid-like gel phase has tightly packed acyl chains, resulting in a rigid environment and slow lateral diffusion rates. In contrast, the liquid-disordered state has high lateral diffusion rates due to the loose packing of acyl chains. The liquid-ordered phase contains highly ordered acyl chains, but retains high lateral diffusion rates. The ordered and dynamic properties of liquid-ordered domains are a result of the presence of cholesterol and sphingolipids [3]. Lipid rafts have been described as the assembly of liquid-ordered domains within the liquid-disordered phase of the plasma membranes [4]. The formation of lipid rafts in compositionally complex plasma membranes under physiological conditions has been supported by studies using giant plasma membrane vesicles (GPMVs) [5,6], plasma membrane spheres (PMS) [7], and even intact eukaryotic cells [8,9]. There are many cellular components and processes associated with and controlled by lipid rafts [10].
Previous studies have shown that the insulin receptor (IR) signaling pathway is susceptible to treatments that can result in the disruption of lipid rafts. Cyclodextrin-mediated cholesterol depletion compromises endogenous IR autophosphorylation [11]. Caveolae have been identified as plasma membrane liquid-ordered microdomains that contain caveolin [12]. Treatment of 3T3-L1 adipocytes with methyl-β-cyclo-dextrin (MβCD) displays dose-dependent effects on the depletion of cholesterol, including the flattening of caveolar invaginations [13]. The disruption of caveolae structures attenuates IR signaling to insulin receptor substrate-1 (IRS-1) and insulin-stimulated glucose transport [14]. A similar effect is observed when using cholesterol oxidase, which converts cholesterol to a steroid that does not support raft formation [13]. Cholesterol depletion in neuron-derived cells decreases phosphorylation of IRS-1 and AKT after treatment with insulin [15]. The sensitivity of the PI3K-AKT pathway to cholesterol depletion has been exploited to heighten the apoptotic response of cancer cells when treated with a combination therapy [16]. Lipid rafts are also involved in the insulin-stimulated migration of GLUT4 to the plasma membrane [17–21]. A similar dependence on lipid rafts and caveolae has been noted for insulin-like growth factor 1 receptor (IGF1R) signaling in 3T3-L1 preadipocytes [22].
Decreasing cholesterol concentrations in membranes could potentially have additional effects on the cell physiology that are not related to raft formation [23,24]. As a result, previous studies involving cholesterol depletion could not establish whether the cellular changes were directly related to lipid raft disruption [21]. To address this issue, we have experimentally manipulated the sterol composition of the plasma membrane in human embryonic kidney (HEK) 293T cells expressing IR. We have investigated the abilities of various sterols (with different raft-forming propensities) to support IR activation. Previous studies have shown that by carrying out MβCD-catalyzed lipid exchange, cholesterol can be removed and replaced with other sterols [25]. The removal of cholesterol resulted in loss of activation as measured by receptor autophosphorylation. IR autophosphorylation was recovered when cholesterol or other lipid raft supporting sterols were substituted. Sterols unable, or only weakly able, to support lipid raft formation did not restore autophosphorylation activity of IR. We observed similar effects for IGF1R after sterol depletion and cholesterol replacement. These data support the notion that the ability of sterols to form ordered domains in the plasma membrane is sufficient for them to support IR activity.
2. Materials and methods
2.1. Expression of receptors in stable HEK 293T cells
Cells stably expressing IR and IGF1R were generated and maintained as previously described [26]. Briefly, human IR and IGF1R cDNA were ligated into a modified mammalian expression vector (pCMV). Streptavidin-binding peptide (SBP)-tags [27] were inserted at the C-termini of the β-subunits of the receptors. To generate stably expressing HEK 293T cells, pCMV-IR or pCMV-IGF1R were cotransfected with a vector providing resistance to puromycin (pBABE-PURO [28]). Single colonies arising from puromycin selection were screened for receptor expression by anti-SBP Western blotting. The colonies with the highest expression were expanded and maintained in media containing puromycin.
HEK 293T cells stably expressing EGFR were generated using the same methodology. Human EGFR cDNA (matching NCBI reference sequence NM_005228.4) was cloned into the pCMV vector as an AscI/NotI fragment amplified from an expression vector containing EGFR, a kind gift from Dr. Dafna Bar-Sagi [29]. The SBP-tag was added immediately after the last coding residue.
2.2. Preparation of media for cholesterol depletion and sterol replacement
Cholesterol depletion media contained sterile-filtered (0.2 μm) 10 mM MβCD in 1× Dulbecco’s Modification of Eagle’s Medium (DMEM, Corning, 10–013-CV). Sterol replacement media contained 2.5 mM MβCD and a range of sterol concentrations (0.04 mM–0.6 mM), depending on the assay (unless indicated otherwise, a concentration of 0.2 mM sterol was used). To prepare sterol replacement media, various sterols in chloroform were added to sterile glass test tubes and dried under a stream of nitrogen gas. MβCD in 1 mL DMEM was added to the dried sterols. The solutions were mixed for 10 min in a shaking incubator at 250 rpm and 37 °C. After 10 min, solutions were sonicated in an ultrasonic bath for 1 min and mixed in a shaking incubator for 1–2 h at 37 °C before adding DMEM to the desired volume. The solutions were sterile filtered before use with the HEK 293T cells.
The following sterols were used for preparing the sterol replacement media: cholesterol from Avanti Polar Lipids; ergosterol, allocholesterol, dihydrocholesterol, and 7-dehydrocholesterol from Sigma Aldrich; 4-cholesten-3-one, coprostanol, desmosterol, epicholesterol, lathosterol, and lanosterol from Steraloids. The sterols generally appeared as single bands on thin layer chromatography, but lanosterol was purchased as a mixture with ~65% lanosterol, the remainder likely being dihydrolanosterol.
2.3. Cholesterol depletion and sterol substitution
HEK 293T cells stably expressing the appropriate receptor were seeded at a density of 1 × 106 cells in a 10 cm tissue culture dish with 10 mL standard media consisting of DMEM supplemented with 5% fetal bovine serum (VWR Life Science Seradigm) and 1× antibiotic-antimycotic solution (Corning). Three days after seeding, media was removed and cells were resuspended in 10 mL of 1× phosphate-buffered saline (PBS) (Corning, 0.144 g/L KH2PO4, 9 g/L NaCl, 0.795 g/L Na2HPO4). Cells were centrifuged at 1000 ×g for 5 min at 4 °C in a swinging-bucket centrifuge, and washed three times with PBS. The cell pellet was gently resuspended in 10 mL serum-free antimycotic-free DMEM media, and 3 mL of the cell suspension was seeded into a 60 mm tissue culture dish. All incubation steps were performed in a tissue culture incubator at 37 °C and 5% CO2.
Cells were allowed to adhere for 1 h before media was removed and replaced with cholesterol depletion media for an additional hour. Next, cholesterol depletion media was removed and replaced with DMEM (for cholesterol depleted cells) or sterol replacement media containing the sterol of choice. After 1 h, the cells were resuspended and divided into two microcentrifuge tubes. Cells were pelleted by centrifugation for 5 min at 1000 ×g, gently washed with PBS three times, and resuspended in 1 mL PBS for autophosphorylation assays.
2.4. Autophosphorylation assay with HEK 293T cells
Resuspended cells were incubated with or without 10 nM ligand (insulin, IGF-1, or EGF) for 5 min on an end-over-end rocker. Cells were pelleted by centrifugation at 16,000 ×g for 1 min and lysed on ice for 10 min with 150 μL lysis buffer (50 mM Tris, pH 7.4, 200 mM NaCl, 1 mM EDTA, 1% (v/v) Triton X-100, 1% (w/v) sodium deoxycholate, 1 mM activated Na3VO4, 10 μg/mL leupeptin, 10 μg/mL aproptinin). Lysates were centrifuged for 2 min at 14,000 ×g (4 °C) and protein concentrations were determined by Bradford assay [30]. Aliquots were removed and mixed with 25 μL 5× Laemmli buffer for analysis by Western blotting.
