

Abstract
Background
Duckweeds (Lemnaceae) are aquatic plants distributed all over the world. The chloroplast genome, as an efficient solar-powered reactor, is an invaluable resource to study biodiversity and to carry foreign genes. The chloroplast genome sequencing has become routine and less expensive with the delivery of high-throughput sequencing technologies, allowing us to deeply investigate genomics and transcriptomics of duckweed organelles.
Results
Here, the complete chloroplast genome of Spirodela polyrhiza 7498 (SpV2) is assembled by PacBio sequencing. The length of 168,956 bp circular genome is composed of a pair of inverted repeats of 31,844 bp, a large single copy of 91,210 bp and a small single copy of 14,058 bp. Compared to the previous version (SpV1) assembled from short reads, the integrity and quality of SpV2 are improved, especially with the retrieval of two repeated fragments in ycf2 gene. There are a number of 107 unique genes, including 78 protein-coding genes, 25 tRNA genes and 4 rRNA genes. With the evidence of full-length cDNAs generated from PacBio isoform sequencing, seven genes (ycf3, clpP, atpF, rpoC1, rpl2, rps12 and ndhA) are detected to contain type-II introns. The ndhA intron has 50% more sequence divergence than the species-barcoding marker of atpF-atpH, showing the potential power to discriminate close species. A number of 37 RNA editing sites are recognized to have cytosine (C) to uracil (U) substitutions, eight of which are newly defined including six from the intergenic regions and two from the coding sequences of rpoC2 and ndhA genes. In addition, nine operon classes are identified using transcriptomic data. It is found that the operons contain multiple subunit genes encoding the same functional complexes comprising of ATP synthase, photosynthesis system, ribosomal proteins, et.al., which could be simultaneously transcribed and coordinately translated in response to the cell stimuli.
Conclusions
The understanding of the chloroplast genomics and the transcriptomics of S.polyrhiza would greatly facilitate the study of phylogenetic evolution and the application of genetically engineering duckweeds.
Keywords: Duckweeds, Chloroplast genome, PacBio, Intron, RNA editing, Operon
Background
Lemnaceae (duckweeds) are the fastest growing plants including five genera of Spirodela, Landoltia, Lemna, Wolffiella and Wolffia. They are phylogenetically located at the early-diverging monocots of the Alismatale order. Duckweeds have ecological and economical merits as wastewater treatment, animal feed and biofuel. The morphology is extremely simplified and small, resulting in the difficulty of species or ecotypes identification [1, 2]. The chloroplast genome has dual characteristics of sequence variation and conservation, which are widely applied in the studies of population genetics and phylogenetic relationships. The entire chloroplast genomes show the potential to serve as a plant super-barcode to distinguish closely related species such as in Conyza (in the family of Asteraceae) [3, 4] and Theobroma cacao (in the family of Malvaceae) [5]. The chloroplast genome is one of the three genetic systems including nuclei, mitochondria, and plastids in plants that possesses both eukaryote-like introns and prokaryote-like operons [6]. One broad hypothesis is that the chloroplast is derived from an initial engulfment and integration of a free-living cyanobacterium into a host cell around 1.5 billion years ago [7]. Group I and II introns in chloroplasts and mitochondria are a large class of self-catalytic ribozymes either with or without assistance from proteins for vivo splicing. In particular, group II introns have the ability of retrotransposition through intron-encoded reverse transcriptase activities [8]. Although most ancestral genes were transferred into the host nucleus during chloroplast evolution, modern chloroplast genomes possess common structural features with a size of ~ 107–218 kb and are compacted with a gene content of ~ 100–120 genes [9]. The chloroplast is also a vital organelle for plants, playing a crucial role by converting solar energy to carbohydrates through photosynthesis, and promoting their growth and starch accumulation.
With the rapid development of sequencing technology, it is easier and cheaper to obtain the complete genomes including nuclei, mitochondria and chloroplast [10]. In 2008, the first duckweed chloroplast genome (L.minor) was sequenced by Sanger sequencing [11]. Another three chloroplast genomes (S.polyrhiza 7498, W.lingulate 7289, and W.australiana 7733) were sequenced by using the SOLiD platform generating short reads (~ 50 bp) and assembled in 2011 [12]. The recent eight species covered the genera of Landoltia, Lemna and Wolffia were assembled by using the Illumina platform to study duckweed phylogeny [13]. In the meanwhile, the duckweed nuclear genomes have become more complete with the expansion of sequencing technology. The Spirodela nuclear genomes were generated by physical mapping and short-read DNA sequencing strategies [14, 15]. The Spirodela genome has continued to be improved by integrating the evidences from cytogenomic, optical mapping and Nanopore sequences [16]. Long-read sequencing, such as SMRT (Single Molecule Real-Time) technology emerged in 2009 [17] has been widely applied in sequencing the chloroplast genomes with the improved contiguity and accuracy. Still, no duckweed chloroplast genomes based on long-read sequencing have been reported. The studies of annotating chloroplast genome and gene structure at the transcriptomic and post-transcriptomic levels were limited, which were involved in a series of RNA regulation and process, such as RNA splicing, 5′- and 3′-end modification, and RNA editing and turnover [18]. Most previous studies relied on the sequence alignment and computer prediction to determine the intron boundary and the possible RNA editing sites, which need to be confirmed by PCR and sequenced one by one [19, 20]. With the high-throughput RNA-seq data with a read length of 75 bp, 66 RNA editing in Spirodela chloroplast genome were defined at the genome-wide level [21]. However, such short reads of 75 bp were impossible to accurately set intron and exon boundaries, as well as to distinguish the operons without the full-length cDNA sequences.
Here, we initiated a project that was originally designed as the nuclear genome sequencing and annotation by using long PacBio reads [22]. Since the raw reads were generated from the total DNA and RNA, we took advantage of such data to study chloroplast genomics and transcriptomics. In this study, we improved and validated the chloroplast genome of S.polyrhiza assembled by PacBio sequencing reads with retrieval of two repeated fragments compared with the last version. The integration of full-length cDNAs from isoform sequencing allowed us to discover new RNA editing sites, to detect introns, and to define poly-cistrons similar to prokaryotic transcripts in Spirodela chloroplast. The understanding of the chloroplast genomics and the transcriptomics of S.polyrhiza would facilitate the study of phylogenetic evolution and the application of genetically engineering the solar reactor of chloroplasts.
