

Abstract
Lipid droplets (LDs) are ubiquitous organelles that store neutral lipids for energy or membrane synthesis and act as hubs for metabolic processes. Cells generate LDs de novo, converting cells to emulsions with LDs constituting the dispersed oil phase in the aqueous cytoplasm. Here we review our current view of LD biogenesis. We present a model of LD formation from the ER in distinct steps and highlight the biology of proteins that govern this biophysical process. Areas of incomplete knowledge are identified, as are connections with physiology and diseases linked to alterations in LD biology.
Keywords: biogenesis, endoplasmic reticulum, lipid droplets, neutral lipids, seipin, sterol esters, triglycerides, triacylglycerol
Introduction
Lipid droplets (LDs), organelles of lipid storage, are found in most eukaryotic cells and in some prokaryotes. LDs are found primarily in the cytoplasm, but in some cell types, the nucleus also contains LDs (Ohsaki et al. 2016). These organelles are composed primarily of a neutral lipid core (oil phase), consisting in most cells of triacylglycerols (TGs) and sterol esters (SEs) (see Figure 1).
Figure 1.

Lipid droplets (LDs): cellular organelles for neutral lipid storage. (a) Fluorescence microscopy image showing LDs (green, stained with BODIPY 493/503) in mammalian SUM159 cells. This cell was transfected with the LD marker mCherry-LiveDrop (red). (b) Electron micrograph showing a monolayer-bound LD in Drosophila S2 cells. The ribosome-studded ER bilayer can be seen in close proximity. Image courtesy of Yi Guo (Mayo Clinic, Rochester, MN). (c) Schematic representation of the structural composition of an LD. Colored objects represent LD surface-bound proteins localized to the phospholipid monolayer. Triacylglycerols and sterol esters are found in the neutral lipid core.
The neutral lipids serve as lipid reservoirs for energy generation or membrane synthesis. The process of neutral lipid synthesis and storage in LDs is also important for protecting cells from lipotoxicity due to the buildup of excess lipids, such as fatty acids, toxic glycerolipids, and sterols, in cell membranes (Devries-Seimon et al. 2005, Koliwad et al. 2010, Listenberger et al. 2003). LDs also serve as localization sites for some proteins (Kory et al. 2016), such as enzymes related to LD metabolism (e.g., specific enzymes of neutral lipid synthesis or lipolysis), or, in some instances, for proteins (e.g., histones) that utilize LDs as a platform for storage (Z. Li et al. 2012).
During the past decade, the cell biology of LDs has been a focus of intensive research. It has become clear that LDs are dynamic organelles, integral to cell metabolism, with a fascinating cycle of biogenesis and consumption. Many interesting questions, such as those pertaining to the mechanisms of LD formation, protein targeting to the surfaces of LDs, and the behavior of lipids or proteins during LD consumption, are being investigated. The focus of this article is to review progress in our understanding of LD biogenesis. Other recent reviews highlight additional features of LD cell biology (Barneda & Christian 2017, Gao & Goodman 2015, Kory et al. 2016, Krahmer et al. 2013a, Roingeard & Melo 2017, Thiam & Foret 2016, Thiam et al. 2013b, Walther & Farese 2012, Welte 2015, Wilfling et al. 2014a).
THE BIOPHYSICS OF OIL DROPLETS IN CELLS
The presence of LDs makes cells exist as emulsions, with the oil or neutral lipid constituting the dispersed phase in the continuous phase of the aqueous cytoplasm. When confronted with excess lipids, such as fatty acids or sterols, cells synthesize neutral lipid oils that then form droplets that are dispersed in the aqueous phase. Because LDs form a separate phase, LD biology in general follows the principles of emulsion physics (Thiam & Foret 2016, Thiam et al. 2013b). One such principle is that emulsions, such as LDs in cells, are metastable; i.e., the emulsion droplets are stable and exist on a timescale that is relevant to cell biology, but they would coalesce to form a continuous oil phase if left to themselves. The driving force for coalescence of LDs is that the system moves to minimize surface tension (i.e., the energy cost for the unfavorable molecular interactions at the interphase between the oil and aqueous medium).
