

Abstract
Background
N6-methyladenosine (m6A) is the most prevalent post-transcriptional modification of eukaryotic mRNA. It has been reported that there is a stimulus-dependent regulation of m6A in the mammalian central nervous system in response to sensory experience, learning, and injury. The mRNA m6A methylation pattern in rat cortex after traumatic brain injury (TBI) has not been investigated.
Results
In this study, we conducted a genome-wide profiling of mRNA m6A methylation in rat cortex via methylated RNA immunoprecipitation sequencing (MeRIP-Seq). After TBI, the expressions of METTL14 and FTO were significantly down-regulated in rat cerebral cortex. Using MeRIP-Seq, we identified a total of 2165 significantly changed peaks, of which 1062 were significantly up-regulated and 1103 peaks were significantly down-regulated. These m6A peaks were located across 1850 genes. The analysis of both m6A peaks and mRNA expression revealed that there were 175 mRNA significantly altered methylation and expression levels after TBI. Moreover, it was found that functional FTO is necessary to repair neurological damage caused by TBI but has no effect on the spatial learning and memory abilities of TBI rats by using FTO inhibitor FB23–2.
Conclusion
This study explored the m6A methylation pattern of mRNA after TBI in rat cortex and identified FTO as possible intervention targets in the epigenetic modification of TBI.
Keywords: m6A methylation, Epigenetic modification, FTO, Traumatic brain injury, Rat cortex
Background
More than 5.3 million people in the United States suffer from disability caused by traumatic brain injury (TBI) each year, and 1.7 million people sustain brain trauma, resulting in more than 60 billion in economic losses [1]. The causes of TBI include blast injury, sports injury, and penetration of the skull. TBI patients suffer from both short-term and long-term effects of their injuries. More than half of the severe TBI survivors are severely disabled within one year after injury, and some patients have devastating neurological disorders, including cognitive or memory impairment, and motor, sensory or emotional function obstacles [2]. In addition, for some patients with mild repetitive TBI, such as athletes in contact sports may develop chronic traumatic encephalopathy in later life [3]. The pathophysiological mechanism of TBI has two aspects, the primary injury caused by direct mechanical force and the secondary injury of neurons. The mechanical force of the impact directly impairs the axon and a part of a neuron. The mechanism involved in secondary injury is more complicated [3–5]. It includes delayed cell death (such as the newly discovered ferroptosis [6]), brain edema, metabolic defects [7], and blood-brain barrier damage caused by biochemical and physiological changes after trauma [8].
Recently, a series of studies have found that epigenetic changes play important roles in TBI-induced pathophysiological responses such as DNA methylation, chromatin post-translational modifications, and miRNA regulation of gene expression [5, 9, 10]. Epigenetics refers to the up- or down-regulation of gene or protein expression without changing the DNA sequence [11]. Bailey and colleagues demonstrated that exposure to simulated free-field explosions resulted in a significant increase in ten-eleven translocation methylcytosine dioxygenase in the prefrontal cortex and hippocampus of male rats [9]. The TET protein family has been proposed as a viable mechanism for active demethylation, and thus these changes may be associated with a higher degree of hypomethylation in the rat brain [12, 13]. GADD45 has been identified as a mediator of demethylation of active DNA. In a combined spinal and neurological injury model, systemic folate therapy increases the methylation of the GADD45a promoter at 12 of its 18 CpG sites, restoring its methylation level to baseline and promoting spinal cord regeneration [14, 15]. TBI induced extensive changes in the DNA methylation patterns of rat hippocampus and the differentially methylated gene sites within 10 kb distance [16]. Another study investigated DNA methylation alterations after TBI in the rat frontal cortex using the brain blast-induced injury model and found that these differentially methylated genes were enrichet in cell death, survival, and nervous system development and function [17].
N6-methyladenosine (m6A) is the most prevalent post-transcriptional modification of eukaryotic mRNA and long non-coding RNA [18]. Adenosine in cellular RNA can be chemically modified by adding a methyl group at the N6 position of the adenine base, thereby producing a m6A adenosine, which is a nucleoside and a part of ribose (ribofuranose) and adenine. The composition is linked in the middle by a β-N9-glycosidic bond [19]. There are three basic mechanisms in m6A methylation: “writers” are methyltransferases including METTL3 and some related proteins like METTL14; “readers” are m6A binding proteins involved in the translation process; “erasers” are demethylases, including ALKBH5 and FTO [20, 21]. According to previous studies, m6A methylation was of great significance in the maintenance and differentiation of embryonic stem cells [22], the development and maintenance of acute myeloid leukemia [23], and the self-renewal of leukemia stem cells/initiating cells [24]. Bioinformatics analysis indicated that the neuron subtype-specific gene region was enriched for m6A. At the level of a single neuron, m6A-modified RNA and its interactions were spread over specific structures such as axons, dendrites, presynaptic nerve endings, and dendritic spines [25]. It has been demonstrated that there is a stimulus-dependent regulation of m6A in the mammalian central nervous system in response to sensory experience, learning, and injury [26, 27]. METTL3 knockdown results in a prolonged cell cycle of cortical neural progenitor cells and decreased differentiation of radial glial cells. Knockout of METTL14 in mouse embryos leads to prolongation of cortical neurogenesis to the postnatal stage. These data indicate a close relationship between m6A and nerve cells [28]. Damage to nerve cells in various regions of the adult nervous system can strongly induce regeneration. This recovery process relies on rapid de novo gene transcription, as well as protein synthesis. Using a peripheral sciatic nerve injury model, up-regulation of m6A in dorsal root ganglion neurons was reported to occur within three days after injury [29].
Compared with DNA modifications in TBI, the roles of RNA methylation have not been elucidated to date. In order to explore the epigenetic modifications of RNA in TBIand their functions in neurogenesis after TBI, we analyzed the N6-methyladenosine methylation profiles in the cortex of controlled cortical injury (CCI) rats. The CCI model in rats is a widely used TBI model [30]. In addition, we also analyzed the differential expressions of mRNA after TBI. Analysis of RNA and m6A methylation levels, we have identified potential novel targets that can provide a basis for further intervention in TBI.
Methods
Animals
Male Sprague-Dawley rats (aged 10 weeks, each weighting approximately 250–300 g) were purchased from Vital River Laboratory Animal Technology Co. Ltd. (Vital River, China). Each rat was housed in a regular environment of 12 h light/dark cycle with free access to water and food for one week in the Animal Experimental Center of Zhongnan Hospital of Wuhan University. The weight of each animal was controlled (300–350 g) during experiments. The procedures of this study were carried out in accordance with the guidelines of the Chinese Council on Animal Protection, and approved by the Institutional Animal Care and Use Committee of Wuhan University (IACUC: 2019010). We made every effort to reduce the suffering of animals in the experiments. The anesthesia method was intraperitoneal injection of 5% pentobarbital (50 mg/kg). In MeRIP-Seq experiments, 18 rats were randomly divided into 6 groups, and each group had 3 rats. Half of the groups were randomly assigned as TBI groups and the other half were sham operation groups (i.e., only cranial incision was performed). The MeRIP-Seq required at least 100 μg RNA in each sample and ~ 50 μg RNA in each rat cortex; therefore, the RNA of 3 rats were pooled as one sample for MeRIP-Seq and there were 3 samples for TBI and Sham groups, respectively.
