

Abstract
The threat of Hepatocellular Carcinoma (HCC) is a growing problem, with incidence rates anticipated to near double over the next two decades. The increasing burden makes discovery of novel diagnostic, prognostic, and therapeutic biomarkers distinguishing HCC from underlying cirrhosis a significant focus. In this study, we analyzed tissue and serum samples from 40 HCC cases and 25 patients with liver cirrhosis (CIRR) to better understand the mechanistic differences between HCC and CIRR. Through pathway and network analysis, we are able to take a systems biology approach to conduct multi-omic analysis of transcriptomic, glycoproteomic, and metabolomic data acquired through various platforms. As a result, we are able to identify the FXR/RXR Activation pathway as being represented by molecules spanning multiple molecular compartments in these samples. Specifically, serum metabolites deoxycholate and chenodeoxycholic acid and serum glycoproteins C4A/C4B, KNG1, and HPX are biomarker candidates identified from this analysis that are of interest for future targeted studies. These results demonstrate the integrative power of multi-omic analysis to prioritize clinically and biologically relevant biomarker candidates that can increase understanding of molecular mechanisms driving HCC and make an impact in patient care.
Keywords: multi-omic, network and pathway analysis, HCC, metabolomics, transcriptomics, glycoproteomics
I. Introduction
Hepatocellular Carcinoma (HCC) is the most common primary malignancy of the liver and the third leading cause of cancer deaths worldwide [1]. Liver resection and transplantation serve as the only potentially curative therapies. However, these options have limited applicability due to lack of resources and restriction to use in early stages [2]. HCC often develops from preliminary liver cirrhosis (CIRR) and therefore tends to be relatively asymptomatic during initial stages. [3]. As a result, HCC is an aggressive cancer and often diagnosed at advanced stages [4]. There is an unmet need to identify novel biomarkers for early detection of HCC due to low sensitivity (40–64%) and common misinterpretation of current diagnostic biomarkers for HCC, such as AFP values [5, 6]. Enhanced understanding of mechanistic differences between HCC and CIRR is important to identify distinguishing features that may be of interest as potential biomarker candidates.
Many obstacles stand in the way of identifying alternative biomarkers for HCC. The process of finding actionable biomarkers is long and costly and potential candidates must be carefully selected. Statistical and bioinformatics analysis of patient derived omics data serves as the initial step in the biomarker discovery pipeline [7]. Recent technological advances in high-throughput sequencing have led to mass multi-omics data acquisition, heightening the difficulty of honing-in on relevant molecular targets. Multi-omics approaches increase reproducibility of results to put forth candidates with confidence for future targeted analysis to improve biomarker robustness. In this paper, we present transcriptomic, glycoproteomic, and metabolomic data acquired by analysis of liver tissues and serum from HCC and CIRR patients. Our aim is to use HCC as an example to present a multi-omic framework implementing pathway and network analysis to prioritize biologically and clinically meaningful molecular molecules.
II. Methods
A. Samples Analyzed
Human liver tissue and serum from 65 adult patients recruited at MedStar Georgetown University Hospital through a protocol approved by the Georgetown IRB were included in this study and multi-omic analysis. All subjects provided informed consent forms and HIPAA authorization forms. Table I provides the characteristics of the 40 HCC cases and 25 patients with CIRR whose samples were analyzed by various platforms to acquire multi-omic data. Of the 40 HCC cases, 25% (10 HCC cases) have cirrhotic liver tissue adjacent to the tumor tissue.