2.5. Western blotting
Samples were analyzed by SDS-PAGE on 8% acrylamide gels and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore). Primary antibodies used were Anti-SBP-tag (Millipore, MAB10764, Clone 20), Anti-pYpY (IR phosphorylated Tyr1162/1163 and IGF1R phosphorylated Tyr1135/1136, Calbiochem, 407707–10T), Anti-pY845 (Phospho-EGFR Tyr845, Cell Signaling, #2231), and Anti-pY1068 (Phospho-EGFR Tyr1068, Cell Signaling, #2234). Secondary antibodies used were HRP conjugated rabbit IgG (GE Healthcare Life Sciences) or HRP conjugated mouse IgG (GE Healthcare Life Sciences). Primary antibodies were diluted 1:1000 in Tris-Buffered Saline and Tween 20 (TBST, 20 mM Tris, pH 7.6, 137 mM NaCl, and 0.1% v/v Tween-20) and secondary antibodies were diluted 1:10,000 in TBST. Membranes were incubated with primary antibodies overnight, followed by 1 h incubations with secondary antibodies. Membranes were thoroughly washed in TBST before treatment with Western blotting substrate (Thermo Scientific Pierce ECL) and exposed to autoradiographic film (BioExcell).
2.6. Isolation of lipids and proteins from cells
The sterol substituted cells were dislodged by pipetting from the tissue culture plates and pelleted by centrifugation at 1000 ×g for 5 min in borosilicate glass tubes. After removal of the supernatant solution, a 1 mL mixture of hexanes and isopropanol (Fisher Scientific) (2:1 v/v) was added to the pelleted cells and samples were vortexed. The glass tubes were rocked at room temperature for 30 min with vortexing every 10 min. After centrifugation at 2000 rpm (805 ×g) for 5 min, the lipid-containing supernatants were dried under nitrogen gas in a fresh glass tube. The dried lipids were stored at −20 °C and used for HP-TLC analysis. The cell residue remaining after centrifugation was used for protein determination. For protein analysis, samples were air dried, then incubated with 1 N NaOH for 1 h at room temperature, with vortexing every 10 min. The solutions were stored at −20 °C and used for protein quantitation by the Bradford assay.
2.7. High performance thin layer chromatography (HP-TLC)
The dried lipids from sterol substituted cells were dissolved in 200 μL of methanol/chloroform (1:1 v/v). A 20 μL aliquot was then loaded on a HP-TLC plate (Silica Gel 60, VWR International) along with sterol standards. After air drying for 5 min, plates were placed in a glass tank containing hexane/ethyl acetate (3:2 v/v). When the solvent front reached the tops of the HP-TLC plates, they were removed from the tank, dried, and sprayed with a solution of 3% (w/v) cupric acetate and 8% phosphoric acid (v/v) dissolved in water. The plate was air dried again and heated at 180–200 °C in an oven until the charred sterol bands clearly appeared. The intensities of sterol bands were analyzed by densitometry using the ImageJ program [31], and the sterol amounts were quantitated by comparison to a standard curve on the same HP-TLC plate using the SlideWrite Plus program (Advanced Graphics Software, Inc., Encinitas, CA). Sterol amounts were normalized to the protein levels in the tubes from which the sterols were purified.
2.8. Fluorescence microscopy
Untreated, cholesterol depleted, and sterol replaced HEK 293T cells were prepared as described above, but in 35 mm dishes with 10 mm circular glass wells. Cells were fixed with 1% (w/v) paraformaldehyde in PBS for 1 min, washed twice with 1× PBS, and incubated with mouse anti-IR α-subunit antibody (MAB1138, 1:100 dilution in PBS) for 1 h. After removal of the antibody solutions, cells were washed twice with 1× PBS, and incubated with fluorescein (FITC) AffiniPure Goat Anti-Mouse IgG (Catalogue number 115-095-062, 1:100 dilution in PBS) for 1 h. Cells were then stained with Hoechst stain (Sigma 14530, 10 mg/mL diluted 1:10,000 in PBS) for 5 min, and washed twice with 1× PBS. Cells were overlaid with PBS and imaged using a Zeiss Axiovert 200M inverted microscope (Carl Zeiss) and AxioVision Microscope Software.
2.9. Flow cytometry
Untreated, cholesterol depleted, and sterol replaced HEK 293T cells were prepared as described above in 60 mm dishes. Cells were washed with Blocking Buffer (0.5% BSA, 2 mM EDTA in PBS) and incubated with CD220-PE (130-103-717, Miltenyi Biotec) or REA Control (S)-PE (130-113-438, Miltenyi Biotec) for 10 min. Cells were washed in Blocking Buffer, fixed in 1% paraformaldehyde in PBS for 15 min on ice, and washed in PBS. Flow cytometry analysis was performed on a BD FACS Calibur.
3. Results
We previously reported that stable expression of SBP-tagged IR in HEK 293T cells results in the production of active α2β2 holoreceptor [26]. Insulin treatment of these cells leads to a strong increase in IR autophosphorylation of the activation loop (Y1158, Y1162, and Y1163) (Fig. 1). We confirmed that our ability to detect autophosphorylated IR by Western blotting was linear with respect to protein concentration (Supplementary Fig. S1). Autophosphorylation of these tyrosine residues triggers a conformational change that stimulates kinase activity and downstream signaling [32]. Sterol depletion by MβCD decreases insulin responsiveness in a time-dependent fashion; autophosphorylation is nearly abolished after 1 h of incubation with 10 mM MβCD (Fig. 1). For the cholesterol depletion assays described below, we used 1 h incubations with 10 mM MβCD to achieve maximum suppression of IR autophosphorylation. We carried out dose-response studies of IR activation in cholesterol-depleted and control cells. The cholesterol-depleted cells did not show IR activation, even at insulin concentrations 1000-fold higher than concentrations that activated IR in control cells (Supplementary Fig. S2).
Fig. 1.

Cholesterol depletion by MβCD results in decreased IR autophosphorylation. Western blot of untreated cells (ctrl), or cells incubated with cholesterol depletion media containing MβCD for various times. Bands were quantified by densitometry, and the signal for pYpY was divided by the signal for SBP-tag to give intensity values shown below each lane. The controls and 60 min depletion conditions were carried out 10 times with similar results.
Next, we tested whether IR activity could be restored by replacement with exogenous cholesterol. After depleting cells of sterols as described above, we incubated them with MβCD pre-loaded with cholesterol. This treatment restored IR autophosphorylation (Fig. 2). Neither sterol depletion nor cholesterol replacement led to an increase in basal (unstimulated) IR activity, suggesting that these treatments did not disrupt autoinhibitory regulation of IR (Supplementary Fig. S3). To rule out the possibility that MβCD treatment radically altered the plasma membrane localization of IR, we carried out fluorescence microscopy and flow cytometry of HEK 293T cells using anti-IR antibodies. IR was present at the plasma membrane at unaltered levels during all stages of the cholesterol substitution protocol (Figs. 3 and 4).
Fig. 2.
