

Abstract
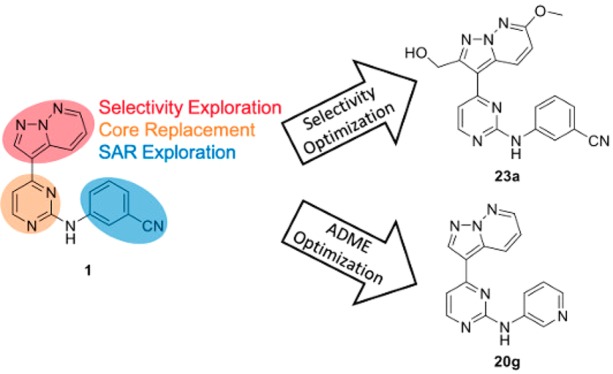
From a high-throughput screen of 42 444 known human kinases inhibitors, a pyrazolo[1,5-b]pyridazine scaffold was identified to begin optimization for the treatment of human African trypanosomiasis. Previously reported data for analogous compounds against human kinases GSK-3β, CDK-2, and CDK-4 were leveraged to try to improve the selectivity of the series, resulting in 23a which showed selectivity for T. b. brucei over these three human enzymes. In parallel, properties known to influence the absorption, distribution, metabolism, and excretion (ADME) profile of the series were optimized resulting in 20g being progressed into an efficacy study in mice. Though 20g showed toxicity in mice, it also demonstrated CNS penetration in a PK study and significant reduction of parasitemia in four out of the six mice.
Introduction
Human African trypanosomiasis (HAT), or sleeping sickness as it is commonly referred to, is a parasitic disease caused by two subspecies of Trypanosoma brucei.1 Despite sharing a life cycle and causing similar symptoms in patients, these two subspecies progress at vastly different rates. T. b. gambiense is more common (98% of cases), and infection takes an average of 3 years to run its course (ending in fatality), while T. b. rhodesiense is more virulent and infections can cause death within weeks to months of infection.2−4 There were 1446 documented cases of HAT in 2017, with an estimated 13 million people living in areas of moderate to high risk of infection.1,2 Fatal if left untreated, HAT has two stages; in the first stage T. brucei resides and multiplies in the blood and lymphatic systems. Infections often are not diagnosed due to nondescript symptoms, commonly associated with the flu.1,3 In the second stage, the parasite crosses the blood–brain barrier (BBB) to infect the central nervous system (CNS), and those infected present more readily diagnosable symptoms such as disruptions to sleeping patterns and cognitive dysfunction and can become comatose.1
There are currently two approved therapies for stage 1 HAT, pentamidine for T. b. gambiense infections and suramin for T. b. rhodesiense infections; both are ineffective against stage 2 of the disease.2
In the past, the two principal treatments for stage 2 of the disease were eflornithine for T. b. gambiense and melarsoprol for T. b. rhodesiense; the mechanisms and drawbacks of each is discussed in a thorough review by Barret et al.5 In 2009, a nifurtimox–eflornithine combination therapy (NECT) was approved for treatment of T. b. gambiense infections.6 NECT requires 14 iv infusions of eflornithine (400 mg kg–1 day–1) over 7 days as well as oral nifurtimox 3 times a day for 10 days.1,2,7,8 Fexinidazole, a nitroaromatic compound which recently concluded a phase II/III clinical trial for treatment of T. b. gambiense infections, demonstrated a 91% cure rate after 10 days of treatment, compared to 97% for NECT therapies.9 This lower cure rate was deemed acceptable as fexinidazole has a more manageable dosing regimen due to it being orally bioavailable; thus fexinidazole was approved for distribution in 2019 by the European Medicines Agency, becoming the first oral therapy for HAT.3,10,11 Notably, T. b. brucei cell lines which were resistant to nifurtimox (also a nitroaromatic compound) were found to be resistant to fexinidazole.12 This cross-resistance could imply similar mechanisms of action, meaning T. b. gambiense strains resistant to NECT could also be resistant to fexinidazole.12,13 Acoziborole (SCYX-7158), which recently had a successful phase I clinical trial could prove to be the first, single-dose, oral therapy for HAT; however, it is not expected to conclude phase II/III trials until 2020.14 While promising, it should be noted that treatments for infectious diseases fail at a higher rate in the clinic than other drug discovery programs.15 Should resistance to NECT/fexinidazole emerge or acoziborole fail in clinical trials, there would be few remaining treatment options for HAT.
Despite the need for treatment options, there has been little investment from the pharmaceutical industry due, in-part, to a lack of financial incentive, which led to the designation of HAT as a neglected tropical disease by the World Health Organization.16,17 With the lack of investment there has been an increasing effort in academic settings and in industrial–academic partnerships to aid the drug discovery process.18 Traditional drug discovery programs can be time-consuming and costly, often starting from a high-throughput screen (HTS) and going through target validation, hit-to-lead optimization, and in vivo testing before advancing to clinical trials.17 We hypothesize that employing one of a variety of drug repurposing strategies can help to shorten the timelines associated with this process.17
T. b. brucei expresses 176 kinases (156 which are described as eukaryotic protein kinases). Many of these are crucial to survival of the parasite, and several have human orthologs which have been pursued in drug discovery.19−22 With this in mind, we undertook a lead repurposing study against T. b. brucei using a biased library of known human kinase inhibitors.18 Starting from a library of 42 444 kinase inhibitors, we identified 797 compounds that showed submicromolar activity against T. b. brucei, as well as >100-fold selectivity over human HepG2 cells (used as a toxicity counter screen). Compounds were clustered based on structural similarity and scored based on a variety of properties: their activity against T. b. brucei, rate and reversibility of action in parasite growth inhibition, and predicted CNS penetration as predicted by the multiparameter optimization (MPO) score. These properties were folded into a composite score to help prioritize starting points for medicinal chemistry optimization.18,23 One of these clusters contained a pyrazolo[1,5-b]pyridazine; a representative compound is shown in Table 1. Its properties are listed, including those known to influence drug absorption, distribution, metabolism, and excretion (ADME), such as human liver microsome (HLM) and rat hepatocyte (RH) intrinsic clearance (Clint), thermodynamic aqueous solubility (aq sol.), and human plasma protein binding (PPB).
Table 1. Representative Hit from the T. b. brucei HTS18,a.
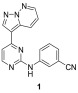
| targeted properties | 1 | |
|---|---|---|
| T. b. brucei pEC50 | >7.5 | 8.2 ± 0.02 |
| MRC5 pTC50 | <5 | <4.3 ± 0.00 |
| HepG2 pTC50 | <5 | <4.0 ± 0.00 |
| aq sol. (μM) | >10 | <0.1 |
| MW | ≤360 | 310 |
| cLogP | ≤3 | 2.8 |
| cLogD | ≤2 | 3.6 |
| MPO score | ≥4 | 5.1 |
| RH Clint* | ≤9 | 66 |
| HLM Clint** | ≤5 | 210 |
| PPB (%) | ≤95 | 98 |
aq sol. = aqueous solubility. MW = molecular weight. MPO = multiparameter optimization. RH = rat hepatocyte. HLM = human liver microsome. PPB = plasma protein binding. ∗ = (μL/min)/106 cells. ∗∗ = (μL/min)/mg protein.
On the basis of the properties of 1, this class of pyrazolo[1,5-b]pyridazines was considered a promising starting point for treatment of both stages 1 and 2 of HAT according to the published target product profile.24,25 This scaffold had already been investigated for activity against serine/threonine kinase (Stk1) in Staphylococcus aureus and for three human kinases: glycogen synthase kinase 3β (GSK-3β) and cyclin dependent kinases (CDK) 2 and 4.26−28 This was unsurprising as the compound screening set used in the HTS had been selected from a library of known kinase inhibitors, and homologs of both GSK-3 and CDK have been identified in T. brucei as potential targets for drug discovery.21,29
With this information, we set out to answer the following questions. First, would we be able to discern a difference between the series activity against T. b. brucei cells and human enzymes GSK-3β, CDK-2, and CDK-4 by utilizing available data for compounds in this class? Second, would understanding this difference enable us to design compounds with improved activity against T. b. brucei and reduced potency against these human kinases? Third, would compounds from this class demonstrate efficacy in mouse models of trypanosome infection? While we used known human kinase inhibitor data to help guide our molecular design, we did not infer anything about the parasitic target from this information, as previous work optimizing for activity against T. brucei GSK-3 has demonstrated antiparasitic activity to have arisen from a multitarget effect.30
Results
Selectivity
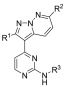
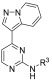
In order to assess the difference in compound potency between T. b. brucei and GSK-3β, CDK-2, and CDK-4, we evaluated previously reported compounds from the original HTS (and synthesized a set of previously reported analogs) with potency data against the three human kinases to see if we could find any difference in the selectivity profiles.26,31 A synthetic strategy was devised (Scheme 1) that would allow for late stage derivatization at the three positions of principal interest (R1–R3, as shown in 8). Starting from 2,4-dichloropyrimidine (5), a Sonogashira coupling was employed with trimethylsilylacetylene. Subsequent silyl deprotection with potassium hydroxide yielded 2-chloro-4-ethynylpyrimidine 6b. Pyridazine 4 was reacted with hydroxylamine-O-sulfonic acid (HOSA) to generate a 1-aminopyridizinium ion in situ, which was then reacted with 6b in a [3 + 2] cycloaddition reaction to form the 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine intermediate 7. Subsequent nucleophilic aromatic substitution or palladium-mediated cross-coupling provided the corresponding product 8.32
Scheme 1. Synthesis of Pyrazolo[1,5-b]pyridazine Analogs.

Reagents and conditions: (a) POCl3, 80 °C, 1 h, 70%; (b) NaOtBu, MeOH or morpholine, n-BuOH, 80 °C, 12 h, 60–100%; (c) HCCR1, CuI, Pd(PPh3)2Cl2, TEA, THF, 50 °C, 12 h, 19–61%; (d) KOH, MeOH, rt, 30 min, 94%; (e) 4, hydroxylamine-O-sulfonic acid, KOH, NaHCO3, H2O, DCM, 70 °C → rt, 12 h, 40–84%; (f) NH2R3, n-BuOH, 150 °C, 2 h, 11–90%; (g) NH2R3, Pd(OAc)2, BINAP or xantphos, Cs2CO3, dioxane, 160 °C, 1 h, 6–70%.
A previously reported crystal structure of a pyrazolo[1,5-b]pyridazine, 9 (Figure 1a), bound into CDK-2 (PDB code 3EID) (Figure 1b and 1c), indicated that substitution at the R1 and R2 positions gave the best chance to improve selectivity, due to the increased interaction with the binding pocket at these positions, while the R3 position was solvent exposed, and the binding pocket would be less sterically restrictive of the molecule.31 Previously reported docking of a pyrazolo[1,5-b]pyridazine into a GSK-3β crystal structure (PDB code 1GNG) predicted a similar binding mode.26,31 Available IC50 data against all three enzymes indicated that substitution at any of these three positions could cause significant shifts in potency, but the largest decreases in potency came with substitution at the R1 and R2 positions. Armed with the published human kinase selectivity data, we compared compounds from the HTS and resynthesized a selected set of analogs from the literature. We sought to identify which substitutions could provide analogs with divergent activity for T. b. brucei over GSK-3β, CDK-2, and CDK-4 (Table 2).
Figure 1.

Previously reported crystal structure of pyrazolo[1,5-b]pyridazine compound, (R)-1-(dimethylamino)-3-(4-((4-(6-morpholinopyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)phenoxy)propan-2-ol, 9 (green, stick representation), bound in human CDK-2 (gray, surface/wire representation) (PDB code 3EID):31 (a) structure of 9; (b) binding mode of 9 within the binding pocket, highlighting key hinge binding interactions with LEU-83; (c) binding position of 9 showing ligand–surface interactions within 5 Å, displaying position of R1 inside the binding pocket.
Table 2. Pyrazolo[1,5-b]pyridazine Analogs with Published Human Kinase Data, Compared with Whole Cell T. b. brucei Potency Dataa.

Compounds without published data against a human kinase data were left blank. *Standard error of mean (SEM) within 0.17.
Substitution at the R1 position caused varying shifts in potency between T. b. brucei and the human kinases of interest. In general, substitution at this position led to a significant loss in potency against CDK-2, an observation that is consistent with the literature.26 While substituting R1 with 4-(trifluoromethyl)phenyl (10f) caused a 100-fold decrease in potency against T. b. brucei (compared with 10e), there was a much greater impact on the potency against GSK-3β and CDK-2, both of which exhibited a >1000-fold decrease. Similarly, substitution with 4-methoxyphenyl (10q) caused a 10-fold decrease in the potency against T. b. brucei (compare with 10o); however the potency against GSK-3β and CDK-2 was decreased by >10 000-fold. Replacement of the phenyl ring with a (smaller) cyclopropyl group (10n) also resulted in a slight decrease in the potency against T. b. brucei relative to the unsubstituted analog (10m). This loss in potency was again much less dramatic than observed against GSK-3β and CDK-2 (between 3.5 and 4 log units). This indicated that while small substituents were tolerated, there was a sharp decrease in activity against T. b. brucei as R1 became larger. In sum, we were able to demonstrate the potential of compounds with T. b. brucei activity that is divergent from human GSK-3β and CDK-2 activity particularly when substituted at R1 or R2.
Substitution at the R2 position with a morpholine group (10d) led to an equipotent compound against T. b. brucei relative to the unsubstituted analog (10c), with a concomitant and significant decrease in activity against both GSK-3β and CDK-4 (∼10× against both). The presence of a methoxy at the R2 position (10j) led to a dramatic decrease in activity of the series against T. b. brucei; it also contributed to a complete loss of activity against CDK-4.
Substitution at R3 caused a significant shift in the potency of the series against T. b. brucei. Analogs with an alkyl attachment at the R3 position (10a, 10b, 10c, 10d) showed moderate inhibition of T. b. brucei, though aryl substituents (summarized in Figure 2) caused a noticeable increase in potency against T. b. brucei (typified by 10e). While these changes caused significant shifts in the activity of the compounds, they did not provide any tangible selectivity improvement against the three human kinases. Substitution of the aryl ring at the 3-position with electron withdrawing groups (EWG) yielded the most potent compounds against both T. b. brucei and the human enzymes (1, 10h). Electron donating groups (EDG) at the 3-position (10i) did not cause any significant shift in the potency, though disubstitution at the 3- and 5-positions (10k, 10l) caused a noticeable decrease in activity against T. b. brucei. Disubstitution at the 3- and 4-positions of the phenyl ring (10m, 10r) did not seem to cause this shift in potency; this trend implied there were less steric restraints on the 4-position of the R3 ring for activity against both T. b. brucei cells and the human enzymes. This observation is consistent with the crystal structures for human GSK-3β and CDK-2, wherein the 4-position of the ring points out of the binding pocket.26,31
Figure 2.

Preliminary structure–activity relationship (SAR) trends for substitution of aromatic rings at R3 position.
Curious as to the role of the pyrazolo[1,5-b]pyridazine head group in the activity of the cluster, a series of analogs was prepared to elucidate the impact of the nitrogen atom positioning within the head group upon T. b. brucei activity. The pyrazolo[1,5-a]pyridine head was synthesized via Sonogashira reaction using trimethylsilylacetylene (Scheme 2). Following desilylation, a [3 + 2] dipolar cycloaddition was effected using a 1-aminopyridinium ion generated in situ to give 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-a]pyridine (11) in moderate yield. Subsequent nucleophilic aromatic substitution gave the desired products.
Scheme 2. Synthesis of Pyrazolo[1,5-a]pyridine Analogs.

Reagents and conditions. (a) pyridine, hydroxylamine-O-sulfonic acid, KOH, NaHCO3, H2O, DCM, 70 °C → rt, 12 h, 51%; (b) NH2R3, K3PO4, XPhos, Pd2(dba)3, toluene, 110 °C, 12 h, 22%; (c) NH2R3, n-BuOH, 110 °C, 12 h, 40–88%.
As Table 3 shows, replacement with the pyrazolo[1,5-a]pyridine head caused a decrease in potency for all four analogs compared to their matched pyrazolo[1,5-b]pyridazine counterparts. Compound 12a showed the smallest decrease in potency compared to 10c but was still 2-fold less potent. The aromatic substituents at the R3 position were a minimum of 7-fold less active against T. b. brucei (12b compared to 10e), all the way up to 15-fold less active (12d compared to 1). Due to this decrease in activity and no-tangible gain in the ADME profile of the series, we turned our attention to replacing the nitrogen atoms in the pyrimidine ring.
Table 3. Effect of Pyrazolo[1,5-a]pyridine Head on T. b. brucei Potency ADME Propertiesb.

Compound detected only in first sample.
∗ = SEM within 0.05. ∗∗ = (μL/min)/106 cells. ∗∗∗ = (μL/min)/mg protein. nd = not determined.
The crystal structure and docking study for both GSK-3β and CDK-2 indicated that the key hinge binding interaction (highlighted in Figure 1b) with the series occurred between the pyrimidine nitrogen and amine at the R3 position.26,31 If altering or removing this interaction did not cause a significant loss in potency against T. b. brucei, it would likely result in a large selectivity improvement over the human enzymes. As such, we set out to remove or replace the pyrimidine and amine nitrogens. The pyridine analogs (shown in Table 4) were prepared as shown in Scheme 1, starting from the corresponding ethynylpyridine. However, this route proved unsuccessful for the phenyl derivatives, so a new synthetic route was employed, shown in Scheme 3. Starting from pyridazine, a [3 + 2] dipolar cycloaddition was performed with methyl propiolate. The subsequent product was saponified using potassium hydroxide to give the corresponding acid. A Hunsdiecker-like transformation provided the bromide, which was then used to perform a Suzuki-cross coupling to install the phenyl ring.33
Table 4. T. b. brucei Activity of the Pyrimidine Replacement Analogs and the Associated ADME Dataa.
![]()

∗ = SEM within 0.08. ∗∗ = (μL/min)/106 cells. ∗∗∗= (μL/min)/mg protein. ∗∗∗∗ = SEM within 0.02.
Scheme 3. Synthesis of Pyrimidine Replacement Analogs Containing Phenyl Ring.

Reagents and conditions: (a) pyridazine, hydroxylamine-O-sulfonic acid, KOH, NaHCO3, H2O, DCM, 70 °C → rt, 12 h, 71%; (b) NaOH, H2O, 70 °C, 1 h, 37%; (c) NBS, DMF, 25 °C, 3 h, 79%; (d) R(BOH)2, Pd(dppf)Cl2·DCM, K2CO3, 3:1 dioxane/H2O, 130 °C, 1 h, 15–72%; (e) 3-aminobenzonitrile, Pd(OAc)2, xantphos, Cs2CO3, dioxane, 160 °C, 1 h, 47%.
Both pyrimidine nitrogen atoms were deemed crucial for potent activity (Table 4), as removal of either (18d, 18b) resulted in a >10-fold potency loss compared to the original hit (1). Removal of both nitrogen atoms (17) resulted in an additional decrease in potency. Removal of the hydrogen bond donor (HBD) and R3 ring (16a, 18a, 18c) resulted in a total loss of activity against T. b. brucei, though it also corresponded with a substantial increase in the solubility of all three compounds.
Given the crystal structures of GSK-3β and CDK-2, as well as the selectivity gained from the presence of a cyclopropyl group at the R1 position in 10n, we performed a methyl scan around the pyrimidine ring to assess potency changes against T. b. brucei. The 5 and 6 positions on the pyrimidine ring were positioned in a similar part of the binding pocket in the GSK-3β crystal structure as the cyclopropyl group of 10n. Since substitution pointing into the binding pocket of the human enzymes had given selectivity for 10n, we hoped that substitution off of the pyrimidine ring would give us a similar selectivity advantage. By use of the same chemistry shown in Scheme 1, two analogs were prepared from the starting pyrimidines with methyl groups at the desired positions. As shown in Table 5, substitution at the 5-position of the pyrimidine ring (19b) lowered the activity of the series by 10-fold compared to the original hit (1) whereas substitution at the 6-position (19a) resulted in a complete loss of activity. In the published crystal structures and docking models against GSK-3β and CDK-2, these two positions on the pyrimidine ring had pointed into the same part of the binding pocket as R1.26,31 While substitution at this position may have resulted in increased selectivity against the human enzymes, the loss in activity against T. b. brucei indicated that there was no room for substitution on this portion of the scaffold.
Table 5. Analogs for Methyl Scan of Pyrimidinea.
| compd | X | Y | T. b. brucei pEC50a | aq sol. (μM) | log D7.4 | RH Clint** | HLM Clint*** | PPB (%) | MRC5 pTC50 |
|---|---|---|---|---|---|---|---|---|---|
| 19a | -H | -CH3 | <4.4 | 0.8 | 4 | 17 | 56 | 97 | <4.3 |
| 19b | -CH3 | -H | 7.0 | 0.2 | 4 | 80 | 170 | 99 | <4.3 |
∗ = SEM within 0.09. ∗∗ = (μL/min)/106 cells. ∗∗∗ = (μL/min)/mg protein.
ADME Optimization
Armed with the knowledge of the substitutions that were tolerated at the R1 and R2 positions, our attention turned to the exploration of the SAR around the R3 position of our hit compound, 1, with the view to determining which R3 substituents would provide the most favorable combination of potency and ADME properties (Table 6). Compounds 20a and 20b demonstrated the importance of the HBD for potency against T. b. brucei. Removal of the nitrile (10e) did not cause any significant change in the ADME properties of the series, though replacement with a trifluoromethyl (20c) improved the clearance of the series in both RH and HLM, though it also resulted in a 4.4-fold decrease in potency against T. b. brucei. ortho-Methyl substitution of the phenyl ring (10g) provided a substantial improvement in the aqueous solubility; comparison of 10g’s melting point (mp = 177–178 °C) to the melting point of 10e (mp = 207–209 °C) suggested that this increase in solubility was due to the disruption of the crystal packing of the compound. However, 10g showed a decrease in the activity against T. b. brucei. To further explore whether ortho-substitution was a viable route to improve the ADME properties while maintaining potency, we investigated compounds with a fluorine atom at either the 2- or 6- position of the ring (20d, 20e). These analogs did not show any significant improvement in the solubility of the series (compared with 1). Next, a number of heterocyclic replacements for the R3 phenyl group were prepared, including pyridines (20f, 20g, 20h), pyrimidines (20i), and pyridazines (20j). The 3-pyridyl analog (20g) showed a 3.5-fold reduction in the potency compared to 1 but showed improvements in the aqueous solubility and RH Clint (though HLM Clint was still high). As blocking the 4-position of the ring (10s) had previously shown reduced HLM clearance, 20k was prepared, though it demonstrated increased HLM and RH clearance over 20g. The pyrimidine analog (20i) displayed further improvement in solubility and HLM clearance, though this was at a 4.9-fold cost to potency when compared to 20g.
Table 6. T. b. brucei Activity and ADME Properties of Analogs with Substituents at the R3 Positionb.
![]()

Low recovery/low stability.
∗ = SEM within 0.14. ∗∗ = (μL/min)/106 cells. ∗∗∗ = (μL/min)/mg protein. ∗∗∗∗ = SEM within 0.17. nt = not tested. nd = not determined.
In addition to aryl substituents, a variety of alkyl substituents were explored at the R3 position, as increasing the sp3 carbon content has been previously shown to improve the aqueous solubility of a chemical series.34,35 As the steric bulk and lipophilicity of the compounds increased, an increase in potency against T. b. brucei was observed; however this increase in potency typically came with a significant reduction in solubility. Compound 20q was the most active of the compounds against T. b. brucei but demonstrated low solubility (2 μM) and high intrinsic clearance against both RH and HLM. Introduction of a polar group at the 4-position was trialed as this region was known to be solvent exposed in human enzymes and we had observed a similar SAR relationship between T. b. brucei and the human targets at the R3 position.26,31 The introduction of a trans-alcohol (20r) greatly improved the ADME profile while simultaneously maintaining the activity against T. b. brucei (pEC50: 6.8) as seen with 20q.
Comparison of the initial human kinase and T. b. brucei data indicated that substitution at the R1 position provided a promising opportunity to obtain selectivity. We also wanted to evaluate the impact that substitution at this position would have on the ADME properties of the series. Compound 21d demonstrated submicromolar activity against T. b. brucei and an improved ADME profile (Table 7), and the corresponding methyl ether (21e) showed that the presence of the HBD did not have a significant effect on the potency. The secondary alcohol (21f) and the isopropyl (21g) derivatives were equipotent, though a slight boost in potency was observed with the cyclopropyl (21h), suggesting that there are steric limitations. Compound 21h also demonstrated an improvement in the solubility and clearance compared to 1. Additionally, the cyclopropyl group at the R1 position was previously shown to cause a total loss of activity against GSK-3β and CDK-2 (Table 2, 10n).
Table 7. T. b. brucei Activity of Analogs with a Substituent at the R1 Positiona.

∗ = SEM within 0.08. ∗∗ = (μL/min)/106 cells. ∗∗∗ = (μL/min)/mg protein. ∗∗∗∗ = SEM within 0.03. nt = not tested.
Substitution at the R2 position with a morpholine or methoxy had previously shown a reduction in activity versus human CDK-2 and CDK-4; as such, we wanted to assess whether these substituents could also help improve the ADME properties of the series (Table 8). The morpholine was found to have no effect on the potency of the series (22c vs 1), though small improvements in the HLM clearance were observed. The methoxy group was found to lower potency by 1 log unit (22d vs 1) against T. b. brucei but had previously been reported to lead to a total loss in activity against CDK-4 (Table 2, 10j).
Table 8. T. b. brucei Activity and Associated ADME Profile of Analogs with a Substituent at the R2 Positionb.