Characteristics of patient-derived samples
| HCC (N=40) | CIRR (N=25) | p-valuea | ||
|---|---|---|---|---|
| Age | Mean (SD) | 61.2(12.2) | 50.5(12.1) | 0.0013 |
| Gender | Male | 77.5% | 72.0% | 0.7683 |
| White | 40.0% | 64.0% | ||
| Race | Black | 35.0% | 32.0% | 0.4073 |
| Other | 25.0% | 4.0% | ||
| HCV Serology | HCV Ab+ | 40.0% | 40.0% | 1 |
| HBs Ab+ | 25.0% | 48.0% | 0.1015 | |
| HBV Serology | HBs Ag+ | 15.0% | 4.0% | 0.2232 |
| Smoking | Yes | 62.5% | 48.0% | 0.3074 |
| Alcohol | Yes | 45.0% | 48.0% | 1 |
| Stage I | 43.3% | |||
| HCC Stage | Stage II | 23.3% | ||
| Stage III or IV | 33.3% |
p -values were based on comparison between 40 HCC vs. 25 CIRR
B. Multi-omic Data
Transcriptomics (mRNA-seq and miRNA-seq):
RNA samples extracted from the 65 liver tissues were analyzed by Illumina HiSeq 4000 using 150 bp paired-end (PE150) form RNA-seq expression profiling. The mRNA-seq data contained an average of 33M reads per sample. The fastq files were imported into Partek Flow for quality assessment and mRNA-seq data analysis. Alignment was performed using the spliced transcripts alignment to a reference (STAR) algorithm, which applies sequential maximum mappable seed search in uncompressed suffix arrays followed by seed clustering and stitching procedure. The aligned reads were quantified to the transcriptome through an Expectation Maximization (EM) method.
The 65 RNA samples described above were analyzed by Illumina NextSeq 550 platform using 2×150 bp paired-end (PE150) for miRNA-seq expression profiling. The miRNA-seq data acquired were analyzed using the QIAseq miRNA quantification data analysis software. The primary analysis of the data involved calculation of unique molecular index (UMI) and primary miRNA mapping. The secondary analysis involved calculating changes in miRNA expression based on UMI counts. For both mRNA-seq and miRNA-seq data, the TMM (trimmed mean of M-values) method was used for normalization.
Glycoproteomics:
Following the removal of high abundant proteins in sera from 65 subjects using Agilent MARS Hu-14 HPLC column, we performed digestion, purification, and enrichment of glycoproteins by hydrazide chemistry. The samples were then analyzed using nanoUPLC coupled with Triple TOF 6600 Sciex system. The acquired LC-MS/MS data were analyzed using MaxQuant to select glycoproteins with statistically significant change in expression levels between HCC cases and CIRR controls [8].
Metabolomics (GC-MS and LC-MS):
Two platforms (GC-TOF-MS and LC-QTOF-MS) were used for metabolomic analysis of tissue and serum samples from 65 subjects [9]. The tissues were homogenized, and metabolite extraction was performed in a single step. GC-MS metabolites were acquired using Agilent 7890 GC coupled to LECO Pegasus HT, equipped with an electron ionization source and TOF analyzer. ChromaTOF with True Signal Deconvolution package was used for data pre-processing including, calculation, peak finding, deconvolution and identification. LECO’s Statistical Compare software tool was used for alignment of the GC-MS data. Spectral similarity searches against the Fiehn library were performed to determine the identities of the analytes.
LC-MS data were acquired by analysis of the metabolite extracts using Waters ACQUITY UPLC system coupled to a Synapt G2-Si QTOF-MS, operating in positive and negative polarity. Peak detection, alignment, and ion annotation were performed using XCMS [10, 11] Putative metabolite identification was performed using MetaboQuest [http://omicscraft.com/MetaboQuest/].
A. Statistical Analysis
To identify ions/molecules with significant changes in intensity levels, Wilcoxon rank-sum test was used within each omic dataset. The p-values were adjusted using the Benjamini-Hochberg false discovery rate [12]. Each omic dataset was then filtered by identified molecules achieving FDR <0.05 significance value.
B. Network and Pathway Analysis
The multi-omic datasets were integrated through pathway and network analysis for selection of key molecules distinguishing HCC from CIRR. Fig. 1 depicts an overview of the multi-omic analysis performed in this study. A series of exclusion criteria were used to filter identified molecules of interest for subsequent analysis. Selected molecules with FDR <0.05 from each dataset were uploaded to Ingenuity Pathway Analysis (IPA, QIAGEN Inc.) for pathway and network analysis.
Figure 1.