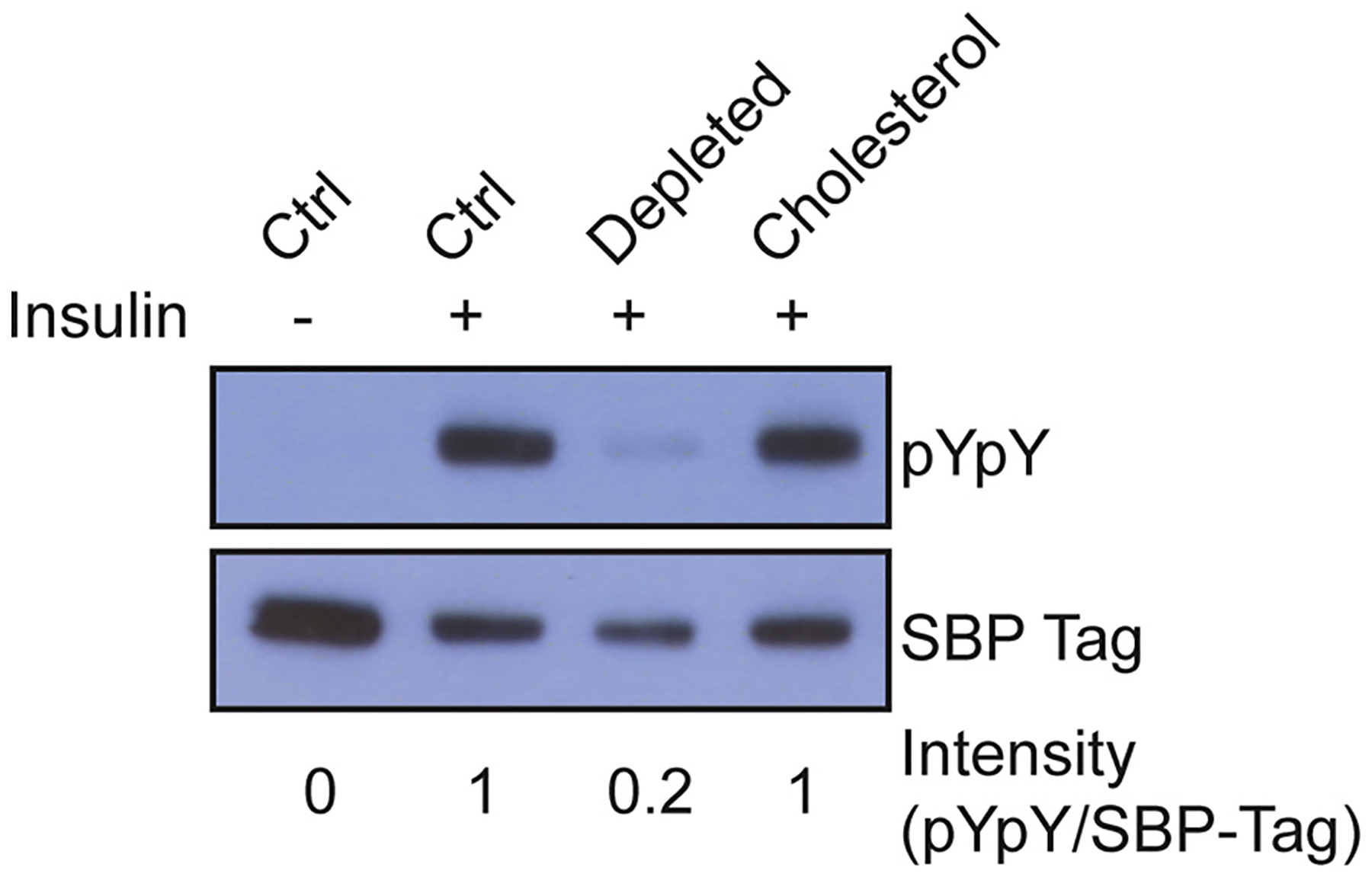
Cholesterol replacement restores IR autophosphorylation. Western blot of untreated cells (ctrl), cells incubated with cholesterol depletion media, or cholesterol depleted cells incubated with MβCD loaded with cholesterol (0.2 mM). Bands were quantified by densitometry, and the signal for pYpY was divided by the signal for SBP-tag to give intensity values shown below each lane. This experiment was carried out 10 times with similar results.
Fig. 3.
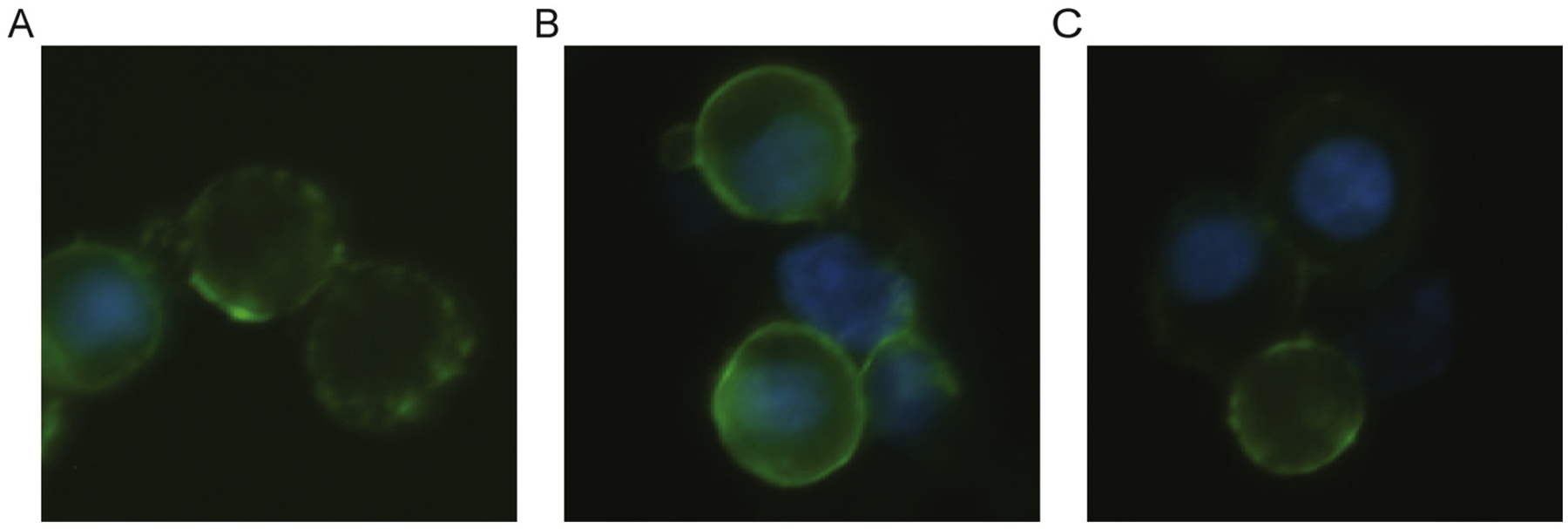
Fluorescence microcopy of HEK 293T cells stably expressing IR. Detection of IR α-subunit (green) by FITC and nucleus (blue) by Hoechst staining in untreated (A), cholesterol depleted (B), and cholesterol substituted (C) cells. The cells stably expressing IR were generated by cotransfection of an IR expression vector and a puromycin resistance vector. This resulted in variable cell-to-cell expression of IR, but plasma membrane localization was consistent.
Fig. 4.
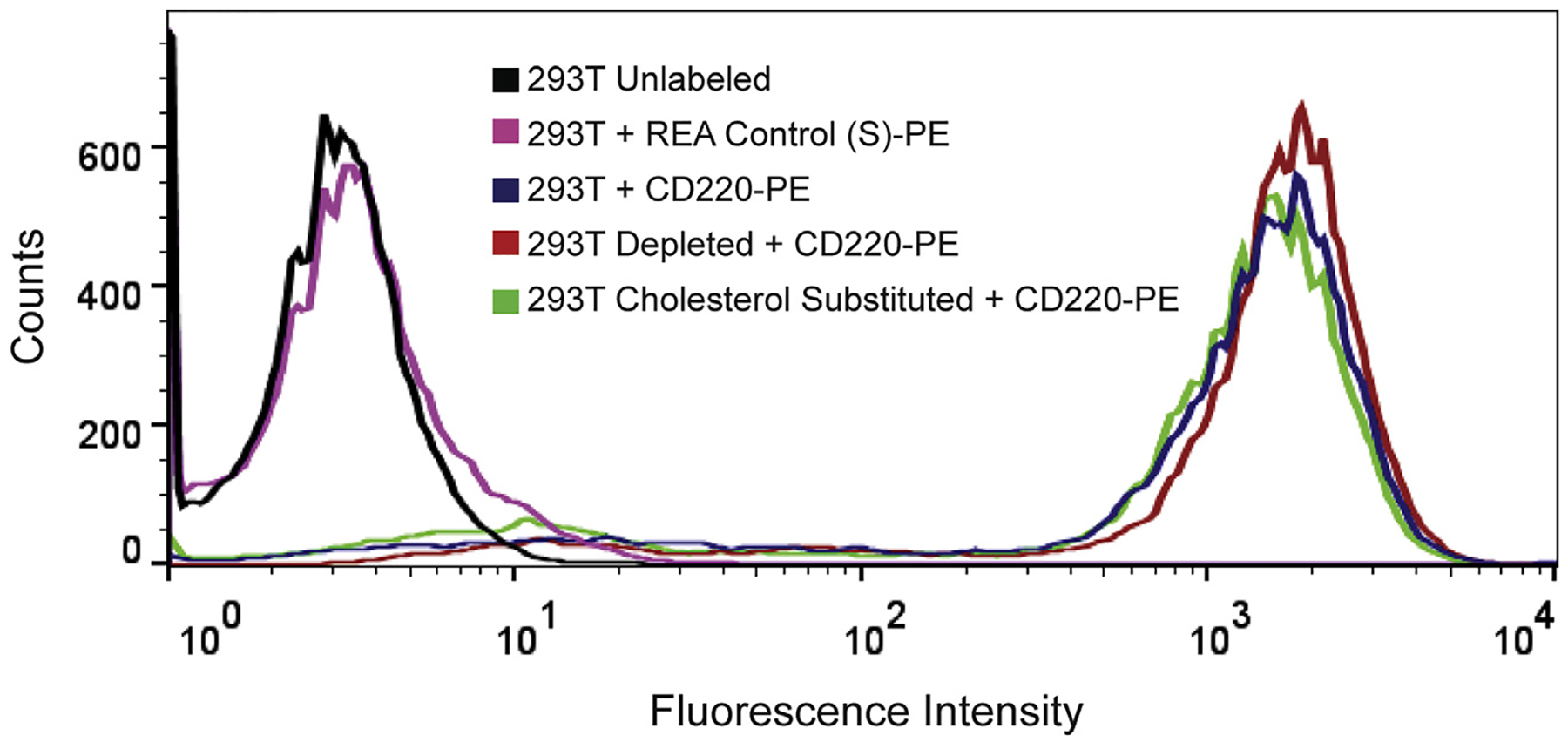
Flow cytometry of HEK 293T cells stably expressing IR. Detection of IR using phycoerythrin (PE) conjugated anti-IR (CD220-PE) in untreated, cholesterol depleted, and cholesterol substituted. Unlabeled cells or cells incubated with an isotype control (REA Control (S)-PE) resulted in no shift in fluorescence intensity.