Animal model of controlled cortical injury
The controlled cortical impact (CCI) was chosen to establish the animal model of TBI as described previously [31, 32]. After intraperitoneal injection of pentobarbital, a 5 mm diameter hole was drilled over the right hemisphere bone equidistant between the lambda and bregma. Then the rats were put on the CCI device (Custom Design & Fabrication, USA). The 3 mm impactor tip was placed at the center of the craniotomy to induce moderate injury at 5 m/s speed, 200 msec dwell time, and 2 mm deformation depth. Any rats with a herniation of dura mater or occlusion of the injury hub were eliminated from the group. Rats were housed in separate cages after surgery and had free access to food and drink for 24 h. After 24 h, rats were fully anesthetized with 5% pentobarbital (50 mg/kg) and the undamaged tissues around the wound were surgically collected and were refrigerated at − 80 °C until RNA extraction.
RNA extraction and quantitative reverse transcription polymerase chain reaction
The total RNA was extracted from the cortical tissues of sham and TBI groups. In quantitative reverse transcription polymerase chain reaction (qRT-PCR) experiments, these were 6 rats each in TBI and Sham groups. The frozen tissue samples were first crushed using a frozen mortar and were ground for two minutes until the samples became powder. Then each sample with 50–100 mg fresh weight was added to 1 mL of TRIzol (Thermo Fisher Scientific, USA) and mixed vigorously. After adding 200 μL of chloroform, shaking by hand for 15 s, and incubating at room temperature for 5 min, centrifugation was carried out at 13,000 rpm, 4 °C for 15 min. The aqueous phase was then transferred to a 1.5 mL RNase-free centrifuge tube. An equal volume of 100% isopropanol was added to the aqueous phase, and after incubating at room temperature for 10 min, it was centrifuged at 13,000 rpm for 15 min at 4 °C. Finally, the supernatant was removed. The precipitate was washed by adding 1 mL of cold 75% ethanol and centrifuged at 13,000 rpm for 5 min at 4 °C. The wash solution was discarded and the RNA pellet was air dried for 5–10 min. Finally, the RNA was dissolved in 50 μL RNase-free water.
To analyze the expression levels of genes encoding m6A methylation enzymes, reverse transcription polymerase chain reaction (qRT-PCR) was performed. A total of 1 μg of RNA was reverse transcribed into cDNA using the ReverTra Ace qPCR RT Kit (TOYOBO, Japan) following the manufacture’s instructions; the cDNA products were diluted 10-fold for further use. Then the PCR was performed with 2 μL diluted cDNA using the ReverTra Ace qPCR RT Kit (TOYOBO, Japan) in the PCR instrument (CFX Connect™, Bio-Rad, USA). Actb was used as the internal control to normalize the expression levels of candidate genes. The primers used in qRT-PCR were designed using the Primer3 website (http://primer3.ut.ee/) and listed in Additional file 1: Table S1.
Methylated RNA immunoprecipitation sequencing (MeRIP-Seq)
The MeRIP-Seq method was used to measure the methylation levels of m6A in RNA. The N6-methyladenine antibodies were used to enrich highly methylated RNA fragments for high-throughput sequencing to detect m6A modifications at the entire transcriptome range as previously described Briefly, after adding 250 μL of fragmentation buffer (10 mM ZnCl2, 10 mM Tris-HCl pH 7.0) to the 50 μL of isolated 2 × poly-A RNA to a final volume of 300 μL, intact poly-A purified RNA was denatured at 95 °C for 5 min using a thermocycler. The fragmentation reaction was stopped by adding 50 μL of Stop Buffer (0.5 M EDTA) to a final volume of 350 μL and immediately put on ice. The fragmented RNA was allowed to bind with m6A-Dynabeads at room temperature while rotating at 7 rpm (tail flip) for 1 h. The m6A-Dynabeads-RNA complex was resuspended in 500 μL of m6A binding buffer and incubated for 3 min at room temperature. The supernatant was removed after placing the beads in the magnet. The next step was to add 125 μL of 42 °C pre-heated Elution Buffer to the m6A-Dynabead complexes and incubate at 42 °C for 5 min. Then, the m6A positive RNA was purified by adding 500 μL of acid phenol-chloroform to the sample collected in the previous step. We took 100 ng of RNA for each sample (100 ng of input and 100 ng of post m6A-IP positive fraction) for library construction utilizing the Stranded mRNA-seq Kit (Illumina, USA). After the sequencing library was established, all samples were sequenced by Illumina Hiseq X10 (OE Biotech, China).
Behavioral testing: modified neurological severity score and Morris water maze
FTO inhibitor FB23–2 (MedChemExpress, USA) was used to block FTO demethylase function by intraperitoneal injection (1.4 mg/kg) 15 min after CCI [33]. The 25 rats were randomly divided into 4 groups: TBI + FB23–2 (n = 7), TBI + DMSO (n = 8), Sham+FB23–2 (n = 5), and Sham+DMSO (n = 5). A modified neurological severity score (mNSS) consisting of motor, sensory, balance, and reflex tests, was used to estimate the neurological deficits in rats [34]. The higher the mNSS score, the greater the neurological dysfunction. We use the Morris water maze (MWM) test to assess spatial learning ability [35]. The experimental training phase was carried out continuously for 5 days, with training 4 times a day. During training, the rats were placed into the pool from the four water entry points facing the pool wall, and the time required for the rats to enter the water to find the underwater hidden platform and stand on it was recorded as the incubation period. The rats were allowed to stand on the platform for 15 s after locating it. If the rat failed to find the platform within 60 s after entering the water, it was gently dragged onto the platform from the water, staying for 15 s before removal for the next training. At day 6 post CCI, we recorded the escape latency of each rat.
Data processing and bioinformatics analyses
Raw data (raw reads) of fastq format were first processed using the Trimmomatic software [36]. Clean data (clean reads) were obtained by removing reads containing adapter, reads containing ploy-N, and low-quality reads from raw data. We then randomly extracted 250,000 paired reads from clean data, using the BLASTn software to align with the NT database (https://ftp.ncbi.nih.gov/blast/db) and take the best result with e-value <1e-10 and coverage > 80%. Meanwhile, the SortMeRNA software was used to discard ribosomal RNA (rRNA) reads and the remaining clean reads were mapped to the reference genome using HISAT2 with default parameters. Unique reads with high mapping quality were retained and potential PCR duplicates were removed using Picard [37, 38].
To access the quality of methylated RNA immunoprecipitation sequencing data, the Trumpet R package was used for evaluation of m6A-seq data quality [39]. The m6A-enriched peaks in each m6A immunoprecipitation sample were identified via MeTDiff peak calling software with the corresponding input sample serving as control. The MeTDiff was run with options (FRAGMENT_LENGTH = 200, PEAK_CUTOFF_PVALUE = 0.01, and PEAK_CUTOFF_FDR = 0.05) for peak detection [40]. Called peaks were annotated by intersection with gene architecture using ChIPseeker [41]. Differential analysis for m6A-Seq identified differences in RNA methylome in a case-control study and the differential peaks were detected by MeTDiff with parameter (FRAGMENT_LENGTH = 200, PEAK_CUTOFF_PVALUE = 0.01, DIFF_PEAK_CUTOFF_FDR = 0.05, and PEAK_CUTOFF_FDR = 0.05), then differential peaks were annotated by ChIPseeker.
GO enrichment and KEGG pathway enrichment analyses of peaks and differentially expressed peaks were performed respectively using R based on the hypergeometric distribution. The statistics in this study were analyzed using SPSS Version 25.0 software (IBM, USA). The network of m6A peak and mRNA expression were constructed using the Cytoscape Version 3.7.2 software.