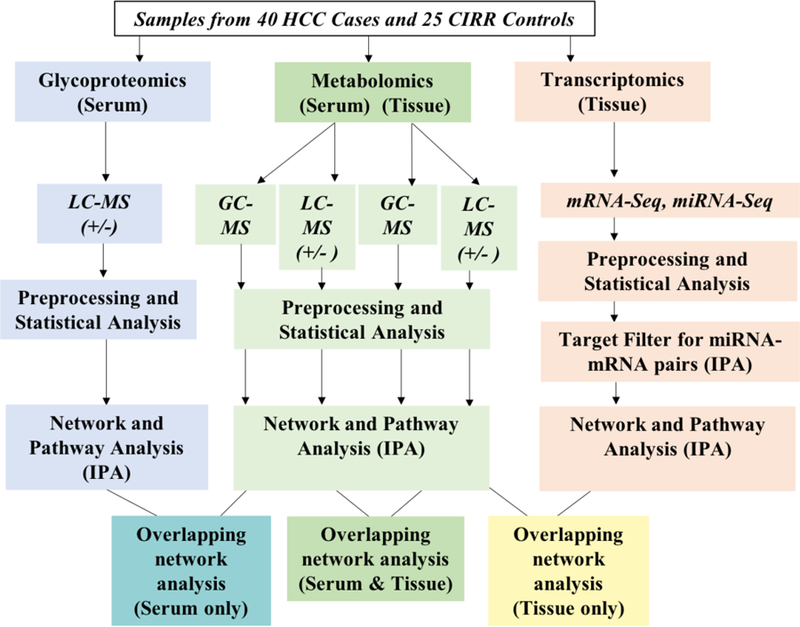
Overview of multi-omic analysis.
The microRNA Target Filter in IPA was used to pair statistically significant miRNA with mRNA targets using experimentally validated interactions from TarBase and miRecords and predicted interactions from TargetScan. Pairs were further filtered to include only reciprocal and dual-upregulated pairs that have been experimentally verified or predicted to associate with high confidence.
III. Results and Discussion
The multi-omic analysis performed in this study serves to emphasize a subset of inter-related transcriptomic, glycoproteomic, and metabolomic molecules that strongly distinguish HCC from CIRR across several physiological levels. Combining molecules that are strongly intertwined across multiple compartments increases reproducibility and provides a more effective and accurate means for cancer biomarker discovery. Table II presents the number of molecules from each omic dataset that were included for pathway analysis. The power of pathway analysis as a systems-biology approach lies in its ability to integrate individually processed omics data to offer improved biological insights [13].
Molecules included for pathway and network analysis based on statistical and identification filters.
| Transcriptomics | Metabolomics | Glycoproteomics | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| miRNA -Seq | mRNA -Seq | GC-MS | LC-X1S (POS+NEG) | LC-MS | |||||||||
| Comparisons | Total Detected | FDR <0.05 | Total Detected | FDR <0.05 | miRNA-mRNA pairs | Total Detected | Compounds with ID | FDR <0.05a | Total Detected | FDR <0.05 | PubChem CIDb | Total Detected | FDR <0.05 |
| Tissue | 2548 | 344 | 11313 | 500 | 1294 | 728 | 250 | 58 | 2881 | 555 | 255 | ||
| Serum | 579 | 149 | 66 | 2596 | 1592 | 639 | 19 | 2 | |||||
number of metabolites with an ID that are also FDR <0.05
Number of metabolites with FDR<0.05 and at least one putative identification with PubChemID
Despite the benefit of multi-omic analysis, data from single-omics studies still have unique features that need to be taken into consideration separately during initial identification and prioritization steps. For instance, recent findings on the relevance of paired miRNA-mRNA regulation in HCC make combined analysis of individual mRNA and miRNA expression profiles of interest for more realistic application [14]. When considering the various scenarios for miRNA-mRNA regulation, only reciprocal or dual-upregulated pairs have been characterized as possible existing relationships [15]. Therefore, only significant mRNA with miRNA pairs of this nature were included for pathway analysis.
Additionally, unique prioritization was implemented on LC-MS-based metabolites included in this analysis. Besides the challenge due to a large number of peaks with unknown analytes, the presence of multiple putative ID’s per m/z is another significant barrier in untargeted LC-MS-based metabolomics studies [16]. To overcome this barrier, we used filters implemented in IPA to prioritize analytes of interest. For each m/z, only putative IDs having a unique PubChem CID and FDR <0.05 were considered. These ID’s were further filtered for exclusion of exogenous or non-mammalian chemicals/toxicants and other non-metabolite classifications of PubChem CIDs. Higher prioritization was given to putative ID’s classified as endogenous mammalian metabolites that were involved in biological canonical pathways. Likewise, higher emphasis was placed on putative ID’s pulled into networks with other metabolites and other omics biomolecules from our data. This approach helped to narrow down putative LC-MS identification in need of future targeted quantification.