We carried out similar experiments on HEK 293T cells expressing the closely related IGF1R, or on EGFR, a member of a different family of receptor tyrosine kinases. Cholesterol depletion of IGF1R-expressing cells produced similar effects to those seen for IR; sterol depletion reduced ligand responsiveness to undetectable levels (Fig. 5A). For EGFR-expressing cells, ligand responsiveness was greatly reduced, but some autophosphorylation within the kinase domain and at the C-terminus was still observed in sterol depleted cells (Fig. 5B–C). For both IGF1R and EGFR, treatment of the sterol depleted cells with cholesterol-loaded MβCD restored full ligand-stimulated activity (Fig. 5B–C).
Fig. 5.
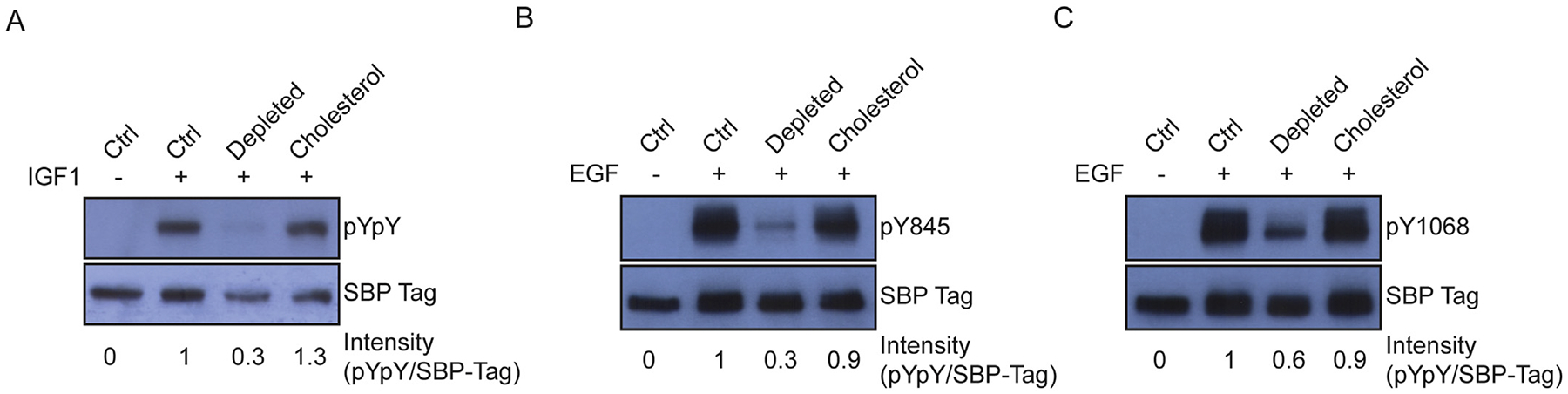
Sterol depletion and cholesterol replacement in HEK 293T cells stably expressing IGF1R (A) or EGFR (B–C). Western blots of untreated cells (ctrl), cells incubated with cholesterol depletion media, or cholesterol depleted cells incubated with MβCD loaded with cholesterol. IR and IGF1R samples were probed for activation-loop phosphorylation (pY1162/pY1163 for IR and pY1135/pY1136 for IGF1R). EGFR samples were probed for kinase domain phosphorylation (pY845) or C-terminal phosphorylation (pY1068). Bands were quantified by densitometry, and the signal for pYpY was divided by the signal for SBP-tag to give intensity values shown below each lane. This experiment was carried out 3 times with similar results.
In the next set of experiments, we carried out the sterol depletion protocol on IR-expressing HEK 293T cells, then replaced the sterols with a panel of exogenous sterols with diverse structures and various propensities to alter membrane order and raft-formation (Fig. 6). Prior studies have shown that substituted sterols are not modified during the time scale of these experiments, and that the substituted sterols alter plasma membrane order about as predicted from artificial lipid vesicle studies, indicative of a substantial plasma membrane localization [33,34]. They can also be removed from cells with a second MβCD exchange step, also indicative of a large degree of plasma membrane localization [33].
Fig. 6.
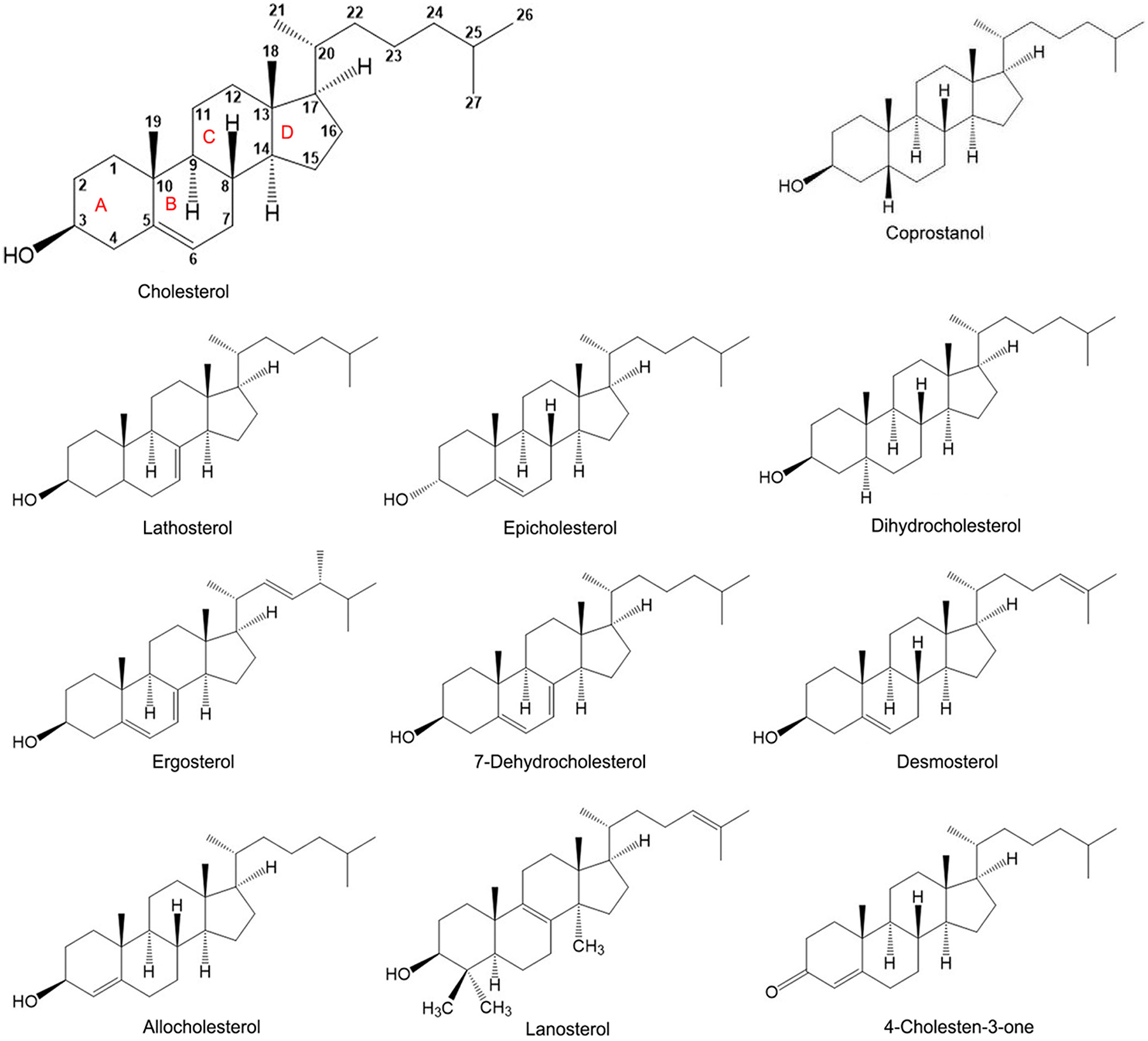
Structure of sterols used in this study.