Results
METTL14 and FTO were both down-regulated after TBI in rat cortex
In order to investigate whether m6A methylation status was changed in rat cortex after TBI, we evaluated the six genes encoding methyltransferases and demethylases by qRT-PCR: METTL3, METTL14, WTAP, VIRMA, FTO, and ALKBH5. The expression of METTL14 (methylation enzyme) and FTO (demethylation enzyme) were both significantly down-regulated in the TBI group compared with the sham group (p < 0.05). The expression of the other four genes including METTL3, WTAP, VIRMA, and ALKBH5 were not significantly changed in the rat cortex after TBI. The expression changes of METTL14 and FTO could lead to a dynamic change in the m6A methylation in the rat cortex after TBI (Fig. 1).
Fig. 1.

Reverse transcription polymerase chain reaction (RT-PCR) of METTL3, METTL14, WTAP, VIRMA, FTO, and ALKBH5. a–b, RT-PCR of demethylases ALKBH5 and FTO, and the FTO is significantly down-regulated after TBI. c–f, RT-PCR of methyltransferases METTL3, METTL14, VIRMA, and WTAP, and the METTL14 is significantly down-regulated after TBI. TBI group (n = 6); Sham group (n = 6). “**” and “***” indicate p < 0.01 and p < 0.001, respectively
Overview of methylated RNA immunoprecipitation sequencing results
To measure the m6A methylation level of the sham and TBI groups, we performed a transcriptome-wide m6A-seq analysis using MeRIP-Seq. We found an average of 11.67 Gb sequencing data in the MeRIP-Seq samples and 13.46 Gb sequencing data of input samples were generated (Additional file 2: Table S2). After removing the adaptor sequence, low-quality reads, and low-quality bases from the 3′ and the 5′ terminals, the average number of clean data was 10.66 Gb in the MeRIP-Seq samples and 12.28 Gb in the input samples (Additional file 2: Table S2). The clean reads of each sample were compared with the rat genome to obtain positional information on the reference genome and an average of 95.43% reads was mapped on the rat genome. The clean reads with multiple alignments to the rat genome were eliminated (5.48%), and the clean reads with unique alignment retained for subsequent analysis (89.95%) (Additional file 3: Table S3).
Topological distribution of m6A peaks
Compared with the sequencing data between MeRIP-Seq samples and their corresponding inputs, m6A peaks were identified, including 11,789 distinct m6A peaks for 4900 transcribed genes from all samples (Additional file 4: Table S4). The chromosomes with the most m6A methylation sites were chromosomes 1, 3, and 10 with 1386, 921, and 905 m6A methylation sites, respectively (Fig. 2a and Additional file 4: Table S4). Specifically, the number of m6A modification sites on the gene ranged from 1 to 19, with 64.31% of genes having one or two methylation sites and 35.69% of genes having 3 or more methylation sites (Additional file 4: Table S4). For instance, Pclo located on chromosome 4 had the maximum number of methylation sites (i.e., 19 sites) (Additional file 4: Table S4). In addition, we analyzed the distribution patterns of m6A methylation peaks in gene structures of mRNA and we found that the peaks were mostly distributed on the exon region, and there was a distinct enrichment peak at the 3′ terminate (near the stop codon) and a enrichment peak at the 5′ terminate (near the start codon). The reads in the input samples were higher than the reads in the IP samples at the CDS region (Fig. 2b). We have chosen three genes to show the m6A methylation pattern. The peak of Trmt10c was located at the CDS region, the peak of Coro7 was located at the CDS and 3' UTR regions, and the peak of Plk was located at the 3' UTR, the CDS, and the 5' UTR regions (Fig. 2c).
Fig. 2.

Topological distribution of m6A peaks. a, The distribution pattern of m6A peaks in different chromosomes. b, The distribution patterns of m6A methylation peaks in gene structures of mRNA. The peaks were mostly distributed on the exon region and there was a distinct enrichment peak at the 3′ terminate (near the stop codon) and an enrichment peak at the 5′ terminate (near the start codon). c, IGV plot shows directly the peaks in the genes of Trmt10c, Coro6 and Plk. The peak of Trmt10c was located at the CDS region, the peak of Coro7 was located at the CDS and 3′ UTR regions, and the peak of Plk was located at the 3′ UTR, the CDS, and the 5′ UTR regions
Significant m6A methylation changes after TBI
To reveal the roles of m6A methylation in TBI, we compared the methylation levels of rat cortex among the TBI group and sham group (Additional file 5: Table S5). An average of 15,305 peaks in the TBI group and an average of 16,714 peaks in the sham group were identified (Fig. 3a). Compared with the input sample, the average logarithmic fold-enrichment of TBI group was 4.11, and the average logarithmic fold-enrichment of sham group was 3.72 (Fig. 3b). The average peak length of TBI group was 3426.14 bp, and the average peak length of sham group was 3831.73 bp (Fig. 3c). A total of 2165 significantly changed peaks were identified (p < 0.01, FDR < 0.05), in which 1062 peaks were significantly up-regulated and 1103 peaks were significantly down-regulated (Fig. 4a). The top 20 differentially methylated mRNA were shown in Table 1. As illustrated above, 1580 out of 4900 genes had peaks that were differentially methylated. The average logarithmic fold-enrichment of all the peaks are 2.53 (Fig. 4b) and the average length of each peak was 4358.7 bp (Fig. 4c). The distribution of p-value among all peaks is shown in Fig. 4d. The peaks with significant changes are mainly distributed in the CDS (49.95%), intron (22.86%), 3′ UTR (19.82%), and 5′ UTR (7.34%), and only 0.02% were found in the intergenic region (Fig. 5a). Many peaks with significant changes spanned multiple genes structures; 556 peaks distributed in the CDS and 3′ UTR regions, 532 peaks in the CDS and intron regions, and 495 peaks in the CDS region (Fig. 5b). Three representative genes with significantly changed peaks are presented in Fig. 5c. The m6A level of CD14, Dll4, and Sox7 were significantly up-regulated after TBI by 3.63 fold, 2.10 fold, and 3.14 fold, respectively.
Fig. 3.

Number of peaks, fold enrichment of peaks and length of peaks in each group after TBI. a, The peak number of each group. An average of 15,305 peaks in the TBI group and an average of 16,714 peaks in the sham group were identified. b, The enrichment of each group. The average logarithmic fold-enrichment of TBI group was 4.11, and the average logarithmic fold-enrichment of sham group was 3.72. c, The peak length of each group. The average peak length of TBI group was 3426.14 bp, and the average peak length of sham group was 3831.73 bp
Fig. 4.

Peak number, fold enrichment, peak length and p-value in the TBI and Sham groups. a, A total of 2165 significantly changed peaks were identified (p < 0.01, FDR < 0.05). b, The average logarithmic fold-enrichment of the peaks were 2.53. c, The average length of each peak was 3.64 (log10). D, The distribution of p-value of all peaks
Table 1.