Low MS/MS signal.
∗ = SEM within 0.13. ∗∗ = (μL/min)/106 cells. ∗∗∗ = (μL/min)/mg protein. nd = not determined. nt = not tested.
Given the observed trends in selectivity, activity, and ADME, we set out to make a series of combination analogs (Table 9). For the R1 position we examined the cyclopropyl group and primary alcohol as these groups were small enough to not significantly impact the potency of the molecule and could potentially provide a selectivity advantage. We selected three different R3 groups: 3-aminobenzonitrile, 3-aminopyridine, and trans-4-aminocyclohexanol based on the observed potency and ADME properties they displayed.
Table 9. T. b. brucei Activity and ADME Properties for a Series of Combination Analogs, Combining Various R1, R2, and R3 Substituentsc.

Poor MS response.
No compound detected.
∗ = SEM within 0.13. ∗∗ = (μL/min)/106 cells. ∗∗∗= (μL/min)/mg protein. nd = not determined. nt = not tested.
The series of matched analogs detailed in Table 9 gave further insight into the SAR of the cluster. At the R1 position, analogs with a cyclopropyl group (23c, 23g, 23l) were more potent than the matched pair analogs containing a primary alcohol (23b, 23f, 23m, respectively). In addition, comparison of 23g and 23l to their matched pair analogs with no substitution at the R1 position (23e and 23k, respectively) showed no decrease in potency. Interestingly this trend was not observed with compounds containing a morpholine group at the R2 position, as 23h was over 10-fold less active than its unsubstituted matched pair 23i. It should also be noted that the unsubstituted analogs at R1 showed the lowest clearance against both RH and HLM, indicating that R1 substituents may be a site of metabolism.
At the R2 position, analogs with a methoxy group 23a and 23g were more potent than their morpholine matched pair analogs 23b and 23h, respectively. The reverse of this trend was seen with 23k compared to 23j (and 22c vs 22d previously). This reversal in trends seems to further indicate that substitution at the R1 position could have an impact on the compound–target interactions at the R2 position. Analogs with substitution at R1 appear to be more potent with a methoxy group at R2, while analogs without substitution at R1 were more potent with the morpholine. Compound 23g, with a methoxy group at R2, showed an increased potency in comparison to the analog with no substitution, 23d. Compound 23d was equipotent against T. b. brucei with the morpholine derivative, 23h. This increase in potency from the introduction of a methoxy group can also be observed comparing 21d (Table 7) to 23a, where 23a was 4-fold more active. Overall the morpholine group was better for controlling the clearance of the series with matched analogs (23h and 23j) containing the morpholine showing lower RH and HLM clearance than the methoxy counterparts (23g and 23k), though all R2 analogs showed higher clearance and were less soluble than the unsubstituted analogs (23d and 20r).
A subset of these combination molecules was sent for single point inhibition assays against all three human kinases (Table 10). Analogs were prioritized based on their predicted selectivity. As expected, 23i and 23e displayed the highest percent inhibition of all three human enzymes, reaffirming the need for substitution at R1 to provide selectivity. This was also evident when examining the selectivity trend for CDK-4, as any substitution at R1 caused a significant decrease in the activity against the enzyme. At R1 the primary alcohol (23a, 23b) was generally less active than the cyclopropyl (23d, 23g, 23h) against CDK-2. The morpholine group did appear to decrease affinity for CDK-2 provided there was substitution at R1 (23h, 23b). For GSK-3β the cyclopropyl group at R1 provided the greatest decrease in percent inhibition (23d, 23h); however the morpholine at R2 seemed to increase potency against the human enzyme. The most selective of these compounds, 23a, displayed less than 50% inhibition against all three targets; however due to a poor ADME profile, it was not considered for further evaluation.
Table 10. % Inhibition of Selected Analogs at 1 μM against Human Kinases CDK-2, CDK-4, and GSK-3βa.
| compd | T. b. brucei pEC50* | CDK-2 cyclin A (%) | CDK-2 cyclin E (%) | CDK-4 cyclin D3 (%) | GSK-3β (%) |
|---|---|---|---|---|---|
| 23a | 7.2 | 47 | 29 | 1 | 49 |
| 23b | 6.8 | 46 | 54 | 32 | 75 |
| 23d | 6.8 | 76 | 70 | 21 | 21 |
| 23e | 7.2 | 84 | 84 | 69 | 89 |
| 23g | 7.2 | 84 | 77 | 25 | 65 |
| 23h | 6.8 | 67 | 49 | 26 | 34 |
| 23i | 7.9 | 89 | 87 | 90 | 81 |
∗ = SEM within 0.13.
With evidence of what was required to obtain selectivity in the series, we set out to determine whether the pyrazolo[1,5-b]pyridazine head would be efficacious in an animal model of infection. On the basis of the observed SAR and ADME data, summarized in Figure 3, we prioritized 20r and 20g for further investigation. Further studies showed that both 20r and 20g were stable in mouse plasma (Table S3, Supporting Information), were not substrates of the cytochrome P450 isozyme 3A4, and did not induce testosterone metabolism in CYP450 3A4 (Table S4, Supporting Information). Both compounds were also found to be permeable in Caco-2 cells and were not actively effluxed (Table S5, Supporting Information).
Figure 3.

Summary of the SAR and structure−property relationships (SPR) for the pyrazolo[1,5-b]pyridazines.
A small number of compounds were assessed to see if they were still fast acting and parasiticidal, including 20r and 20g (Table S6, Supporting Information). Using a set of criteria previously disclosed,18 inhibitory properties of both compounds were assessed at set time points (Figure 4); we defined fast-acting compounds as having pEC50 ≥ 6 after 18 h of incubation. Given this definition, 20g was fast-acting and 20r was slow acting. In parallel, washout assays were performed to assess whether the compounds were cidal or static. The compounds were incubated for 18 h with T. b. brucei and then washed out, and the parasite was incubated for 72 h before the wells were assayed to determine pEC50 values. Compound 20g was deemed cidal as it demonstrated pEC99 ≥ 6 following the washout assay, whereas 20r was not cidal.
Figure 4.

Rate of action of 20g and 20r, comparing pEC50 values against T. b. brucei following defined periods of incubation with the compound. Compounds with pEC50 ≥ 6 at 18 h were deemed fast acting (shown with a darkened line).
Both 20g and 20r were advanced to pharmacokinetic (PK) studies in mice, 20r for its superior ADME profile, and 20g for its rate of action, cidal activity, and high potency. Although neither compound contained structural features that were indicative of selectivity over human GSK-3β, CDK-2, and CDK-4, we theorized that their ADME profiles and activity gave the highest probability of providing proof of concept efficacy. With the knowledge that inhibitors of GSK-3β have been shown to have no negative effect in in vivo studies and lack of activity observed against human MRC5 cells for either 20g and 20r, we believed that there may be an acceptable therapeutic window.19,36
Following a 10 mg/kg intraperitoneal dose (ip) into female NMRI mice, 20r (Figure S2, Supporting Information) showed low whole blood exposure, with levels above the EC50 value (152 nM or 47 ng/mL) for <4 h following ip administration. In addition, the compound levels were below the lower limit of quantitation in the CNS after 4 h. We therefore deprioritized 20r for efficacy studies.
Compound 20g demonstrated much better BBB penetration with similar concentrations in both the blood and brain (Figure 5). In addition, although the compound displayed a short half-life (t1/2 = 0.68 h), blood and brain concentrations were >10 × EC50 value (20 nM or 6.1 ng/mL) for the entire duration of the study. Given the window for activity, as well as BBB penetration, 20g was taken forward into an efficacy study.
Figure 5.

Peripheral blood levels of 20g after ip administration to female NMRI mice (n = 3) at a target dose of 10 mg/kg in 1% DMSO, 20% Captisol in water. Individual values for each time point are represented in the plot.
For the efficacy study, mice were infected with T. b. brucei STIB795 and 3 days postinfection 20g was administered at 10 mg kg–1 day–1 ip for 5 consecutive days with daily monitoring of parasitemia levels. A significant reduction was observed in parasitemia for four of six mice, resulting in parasite concentrations dropping below detectable levels by day 4 of treatment (Table S10, Supporting Information). Despite the reduction of parasitemia, 20g showed a negligible improvement in the survival rate of the mice compared with the control group, likely due to the toxicity of the compound, as mice presented with conjunctivitis, lacrimation, facial inflammation, in addition to an inflamed liver and kidneys (Figure 6).
Figure 6.

Effect of 20g on trypanosomes in a mouse efficacy model of HAT. Percent (%) survival rate of mice dosed ip at 10 mg kg–1 day–1, compared to the control vehicle.
Due to the toxicity observed from the efficacy study, we deemed it pertinent to examine the wider kinase selectivity of 20g, profiling it against a panel of human kinases (Figure 7). Of the 47 kinases 20g was profiled against, 14 showed greater than 50% inhibition at 1 μM. Unsurprisingly, given the lack of substitution around R1 and R2, 20g still showed potent inhibition of GSK-3β, CDK-2, and CDK-4. Further efforts in the optimization of this series would need to address this toxicity. Given the improvement in the selectivity of 23a, a more thorough investigation of substituents at R1 and R2 is warranted. The difference in toxicity, shown in Table S11, Supporting Information, against L6 (20g pTC50 > 6.2, 23a pTC50 < 4.3) and THP-1 (20g pTC50 > 6.2, 23a pTC50 < 4.3) indicates that these may be better cell lines to use when optimizing for toxicity.
Figure 7.