Canonical pathways derived from filtered molecules for each omic dataset were compared for overlap. The top 10 significant (p<0.05) pathways from each single-omics study are provided in Table III. In tissue, 104 significant (p<0.05) pathways were identified from transcriptomics data and compared to 31 significant pathways from metabolomics data. Of these, two pathways (FXR/RXR Activation and Sirtuin Signaling) were found to overlap across omics in tissue. In serum, 16 significant pathways (p<0.05) were identified from metabolomics data and compared to four significant pathways identified from glycoproteomics data. In serum only one pathway (FXR/RXR Activation) was found to overlap. Although circulating serum biomarkers are useful for non-invasive clinical diagnostic purposes, tissue omics data still offers insight into the molecular mechanisms contributing to HCC. For instance, five significant (p<0.05) pathways (FXR/RXR Activation, Sorbitol Degradation I, GABA Receptor Signaling, tRNA Charging, and Tyrosine Degradation I) were found to overlap between serum and tissue metabolomics data. Combining omics data at a pathway level enables overlay between serum and tissue data to give weight to serum biomarkers that arise downstream from integral processes contributing to the pathogenesis of HCC.
Top 10 pathways for each omics dataset (p<0.05) from the 40 (hcc) vs. 25 (cirr) analyses
| Scrum | Tissue | ||
|---|---|---|---|
| Metabolomics (GC-MS + LC-MS) | Glvcoproteomics | Transcriptomics (miRNA-mRNA pairs) | Metabolomics (GC-MS + LC-MS)* |
| tRNA Charging | Complement System | Sirtuin Signaling Pathway | Purine Ribonucleosidcs Degradation to Ribose-1-phosphate |
| (S)-reticulinc Biosynthesis 11 | LXR/RXR Activation | LXR/RXR Activation | Glutamate Degradation II |
| FXR/RXR Activation | FXR/RXR Activation | Complement System | Sirtuin Signaling Pathway |
| Tyrosine Degradation I | Acute Phase Response Signaling | Granzvme A Signaling | Uracil Degradation II (Reductive) |
| Arginine Degradation VI | NER Pathway | Lactose Degradation III | |
| Sorbitol Degradation I | FXR/RXR Activation | Sucrose Degradation V (Mammalian) | |
| Phosphatidylethanolamine Biosyntliesis III | DNA Methylation and Transcriptional Repression Signaling | Adenine and Adenosine Salvage III | |
| Glycine Biosynthesis I | Transcriptional Regulatory Network in Embryonic Stem Cells | Adenosine Nucleotides Degradation II | |
| 4-hydroxyphenvlpyruvate Biosynthesis | Nicotine Degradation II | Sorbitol Degradation I | |
| Superpathway of Serine and Glycine Biosynthesis I | Xenobiotic Metabolism Signaling | Aspartate Biosynthesis | |
FXR /RXR Activation pathway is also p<0.05 in tissue metabolomics
FXR/RXR Activation was the only significant pathway depicted by all omics data in both tissue and serum when comparing HCC to CIRR. The molecules comprising this pathway from each level of multi-omic analysis in serum were studied in detail and integrated by network analysis as depicted in Fig. 2. The farnesoid X receptor (FXR) and retinoid X receptor (RXR) are nuclear receptors that play a key role in maintaining the homeostasis of liver metabolism [17]. FXR binds targets as a heterodimer with RXR and manages expression of genes involved in bile acid homeostasis, lipid and glucose metabolism, and inflammation [18]. Recently, downregulation of FXR in HCC has been linked to carcinogenesis through lack of negative feedback on NF-kB mediated inflammation and suppression of Wnt/β-catenin and JNK signaling pathways [19, 20, 17]. Decreased FXR expression has been associated with increased inflammation and proliferation, as well as dysregulated bile acid (BA) levels contributing to hepatotoxicity.