Results
Chloroplast genome assembly, validation and annotation
The last version of the complete chloroplast genome of S.polyrhiza 7498 (SpV1) was sequenced on a SOLiD platform and published in 2011 (GenBank accession number: JN160603) [12]. Because of the limitations of the second-generation sequencing technology with short reads (50 bp), the assembly of SpV1 was tedious and challenging to resolve boundaries of IR regions, resulting in 3 genomic breakage and 52 small gaps (Table 1). Here, the total DNA originated from nuclei, mitochondrion and chloroplasts was prepared from the whole duckweed tissue using CTAB method [23]. The high-quality DNA was sequenced on the PacBio platform, generating long reads with the mean length of 10,789 bp. After bioinformatic filtering, a total of 239,086 high-quality long reads were selected to be chloroplast related sequences, which were used to run the chloroplast genome de novo assembly. A single circular strand genome with a size of 168,956 bp (GenBank accession number: MN419335) was directly constructed by using a long-read based bioinformatic pipeline (Additional file 1: Figure S1) [24] without any manual correction and sequence collapses, skipping further PCR amplification and capillary electrophoresis (CE) sequencing to fill unassembled gaps. In contrast, SpV1 was assembled from short reads with a read length of 50 bp, resulting in 52 contigs and 3 scaffolds (Table 1). The broken scaffolds were manually ordered based on other chloroplast genomes. A number of 52 pairs of primers were designed to close the gaps and to reach the final genome with tremendous efforts [12]. The chloroplast genome with long-read assembly exhibited the typical quadripartite structure, a pair of inverted repeat regions (IRs) of 31,844 bp separated by a large single copy (LSC) of 91,210 bp and a small single copy (SSC) of 14,058 bp (Fig. 1). The GC content was 40.06, 33.47 and 30.17%, respectively, and the overall GC content was 35.68%. The sequence similarity between SpV2 and SpV1 was 99.9% (Fig. 2), indicating high accuracy of the assembled genome. The chloroplast genome was annotated as 107 unique genes, including 78 protein-coding genes, 25 tRNAs and 4 rRNAs. There were 19 genes, including seven protein-coding genes, eight tRNAs and four rRNAs in the IR regions (Additional file 1: Table S1). A coverage plot was demonstrated by re-mapping the PacBio reads to the chloroplast genome, showing an even distribution across the genome with a mean coverage of 7837 times (Fig. 2).
Table 1.
The comparative statistics of the chloroplast genome assembly of S.polyrhiza 7498 generated from long reads of PacBio and short reads of SOLiD platform
| Category | PacBio | SOLiD |
|---|---|---|
| Number of selected readsa | 239,086 | 19,906,092 |
| Total nucleotides (selected data) (bp)a | 2,579,414,638 | 995,304,600 |
| Mean read length (selected data) (bp)a | 10,789 | 50 |
| Number of scaffolds | 1 | 3 |
| Number of genome gaps | 0 | 52 |
| Total genome coverage | 7837 | 5474 |
| Genome Size (bp) | 168,956 | 168,788 |
| LSC (bp) | 91,210 | 91,222 |
| SSC (bp) | 14,058 | 14,056 |
| IR (bp) | 31,844 | 31,755 |
| GC content (%) | 35.68 | 35.69 |
| GenBank ID | MN419335 | JN160603 |
aOnly the selected chloroplast-related PacBio reads and SOLiD reads are counted
Fig. 1.

Gene map of the chloroplast genome of S.polyrhiza 7498. Genes are labelled based on the annotation data. Genes are color-coded in different functional groups. The middle circle indicates a quadripartite structure. The darker area in the inner circle indicates the GC content
Fig. 2.

Sequencing coverage and genome comparison. a The x-axis shows the chloroplast genome of S.polyrhiza. The y-axis indicates the sequencing depth across the genome. b The sequence alignment of two versions of S.polyrhiza 7498 chloroplast genomes. The lines indicate the genome collinearity and IR regions
Ycf2 was a large functional gene encoding 2310 amino acids in chloroplast IR regions. We retrieved two extra fragments of 45 bp and 48 bp which were located at 2599 and 5065 bp within ycf2 gene compared to the previous version (Fig. 3). Surprisingly, the recovered sequences were the copies of the downstream nucleotides, which could be a failure of genome assembly in SpV1 due to short reads of second-generation sequencing. Such limitation could be easily conquered by the nature of PacBio long reads with the spanning of the ambiguous repeats.
Fig. 3.

The comparison of ycf2 gene in SpV1 and SpV2. The ycf2 gene in SpV2 are 6930 bp, containing two sets of repeats labelled with green and blue arrow, while one copy of repeats is missing in SpV1 due to the limitation of short-read assembly
Intron identification
The full-length cDNAs generated by PacBio isoform sequencing allowed us to define the chloroplast transcript structures. Here, we defined nine type-II introns within seven genes (ycf3, clpP, atpF, rpoC1, rpl2, rps12 and ndhA), and the gene of ycf3 and clpP contained 2 introns (Additional file 1: Table S2). We found that the length of introns was extremely conserved in plant species, except the genes of clpP and rpoC1 in Poaceae were absent of introns. Previous research has revealed that the intron loss of rpoC1 and clpP genes occurred before grasses species differentiation [25]. We found that the early-diverging monocot of Amborella had the longest atpF introns (1825 bp), whereas the dicot of tobacco had the shortest one (1250 bp), indicating that introns might play roles in genomic diversity during the chloroplast evolution (Fig. 4). To assess the degree of DNA polymorphism between introns, sequence divergences in four duckweed species were calculated with the overall mean distance respectively. The region of ndhA intron showed the highest genetic distance, while the non-coding intron in the rps12 gene was the most conserved one (Table 2). The ndhA intron had 50% more polymorphism compared to the proposed species barcode marker of atpF-atpH [26], showing sufficient genetic distance and potential to discriminate close species.
Fig. 4.