The top 20 significantly changed m6A methylation peak based on p-value
| mRNA | Chromosome | Peak region | Peak start | Peak end | Lg (p-value) | Log2 (fold-change) | Up/Down |
|---|---|---|---|---|---|---|---|
| Sp4 | 6 | 3′ UTR | 146,193,411 | 146,193,560 | −434 | −1.90 | Down |
| Mycbp2 | 15 | Exon | 87,493,207 | 87,494,725 | −434 | −3.83 | Down |
| Pias4 | 7 | Exon | 11,414,644 | 11,423,227 | −8.36 | −2.14 | Down |
| Fbxo32 | 7 | 3′ UTR | 98,065,124 | 98,067,070 | −8.13 | −1.43 | Down |
| Crhbp | 2 | 5′ UTR | 25,152,370 | 25,153,284 | −7.28 | −1.61 | Down |
| Kcnj11 | 1 | 3′ UTR | 102,103,938 | 102,104,138 | −7.15 | −1.83 | Down |
| Amer3 | 9 | Exon | 41,096,883 | 41,097,424 | −7.00 | −1.82 | Down |
| Spg7 | 19 | 3′ UTR | 55,913,654 | 55,913,954 | −6.85 | −2.99 | Down |
| Slc2a13 | 7 | 5′ UTR | 132,757,358 | 132,757,508 | −6.62 | −2.47 | Down |
| Trpc6 | 8 | Exon | 6,872,050 | 6,872,448 | −6.62 | −1.50 | Down |
| Slfn4 | 10 | 5′ UTR | 70,428,465 | 70,428,814 | −7.21 | 1.81 | Up |
| Mdfic | 4 | 3′ UTR | 42,280,721 | 42,280,870 | −6.79 | 4.17 | Up |
| Prss35 | 8 | 3′ UTR | 94,438,220 | 94,439,065 | −6.27 | 1.05 | Up |
| Ppp1r3b | 16 | 3′ UTR | 60,418,476 | 60,418,677 | −6.18 | 3.14 | Up |
| Nlgn3 | X | Exon | 71,221,689 | 71,222,484 | −6.18 | 1.03 | Up |
| Hcn1 | 2 | Exon | 50,495,344 | 50,499,750 | −6.13 | 1.12 | Up |
| Rps6kb1 | 10 | 5′ UTR | 73,854,215 | 73,865,364 | −6.08 | 1.49 | Up |
| Gpkow | X | 3′ UTR | 15,638,954 | 15,640,719 | −6.02 | 2.39 | Up |
| Prelid3a | 18 | 3′ UTR | 63,213,663 | 63,215,734 | −5.87 | 2.87 | Up |
| Pprc1 | 1 | Exon | 265,818,641 | 265,818,890 | −5.80 | 4.01 | Up |
Fig. 5.

The distribution of significantly changed peaks after TBI. a, Sector graph shows the ratio of peaks in each region. The peaks with significant changes were mainly distributed in the exon (49.95%), CDS (22.86%), 3′ UTR (19.82%), and 5′ UTR (7.34%) and only 0.02% were found in the intergenic region. b, Showing the exact distribution pattern of significantly changed peaks post-TBI. There were 556 peaks distributed in the CDS and 3′ UTR regions, 532 peaks in the CDS and intron regions, and 495 peaks in the CDS region. c, Three representative genes with significantly changed peaks. The m6A level of CD14, Dll4, and Sox7 were significantly up-regulated after TBI by 3.63 fold, 2.10 fold, 3.14 fold, respectively
GO and KEGG analyses revealed the biological information behind m6A methylation
To clarify the function of RNA m6A methylation in TBI, we subjected the genes with significantly changed m6A methylation level to gene functional enrichment analysis using the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). GO aims to establish a unified paradigm to explain the role of eukaryotic genes and proteins in cells. GO analyses showed m6A methylation was related to three parts of biological information. Molecular function: protein binding, poly(A) RNA binding, chromatin binding; cellular component: nucleoplasm, nucleus, cytoplasm; biological process: axonal fasciculation, cerebellum development, in utero embryonic development (Fig. 6a). Through KEGG analyses, we annotated the genes with significantly changed m6A methylation levels. These genes were mostly enriched in the pathway including renal cell carcinoma, microRNAs in cancer, and proteoglycans in cancer (Fig. 6b).
Fig. 6.
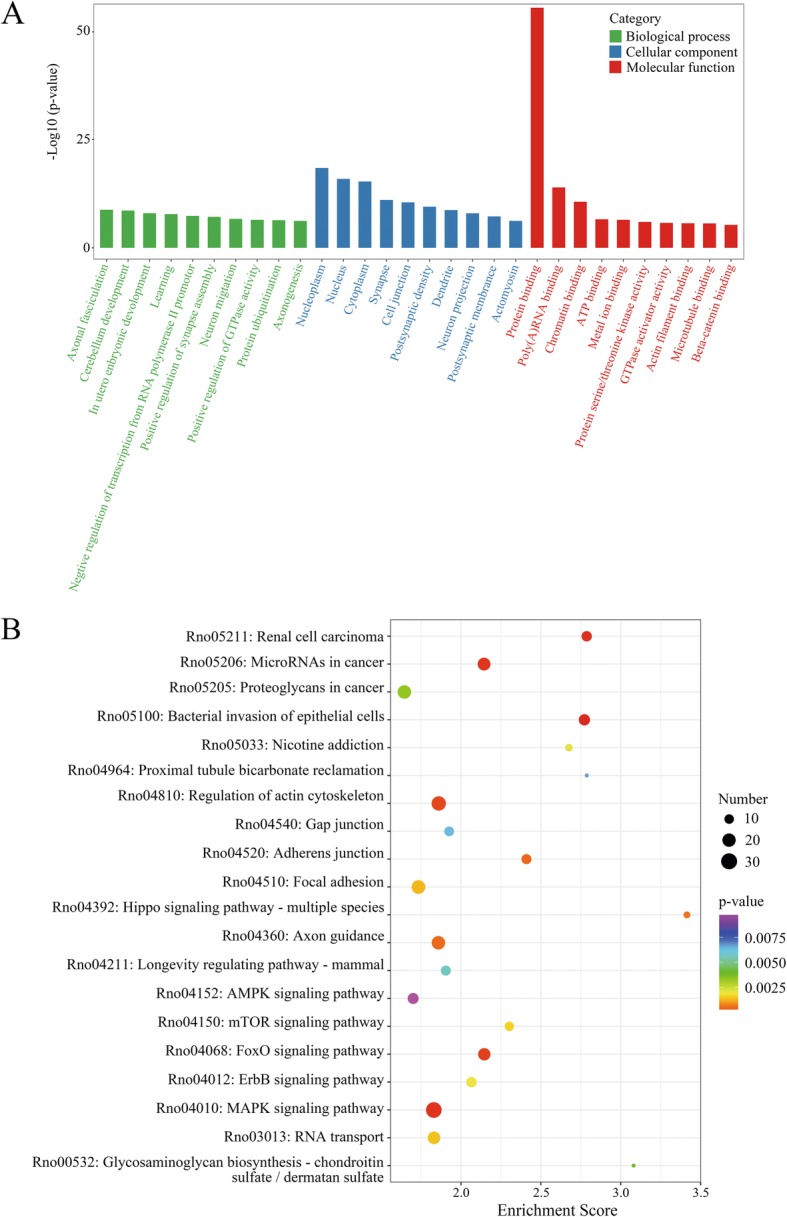
GO and KEGG analyses revealed the biological information behind mRNA m6A methylation. a, The top 10 enriched GO terms of the m6A peaks. b, The top 20 enriched pathways of m6A peaks
Gene expression profiles after TBI
In order to further clarify the relationship between m6A methylation level and gene expression, we measured the level of mRNA changes in rat cerebral cortex after TBI using the RNA sequencing data of input experiments (Additional file 6: Table S6). Figure 7a shows the principal component analysis of the TBI and sham groups. It was found that the confidence ellipses of samples among sham and TBI groups were separated from each other, indicating that the gene expression patterns were similar within the same treatment group, but significantly different between sham and TBI groups (Fig. 7a). It was found that a total of 428 mRNAs increased and 280 mRNAs decreased expression (p < 0.05, log2FC > 1) in rat cerebral cortex after TBI (Fig. 7b). The top 20 differentially changed mRNA are shown in Table 2. The heatmap shows the relative expression level of the three TBI IPs and the three sham samples in the same group have similar presentation (Fig. 7c). The volcano plot (Fig. 7d) shows the significantly up-regulated and down-regulated mRNA after TBI. We marked the top 5 most significantly up-regulated genes (Il1r2, Fosl1, Ptx3, Hsbp8, and Msn) and top 5 down-regulated genes (Hdac9, Tril, Fam107a, Pcdh20, and Prom1) in the volcano plot.