Figure 2.

IPA network generated connecting serum glycoproteins and metabolites from our data with other interacting molecules comprising the FXR/RXR Activation pathway. Green molecules are downregulated and red molecules are upregulated.
Molecules found to be involved in the FXR/RXR Activation pathway from our serum glycoproteomics and metabolomics data can be found listed below in Table IV. These molecules may be of interest for future targeted identification and biomarker validation. For serum glycoproteomics and metabolomics, molecules C4A/C4B, HPX, KNG1, chenodeoxycholic acid and deoxycholate are of particular interest. In our previous work, these highlighted glycopeptides and metabolites were also investigated as potential biomarkers for HCC [8, 21, 22]. In addition, several other molecules comprising the FXR/RXR Activation pathway were emphasized in our previous analysis as well, including taurocholic acid, ApoB, ApoA1, ApoCII/III, bile acid, and cAMP [23, 24].
Serum molecules comprising the FXR RXR Activation Pathway
| Serum Molecules | |
|---|---|
| Metabolomics (GC-MS + LC-MS) | Glycoproteomics |
| Chenodeoxycholic acid (CID:10133)* | Complement Component (C4A/C4B)* (Uniprot:P0C0L4/P0C0L5) |
| Deoxycholate (CID:222528)* | Hemopexin (HPX) (Uniprot:P02790) |
| D-glucose (CID:5793)* | Kininogen-1 precursor (KNG1) (Uniprot:P01042) |
| xylitol (KEGG:D00061) | Apolipoprotein B (APOB) (Uniprot:P04114) |
| Inter-alpha-trypsin Inhibitor Heavy Chain H4 (ITIH4) (Uniprot:Q14624) | |
| Complement Component C9 (C9) (Uniprot:P02748) | |
| Alpha-2-HS-Glycoprotein (AHSG) (Uniprot:P02765) | |
| Clusterin (CLU) (Uniprot:P10909) | |
Molecules with FDR<0.05
These candidates for potential serum biomarkers differentiating HCC from CIRR are also well-established in the literature. For emphasized glycoproteins, KNG1 has been reported as a biomarker for sorafenib-resistant HCC [25]. KNG1 is overexpressed in HCC and plays a role in coagulation, inflammation, apoptosis, metastasis, and cholesterol metabolism [26]. Fucosylation patterns of HPX have been studied in the literature as well as potential liver-specific N-glycan changes thought to distinguish HCC from CIRR [27, 28]. C4A has been reported as a potential biomarker in combination with CP, FGA, and PON1 for HCV-infected alcoholic HCC patients [29]. C4A/C4B are glycoproteins involved in the classical or lectin pathways of the complement system. C4A/C4B upregulation is thought to contribute to HCC development through inflammatory and immunosuppressive mechanisms [30].
Dysregulated levels of BAs including metabolites chenodeoxycholic acid and deoxycholate have been implicated in the pathogenesis of HCC through mechanisms leading to increased inflammation [31]. Accrual of BAs can dysregulate mitochondrial function and cause hepatotoxicity and cell death through unrestrained formation of reactive oxygen species (ROS). Deoxycholate and Chenodeoxycholic acid have also been reported to induce oncogene c-myc [32]. Recent work has connected bile acid metabolism to liver cancer immunosurveillance [33]. Specifically, metabolism of primary into secondary bile acids by commensal gut microbiota has been implicated in immunosuppression [33].
Taken as a whole, identifying multi-omic biomarker candidates inter-related at the pathway level can aid in reproducibility by increasing the likelihood of detection and applicability in the face of patient heterogeneity. For instance, the glycoproteomics data for this cohort was initially excluded due to limitations in feature identification. Excluding glycoproteomics, the FXR/RXR Activation pathway was still the only pathway to overlap across tissue and serum transcriptomics and metabolomics data. However, inclusion of significant glycoproteins that were identified only reinforced the relevance of this pathway to explain mechanistic differences between HCC and CIRR. Therefore, it was still possible to utilize the identified glycoproteins through this integrative multi-omic approach at a pathway level. Future work will focus on targeted identification of the molecules of interest presented in this study. Further, additional cohorts comparing HCC cases to cirrhotic controls can be analyzed using this multi-omic framework to better identify early diagnosis biomarker candidates. This can lead to improved diagnosis, while also providing insight into molecular mechanisms driving the pathogenesis of HCC.