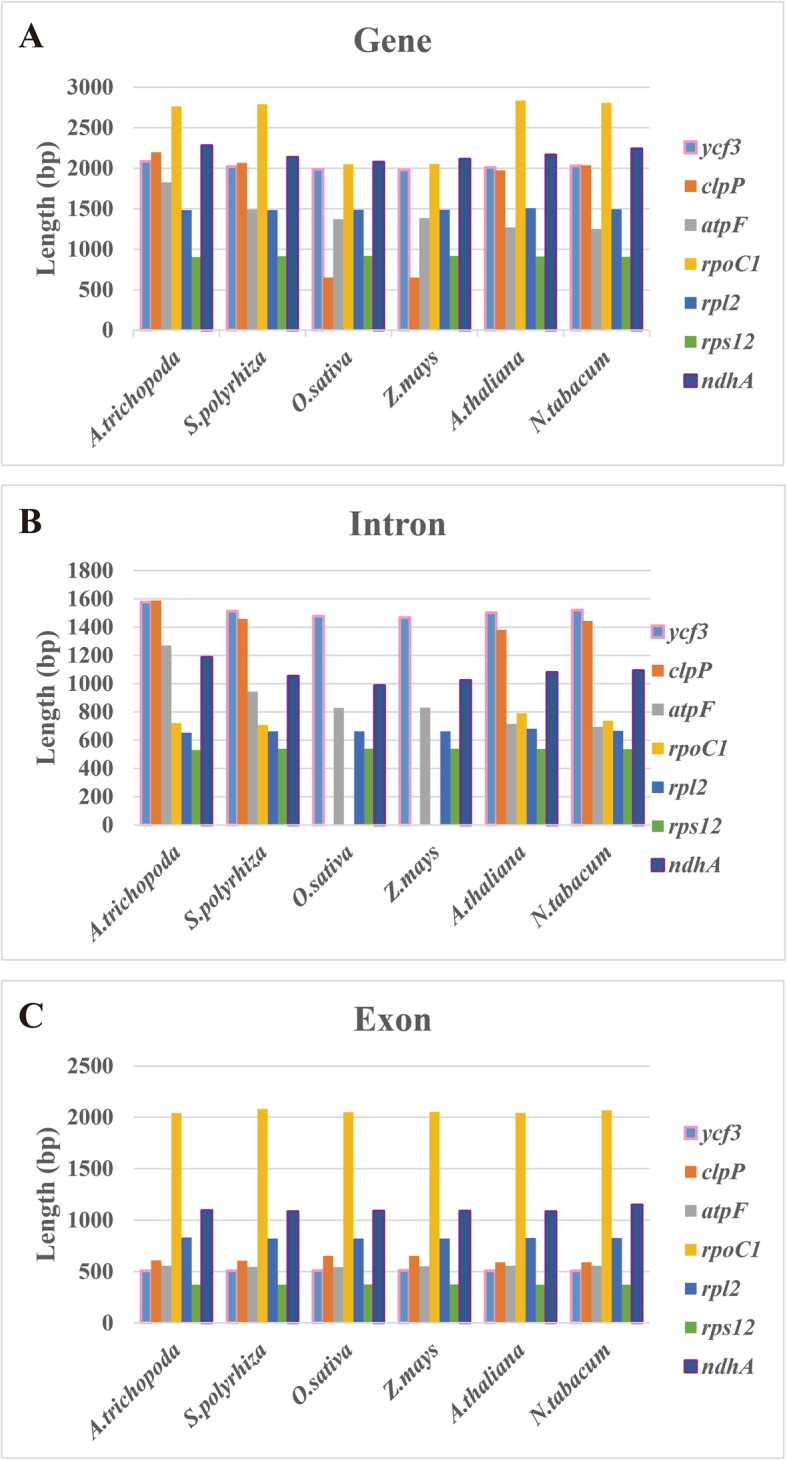
Intron comparison of seven genes in plants. a, b and c display the length of genes, introns and exons within six plant species, respectively. Their sequences are downloaded from A.trichopoda (NC_005086.1), S.polyrhiza 7498 (MN419335), O.sativa (NC_001320.1), Z.mays (NC_001666.2), A.thaliana (NC_000932.1) and N.tabacum (NC_001879.2). The X axis indicates species and Y axis shows sequence length (bp)
Table 2.
Measurement of intron divergences between duckweed species
| Gene | Aligned Length (bp) | Base Variable | Overall Mean Distance |
|---|---|---|---|
| atpF-atpHa | 493 | 85 | 0.0960 |
| rbcLb | 1461 | 92 | 0.0366 |
| atpF | 949 | 147 | 0.1089 |
| rpoC1 | 740 | 94 | 0.0716 |
| rps12 | 540 | 5 | 0.0053 |
| rpl2 | 664 | 8 | 0.0071 |
| ndhA | 1091 | 235 | 0.1413 |
| ycf3_1 | 778 | 72 | 0.0551 |
| ycf3_2 | 827 | 72 | 0.0503 |
| clpP_1 | 868 | 122 | 0.0875 |
| clpP_2 | 688 | 94 | 0.0861 |
Aligned length are longer than the original sequence length because of the addition of the aligned gaps. Base variation is the base polymorphism excluding insertions or deletions. The controls of the intergenic region of atpF-atpHa and the coding sequence of rbcLb are also included. The duckweed species include S.polyrhiza (MN419335), L.minor (DQ400350), W.ligulata (JN160604) and W.australiana (JN160604)
RNA editing analysis
After a chloroplast mRNA molecule is transcribed, it usually undergoes RNA editing, a process of C-to-U conversion at specific sites to regulate gene expression and translation in chloroplasts. Here, with isoform sequences, we defined 37 RNA editing sites, including 30 sites that occurred in protein-coding sequences, one in intron and six in non-coding regions (Additional file 1: Table S3). The RNA editing efficiency had a range of 21 to 100% with a median value of 93%. In 2011, the study using Illumina short reads was able to define 66 editing sites [21], 29 of which were overlapped with this study. Combined with known and newly discovered RNA editing sites, there were 74 in total, 62 of which occurred in gene regions, whereas the Ndh gene showed the most heavily edited sites (33 sites) (Additional file 1: Figure S2). The eight newly defined editing events contained two from the coding regions of rpoC2 and ndhA genes and six from the location of intergenic regions (Additional file 1: Table S3). The event of RNA editing in Spirodela rpoC2 was consistent with rice and tobacco, whereas the C-to-U conversion in ndhA made Spirodela keep the conserved amino acid of L as other plants (Additional file 1: Figure S3).