Fig. 7.

mRNA changes after traumatic brain injury. a, Principal component analysis of the TBI group and the sham group. The confidence ellipses of samples among sham and TBI groups were separate from each other. b, The expression level of mRNA post-TBI. A total of 428 mRNA expression increased and 280 mRNA expression decreased (p < 0.05, log2FC > 1) in rat cerebral cortex after TBI. c, Heat map shows the relative expression level of the three TBI IP and the three sham IP. d, Volcano plot shows the significantly up-regulated and down-regulated mRNA after TBI
Table 2.
The top 20 differently expressed mRNA based on p-value
| mRNA | Chromosome | Gene start (bp) | Gene end (bp) | p-value | Log2 (fold-change) | Up/Down |
|---|---|---|---|---|---|---|
| Il1r2 | 9 | 46,840,992 | 46,881,264 | 1.86E-22 | 5.18 | Up |
| Fosl1 | 1 | 220,826,560 | 220,835,066 | 1.92E-20 | 3.11 | Up |
| Ptx3 | 2 | 158,097,843 | 158,103,653 | 1.91E-15 | 2.25 | Up |
| Hspb8 | 12 | 45,905,371 | 45,920,013 | 1.77E-14 | 1.54 | Up |
| Msn | X | 65,226,748 | 65,295,810 | 2.22E-14 | 1.30 | Up |
| Cebpd | 11 | 89,008,008 | 89,009,146 | 6.16E-13 | 1.43 | Up |
| Lif | 14 | 84,482,674 | 84,500,642 | 9.96E-13 | 3.46 | Up |
| Emp1 | 4 | 169,147,243 | 169,181,966 | 1.82E-12 | 1.95 | Up |
| Csf2rb | 7 | 119,554,354 | 119,568,776 | 9.85E-12 | 1.37 | Up |
| Cd44 | 3 | 92,696,313 | 92,783,658 | 1.13E-11 | 2.24 | Up |
| Hdac9 | 6 | 53,488,883 | 54,059,119 | 3.45E-11 | −1.72 | Down |
| Tril | 4 | 83,967,696 | 83,972,540 | 9.06E-11 | −1.42 | Down |
| Fam107a | 15 | 18,399,515 | 18,415,965 | 6.17E-10 | −1.03 | Down |
| Pcdh20 | 15 | 71,772,777 | 71,779,033 | 4.29E-09 | −1.33 | Down |
| Prom1 | 14 | 71,533,063 | 71,637,417 | 7.08E-09 | −2.53 | Down |
| Slc38a3 | 8 | 116,406,241 | 116,422,366 | 1.19E-08 | −1.07 | Down |
| Itm2a | X | 78,490,866 | 78,496,847 | 1.66E-08 | −1.76 | Down |
| RGD1559896 | 19 | 57,396,606 | 57,422,093 | 9.56E-08 | −1.17 | Down |
| Clic6 | 11 | 32,655,616 | 32,699,382 | 4.06E-07 | −2.72 | Down |
| Nat8f3 | 4 | 117,490,035 | 117,490,721 | 5.63E-07 | −2.39 | Down |
Combined analysis of m6A methylation and gene expression after TBI
Combined analysis of m6A methylation and RNA expression levels used peaks with log2 foldchange > 0.5, p < 0.01 and the mRNA with log2 foldchange > 0.5, p < 0.05. As a result, there were 175 mRNAs where their m6A peaks and mRNA levels both changed significantly, among which the levels of 40 mRNAs were both up-regulated and the levels of 41 mRNAs were both down-regulated. Moreover, there were 47 genes with up-regulated mRNA expression and down-regulated m6A peaks and 47 genes with down-regulated mRNA expression and up-regulated m6A peaks (Additional file 7: Table S7). The relationship between m6A methylation and mRNA expression are shown in the quadrant graph and Venn diagram (Fig. 8a and b). Last, we conducted the protein-protein interaction network to show the connection between the proteins encoded by the 175 genes (Fig. 8c).
Fig. 8.

Joint analysis of m6A methylation and mRNA expression after TBI. a, b, Four quadrant graph and venn diagram shows the relationship between mRNA m6A methylation and its mRNA expression. c, The protein-protein interaction network shows the connection between the proteins of the genes used in the combined analysis
FTO inhibitor FB23–2 increases the mNSS score of rats after TBI
In order to investigate the roles of m6A demethylase FTO in controlling rat neurological function after TBI, we performed the mNSS and MWM tests in the four groups: TBI + FB23–2, TBI + DMSO, Sham+FB23–2, and Sham+DMSO (Additional file 8: Tables S8 and Additional file 9: Table S9). The mNSS scores of sham+FB23–2 and the sham+DMSO group for four consecutive days showed no difference, indicating that FB23–2 has no effect on neurological function in normal rats. All TBI groups (i.e., TBI + FB23–2 and TBI + DMSO) scored significantly higher on the mNSS than the sham groups (i.e., Sham+FB23–2 and Sham+DMSO) for four consecutive days, indicating that CCI caused damage to neurological function. Interestingly, inhibition of FTO exacerbated damage to neurological function caused by TBI. Specifically, in the first day post-CCI, the mean score of TBI + FB23–2 group was 10, which was significantly higher than the TBI + DMSO group (7.875; p = 0.0135); On the second day post-CCI, the mean score of the TBI + FB23–2 group was 8.857, which was significantly higher than the TBI + DMSO group (6.875, p = 0.025). The trend continued on the third (p = 0.0018) and fourth (p = 0.0002) day post-CCI with the TBI + FB23–2 group scoring 10.14 and 8.71, while the TBI + DMSO group scored 6.75 and 4.13, respectively (Fig. 9a). However, the results of MWM test indicated that the spatial learning and memory abilities of TBI + FB23–2 and TBI + DMSO group showed no difference, indicating that inhibition of FTO don’t affect the spatial learning and memory of CCI rats (Fig. 9b and c).
Fig. 9.

Effects of FTO inhibitior FB23–2 on neurological functions and cognitive and learning abilities in TBI. a, the score of four groups in mNSS: TBI + FB23–2 (n = 7), TBI + DMSO (n = 8), Sham+FB23–2 (n = 5), and Sham+DMSO (n = 5). b and c, the escape latency time and target quadrant time of four groups in MWM, respectively: TBI + FB23–2 (n = 7), TBI + DMSO (n = 8), Sham+FB23–2 (n = 5), and Sham+DMSO (n = 5). “*”, “**”, and “***” indicate p < 0.05, p < 0.01, and p < 0.001 in TBI vs Sham, respectively; “#”, “##”, and “###” indicate p < 0.05, p < 0.01, and p < 0.001 in FB23–2 vs DMSO. Error bars represent SD. NS indicates p > 0.05
Discussion
An increasing amount of research has focused on the role of RNA m6A methylation in neurobiology and neurological diseases [24]. The transcriptome-wide profiling of RNA m6A in the prefrontal cortex of mice that received behavioral training revealed that RNA m6A methylation may be associated with memory consolidation [27]. It has been found that heterogeneity of RNA m6A methylation exist in the cerebellum and cortex of mice (methylation level and methylation site) through m6A-immunoprecipitation [29]. However, the RNA m6A methylation in rat cortex was not reported. Using MeRIP-Seq, we obtained a panorama of m6A methylation in rat cortex after TBI, which added more information to the role of m6A methylation in TBI epigenetics.