In cells, LD stability is modulated by the presence of lipid or protein surfactants at the interface of the oil and water phases. Surfactants lower the LD surface tension. The major surfactants for LDs constitute a surface monolayer of amphiphilic molecules, such as phospholipids, with the polar head groups oriented toward the aqueous cytosol and the hydrophobic acyl chains oriented toward the neutral lipid cores, thus allowing for preferred molecular interactions of molecules in each phase at the interface. Of the different phospholipids, phosphatidylcholine (PC) appears to be particularly important as an optimal biological surfactant to lower surface tension (Krahmer et al. 2011, Moessinger et al. 2014, Thiam & Foret 2016, Thiam et al. 2013b). When the levels of surfactants, such as PC, are inadequate, LDs coalesce via fusion to minimize the interface surface and to increase surface coverage by surfactant. This is likely how giant LDs (>2 μm diameter) are formed (Krahmer et al. 2011), for example, in liver steatosis. Similarly, when lipids with worse surfactant properties, such as phosphatidic acid, accumulate on LDs, they can induce LD coalescence (Fei et al. 2011).
In addition to coalescing, oil droplets in an emulsion can combine to form larger droplets via diffusion of neutral lipids from a smaller droplet to a larger droplet in a process termed Ostwald ripening (Thiam et al. 2013b). In this process, single molecules diffuse from one LD to another through the aqueous solution. Because small LDs have higher surface-to-volume ratios than do larger LDs (a measure reflected in their Laplace pressure), a larger fraction of their oil molecules is close to the aqueous environment and can diffuse into it. Due to this different likelihood of solubilization for LDs of different sizes, Ostwald ripening results in a net transfer of oil molecules from smaller, less stable LDs to larger, more stable LDs. However, because TGs are only very poorly soluble in water, this process in cells is likely aided by specific protein machinery (discussed below). The combination of two LDs to form a single LD by fusion or diffusion illustrates how LDs behave according to the principles of biophysics and how the cell regulates these processes via proteins.
BIOCHEMISTRY AND GENETICS UNCOVER PROTEINS REQUIRED FOR NORMAL CELLULAR LIPID DROPLETS
Although the principles of emulsion physics apply to LD biology, a central hypothesis that drives the field of LD cell biology is that cells utilize specific protein machinery to catalyze and/or regulate these biophysical processes. Crucial tasks for the field are, thus, to identify the important proteins that govern LD biology and to elucidate their functions and means of regulation. With the goal of identifying such proteins, unbiased screens have played a key role. These include genetic screens, carried out in yeast, fly, and mammalian cells, to identify genes and proteins that are functionally involved in LD biology (Beller et al. 2008, Fei et al. 2008, Gronke et al. 2005, Guo et al. 2008, Z. Liu et al. 2014, Szymanski et al. 2007, Whittaker et al. 2010) and unbiased quantitative assays of proteins for components of LD organelles in these types of cells (Beller et al. 2006, Brasaemle et al. 2004, Cermelli et al. 2006, Currie et al. 2014, Ding et al. 2012, Khor et al. 2014, Krahmer et al. 2013b, Liu et al. 2004, Schmidt et al. 2013, Zhang et al. 2012). Such LD proteomes have been determined by organelle purification and mass spectrometry–based proteomics. Many such studies identified core components of LDs, such as enzymes of lipid synthesis and degradation and structural or regulatory proteins, such as perilipins (Kimmel & Sztalryd 2016). However, due to the challenges of obtaining pure preparations of LDs, combined with the ever-increasing sensitivity of mass spectrometry, many LD proteome lists are populated by protein contaminants. Techniques such as protein correlation profiling have been used in numerous cell types to minimize false positive results and to identify LD proteins that are enriched in the LD fraction (Currie et al. 2014, Krahmer et al. 2013b).
The core LD protein machinery may be similar in many cell types. However, some cell types, such as adipocytes or hepatocytes, are highly specialized cells for storing neutral lipids and have specific proteins for regulating certain LD processes. For example, perilipin1 is found predominantly in adipocytes, where it plays a key role in regulating fasting-induced lipolysis (Brasaemle et al. 2000, Kimmel & Sztalryd 2016, Paul et al. 2008).