Operon classification
An operon, i.e., poly-cistronic mRNA is a messenger RNA that could efficiently encode more than one protein. Such a phenomenon is typical in prokaryotic organisms, including chloroplast due to its origin of cyanobacteria [27]. The coding sequences within an operon is usually grouped and regulated together controlled by a regulatory region of a promoter and an operator. These protein products have a related function of either subunit of building a final complex protein or participating in a common biological process. Thanks to the isoform sequencing with a read length of 10 Kb, we could investigate the operon structures based on the full-length transcripts. Here, we identified nine operons after we mapped transcripts against the genome with a deep coverage (Table 3 and Fig. 5). The operons included gene clusters that encoded different functional groups, such as ATP synthase, RNA polymerase, photosystem II, photosystem I, cytochrome complex, NADH dehydrogenase, ribosome proteins, which are involved in the process of photosynthesis and respiration. It was reported that the psbB operon contained genes for the PSII (psbB, psbT, psbH) and cytochrome (petB and petD) complexes, which are required during chloroplast biogenesis [28]. The enzyme of plastid-encoded RNA polymerase (PEP) was composed core subunits (including the plastid genes of rpoA, rpoB, rpoC1 and rpoC2) and mainly responsible for the transcription of photosynthesis genes [29, 30]. Like in bacteria and other plants, rpoA gene encoding a α-subunit of PEP was found in a gene cluster comprising of ribosomal protein genes in Spirodela. The gene cluster of rpoB, rpoC1 and rpoC2, encoding the β, β′ and β″ subunits of PEP formed a separate operon (Table 3 and Fig. 5). The operon of NADH dehydrogenase was composed of four genes, mainly involved in electron transport around photosystem I and chloro-respiration. All operons in Spirodela had great homology with Z.mays and the largest ribosomal protein operon ‘rpl22-rps3-rpl16-rpl14-rps8-rpl36-rps11-rpoA’ was consistent with Cyanophora paradoxa and Spinacia oleracea, where it was called S10 (or spc-like) operon [31, 32]. As we knew, the size of the chloroplast genome was compact, but it played a critical role in photosynthesis in the survival of plants. The pattern of co-transcription in the chloroplast of duckweed may enhance the work efficiency of transcription-translation factors like RNA polymerase.
Table 3.
The defined operons in SpV2
| Operon | Genes | Functions | Length | Genome Position |
|---|---|---|---|---|
| Atp_1 | atpI+atpH+atpF+atpA | ATP synthase | 5,758 | 17,612-12,186 |
| Atp_2 | atpB+atpE | ATP synthase | 2,141 | 60,381-58,481 |
| Psb_1 | psbD+psbC+psbZ | PSII | 3,398 | 37,462-40,616 |
| Psb_2 | psbB+psbT+psbH+petB+petD | PSII; Cytochrome complex | 5,689 | 78,885-84,218 |
| Psa | psaA+psaB | PSI | 4,818 | 46,372-41,890 |
| Ndh | rps15+ndhH+ndhA+ndhI | NADH dehydrogenase | 4,611 | 137,464-133,111 |
| Rpl_1 | rpl23+rpl2+rps19 | Ribosomal proteins | 2,319 | 92,997-90,876 |
| Rpo | rpoB+rpoC1+rpoC2+rps2 | RNA polymerase; Ribosomal protein | 11,837 | 29,112-17,867 |
| Rpl_2 | rpl22+rps3+rpl16+rpl14+rps8 +rpl36+rps11+rpoA | Ribosomal proteins | 6,257 | 90,586-84,434 |
aThe length of operon is counted in bp. The column of operon is named with the abbreviation of gene family. The connections of genes are indicated by a plus sign. The gene order in the operon is based on the full-length transcript. Genome Position means the location of operon in the new version of S.polyrhiza 7498 chloroplast genome. PSII means photosystem II and PSI is photosystem I
Fig. 5.

The defined operon structure in S.polyrhiza chloroplast. The genes are shown in thick lines with exon in black and intron in grey. The gene names are given on the top of lines. The gene order is based on the physical genome location
Discussion
Third generation sequencing (TGS) technology facilitates chloroplast genomic and Transcriptomic analysis
Compared with second-generation sequencing technologies featured with short reads of 150~300 bp, third-generation sequencing (TGS) has a striking advantage of long reads up to 500 Kb like Nanopore. The long reads could manage repeat regions by using unique flanking sequences and improve genome assembly which can fill potential gaps. Still, the genome completeness depends on the complexity of targeted genomes and the length and quality of sequencing data [10]. With the announcement of the launch of PacBio Sequel II system, it generates 8-times more data and makes sequencing more affordable. No matter how hard scientists try to remove organellar DNA from the total DNA (including nuclear, mitochondria and chloroplast DNA), chloroplast genome still can be assembled from the left “purified” DNA as a side project of the whole genome sequencing study due to its high copy number [33]. Our trial confirmed that two pairs of repeats in the coding sequence of ycf2 gene were filled in the assembly of the chloroplast genome of S.polyrhiza. The phylogenetic analysis suggested that ycf2 gene was evolved from the membrane-bound AAA-protease FtsH of the ancestral endosymbiont [34]. It can be found both in non-green (Epifagus virginiana) and green plants, but was absent in the grass family, indicating that its function was not essential for photosynthesis. The knock-out experiment in tobacco showed that ycf2 gene was indispensable for plant cell survival and probably related to ATPase metabolic process [35]. The nucleotide sequences of ycf2 were rich in diversity [36] and repeats [37]. Here, we retrieved two repeat copies in the ycf2 gene, which were also shown in Nicotiana tabacum and Arabidopsis thaliana, indicating the essential structure in gene function [35].
Post-transcriptional control is important for the regulation of gene expression. The gene structures of introns and operons remained unknown, although some RNA editing sites were detected by using high-throughput RNA-seq [21]. Given the power of obtaining full-length transcripts without assembly from PacBio isoform sequencing (Iso-Seq), it is advantageous for gene annotation, identification of introns, RNA editing and operons in chloroplasts. An accurate and intact genome, as well as the well-defined annotation, will be beneficial to phylogenetic classification and to subsequently molecular studies.