Compared with the sham group, the TBI group showed lower METTL14 and FTO expression levels (Fig. 1). As one of the methyltransferases, METTL14 can form a complex with METTL3 to catalyze m6A methylation on RNA [19]. On the contrary, FTO, which had been considered a demethylase, could mediate m6A demethylation of RNA [42]. As a result of METTL14 and FTO down-regulation in TBI, m6A methylation resulted in both increased peaks (1062) and decreased peaks (1103) after TBI (Fig. 4a). Previous studies focused on the methylation level of m6A in mouse hippocampus after brain injury found that the expression of METTL3 was decreased but the expression of other enzymes (METTL14, FTO, WTAP, and ALKBH5) were not significantly changed [43]. The differences of m6A-related methyltransferases and demethylases between mouse hippocampus and rat cerebral cortex might be caused by interspecies differences or different organ sources. The differently methylated m6A peaks in TBI were mostly distributed at the 3′ UTR near stop codons and the 5′ UTR near start codons, which was consist with previous studies in both human and mouse [44–46]. Specifically, m6A distribution was highly enriched for 3’UTR (> 40%) mRNA, especially at the beginning of the last exon (within 150–400 NT), where it rose sharply (6-fold) [47].
The m6A methylation levels of several genes related to pathophysiological processes of TBI were significantly changed. The expression of CD14 was an important part of the response by the central nervous system in acute inflammation after trauma [48]. There were more CD14+ cells in vascular peripheral and brain parenchyma after TBI within 1–2 days. Dll4 was a protein- coding gene involved in the Notch signaling pathway as Notch ligand activates Notch1 and Notch4 [49, 50]. During spinal cord neurogenesis, Dll4 inhibits V2a interneuron fate [51]. Recent studies have shown that Dll4 and Notch signaling are involved in sprouting angiogenesis and artery formation, and the over expression of Dll4 inhibits the vascular response [52, 53]. Interestingly, the expression level of Dll4 and the m6A methylation level of Dll4 were both up-regulated, which made the function of Dll4 in TBI become unpredictable. Bcl2 is part of the intrinsic apoptotic pathway and ceramide signaling pathway (affecting growth, proliferation, differentiation, apoptosis, and injury). The m6A peak of Bcl2 was down-regulated after TBI, and the mRNA was also down-regulated.
A previous study suggested that m6A modification induced mRNA instability [54]. In order to better understand the roles of m6A methylation in TBI, we screened all the differently expressed peaks combined with the differently expressed mRNA. As a result, we identified 175 mRNA whose m6A peak and mRNA level both changed significantly, which could be divided into four outcomes: mRNA and m6A peaks both up-regulated (40), mRNA and m6A peak both down-regulated (41), the m6A peak up-regulated and mRNA peak down-regulated (47), and the m6A peak down-regulated and mRNA peak up-regulated (47). In our results, we didn’t observe a significant correlation between m6A methylation and mRNA expression. Previous research showed that m6A can both promote and inhibit of translation [55]. YTHDF2-mediated degradation controls the lifetime of target transcripts, while YTHDF1-mediated translation promotes translation efficiency. Thus the exact protein level of these m6A methylated mRNA could not be foretold, which remains to be further researched.
To verify the roles of m6A demethylase FTO in TBI rats, we inhibited the demethylation functions of FTO by using FTO inhibitor FB23–2, and then performed behavioral experiments. FB23–2 can inhibit the m6A demethylase FTO, and it has been confirmed in leukemia studies that the degree of m6A methylation is reduced after intraperitoneal injection of FTO [33]. In the mNSS experiments, the neurological function of rats after TBI presents a tendency to restore itself, which is in agreement with previous studies [56]. However, the self-healing phenomenon was not found in FB23–2-injected TBI rats, indicating that functional FTO is necessary to repair neurological damage caused by TBI. Corroborating this, we detected FTO mRNA in rat cerebral cortex after TBI by qRT-PCR. Taken together, we believe that FTO plays an important role in the maintenance of neurological function in TBI rats. Besides, the escape latency and the time spent in target quadrant post-CCI of TBI + FB23–2 group and TBI + DMSO group showed no difference in MWM test, indicating that FB23–2 has no effect on the spatial learning and memory abilities of TBI rats. Based on above results, the inhibition of FTO result in the exacerbate the neurological damage, and this result may imply that the increase of FTO expression level and enhancement of FTO function could be a solution to accelerate neurological repair after TBI. And the We suggested that adenovirus transfection of construct that increased FTO expression to further verify the role of FTO in TBI. This study also found that METTL14 is reduced after TBI, but unfortunately there is currently no effective METTL14 inhibitor, and its role in TBI cannot be confirmed in our study, which also remains to be further elucidated.
The m6A methylation of mRNA after TBI has not been studied. The animal experiment results showed that the m6A methylation level changed significantly after TBI in rat cortex. These results in a rat TBI model might have meaningful correspondence to m6A related epigenetic changes in human studies [57]. Though we found that functional FTO is necessary to repair neurological damage caused by TBI, one limitation to our study was that it is not definitely clear the downstream targets of FTO. Future research could perform MeRIP-Seq on TBI + FB23–2 and TBI + DMSO rats to find out the direct target genes of FTO in TBI. Notably, FTO has been been found to be involved in leukemia, DNA damage repair, and heat stress [58].
Conclusion
Our study showed that the METTL14 and FTO were down-regulated after TBI in rat cortex. Through MeRIP-Seq, we identified the m6A methylation levels were significantly changed in 1580 mRNA. The following joint analysis of m6A peaks and mRNA expression revealed that there were 175 mRNA significantly changed after TBI. These genes may be the key genes to interfere in the epigenetic regulation of TBI. Moreover, we found that FTO played an important role in the maintenance of neurological function in TBI rats and could be a intervention target for alleviating damage caused by TBI.
Supplementary information
Additional file 1: Table S1. PCR primers used in this study
Additional file 2: Table S2. The detailed information of raw data
Additional file 3: Table S3. Statistical analysis of reads mapped in reference genome
Additional file 4: Table S4. The m6A peak number on each gene
Additional file 5: Table S5. The detailed information of significantly changed m6A peaks
Additional file 6: Table S6. The detailed information of significantly changed mRNA
Additional file 7: Table S7. The detailed information of conjoint analysis between mRNA expression and m6A methylation
Additional file 8: Table S8. The raw data of mNSS tests
Additional file 9: Table S9. The raw data of MWM tests
Acknowledgements
The authors acknowledge the members of Emergency Center, Zhongnan Hospital of Wuhan University. We thank LetPub (www.letup.com) for its linguistic assistance during the preparation of the manuscript.
Abbreviations
- CCI
Controlled cortical injury
- m6A
N6-methyladenosine
- MeRIP-Seq
Methylated RNA immunoprecipitation sequencing
- qRT-PCR
Quantitative reverse transcription polymerase chain reaction
- TBI
Traumatic brain injury
Authors’ contributions
YZ and XJ conceived of and designed the research plans. JY, YZ, HM, RZ, RL, and PW established the CCI model and isolated RNA samples from cerebral cortex tissues. JY and YZ completed the data processing, normalization, and bioinformatics analyses. JY wrote the article with contributions from all the authors. YZ and XJ supervised and fixed the writing. All authors have read and approved the manuscript.