IV. Conclusion
Early detection and diagnosis of HCC are essential to improve patient prognosis and make curative therapy through transplantation a possibility. There is a need to identify biomarkers with greater sensitivity and specificity compared to AFP that can distinguish between HCC and CIRR in diverse patient populations. In this study, an integrative analysis was conducted of transcriptomics, glycoproteomics, and metabolomics data acquired by analysis of tissue and serum samples from 40 HCC cases and 25 patients with CIRR. Through this analysis, we identified metabolites deoxycholate and chenodeoxycholic acid and glycoproteins C4A/C4B, KNG1, and HPX in serum as potential biomarker candidates for future targeted study. These biomarker candidates identified through multi-omic pathway and network analysis are all part of the FXR/RXR Activation pathway and span across multiple tiers of biological data. We hypothesize that this heightens the clinical applicability and biological relevance of these serum molecules through being linked to molecular mechanisms driving the pathogenesis of HCC in tissue.
Acknowledgment
The authors wish to thank the shared resources at Georgetown University, especially the Genomics & Epigenomics Shared Resource (GESR), the Proteomics & Metabolomics Shared Resource (PMSR), and the Histopathology & Tissue Shared Resource (HTSR).
The work presented in this paper is supported by NIH grants U01CA185188 and R01GM123766 awarded to H.W.R.
REFERENCES
- [1].Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. “Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries.” CA Cancer J Clin 2018;68(6):394–424. [DOI] [PubMed] [Google Scholar]
- [2].Pinter M, Peck‐Radosavljevic M. “Review article: systemic treatment of hepatocellular carcinoma.” Aliment Pharmacol Ther 2018;48:598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Pinter M, Trauner M, Peck-radosavljevic M, Sieghart W. “Cancer and liver cirrhosis: implications on prognosis and management.” ESMO Open 2016;1(2):e000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Daher S, Massarwa M, Benson AA, Khoury T. “Current and Future Treatment of Hepatocellular Carcinoma: An Updated Comprehensive Review.” J Clin Transl Hepatol 2018;6(1):69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chauhan R, Lahiri N. “Tissue-and Serum-Associated Biomarkers of Hepatocellular Carcinoma.” Biomark Cancer 2016;8(Suppl 1):37–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lou J, Zhang L, Lv S, Zhang C, Jiang S. “Biomarkers for Hepatocellular Carcinoma.” Biomark Cancer 2017;9:1–9.d [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Selleck MJ, Senthil M, Wall NR. “Making Meaningful Clinical Use of Biomarkers.” Biomark Insights 2017;12:1177271917715236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Di Poto C, Wang M, Su S, Ma J, Ressom HW. “Identification of Glycoprotein Biomarkers for Hepatocellular Carcinoma.” Abstract for a poster presentation at ASMS 2017 Meeting, June 4–8, 2017, Indianapolis, IN. [Google Scholar]
- [9].Ferrarini A, Di Poto C, Varghese RS, Ressom HW. “Tracking Aberrant Pathways in Hepatocellular Carcinoma Using Metabolomics: From Tissue Alterations to Blood Biomarkers.” Abstract for a poster presentation at Metabolomics 2015, June 29-July 2, 2015, Burlingame, CA. [Google Scholar]
- [10].Smith CA, Want EJ, O’maille G, Abagyan R, Siuzdak G. “XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification.” Anal Chem 2006;78(3):779–87. [DOI] [PubMed] [Google Scholar]
- [11].Mahieu NG, Genenbacher JL, Patti GJ. “A roadmap for the XCMS family of software solutions in metabolomics.” Curr Opin Chem Biol 2016;30:87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Green GH, Diggle PJ. “On the operational characteristics of the Benjamini and Hochberg False Discovery Rate procedure.” Stat Appl Genet Mol Biol 2007;6:Article27. [DOI] [PubMed] [Google Scholar]