Introns and molecular evolution
Although an intron is a piece of non-coding DNA, there are many important implications for plant physiological activities and modern botanical applications. Introns are a group of self-catalytic ribozymes that could splice their own excision from mRNA, tRNA and rRNA precursors [38]. Introns help to infer phylogenetic relationships, better than the conserved genes such as rbcL due to their rapidly evolving noncoding sequences. Duckweeds represent the early-diverging monocot of the phylogenetic tree with their small and simple plant bodies, which is challenging to identify species by merely counting on morphology for non-experts. The method of DNA barcode of chloroplast markers alleviates such a situation by using PCR amplification and sequence variation. The overall polymorphisms of intergenic regions and introns are higher than the most coding DNA, providing valuable information to distinguish plant lineages. The atpF-atpH noncoding spacer was proposed as the best DNA barcoding marker for species-level identification of duckweeds [26]. Still, five out of 19 species failed to be separated from other sister species. Searching for more loci with enough variability would help to increase the discriminable resolution when they are combined with known markers. It was found that chloroplast introns showed the power of species identification with the sequence variability and the presence of highly conserved sequences in the flanking regions, which were suitable to design universal primers for DNA barcoding. The ndhA intron, together with the marker of psbE-psbL could distinguish Fagopyrum between species and subspecies [39]. Here, the comparison of nucleotide divergence and genetic distance between duckweed chloroplast coding sequences, intergenic regions and intron sequences offer scientists more markers to understand species phylogenetic relationship and plant evolution. Still, it is necessary to verify the potential of the utilization of ndhA intron itself or with other markers to distinguish intra- and inter-species in duckweeds.
RNA editing and its evolution
RNA editing is a post-transcriptional modification that broadly exists in land plants from hornworts and ferns to seed plants. We could not detect RNA editing sites in the Spirodela chloroplast genome all at once only using one technique. With deep sequencing and various sequencing platforms, we expect more and more editing sites would be uncovered, especially for GC biased or very lowly expressed transcripts. Short reads generated by second-generation sequencing were able to define 66 editing sites [21]. Here, long reads using PacBio isoform sequencing identified 37 RNA editing sites. Excluding overlapped sites from two platforms, there are 74 RNA editing in Spirodela, more than twice of those in rice (35 sites) and maize (26 sites) [40]. The early-branching flowering plant of Amborella trichopoda was found to have 138 sites of RNA editing. It was proposed that early-branching flowering plants carried more abundant chloroplast RNA editing, whereas there was a tremendous decrease in RNA editing frequencies during flowering plant evolution [41]. To re-establish evolutionarily conserved amino acids and to maintain protein functions, Spirodela, as an early-diverging monocot shared many conserved editing sites with other plants, such as the ndh gene family of ndhA, ndhB, ndhD and ndhF. Spirodela also presents some species-specific RNA editing compared to its relatives, as some sites in ndhB [21].
Operon and chloroplast photosynthetic reactor
In nuclear transgenic plants, the expression of multiple genes is time-consuming and extremely laborious with the requirement of putting one gene at a time and with subsequent backcrosses to select complete pathways with multi-subunit proteins, which is also compounded by variable expression levels. However, most chloroplast genes of plants are co-regulated and co-transcribed [42]. Such knowledge about operon structures would enable engineering new pathways in a simulated operon via a single transformation event into the chloroplast genome. It was reported that an artificially foreign pathway including seven genes was engineered into the tobacco chloroplast genomes [35]. Large amounts of foreign protein accumulation were observed in these transgenic lines, showing that the chloroplast posttranscriptional machinery can efficiently detect and translate genes in operons [43]. The Bacillus thuringiensis (Bt) cry2Aa2 operon was introduced into chloroplasts, resulting in 45% of the total soluble proteins in mature leaves and 100% of the observation of insect mortality after consuming the transgenic plants [44]. Understanding the operon information in duckweed chloroplasts lays the foundation and makes expressing foreign multiple proteins possible in terms of its rapid growth and biomass accumulation, facilitating duckweeds into an efficient photosynthetic reactor to produce pharmaceutical proteins or other foreign pathways.
Conclusions
Here, a single circular strand genome with a size of 168,956 bp is directly constructed by using a long-read assembly without any manual correction and sequence collapses, skipping further PCR amplification to fill unassembled gaps. With the evidence of full-length cDNA generated from PacBio isoform sequencing, we accurately detect nine introns, 37 RNA editing sites and nine operons. We propose that the ndhA intron could be a potential species-barcoding marker with the sufficient genetic distance to discriminate close species, given its sequence divergence higher than the known atpF-atpH marker. In addition, the identified operon classes that encode the same functional complexes would lay the foundation of genetically engineering high protein, starch or oil duckweeds.
Methods
Plant DNA preparation and genome sequencing
Spirodela polyrhiza 7498 was originally collected by Dr. Elias Landolt from North Carolina, USA and kept in Wenqin Wang’s lab and Rutgers Duckweed Stock Cooperative (http://www.ruduckweed.org), which are publicly available. No permissions were necessary to collect the samples. The DNA was prepared from whole plant tissue using CTAB method [23]. Spirodela polyrhiza 7498 was sterilely cultured and fifty micrograms of high-molecular-weight total DNA was extracted. A 20-kb insert SMRTbell library was constructed and sequenced using the PacBio Sequel platform (Pacific Biosciences, Frasergen, Wuhan, China).
PacBio isoform sequencing
The total RNA was isolated from the samples treated by multiple conditions (37 °C, 0 °C, desiccation, pH value of 9, UV exposure, 20 mg/l CuCl2, 300 mg/1 KNO3, 250 nM ABA, 10 mM kinetin, 300 mM mannitol) using TRIzol reagent (Invitrogen) and the RNeasy Mini kit after DNase I digestion (Qiagen). The extracted RNAs were evenly pooled. The library was constructed using a Clontech SMARTer PCR cDNA Synthesis Kit (Clontech) and sequenced on PacBio isoform sequencing (Iso-Seq) platform.