Percent (%) inhibition at 1 μM 20g against human kinases. Line is drawn to show 50% inhibition of enzymes. Kinases showing significant inhibition (above 50%) are listed on the X-axis. We define significant inhibition as >50% inhibition at 1 μM (red line), weak to moderate inhibition to be between 25% (green line) and 50%, and insignificant inhibition to be <25%. For all values of inhibition see Table S9, Supporting Information.
A selection of compounds from this cluster was also screened against T. cruzi (Table S11, Supporting Information), Leishmania donovani (Table S11, Supporting Information), and adults of the flatworm pathogen, Schistosoma mansoni (Table S12, Supporting Information). Compound 10e was the most active compound against T. cruzi, although this was accompanied by poor selectivity versus L6 cells. While S1a was the most active against L. donovani and had a good selectivity profile. Further optimization to understand the SAR versus T. cruzi and L. donovani is warranted. Against S. mansoni, most compounds were inactive, although 10s and S1k showed similar activities characterized by a marked decrease in movement and an inability by the parasite to adhere to the bottom of the floor of the assay well with its oral and ventral suckers. Apart from these two compounds, this cluster does not warrant further investigation as a source of antischistosomal agents.
Conclusions
Starting from a screen of 42 444 known kinase inhibitors, we set out to optimize a pyrazolo[1,5-b]pyridazine scaffold for the treatment of HAT. Using published SAR data against known human kinases GSK-3β, CDK-2, and CDK-4, we were able to design an analog, 23a, which showed selectivity against all three human targets. As 23a did not have an ADME profile we deemed acceptable, 20g was taken forward into a mouse model to determine whether the series as a whole had potential to be effective in vivo. Compound 20g was selected because it was fast acting, parasiticidal, had desirable ADME properties close to our targeted values, and showed good BBB penetration in the PK study. Compound 20g demonstrated acceptable brain concentrations and levels in excess of 10 × EC50 for 4 h in the PK study and demonstrated a reduction in parasitemia in four out of the six mice tested. Although 20g’s toxicity in vivo did not warrant further investigation, it highlights the promise of this series as anti-HAT agents.
Experimental Section
Strains and Media
Bloodstream Trypanosoma brucei brucei Lister 427 was cultured in Hirumi’s modified Iscove’s medium (HMI-9), supplemented with 10% heat-inactivated FBS, at 37 °C and 5% CO2 in T-25 vented flask (Corning).
MRC5-SV2 cell line (SV40-transformed human lung fibroblast cell line) was cultured in DMEM medium supplemented with 10% FBS at 37 °C and 5% CO2 in T-75 vented flask (Corning).
The T. cruzi Tulahuen C4 strain, expressing the β-galactosidase gene (LacZ) and L6 rat skeletal muscle cells, used as host cells, were cultured in RPMI-1640 supplemented with 10% iFBS, 2 mM l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C and 5% CO2.
Leishmania donovani MHOM/ET/67/HU3 cells with the luciferase gene integrated into the parasite genome were grown at 28 °C in RPMI 1640-modified medium (Invitrogen) supplemented with 20% FBS with 100 mg/mL of hygromycin B.37
The human myelomonocytic cell line THP-1 was grown at 37 °C and 5% CO2 in RPMI-1640 supplemented with 10% iFBS, 2 mM glutamate, 100 U/mL penicillin, and 100 mg/mL streptomycin. 3 ×104 THP-1 cells per well in 96-well plates were differentiated to macrophages with 20 ng/mL of PMA treatment for 48 h followed by 24 h of culture in fresh medium.
Maintenance of the Schistosoma mansoni life cycle (NMRI isolate), the preparation of adult worms (≥42-days-old), and phenotypic screens with test compounds were as described.38−40
Preparation of Compound Plates
For dose–response experiments, compound plates were prepared for each analogue by serial 3-fold dilutions in 100% DMSO. Five concentration points (mammalian cytotoxicity) or ten concentration points (parasite growth inhibition) were made in a 96-well transparent Nunclon plate. Internal control (pentamidine) is included randomly for Trypanosoma brucei assay plates.
Trypanosoma brucei Growth Inhibition Assay
In order to determine the T. b. brucei EC50 values, an amount of 4 μL per well from compound master plates was dispensed into a new plate and an amount of 96 μL of HMI-9 per well was added to generate a 4% DMSO intermediate plate. Mid-log phase growth T. b. brucei was diluted to a working cell density of 2750 cells/mL and 90 μL/well dispensed into 96-well flat-bottom transparent assay plates (Nunc) where an amount of 10 μL/well from intermediate plates was added. Final top concentration of compounds was 40 μM in 0.4% DMSO per well.
Assay plates were incubated for 72 h at 37 °C and 5% CO2. Four hours prior to the end of the incubation, 20 μL of a 440 μM resazurin solution in prewarmed HMI-9 was added to each well and incubated for another 4 h. Fluorescence was then measured in an Infinite F200 plate reader (Tecan) at 550 nm (excitation filter) and 590 nm (emission filter). A four-parameter equation was employed to fit the dose–response curves and determine the EC50 using the SigmaPlot 13.0 software. Assays were performed in duplicate at least twice to achieve a minimal n = 3 per dose response.
Rate of Action Assays
Mid-log T. brucei brucei cultures were diluted to the required cell density, according to the different incubation time points described. An amount of 90 μL per well was dispensed in final assay plates Nunclon 96-well flat bottom Solid White, and an amount of 10 μL of intermediate plates was added to each well, as described before. Four sets of assay plates were arranged to assay in order to be sequentially stopped at each indicated time point. Top and bottom rows were dismissed for compound assay, to reduce evaporation effects.
Plates were incubated at 37 °C and 5% CO2 for the indicated time points; incubation was stopped by addition of 10 μL of prewarmed Cell Titer Glo reagent (Promega), and after shaking the plates were incubated at room temperature for 10 min to allow the signal to settle. Plate luminescence was read on an Infinite F200 plate reader (Tecan), and raw data were processed and analyzed as previously described.
Reversibility Assays
Mid-log T. brucei brucei cultures were adjusted to a working density of 3500 cell/mL. An amount of 90 μL per well was dispensed in final assay plates 96-well transparent Nunclon plates, and an amount of 10 μL of intermediate plates was added to each well, as described before. Top and bottom rows were dismissed for compound assay, to reduce evaporation effects.
Plates were incubated at 37 °C and 5% CO2 for 18 h; once drug exposure was finished, plates were spun for 5 min at 500 rpm and room temperature to allow cells to settle at the well bottoms. 95 μL of media was replaced with fresh prewarmed media, and the process was repeated 3 times. Finally, an amount of 10 μL of each well was seeded in a new microtiter plate with 90 μL of fresh prewarmed media, in duplicate. Plates were incubated for 72 h, and viability was determined by resazurin reduction as previously described.
Cytotoxicity Assay in MRC5 Cells
Intermediate plates were made as described, adding 95 μL of DMEM complete media to 5 μL of compound per well setting a 5% DMSO amount.
The log-phase MRC5 cells were removed from a T-75 TC flask using TrypLE Express (Thermo) and dispersed by gentle pipetting. Cell density was adjusted to working concentration in prewarmed DMEM medium: 25 000 cells in 90 μL of culture were plated in 96-well transparent Nunclon plates and allowed to settle for 24 h at 37 °C and 5% CO2. After settling incubation, an amount of 10 μL of fresh made intermediate plate was added per well: final maximal concentration for compounds was 50 μM in 0.5% DMSO per well. Plates were incubated for 48 h at 37 °C and 5% CO2. 4 h prior to fluorescence measurement, 20 μL of 500 μM resazurin solution was added. Fluorescence was read in an Infinite F200 plate reader (Tecan) at 550 nm (excitation filter) and 590 nm (emission filter).
A four-parameter equation was used to fit the dose–response curves and for determination of EC50 by SigmaPlot 13.0 software. Assays were performed in duplicate at least twice for positive compounds, to achieve a minimal n = 3 per dose response.
β-d-Galactosidase Transgenic T. cruzi Assay
A Thermo Scientific Multidrop Combi dispenser (MTX Lab Systems, Vienna, VA) was used to dispense 90 μL of T. cruzi amastigote-infected L6 cell culture (4 × 103 infected L6 cells per well) into 96-well Corning assay plates (Corning Inc., Corning, NY) already containing 10 μL of the compounds to be screened and controls. The plates were incubated at 37 °C for 96 h. Then, 30 μL of 100 μM CPRG and 0.1% NP40 diluted with PBS were added to each well, and the plates were incubated for 4 h at 37 °C in the dark. Absorbance at 585 nm was measured in a Vmax kinetic microplate reader (Molecular Probes). Compound activities were normalized using the in-plate negative (benznidazole at 10 μg/mL) and positive (0.2% DMSO) growth controls.
Resazurin-Based L6 Assay
An amount of 100 μL per well of culture medium containing the compounds and controls was added to L6 cells previously cultured (4 × 103 L6 cells per well). After 72 h at 37 °C the medium was exchanged and the viable cell number was determined by resazurin (Sigma-Aldrich) reduction. 20 μL of resazurin (1.1 mg/mL) was added to each well and incubated in the dark for 2 h at 37 °C. Cell viability was estimated by measuring the final fluorescence at 570–590 nm in an Infinite F200 plate reader (Tecan).
Cytotoxicity Assay in THP-1
Cellular toxicity of all compounds was determined using the colorimetric MTT-based assay after incubation at 37 °C for 72 h in the presence of increasing concentrations of compounds (final maximal concentration was 50 μM in 0.5% DMSO per well).41 The results are expressed as EC50 values, the concentration of compound that reduces cell growth by 50% versus untreated control cells. Assays were performed in duplicate at least twice to achieve a minimal n = 3 per dose response.
Determination of EC50 in L. donovani
Macrophage-differentiated THP-1 cells were infected at a macrophage/parasite ratio of 1/10 with stationary L. donovani promastigotes for 24 h at 35 °C and 5% CO2, and extracellular parasites were removed by washing with PBS. Infected cell cultures were then incubated with different compounds concentrations at 37 °C for 72 h. Luminescence was measured using the Promega kit luciferase assay system (Promega, Madison, WI). Assays were performed in duplicate at least twice to achieve a minimal n = 3 per dose response.
ADME Experiment Protocols. Aqueous pH 7.4 Solubility
Compounds are dried down from 10 mM DMSO solutions using centrifugal evaporation technique. Phosphate buffer (0.1 M, pH 7.4) was added and StirStix inserted in the glass vials, and shaking is then performed at a constant temperature of 25 °C for 20–24 h. This step is followed by double centrifugation with a tip wash in between, to ensure that no residues of the dried compound are interfering. The solutions are diluted before analysis and quantification using LC/MS/MS is performed.
log D7.4
The distribution coefficient between 1-octanol and aqueous buffer, log D, at pH 7.4, is based on the traditional shake flask technique but with the modification of measuring compounds in mixtures of 10 at a time using UPLC with quantitative MS to measure the relative octanol and aqueous concentrations of compounds. The buffer solution used is 10 mM sodium phosphate (pH 7.4). The method has been validated for log D7.4 ranging from −2 to 5.0.
Human Plasma Protein Binding (PPB)
PPB is determined using equilibrium dialysis (RED device) to separate free from bound compound. The amount of compound in plasma (1 mM initial concentration in DMSO) and in dialysis buffer (pH 7.4 phosphate buffer) is measured by LC/MS/MS after 18 h in a dialysis chamber at 37 °C. Sample levels are quantified using a seven-point calibration curve in plasma. The fraction unbound (fu, %) is reported.
Human Liver Microsomal Clint
In vitro intrinsic clearance was determined from human liver microsomes using a standard approach.42 Following incubation and preparation, the samples are analyzed using LC/MS/MS. Refined data are uploaded to IBIS and are displayed as Clint (intrinsic clearance) in (μL/min)/mg protein.
Rat Hepatocyte Clint
In vitro intrinsic clearance was determined from rat han Wistar hepatocytes at a final concentration of 1 million cells/mL. The cells were preincubated at 37 °C for 15 min before addition of test compound (as a 50 μM stock solution) to a final concentration of 1 μM. The samples were analyzed using LC/MS/MS at predefined time points. Refined data were uploaded to IBIS and are displayed as Clint (intrinsic clearance) (μL/min)/1 million cells.
Calculated log P and log D Values
Both log P and log D predictions are based on a modified version of the method43 where the predicted partition coefficients are composed of the molecules’ atomic increments.
Matrix Stability
For stability in mouse matrix, CD-1 is determined at 37 °C. The assay is performed by adding 500 μM compound solution to the matrix to achieve a final concentration of 2 μM. Following incubation for 0, 0.25, 0.5, 1, 2, and 4 h, the samples are quenched and analyzed using LC/MS to determine the percentage of drug remaining.
Caco-2 Permeability
The Caco-2 permeability of test compounds is measured using an established protocol at Charles River Labs. Concentration of test compounds is determined by measuring the peak area rations at T = 0 and 2 h on the apical and basolateral sides of the basal plate.
CYP Induction
CYP induction is measured using established protocols at Charles River Labs. The assay is performed by adding the test compound to the positive or negative controls, incubating for 3 h at 37 °C, following determination of the fold-induction relative to the vehicle control by LC/MS/MS analysis.
CYP Inhibition
CYP inhibition is measured using established protocols at Charles River Labs. A 50 mM stock solution was prepared and serially diluted (1.5-fold and 4-fold). This was then combined with a buffer/cofactor/substrate solution before combining with the human liver microsomes. The resulting mixture was incubated at 37 °C for 30 min with gentle shaking before the percentage inhibition was calculated relative to zero inhibition by LC/MS/MS analysis.
For all experiments, compounds were marked as not tested if they were not run in an assay and marked as not determined if they were run in an assay, but the result was inconclusive.
Pharmacokinetics of 20r and 20g Following ip Administration to Female NMRI. Estimation of Brain Penetration
Compounds 20r and 20g were dosed to two groups of NMRI female mice animals (groups 1 and 2, n = 3; groups 3 and 4, n = 6) by intraperitoneal (ip) route. The dosing volume was 10 mL/kg for a total dose of 10 mg/kg, and dosing formulation was prepared in 1% (v/v) dimethyl sulfoxide (DMSO)/99% (v/v) 20% (w/v) sulfobutyl ether-β-cyclodextrin (SBE-β-CD) (Captisol) in water. Food and tap water was available ad libitum.
For groups 1 and 2 and following ip dosing of each compound, blood samples were collected from the tail vein into capillary tubes containing K2EDTA at the following time-points: 0.0833, 0.25, 0.5, 1, 2, 4, 6, 8, and 24 h postdose.
In order to obtain simultaneous blood and brain samples, mice included in groups 3 and 4 were placed under terminal anesthetic (isoflurane) and blood samples (0.3 mL) collected from the retro-orbital sinus into K2EDTA tubes at 0.5 h (n = 3) and 4 h (n = 3) after intraperitoneal administration of 20r (group 3) and 20g (group 4). Immediately following blood sample collection death was confirmed by cervical dislocation and the brain removed.
Aliquots of each blood sample were diluted with an equal volume of water. Mouse brain samples were weighed, water was added at a 1/2 w/v ratio (brain/water), and then the samples were homogenized. Both blood and brain samples were stored in a freezer set to maintain a temperature of −80 °C until analysis.
Diluted blood and brain homogenates were processed under standard liquid–liquid extraction procedures using acetonitrile containing internal standard and analyzed by LC/MS/MS.
Noncompartmental analysis was performed using Phoenix pharmacokinetic software version 1.4 (Certara), and the main pharmacokinetic parameters were estimated.
Mouse Efficacy Study of Acute HAT
Six female NMRI mice (35–40 g weight from Charles River Laboratories) per group were infected by intraperitoneal (ip) injection with 104 bloodstream forms of T. b. brucei (STIB795) strain in 0.2 mL of TDB glucose on day 0. After 3 days, the infected mice were divided into two groups: control (infected mice treated with vehicle) and 20g (infected mice treated with 10 mg kg–1 day–1). On day 3, all mice received a 0.2 mL ip injection with 10 mg kg–1 day–120g or 5% DMSO, 20% Captisol for control group. This treatment was administered for 5 consecutive days. Parasitemia (measured as parasites per milliliter) was individually checked by direct microscopic counting of parasites in a Neubauer chamber using 2 μL of blood from mouse tail, suspending in 100 μL of TDB glucose. Mortality was checked daily up to 30 days after infection and expressed as a percentage of cumulative mortality.
Chemistry Experimental. General Methods
All starting materials were commercially obtained and were used without further purification, unless specified. Reaction solvents were purified by passage through alumina columns on a purification system manufactured by Innovative Technology (Newburyport, MA). NMR spectra were obtained on Varian NMR systems, operating at 400 or 500 MHz for 1 H acquisitions. LCMS analysis was performed using a Waters AllianceHT e2795 reverse phase HPLC (Waters SunFire C18 4.6 mm × 50 mm, 3.5 μm column), with a Waters 2996 photodiode array detector scanning from 210 to 600 nm, and Waters Micromass ZQ detector (electrospray ionization). Where required, final compounds were purified by preparative reverse phase HPLC (columns Waters Symmetry RP8 30 mm × 50 mm, 5 μm column, or OBD RP18 30 mm × 50 mm, 5 μm), with a single wavelength UV–visible detector and Waters Micromass ZQ (electrospray ionization). All final compounds have purities greater than 95% based upon LCMS analysis. Melting points were obtained on a Thermo Fisher Mel-Temp apparatus. 1H NMR spectra were obtained with Varian NMR systems, operating at either 400 or 500 MHz at room temperature, using solvents from Cambridge Isotope Laboratories. Chemical shifts (δ, ppm) are reported relative to the solvent peak (CDCl3, 7.26 [1H]; DMSO-d6, 2.50 [1H]; acetone-d6, 2.05; or CD3OD, 3.31 [1H]). Data for 1H NMR spectra are reported as follows: chemical shift (ppm), multiplicity (s for singlet, d for doublet, t for triplet, dd for doublet of doublet, m for multiplet), coupling constant (Hz), and integration.
Chemical Synthesis and Characterization. 3-((4-(Pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (1)
In a vial 7a (20 mg, 86 μmol), K3PO4 (26 mg, 121 μmol), XPhos (6 mg, 13 μmol), bis(dibenzylideneacetone)palladium(0) (4 mg, 4.3 μmol), and 3-aminobenzonitrile (20 mg, 173 μmol) were combined. The vial was sealed, evacuated, and backfilled with N2 three times. Degassed and anhydrous toluene (1 mL, dried over molecular sieves and stored under N2) was added to a sealed vial. The reaction was left stirring overnight at 101 °C. The reaction mixture which was heterogeneous was red at room temperature, turned brown and almost clear upon heating. After 16 h, the reaction mixture was evaporated to dryness to get a slurry which was extracted with DCM and washed with water and then dried over sodium sulfate, decanted, and concentrated to get the crude product which was purified by column chromatography using EtOAc/hex 40–100% 12 CV to afford the title compound as a colorless solid (15 mg, 55%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.96 (s, 1 H), 9.11–9.18 (m, 1 H), 8.93 (s, 1 H), 8.64 (dd, J = 4.4, 2.0 Hz, 1 H), 8.55 (d, J = 5.4 Hz, 1 H), 8.38 (br s, 1 H), 7.93–7.98 (m, 1 H), 7.55 (t, J = 8.1 Hz, 1 H), 7.46–7.51 (m, 2 H), 7.43 (d, J = 7.8 Hz, 1 H). LC–MS (m/z): 314.1 [M + H]+.
3-Chloropyridazine (3)
Compound 2 (470 mg, 4.7 mmol) and POCl3 (4.5 mL, 49 mmol) were heated at 80 °C for 2 h. The heterogeneous mixture slowly turned clear and then a dark red/brown color. The reaction mixture was then cooled to room temperature, poured into water, neutralized slowly with sat. NaHCO3, and extracted with DCM. The organic layers were washed with water, brine, dried over sodium sulfate, decanted, and concentrated to afford the crude compound as a brown solid (360 mg, 64%). 1H NMR (500 MHz, methanol-d4) δ ppm 9.16 (dd, J = 4.9, 1.5 Hz, 1 H), 7.84 (dd, J = 8.8, 1.5 Hz, 1 H), 7.74 (dd, J = 8.8, 4.9 Hz, 1 H). LC–MS (m/z): 114.4 (35Cl), 116.5 (37Cl) [M + H]+.
4-(Pyridazin-3-yl)morpholine (4a)
To a solution of 3 (360 mg, 3.1 mmol) in n-BuOH (5 mL) was added morpholine (0.62 mL, 7.8 mmol). The reaction mixture was heated to 80 °C for 16 h, then cooled to room temperature. Ethyl acetate was added to the reaction mixture, and a solid precipitated from solution. The precipitate was filtered and filtrate was concentrated in vacuo, then purified by column chromatography using MeOH/EtOAc, 0–20%, 12 CV to afford the product as a pale-yellow solid (430 mg, 90%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.57 (dd, J = 4.4, 1.0 Hz, 1 H), 7.39 (dd, J = 9.3, 4.4 Hz, 1 H), 7.25 (dd, J = 9.3, 1.0 Hz, 1 H), 3.71 (t, J = 4.9 Hz, 4 H), 3.52 (t, J = 4.9 Hz, 4 H). LC–MS (m/z): 165.8 [M + H]+.
3-Methoxypyridazine (4b)
Compound 3 (2.4 g, 21 mmol) was dissolved in MeOH (150 mL), sodium tert-butoxide (2.01 g, 21 mmol) was added, and the reaction was heated to 65 °C for 16 h. All volatiles were removed in vacuo and then the reaction mixture was taken up in EtOAc and MeOH and filtered to give the product in the filtrate. All volatiles were removed in vacuo to obtain the desired compound as a red oil (2.30 g, 99%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.87 (dd, J = 4.4, 1.5 Hz, 1 H), 7.59 (dd, J = 8.8, 4.4 Hz, 1 H), 7.19 (dd, J = 8.8, 1.5 Hz, 1 H), 4.00 (s, 3 H). LC–MS (m/z): 110.5 [M + H]+.
2-Chloro-4-((trimethylsilyl)ethynyl)pyrimidine (6a)