IR autophosphorylation activity was restored by ergosterol, dihydrocholesterol, 7-dehydrocholesterol, desmosterol, and lathosterol, but not substantially restored by lanosterol, allocholesterol, 4-cholesten-3-one, or epicholesterol (Fig. 7A–B). As was the case with cholesterol replacement, sterol replacement did not cause any elevation of basal IR activity (Supplementary Fig. S4). Additional sterol substitution assays showed weak restoration of IR autophosphorylation of allocholesterol and epicholesterol, but not with lanosterol, coprostanol, or 4-cholesten-3-one (Fig. 8A).
Fig. 7.
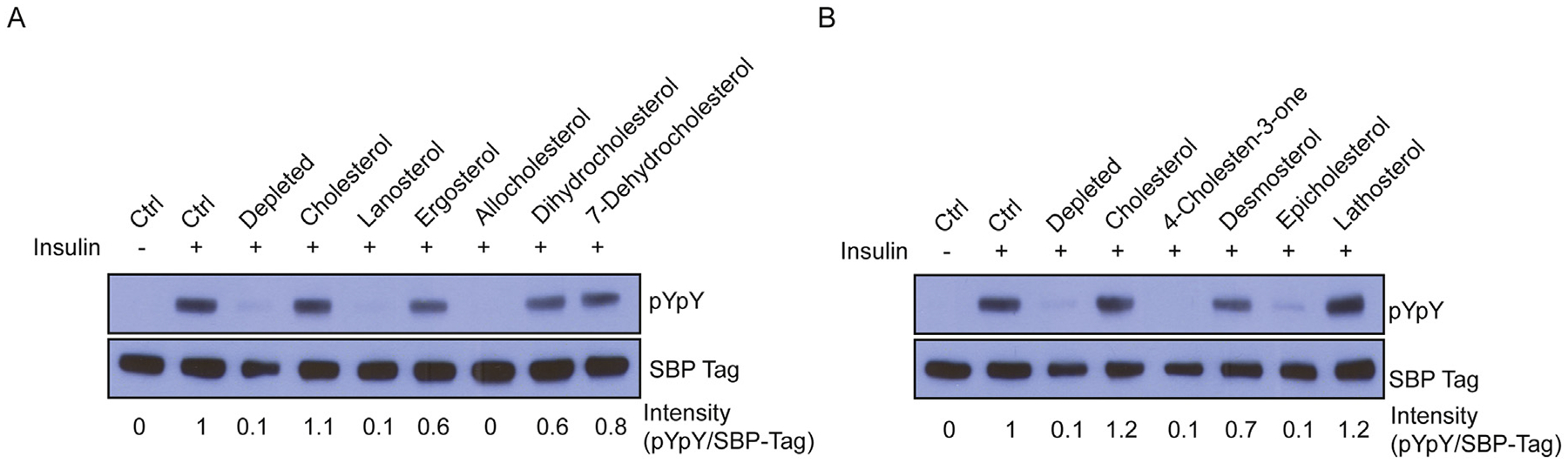
Effect of sterol replacement upon IR activity. (A-B) Western blots of untreated cells (ctrl), cells incubated with cholesterol depletion media, or cholesterol depleted cells incubated with sterol loaded MβCD (0.2 mM cholesterol, 0.4 mM for all other sterols). Bands were quantified by densitometry, and the signal for pYpY was divided by the signal for SBP-tag to give intensity values shown below each lane. This experiment was carried out at least 3 times with similar results for each sterol.
Fig. 8.
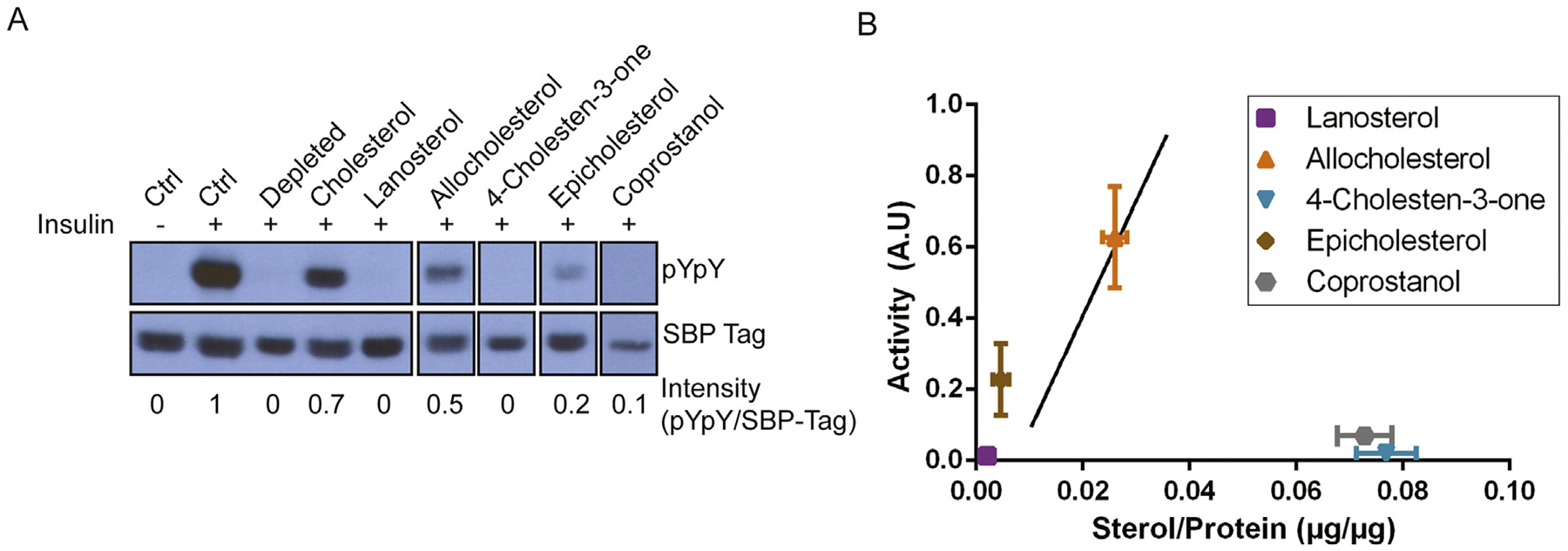
Additional analysis of the effect of cholesterol and sterols on IR autophosphorylation. (A) Western blots of untreated cells (ctrl), cholesterol depleted cells, or cholesterol depleted cells incubated with MβCD loaded with sterols (0.2 mM cholesterol, 0.4 mM for all other sterols). Bands were quantified by densitometry, and the signal for pYpY was divided by the signal for SBP-tag to give intensity values shown below each lane. This experiment was carried out 3 times with similar results. (B) Relationship of autophosphorylation activity to levels of sterols isolated from the sterol substituted cells. The solid line reproduces the cholesterol standard curve from Fig. 9 and error bars represent standard deviation. Sterol incorporation was quantified by HP-TLC and protein concentration was measured by Bradford assay.
Failure of a sterol to support IR activation could be due to a change in the membrane environment of IR, or alternatively to a lack of sterol incorporation. To distinguish between these possibilities, we carried out HP-TLC analysis of cells after cholesterol replacement and established a concentration dependent relationship between cholesterol incorporation and IR autophosphorylation activity (Fig. 9).
Fig. 9.