Genome assembly and annotation
The PacBio raw reads were corrected into preads by Falcon (version 0.3.0) which was used in the downstream steps. The complete chloroplast genome of Arabidopsis thaliana (NC_000932.1) was downloaded from NCBI as a reference genome. The program BLASR (version 5.3.1) [45] was used to fish out the relevant chloroplast reads. The chloroplast genome of S.polyrhiza was assembled using a Perl-based software named “Organelle_PBA” (https://github.com/aubombarely/Organelle_PBA) [24]. The genome assembly pipeline generated by us was submitted to Github (https://github.com/Yating-zhang/chloroplast_Pacbio). The short-read and long-read based chloroplast genomes were aligned by BLASTN and the coverage plot was shown by using Samtools (version 1.7) [46]. The chloroplast genome was annotated with the tool GeSeq (https://chlorobox.mpimp-golm.mpg.de/geseq.html) [47] using the default parameters. The genome features were further manually curated by using transcript sequences to determine the boundary of introns and exons, and start codons as well. A circular map of the annotated genome was illustrated by using Organellar Genome DRAW (OGDRAW) (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html) [48].
Comparative genome analysis
The whole genome comparison between SpV1 and SpV2 was conducted by LASTZ alignment (version 1.02.00, http://www.bx.psu.edu/~rsharris/lastz/) and visualized in Geneious Prime® 2019.1.1. The chloroplast genomes of Amborella trichopoda (NC_005086.1), Nicotiana tabacum (NC_001879.2), Arabidopsis thaliana (NC_000932.1), Oryza sativa Japonica Group (NC_001320.1), Zea mays (NC_001666.2), Lemna minor (DQ400350), Wolffiella lingulata 7289 (JN160604) and Wolffia australiana 7733 (JN160604) were downloaded from NCBI GenBank and were aligned with SpV2 by Mauve (version 2.4.0) multiple genome alignment program.
Intron, RNA editing and operon analysis
The full-length transcript generated by PacBio isoform sequencing were mapped to the previous and new chloroplast genome of S.polyrhiza by Gmap (version 2017-11-15) [49]. The RNA editing sites, introns and operons were identified through the mapped data and visualized under IGV (Integrative Genomics Viewer) (version 2.5.0) [50]. The four duckweed chloroplast genomes (S.polyrhiza (MN419335), L.minor (DQ400350), W.ligulata (JN160604) and W.australiana (JN160604)) were aligned and the genetic distance were computed by MEGA (version 6.06) [51]. The pairwise distance was calculated by the sequence polymorphism normalized by the sequence length.
Supplementary information
Additional file 1: Table S1. Annotated gene list in the chloroplast of SpV2. Table S2. A comparison of detected introns among model plants. Table S3. The list of RNA editing sites in SpV2. Figure S1. Bioinformatic pipeline of chloroplast genome assembly and annotation. Details are described under Methods. Figure S2. The distribution of RNA editing events in the chloroplast genes of S.polyrhiza. Graph shows the number of currently detected RNA editing sites in protein coding genes. Figure S3. Alignments of editing sites in rpoC2 and ndhA genes. The sequences included RNA editing sites are shown before RNA editing. The amino acid is in orange and substitutions are marked with arrows. The start, RNA editing and end locations are listed above the alignment. All aligned sequences are antisense from the reference except rpoC2 and rpoC1 gene of rice.
Acknowledgements
We thank Qiong Wang and other lab members from Yongrui Wu’s lab (Shanghai Institutes for Biological Sciences, Chinese Academy of Science) for their technical help and collaboration.
Abbreviations
- BLAST
Basic local alignment search tool
- PacBio
Pacific Biosciences
- RNA-seq
RNA sequencing; Iso-Seq: isoform sequencing
- SOLiD
Sequencing by Oligonucleotide Ligation and Detection
Authors’ contributions
WW conceived the project, YZ designed experiments and analyzed the data, DA prepared DNA and sequencing data, CL provided bioinformatic analysis, ZZ performed the experiments, YZ and WW drafted the manuscript, all authors approved the manuscript.
Funding
The study was supported by the National Natural Science Foundation of China Grants (31670366 to Wenqin Wang). The funders had no role in designing experiment, data collecting and analysis, and draft manuscript.
Availability of data and materials
The chloroplast genome assembly and annotation (SpV2) were deposited in GenBank under the accession number of MN419335.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12864-020-6499-y.