In a vial 5 (500 mg, 3.36 mmol), dichlorobistriphenylphosphine palladium(II) PdCl2(PPh3)2 (47 mg, 67 μmol), and copper(I) iodide (19 mg, 100 μmol) were added. The vial was evacuated and backfilled with N2 three times. To this, a degassed mixture of triethylamine (1.40 mL, 10.1 mmol) and THF (20 mL) was added followed by ethynyltrimethylsilane (0.525 mL, 3.69 mmol). The resultant solution was stirred at 50 °C overnight. The reaction mixture turned from yellow/orange in color to dark brown after addition of TMS-acetylene. Upon completion, reaction mixture was concentrated to give a dark slurry which was extracted with DCM and washed with water (3 × 50 mL) followed by brine, and the organic layer was dried using Na2SO4, decanted, and concentrated to give the crude product. Column purification was performed using EtOAc/hex, 5% to 50% 12 CV, 50–100% 5 CV to afford the product as a white solid (425 mg, 61%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.55 (d, J = 4.9 Hz, 1 H), 7.28 (d, J = 4.9 Hz, 1 H), 0.23 (s, 9 H). LC–MS (m/z): 210.9 (35Cl), 212.9 (37Cl) [M + H]+.
2-Chloro-4-ethynylpyrimidine (6b)
A solution of 6a (390 mg, 1.85 mmol) in methanol (10 mL) was treated with half of a stock solution of KOH (0.519 mg, 9.25 μmol) in 1 mL of MeOH. After 30 min, another half of KOH solution was added and the mixture was stirred at room temperature; total consumption of the reactant was observed. The mixture was concentrated to get the crude product as buff colored solid. Column purification was done using EtOAc/hexane 10–100%, 15 CV to yield the desired compound as a buff solid (200 mg, 78%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.64 (d, J = 4.9 Hz, 1 H), 7.37 (d, J = 4.9 Hz, 32 H), 3.48 (s, 1 H). LC–MS (m/z): 138.6 (35Cl), 140.6 (37Cl) [M + H]+.
2-Chloro-4-((4-(trifluoromethyl)phenyl)ethynyl)pyrimidine (6c)
In a vial 5 (200 mg, 1.34 mmol), dichlorobistriphenylphosphine palladium(II) PdCl2(PPh3)2 (19 mg, 27 μmol), triethylamine (0.6 mL, 4.03 mmol), and copper(I) iodide (8 mg, 40 μmol) were added followed by 1-ethynyl-4-(trifluoromethyl)benzene (0.244 mL, 1.48 mmol). A degassed mixture of triethylamine (0.6 mL, 4.03 mmol) and THF (5 mL) was then added. The resultant solution was stirred at 50 °C overnight. After completion of reaction column purification was done using EtOAc/hex, 5% to 50% 12 CV, 50–100% 5 CV to isolate the desired compound as an orange solid (191 mg, 50%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.89 (d, J = 5.4 Hz, 1 H), 7.85–7.95 (m, 5 H). LC–MS (m/z): 283.0 (35Cl), 285.0 (37Cl) [M + H]+.
2-Chloro-4-(cyclopropylethynyl)pyrimidine (6d)
In a reaction vial 5 (1.0 g, 6.7 mmol), Pd(PPh3)2Cl2 (94 mg, 134 μmol), and CuI (38 mg, 201 μmol) were added. A degassed solution of triethylamine (2.81 mL, 20.1 mmol) and THF (12 mL) was added under N2 to the reaction vial, followed by ethynylcyclopropane (625 μL, 7.38 mmol). The reaction mixture was heated to 50 °C for 16 h. The reaction mixture was then filtered through Celite and washed with EtOAc. The reaction mixture was concentrated in vacuo and then subjected to column purification EtOAc/hexanes 0–25%, to afford the desired compound as an orange solid (1.03 g, 86%). 1H NMR (400 MHz, chloroform-d) δ ppm 8.52 (d, J = 5.1 Hz, 1 H), 7.20 (d, J = 5.1 Hz, 1 H), 1.47–1.56 (m, 1 H), 0.92–1.06 (m, 4 H). LC–MS (m/z): 178.9 (35Cl), 180.8 (37Cl) [M + H]+.
2-Chloro-4-((4-methoxyphenyl)ethynyl)pyrimidine (6e)
In a vial, 5 (0.3 g, 2.01 mmol), dichlorobistriphenylphosphine palladium(II) PdCl2(PPh3)2 (28 mg, 40 μmol), and copper(I) iodide (11.5 mg, 60 μmol) were added, followed by a degassed mixture of triethylamine (0.84 mL) and THF (5 mL), and then 1-ethynyl-4-methoxybenzene (293 μL, 2.2 mmol) was added. The resultant solution was stirred at 50 °C overnight. The yellow/orange reaction mixture turned dark brown after addition of TMS-acetylene and on heating. The mixture was evaporated to dryness, and then the slurry obtained was dissolved in DCM. The organic layer was washed with water and brine, then dried over sodium sulfate, decanted, and concentrated to get the crude product which was purified by column chromatography using EtOAc/hex, 5% to 50% 12 CV, 50–100% 5 CV to afford the title compound as a brown solid (0.4 g, 86%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.59 (d, J = 5.4 Hz, 1 H), 7.56–7.60 (m, 2 H), 7.36 (d, J = 5.4 Hz, 1 H), 6.91–6.95 (m, 2 H), 3.86 (s, 3 H). LC–MS (m/z): 245.0 (35Cl), 247.0 (37Cl) [M + H]+.
4-(2-Chloropyrimidin-4-yl)-2-methylbut-3-yn-2-ol (6f)
In a reaction vial 5 (2 g, 13.36 mmol), 2-methylbut-3-yn-2-ol (1.56 mL, 16 mmol), Pd(PPh3)2Cl2 (94 mg, 134 μmol), and CuI (51 mg, 269 μmol) were added. A degassed solution of TEA (9.4 mL) and THF (50 mL) was added under N2 to the reaction vial. The reaction mixture was heated to 60 °C for 16 h. The reaction was then poured into water and extracted with DCM. The organic layer was washed with water and brine, then dried over sodium sulfate, decanted, concentrated to get the crude product, which was purified by column chromatography using EtOAc/hex 30–100% 15 CV affording the desired compound as a yellow solid (1.60 g, 60%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.78 (d, J = 5.4 Hz, 1 H), 7.61 (d, J = 5.4 Hz, 1 H), 5.79 (s, 1 H), 1.48 (s, 6 H). LC–MS (m/z): 196.9 (35Cl), 198.9 (37Cl) [M + H]+.
3-(2-Chloropyrimidin-4-yl)prop-2-yn-1-ol (6g)
In a reaction vial 5 (1.0 g, 6.7 mmol), Pd(PPh3)2Cl2 (94 mg, 134 μmol), and CuI (38 mg, 201 μmol) were added. A degassed solution of triethylamine (3.74 mL, 26.9 mmol) and THF (20 mL) was added under N2 to the reaction vial, followed by propargyl alcohol (464 μL, 8.06 mmol). The reaction mixture was heated to 50 °C for 16 h. The reaction was then filtered through Celite and washed with methanol. The reaction mixture was concentrated in vacuo, then subjected to column purification with MeOH/DCM 0–5%, to yield the desired compound as a brown solid (420 mg, 37%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.79 (d, J = 5.4 Hz, 1 H), 7.64 (d, J = 5.4 Hz, 1 H), 5.60 (d, J = 5.9 Hz, 1 H), 4.39 (t, J = 5.9 Hz, 2 H). LC–MS (m/z): 168.8 (35Cl), 170.7 (37Cl) [M + H]+.
2-Chloro-4-(3-methoxyprop-1-yn-1-yl)pyrimidine (6h)
In a reaction vial 5 (500 mg, 3.36 mmol), Pd(PPh3)2Cl2 (47 mg, 67 μmol), and CuI (19 mg, 101 μmol) were added. A degassed solution of triethylamine (1.87 mL, 13.4 mmol) and THF (10 mL) was added under N2 to the reaction vial, followed by 3-methoxyprop-1-yne (340 μL, 4.03 mmol). The reaction mixture was heated to 50 °C for 16 h. The reaction mixture was then filtered through Celite and concentrated in vacuo and subjected to column purification EtOAc/hexanes 10–50%, to afford the title compound as a yellow solid (183 mg, 30%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.62 (d, J = 5.4 Hz, 1 H), 7.33 (d, J = 5.4 Hz, 1 H), 4.37 (s, 2 H), 3.47 (s, 3 H). LC–MS (m/z): 182.8 (35Cl), 184.8 (37Cl) [M + H]+.
4-(2-Chloropyrimidin-4-yl)but-3-yn-2-ol (6i)
In a reaction vial 5 (500 mg, 3.4 mmol), Pd(PPh3)2Cl2 (47 mg, 67 μmol), and CuI (19 mg, 100 μmol) were added. A degassed solution of triethylamine (1.9 mL, 13 mmol) and THF (10 mL) was added under N2 to the reaction vial, followed by but-3-yn-2-ol (320 μL, 4.0 mmol). The reaction mixture was heated to 50 °C for 16 h. The reaction mixture was then filtered through Celite and washed with MeOH, then concentrated in vacuo and subjected to column purification EtOAc/hexanes 20–50%, to yield the desired compound as a red oil (292 mg, 48%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.61 (d, J = 4.9 Hz, 1 H), 7.31 (d, J = 4.9 Hz, 1 H), 4.76–4.85 (m, 1 H), 1.59 (d, J = 6.4 Hz, 3 H). LC–MS (m/z): 182.9 (35Cl), 184.8 (37Cl) [M + H]+.
2-Chloro-4-(3-methylbut-1-yn-1-yl)pyrimidine (6j)
In a reaction vial 5 (500 mg, 3.36 mmol), Pd(PPh3)2Cl2 (47 mg, 67 μmol), and CuI (19 mg, 101 μmol) were added. A degassed solution of triethylamine (1.87 mL, 13.4 mmol) and THF (10 mL) was added under N2 to the reaction vial, followed by 3-methylbut-1-yne (411 μL, 4.03 mmol). The reaction mixture was heated to 50 °C for 16 h. The reaction mixture was then filtered through Celite and washed with EtOAc and methanol, then concentrated in vacuo and subjected to column purification EtOAc/hexanes 0–25%, to afford the title compound as a yellow solid (487.1 mg, 80%). 1H NMR (400 MHz, chloroform-d) δ ppm 8.55 (d, J = 5.1 Hz, 1 H), 7.25 (d, J = 5.1 Hz, 1 H), 2.76–2.94 (m, 1 H), 1.30 (d, J = 7.3 Hz, 6 H). LC–MS (m/z): 180.9 (35Cl), 182.8 (37Cl) [M + H]+.
3-(2-Chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (7a)
(Aminooxy)sulfonic acid (906 mg, 8.01 mmol) was dissolved in water (5 mL). To the clear colorless solution 2.5 M sodium bicarbonate was added until effervescence ceased (pH ∼ 5). The clear colorless solution was heated at 70 °C, and pyridazine (0.385 mL, 5.34 mmol) was added, and then the reaction was stirred at 70 °C for 2 h. The reaction mixture then cooled to room temperature, neutralized with 2.5 M sodium bicarbonate (until effervescence ceased), and then a solution of 6b (185 mg, 1.34 mmol) in DCM (10 mL) was added followed by KOH (22 mg, 3.9 mmol). Reaction mixture was stirred overnight at room temperature. After completion the reaction mixture was extracted in DCM, washed with water and brine, then dried over sodium sulfate, decanted, and concentrated to get the crude product. Column purification was performed using EtOAc/hex 30–100% 15 CV to afford the compound as a red solid (260 mg, 84%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.04 (s, 1 H), 8.90 (dd, J = 8.8, 2.0 Hz, 1 H), 8.73 (d, J = 5.4 Hz, 1 H), 8.69 (dd, J = 4.4, 2.0 Hz, 1 H), 8.07 (d, J = 5.4 Hz, 1 H), 7.58 (dd, J = 8.8, 4.4 Hz, 1 H). LC–MS (m/z): 231.9 (35Cl), 234.0 (37Cl) [M + H]+.
4-(3-(2-Chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazin-6-yl)morpholine (7b)
(Aminooxy)sulfonic acid (421 mg, 3.72 mmol) was dissolved in 3 mL of water, and to the clear solution was added 2.5 M sodium bicarbonate (376 mg, 4.47 mmol) until effervescence ceased (pH ∼ 5). The clear colorless solution was heated at 70 °C, and 4a (462 mg, 2.79 mmol) was added and the reaction stirred at 70 °C for 2 h. The reaction mixture was then cooled to room temperature, neutralized with 2.5 M sodium bicarbonate (until effervescence ceased). A solution of 6b (129 mg, 931 μmol) in DCM (10 mL) was added followed by KOH (104 mg, 1.9 mmol), and the reaction mixture was stirred at room temperature for 16 h. DCM layer turned dark pink. Reaction mixture was then extracted into DCM, and the organic layer was washed with water and brine, then dried over sodium sulfate, decanted, and concentrated to get the crude product, which was purified by column chromatography using EtOAc/hex, 20–100% 15 CV, to obtain the desired compound as a white solid (160 mg, 5%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.77 (d, J = 9.8 Hz, 1 H), 8.51 (d, J = 5.4 Hz, 1 H), 8.29 (s, 1 H), 7.45 (d, J = 5.4 Hz, 1 H), 7.03 (d, J = 9.8 Hz, 1 H), 3.88 (t, J = 4.9 Hz, 4 H), 3.62 (t, J = 4.9 Hz, 4 H). LC–MS (m/z): 317.0 (35Cl), 318.9 (37Cl) [M + H]+.
3-(2-Chloropyrimidin-4-yl)-6-methoxypyrazolo[1,5-b]pyridazine (7c)
(Aminooxy)sulfonic acid (1.90 g, 16.8 mmol) was dissolved in 4 mL of water, and to the clear solution was added saturated sodium bicarbonate until effervescence ceased (pH ∼ 6). The clear colorless solution was heated at 70 °C, and 4b (1.24 g, 11.2 mmol) was added in 20 mL of water, and the reaction mixture was stirred at 70 °C for 2 h. The reaction mixture was then cooled to room temperature, neutralized with saturated sodium bicarbonate (until effervescence ceased), and then a solution of 6b (622 mg, 4.49 mmol) in DCM (80 mL) and KOH (252 mg, 4.49 mmol) were added, and the reaction mixture was stirred at room temperature for 16 h. The reaction mixture was diluted with DCM and water and extracted. The organic layer was washed with water and brine, then dried over sodium sulfate, filtered, and evaporated to get the crude product which was purified by column chromatography using EtOAc/hex 20–50% to provide the desired product as a brown solid (496 mg, 42%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.82 (s, 1 H), 8.73 (d, J = 9.3 Hz, 1 H), 8.70 (d, J = 5.4 Hz, 1 H), 8.01 (d, J = 5.4 Hz, 1 H), 7.27 (d, J = 9.3 Hz, 1 H), 4.02 (s, 3 H). LC–MS (m/z): 262.0 (35Cl), 264.1 (37Cl) [M + H]+.
3-(2-Chloropyrimidin-4-yl)-2-(4-(trifluoromethyl)phenyl)pyrazolo[1,5-b]pyridazine (7d)
(Aminooxy)sulfonic acid (559 mg, 4.95 mmol) was dissolved in 5 mL of water, and to the clear solution was added 2.5 M sodium bicarbonate until effervescence ceased (pH ∼ 5). The clear colorless solution was heated to 70 °C, and pyridazine (0.24 mL, 3.30 mmol) was added, and the reaction mixture was stirred at 70 °C for 2 h. Reaction mixture was cooled to room temperature, neutralized with 2.5 M sodium bicarbonate (until effervescence ceased), then solution of 6c (223 mg, 824 μmol) in DCM (4 mL) and KOH (138 mg, 2.46 mmol) were added, and the reaction mixture was then stirred overnight at room temperature. After 16 h the reaction mixture was extracted in DCM, washed the DCM layer with water, brine and dried over sodium sulfate, decanted, and concentrated to get the crude product which was purified by column chromatography using EtOAc/hex, 10–100% 15 CV to afford the title compound as a bronze solid (150 mg, 47%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.78 (dd, J = 9.3, 2.0 Hz, 1 H), 8.74 (dd, J = 4.4, 2.0 Hz, 1 H), 8.62 (d, J = 5.4 Hz, 1 H), 7.91 (s, 4 H), 7.60 (dd, J = 9.3, 4.4 Hz, 1 H), 7.23 (d, J = 5.4 Hz, 1 H). LC–MS (m/z): 314.9 (35Cl), 317.0 (37Cl) [M + H]+.
3-(2-Chloropyrimidin-4-yl)-2-cyclopropylpyrazolo[1,5-b]pyridazine (7e)
(Aminooxy)sulfonic acid (1.63 g, 14.4 mmol) was dissolved in 4 mL of water, and saturated sodium bicarbonate was added to the clear solution until effervescence ceased (pH ∼ 6). The clear colorless solution was heated to 70 °C, and pyridazine (693 μL, 9.60 mmol) was added, and the reaction mixture was stirred at 70 °C for 2 h. The reaction mixture was then cooled to room temperature, neutralized with saturated sodium bicarbonate (until effervescence ceased), then a solution of 6d (429 mg, 2.4 mmol) in DCM (7 mL) was added followed by KOH (135 mg, 2.4 mmol), and the reaction was stirred for 16 h at room temperature. Partial conversion to product was observed, so additional KOH (270 mg, 4.8 mmol) was added. The reaction was stirred for another 8 h. After 8 h another 2 equiv of KOH (270 mg, 4.8 mmol) was added, and the reaction continued stirring for an additional 16 h. The reaction mixture was diluted with DCM and washed with water and brine, then dried over magnesium sulfate, filtered, and evaporated to get the crude product which was purified by column chromatography using EtOAc/hex 20–50% and providing the desired compound as a pale-yellow solid (472.5 mg, 72%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.92 (dd, J = 8.8, 2.0 Hz, 1 H), 8.61 (d, J = 5.4 Hz, 1 H), 8.38 (dd, J = 4.4, 2.0 Hz, 1 H), 7.87 (d, J = 5.4 Hz, 1 H), 7.24 (dd, J = 8.8, 4.4 Hz, 1 H), 2.23–2.31 (m, 1 H), 1.29–1.35 (m, 2 H), 1.17–1.23 (m, 2 H). LC–MS (m/z): 272.1 (35Cl), 274.1 (37Cl) [M + H]+.
3-(2-Chloropyrimidin-4-yl)-2-(4-methoxyphenyl)pyrazolo[1,5-b]pyridazine (7f)
(Aminooxy)sulfonic acid (1.11 g, 9.81 mmol) was dissolved in 5 mL of water, and to the clear solution 2.5 M sodium bicarbonate was added until effervescence ceased (pH ∼ 5). The clear colorless solution was heated to 70 °C, and pyridazine (0.471 mL, 6.54 mmol) was added, then the reaction mixture was stirred at 70 °C for 2 h. The reaction cooled to room temperature, neutralized with 2.5 M sodium bicarbonate (until effervescence ceased), and then a solution of 6e (0.4 g, 1.63 mmol) in DCM (20 mL) was added followed by KOH (275 mg, 4.9 mmol). The reaction mixture was stirred at room temperature for 16 h, then diluted with DCM, washed with water and brine, then dried over sodium sulfate, decanted, and concentrated to get the crude product, which was purified by column chromatography using EtOAc/hex, 10–100% 15 CV to provide the title compound as an off-white solid (35 mg, 46% based on recovered starting material). Recovered 345 mg of unreacted starting material. 1H NMR (500 MHz, chloroform-d) δ ppm 8.96 (dd, J = 8.8, 2.0 Hz, 1 H), 8.46 (dd, J = 4.9, 2.0 Hz, 1 H), 8.34 (d, J = 5.4 Hz, 1 H), 7.58–7.61 (m, 2 H), 7.13 (d, J = 5.4 Hz, 1 H), 7.02–7.05 (m, 2 H), 6.89–6.95 (m, 1 H), 3.91 (s, 3 H). LC–MS (m/z): 338.0 (35Cl), 339.9 (37Cl) [M + H]+.
2-(3-(2-Chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazin-2-yl)propan-2-ol (7g)
(Aminooxy)sulfonic acid (280 mg, 2.47 mmol) was dissolved in 5 mL of water, and to the clear solution 2.5 M sodium bicarbonate was added until effervescence ceased (pH ∼ 5). The clear colorless solution was heated to 70 °C, and pyridazine (119 μL, 1.65 mmol) was added, and the reaction mixture was stirred at 70 °C for 2 h. The reaction was then cooled to room temperature, neutralized with 2.5 M sodium bicarbonate (until effervescence ceased), and then a solution of 6f (81 mg, 412 μmol) in DCM (10 mL) was added followed by KOH (69 mg, 1.2 mmol), and the reaction mixture was stirred at room temperature. After 16 h the reaction mixture was diluted with DCM and washed with water and brine, then dried over sodium sulfate, decanted, and concentrated to get the crude product which was purified by column chromatography using EtOAc/hex 30–100% 15 CV to afford the title compound as a brown solid (47 mg, 40%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.77 (d, J = 5.4 Hz, 1 H), 8.73 (dd, J = 9.3, 1.5 Hz, 1 H), 8.62 (dd, J = 4.4, 1.5 Hz, 1 H), 8.54 (d, J = 5.4 Hz, 1 H), 7.49 (dd, J = 9.3, 4.4 Hz, 1 H), 5.95 (s, 1 H), 1.64 (s, 6 H). LC–MS (m/z): 290.0 (35Cl), 292.1 (37Cl) [M + H]+.
(3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazin-2-yl)methanol (7h)
(Aminooxy)sulfonic acid (450 mg, 4.0 mmol) was dissolved in 3 mL of water, and to the clear solution 2.5 M sodium bicarbonate was added until effervescence ceased (pH ∼ 5). The clear colorless solution was heated to 70 °C, and pyridazine (190 μL, 2.7 mmol) was added, and the reaction mixture was stirred at 70 °C for 2 h. The reaction was then cooled to room temperature, neutralized with 2.5 M sodium bicarbonate (until effervescence ceased), and then a solution of 6g (110 mg, 670 μmol) in DCM (5 mL) was added followed by KOH (40 mg, 670 μmol), and the reaction mixture was stirred at room temperature. After 16 h the reaction mixture was diluted with DCM and washed with water and brine, then dried over sodium sulfate. The reaction was filtered and evaporated in vacuo, then subjected to column purification using MeOH/DCM 2% to afford the title compound as an orange solid (34 mg, 20%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.87 (dd, J = 8.8, 2.0 Hz, 1 H), 8.78 (d, J = 5.4 Hz, 1 H), 8.67 (dd, J = 4.4, 2.0 Hz, 1 H), 8.05 (d, J = 5.4 Hz, 1 H), 7.56 (dd, J = 8.8, 4.4 Hz, 1 H), 5.76 (t, J = 5.4 Hz, 1 H), 4.89 (d, J = 5.4 Hz, 1 H). LC–MS (m/z): 262.0 (35Cl), 264.0 (37Cl) [M + H]+.
3-(2-Chloropyrimidin-4-yl)-2-(methoxymethyl)pyrazolo[1,5-b]pyridazine (7i)
(Aminooxy)sulfonic acid (680 mg, 6.01 mmol) was dissolved in 5 mL of water, and saturated sodium bicarbonate was added to the clear solution until effervescence ceased (pH ∼ 6). The clear colorless solution was heated at 70 °C, and pyridazine (289 μL, 4.01 mmol) was added, and the reaction was stirred at 70 °C for 2 h. Reaction mixture was then cooled to room temperature, neutralized with saturated sodium bicarbonate (until effervescence ceased), and then a solution of 6h (183 mg, 1.0 mmol) in DCM (10 mL) was added followed by KOH (112 mg, 2.0 mmol), and the reaction was stirred for 16 h at room temperature. The reaction mixture was extracted in DCM washed with water and brine, then dried over magnesium sulfate, filtered, and evaporated to get the crude product which was purified by column chromatography using EtOAc/hex 20–50% to yield the title compound as a white solid (229 mg, 83%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.86 (dd, J = 9.3, 2.0 Hz, 1 H), 8.79 (d, J = 5.4 Hz, 1 H), 8.69 (dd, J = 4.4, 2.0 Hz, 1 H), 7.85 (d, J = 5.4 Hz, 1 H), 7.57 (dd, J = 9.3, 4.4 Hz, 1 H), 4.86 (s, 2 H), 3.37 (s, 3 H). LC–MS (m/z): 276.0 (35Cl), 278.0 (37Cl) [M + H]+.
1-(3-(2-Chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazin-2-yl)ethan-1-ol (7j)
(Aminooxy)sulfonic acid (1.1 g, 9.6 mmol) was dissolved in 5 mL of water, and saturated sodium bicarbonate was added to the clear solution until effervescence ceased (pH ∼ 6). The clear colorless solution was heated at 70 °C, and pyridazine (462 μL, 6.4 mmol) was added, and the reaction mixture was stirred at 70 °C for 2 h. Reaction mixture was then cooled to room temperature, neutralized with saturated sodium bicarbonate (until effervescence ceased), and then a solution of 6i (292 mg, 1.6 mmol) in DCM (15 mL) was added followed by KOH (180 mg, 3.2 mmol), and the reaction was stirred for 16 h at room temperature. The reaction mixture was diluted with DCM and washed with water and brine, then dried over magnesium sulfate, filtered, and evaporated to get the crude product which was purified by column chromatography using EtOAc/hex 45–80% to provide the title compound as a yellow solid (290 mg, 66%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.81 (d, J = 9.3 Hz, 1 H), 8.76 (d, J = 5.4 Hz, 1 H), 8.63–8.67 (m, 1 H), 8.18 (d, J = 5.4 Hz, 1 H), 7.52 (dd, J = 9.3, 4.4 Hz, 1 H), 5.76 (d, J = 6.4 Hz, 1 H), 5.25 (quin, J = 6.4 Hz, 1 H), 1.62 (d, J = 6.4 Hz, 3 H). LC–MS (m/z): 276.0 (35Cl), 278.0 (37Cl) [M + H]+.
3-(2-Chloropyrimidin-4-yl)-2-isopropylpyrazolo[1,5-b]pyridazine (7k)
(Aminooxy)sulfonic acid (1.83 g, 16.2 mmol) was dissolved in 5 mL of water, and saturated sodium bicarbonate was added to the clear solution until effervescence ceased (pH ∼ 6). The clear colorless solution was heated to 70 °C, and pyridazine (778 μL, 10.8 mmol) was added, and the reaction mixture was stirred at 70 °C for 2 h. The reaction mixture was then cooled to room temperature, neutralized with saturated sodium bicarbonate (until effervescence ceased), and then a solution of 6j (487 mg, 2.7 mmol) in DCM (7 mL) was added followed by KOH (151 mg, 2.70 mmol), and the reaction was stirred for 16 h at room temperature, at which point more KOH (302 mg, 5.4 mmol) was added and stirring was continued for another 3 h. The reaction mixture was diluted with DCM, washed with water and brine, then dried over magnesium sulfate, filtered, and evaporated to get the crude product which was purified by column chromatography using EtOAc/hex 20–50% to afford the title compound as a yellow solid (471 mg, 64%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.87 (d, J = 9.3 Hz, 1 H), 8.60 (d, J = 5.4 Hz, 1 H), 8.39 (d, J = 4.4 Hz, 1 H), 7.47 (d, J = 5.4 Hz, 1 H), 7.24 (dd, J = 9.3, 4.4 Hz, 1 H), 3.54–3.64 (m, 1 H), 1.51 (d, J = 6.4 Hz, 6 H). LC–MS (m/z): 274.1 (35Cl), 276.0 (37Cl) [M + H]+.
(3-(2-Chloropyrimidin-4-yl)-6-methoxypyrazolo[1,5-b]pyridazin-2-yl)methanol (7l)