Funding
This study was supported by the Emergency Diagnostic & Therapeutic Center of Central China, the Zhongnan Hospital of Wuhan University Science, Technology and Innovation Seed Fund (cxpy2017039, WJ2019H010/znpy2018043), and the Fundamental Research Funds for the Central Universities (2042019kf0149). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
All of the datasets in this study can be obtained by reasonable request to the corresponding authors.
Ethics approval and consent to participate
All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of Wuhan University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jiangtao Yu and Yuxian Zhang contributed equally to this work.
Contributor Information
Jiangtao Yu, Email: yjt@whu.edu.cn.
Yuxian Zhang, Email: zhangyuxian@whu.edu.cn.
Haoli Ma, Email: mahaoli@whu.edu.cn.
Rong Zeng, Email: zengrr@whu.edu.cn.
Ruining Liu, Email: rn.liu@whu.edu.cn.
Pengcheng Wang, Email: dtzy8912@whu.edu.cn.
Xiaoqing Jin, Email: redjin@whu.edu.cn.
Yan Zhao, Email: doctoryanzhao@whu.edu.cn.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13041-020-0554-0.
References
- 1.Taylor CA, Bell JM, Breiding MJ, Xu L. Traumatic brain injury-related emergency department visits, hospitalizations, and deaths - United States, 2007 and 2013. MMWR Surveill Summ (Washington, DC : 2002) 2017;66(9):1–16. doi: 10.15585/mmwr.ss6609a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Azouvi P, Arnould A, Dromer E, Vallat-Azouvi C. Neuropsychology of traumatic brain injury: An expert overview. Rev Neurol (Paris) 2017;173(7–8):461–472. doi: 10.1016/j.neurol.2017.07.006. [DOI] [PubMed] [Google Scholar]
- 3.Dixon KJ. Pathophysiology of traumatic brain injury. Phys Med Rehabil Clin N Am. 2017;28(2):215–225. doi: 10.1016/j.pmr.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 4.McGinn MJ, Povlishock JT. Pathophysiology of traumatic brain injury. Neurosurg Clin N Am. 2016;27(4):397–407. doi: 10.1016/j.nec.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Wong VS, Langley B. Epigenetic changes following traumatic brain injury and their implications for outcome, recovery and therapy. Neurosci Lett. 2016;625:26–33. doi: 10.1016/j.neulet.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao X, Jiang Y, Liang W, Wang Y, Cao S, Yan H, Gao L, Zhang L. miR-212-5p attenuates ferroptotic neuronal death after traumatic brain injury by targeting Ptgs2. Mol Brain. 2019;12(1):78. doi: 10.1186/s13041-019-0501-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu Z, Lv XA, Dai Q, Ge YQ, Xu J. Acute upregulation of neuronal mitochondrial type-1 cannabinoid receptor and it's role in metabolic defects and neuronal apoptosis after TBI. Mol Brain. 2016;9(1):75. doi: 10.1186/s13041-016-0257-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White TE, Surles-Zeigler MC, Ford GD, Gates AS, Davids B, Distel T, LaPlaca MC, Ford BD. Bilateral gene interaction hierarchy analysis of the cell death gene response emphasizes the significance of cell cycle genes following unilateral traumatic brain injury. BMC Genomics. 2016;17:130. doi: 10.1186/s12864-016-2412-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puccio AM, Alexander S. Chapter 4 genomics, transcriptomics, and epigenomics in traumatic brain injury research. Annu Rev Nurs Res. 2015;33:75–109. doi: 10.1891/0739-6686.33.75. [DOI] [PubMed] [Google Scholar]
- 10.Zhong J, Jiang L, Cheng C, Huang Z, Zhang H, Liu H, He J, Cao F, Peng J, Jiang Y, et al. Altered expression of long non-coding RNA and mRNA in mouse cortex after traumatic brain injury. Brain Res. 1646;2016:589–600. doi: 10.1016/j.brainres.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 11.Karuppagounder SS, Kumar A, Shao DS, Zille M, Bourassa MW, Caulfield JT, Alim I, Ratan RR. Metabolism and epigenetics in the nervous system: Creating cellular fitness and resistance to neuronal death in neurological conditions via modulation of oxygen-, iron-, and 2-oxoglutarate-dependent dioxygenases. Brain Res. 2015;1628(Pt B):273–287. doi: 10.1016/j.brainres.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bailey ZS, Grinter MB, De La Torre CD, VandeVord PJ. Blast induced neurotrauma causes overpressure dependent changes to the DNA methylation equilibrium. Neurosci Lett. 2015;604:119–123. doi: 10.1016/j.neulet.2015.07.035. [DOI] [PubMed] [Google Scholar]
- 13.Mateen BA, Hill CS, Biddie SC, Menon DK. DNA methylation: basic biology and application to traumatic brain injury. J Neurotrauma. 2017;34(16):2379–2388. doi: 10.1089/neu.2017.5007. [DOI] [PubMed] [Google Scholar]
- 14.Iskandar BJ, Rizk E, Meier B, Hariharan N, Bottiglieri T, Finnell RH, Jarrard DF, Banerjee RV, Skene JH, Nelson A, et al. Folate regulation of axonal regeneration in the rodent central nervous system through DNA methylation. J Clin Invest. 2010;120(5):1603–1616. doi: 10.1172/JCI40000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Befort K, Karchewski L, Lanoue C, Woolf CJ. Selective up-regulation of the growth arrest DNA damage-inducible gene Gadd45 alpha in sensory and motor neurons after peripheral nerve injury. Eur J Neurosci. 2003;18(4):911–922. doi: 10.1046/j.1460-9568.2003.02827.x. [DOI] [PubMed] [Google Scholar]
- 16.Meng Q, Zhuang Y, Ying Z, Agrawal R, Yang X, Gomez-Pinilla F. Traumatic brain injury induces genome-wide Transcriptomic, Methylomic, and network perturbations in brain and blood predicting neurological disorders. EBioMedicine. 2017;16:184–194. doi: 10.1016/j.ebiom.2017.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haghighi F, Ge Y, Chen S, Xin Y, Umali MU, De Gasperi R, Gama Sosa MA, Ahlers ST, Elder GA. Neuronal DNA methylation profiling of blast-related traumatic brain injury. J Neurotrauma. 2015;32(16):1200–1209. doi: 10.1089/neu.2014.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao BS, Nachtergaele S, Roundtree IA, He C. Our views of dynamic N(6)-methyladenosine RNA methylation. RNA. 2018;24(3):268–272. doi: 10.1261/rna.064295.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer KD, Jaffrey SR. Rethinking m(6)a readers, writers, and erasers. Annu Rev Cell Dev Biol. 2017;33:319–342. doi: 10.1146/annurev-cellbio-100616-060758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu B, Li L, Huang Y, Ma J, Min J. Readers, writers and erasers of N(6)-methylated adenosine modification. Curr Opin Struct Biol. 2017;47:67–76. doi: 10.1016/j.sbi.2017.05.011. [DOI] [PubMed] [Google Scholar]
- 21.Visvanathan Abhirami, Somasundaram Kumaravel. mRNA Traffic Control Reviewed: N6-Methyladenosine (m6 A) Takes the Driver's Seat. BioEssays. 2017;40(1):1700093. doi: 10.1002/bies.201700093. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16(2):191–198. doi: 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, Shi H, Skibbe J, Shen C, Hu C, et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m(6)A Modification. Cell Stem Cell. 2018;22(2):191–205. doi: 10.1016/j.stem.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du K, Zhang L, Lee T, Sun T. m(6)a RNA methylation controls neural development and is involved in human diseases. Mol Neurobiol. 2019;56(3):1596–1606. doi: 10.1007/s12035-018-1138-1. [DOI] [PubMed] [Google Scholar]