References
- 1.Les DH, Crawford DJ, Landolt E, Gabel JD, Kimball RT. Phylogeny and systematics of Lemnaceae, the duckweed family. Syst Bot. 2002;27(2):221–240. [Google Scholar]
- 2.Stomp AM. The duckweeds: A valuable plant for biomanufacturing. Biotechnol Annu Rev. 2005;11(05):69–99. doi: 10.1016/S1387-2656(05)11002-3. [DOI] [PubMed] [Google Scholar]
- 3.Wang A, Wu H, Zhu X, Lin J. Species identification of Conyza bonariensis assisted by chloroplast genome sequencing. Front Genet. 2018;9:374. doi: 10.3389/fgene.2018.00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dodsworth S. Genome skimming for next-generation biodiversity analysis. Trends Plant Sci. 2015;20(9):525–527. doi: 10.1016/j.tplants.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 5.Kane N, Sveinsson S, Dempewolf H, Yang JY, Zhang D, Engels JM, Cronk Q. Ultra-barcoding in cacao (Theobroma spp.; Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. Am J Bot. 2012;99(2):320–329. doi: 10.3732/ajb.1100570. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharyya D, Chakraborty S. Chloroplast: the Trojan horse in plant-virus interaction. Mol Plant Pathol. 2018;19(2):504–518. doi: 10.1111/mpp.12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gray MW. The evolutionary origins of organelles. Trends Genet. 1989;5(9):294–299. doi: 10.1016/0168-9525(89)90111-X. [DOI] [PubMed] [Google Scholar]
- 8.Bonen L. Cis- and trans-splicing of group II introns in plant mitochondria. Mitochondrion. 2008;8(1):26–34. doi: 10.1016/j.mito.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 9.Daniell H, Lin CS, Yu M, Chang WJ. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 2016;17(1):134. doi: 10.1186/s13059-016-1004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Changsheng, Lin Feng, An Dong, Wang Wenqin, Huang Ruidong. Genome Sequencing and Assembly by Long Reads in Plants. Genes. 2017;9(1):6. doi: 10.3390/genes9010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mardanov AV, Ravin NV, Kuznetsov BB, Samigullin TH, Antonov AS, Kolganova TV, Skyabin KG. Complete sequence of the duckweed (Lemna minor) chloroplast genome: structural organization and phylogenetic relationships to other angiosperms. J Mol Evol. 2008;66(6):555–564. doi: 10.1007/s00239-008-9091-7. [DOI] [PubMed] [Google Scholar]
- 12.Wang W, Messing J. High-throughput sequencing of three Lemnoideae (duckweeds) chloroplast genomes from total DNA. PLoS One. 2011;6(9):1–9. doi: 10.1371/journal.pone.0024670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding Y, Fang Y, Guo L, Li Z, He K, Zhao Y, Zhao H. Phylogenic study of Lemnoideae (duckweeds) through complete chloroplast genomes for eight accessions. Peer J. 2017;5:e4186. doi: 10.7717/peerj.4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W, Haberer G, Gundlach H, Glasser C, Nussbaumer T, Luo MC, Lomsadze A, Borodovsky M, Kerstetter RA, Shanklin J, et al. The Spirodela polyrhiza genome reveals insights into its neotenous reduction fast growth and aquatic lifestyle. Nat Commun. 2014;5:3311. doi: 10.1038/ncomms4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michael TP, Bryant D, Gutierrez R, Borisjuk N, Chu P, Zhang H, Xia J, Zhou J, Peng H, El Baidouri M, et al. Comprehensive definition of genome features in Spirodela polyrhiza by high-depth physical mapping and short-read DNA sequencing strategies. Plant J. 2017;89(3):617–635. doi: 10.1111/tpj.13400. [DOI] [PubMed] [Google Scholar]
- 16.Hoang PNT, Michael TP, Gilbert S, Chu P, Motley ST, Appenroth KJ, Schubert I, Lam E. Generating a high-confidence reference genome map of the greater duckweed by integration of cytogenomic, optical mapping, and Oxford Nanopore technologies. Plant J. 2018;96(3):670–684. doi: 10.1111/tpj.14049. [DOI] [PubMed] [Google Scholar]
- 17.Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., Peluso P., Rank D., Baybayan P., Bettman B., Bibillo A., Bjornson K., Chaudhuri B., Christians F., Cicero R., Clark S., Dalal R., deWinter A., Dixon J., Foquet M., Gaertner A., Hardenbol P., Heiner C., Hester K., Holden D., Kearns G., Kong X., Kuse R., Lacroix Y., Lin S., Lundquist P., Ma C., Marks P., Maxham M., Murphy D., Park I., Pham T., Phillips M., Roy J., Sebra R., Shen G., Sorenson J., Tomaney A., Travers K., Trulson M., Vieceli J., Wegener J., Wu D., Yang A., Zaccarin D., Zhao P., Zhong F., Korlach J., Turner S. Real-Time DNA Sequencing from Single Polymerase Molecules. Science. 2009;323(5910):133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 18.Stern DB, Goldschmidt-Clermont M, Hanson MR. Chloroplast RNA metabolism. Annu Rev Plant Biol. 2010;61:125–155. doi: 10.1146/annurev-arplant-042809-112242. [DOI] [PubMed] [Google Scholar]
- 19.Jung J, Kim JI, Jeong Y-S, Yi G. AGORA: organellar genome annotation from the amino acid and nucleotide references. Bioinformatics (Oxford, England) 2018;34(15):2661–2663. doi: 10.1093/bioinformatics/bty196. [DOI] [PubMed] [Google Scholar]
- 20.Lo Giudice C, Hernandez I, Ceci LR, Pesole G, Picardi E. RNA editing in plants: a comprehensive survey of bioinformatics tools and databases. Plant Physiol Biochem. 2019;137:53–61. doi: 10.1016/j.plaphy.2019.02.001. [DOI] [PubMed] [Google Scholar]
- 21.Wang W, Zhang W, Wu Y, Maliga P, Messing J. RNA editing in chloroplasts of Spirodela polyrhiza, an aquatic Monocotelydonous species. PLoS One. 2015;10(10):e0140285. doi: 10.1371/journal.pone.0140285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.An D, Zhou Y, Li C, Xiao Q, Wang T, Zhang Y, Wu Y, Li Y, Chao D-Y, Messing J, et al. Plant evolution and environmental adaptation unveiled by long-read whole-genome sequencing of Spirodela. PNAS. 2019;116(38):18893–18899. doi: 10.1073/pnas.1910401116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murray MG, Thompson WF. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980;8(19):4321–4325. doi: 10.1093/nar/8.19.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soorni A, Haak D, Zaitlin D, Bombarely A. Organelle_PBA, a pipeline for assembling chloroplast and mitochondrial genomes from PacBio DNA sequencing data. BMC Genomics. 2017;18(1):1–8. doi: 10.1186/s12864-016-3412-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guisinger MM, Chumley TW, Kuehl JV, Boore JL, Jansen RK. Implications of the plastid genome sequence of typha (typhaceae, poales) for understanding genome evolution in poaceae. J Mol Evol. 2010;70(2):149–166. doi: 10.1007/s00239-009-9317-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang W, Wu Y, Yan Y, Ermakova M, Kerstetter R, Messing J. DNA barcoding of the Lemnaceae, a family of aquatic monocots. BMC Plant Biol. 2010;10:205. doi: 10.1186/1471-2229-10-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghulam MM, Courtois F, Lerbs-Mache S, Merendino L. Complex processing patterns of mRNAs of the large ATP synthase operon in Arabidopsis chloroplasts. PLoS One. 2013;8(11):e78265. doi: 10.1371/journal.pone.0078265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dixit R, Trivedi PK, Nath P, Sane PV. Organization and post-transcriptional processing of the psb B operon from chloroplasts of Populus deltoides. Curr Genet. 1999;36(3):165–172. doi: 10.1007/s002940050487. [DOI] [PubMed] [Google Scholar]