(Aminooxy)sulfonic acid (909 mg, 8.0 mmol) was dissolved in 4 mL of water, and saturated sodium bicarbonate was added to the clear solution until effervescence ceased (pH ∼ 6). The clear colorless solution was heated at 70 °C, and 4b (590 mg, 5.36 mmol) was added, then the reaction was stirred at 70 °C for 2 h. The reaction mixture was then cooled to room temperature, neutralized with saturated sodium bicarbonate (until effervescence ceased), and then a solution of 6g (452 mg, 2.68 mmol) in DCM (15 mL) was added followed by KOH (150 mg, 2.7 mmol), and the reaction mixture was stirred for 16 h at room temperature. The reaction mixture was diluted with DCM and washed with water and brine, then dried over sodium sulfate, filtered, and evaporated to get the crude product which was purified by column chromatography using EtOAc/hex 25–50% to afford the title compound as an orange solid (265 mg, 34%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.76 (d, J = 5.4 Hz, 1 H), 8.72 (d, J = 9.8 Hz, 1 H), 8.01 (d, J = 5.4 Hz, 1 H), 7.25 (d, J = 9.8 Hz, 1 H), 5.69 (t, J = 5.4 Hz, 1 H), 4.81 (d, J = 5.4 Hz, 2 H), 4.00 (s, 3 H). LC–MS (m/z): 292.0 (35Cl), 294.0 (37Cl) [M + H]+.
(3-(2-Chloropyrimidin-4-yl)-6-morpholinopyrazolo[1,5-b]pyridazin-2-yl)methanol (7m)
(Aminooxy)sulfonic acid (590 mg, 5.2 mmol) was dissolved in 5 mL of water, and saturated sodium bicarbonate was added to the clear solution until effervescence ceased (pH ∼ 6). The clear colorless solution was heated to 70 °C, and 4a (570 mg, 3.5 mmol) was added, and the reaction mixture was stirred at 70 °C for 2 h. Reaction mixture was then cooled to room temperature, neutralized with saturated sodium bicarbonate (until effervescence ceased), and then a solution of 6g (290 mg, 5.2 mmol) in DCM (15 mL) was added followed by KOH (98 mg, 1.7 mmol), and the reaction mixture was stirred for 16 h at room temperature. The reaction mixture was diluted with DCM and washed with water and brine, then dried over sodium sulfate, filtered, and evaporated to get the crude product which was purified by column chromatography run isocratically with 100% EtOAc, to afford the title compound as a yellow solid (88.1 mg, 15%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.71 (d, J = 5.4 Hz, 1 H), 8.58 (d, J = 9.8 Hz, 1 H), 7.97 (d, J = 5.4 Hz, 1 H), 7.52 (d, J = 9.8 Hz, 1 H), 5.62 (t, J = 5.4 Hz, 1 H), 4.78 (d, J = 5.4 Hz, 2 H), 3.76 (t, J = 4.9 Hz, 4 H), 3.52 (t, J = 4.9 Hz, 4 H). LC–MS (m/z): 347.1 (35Cl), 349.0 (37Cl) [M + H]+.
3-(2-Chloropyrimidin-4-yl)-2-cyclopropyl-6-methoxypyrazolo[1,5-b]pyridazine (7n)
(Aminooxy)sulfonic acid (380 mg, 3.36 mmol) was dissolved in 4 mL of water, and to the clear solution saturated sodium bicarbonate was added until effervescence ceased (pH ∼ 6). The clear colorless solution was heated to 70 °C, and 4b (247 mg, 2.24 mmol) was added, and the reaction mixture was stirred at 70 °C for 2 h. The reaction mixture was then cooled to room temperature, neutralized with saturated sodium bicarbonate (until effervescence ceased), and then a solution of 2-chloro-4-(cyclopropylethynyl)pyrimidine (200 mg, 1.1 mmol) in DCM (10 mL) was added followed by KOH (63 mg, 1.1 mmol), and the reaction was stirred for 16 h at room temperature. Partial conversion to product was observed, so additional KOH (63 mg, 1.1 mmol) was added, and the reaction was stirred for another 24 h. The reaction mixture was diluted with DCM and washed with water and brine, then dried over sodium sulfate, filtered, and evaporated to get the crude product which was purified by column chromatography using EtOAc/hex 10–30% to yield the desired compound as a white solid (99.1 mg, 29%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.73 (d, J = 5.4 Hz, 1 H), 8.63 (d, J = 9.8 Hz, 1 H), 7.99 (d, J = 5.4 Hz, 1 H), 7.20 (d, J = 9.8 Hz, 1 H), 3.98 (s, 3 H), 2.40–2.46 (m, 1 H), 1.09–1.14 (m, 2 H), 1.00–1.05 (m, 2 H). LC–MS (m/z): 302.1 (35Cl), 304.1 (37Cl) [M + H]+.
4-(3-(2-Chloropyrimidin-4-yl)-2-cyclopropylpyrazolo[1,5-b]pyridazin-6-yl)morpholine (7o)
(Aminooxy)sulfonic acid (439 mg, 3.89 mmol) was dissolved in 4 mL of water, and saturated sodium bicarbonate was added to the clear solution until effervescence ceased (pH ∼ 5). The clear colorless solution was heated to 70 °C, and 4a (428 mg, 2.59 mmol) was added, and the reaction mixture was stirred at 70 °C for 2 h. The reaction mixture was then cooled to room temperature, neutralized with saturated sodium bicarbonate (until effervescence ceased), and then a solution of 6d (231 mg, 1.3 mmol) in DCM (20 mL) was added followed by KOH (73 mg, 1.3 mmol), and the reaction was stirred for 16 h at room temperature. The reaction mixture was diluted with DCM and washed with water and brine, then dried over magnesium sulfate, filtered, and evaporated to get the crude product which was purified by column chromatography using EtOAc/hex 10–50% to provide the desired compound as a yellow solid (183.7 mg, 40%). 1H NMR (400 MHz, chloroform-d) δ ppm 8.71 (d, J = 9.5 Hz, 1 H), 8.55 (d, J = 5.1 Hz, 1 H), 7.84 (d, J = 5.1 Hz, 1 H), 6.98 (d, J = 9.5 Hz, 1 H), 3.86 (t, J = 5.1 Hz, 4 H), 3.58 (t, J = 5.1 Hz, 4 H), 2.13–2.23 (m, 1 H), 1.20–1.28 (m, 2 H), 1.08–1.18 (m, 2 H). LC–MS (m/z): 357.1 (35Cl), 359.0 (37Cl) [M + H]+.
N-(4-Chlorobenzyl)-4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (10a)
In a reaction vial 7a (20 mg, 86 μmol) was stirred in n-BuOH (1 mL), and 4-chlorobenzylamine (25 mg, 170 μmol) was added. The reaction mixture was stirred at 115 °C for 16 h. The solution changed from a red/brown color to clear yellow on heating. Upon completion of the reaction, the mixture was concentrated and taken up in DCM and washed with water and brine, then dried over sodium sulfate, decanted, and concentrated to get the crude product, which was purified by column chromatography using EtOAc/hexane 20–100%, 12 CV to yield the product as a colorless solid (7 mg, 24%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.46 (s, 1 H), 8.35 (dd, J = 4.2, 1.7 Hz, 1 H), 8.31 (d, J = 5.4 Hz, 1 H), 7.30–7.41 (m, 4 H), 6.99–7.07 (m, 1 H), 6.94 (d, J = 5.4 Hz, 1 H), 5.54–5.68 (m, 1 H), 4.70 (d, J = 5.9 Hz, 2 H). LC–MS (m/z): 337.0 (35Cl), 339.1 (37Cl) [M + H]+.
N-(3-Morpholinopropyl)-4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (10b)
In a microwave vial containing 7a (30 mg, 129 μmol), EtOH (1 mL) was added followed by 3-morpholinopropan-1-amine (37 mg, 260 μmol), and the reaction mixture was subjected to microwave irradiation at 150 °C for 15 min. Upon completion of the reaction, the reaction mixture was concentrated and taken up in DCM which was washed with water and brine, then dried over sodium sulfate, decanted, and concentrated to get the crude product, which was purified by column chromatography using EtOAc/MeOH 0–20%, 12 CV to yield the product as a dark yellow solid (15 mg, 34%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.97 (dd, J = 9.3, 2.0 Hz, 1 H), 8.45 (s, 1 H), 8.37 (dd, J = 4.4, 2.0 Hz, 1 H), 8.27 (d, J = 4.9 Hz, 1 H), 7.16 (dd, J = 9.3, 4.4 Hz, 1 H), 6.86 (d, J = 4.9 Hz, 1 H), 6.01 (br s, 1 H), 3.79 (t, J = 4.2 Hz, 4 H), 3.59 (q, J = 6.3 Hz, 2 H), 2.57–2.65 (m, 6 H), 1.91 (quin, J = 6.4 Hz, 2 H). LC–MS (m/z): 340.1 [M + H]+.
N-Cyclopropyl-4-(6-morpholinopyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (10d)
In a vial 7b (15 mg, 47 μmol) and cyclopropylamine (1.0 mL, 14 mmol) were stirred in 1 mL of n-BuOH. Reaction mixture was subjected to microwave irradiation at 150 °C for 1 h. The reaction mixture was then evaporated to dryness and column purification performed using EtOAc/hex 50–100% 12 CV followed by EtOAc/MeOH 1–20% 5 CV to afford the title compound as a brown solid (8.9 mg, 55%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.83–8.91 (m, 1 H), 8.28 (d, J = 4.9 Hz, 1 H), 8.23 (s, 1 H), 6.92 (d, J = 9.3 Hz, 1 H), 6.90 (d, J = 4.9 Hz, 1 H), 5.38 (br s, 1 H), 3.87 (t, J = 4.9 Hz, 4 H), 3.59 (t, J = 4.9 Hz, 4 H), 2.82–2.87 (m, 1 H), 0.85–0.90 (m, 2 H), 0.62–0.66 (m, 2 H). LC–MS (m/z): 338.1 [M + H]+.
N-Phenyl-4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (10e)
An oven-dried flask containing 7a (10 mg, 43 μmol) in DMSO (0.1 mL) was heated until the solution became clear, then n-BuOH (0.3 mL) and aniline (8 μL, 86 μmol) were added to the flask. The reaction mixture was heated to 120 °C for 16 h. After completion of reaction, the mixture was evaporated to dryness and column purification was performed using EtOAc/hex 20–100% 15 CV to obtain the product as an orange solid (10 mg, 80%). Mp = 207–209 °C. 1H NMR (500 MHz, chloroform-d) δ ppm 8.91 (dd, J = 9.3, 2.0 Hz, 1 H), 8.52 (s, 1 H), 8.43 (d, J = 5.4 Hz, 1 H), 8.40 (dd, J = 4.4, 2.0 Hz, 1 H), 7.61–7.65 (m, 2 H), 7.37–7.43 (m, 2 H), 7.18 (br s, 1 H), 7.11–7.16 (m, 2 H), 7.08 (d, J = 5.37 Hz, 1 H). LC–MS (m/z): 289.3 [M + H]+.
N-Phenyl-4-(2-(4-(trifluoromethyl)phenyl)pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (10f)
To an oven-dried flask containing a solution of 7d (20 mg, 53 μmol) in n-BuOH (1 mL), aniline (10 μL, 106 μmol) was added, and the reaction was heated to 110 °C for 16 h. After completion of reaction, reaction mixture was evaporated to dryness and column purification was performed using EtOAc/hex, 20–100% 12 CV to afford the title compound as a colorless solid (20 mg, 87%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.69 (dd, J = 9.3, 2.0 Hz, 1 H), 8.43 (dd, J = 4.4, 2.0 Hz, 1 H), 8.26 (d, J = 5.4 Hz, 1 H), 7.87 (d, J = 8.3 Hz, 2 H), 7.74 (d, J = 8.3 Hz, 2 H), 7.59–7.62 (m, 2 H), 7.33–7.37 (m, 2 H), 7.09–7.16 (m, 2 H), 6.60 (d, J = 5.4 Hz, 1 H). LC–MS (m/z): 433.1 [M + H]+.
4-(Pyrazolo[1,5-b]pyridazin-3-yl)-N-(o-tolyl)pyrimidin-2-amine (10g)
To an oven-dried flask containing o-toluidine (19 μL, 173 μmol) and 7a (20 mg, 86 μmol), n-BuOH was added. The reaction mixture was heated at 100 °C for 16 h. After completion of reaction, the mixture was evaporated to dryness and column purification was performed using EtOAc/hex 20–100% 15 CV to yield the title compound as an off-white solid (7 mg, 27%). Mp = 177–178 °C. 1H NMR (500 MHz, chloroform-d) δ ppm 8.64 (dd, J = 9.3, 2.0 Hz, 1 H), 8.49 (s, 1 H), 8.40 (d, J = 5.4 Hz, 1 H), 8.35 (dd, J = 4.4, 2.0 Hz, 1 H), 7.82 (d, J = 7.8 Hz, 1 H), 7.28–7.32 (m, 2 H), 7.16 (td, J = 7.3, 1.5 Hz, 1 H), 7.01–7.05 (m, 2 H), 6.86 (br s, 1 H), 2.36 (s, 3 H). LC–MS (m/z): 303.3 [M + H]+.
N-(3-Nitrophenyl)-4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (10h)
In a vial 7a (50 mg, 216 μmol) and 3-nitroaniline (52 mg, 540 μmol) were stirred in n-BuOH (1.5 mL), and the reaction mixture was subjected to microwave irradiation at 150 °C for 2 h. After completion of reaction, the mixture was filtered and washed with MeOH. The precipitate was purified using column chromatography using EtOAc/hex, 50–100%, to yield the title compound as a yellow solid (52 mg, 72%). 1H NMR (500 MHz, DMSO-d6) δ ppm 10.14 (s, 1 H), 9.15–9.21 (m, 1 H), 8.98 (s, 1 H), 8.92 (t, J = 2.4 Hz, 1 H), 8.66 (dd, J = 4.4, 2.0 Hz, 1 H), 8.59 (d, J = 5.4 Hz, 1 H), 8.11 (dd, J = 8.3, 2.0 Hz, 1 H), 7.84 (dd, J = 8.3, 2.0 Hz, 1 H), 7.64 (t, J = 8.3 Hz, 1 H), 7.53 (d, J = 5.4 Hz, 1 H), 7.49 (dd, J = 8.8, 4.4 Hz, 1 H). LC–MS (m/z): 334.0 [M + H]+.
N-(3-Methoxyphenyl)-4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (10i)
To an oven-dried flask containing 3-methoxyaniline (22 μL, 194 μmol), a solution of 7a (30 mg, 130 μmol) and acetic acid (1 mL) in dioxane (2 mL) was added. The reaction mixture was stirred at 110 °C for 23 h and monitored by TLC. The reaction mixture appeared dark red/brown, and some solid particles were observed. Upon completion, the mixture was extracted with DCM (3 × 100 mL). The combined organic phase was washed with brine and dried over sodium sulfate. Column purification was performed by preparative reverse phase HPLC using 5–100% ACN/water system to afford the pure product as white solid (15 mg, 36%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.57 (s, 1 H), 9.17–9.24 (m, 1 H), 8.90 (s, 1 H), 8.62 (dd, J = 4.4, 2.0 Hz, 1 H), 8.49 (d, J = 5.4 Hz, 1 H), 7.43–7.49 (m, 2 H), 7.39 (d, J = 5.4 Hz, 1 H), 7.33 (d, J = 8.3 Hz, 1 H), 7.23 (t, J = 8.3 Hz, 1 H), 6.58 (dd, J = 7.8, 2.4 Hz, 1 H), 3.76 (s, 3 H). LC–MS (m/z): 319.0 [M + H]+.
4-(2-Cyclopropylpyrazolo[1,5-b]pyridazin-3-yl)-N-(3,4-difluorophenyl)pyrimidin-2-amine (10n)
In a vial, 7e (30 mg, 110 μmol) and 3,4-difluoroaniline (21 mg, 170 μmol) were dissolved in n-BuOH (1.5 mL). The vial was sealed, and the reaction was subjected to microwave irradiation at 150 °C for 2 h. The reaction was cooled to room temperature, then filtered and washed with diethyl ether. The precipitate was taken up in DCM and washed with saturated sodium bicarbonate. The organic layer was dried over sodium sulfate, filtered, and evaporated in vacuo to yield the product (22 mg, 55%). 1H NMR (400 MHz, chloroform-d) δ ppm 8.66 (dd, J = 8.8, 1.5 Hz, 1 H), 8.48 (d, J = 5.1 Hz, 1 H), 8.32 (dd, J = 4.4, 1.5 Hz, 1 H), 7.86 (dd, J = 8.1, 2.2 Hz, 1 H), 7.36 (d, J = 5.1 Hz, 1 H), 7.09–7.15 (m, 3 H), 2.32–2.42 (m, 1 H), 1.29–1.35 (m, 2 H), 1.13–1.21 (m, 2 H). LC–MS (m/z): 365.0 [M + H]+.
N-(4-(tert-Butyl)phenyl)-4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (10s)
To an oven-dried flask was added 4-tertbutylaniline (28 μL, 173 μmol) to a solution of 7a (20 mg, 86 μmol) in n-BuOH. The reaction mixture was heated to 100 °C for 16 h. After completion of reaction, the mixture was evaporated to dryness and column purification was performed using EtOAc/hex 20–100% 15 CV to afford the title compound as a yellow solid (16 mg, 53%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.83 (dd, J = 8.8, 2.0 Hz, 1 H), 8.51 (s, 1 H), 8.39 (dd, J = 4.4, 2.0 Hz, 1 H), 8.36 (d, J = 5.4 Hz, 1 H), 7.70 (br s, 1 H), 7.49–7.54 (m, 2 H), 7.40–7.45 (m, 2 H), 7.09 (dd, J = 8.8, 4.4 Hz, 1 H), 7.05 (d, J = 5.4 Hz, 1 H), 1.37 (s, 9 H). LC–MS (m/z): 345.4 [M + H]+.
3-(2-Chloropyrimidin-4-yl)pyrazolo[1,5-a]pyridine (11)
(Aminooxy)sulfonic acid (490 mg, 4.33 mmol) was dissolved in 2 mL of water, and to the clear solution was added 2.5 M sodium bicarbonate until effervescence ceased (pH ∼ 5). The clear colorless solution was heated to 70 °C, and pyridine (0.233 mL, 2.89 mmol) was added, and the reaction mixture was stirred at 70 °C for 2 h. The reaction was cooled to room temperature, neutralized with 2.5 M sodium bicarbonate (until effervescence ceased), and then solution of 6b (185 mg, 1.34 mmol) in DCM (10 mL) was added followed by KOH (100 mg, 1.78 mmol). The reaction was stirred for 16 h at room temperature. The reaction mixture was then diluted with DCM and washed with water and brine, then dried over sodium sulfate, decanted, and concentrated to get the crude product, which was purified by column chromatography using EtOAc/hex, 50–100% 12 CV, followed by EtOAc/MeOH 0–20% 5 CV to obtain the desired compound as a beige solid (85 mg, 51%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.95 (s, 1 H), 8.90 (d, J = 6.8 Hz, 1 H), 8.62 (d, J = 5.4 Hz, 1 H), 8.49 (d, J = 8.8 Hz, 1 H), 7.97 (d, J = 5.4 Hz, 1 H), 7.62–7.68 (m, 1 H), 7.19 (td, J = 6.8, 1.5 Hz, 1 H). LC–MS (m/z): 230.9 (35Cl), 232.9 (37Cl) [M + H]+.
N-Cyclopropyl-4-(pyrazolo[1,5-a]pyridin-3-yl)pyrimidin-2-amine (12a)
In a vial 11 (10 mg, 43 μmol), cyclopropylamine (6 μL, 87 μmol), and DIPEA (16 μL, 87 μmol) were stirred in n-BuOH and subjected to microwave irradiation at 150 °C for 1 h. The reaction mixture was evaporated to dryness and purified by column chromatography using EtOAc/hex, 40–100%, 7 CV, followed by MeOH/EtOAc, 0–20% 5 CV to yield the title compound as a colorless solid (8 mg, 66%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.69–8.79 (m, 1 H), 8.52 (d, J = 6.8 Hz, 1 H), 8.42 (s, 1 H), 8.28 (d, J = 4.9 Hz, 1 H), 7.33 (ddd, J = 8.8, 6.8, 1.0 Hz, 1 H), 6.87–6.95 (m, 2 H), 2.82–2.92 (m, 1 H), 0.84–0.93 (m, 2 H), 0.60–0.68 (m, 2 H). LC–MS (m/z): 252.0 [M + H]+.
N-Phenyl-4-(pyrazolo[1,5-a]pyridin-3-yl)pyrimidin-2-amine (12b)
In a vial were added 11 (10 mg, 43 μmol) and aniline (8 μL, 87 mmol), and the mixture was stirred in n-BuOH at 110 °C for 16 h. The reaction mixture was evaporated to dryness and purified by column chromatography using EtOAc/hex, 40–100% 12 CV followed by EtOAc/MeOH 1–20% 5 CV to afford the title compound as a beige solid (11 mg, 88%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.51–8.57 (m, 2 H), 8.46 (s, 1 H), 8.35–8.40 (m, 1 H), 7.67 (d, J = 7.3 Hz, 2 H), 7.36–7.41 (m, 2 H), 7.28–7.35 (m, 2 H), 7.10 (t, J = 7.3 Hz, 1 H), 7.04 (d, J = 5.4 Hz, 1 H), 6.89–6.95 (m, 1 H). LC–MS (m/z): 288.0 [M + H]+.
N-(3-Methoxyphenyl)-4-(pyrazolo[1,5-a]pyridin-3-yl)pyrimidin-2-amine (12c)
In a vial 11 (20 mg, 87 μmol) and 3-anisidine (20 μL, 0.173 mmol) were stirred in n-BuOH at 110 °C for 16 h. The reaction mixture was then evaporated to dryness and column purification was performed using EtOAc/hex 40–100% 12 CV followed by EtOAc/MeOH 1–20% 5 CV to afford the title compound as a beige solid (11 mg, 40%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.49 (s, 1 H), 8.81–8.86 (m, 2 H), 8.77 (d, J = 8.8 Hz, 1 H), 8.41 (d, J = 5.4 Hz, 1 H), 7.46–7.52 (m, 2 H), 7.30–7.37 (m, 2 H), 7.22 (t, J = 8.1 Hz, 1 H), 7.11 (t, J = 6.8 Hz, 1 H), 6.57 (dd, J = 8.3, 2.2 Hz, 1 H), 3.76 (s, 3 H). LC–MS (m/z): 318.0 [M + H]+.
3-((4-(Pyrazolo[1,5-a]pyridin-3-yl)pyrimidin-2-yl)amino)benzonitrile (12d)
In a vial 11 (20 mg, 87 μmol), K3PO4 (37 mg, 173 μmol), XPhos (6 mg, 13 μmol), tris(dibenzylideneacetone)palladium(0) (4.0 mg, 4.3 μmol), and 3-aminobenzonitrile (20.5 mg, 173 μmol) were added. The vial was evacuated, then backfilled with N2 three times. Degassed anhydrous toluene (1 mL) was added to a sealed vial. The reaction mixture was degassed for another 5 min and left stirring at 110 °C for 16 h. The heterogeneous red reaction mixture slowly turned yellow and clear with some floating particles. The reaction mixture was evaporated to dryness and purified by using preparative reverse phase HPLC to afford the title compound as a pale-yellow solid (6 mg, 22%). 1H NMR (500 MHz, chloroform-d) δ ppm 9.19 (s, 1 H), 8.86 (d, J = 7.0 Hz, 1 H), 8.85 (s, 1 H), 8.71 (d, J = 9.0 Hz, 1 H), 8.53 (s, 1 H), 8.48 (d, J = 5.0 Hz, 1 H), 8.43 (t, J = 2.0 Hz, 1 H), 7.96 (dt, J = 8.0, 1.5 Hz, 1 H), 7.56–7.52 (m, 2 H), 7.42–7.41 (m, 2 H), 7.13 (td, J = 7.0, 1.5 Hz, 1 H). LC–MS (m/z): 313.0 [M + H]+.
Methyl Pyrazolo[1,5-b]pyridazine-3-carboxylate (14a)
(Aminooxy)sulfonic acid (8.1 g, 71 mmol) was dissolved in 13 mL of water, and to the clear colorless solution was added saturated sodium bicarbonate until effervescence ceased (pH ∼ 6). The clear colorless solution was heated to 70 °C, and pyridazine (3.4 mL, 48 mmol) was added dropwise, and the reaction was stirred at 70 °C for 2 h. The reaction was cooled to room temperature, neutralized with sat. sodium bicarbonate (until effervescence ceased), and then a solution of 13 (1.1 mL, 12 mmol) in DCM (50 mL) was added followed by KOH (1.3 g, 24 mmol). The reaction was stirred for 16 h at room temperature. After completion of reaction, the reaction mixture was diluted with DCM, washed with water, brine, and dried over magnesium sulfate, filtered, and evaporated to obtain the desired compound as a tan solid (1.5 g, 71%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.56 (dd, J = 9.0, 1.7 Hz, 1 H), 8.49 (s, 1 H), 8.44 (dd, J = 4.2, 1.7 Hz, 1 H), 7.25–7.29 (m, 1 H), 3.94 (s, 3 H). LC–MS (m/z): 177.9 [M + H]+.
Pyrazolo[1,5-b]pyridazine-3-carboxylic Acid Hydrochloride (14b)
In a round-bottom flask 14a (1.0 g, 5.6 mmol) was dissolved in water (15 mL), sodium hydroxide (677 mg, 16.9 mmol) was added, and the reaction was heated to 70 °C for 1 h. The reaction was cooled to room temperature, and 1 M HCl was added, which caused a precipitate to form. The precipitate was filtered to yield the desired product as a white solid (414 mg, 37%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.62 (dd, J = 4.4, 2.0 Hz, 1 H), 8.52 (dd, J = 9.3, 2.0 Hz, 1 H), 8.47 (s, 1 H), 7.48 (dd, J = 9.3, 4.4 Hz, 1 H). LC–MS (m/z): 163.8 [M + H]+.
3-Bromopyrazolo[1,5-bpyridazine (15)
In a round-bottom flask 14b (410 mg, 2.1 mmol) was dissolved in anhydrous DMF (10 mL), N-bromosuccinimide (520 mg, 2.9 mmol) was added, and the reaction was stirred at room temperature for 3 h. The reaction was poured into water and extracted with EtOAc, then the organic layer was washed three times with 1 M LiCl, followed by brine, then dried over magnesium sulfate, filtered, and evaporated in vacuo to yield the compound as a white solid (327 mg, 80%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.51 (dd, J = 4.4, 2.0 Hz, 1 H), 8.27 (s, 1 H), 8.19 (dd, J = 8.8, 2.0 Hz, 1 H), 7.30 (dd, J = 8.8, 4.4 Hz, 1 H). LC–MS (m/z): 197.8 (79Br), 199.8 (81Br) [M + H]+.
3-Phenylpyrazolo[1,5-b]pyridazine (16a)
In a vial 15 (42.1 mg, 213 μmol) was added to 1.5 mL of 3:1 mixture of degassed dioxane/water, 0.25 mL of DCM was added followed by phenyl boronic acid (31.1 mg, 255 μmol), Pd(dppf)2Cl2 (9 mg, 11 μmol), and potassium carbonate (88 mg, 628 μmol). The reaction mixture was subjected to microwave irradiation and heated to 130 °C for 1 h. The reaction was diluted with DCM and water and extracted 3 times into DCM, washed with brine, dried over magnesium sulfate, filtered, and evaporated in vacuo. Column purification was performed using EtOAc/hex 0–50% to afford the product as a white solid (6.2 mg, 15%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.31 (dd, J = 4.4, 2.0 Hz, 1 H), 8.24 (s, 1 H), 8.21 (dd, J = 8.8, 2.0 Hz, 1 H), 7.56–7.60 (m, 2 H), 7.47–7.51 (m, 2 H), 7.34–7.38 (m, 1 H), 7.05 (dd, J = 8.8, 4.4 Hz, 1 H). LC–MS (m/z): 196.0 [M + H]+.
3-(3-Chlorophenyl)pyrazolo[1,5-b]pyridazine (16b)
In a sealed vial 15 (100 mg, 505 μmol), 28 (103 mg, 656 μmol), potassium acetate (209 mg, 1.51 mmol), and PdCl2(dppf)·CH2Cl2 (21 mg, 25 mmol) were stirred in 4 mL of 3:1 mixture of degassed dioxane/water. The reaction was evacuated and backfilled with N2 three times, then subjected to microwave irradiation at 130 °C for 1 h. The reaction was cooled and filtered through Celite and washed with EtOAc, then concentrated in vacuo. Column purification was performed using EtOAc/hex 25–50% to yield the desired compound as a yellow solid (83 mg, 72%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.34 (dd, J = 4.4, 2.0 Hz, 1 H), 8.23 (s, 1 H), 8.19 (dd, J = 9.3, 2.0 Hz, 1 H), 7.55–7.57 (m, 1 H), 7.44–7.48 (m, 1 H), 7.41 (t, J = 7.8 Hz, 1 H), 7.31–7.35 (m, 1 H), 7.09 (dd, J = 9.3, 4.4 Hz, 1 H). LC–MS (m/z): 230.2 (35Cl), 232.0 (37Cl) [M + H]+.
3-((3-(Pyrazolo[1,5-b]pyridazin-3-yl)phenyl)amino)benzonitrile (17)
In a sealed vial 16b, 3-aminobenzonitrile (51 mg, 435 μmol), cesium carbonate (212 mg, 653 μmol), palladium(II) acetate (2.4 mg, 10.9 μmol), and Xantphos (12.6 mg, 21.7 μmol) were stirred in anhydrous dioxane (2 mL), and the reaction mixture was evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 160 °C for 1 h. The reaction was filtered through Celite and washed with EtOAc, then evaporated to dryness. Column purification was performed using EtOAc/hex 20–50%, to afford the title compound as a yellow solid (31.9 mg, 47%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.32 (dd, J = 4.4, 2.0 Hz, 1 H), 8.21 (s, 1 H), 8.18 (dd, J = 8.8, 2.0 Hz, 1 H), 7.43 (t, J = 7.8 Hz, 1 H), 7.33–7.38 (m, 2 H), 7.27–7.29 (m, 2 H), 7.24–7.25 (m, 1 H), 7.17–7.21 (m, 1 H), 7.10 (dd, J = 7.8, 2.0 Hz, 1 H), 7.07 (dd, J = 8.8, 4.4 Hz, 1 H), 5.94 (br s, 1 H). LC–MS (m/z): 312.1 [M + H]+.