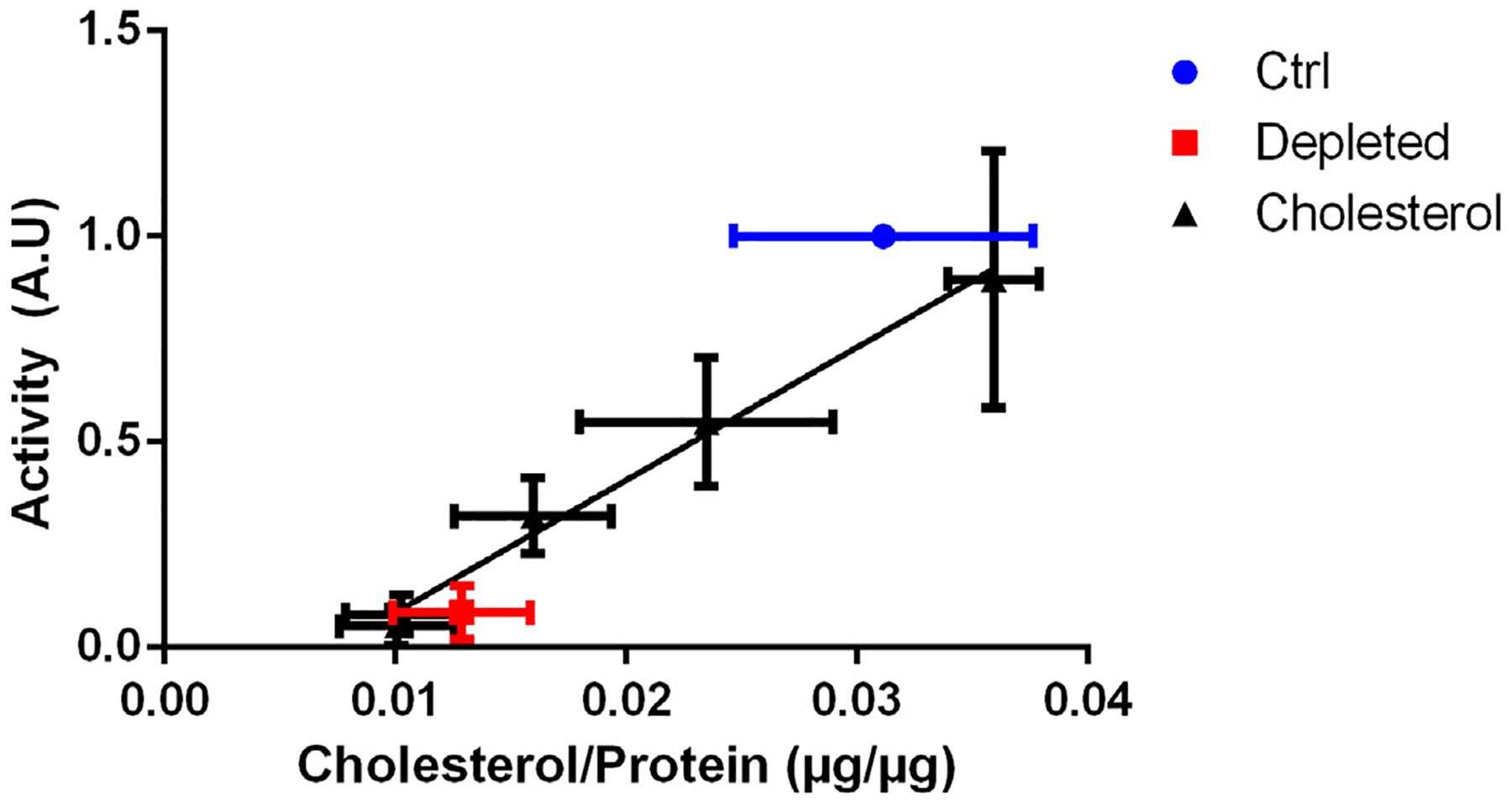
Cholesterol concentration dependence of IR autophosphorylation. Quantification of cholesterol isolated from plasma membranes of untreated (ctrl), cholesterol depleted, or cholesterol substituted cells. Quantification of cholesterol by HP-TLC analysis and quantification of protein levels by Bradford assay. Autophosphorylation activity was normalized to insulin-stimulated untreated (ctrl) cells. The error bars represent standard deviation (n = 4) and the solid line is the best linear fit to the cholesterol standards (r2 = 0.82).
HP-TLC analysis showed that coprostanol and 4-cholesten-3-one were incorporated at significant levels, but failed to restore IR autophosphorylation (Fig. 8B). In contrast, lanosterol was not readily incorporated into the cells, which could explain its inability to restore activity. Similarly, experiments with 4,6-cholestadien-3β-ol and pregnenolone, showed that they did not restore IR activity, but did not incorporate into cells at significant levels (data not shown). Allocholesterol, to the extent incorporated, restored IR activity to a similar extent as cholesterol. Epicholesterol was minimally incorporated, but restored IR autophosphorylation to an extent consistent with its being able to support activity as well as cholesterol when at the same concentration in cells. The substitution/activity data are summarized in Table 1.
Table 1.
The ability of sterols with varying lipid raft supporting propensities [35–40] to restore IR autophosphorylation.
| Sterol | Lipid rafts | IR autophosphorylation |
|---|---|---|
| Cholesterol | Strong | Yes |
| Ergosterol | Strong | Yes |
| Dihydrocholesterol | Strong | Yes |
| 7-Dehydrocholesterol | Strong | Yes |
| Lathosterol | Strong | Yes |
| Desmosterol | Intermediatea | Yes |
| Allocholesterol | Intermediate | Yesb |
| Epicholesterol | Intermediate | Yesb |
| Lanosterol | Intermediate | Noc |
| 4-Cholesten-3-one | Weak | No |
| Coprostanol | Weak | No |
It should be noted that results using asymmetric lipid vesicles indicate desmosterol may actually have strong raft supporting abilities (J. St. Clair Wellman, and E.L., unpublished results).
Allocholesterol and epicholesterol restore activity upon sufficient incorporation.
Lanosterol had minimal activity, but also poor or no incorporation.
4. Discussion
In this study, we demonstrate that IR expressed in HEK 293T decreased autophosphorylation after removal of cholesterol. This is consistent with the findings of Vainio et al., which showed a lack of autophosphorylation of IR after the removal of cholesterol in hepatoma cells [11]. In contrast, others have reported that cholesterol depletion did not affect insulin-stimulated tyrosine-specific IR autophosphorylation in 3T3-L1 adipocytes. In the latter experiments, autophosphorylation was performed in caveolae fractions, rather than in whole cells [13,14]. It is conceivable that the caveolae isolation process could have altered the IR conformation, or changed the final lipid bilayer composition in such a way as to affect IR regulation.
Here, we have taken advantage of methodology to manipulate the sterol composition of intact cell membranes. IR autophosphorylation was restored to cholesterol-depleted cells by substitution of cholesterol, ergosterol, dihydrocholesterol, 7-dehydrocholesterol, desmosterol, lathosterol, allocholesterol, and (to a lesser extent) epicholesterol. These sterols are known to support the formation of lipid rafts to at least some degree [35–40]. IR autophosphorylation activity was not substantially restored by lanosterol, which has intermediate lipid raft supporting abilities, or coprostanol, 4-cholesten-3-one, which have weak to no raft-supporting ability [37–40]. A caveat is that the low activity with lanosterol may reflect poor incorporation. It should be noted that the levels of sterol incorporation were generally similar to previous experiments performed in mammalian MDA-MB-231 breast cancer cells [33].
Sterol depletion of IGF1R-expressing cells decreased receptor autophosphorylation, and exogenous cholesterol restored autophosphorylation in a similar manner as for IR. These observations are in agreement with previous studies involving cholesterol depletion and IGF1R signaling [22,41,42].