- [13].Wang J, Zhang Y, Marian C, Ressom HW. “Identification of aberrant pathways and network activities from high-throughput data.” Brief Bioinformatics 2012;13(4):406–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Oliveto S, Mancino M, Manfrini N, Biffo S. “Role of microRNAs in translation regulation and cancer.” World J Biol Chem 2017;8(1):45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tian XJ, Zhang H, Zhang J, Xing J. “Reciprocal regulation between mRNA and microRNA enables a bistable switch that directs cell fate decisions.” FEBS Lett 2016;590(19):3443–3455. [DOI] [PubMed] [Google Scholar]
- [16].Chaleckis R, Meister I, Zhang P, Wheelock CE. “Challenges, progress and promises of metabolite annotation for LC-MS-based metabolomics.” Curr Opin Biotechnol 2018;55:44–50. [DOI] [PubMed] [Google Scholar]
- [17].Wang YD, Chen WD, Li C, et al. “Farnesoid X receptor antagonizes JNK signaling pathway in liver carcinogenesis by activating SOD3.” Mol Endocrinol 2015;29(2):322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rudraiah S, Zhang X, Wang L. “Nuclear Receptors as Therapeutic Targets in Liver Disease: Are We There Yet?” Annu Rev Pharmacol Toxicol 2016;56:605–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. “Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response.” Hepatology 2008;48(5):1632–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liu X, Zhang X, Ji L, Gu J, Zhou M, Chen S. “Farnesoid X receptor associates with β-catenin and inhibits its activity in hepatocellular carcinoma.” Oncotarget 2015;6(6):4226–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tsai TH, Song E, Zhu R, et al. “LC-MS/MS-based serum proteomics for identification of candidate biomarkers for hepatocellular carcinoma.” Proteomics 2015;15(13):2369–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang M, Yu G, Ressom HW. “Integrative Analysis of Proteomic, Glycomic, and Metabolomic Data for Biomarker Discovery.” IEEE J Biomed Health Inform 2016;20(5):1225–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Di Poto C, He S, Varghese RS, et al. “Identification of race-associated metabolite biomarkers for hepatocellular carcinoma in patients with liver cirrhosis and hepatitis C virus infection.” PLoS ONE 2018;13(3):e0192748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Di Poto C, Ferrarini A, Zhao Y, et al. “Metabolomic Characterization of Hepatocellular Carcinoma in Patients with Liver Cirrhosis for Biomarker Discovery.” Cancer Epidemiol Biomarkers Prev 2017;26(5):675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Huang D, Yuan W, Li H, Li S, Chen Z, Yang H. “Identification of key pathways and biomarkers in sorafenib-resistant hepatocellular carcinoma using bioinformatics analysis.” Exp Ther Med 2018;16(3):1850–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang J, Wang X, Lin S, et al. “Identification of kininogen-1 as a serum biomarker for the early detection of advanced colorectal adenoma and colorectal cancer.” PLoS ONE 2013;8(7):e70519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Debruyne EN, Vanderschaeghe D, Van vlierberghe H, Vanhecke A, Callewaert N, Delanghe JR. “Diagnostic value of the hemopexin N-glycan profile in hepatocellular carcinoma patients.” Clin Chem 2010;56(5):823–31. [DOI] [PubMed] [Google Scholar]
- [28].Benicky J, Sanda M, Pompach P, Wu J, Goldman R. “Quantification of fucosylated hemopexin and complement factor H in plasma of patients with liver disease.” Anal Chem 2014;86(21):10716–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ferrín G, Rodríguez-perálvarez M, Aguilar-melero P, et al. “Plasma protein biomarkers of hepatocellular carcinoma in HCV-infected alcoholic patients with cirrhosis.” PLoS ONE 2015;10(3):e0118527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Qin X, Gao B. “The complement system in liver diseases.” Cell Mol Immunol 2006;3(5):333–4. [PubMed] [Google Scholar]
- [31].Sun L, Beggs K, Borude P, et al. “Bile acids promote diethylnitrosamine-induced hepatocellular carcinoma via increased inflammatory signaling.” Am J Physiol Gastrointest Liver Physiol 2016;311(1):G91–G104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xie G, Wang X, Huang F, et al. “Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis.” Int J Cancer 2016;139(8):1764–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ma C, Han M, Heinrich B, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018;360(6391). [DOI] [PMC free article] [PubMed] [Google Scholar]