3-(Pyridin-2-yl)pyrazolo[1,5-b]pyridazine (18a)
In a vial, (aminooxy)sulfonic acid (658 mg, 5.82 mmol) was dissolved in 1 mL of water, and to the clear colorless solution saturated sodium bicarbonate was added until effervescence ceased (pH ∼ 6). The clear colorless solution was heated to 70 °C, and pyridazine (0.280 mL, 3.88 mmol) was added dropwise, and the reaction was stirred at 70 °C for 2 h. Reaction mixture then cooled to room temperature, neutralized with saturated sodium bicarbonate (until effervescence ceased), and then a solution of 2-ethynylpyridine (100 mg, 0.716 μmol) in DCM (8 mL) was added followed by 1,8-diazabicyclo[5.4.0]undec-7-ene (0.145 mL, 970 μmol). The reaction mixture was subjected to microwave irradiation at 100 °C for 30 min, then 120 °C for 30 min. The reaction was then left stirring at 40 °C for 72 h After completion, the reaction mixture was extracted in DCM, washed with water, brine, and dried over sodium sulfate, filtered, and evaporated to get the crude product. Column purification was performed using EtOAc/hex 20–50%, followed by a second column purification using MeOH/DCM 0–5% to afford the title compound as an orange solid (7.1 mg, 4%). 1H NMR (500 MHz, chloroform-d) δ ppm 9.03 (dd, J = 9.3, 2.0 Hz, 1 H), 8.65–8.69 (m, 1 H), 8.47 (s, 1 H), 8.37 (dd, J = 4.4, 2.0 Hz, 1 H), 7.76 (td, J = 7.3, 2.0 Hz, 1 H), 7.68–7.72 (m, 1 H), 7.18 (ddd, J = 7.3, 4.9, 1.0 Hz, 1 H), 7.15 (dd, J = 9.3, 4.4 Hz, 1 H). LC–MS (m/z): 196.9 [M + H]+.
3-((6-(Pyrazolo[1,5-b]pyridazin-3-yl)pyridin-2-yl)amino)benzonitrile (18b)
In a sealed vial 29 (27.5 mg, 119 μmol), cesium carbonate (117 mg, 358 μmol), 3-aminobenzonitrile (28 mg, 238 μmol), palladium(II) acetate (1.3 mg, 6.0 μmol), and Xantphos (6.9 mg, 11.9 μmol) were stirred in anhydrous dioxane (1.5 mL), the reaction mixture was evacuated and backfilled with N2 three times. The reaction mixture was then subjected to MW irradiation at 160 °C for 1 h. The reaction was filtered through Celite and washed with EtOAc, then evaporated to dryness. Column purification was performed using EtOAc/hex 20–50% to afford the product as a yellow solid (16.0 mg, 43%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.84 (dd, J = 9.3, 2.0 Hz, 1 H), 8.45 (s, 1 H), 8.36 (dd, J = 4.4, 2.0 Hz, 1 H), 8.09 (s, 1 H), 7.65 (t, J = 7.8 Hz, 1 H), 7.52–7.57 (m, 1 H), 7.44 (t, J = 7.8 Hz, 1 H), 7.32–7.36 (m, 1 H), 7.15 (dd, J = 9.3, 4.4 Hz, 1 H), 6.66 (d, J = 7.8 Hz, 1 H), 6.54–6.60 (m, 1 H). LC–MS (m/z): 313.1 [M + H]+.
3-(Pyridin-4-yl)pyrazolo[1,5-b]pyridazine (18c)
(Aminooxy)sulfonic acid (486 mg, 4.30 mmol) was dissolved in 2 mL of water, to the clear colorless solution saturated sodium bicarbonate was added until effervescence ceased (pH ∼ 6). The clear colorless solution was heated to 70 °C, and pyridazine (0.207 mL, 2.87 mmol) was added dropwise, then the reaction was stirred at 70 °C for 2 h. The reaction mixture was then cooled to room temperature, and neutralized with saturated sodium bicarbonate (until effervescence ceased), then a solution of 4-ethynylpyridine HCl (100 mg, 0.716 μmol) in DCM (5 mL) was added followed by KOH (40 mg, 716 μmol). The reaction mixture was stirred at room temperature for 16 h. After completion of reaction the reaction mixture was diluted with DCM, washed with water, brine and dried over sodium sulfate, filtered, and evaporated to get the crude product. Column purification was performed using EtOAc/hex 50–100% to yield the product as an off-white solid (16 mg, 11%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.68 (dd, J = 4.4, 2.0 Hz, 2 H), 8.39 (dd, J = 4.4, 2.0 Hz, 1 H), 8.35 (s, 1 H), 8.27 (dd, J = 9.3, 2.0 Hz, 1 H), 7.50 (dd, J = 4.4, 2.0 Hz, 2 H), 7.17 (dd, J = 9.3, 4.4 Hz, 1 H). LC–MS (m/z): 196.9 [M + H]+.
3-((4-(Pyrazolo[1,5-b]pyridazin-3-yl)pyridin-2-yl)amino)benzonitrile (18d)
In a vial 30 (50 mg, 217 μmol), cesium carbonate (212 mg, 650 μmol), palladium(II) acetate (2.43 mg, 11 μmol), Xantphos (12.5 mg, 22 μmol), and 3-aminobenzonitrile (25.6 mg, 217 μmol) were added followed by anhydrous dioxane (1.5 mL) and the vial was sealed. The reaction mixture was evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 160 °C for 1 h, then filtered through Celite and washed with EtOAc, and evaporated to dryness. Column purification was performed using MeOH/DCM 2–10% followed by reverse phase column purification using ACN/water 5–95% with 0.1% formic acid, to afford the title compound as a yellow solid (6.6 mg, 10%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.39 (dd, J = 4.4, 2.0 Hz, 1 H), 8.30–8.36 (m, 2 H), 8.24 (dd, J = 8.8, 2.0 Hz, 1 H), 7.99–8.04 (s, 1 H), 7.64 (d, J = 7.8 Hz, 1 H), 7.43 (t, J = 7.8 Hz, 1 H), 7.31 (d, J = 7.8 Hz, 1 H), 7.17 (dd, J = 8.8, 4.4 Hz, 1 H), 7.07 (d, J = 5.4 Hz, 1 H), 6.98 (s, 1 H), 6.84 (br s, 1 H). LC–MS (m/z): 313.1 [M + H]+.
3-((4-Methyl-6-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (19a)
In a vial 26 (30 mg, 122 μmol) and 3-aminobenzonitrile (29 mg, 244 μmol) were stirred in n-BuOH (1.5 mL) and subjected to microwave irradiation at 150 °C for 2 h. Reaction was filtered and washed with diethyl ether, the precipitate was taken up in DCM and washed with a saturated sodium bicarbonate solution and brine, then dried over magnesium sulfate, filtered, and evaporated in vacuo, to yield the product as a white solid (19.8 mg, 50%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.89 (s, 1 H), 9.05–9.11 (m, 1 H), 8.87 (s, 1 H), 8.61 (dd, J = 4.4, 2.0 Hz, 1 H), 8.34–8.37 (m, 1 H), 7.97 (d, J = 7.8 Hz, 1 H), 7.53 (t, J = 7.8 Hz, 1 H), 7.45 (dd, J = 9.3, 4.4 Hz, 1 H), 7.38–7.42 (m, 2 H), 2.43 (s, 3 H). LC–MS (m/z): 328.2 [M + H]+.
3-((5-Methyl-4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (19b)
In a vial 29 (35 mg, 140 μmol), 3-aminobenzonitrile (34 mg, 430 μmol), cesium carbonate (139 mg, 427 μmol), palladium(II) acetate (1.6 mg, 7.1 μmol), and Xantphos (8.2 mg, 14 μmol) were stirred in 1.5 mL of anhydrous dioxane. The reaction mixture was sealed and then evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 130 °C for 1 h. The reaction was filtered through Celite and washed with EtOAc, then evaporated to dryness. Column purification was performed using MeOH/DCM 0–5%, followed by another column purification using MeOH/DCM 1–3% to afford the title compound as a white solid (7.4 mg, 16%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.87 (s, 1 H), 9.11 (dd, J = 9.3, 2.0 Hz, 1 H), 8.71 (s, 1 H), 8.66 (dd, J = 4.4, 2.0 Hz, 1 H), 8.46–8.49 (m, 1 H), 8.36 (t, J = 2.0 Hz, 1 H), 7.93–7.98 (m, 1 H), 7.51 (t, J = 7.8 Hz, 1 H), 7.46 (dd, J = 9.3, 4.4 Hz, 1 H), 7.36–7.40 (m, 1 H), 2.46 (s, 3 H). LC–MS (m/z): 328.1 [M + H]+.
3-((4-(Pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)oxy)benzonitrile (20a)
3-Hydroxybenzonitrile (8 mg, 65 μmol) in anhydrous DMF was stirred with potassium carbonate (13 mg, 97 μmol) at room temperature under N2 for 30 min, then 7a (15 mg, 65 μmol) was added and the reaction mixture was stirred at 80 °C for 16 h. After completion of reaction, the reaction mixture was dumped in water and extracted with EtOAc. Organic layer was then washed with water, brine and dried over sodium sulfate, decanted and concentrated to get the crude product which was purified by column chromatography using acetone/hex 20–50% 15 CV then 50–100% 5 CV to afford the title compound as a white solid (11 mg, 51%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.56 (d, J = 5.4 Hz, 1 H), 8.54 (s, 1 H), 8.39–8.45 (m, 2 H), 7.59–7.66 (m, 3 H), 7.53–7.57 (m, 1 H), 7.40 (d, J = 5.4 Hz, 1 H), 7.11 (dd, J = 8.8, 4.4 Hz, 1 H). LC–MS (m/z): 315.0 [M + H]+.
3-(Methyl(4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (20b)
In a vial 7a (100 mg, 430 μmol), cesium carbonate (420 mg, 1.3 mmol), 3-(methylamino)benzonitrile (110 mg, 860 μmol), palladium(II) acetate (4.8 mg, 22 μmol), and Xantphos (27 mg, 43 μmol) were added in 5 mL of anhydrous dioxane. The reaction mixture was sealed then evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 160 °C for 1 h. The reaction was filtered through Celite and washed with EtOAc, then evaporated to dryness. Column purification was done using MeOH/DCM 0–4%, followed by isocratic column purification 80% EtOAc/hexanes affording the title compound as an off-white solid (18.8 mg, 13%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.88 (s, 1 H), 8.58 (dd, J = 4.4, 2.0 Hz, 1 H), 8.45 (d, J = 5.4 Hz, 1 H), 8.38 (dd, J = 9.3, 2.0 Hz, 1 H), 8.00 (s, 1 H), 7.83 (d, J = 7.8 Hz, 1 H), 7.75 (d, J = 7.8 Hz, 1 H), 7.66 (t, J = 7.8 Hz, 1 H), 7.38 (d, J = 5.4 Hz, 1 H), 7.30 (dd, J = 9.3, 4.4 Hz, 1 H), 3.59 (s, 3 H). LC–MS (m/z): 328.2 [M + H]+.
4-(Pyrazolo[1,5-b]pyridazin-3-yl)-N-(3-(trifluoromethyl)phenyl)pyrimidin-2-amine (20c)
In a vial 7a (50 mg, 216 μmol), 3-(trifluoromethyl)aniline (67 μL, 540 μmol) were stirred in n-BuOH (1.5 mL), and the reaction mixture was subjected to microwave irradiation at 150 °C for 2 h. After completion the reaction was evaporated to dryness and column purification was performed using EtOAc/hex, 33–66% to afford the title compound as a white solid (52 mg, 68%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.95 (s, 1 H), 9.10–9.18 (m, 1 H), 8.93 (s, 1 H), 8.64 (dd, J = 4.4, 2.0 Hz, 1 H), 8.55 (d, J = 5.4 Hz, 1 H), 8.30 (s, 1 H), 7.97 (d, J = 8.3 Hz, 1 H), 7.57 (t, J = 8.3 Hz, 1 H), 7.43–7.50 (m, 2 H), 7.33 (d, J = 8.3 Hz, 1 H). LC–MS (m/z): 357.0 [M + H]+.
2-Fluoro-3-((4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (20d)
In a vial 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine 7a (40 mg, 173 μmol), cesium carbonate (169 mg, 518 μmol), 3-amino-2-fluorobenzonitrile (47 mg, 345 μmol), palladium(II) acetate (2 mg, 8.6 μmol), and Xantphos (10 mg, 17 μmol) were added in anhydrous dioxane (1.5 mL), the reaction mixture was sealed then evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 160 °C for 1 h. The reaction was filtered through Celite and washed with EtOAc, then evaporated to dryness. The reaction mixture was taken up in DCM and water, and extracted 2X into DCM, then washed with brine, dried with Na2SO4, filtered, and evaporated in vacuo. Column purification was performed using EtOAc/hex 25–100% followed by reverse phase column purification using ACN/water 5–95% with 0.1% ammonium hydroxide, to afford the title compound as a white solid (5.0 mg, 9%). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.62 (s, 1 H), 8.94–9.01 (m, 1 H), 8.90 (s, 1 H), 8.56–8.63 (m, 1 H), 8.45 (d, J = 5.1 Hz, 1 H), 8.19 (t, J = 7.3 Hz, 1 H), 7.65 (t, J = 7.3 Hz, 1 H), 7.43 (s, 3 H). LC–MS 332.0 (m/z): [M + H]+.
4-Fluoro-3-((4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (20e)
In a vial 7a (40 mg, 173 μmol), cesium carbonate (169 mg, 518 μmol), 3-amino-4-fluorobenzonitrile (47 mg, 345 μmol), palladium(II) acetate (2 mg, 8.6 μmol), and Xantphos (10 mg, 17 μmol) were added with anhydrous dioxane (1.5 mL), the reaction mixture was sealed, then evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 160 °C for 1 h. The reaction was filtered through Celite and washed with EtOAc, then evaporated to dryness. Column purification was done using EtOAc/hex 25–100%, followed by MeOH/DCM 4% with 5% ammonium hydroxide, followed by reverse phase column purification using ACN/water 5–95% with 0.1% ammonium hydroxide, to afford the title compound as a white solid (5.0 mg, 8.7%). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.60 (s, 1 H), 9.11 (d, J = 8.8 Hz, 1 H), 8.93 (s, 1 H), 8.63 (dd, J = 4.4, 2.2 Hz, 1 H), 8.49–8.58 (m, 2 H), 7.62–7.67 (m, 1 H), 7.49–7.59 (m, 2 H), 7.43 (dd, J = 8.8, 4.4 Hz, 1 H). LC–MS (m/z): 332.0 [M + H]+.
4-(Pyrazolo[1,5-b]pyridazin-3-yl)-N-(pyridin-2-yl)pyrimidin-2-amine (20f)
In a vial 7a (40 mg, 173 μmol), cesium carbonate (169 mg, 518 μmol), 2-aminopyridine (33 mg, 350 μmol), palladium(II) acetate (2 mg, 8.6 μmol), and Xantphos (10 mg, 17 μmol) were added to anhydrous dioxane (1.5 mL), the reaction mixture was sealed then evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 160 °C for 1 h. The reaction was filtered through Celite and washed with EtOAc, then evaporated to dryness. Column purification was performed using EtOAc/hex 50–100%, to afford the title compound as a pale-yellow solid (20.2 mg, 41%).1H NMR (400 MHz, chloroform-d) δ ppm 9.00 (dd, J = 8.8, 2.2 Hz, 1 H), 8.54 (s, 1 H), 8.51 (d, J = 5.1 Hz, 1 H), 8.43 (dd, J = 4.4, 2.2 Hz, 1 H), 8.40 (d, J = 8.1 Hz, 1 H), 8.34 (d, J = 5.1 Hz, 1 H), 7.96 (br s, 1 H), 7.69–7.76 (m, 1 H), 7.22 (dd, J = 8.8, 4.4 Hz, 1 H), 7.16 (d, J = 5.1 Hz, 1 H), 6.98 (dd, J = 8.1, 5.1 Hz, 1 H). LC–MS (m/z): 290.1 [M + H]+.
4-(Pyrazolo[1,5-b]pyridazin-3-yl)-N-(pyridin-3-yl)pyrimidin-2-amine (20g)
In a vial 7a (50 mg, 216 μmol), cesium carbonate (140 mg, 431 μmol), palladium(II) acetate (0.9 mg, 4 μmol), and Xantphos (4 mg, 7 μmol) were dissolved in 2 mL of anhydrous dioxane, the reaction mixture was sealed then evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 160 °C for 1 h. The reaction was filtered through Celite and washed with EtOAc, then concentrated in vacuo. Reverse phase preparative HPLC purification was performed using ACN/water 5–95% with 0.1% formic acid, to afford the title compound as a white solid (9.1 mg, 15%), isolated as a 0.5 eq formic acid solvate. 1H NMR (500 MHz, DMSO-d6) δ ppm 9.79 (s, 1 H), 9.12–9.18 (m, 1 H), 8.92 (s, 1 H), 8.89 (d, J = 2.0 Hz, 1 H), 8.63 (dd, J = 4.4, 2.0 Hz, 1 H), 8.51 (d, J = 5.4 Hz, 1 H), 8.41 (br s, 1 H), 8.24 (d, J = 8.3 Hz, 1 H), 8.20 (d, J = 4.9 Hz, 1 H), 7.48 (dd, J = 8.8, 4.4 Hz, 1 H), 7.44 (d, J = 5.4 Hz, 1 H), 7.37 (dd, J = 8.3, 4.9 Hz, 1 H). LC–MS (m/z): 290.2 [M + H]+.
4-(Pyrazolo[1,5-b]pyridazin-3-yl)-N-(pyridin-4-yl)pyrimidin-2-amine (20h)
In a vial 7a (30 mg, 130 μmol) and 4-aminopyridine (18 mg, 190 μmol) were stirred in n-BuOH (1.5 mL) and heated to 150 °C for 2 h. The reaction was concentrated in vacuo. Column purification was performed using EtOAc/hexanes 50–75% then changed to MeOH/DCM 0–20%, a second column was run using an isocratic method MeOH/DCM 30%, to afford the title compound as a yellow solid (5.2 mg, 14%). 1H NMR (400 MHz, methanol-d4) δ ppm 9.39 (d, J = 7.3 Hz, 2 H), 9.05 (d, J = 9.5 Hz, 1 H), 8.93 (s, 1 H), 8.85 (d, J = 5.1 Hz, 1 H), 8.61 (d, J = 4.4 Hz, 1 H), 8.03 (d, J = 5.1 Hz, 1 H), 7.55 (dd, J = 9.5, 4.4 Hz, 1 H), 7.09 (d, J = 7.3 Hz, 2 H). LC–MS (m/z): 290.1 [M + H]+.
4-(Pyrazolo[1,5-b]pyridazin-3-yl)-N-(pyrimidin-5-yl)pyrimidin-2-amine (20i)
In a vial 7a (50 mg, 220 μmol), 5-aminopyrimidine (205 mg, 2.2 mmol), palladium(II) acetate (2.4 mg, 11 μmol), Xantphos (12.5 mg, 21.6 μmol), and cesium carbonate (211 mg, 648 μmol) were stirred in anhydrous dioxane (5 mL). The reaction mixture was sealed then evacuated and backfilled with N2 three times. The reaction was then subjected to microwave irradiation at 160 °C for 1.5 h. The reaction was filtered through Celite and washed with EtOAc, then evaporated to dryness. Column purification was performed using MeOH/DCM 0–5%, followed by reverse phase preparative HPLC column purification using ACN/water 5–70% with 0.1% formic acid, to afford the title compound as a white solid (3.6 mg, 6%). 1H NMR (500 MHz, chloroform-d) δ ppm 9.14 (s, 2 H), 8.97 (s, 1 H), 8.85 (dd, J = 8.8, 2.0 Hz, 1 H), 8.56 (s, 1 H), 8.50 (d, J = 5.4 Hz, 1 H), 8.44 (dd, J = 4.9, 2.0 Hz, 1 H), 7.25 (dd, J = 8.8, 4.4 Hz, 1 H), 7.20 (d, J = 5.4 Hz, 1 H), 7.05 (br s, 1 H). LC–MS (m/z): 291.2 [M + H]+.
N-(4-(Pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)pyridazin-4-amine (20j)
In a vial 7a (50 mg, 216 μmol) and 4-aminopyridazine (41 mg, 432 μmol) were stirred in n-BuOH (1.5 mL) and subject to microwave irradiation to 120 °C for 30 min. The reaction was concentrated in vacuo and column purification was performed MeOH/DCM 2–20% followed by reverse phase preparative HPLC purification using ACN/water 5–50% with 0.1% formic acid, to afford the title compound as a yellow solid (5.5 mg, 11%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.21–9.26 (m, 1 H), 9.13 (dd, J = 8.78, 2.0 Hz, 1 H), 9.10 (s, 1 H), 8.87 (d, J = 5.4 Hz, 1 H), 8.69 (dd, J = 4.4, 2.0 Hz, 1 H), 8.35–8.41 (m, 2 H), 8.05 (d, J = 5.4 Hz, 1 H), 7.58 (dd, J = 8.8, 4.4 Hz, 1 H), 6.84–6.89 (m, 1 H). LC–MS (m/z): 291.1 [M + H]+.
N-(6-Methylpyridin-3-yl)-4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (20k)
In a vial 7a (50 mg, 220 μmol), 3-amino-6-methylpyridine (47 mg, 430 μmol), cesium carbonate (211 mg, 648 μmol), palladium(II) acetate (2.4 mg, 11 μmol), and Xantphos (12.5 mg, 21.6 μmol) were stirred in anhydrous dioxane (1.5 mL). The reaction mixture was sealed then evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 160 °C for 1 h. The reaction was filtered through Celite and washed with EtOAc, then evaporated to dryness. Reverse phase preparative HPLC column purification was performed using ACN/water 5–70% with 0.1% formic acid. The compound was then taken up in DCM and stirred with resin-bound silicon carbonate for 12 h then filtered and evaporated in vacuo, to afford the title compound as an off-white solid (25.9 mg, 40%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.64 (s, 1 H), 9.09–9.16 (m, 1 H), 8.90 (s, 1 H), 8.73 (d, J = 2.4 Hz, 1 H), 8.62 (dd, J = 4.4, 2.0 Hz, 1 H), 8.48 (d, J = 5.4 Hz, 1 H), 8.11 (dd, J = 8.8, 2.4 Hz, 1 H), 7.47 (dd, J = 8.8, 4.4 Hz, 1 H), 7.40 (d, J = 5.4 Hz, 1 H), 7.22 (d, J = 8.8 Hz, 1 H), 2.44 (s, 3 H). LC–MS (m/z): 304.2 [M + H]+.
3-((4-(Pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)propan-1-ol (20l)
To an oven-dried flask 3-aminopropan-1-ol (7 mg, 90 μmol) and 7a (10 mg, 43 μmol) were added in n-BuOH. The reaction mixture was stirred at 110 °C for 16 h. After completion of reaction mixture was evaporated to dryness and the solid obtained was titrated with cold methanol, filtered and dried under high vacuum affording the title compound as a solid (6 mg, 68%). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.01–9.25 (m, 1 H), 8.82 (s, 1 H), 8.58 (d, J = 5.1 Hz, 1 H), 8.27 (d, J = 4.4 Hz, 1 H), 7.41 (dd, J = 8.8, 4.4 Hz, 1 H), 7.19 (br s, 1 H), 7.10 (d, J = 5.1 Hz, 1 H), 4.53 (br s, 1 H), 3.37–3.60 (m, 4 H), 1.64–1.82 (m, 2 H). LC–MS (m/z): 271.1 [M + H]+.
N-(4-(Pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)acrylamide (20m)
To the solution of (10 mg, 47 μmol, prepared by refluxing 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine in liq NH3, added triethylamine (10 μL, 71 μmol) in 1 mL THF followed by acryloyl chloride (6 μL, 71 μmol), and reaction mixture was stirred at 50 °C overnight. The reaction mixture then evaporated to dryness and taken up in DCM, washed with water and brine then dried over sodium sulfate, decanted, and concentrated to get the crude product which was purified by column chromatography using EtOAc/hex 30–100% 12 CV to afford the title compound as a solid (6 mg, 48%). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.93 (s, 1 H), 9.71 (d, J = 8.8 Hz, 1 H), 8.97 (s, 1 H), 8.57–8.70 (m, 2 H), 7.71 (d, J = 5.1 Hz, 1 H), 7.53 (dd, J = 8.8, 4.4 Hz, 1 H), 6.71 (dd, J = 16.9, 10.3 Hz, 1 H), 6.40 (d, J = 16.9 Hz, 1 H), 5.85 (d, J = 10.3 Hz, 1 H). LC–MS (m/z): 267.1 [M + H]+.
4-(Pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (20n)
In a vial 10a (34.3 mg, 103 μmol) was dissolved in TFA (2 mL) and stirred at 80 °C for 20 min. The reaction was quenched with saturated sodium bicarbonate and filtered. The precipitate was purified using column purification using MeOH/DCM 5% to afford the title compound as a white solid (8.1 mg, 37%). 1H NMR (400 MHz, chloroform-d) δ ppm 8.99 (d, J = 8.8 Hz, 1 H), 8.49 (s, 1 H), 8.41 (d, J = 4.4 Hz, 1 H), 8.32 (d, J = 5.1 Hz, 1 H), 7.20 (dd, J = 8.8, 4.4 Hz, 1 H), 6.99 (d, J = 5.1 Hz, 1 H), 5.02 (br s, 2 H). LC–MS (m/z): 213.0 [M + H]+.
N-Methyl-4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (20o)
In a vial 7a (30 mg, 130 μmol) and methylamine (2.0 M solution in THF) (129.5 μL, 259 μmol) were stirred in n-BuOH and subjected to microwave irradiation at 150 °C for 2 h. The reaction was concentrated in vacuo, then subjected to column purification using EtOAc/hexanes 50–100% to afford the title compound as an off-white solid (22.8 mg, 78%). 1H NMR (400 MHz, methanol-d4) δ ppm 9.12 (dd, J = 8.8, 2.2 Hz, 1 H), 8.65 (s, 1 H), 8.49 (dd, J = 4.4, 2.2 Hz, 1 H), 8.22 (d, J = 5.1 Hz, 1 H), 7.36 (dd, J = 8.8, 4.4 Hz, 1 H), 7.07 (d, J = 5.1 Hz, 1 H), 3.02 (s, 3 H). LC–MS (m/z): 227.0 [M + H]+.
N-Isopropyl-4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (20p)
In a vial 7a (50 mg, 220 μmol) and isopropylamine (35 μL, 430 μmol) were stirred in n-BuOH (1.5 mL), and the reaction mixture was subjected to microwave irradiation at 150 °C for 2 h. After completion of reaction mixture was evaporated to dryness. Column purification performed using reverse phase preparative HPLC using 5–35% ACN/water system, affording the title compound as a yellow solid (7.0 mg, 15%). 1H NMR (500 MHz, methanol-d4) δ ppm 9.00–9.13 (m, 1 H), 8.88 (s, 1 H), 8.64 (dd, J = 4.4, 2.0 Hz, 1 H), 8.16 (d, J = 6.4 Hz, 1 H), 7.57 (dd, J = 8.8, 4.4 Hz, 1 H), 7.38 (d, J = 6.4 Hz, 1 H), 4.89–4.90 (m, 1 H), 1.40 (d, J = 6.35 Hz, 6 H). LC–MS (m/z): 255.1 [M + H]+.
N-Cyclohexyl-4-(pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (20q)
In a vial 7a (50 mg, 216 μmol) and cyclohexylamine (49 μL, 432 μmol) were stirred in 1.5 mL of n-BuOH, and the reaction was subjected to microwave irradiation at 150 °C for 2 h, the reaction was then conventionally heated to 50 °C for 12 h, and then subjected to microwave irradiation at 150 °C for 30 min. The reaction was filtered and washed with diethyl ether to give the crude product in the precipitate. Reverse phase preparative HPLC purification was performed using ACN/water 5–95% with 0.1% formic acid to afford the title compound as a white solid (26.2 mg, 41%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.96 (dd, J = 8.8, 2.0 Hz, 1 H), 8.46 (s, 1 H), 8.38 (dd, J = 4.4, 2.0 Hz, 1 H), 8.23–8.32 (m, 1 H), 7.16 (dd, J = 8.8, 4.5 Hz, 1 H), 6.86 (d, J = 5.4 Hz, 1 H), 5.12 (br s, 1 H), 3.84–3.95 (m, 1 H), 2.09–2.19 (m, 2 H), 1.76–1.87 (m, 2 H), 1.64–1.73 (m, 1 H), 1.41–1.53 (m, 2 H), 1.22–1.38 (m, 3 H). LC–MS (m/z): 295.1 [M + H]+.
4-((4-(Pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)cyclohexan-1-ol (20r)
In a vial 7a (0.040 g, 173 μmol), 4-aminocyclohexanol (0.040 g, 347 μmol) were stirred in n-BuOH (1.5 mL), and the reaction mixture was subjected to microwave irradiation at 150 °C for 2 h. After completion of reaction mixture was filtered and washed with diethyl ether, the filtrate was evaporated to dryness and column purification was performed using MeOH/DCM 2–4% to yield the title compound as a white solid (6.0 mg, 11%). 1H NMR (500 MHz, methanol-d4) δ ppm 9.04–9.13 (m, 1 H), 8.64 (s, 1 H), 8.49 (dd, J = 4.4, 2.0 Hz, 1 H), 8.21 (d, J = 5.4 Hz, 1 H), 7.36 (dd, J = 8.8, 4.4 Hz, 1 H), 7.04 (d, J = 5.4 Hz, 1 H), 3.76–3.87 (m, 1 H), 3.58–3.67 (m, 1 H), 2.09–2.20 (m, 2 H), 1.98–2.08 (m, 2 H), 1.35–1.53 (m, 4 H). LC–MS (m/z): 311.1 [M + H]+.