Depletion of sterols from EGFR-expressing cells reduced (but did not eliminate) receptor autophosphorylation. Other groups have observed ligand independent activation of EGFR after cholesterol depletion with MβCD and disruption of lipid rafts with filipin III [43]. Similar results have been reported in COS-1 and NIH-3T3 cells [44,45]. Our conflicting results are likely due to the differences between the cell lines. In A549 cells, cholesterol depletion inhibits EGFR autophosphorylation, whereas EGFR autophosphorylation increases upon cholesterol depletion in A431 cells (P.S., E.L., W.T.M., unpublished data). Interestingly, it was previously shown that replenishment with higher than normal levels of cholesterol inhibited EGFR autophosphorylation [45], also consistent with a complex role for cholesterol in EGFR function. Other differences between our studies and previous ones include the use of different phospho-antibodies (Tyr845, Tyr1068 vs. Tyr1173, global anti-phosphotyrosine), the type of expression system used for EGFR expression (cells stably expressing receptor vs. endogenous receptor), and levels of EGFR expression (high expression in stable cells vs. lower levels of endogenous receptor). Additional studies will be required to resolve the dependence of EGFR on cholesterol under these different conditions.
The use of cholesterol-lowering statins has been linked to the development of new onset diabetes [46]. The known sensitivity of the IR signaling pathway to cholesterol depletion has sparked interest in elucidating possible mechanisms for this correlation. These studies have produced conflicting results, depending on the type of statin and dosing [47]. One proposed mechanism is the direct inhibition of L type Ca2+ channels by simvastatin in β-cells, which prevents glucose-induced insulin secretion [48]. However, studies with a squalene epoxidase inhibitor (NB598) to simulate chronic cholesterol depletion did not appear to have an effect on insulin secretion [49]. Lovastatin has been shown to inhibit phosphorylation of the IR β subunit and prevent downstream signaling [50]. Atorvastatin has been reported to down-regulate IR and IRS-1 levels in adipocytes [51], but others have shown no change in mRNA levels, protein expression, or phosphorylation of IR in adipocytes with atorvastatin or pravastatin treatment [52]. Atorvastatin resulted in decreased expression of caveolin-1and GLUT4 translocation [51,52]. In mice, insulin-deficiency is correlated with a decrease in the synthesis of cholesterol in the neuronal and glial cells [15]. Our results are consistent with a direct effect on IR activity by lowered cholesterol. Thus, although the benefits of statins in reducing cardiovascular events have been clearly demonstrated, it is possible that long-term reduction of cholesterol by statins could alter the plasma membrane environment and contribute to decreased insulin sensitivity and new-onset diabetes.
In this report, we have shown that cholesterol depletion by MβCD inhibits IR autophosphorylation, and lipid-raft supporting sterols restore IR activation. These data suggest the potential importance of lipid rafts for proper ligand-stimulated activity in the plasma membrane. It should be noted that not all biological processes involving the plasma membrane are as permissive as IR receptor phosphorylation in terms of sterol structure. Studies of endocytosis and uptake of Yersinia show a requirement for a more restricted range of sterols [33,34].
It is not known whether there is any direct interaction between cholesterol and the transmembrane region of IR. In this regard, it is important to note that the presence of cholesterol is not absolutely required for insulin-stimulated autophosphorylation of IR in vitro. We have previously demonstrated that detergent-solubilized IR or IGF1R show increased autophosphorylation and substrate phosphorylation when stimulated with their cognate ligands [26]. Therefore, we postulate that indirect effects of cholesterol on the surrounding lipid bilayer are likely to explain the importance of sterol on IR/IGF1R function.
Removal of cholesterol could potentially impact IR/IGF1R signaling by a number of mechanisms. One might be a direct effect of the loss of ordered membrane domains. Lipid domains could support IR phosphorylation by concentrating IR and other proteins such as to increase interactions with proteins that enhance phosphorylation, or act by segregating IR into different domains than proteins that inhibit phosphorylation. There might also be more direct effects that do not involve modulation of interactions of IR with other proteins. Cholesterol has a condensing effect on lipid bilayers, tightening lipid packing and resulting in increased bilayer thickness along with inducing other properties characteristic of a liquid ordered domain [53]. Thus, cholesterol depletion would likely result in a decrease of the local plasma membrane thickness. This may position the transmembrane domains of IR in a configuration or change their tilt in a fashion that is unfavorable for trans-autophosphorylation and receptor activation, even upon ligand binding. Alternatively, the lipid environment without cholesterol may not be conducive to propagating the necessary conformational changes from the extracellular ectodomain to the intracellular kinase domains upon insulin/IGF1 binding.
Supplementary Material
Funding sources
This work has been supported by Department of Veterans Affairs Merit Review Program BX002292 to W.T.M. and NIH grant GM 122493 to E.L.
Footnotes
Transparency document
The Transparency document associated with this article can be found, in online version.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbamem.2019.01.009.
References
- [1].van Meer G, Voelker DR, Feigenson GW, Membrane lipids: where they are and how they behave, Nat. Rev. Mol. Cell Biol 9 (2008) 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Massey JB, Interaction of ceramides with phosphatidylcholine, sphingomyelin and sphingomyelin/cholesterol bilayers, Biochim. Biophys. Acta 1510 (2001) 167–184. [DOI] [PubMed] [Google Scholar]
- [3].Simons K, Sampaio JL, Membrane organization and lipid rafts, Cold Spring Harb. Perspect. Biol 3 (2011) a004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Brown DA, London E, Functions of lipid rafts in biological membranes, Annu. Rev. Cell Dev. Biol 14 (1998) 111–136. [DOI] [PubMed] [Google Scholar]
- [5].Baumgart T, Hammond AT, Sengupta P, Hess ST, Holowka DA, Baird BA, Webb WW, Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles, Proc. Natl. Acad. Sci. U. S. A 104 (2007) 3165–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Levental I, Grzybek M, Simons K, Raft domains of variable properties and compositions in plasma membrane vesicles, Proc. Natl. Acad. Sci. U. S. A 108 (2011) 11411–11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lingwood D, Ries J, Schwille P, Simons K, Plasma membranes are poised for activation of raft phase coalescence at physiological temperature, Proc. Natl. Acad. Sci. U. S. A 105 (2008) 10005–10010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Toulmay A, Prinz WA, Direct imaging reveals stable, micrometer-scale lipid domains that segregate proteins in live cells, J. Cell Biol 202 (2013) 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Stone MB, Shelby SA, Nunez MF, Wisser K, Veatch SL, Protein sorting by lipid phase-like domains supports emergent signaling function in B lymphocyte plasma membranes, elife 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Head BP, Patel HH, Insel PA, Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function: membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling, Biochim. Biophys. Acta 1838 (2014) 532–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vainio S, Heino S, Mansson JE, Fredman P, Kuismanen E, Vaarala O, Ikonen E, Dynamic association of human insulin receptor with lipid rafts in cells lacking caveolae, EMBO Rep. 3 (2002) 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Martinez-Outschoorn UE, Sotgia F, Lisanti MP, Caveolae and signalling in cancer, Nat. Rev. Cancer 15 (2015) 225–237. [DOI] [PubMed] [Google Scholar]
- [13].Gustavsson J, Parpal S, Karlsson M, Ramsing C, Thorn H, Borg M, Lindroth M, Peterson KH, Magnusson KE, Stralfors P, Localization of the insulin receptor in caveolae of adipocyte plasma membrane, FASEB J. 13 (1999) 1961–1971. [PubMed] [Google Scholar]
- [14].Parpal S, Karlsson M, Thorn H, Stralfors P, Cholesterol depletion disrupts caveolae and insulin receptor signaling for metabolic control via insulin receptor substrate-1, but not for mitogen-activated protein kinase control, J. Biol. Chem 276 (2001) 9670–9678. [DOI] [PubMed] [Google Scholar]