- 29.Borner T, Aleynikova AY, Zubo YO, Kusnetsov VV. Chloroplast RNA polymerases: role in chloroplast biogenesis. Biochim Biophys Acta. 2015;1847(9):761–769. doi: 10.1016/j.bbabio.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Chevalier F, Ghulam MM, Rondet D, Pfannschmidt T, Merendino L, Lerbs-Mache S. Characterization of the psbH precursor RNAs reveals a precise endoribonuclease cleavage site in the psbT/psbH intergenic region that is dependent on psbN gene expression. Plant Mol Biol. 2015;88(4–5):357–367. doi: 10.1007/s11103-015-0325-y. [DOI] [PubMed] [Google Scholar]
- 31.Michalowski CB, Pfanzagl B, Loffelhardt W, Bohnert HJ. The cyanelle S10 spc ribosomal protein gene operon from Cyanophora paradoxa. Mol Gen Genet. 1990;224(2):222–231. doi: 10.1007/BF00271555. [DOI] [PubMed] [Google Scholar]
- 32.Zhou DX, Quigley F, Massenet O, Mache R. Cotranscription of the S10- and spc-like operons in spinach chloroplasts and identification of three of their gene products. Mol Gen Genet. 1989;216(2–3):439–445. doi: 10.1007/BF00334388. [DOI] [PubMed] [Google Scholar]
- 33.Lutz KA, Wang W, Zdepski A, Michael TP. Isolation and analysis of high quality nuclear DNA with reduced organellar DNA for plant genome sequencing and resequencing. BMC Biotechnol. 2011;11:54. doi: 10.1186/1472-6750-11-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kikuchi S, Asakura Y, Imai M, Nakahira Y, Kotani Y, Hashiguchi Y, Nakai Y, Takafuji K, Bédard J, Hirabayashi-Ishioka Y, et al. A Ycf2-FtsHi Heteromeric AAA-ATPase Complex Is Required for Chloroplast Protein Import. Plant Cell. 2018;30(11):2677–2703. doi: 10.1105/tpc.18.00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drescher A, Ruf S, Calsa T, Jr, Carrer H, Bock R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 2000;22(2):97–104. doi: 10.1046/j.1365-313x.2000.00722.x. [DOI] [PubMed] [Google Scholar]
- 36.Xu L, Xing Y, Wang B, Liu C, Wang W, Kang T. Plastid genome and composition analysis of two medical ferns: Dryopteris crassirhizoma Nakai and Osmunda japonica Thunb. Chin Med. 2019;14:9–9. doi: 10.1186/s13020-019-0230-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan C, Du J, Gao L, Li Y, Hou X. The complete chloroplast genome sequence of watercress (Nasturtium officinale R. Br.): genome organization, adaptive evolution and phylogenetic relationships in Cardamineae. Gene. 2019;699:24–36. doi: 10.1016/j.gene.2019.02.075. [DOI] [PubMed] [Google Scholar]
- 38.Ostersetzer O, Cooke AM, Watkins KP, Barkan A. CRS1, a chloroplast group II intron splicing factor, promotes intron folding through specific interactions with two intron domains. Plant Cell. 2005;17(1):241–255. doi: 10.1105/tpc.104.027516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang Yue, Li Zhiqiang, Wang Chenglong, Zou Chenyan, Wen Wen, Shao Jirong, Zhu Xuemei. psbE-psbL and ndhA Intron, the Promising Plastid DNA Barcode of Fagopyrum. International Journal of Molecular Sciences. 2019;20(14):3455. doi: 10.3390/ijms20143455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brennicke A, Takenaka M, Härtel B, Zehrmann A, Verbitskiy D. RNA editing in plants and its evolution. Annu Rev Genet. 2013;47(1):335–352. doi: 10.1146/annurev-genet-111212-133519. [DOI] [PubMed] [Google Scholar]
- 41.Hein A, Polsakiewicz M, Knoop V. Frequent chloroplast RNA editing in early-branching flowering plants: pilot studies on angiosperm-wide coexistence of editing sites and their nuclear specificity factors. BMC Evol Biol. 2016;16:23. doi: 10.1186/s12862-016-0589-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bogorad L. Engineering chloroplasts: an alternative site for foreign genes, proteins, reactions and products. Trends Biotechnol. 2000;18(6):257–263. doi: 10.1016/S0167-7799(00)01444-X. [DOI] [PubMed] [Google Scholar]
- 43.Quesada-Vargas T, Ruiz ON, Daniell H. Characterization of heterologous multigene operons in transgenic chloroplasts: transcription, processing, and translation. Plant Physiol. 2005;138(3):1746–1762. doi: 10.1104/pp.105.063040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Cosa B, Moar W, Lee SB, Miller M, Daniell H. Overexpression of the Bt cry2Aa2 operon in chloroplasts leads to formation of insecticidal crystals. Nature Biotechnol. 2001;19(1):71–74. doi: 10.1038/83559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chaisson MJ, Tesler G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics. 2012;13(238):1–17. doi: 10.1186/1471-2105-13-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. GeSeq - Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017;45(Web Server issue):W6–W11. doi: 10.1093/nar/gkx391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lohse M, Drechsel O, Bock R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2007;52(5–6):267–274. doi: 10.1007/s00294-007-0161-y. [DOI] [PubMed] [Google Scholar]
- 49.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21(9):1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 50.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinformatics. 2013;14(2):178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Annotated gene list in the chloroplast of SpV2. Table S2. A comparison of detected introns among model plants. Table S3. The list of RNA editing sites in SpV2. Figure S1. Bioinformatic pipeline of chloroplast genome assembly and annotation. Details are described under Methods. Figure S2. The distribution of RNA editing events in the chloroplast genes of S.polyrhiza. Graph shows the number of currently detected RNA editing sites in protein coding genes. Figure S3. Alignments of editing sites in rpoC2 and ndhA genes. The sequences included RNA editing sites are shown before RNA editing. The amino acid is in orange and substitutions are marked with arrows. The start, RNA editing and end locations are listed above the alignment. All aligned sequences are antisense from the reference except rpoC2 and rpoC1 gene of rice.
Data Availability Statement
The chloroplast genome assembly and annotation (SpV2) were deposited in GenBank under the accession number of MN419335.