3-((4-(2-(4-Methoxyphenyl)pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (21a)
In a vial were added 7f (20 mg, 59 μmol), anhydrous K3PO4 (25 mg, 118 μmol), XPhos (4 mg, 9 μmol), tris(dibenzylideneacetone)palladium(0) (3 mg, 3.0 μmol), and 3-aminobenzonitrile (14 mg, 118 μmol). The vial was sealed with a stir bar and evacuated and backfilled with N2 three times. Degassed anhydrous toluene (1 mL, dried over molecular sieves and stored under N2) was added to a sealed vial and the reaction mixture was degassed with nitrogen for an additional 5 min. The reaction was stirred at 101 °C for 16 h. The reaction mixture was evaporated to dryness and then extracted with DCM and washed with water, and brine then dried over sodium sulfate, decanted and concentrated to get the crude product. Purification was done by column chromatography using EtOAc/hex 40–100% 12 CV to yield the title compound as a yellow solid (3 mg, 12%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.71 (dd, J = 9.0, 1.5 Hz, 1 H), 8.40 (dd, J = 4.5, 2.0 Hz, 1 H), 8.31 (s, 1 H), 8.27 (d, J = 5.0 Hz, 1 H), 7.66 (d, J = 8.0 Hz, 1 H), 7.63 (d, J = 9.0 Hz, 2 H), 7.414 (t, J = 8.0 Hz, 1 H), 7.36–7.32 (m, 2 H), 7.20 (dd, J = 9.0, 4.5 Hz, 1 H), 7.00 (d, J = 9.0 Hz, 2 H), 6.76 (d, J = 5.5 Hz, 1 H), 3.89 (s, 3 H). LC–MS (m/z): 420.0 [M + H]+.
3-((4-(2-(4-(Trifluoromethyl)phenyl)pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (21b)
In a vial 7d (15 mg, 40 μmol), cesium carbonate (39 mg, 120 μmol), 3-aminobenzonitrile (9 mg, 80 μmol), palladium(II) acetate (0.5 mg, 4.0 μmol), and Xantphos (2.3 mg, 4 μmol) were added in 1.5 mL of anhydrous dioxane. The reaction mixture was sealed then evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 160 °C for 1 h. The reaction was filtered through Celite and washed with EtOAc, then evaporated to dryness. Column purification was performed using EtOAc/hexanes 25–100%, to yield the title compound as a pale-yellow solid (11.7 mg, 64%). 1H NMR (500 MHz, DMSO-d6) δ ppm 10.06 (s, 1 H), 8.80–8.85 (m, 1 H), 8.69 (dd, J = 4.4, 2.0 Hz, 1 H), 8.50 (d, J = 5.4 Hz, 1 H), 8.19 (s, 1 H), 7.89 (d, J = 8.3 Hz, 2 H), 7.82–7.87 (m, 3 H), 7.49 (dd, J = 8.8, 4.4 Hz, 1 H), 7.41 (t, J = 7.8 Hz, 1 H), 7.36 (d, J = 7.8 Hz, 1 H), 6.79 (d, J = 5.37 Hz, 1 H). LC–MS (m/z): 458.1 [M + H]+.
3-((4-(2-(2-Hydroxypropan-2-yl)pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (21c)
In a vial 7g (71 mg, 170 μmol), and 3-aminobenzonitrile (87 mg, 740 μmol) were stirred in n-BuOH (2 mL) and the reaction mixture was subjected to microwave irradiation at 150 °C for 2 h. After completion of reaction mixture was evaporated to dryness and column purification was done using MeOH/DCM 0–5%, followed by column purification 25–100% EtOAc/hexanes, followed by preparative reverse phase HPLC purification 30–100% ACN/water, affording the title compound as a white solid (2.9 mg, 4%). 1H NMR (500 MHz, methanol-d4) δ ppm 8.54–8.60 (m, 2 H), 8.47 (dd, J = 4.4, 1.5 Hz, 1 H), 8.26 (s, 1 H), 7.89 (d, J = 8.3 Hz, 1 H), 7.51 (d, J = 5.4 Hz, 1 H), 7.47 (t, J = 8.3 Hz, 1 H), 7.31–7.38 (m, 2 H), 1.69 (s, 6 H). LC–MS (m/z): 372.1 [M + H]+.
3-((4-(2-(Hydroxymethyl)pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (21d)
In a vial 7h (33.5 mg, 128 μmol) and 3-aminobenzonitrile (37.8 mg, 320 μmol) were stirred in n-BuOH (2 mL) and subjected to microwave irradiation at 150 °C for 1 h. The reaction mixture was filtered, and the precipitate was washed with MeOH. The precipitate was found to contain the desired product as a brown solid (8 mg, 18%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.96 (s, 1 H), 9.02–9.09 (m, 1 H), 8.58–8.64 (m, 2 H), 8.38 (s, 1 H), 7.96 (dd, J = 8.3, 2.0 Hz, 1 H), 7.49–7.56 (m, 2 H), 7.40–7.47 (m, 2 H), 5.64 (br s, 1 H), 4.87 (s, 2 H). LC–MS (m/z): 344.2 [M + H]+.
3-((4-(2-(Methoxymethyl)pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (21e)
In a vial were added 7i (100 mg, 362 μmol) and 3-aminobenzonitrile (85.7 mg, 725 μmol) with 1.5 mL of n-BuOH. The reaction mixture was subjected to microwave irradiation at 150 °C for 2 h. The reaction was cooled to room temperature, filtered, and washed with diethyl ether to yield the desired product in the precipitate as an off-white solid (116.8 mg, 90%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.98 (s, 1 H), 9.03–9.09 (m, 1 H), 8.65 (dd, J = 4.4, 2.0 Hz, 1 H), 8.62 (d, J = 5.4 Hz, 1 H), 8.38 (s, 1 H), 7.94–7.99 (m, 1 H), 7.53 (t, J = 8.1 Hz, 1 H), 7.46 (dd, J = 8.8, 4.4 Hz, 1 H), 7.42 (d, J = 7.3 Hz, 1 H), 7.33 (d, J = 5.4 Hz, 1 H), 4.84 (s, 2 H), 3.37 (s, 3 H). LC–MS (m/z): 358.1 [M + H]+.
3-((4-(2-(1-Hydroxyethyl)pyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (21f)
In a vial were added 7j (100 mg, 363 μmol) and 3-aminobenzonitrile (86 mg, 725 μmol) with 1.5 mL of n-BuOH. The reaction mixture was subjected to microwave irradiation at 150 °C for 2 h. The reaction was then concentrated in vacuo. Column purification was performed using EtOAc/hex 45–100% to afford the title compound as a white solid (95.5 mg, 74%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.96 (s, 1 H), 8.93 (d, J = 8.8 Hz, 1 H), 8.58–8.63 (m, 2 H), 8.38 (s, 1 H), 7.97 (d, J = 8.3 Hz, 1 H), 7.59 (d, J = 5.4 Hz, 1 H), 7.51 (t, J = 8.3 Hz, 1 H), 7.39–7.44 (m, 2 H), 5.63 (d, J = 6.4 Hz, 1 H), 5.28 (quin, J = 6.4 Hz, 1 H), 1.62 (d, J = 6.4 Hz, 3 H). LC–MS (m/z): 358.1 [M + H]+.
3-((4-(2-Isopropylpyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (21g)
In a vial 7k (100 mg, 365 μmol) and 3-aminobenzonitrile (86 mg, 731 μmol) were stirred in 1.5 mL of n-BuOH. The reaction mixture was subjected to microwave irradiation and heated to 150 °C for 2 h. The reaction mixture was concentrated in vacuo. Column purification was performed using 1–5% MeOH in DCM, followed by reverse phase preparative HPLC purification using ACN/water 5–95% with 0.1% formic acid, providing the title compound as a tan solid (36.8 mg, 28%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.98 (s, 1 H), 8.75–8.82 (m, 1 H), 8.59 (d, J = 5.4 Hz, 1 H), 8.55 (d, J = 4.4 Hz, 1 H), 8.37 (s, 1 H), 7.97 (d, J = 8.3 Hz, 1 H), 7.51 (t, J = 8.3 Hz, 1 H), 7.37–7.43 (m, 2 H), 7.20 (d, J = 5.4 Hz, 1 H), 3.79 (spt, J = 6.8 Hz, 1 H), 1.38 (d, J = 6.8 Hz, 6 H). LC–MS (m/z): 356.1 [M + H]+.
3-((4-(2-Cyclopropylpyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (21h)
In a vial 7e (100 mg, 368 μmol) and 3-aminobenzonitrile (87 mg, 736 μmol) were stirred in 1.5 mL of n-BuOH. The reaction mixture was subjected to microwave irradiation at 150 °C for 2 h. The reaction was filtered and washed with diethyl ether to give the crude product in the precipitate. Column purification was performed using MeOH/DCM 0–3% followed by reverse phase purification using ACN/water 5–95% with 0.1% formic acid to afford the title compound as a white solid (46.1 mg, 35%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.68 (dd, J = 8.8, 2.0 Hz, 1 H), 8.52 (d, J = 5.4 Hz, 1 H), 8.34–8.35 (m, 1 H), 8.33 (dd, J = 4.4, 2.0 Hz, 1 H), 7.65–7.70 (m, 1 H), 7.41–7.45 (m, 3 H), 7.34 (d, J = 7.8 Hz, 1 H), 7.16 (dd, J = 8.8, 4.4 Hz, 1 H), 2.31–2.39 (m, 1 H), 1.30–1.35 (m, 2 H), 1.15–1.20 (m, 2 H). LC–MS (m/z): 354.1 [M + H]+.
4-(6-Morpholinopyrazolo[1,5-b]pyridazin-3-yl)-N-phenylpyrimidin-2-amine (22a)
In a reaction vial were added 7b (20 mg, 63 μmol) and aniline (12 μL, 126 μmol) in n-BuOH (1 mL), the reaction mixture was subjected to microwave irradiation at 150 °C for 1 h. Then the reaction mixture was evaporated to dryness and column purification done using EtOAc/hex 50–100% 12 CV followed by EtOAc/MeOH 1–20% 5 CV to afford the title compound as a yellow/brown solid (10 mg, 42%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.65 (d, J = 9.8 Hz, 1 H), 8.37 (d, J = 5.4 Hz, 1 H), 8.26 (s, 1 H), 7.62 (d, J = 7.8 Hz, 2 H), 7.38 (t, J = 7.8 Hz, 2 H), 7.25 (br s, 1 H), 7.11 (t, J = 7.8 Hz, 1 H), 7.02 (d, J = 5.4 Hz, 1 H), 6.87 (d, J = 9.8 Hz, 1 H), 3.86 (t, J = 4.4 Hz, 4 H), 3.59 (t, J = 4.4 Hz, 4 H). LC–MS (m/z): 374.0 [M + H]+.
N-(3-Methoxyphenyl)-4-(6-morpholinopyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (22b)
In a vial 7b (0.020 g, 63.14 μmol) and m-anisidine (15 μL, 126 μmol) were stirred in n-BuOH. The reaction mixture was subjected to microwave irradiation at 150 °C for 1 h. On cooling solid precipitation seen, which was filtered and washed with diethyl ether and air-dried to afford the title compound as a dark brown solid (14 mg, 55%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.70 (d, J = 9.8 Hz, 1 H), 8.38 (d, J = 5.4 Hz, 1 H), 8.27 (s, 1 H), 7.36 (t, J = 2.4 Hz, 1 H), 7.27–7.31 (m, 1 H), 7.10–7.15 (m, 2 H), 7.03 (d, J = 5.4 Hz, 1 H), 6.89 (d, J = 9.8 Hz, 1 H), 6.66 (dd, J = 8.3, 2.4 Hz, 1 H), 3.87 (t, J = 4.9 Hz, 4 H), 3.83 (s, 3 H), 3.60 (t, J = 4.9 Hz, 4 H). LC–MS (m/z): 404.0 [M + H]+.
3-((4-(6-Morpholinopyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (22c)
In a vial 7b (20 mg, 63 μmol), K3PO4 (27 mg, 126 μmol), XPhos (5 mg, 9.5 μmol), tris(dibenzylideneacetone)palladium(0) (3 mg, 3.2 μmol), and m-aminobenzonitrile (15 mg, 126 μmol) were added. The vial was sealed with a stir bar and then evacuated and backfilled with N2 three times, 1 mL of n-BuOH was added to a sealed vial. The reaction was left stirring at 101 °C for 16 h. Solid precipitation was seen in the reaction vial, which was filtered and washed with diethyl ether. Column purification was undertaken on the precipitate using EtOAc/hex 70–100% 5 CV followed by MeOH/EtOAc 1–20% 10 CV to afford the title compound as a white solid (6.3 mg, 25%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.68 (d, J = 9.8 Hz, 1 H), 8.39–8.45 (m, 2 H), 8.28 (s, 1 H), 7.56 (ddd, J = 7.8, 2.4, 1.0 Hz, 1 H), 7.45 (t, J = 7.8 Hz, 1 H), 7.36 (dt, J = 7.8, 1.0 Hz, 1 H), 7.32 (br s, 1 H), 7.12 (d, J = 5.4 Hz, 1 H), 7.06 (d, J = 9.8 Hz, 1 H), 3.87 (t, J = 4.9 Hz, 4 H), 3.61 (t, J = 4.9 Hz, 4 H). LC–MS (m/z): 399.0 [M + H]+.
3-((4-(6-Methoxypyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (22d)
In a vial 3- 7c (15 mg, 57 μmol) and 3-aminobenzonitrile (34 mg, 287 μmol) were stirred in n-BuOH (1.5 mL) and subjected to microwave irradiation at 150 °C for 2 h. The reaction mixture was then heated to 110 °C using conventional heating for an additional 96 h. The mixture was then concentrated, and column purification was undertaken using EtOAc/hex, 25–33% to yield the title compound as a white solid (7.0 mg, 37%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.74 (d, J = 9.8 Hz, 1 H), 8.45 (d, J = 5.4 Hz, 1 H), 8.32–8.36 (m, 2 H), 7.62–7.67 (m, 1 H), 7.45 (t, J = 7.8 Hz, 1 H), 7.36 (d, J = 7.8 Hz, 1 H), 7.20 (br s, 1 H), 7.14 (d, J = 5.4 Hz, 1 H), 6.95 (d, J = 9.8 Hz, 1 H), 4.11 (s, 3 H). LC–MS (m/z): 344.1 [M + H]+.
3-((4-(2-(Hydroxymethyl)-6-methoxypyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (23a)
In a vial 7l (20 mg, 69 μmol) and 3-aminobenzonitrile (24 mg, 206 μmol) were added to n-BuOH (1 mL), and the reaction was heated to 150 °C for 2 h, then 180 °C for 3 h. The reaction was filtered and washed with diethyl ether to yield the desired product in the precipitate as a white solid (7.2 mg, 28%). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.96 (s, 1 H), 8.93 (d, J = 9.5 Hz, 1 H), 8.59 (d, J = 5.1 Hz, 1 H), 8.38 (t, J = 1.5 Hz, 1 H), 7.93–7.98 (m, 1 H), 7.47–7.56 (m, 2 H), 7.41 (dt, J = 7.3, 1.5 Hz, 1 H), 7.14 (d, J = 9.5 Hz, 1 H), 5.59 (t, J = 5.1 Hz, 1 H), 4.80 (d, J = 5.1 Hz, 2 H), 4.00 (s, 3 H). LC–MS (m/z): 374.1 [M + H]+.
3-((4-(2-(Hydroxymethyl)-6-morpholinopyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (23b)
In a vial 7m (30 mg, 87 μmol) and 3-aminobenzonitrile (20 mg, 173 μmol) were stirred in 1.5 mL of n-BuOH, and the reaction was subjected to microwave irradiation at 150 °C for 3 h. The reaction was filtered and washed with diethyl ether to give the crude product in the precipitate. Column purification was undertaken using MeOH/DCM 2–30% to afford the title compound as a white solid (20.8 mg, 56%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.94 (s, 1 H), 8.76 (d, J = 9.8 Hz, 1 H), 8.56 (d, J = 5.4 Hz, 1 H), 8.36 (s, 1 H), 7.95–7.99 (m, 1 H), 7.53 (t, J = 8.3 Hz, 1 H), 7.46 (d, J = 5.4 Hz, 1 H), 7.36–7.42 (m, 2 H), 5.52 (t, J = 4.9 Hz, 1 H), 4.76 (d, J = 4.9 Hz, 2 H), 3.76 (t, J = 4.9 Hz, 4 H), 3.52 (t, J = 4.9 Hz, 4 H). LC–MS (m/z): 429.1 [M + H]+.
3-((4-(2-Cyclopropyl-6-morpholinopyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)benzonitrile (23c)
In a vial 7o (25 mg, 70 μmol), cesium carbonate (69 mg, 220 μmol), 3-aminopyridine (17 mg, 140 μmol), palladium(II) acetate (0.8 mg, 3.5 μmol), and Xantphos (4.1 mg, 7.0 μmol) were stirred in anhydrous dioxane (2 mL), the reaction mixture was sealed then evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 160 °C for 1 h. The reaction was filtered through Celite and washed with EtOAc, then evaporated to dryness. Column purification was performed using EtOAc/hex 25–75%, to afford the title compound as an orange solid (21.5 mg, 70%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.51 (d, J = 9.8 Hz, 1 H), 8.46 (d, J = 5.4 Hz, 1 H), 8.39 (s, 1 H), 7.62 (dd, J = 8.3, 2.0 Hz, 1 H), 7.40–7.49 (m, 2 H), 7.34 (d, J = 7.8 Hz, 1 H), 7.22–7.25 (s, 1 H), 6.95 (d, J = 9.8 Hz, 1 H), 3.85 (t, J = 4.4 Hz, 4 H), 3.57 (t, J = 5.4 Hz, 4 H), 2.22–2.29 (m, 1 H), 1.23–1.28 (m, 2 H), 1.08–1.14 (m, 2 H) LC–MS (m/z): 439.1 [M + H]+.
4-(2-Cyclopropylpyrazolo[1,5-b]pyridazin-3-yl)-N-(pyridin-3-yl)pyrimidin-2-amine (23d)
In a vial 3-(2-chloropyrimidin-4-yl)-2-cyclopropylpyrazolo[1,5-b]pyridazine 7e (30 mg, 110 μmol), cesium carbonate (108 mg, 331 μmol), 3-aminopyridine (21 mg, 220 μmol), palladium(II) acetate (1.2 mg, 5.5 μmol), and Xantphos (6.4 mg, 11 μmol) were stirred in anhydrous dioxane (1.5 mL), the reaction mixture was sealed then evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 160 °C for 1 h. The reaction was filtered through Celite and washed with EtOAc, then evaporated to dryness. Reverse phase column purification was done using ACN/water 5–95% with 0.1% formic acid, to afford the title compound as a white solid (17.0 mg, 47%). 1H NMR (500 MHz, methanol-d4) δ ppm 8.92 (d, J = 2.4 Hz, 1 H), 8.89 (dd, J = 8.8, 2.0 Hz, 1 H), 8.52 (d, J = 5.4 Hz, 1 H), 8.40 (dd, J = 4.4, 2.0 Hz, 1 H), 8.32 (ddd, J = 8.3, 2.4, 1.5 Hz, 1 H), 8.17 (dd, J = 4.6, 1.5 Hz, 1 H), 7.47 (d, J = 5.4 Hz, 1 H), 7.39 (dd, J = 8.3, 4.6 Hz, 1 H), 7.28 (dd, J = 8.8, 4.4 Hz, 1 H), 2.45–2.53 (m, 1 H), 1.11–1.23 (m, 4 H). LC–MS (m/z): 330.1 [M + H]+.
4-(6-Methoxypyrazolo[1,5-b]pyridazin-3-yl)-N-(pyridin-3-yl)pyrimidin-2-amine (23e)
In a vial 7c (30 mg, 115 μmol), cesium carbonate (112 mg, 344 μmol), 3-aminopyridine (22 mg, 230 μmol), palladium(II) acetate (1.3 mg, 5.7 μmol), and Xantphos (6.6 mg, 12 μmol) were stirred in anhydrous dioxane (1.5 mL). The reaction mixture was sealed then evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 160 °C for 1 h. The reaction was filtered through Celite and washed with MeOH, then evaporated to dryness. Reverse phase column purification was performed ACN/water 5–95% with 0.1% formic acid, affording the title compound as a yellow solid (10.3 mg, 28%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.75 (s, 1 H), 9.00 (d, J = 9.8 Hz, 1 H), 8.88 (d, J = 2.9 Hz, 1 H), 8.71 (s, 1 H), 8.49 (d, J = 5.4 Hz, 1 H), 8.23 (ddd, J = 8.3, 2.9, 1.5 Hz, 1 H), 8.19 (dd, J = 4.9, 1.5 Hz, 1 H), 7.39 (d, J = 5.4 Hz, 1 H), 7.36 (dd, J = 8.3, 4.9 Hz, 1 H), 7.18 (d, J = 9.8 Hz, 1 H), 4.02 (s, 3 H). LC–MS (m/z): 320.1 [M + H]+.
(6-Methoxy-3-(2-(pyridin-3-ylamino)pyrimidin-4-yl)pyrazolo[1,5-b]pyridazin-2-yl)methanol (23f)
In a vial 7l (30 mg, 103 μmol), cesium carbonate (101 mg, 309 μmol), 3-aminopyridine (19.4 mg, 206 μmol), palladium(II) acetate (1.2 mg, 8.2 μmol), and Xantphos (6.0 mg, 10.3 μmol) were stirred in anhydrous dioxane (1.5 mL). The reaction mixture was sealed then evacuated and backfilled with nitrogen 3×. The reaction mixture was then subjected to MW irradiation at 160 °C for 1.5 h. The reaction was filtered through Celite and washed with MeOH, then evaporated to dryness. Reverse phase column purification was performed ACN/water 5–95% with 0.1% formic acid, to afford the title compound as a yellow solid (15.5 mg, 43%). 1H NMR (500 MHz, DMSO-d6) δ ppm 9.75 (s, 1 H), 8.92 (d, J = 9.8 Hz, 1 H), 8.88 (d, J = 2.4 Hz, 1 H), 8.54 (d, J = 5.4 Hz, 1 H), 8.24 (ddd, J = 8.3, 2.4, 1.5 Hz, 1 H), 8.18 (dd, J = 4.4, 1.5 Hz, 1 H), 7.45 (d, J = 5.4 Hz, 1 H), 7.34 (dd, J = 8.3, 4.4 Hz, 1 H), 7.13 (d, J = 9.8 Hz, 1 H), 5.53–5.58 (m, 1 H), 4.80 (d, J = 2.4 Hz, 2 H), 4.00 (s, 3 H). LC–MS (m/z): 350.2 [M + H]+.
4-(2-Cyclopropyl-6-methoxypyrazolo[1,5-b]pyridazin-3-yl)-N-(pyridin-3-yl)pyrimidin-2-amine (23g)
In a vial 7n (30 mg, 99 μmol), cesium carbonate (97 mg, 298 μmol), 3-aminopyridine (19 mg, 199 μmol), palladium(II) acetate (1.1 mg, 5.0 μmol), and Xantphos (5.8 mg, 10.0 μmol) were stirred in anhydrous dioxane (1.5 mL). The reaction mixture was sealed then evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 160 °C for 1 h. The reaction was filtered through Celite and washed with MeOH, then evaporated to dryness. Reverse phase preparative HPLC purification was performed using ACN/water 5–95% with 0.1% formic acid, to provide the title compound as a yellow solid (7.4 mg, 21%). 1H NMR (500 MHz, methanol-d4) δ ppm 8.90 (d, J = 2.9 Hz, 1 H), 8.77 (d, J = 9.3 Hz, 1 H), 8.49 (d, J = 5.4 Hz, 1 H), 8.31 (ddd, J = 8.3, 2.9, 1.5 Hz, 1 H), 8.17 (dd, J = 4.9, 1.5 Hz, 1 H), 7.45 (d, J = 5.4 Hz, 1 H), 7.39 (dd, J = 8.3, 4.9 Hz, 1 H), 6.94 (d, J = 9.3 Hz, 1 H), 4.04 (s, 3 H), 2.36–2.43 (m, 1 H), 1.11–1.17 (m, 2 H), 1.05–1.10 (m, 2 H). LC–MS (m/z): 360.2 [M + H]+.
4-(2-Cyclopropyl-6-morpholinopyrazolo[1,5-b]pyridazin-3-yl)-N-(pyridin-3-yl)pyrimidin-2-amine (23h)
In a vial 7o (30 mg, 84 μmol), cesium carbonate (82 mg, 252 μmol), 3-aminopyridine (16 mg, 168 μmol), palladium(II) acetate (0.9 mg, 4.2 μmol), and Xantphos (5 mg, 8.4 μmol) were stirred in anhydrous dioxane (1.5 mL), and the reaction mixture was sealed then evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 160 °C for 1 h. The reaction was filtered through Celite and washed with EtOAc, then evaporated to dryness. Reverse phase preparative HPLC purification was performed using ACN/water 5–95% with 0.1% formic acid, to afford the title compound as a pale-yellow solid (11.0 mg, 32%). 1H NMR (500 MHz, methanol-d4) δ ppm 8.91 (d, J = 2.9 Hz, 1 H), 8.62 (d, J = 9.8 Hz, 1 H), 8.46 (d, J = 5.4 Hz, 1 H), 8.29 (ddd, J = 8.3, 2.9, 1.5 Hz, 1 H), 8.16 (dd, J = 4.9, 1.5 Hz, 1 H), 7.43 (d, J = 5.4 Hz, 2 H), 7.40 (dd, J = 8.3, 4.9 Hz, 1 H), 7.17 (d, J = 9.8 Hz, 1 H), 3.83 (t, J = 4.9 Hz, 4 H), 3.55 (t, J = 4.9 Hz, 4 H), 2.32–2.42 (m, 1 H), 1.10–1.15 (m, 2 H), 1.04–1.08 (m, 2 H). LC–MS (m/z): 415.1 [M + H]+.
4-(6-Morpholinopyrazolo[1,5-b]pyridazin-3-yl)-N-(pyridin-3-yl)pyrimidin-2-amine (23i)
In a vial 7b (30 mg, 95 μmol), cesium carbonate (93 mg, 284 μmol), 3-aminopyridine (18 mg, 189 μmol), palladium(II) acetate (1.1 mg, 4.7 μmol), and Xantphos (5.5 mg, 9.5 μmol) were stirred in anhydrous dioxane (1.5 mL), and the reaction mixture was sealed then evacuated and backfilled with N2 three times. The reaction mixture was then subjected to microwave irradiation at 160 °C for 1 h. The reaction was filtered through Celite and washed with EtOAc, then evaporated to dryness. Reverse phase preparative HPLC purification was performed using ACN/water 5–95% with 0.1% formic acid, affording the title compound as a white solid (7.2 mg, 20%). 1H NMR (500 MHz, methanol-d4) δ ppm 8.89–8.93 (m, 1 H), 8.79 (d, J = 9.8 Hz, 1 H), 8.38–8.42 (m, 2 H), 8.26 (ddd, J = 8.3, 2.9, 1.5 Hz, 1 H), 8.15–8.21 (m, 1 H), 7.40 (dd, J = 8.3, 4.9 Hz, 1 H), 7.24–7.29 (m, 2 H), 3.85 (t, J = 4.9 Hz, 4 H), 3.60 (t, J = 4.9 Hz, 4 H). LC–MS (m/z): 375.1 [M + H]+.
4-((4-(6-Morpholinopyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)cyclohexan-1-ol (23j)
In a vial 2–4b (20 mg, 63 μmol), and trans-4-amino-cyclohexanol (36 mg, 316 μmol) were stirred in n-BuOH (2 mL) and heated to 110 °C for 72 h. Reaction mixture was then concentrated and column purification was undertaken using MeOH/DCM, 2–5% to afford the title compound as a white solid (16.3 mg, 65.3%). 1H NMR (500 MHz, methanol-d4) δ ppm 8.82 (d, J = 9.8 Hz, 1 H), 8.37 (s, 1 H), 8.18 (d, J = 5.4 Hz, 1 H), 7.30 (d, J = 9.8 Hz, 1 H), 6.99 (d, J = 5.4 Hz, 1 H), 3.80–3.90 (m, 5 H), 3.56–3.68 (m, 5 H), 2.12–2.20 (m, 2 H), 2.01–2.09 (m, 2 H), 1.38–1.55 (m, 4 H). LC–MS (m/z): 396.2 [M + H]+.
4-((4-(6-Methoxypyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)cyclohexan-1-ol (23k)
In a vial 7c (50 mg, 191 μmol) and trans-4-aminocyclohexanol (44 mg, 392 μmol) were stirred in n-BuOH (1.5 mL) and subjected to microwave irradiation at 150 °C for 2.5 h. The reaction was concentrated in vacuo then subjected to column purification MeOH/DCM 0–5%, to afford the title compound as a yellow solid (38.5 mg, 59%). 1H NMR (500 MHz, methanol-d4) δ ppm 8.88–8.95 (m, 1 H), 8.42 (s, 1 H), 8.18 (d, J = 5.4 Hz, 1 H), 6.98–7.03 (m, 2 H), 4.07 (s, 3 H), 3.75–3.84 (m, 1 H), 3.58–3.66 (m, 1 H), 2.08–2.17 (m, 2 H), 1.99–2.06 (m, 2 H), 1.35–1.52 (m, 4 H). LC–MS (m/z): 341.2 [M + H]+.
4-((4-(2-Cyclopropyl-6-methoxypyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)cyclohexan-1-ol (23l)
In a vial 7n (30 mg, 99 μmol) and trans-4-aminocyclohexanol (23 mg, 199 μmol) were stirred in n-BuOH (1.5 mL) subjected to microwave irradiation at 150 °C for 2 h. The reaction was concentrated in vacuo then subjected to column purification MeOH/DCM 0–10%, to afford the title compound as a white solid (17.3 mg, 66% based on recovered starting material). 1H NMR (500 MHz, methanol-d4) δ ppm 8.69–8.76 (m, 1 H), 8.23 (d, J = 5.4 Hz, 1 H), 7.16 (d, J = 5.4 Hz, 1 H), 6.92 (d, J = 9.3 Hz, 1 H), 4.03 (s, 3 H), 3.75–3.85 (m, 1 H), 3.57–3.65 (m, 1 H), 2.33–2.41 (m, 1 H), 2.07–2.16 (m, 2 H), 1.99–2.05 (m, 2 H), 1.36–1.48 (m, 4 H), 1.09–1.13 (m, 2 H), 1.04–1.07 (m, 2 H). LC–MS (m/z): 381.2 [M + H]+.
4-((4-(2-(Hydroxymethyl)-6-methoxypyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-yl)amino)cyclohexan-1-ol (23m)
In a vial 7l (50 mg, 171 μmol) and trans-4-aminocyclohexanol (39.5 mg, 343 μmol) were stirred in n-BuOH (1.5 mL) and subjected to microwave irradiation at 150 °C for 2 h. The reaction was concentrated in vacuo then subjected to column purification EtOAc/hex 25–100%, to afford the title compound as a yellow solid (33.2 mg, 52%). 1H NMR (500 MHz, methanol-d4) δ ppm 8.70 (d, J = 9.8 Hz, 1 H), 8.26 (d, J = 5.4 Hz, 1 H), 7.09 (d, J = 5.4 Hz, 1 H), 7.00 (d, J = 9.8 Hz, 1 H), 4.91 (s, 2 H), 4.06 (s, 3 H), 3.75–3.83 (m, 1 H), 3.57–3.65 (m, 1 H), 2.07–2.15 (m, 2 H), 2.00–2.03 (m, 2 H), 1.35–1.50 (m, 4 H). LC–MS (m/z): 371.2 [M + H]+.
2-Chloro-4-methyl-6-((trimethylsilyl)ethynyl)pyrimidine (24a)