- [15].Fukui K, Ferris HA, Kahn CR, Effect of cholesterol reduction on receptor signaling in neurons, J. Biol. Chem 290 (2015) 26383–26392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yamaguchi R, Perkins G, Hirota K, Targeting cholesterol with beta-cyclodextrin sensitizes cancer cells for apoptosis, FEBS Lett. 589 (2015) 4097–4105. [DOI] [PubMed] [Google Scholar]
- [17].Chiang SH, Baumann CA, Kanzaki M, Thurmond DC, Watson RT, Neudauer CL, Macara IG, Pessin JE, Saltiel AR, Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of TC10, Nature 410 (2001) 944–948. [DOI] [PubMed] [Google Scholar]
- [18].Gustavsson J, Parpal S, Stralfors P, Insulin-stimulated glucose uptake involves the transition of glucose transporters to a caveolae-rich fraction within the plasma membrane: implications for type II diabetes, Mol. Med 2 (1996) 367–372. [PMC free article] [PubMed] [Google Scholar]
- [19].Voldstedlund M, Tranum-Jensen J, Vinten J, Quantitation of Na+/K(+)-ATPase and glucose transporter isoforms in rat adipocyte plasma membrane by immunogold labeling, J. Membr. Biol 136 (1993) 63–73. [DOI] [PubMed] [Google Scholar]
- [20].Malide D, Ramm G, Cushman SW, Slot JW, Immunoelectron microscopic evidence that GLUT4 translocation explains the stimulation of glucose transport in isolated rat white adipose cells, J. Cell Sci 113 (Pt 23) (2000) 4203–4210. [DOI] [PubMed] [Google Scholar]
- [21].Bickel PE, Lipid rafts and insulin signaling, Am. J. Physiol. Endocrinol. Metab 282 (2002) E1–E10. [DOI] [PubMed] [Google Scholar]
- [22].Huo H, Guo X, Hong S, Jiang M, Liu X, Liao K, Lipid rafts/caveolae are essential for insulin-like growth factor-1 receptor signaling during 3T3-L1 preadipocyte differentiation induction, J. Biol. Chem 278 (2003) 11561–11569. [DOI] [PubMed] [Google Scholar]
- [23].Khatibzadeh N, Spector AA, Brownell WE, Anvari B, Effects of plasma membrane cholesterol level and cytoskeleton F-actin on cell protrusion mechanics, PLoS One 8 (2013) e57147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sarkar P, Chakraborty H, Chattopadhyay A, Differential membrane dipolar orientation induced by acute and chronic cholesterol depletion, Sci. Rep 7 (2017) 4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kim J, London E, Using sterol substitution to probe the role of membrane domains in membrane functions, Lipids 50 (2015) 721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Delle Bovi RJ, Miller WT, Expression and purification of functional insulin and insulin-like growth factor 1 holoreceptors from mammalian cells, Anal. Biochem 536 (2017) 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Keefe AD, Wilson DS, Seelig B, Szostak JW, One-step purification of recombinant proteins using a nanomolar-affinity streptavidin-binding peptide, the SBP-Tag, Protein Expr. Purif 23 (2001) 440–446. [DOI] [PubMed] [Google Scholar]
- [28].Morgenstern JP, Land H, Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line, Nucleic Acids Res. 18 (1990) 3587–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kim HJ, Taylor LJ, Bar-Sagi D, Spatial regulation of EGFR signaling by Sprouty2, Curr. Biol 17 (2007) 455–461. [DOI] [PubMed] [Google Scholar]
- [30].Bradford MM, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem 72 (1976) 248–254. [DOI] [PubMed] [Google Scholar]
- [31].Schneider CA, Rasband WS, Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis, Nat. Methods 9 (2012) 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hubbard SR, Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog, EMBO J. 16 (1997) 5572–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kim JH, Singh A, Del Poeta M, Brown DA, London E, The effect of sterol structure upon clathrin-mediated and clathrin-independent endocytosis, J. Cell Sci 130 (2017) 2682–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kim J, Fukuto HS, Brown DA, Bliska JB, London E, Effects of host cell sterol composition upon internalization of Yersinia pseudotuberculosis and clustered beta1 integrin, J. Biol. Chem 293 (2018) 1466–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].LaRocca TJ, Pathak P, Chiantia S, Toledo A, Silvius JR, Benach JL, London E, Proving lipid rafts exist: membrane domains in the prokaryote Borrelia burgdorferi have the same properties as eukaryotic lipid rafts, PLoS Pathog. 9 (2013) e1003353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Xu X, Bittman R, Duportail G, Heissler D, Vilcheze C, London E, Effect of the structure of natural sterols and sphingolipids on the formation of ordered sphingolipid/sterol domains (rafts). Comparison of cholesterol to plant, fungal, and disease-associated sterols and comparison of sphingomyelin, cerebrosides, and ceramide, J. Biol. Chem 276 (2001) 33540–33546. [DOI] [PubMed] [Google Scholar]
- [37].Megha, Bakht O, London E, Cholesterol precursors stabilize ordinary and ceramide-rich ordered lipid domains (lipid rafts) to different degrees. Implications for the Bloch hypothesis and sterol biosynthesis disorders, J. Biol. Chem 281 (2006) 21903–21913. [DOI] [PubMed] [Google Scholar]
- [38].Xu X, London E, The effect of sterol structure on membrane lipid domains reveals how cholesterol can induce lipid domain formation, Biochemistry 39 (2000) 843–849. [DOI] [PubMed] [Google Scholar]
- [39].Nelson LD, Johnson AE, London E, How interaction of perfringolysin O with membranes is controlled by sterol structure, lipid structure, and physiological low pH: insights into the origin of perfringolysin O-lipid raft interaction, J. Biol. Chem 283 (2008) 4632–4642. [DOI] [PubMed] [Google Scholar]
- [40].Wang J, Megha, London E, Relationship between sterol/steroid structure and participation in ordered lipid domains (lipid rafts): implications for lipid raft structure and function, Biochemistry 43 (2004) 1010–1018. [DOI] [PubMed] [Google Scholar]
- [41].Romanelli RJ, Mahajan KR, Fulmer CG, Wood TL, Insulin-like growth factor-I-stimulated Akt phosphorylation and oligodendrocyte progenitor cell survival require cholesterol-enriched membranes, J. Neurosci. Res 87 (2009) 3369–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hong S, Huo H, Xu J, Liao K, Insulin-like growth factor-1 receptor signaling in 3T3-L1 adipocyte differentiation requires lipid rafts but not caveolae, Cell Death Differ. 11 (2004) 714–723. [DOI] [PubMed] [Google Scholar]
- [43].Lambert S, Vind-Kezunovic D, Karvinen S, Gniadecki R, Ligand-independent activation of the EGFR by lipid raft disruption, J. Invest. Dermatol 126 (2006) 954–962. [DOI] [PubMed] [Google Scholar]
- [44].Chen X, Resh MD, Cholesterol depletion from the plasma membrane triggers ligand-independent activation of the epidermal growth factor receptor, J. Biol. Chem 277 (2002) 49631–49637. [DOI] [PubMed] [Google Scholar]
- [45].Pike LJ, Casey L, Cholesterol levels modulate EGF receptor-mediated signaling by altering receptor function and trafficking, Biochemistry 41 (2002) 10315–10322. [DOI] [PubMed] [Google Scholar]
- [46].Beckett RD, Schepers SM, Gordon SK, Risk of new-onset diabetes associated with statin use, SAGE Open Med. 3 (2015) (2050312115605518). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Brault M, Ray J, Gomez YH, Mantzoros CS, Daskalopoulou SS, Statin treatment and new-onset diabetes: a review of proposed mechanisms, Metabolism 63 (2014) 735–745. [DOI] [PubMed] [Google Scholar]
- [48].Yada T, Nakata M, Shiraishi T, Kakei M, Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L-type Ca2+ channels in rat islet beta-cells, Br. J. Pharmacol 126 (1999) 1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ishikawa M, Okajima F, Inoue N, Motomura K, Kato T, Takahashi A, Oikawa S, Yamada N, Shimano H, Distinct effects of pravastatin, atorvastatin, and simvastatin on insulin secretion from a beta-cell line, MIN6 cells, J. Atheroscler. Thromb 13 (2006) 329–335. [DOI] [PubMed] [Google Scholar]
- [50].McGuire TF, Xu XQ, Corey SJ, Romero GG, Sebti SM, Lovastatin disrupts early events in insulin signaling: a potential mechanism of lovastatin’s anti-mito-genic activity, Biochem. Biophys. Res. Commun 204 (1994) 399–406. [DOI] [PubMed] [Google Scholar]
- [51].Nakata M, Nagasaka S, Kusaka I, Matsuoka H, Ishibashi S, Yada T, Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): implications in glycaemic control, Diabetologia 49 (2006) 1881–1892. [DOI] [PubMed] [Google Scholar]
- [52].Takaguri A, Satoh K, Itagaki M, Tokumitsu Y, Ichihara K, Effects of atorvastatin and pravastatin on signal transduction related to glucose uptake in 3T3L1 adipocytes, J. Pharmacol. Sci 107 (2008) 80–89. [DOI] [PubMed] [Google Scholar]
- [53].Hung WC, Lee MT, Chen FY, Huang HW, The condensing effect of cholesterol in lipid bilayers, Biophys. J 92 (2007) 3960–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.