- 25.Widagdo J, Anggono V. The m6A-epitranscriptomic signature in neurobiology: from neurodevelopment to brain plasticity. J Neurochem. 2018;147(2):137–152. doi: 10.1111/jnc.14481. [DOI] [PubMed] [Google Scholar]
- 26.Weng YL, Wang X, An R, Cassin J, Vissers C, Liu Y, Liu Y, Xu T, Wang X, Wong SZH, et al. Epitranscriptomic m(6)A Regulation of Axon Regeneration in the Adult Mammalian Nervous System. Neuron. 2018;97(2):313–325. doi: 10.1016/j.neuron.2017.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Widagdo J, Zhao QY, Kempen MJ, Tan MC, Ratnu VS, Wei W, Leighton L, Spadaro PA, Edson J, Anggono V, et al. Experience-dependent accumulation of N6-Methyladenosine in the prefrontal cortex is associated with memory processes in mice. J Neurosci. 2016;36(25):6771–6777. doi: 10.1523/JNEUROSCI.4053-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weng YL, Wang X, An R, Cassin J, Vissers C, Liu Y, Liu Y, Xu T, Wang X, Wong SZH, et al. Epitranscriptomic m(6)a regulation of axon regeneration in the adult mammalian nervous system. Neuron. 2018;97(2):313–325. doi: 10.1016/j.neuron.2017.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang M, Lv H, Zhang W, Ma C, He X, Zhao S, Zhang ZW, Zeng YX, Song S, Niu Y, et al. Region-specific RNA m(6)A methylation represents a new layer of control in the gene regulatory network in the mouse brain. Open Biol. 2017;7(9). [DOI] [PMC free article] [PubMed]
- 30.Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39(3):253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- 31.Siebold L, Obenaus A, Goyal R. Criteria to define mild, moderate, and severe traumatic brain injury in the mouse controlled cortical impact model. Exp Neurol. 2018;310:48–57. doi: 10.1016/j.expneurol.2018.07.004. [DOI] [PubMed] [Google Scholar]
- 32.Osier N, Dixon CE. The controlled cortical impact model of experimental brain trauma: overview, research applications, and protocol. Methods Mol Biol. 2016;1462:177–192. doi: 10.1007/978-1-4939-3816-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, Ni T, Zhang ZS, Zhang T, Li C, et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell. 2019;35(4):677–691. doi: 10.1016/j.ccell.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gold EM, Su D, Lopez-Velazquez L, Haus DL, Perez H, Lacuesta GA, Anderson AJ, Cummings BJ. Functional assessment of long-term deficits in rodent models of traumatic brain injury. Regen Med. 2013;8(4):483–516. doi: 10.2217/rme.13.41. [DOI] [PubMed] [Google Scholar]
- 35.Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1(2):848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kopylova E, Noe L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28(24):3211–3217. doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- 38.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang T, Zhang SW, Zhang L, Meng J. Trumpet: transcriptome-guided quality assessment of m(6)A-seq data. BMC Bioinform. 2018;19(1):260. doi: 10.1186/s12859-018-2266-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cui X, Zhang L, Meng J, Rao MK, Chen Y, Huang Y. MeTDiff: a novel differential RNA methylation analysis for MeRIP-Seq data. IEEE/ACM Trans Comput Biol Bioinform. 2018;15(2):526–534. doi: 10.1109/TCBB.2015.2403355. [DOI] [PubMed] [Google Scholar]
- 41.Yu G, Wang LG, He QY. ChIPseeker: an R/bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31(14):2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 42.Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Mao J, Wang X, Lin Y, Hou G, Zhu J, Xie B. Genome-wide screening of altered m6A-tagged transcript profiles in the hippocampus after traumatic brain injury in mice. Epigenomics. 2019. [DOI] [PubMed]
- 44.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 45.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014;8(1):284–296. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, Mele A, Haripal B, Zucker-Scharff I, Moore MJ, Park CY, et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015;29(19):2037–2053. doi: 10.1101/gad.269415.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Janova H, Bottcher C, Holtman IR, Regen T, van Rossum D, Gotz A, Ernst AS, Fritsche C, Gertig U, Saiepour N, et al. CD14 is a key organizer of microglial responses to CNS infection and injury. Glia. 2016;64(4):635–649. doi: 10.1002/glia.22955. [DOI] [PubMed] [Google Scholar]
- 49.Yoneya T, Tahara T, Nagao K, Yamada Y, Yamamoto T, Osawa M, Miyatani S, Nishikawa M. Molecular cloning of delta-4, a new mouse and human notch ligand. J Biochem. 2001;129(1):27–34. doi: 10.1093/oxfordjournals.jbchem.a002832. [DOI] [PubMed] [Google Scholar]
- 50.Brutsch R, Liebler SS, Wustehube J, Bartol A, Herberich SE, Adam MG, Telzerow A, Augustin HG, Fischer A. Integrin cytoplasmic domain-associated protein-1 attenuates sprouting angiogenesis. Circ Res. 2010;107(5):592–601. doi: 10.1161/CIRCRESAHA.110.217257. [DOI] [PubMed] [Google Scholar]
- 51.Del Barrio MG, Taveira-Marques R, Muroyama Y, Yuk DI, Li S, Wines-Samuelson M, Shen J, Smith HK, Xiang M, Rowitch D, et al. A regulatory network involving Foxn4, Mash1 and delta-like 4/Notch1 generates V2a and V2b spinal interneurons from a common progenitor pool. Development (Cambridge, England) 2007;134(19):3427–3436. doi: 10.1242/dev.005868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pitulescu ME, Schmidt I, Giaimo BD, Antoine T, Berkenfeld F, Ferrante F, Park H, Ehling M, Biljes D, Rocha SF, et al. Dll4 and notch signalling couples sprouting angiogenesis and artery formation. Nat Cell Biol. 2017;19(8):915–927. doi: 10.1038/ncb3555. [DOI] [PubMed] [Google Scholar]
- 53.Trindade A, Djokovic D, Gigante J, Mendonca L, Duarte A. Endothelial Dll4 overexpression reduces vascular response and inhibits tumor growth and metastasization in vivo. BMC Cancer. 2017;17(1):189. doi: 10.1186/s12885-017-3171-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science (New York, NY) 2015;347(6225):1002–1006. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- 55.Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng SX, Xu ZW, Yi TL, Sun HT, Yang C, Yu ZQ, Yang XS, Jin XH, Tu Y, Zhang S. iTRAQ-based quantitative proteomics reveals the new evidence base for traumatic brain injury treated with targeted temperature management. Neurotherapeutics. 2018;15(1):216–232. doi: 10.1007/s13311-017-0591-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8(6):e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell. 2017;31(1):127–141. doi: 10.1016/j.ccell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. PCR primers used in this study
Additional file 2: Table S2. The detailed information of raw data
Additional file 3: Table S3. Statistical analysis of reads mapped in reference genome
Additional file 4: Table S4. The m6A peak number on each gene
Additional file 5: Table S5. The detailed information of significantly changed m6A peaks
Additional file 6: Table S6. The detailed information of significantly changed mRNA
Additional file 7: Table S7. The detailed information of conjoint analysis between mRNA expression and m6A methylation
Additional file 8: Table S8. The raw data of mNSS tests
Additional file 9: Table S9. The raw data of MWM tests
Data Availability Statement
All of the datasets in this study can be obtained by reasonable request to the corresponding authors.