In a reaction vial 2,4-dichloro-6-methylpyrimidine (1 g, 6.1 mmol), Pd(PPh3)2Cl2 (86 mg, 123 μmol), and CuI (35 mg, 184 μmol) were added. A degassed solution of triethylamine (2.6 mL, 18.4 mmol) and THF (12 mL) were added under N2 to the reaction vial, followed by ethynyltrimethylsilane (961 μL, 6.75 mmol). The reaction mixture was heated to 50 °C for 16 h. Reaction mixture was then filtered through Celite and washed with EtOAc. The reaction mixture was concentrated in vacuo then subjected to column purification EtOAc/hexanes 0–10%, to yield the desired product as an off-white solid (370 mg, 27%). 1H NMR (500 MHz, chloroform-d) δ ppm 7.16 (s, 1 H), 2.48 (s, 3 H), 0.24 (s, 9 H). LC–MS (m/z): 225.0 (35Cl), 227.0 (37Cl) [M + H]+.
2-Chloro-4-ethynyl-6-methylpyrimidine (24b)
In a vial 24a (370 mg, 1.65 mmol) was stirred in MeOH (10 mL), and KOH (0.46 mg, 8.2 μmol) was added. After 30 min the reaction was stopped and concentrated in vacuo, to yield the product as a pale-yellow solid (240 mg, 96%). 1H NMR (500 MHz, chloroform-d) δ ppm 7.23 (s, 1 H), 3.40 (s, 1 H), 2.55 (s, 3 H). LC–MS (m/z): 152.7 (35Cl), 154.7 (37Cl) [M + H]+.
3-(2-Chloro-6-methylpyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (25)
(Aminooxy)sulfonic acid (889 mg, 7.86 mmol) was dissolved in 7 mL of water, and to the clear solution was added saturated sodium bicarbonate until effervescence ceased (pH ∼ 6). The clear colorless solution was heated to 70 °C, and pyridazine (378 μL, 5.24 mmol) was added and the reaction mixture was stirred at 70 °C for 2 h. The reaction was cooled to room temperature, neutralized with saturated sodium bicarbonate (until effervescence ceased) and then solution of 24b (200 mg, 1.31 mmol) in DCM (20 mL) was added followed by KOH (74 mg, 1.3 mmol) and the reaction was stirred at room temperature for 16 h. The reaction mixture was diluted with DCM and washed with water and brine then dried over sodium sulfate, filtered and evaporated to get the crude product which was purified by column chromatography using EtOAc/hex 30–50% to provide the desired compound as a pink solid (121 mg, 38%). 1H NMR (500 MHz, chloroform-d) δ ppm 9.06 (dd, J = 9.3, 2.0 Hz, 1 H), 8.53 (s, 1 H), 8.46 (dd, J = 4.4, 2.0 Hz, 1 H), 7.40 (s, 1 H), 7.29 (dd, J = 9.3, 4.4 Hz, 1 H), 2.59 (s, 3 H). LC–MS (m/z): 246.1 (35Cl), 248.0 (37Cl) [M + H]+.
2-Chloro-5-methyl-4-((trimethylsilyl)ethynyl)pyrimidine (26a)
In a reaction vial 2,4-dichloro-5-methylpyrimidine (1 g, 6.1 mmol), Pd(PPh3)2Cl2 (86 mg, 123 μmol), and CuI (35 mg, 184 μmol) were added. A degassed solution of triethylamine (2.6 mL, 18.4 mmol) in THF (12 mL) was added under N2 to the reaction vial, followed by ethynyltrimethylsilane (961 μL, 6.75 mmol). The reaction mixture was heated to 50 °C for 16 h. The reaction was then filtered through Celite and washed with EtOAc. The reaction mixture was concentrated in vacuo and subjected to column purification EtOAc/hexanes 0–10%, to provide the compound as a white solid (713 mg, 52%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.46 (s, 1 H), 2.36 (s, 3 H), 0.28 (s, 9 H). LC–MS (m/z): 225.0 (35Cl), 227.0 (37Cl) [M + H]+.
2-Chloro-4-ethynyl-5-methylpyrimidine (26b)
In a vial 26a (713 mg, 3.2 mmol) was stirred in MeOH (20 mL), and KOH (0.9 mg, 16 μmol) was added, and the reaction was stirred at room temperature. After 30 min the reaction was stopped and concentrated in vacuo, then subjected to column purification EtOAc/hex 10–25% to obtain the desired compound as a white solid (361 mg, 75%). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.74 (s, 1 H), 5.13 (s, 1 H), 2.31 (s, 3 H). LC–MS (m/z): 152.8 (35Cl), 154.7 (37Cl) [M + H]+.
3-(2-Chloro-5-methylpyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (27)
(Aminooxy)sulfonic acid (889 mg, 7.86 mmol) was dissolved in 7 mL of water, and saturated sodium bicarbonate was added to the clear solution until effervescence ceased (pH ∼ 6). The clear colorless solution was heated at 70 °C, and pyridazine (378 μL, 5.24 mmol) was added and the reaction mixture was stirred at 70 °C for 2 h. The reaction was then cooled to room temperature, neutralized with saturated sodium bicarbonate (until effervescence ceased) and then a solution of 26b (200 mg, 1.31 mmol) in DCM (20 mL) was added followed by KOH (148 mg, 2.6 mmol) and the reaction was stirred at room temperature for 16 h. The reaction mixture was diluted with DCM and washed with water and brine then dried over sodium sulfate, filtered and evaporated to get the crude product which was purified by column chromatography using MeOH/DCM 0–2% to yield the desired compound as a brown solid (206 mg, 64%). 1H NMR (500 MHz, chloroform-d) δ ppm 9.13 (dd, J = 9.3, 2.0 Hz, 1 H), 8.55 (s, 1 H), 8.50 (dd, J = 4.4, 2.0 Hz, 1 H), 8.45 (s, 1 H), 7.32 (dd, J = 9.3, 4.4 Hz, 1 H), 2.60 (s, 3 H). LC–MS (m/z): 246.1 (35Cl), 248.0 (37Cl) [M+1].
2-(3-Chlorophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (28)
In a vial 3-chloroiodobenzene (52 μL, 420 μmol), bis(pinacolato)diboron (160 mg, 629 μmol), potassium acetate (144 mg, 1.47 mmol), and PdCl2(dppf)·CH2Cl2 (17 mg, 21 mmol) were combined, and the vial was sealed. Anhydrous dioxane (3 mL) was added and the reaction was evacuated and backfilled with N2 three times, then subjected to microwave irradiation at 160 °C for 30 min. The reaction was cooled and filtered through Celite, concentrated in vacuo and taken forward without purification. LC–MS (m/z): 156.7 (35Cl), 158.8 (37Cl) [M + H]+ (boronic acid).
3-(6-Chloropyridin-2-yl)pyrazolo[1,5-b]pyridazine (29)
(Aminooxy)sulfonic acid (296 mg, 2.62 mmol) was dissolved in 2 mL of water, and to the clear solution was added saturated sodium bicarbonate until effervescence ceased (pH ∼ 6). The clear colorless solution was heated to 70 °C, then pyridazine (126 μL, 1.74 mmol) was added, and the reaction mixture was stirred at 70 °C for 2 h. The reaction mixture was then cooled to room temperature, neutralized with saturated sodium bicarbonate (until effervescence ceased) and then solution of 2-chloro-6-ethynylpyridine (120 mg, 872 μmol) in DCM (10 mL) was added followed by KOH (49 mg, 872 μmol) and the reaction mixture was stirred at 40 °C for 72 h, partial conversion to product was observed. Additional KOH (49 mg, 872 μmol) was added and the reaction was stirred for another 24 h at 40 °C. The reaction mixture was diluted with DCM and water and extracted. The organic layer was washed with brine then dried over sodium sulfate, filtered and evaporated to get the crude product which was purified by column chromatography using EtOAc/hex 25% to yield the product as a tan solid (18.5 mg, 15% based on recovered starting material). 1H NMR (400 MHz, chloroform-d) δ ppm 9.00 (dd, J = 8.8, 2.2 Hz, 1 H), 8.44 (s, 1 H), 8.38 (dd, J = 4.4, 2.2 Hz, 1 H), 7.69 (t, J = 8.1 Hz, 1 H), 7.58 (d, J = 8.1 Hz, 1 H), 7.13–7.22 (m, 2 H). LC–MS (m/z): 231.0 (35Cl), 233.0 (37Cl) [M + H]+.
3-(2-Chloropyridin-4-yl)pyrazolo[1,5-b]pyridazine (30)
(Aminooxy)sulfonic acid (2.06 g, 18.2 mmol) was dissolved in 4 mL of water, and saturated sodium bicarbonate was added to the clear solution until effervescence ceased (pH ∼ 6). The clear colorless solution was heated at 70 °C, and pyridazine (875 μL, 12.1 mmol) was added. The reaction mixture heated at 70 °C for 2 h, then cooled to room temperature, neutralized with saturated sodium bicarbonate (until effervescence ceased) and then a solution of 2-chloro-4-ethynylpyridine (417 mg, 3.03 mmol) in DCM (15 mL) and KOH (170 mg, 3.0 mmol) was added then reaction mixture was stirred for 16 h at room temperature. The reaction mixture was diluted with DCM and washed with water and brine then dried over magnesium sulfate, filtered and evaporated to get the crude product which was purified by column chromatography using MeOH/DCM 0–2% followed by an additional column purification using EtOAc/hexanes 25–100% to afford the desired compound as a brown solid (173 mg, 25%). 1H NMR (500 MHz, chloroform-d) δ ppm 8.46 (d, J = 5.4 Hz, 1 H), 8.43 (dd, J = 4.4, 2.0 Hz, 1 H), 8.36 (s, 1 H), 8.28 (dd, J = 9.3, 2.0 Hz, 1 H), 7.53–7.56 (m, 1 H), 7.44 (dd, J = 5.4, 1.5 Hz, 1 H), 7.22 (dd, J = 9.3, 4.4 Hz, 1 H). LC–MS (m/z): 230.9 (35Cl), 233.0 (37Cl) [M + H]+.
Acknowledgments
This work was supported by National Institutes of Health Grants (R01AI114685 (M.P.P. and M.N.), R01AI082577 (M.P.P.), R56AI099476 (M.P.P.), R01AI124046 (M.P.P.), R21AI127594 (M.P.P.), the Spanish Ministerio de Economía, Industria y Competitividad (M.N., Grant SAF2015-71444-P; D.G.P., Grant SAF2016-79957-R., and Subdirección General de Redes y Centros de Investigación Cooperativa (RICET) (M.N., Grant RD16/0027/0019; D.G.P., Grant RD16/0027/0014), Grant RTI2018-097210-B-100 (MINCIU-FEDER) to F.G. C.R.C. acknowledges grant support from the NIH-NIAID (Grant R21AI126296) and the Bill and Melinda Gates Foundation (Grant OPP1171488), as well as the technical assistance of Brian M. Suzuki for screening adult S. mansoni. We are grateful to AstraZeneca for performing the in vitro ADME experiments presented throughout and to Charles River Labs for the in vitro ADME data presented in Tables S3–S5 in the Supporting Information. We thank GSK Tres Cantos open lab foundation for running the PK studies discussed in this publication. An academic license for ChemAxon (https://www.chemaxon.com) is gratefully acknowledged. We thank Dr. Melissa Buskes for help in the preparation of this manuscript.
Glossary
Abbreviations Used
- BBB
blood–brain barrier
- NECT
nifurtimox–eflornithine combination therapy
- iv
intravenous
- HTS
high-throughput screen
- CNS
central nervous system
- MPO
multiparameter optimization
- ADME
absorption, distribution, metabolism, and excretion
- RH
rat hepatocyte
- Clint
intrinsic clearance
- HAT
human African trypanosomiasis
- HLM
human liver microsome
- GSK-3β
glycogen synthase kinase 3β
- CDK
cyclin dependent kinase
- PPB
plasma protein binding
- aq sol.
thermodynamic aqueous solubility
- HBD
hydrogen bond donor
- SAR
structure–activity relationship
- PK
pharmacokinetic
- ip
intraperitoneal.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01741.
Cidality assay results, additional compounds and associated biological data not presented in the manuscript, comparison of T. b. brucei pEC50 inhibition data to literature values for GSK-3β pIC50, CDK-2 pIC50, and CDK-4 pIC50, additional ADME data (including mouse plasma stability, CYP450 inhibition and induction, Caco-2), kinase panel profiling of 20g, blood and brain concentration of 20r as measured in the PK study, parasitemia levels measured during efficacy study, biological data of compound set against T. cruzi, L. donovani, S. mansoni, L6 and THP-1 cells, and HPLC traces of all compounds tested (PDF)
Molecular formula strings and some data (CSV)
The authors declare no competing financial interest.
Notes
⊥ C.C.-O.: current e-mail, carlos.cordon-obras@pasteur.fr.
Supplementary Material
References
- About Sleeping Sickness. https://www.dndi.org/diseases-projects/hat/ (accessed Jan 25, 2018).
- Trypanosomiasis, human African (sleeping sickness). http://www.who.int/mediacentre/factsheets/fs259/en/ (accessed Jan 25, 2018).
- Pollastri M. P. Fexinidazole: A New Drug for African Sleeping Sickness on the Horizon. Trends Parasitol. 2018, 34 (3), 178–179. 10.1016/j.pt.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun R.; Blum J.; Chappuis F.; Burri C. Human African trypanosomiasis. Lancet 2010, 375 (9709), 148–159. 10.1016/S0140-6736(09)60829-1. [DOI] [PubMed] [Google Scholar]
- Barrett M. P.; Boykin D. W.; Brun R.; Tidwell R. R. Human African trypanosomiasis: pharmacological re-engagement with a neglected disease. Br. J. Pharmacol. 2007, 152 (8), 1155–1171. 10.1038/sj.bjp.0707354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco J. R.; Simarro P. P.; Diarra A.; Ruiz-Postigo J. A.; Samo M.; Jannin J. G. Monitoring the use of nifurtimox-eflornithine combination therapy (NECT) in the treatment of second stage gambiense human African trypanosomiasis. Res. Rep. Trop. Med. 2012, 3, 93–101. 10.2147/RRTM.S34399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priotto G.; Kasparian S.; Ngouama D.; Ghorashian S.; Arnold U.; Ghabri S.; Karunakara U. Nifurtimox-eflornithine combination therapy for second-stage Trypanosoma brucei gambiense sleeping sickness: a randomized clinical trial in Congo. Clin. Infect. Dis. 2007, 45 (11), 1435–1442. 10.1086/522982. [DOI] [PubMed] [Google Scholar]
- Priotto G.; Kasparian S.; Mutombo W.; Ngouama D.; Ghorashian S.; Arnold U.; Ghabri S.; Baudin E.; Buard V.; Kazadi-Kyanza S.; Ilunga M.; Mutangala W.; Pohlig G.; Schmid C.; Karunakara U.; Torreele E.; Kande V. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial. Lancet 2009, 374 (9683), 56–64. 10.1016/S0140-6736(09)61117-X. [DOI] [PubMed] [Google Scholar]
- Phase II/III studies show high efficacy and safety of fexinidazole, the first oral treatment for sleeping sickness. DNDi, 2017. [Google Scholar]
- Mesu V. K. B. K.; Kalonji W. M.; Bardonneau C.; Mordt O. V.; Blesson S.; Simon F.; Delhomme S.; Bernhard S.; Kuziena W.; Lubaki J.-P. F.; Vuvu S. L.; Ngima P. N.; Mbembo H. M.; Ilunga M.; Bonama A. K.; Heradi J. A.; Solomo J. L. L.; Mandula G.; Badibabi L. K.; Dama F. R.; Lukula P. K.; Tete D. N.; Lumbala C.; Scherrer B.; Strub-Wourgaft N.; Tarral A. Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: a pivotal multicentre, randomised, non-inferiority trial. Lancet 2018, 391 (10116), 144–154. 10.1016/S0140-6736(17)32758-7. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency recommends fexinidazole, the first all-oral treatment for sleeping sickness. https://www.dndi.org/2018/media-centre/press-releases/ema-recommends-fexinidazole-first-all-oral-treatment-sleeping-sickness/ (accessed Dec 5, 2018).
- Sokolova A. Y.; Wyllie S.; Patterson S.; Oza S. L.; Read K. D.; Fairlamb A. H. Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis. Antimicrob. Agents Chemother. 2010, 54 (7), 2893–2900. 10.1128/AAC.00332-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S.; Wyllie S. Nitro drugs for the treatment of trypanosomatid diseases: past, present, and future prospects. Trends Parasitol. 2014, 30 (6), 289–298. 10.1016/j.pt.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivotal clinical trial to begin for first oral drug candidate specifically developed for sleeping sickness. https://www.dndi.org/2015/media-centre/press-releases/pr-scyx-7158/ (accessed Jan 31, 2018).
- Nwaka S.; Ramirez B.; Brun R.; Maes L.; Douglas F.; Ridley R. Advancing drug innovation for neglected diseases-criteria for lead progression. PLoS Neglected Trop. Dis. 2009, 3 (8), e440 10.1371/journal.pntd.0000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neglected Tropical Diseases. http://www.who.int/neglected_diseases/diseases/en/ (accessed Jan 25, 2018).
- Klug D. M.; Gelb M. H.; Pollastri M. P. Repurposing strategies for tropical disease drug discovery. Bioorg. Med. Chem. Lett. 2016, 26 (11), 2569–2576. 10.1016/j.bmcl.2016.03.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz R.; Luengo-Arratta S. A.; Seixas J. D.; Amata E.; Devine W.; Cordon-Obras C.; Rojas-Barros D. I.; Jimenez E.; Ortega F.; Crouch S.; Colmenarejo G.; Fiandor J. M.; Martin J. J.; Berlanga M.; Gonzalez S.; Manzano P.; Navarro M.; Pollastri M. P. Identification and characterization of hundreds of potent and selective inhibitors of Trypanosoma brucei growth from a kinase-targeted library screening campaign. PLoS Neglected Trop. Dis. 2014, 8 (10), e3253 10.1371/journal.pntd.0003253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oduor R. O.; Ojo K. K.; Williams G. P.; Bertelli F.; Mills J.; Maes L.; Pryde D. C.; Parkinson T.; Van Voorhis W. C.; Holler T. P. Trypanosoma brucei glycogen synthase kinase-3, a target for anti-trypanosomal drug development: a public-private partnership to identify novel leads. PLoS Neglected Trop. Dis. 2011, 5 (4), e1017 10.1371/journal.pntd.0001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo K. K.; Gillespie J. R.; Riechers A. J.; Napuli A. J.; Verlinde C. L.; Buckner F. S.; Gelb M. H.; Domostoj M. M.; Wells S. J.; Scheer A.; Wells T. N.; Van Voorhis W. C. Glycogen synthase kinase 3 is a potential drug target for African trypanosomiasis therapy. Antimicrob. Agents Chemother. 2008, 52 (10), 3710–3717. 10.1128/AAC.00364-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naula C.; Parsons M.; Mottram J. C. Protein kinases as drug targets in trypanosomes and Leishmania. Biochim. Biophys. Acta, Proteins Proteomics 2005, 1754 (1–2), 151–159. 10.1016/j.bbapap.2005.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M.; Worthey E. A.; Ward P. N.; Mottram J. C. Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics 2005, 6, 127. 10.1186/1471-2164-6-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1 (6), 435–449. 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro G.; Chokpaiboon S.; De Muylder G.; Bray W. M.; Nisam S. C.; McKerrow J. H.; Pudhom K.; Linington R. G. Hit-to-lead development of the chamigrane endoperoxide merulin A for the treatment of African sleeping sickness. PLoS One 2012, 7 (9), e46172 10.1371/journal.pone.0046172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Target Product Profile - Sleeping Sickness. https://www.dndi.org/diseases-projects/hat/hat-target-product-profile/ (accessed Jun 20, 2019).
- Tavares F. X.; Boucheron J. A.; Dickerson S. H.; Griffin R. J.; Preugschat F.; Thomson S. A.; Wang T. Y.; Zhou H. N-Phenyl-4-pyrazolo[1,5-b]pyridazin-3-ylpyrimidin-2-amines as Potent and Selective Inhibitors of Glycogen Synthase Kinase 3 with Good Cellular Efficacy. J. Med. Chem. 2004, 47 (19), 4716–4730. 10.1021/jm040063i. [DOI] [PubMed] [Google Scholar]
- Xiao J.; Guo Z.; Guo Y.; Chu F.; Sun P. Inhibitory mode of N-phenyl-4-pyrazolo[1,5-b] pyridazin-3-ylpyrimidin-2-amine series derivatives against GSK-3: molecular docking and 3D-QSAR analyses. Protein Eng., Des. Sel. 2006, 19 (2), 47–54. 10.1093/protein/gzi074. [DOI] [PubMed] [Google Scholar]
- Schaenzer A. J.; Wlodarchak N.; Drewry D. H.; Zuercher W. J.; Rose W. E.; Ferrer C. A.; Sauer J. D.; Striker R. GW779439X and Its Pyrazolopyridazine Derivatives Inhibit the Serine/Threonine Kinase Stk1 and Act As Antibiotic Adjuvants against beta-Lactam-Resistant Staphylococcus aureus. ACS Infect. Dis. 2018, 4, 1508–1518. 10.1021/acsinfecdis.8b00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urich R.; Grimaldi R.; Luksch T.; Frearson J. A.; Brenk R.; Wyatt P. G. The design and synthesis of potent and selective inhibitors of Trypanosoma brucei glycogen synthase kinase 3 for the treatment of human african trypanosomiasis. J. Med. Chem. 2014, 57 (18), 7536–7549. 10.1021/jm500239b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodland A.; Grimaldi R.; Luksch T.; Cleghorn L. A.; Ojo K. K.; Van Voorhis W. C.; Brenk R.; Frearson J. A.; Gilbert I. H.; Wyatt P. G. From on-target to off-target activity: identification and optimisation of Trypanosoma brucei GSK3 inhibitors and their characterisation as anti-Trypanosoma brucei drug discovery lead molecules. ChemMedChem 2013, 8 (7), 1127–1137. 10.1002/cmdc.201300072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens K. L.; Reno M. J.; Alberti J. B.; Price D. J.; Kane-Carson L. S.; Knick V. B.; Shewchuk L. M.; Hassell A. M.; Veal J. M.; Davis S. T.; Griffin R. J.; Peel M. R. Synthesis and evaluation of pyrazolo[1,5-b]pyridazines as selective cyclin dependent kinase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18 (21), 5758–5762. 10.1016/j.bmcl.2008.09.069. [DOI] [PubMed] [Google Scholar]
- Zhang H. Q.; Xia Z.; Vasudevan A.; Djuric S. W. Efficient Pd-catalyzed synthesis of 2-arylaminopyrimidines via microwave irradiation. Tetrahedron Lett. 2006, 47 (28), 4881–4884. 10.1016/j.tetlet.2006.05.036. [DOI] [Google Scholar]
- Hagan J. J.; Ratti E.; Routledge C.. Use of Cyclooxygenase-2 Inhibitors for the Treatment of Depressive Disorders. Patent WO/2005/048999, 2005.
- Ishikawa M.; Hashimoto Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem. 2011, 54 (6), 1539–1554. 10.1021/jm101356p. [DOI] [PubMed] [Google Scholar]
- Bachovchin K. A.; Sharma A.; Bag S.; Klug D. M.; Schneider K. M.; Singh B.; Jalani H. B.; Buskes M. J.; Mehta N.; Tanghe S.; Momper J. D.; Sciotti R. J.; Rodriguez A.; Mensa-Wilmot K.; Pollastri M. P.; Ferrins L. Improvement of Aqueous Solubility of Lapatinib-Derived Analogues: Identification of a Quinolinimine Lead for Human African Trypanosomiasis Drug Development. J. Med. Chem. 2019, 62 (2), 665–687. 10.1021/acs.jmedchem.8b01365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O.; Eldar-Finkelman H. Long-term treatment with novel glycogen synthase kinase-3 inhibitor improves glucose homeostasis in ob/ob mice: molecular characterization in liver and muscle. J. Pharmacol. Exp. Ther. 2006, 316 (1), 17–24. 10.1124/jpet.105.090266. [DOI] [PubMed] [Google Scholar]
- Garcia-Hernandez R.; Gomez-Perez V.; Castanys S.; Gamarro F. Fitness of Leishmania donovani parasites resistant to drug combinations. PLoS Neglected Trop. Dis. 2015, 9 (4), e0003704 10.1371/journal.pntd.0003704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long T.; Rojo-Arreola L.; Shi D.; El-Sakkary N.; Jarnagin K.; Rock F.; Meewan M.; Rascon A. A. Jr.; Lin L.; Cunningham K. A.; Lemieux G. A.; Podust L.; Abagyan R.; Ashrafi K.; McKerrow J. H.; Caffrey C. R. Phenotypic, chemical and functional characterization of cyclic nucleotide phosphodiesterase 4 (PDE4) as a potential anthelmintic drug target. PLoS Neglected Trop. Dis. 2017, 11 (7), e0005680 10.1371/journal.pntd.0005680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst A.; Nguyen T. N.; El-Sakkary N.; Skinner D.; Suzuki B. M.; Buckner F. S.; Gelb M. H.; Caffrey C. R.; Debnath A. Bioactivity of Farnesyltransferase Inhibitors Against Entamoeba histolytica and Schistosoma mansoni. Front. Cell. Infect. Microbiol. 2019, 9 (180), 1–12. 10.3389/fcimb.2019.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdulla M. H.; Ruelas D. S.; Wolff B.; Snedecor J.; Lim K. C.; Xu F.; Renslo A. R.; Williams J.; McKerrow J. H.; Caffrey C. R. Drug discovery for schistosomiasis: hit and lead compounds identified in a library of known drugs by medium-throughput phenotypic screening. PLoS Neglected Trop. Dis. 2009, 3 (7), e478 10.1371/journal.pntd.0000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Perez V.; Manzano J. I.; Garcia-Hernandez R.; Castanys S.; Campos Rosa J. M.; Gamarro F. 4-Amino bis-pyridinium derivatives as novel antileishmanial agents. Antimicrob. Agents Chemother. 2014, 58 (7), 4103–4112. 10.1128/AAC.02481-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konsoula R.; Jung M. In vitro plasma stability, permeability and solubility of mercaptoacetamide histone deacetylase inhibitors. Int. J. Pharm. 2008, 361 (1–2), 19–25. 10.1016/j.ijpharm.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanadhan V. N.; Ghose A. K.; Revankar G. R.; Robins R. K. Atomic Physiochemical Parameters for Three Dimensional Structure Directed Structure-Activity Relationships. 4. Additional Parameters for Hydrophobic and Dispersive Interactions and Their Application for an Automated Superposition of Certain Naturally Occuring Nucleoside Antibiotics. J. Chem. Inf. Model. 1989, 29, 163–172. 10.1021/ci00063a006. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.