OVERVIEW OF LIPID DROPLET FORMATION: CONCEPTUAL STEPS IN THE FORMATION OF AN ORGANELLE
In eukaryotes, LDs are formed de novo from the ER (Choudhary et al. 2015, Jacquier et al. 2011, Kassan et al. 2013). This was long suspected because the enzymes of neutral lipid synthesis are localized in the ER (Buhman et al. 2001), where they generate neutral lipids (TGs and SEs) for LD formation. How, in the next step, cells form emulsion droplets from the synthesized neutral lipid and which proteins function in this biogenesis process are subjects of considerable investigation. Our focus here is to review the major advances in our understanding of these issues. Because most current knowledge is derived from studying the generation of TG-containing LDs, we focus on this process. It remains to be clarified whether the same concepts will apply broadly to the formation of SE-rich LDs.
To facilitate investigation, it is helpful to break down the process of LD formation into discrete conceptual steps. We propose that the steps follow a conventional and logical model of oil formation and budding into the aqueous cytosol; these steps are illustrated in Figure 2. However, this model is far from validated. Other proposed models of LD formation include a model of diffusion of TGs from the ER into formed LDs contained in a membrane cup (Robenek et al. 2004), a model of LDs budding first into the lumen of the ER (Choudhary et al. 2011), and an ER scission model (Ploegh 2007).
Figure 2.

Model of steps of lipid droplet (LD) formation and expansion. Abbreviations: DGAT, diacylglycerol acyltransferase; eLD, expanding LD; iLD, initial LD; TG, triacylglycerol.
Step 1: Triacylglycerol Synthesis Within the ER
In eukaryotes, neutral lipids, such as TGs or SEs, are synthesized predominantly in the ER (Buhman et al. 2001). TGs were discovered in the early 1800s, and the first observations of TG storage in cells were reported by Richard Altmann as early as the 1890s (Altmann 1894). It was not until approximately 1960 that the biochemical reactions yielding TGs were discovered by Kennedy and coworkers (Weiss et al. 1960). This pathway, termed the de novo glycerolipid synthesis pathway, or the Kennedy pathway, is the primary pathway of TG synthesis in most cells. It uses glycerolphosphate and fatty acyl-CoA to generate glycerolipids, such as glycerophospholipids or TGs. A second pathway for TG synthesis, the monoacylglycerol pathway, exists in some cell types, such as intestinal enterocytes and adipocytes, and is involved in the recycling of mono- acylglycerols and diacylglycerols, generated by TG hydrolysis, back to TGs via a reesterification process (Coleman & Haynes 1985). The monoacylglycerol acyltransferase (MGAT) enzymes 1–3 play key roles in this pathway (Cheng et al. 2003, Yen & Farese 2003, Yen et al. 2002).
In either pathway, the final step of TG synthesis is catalyzed by the DGAT (diacylglycerol acyltransferase) enzymes: DGAT1 and DGAT2 (Cases et al. 1998b, 2001; Lardizabal et al. 2001). Thus, these enzymes are central to the process of LD biogenesis. The two DGAT enzymes are an example of convergent evolution at the biochemical level, inasmuch as the enzymes are unrelated in sequence but catalyze the same biochemical reaction. The two enzymes markedly differ in their structures, substrate preferences, subcellular localizations, and physiological roles. DGAT1, a member of the membrane-bound O-acyltransferase gene family (Chang et al. 2011), is local- ized exclusively to the ER, where it exists as dimers or tetramers that appear to be the functional units (Cases et al. 1998b, Cheng et al. 2001, Shockey et al. 2006). Here, the enzyme encounters its substrates, a fatty acyl-CoA and an acyl acceptor, and catalyzes the esterification reaction to generate a neutral lipid. DGAT1 can esterify a variety of substrates, including diacylglycerol, retinol, monoacylglycerol, and long-chain alcohols (Yen et al. 2005), although TG synthesis ap- pears to be its predominant function. DGAT1 has an important role in detoxifying excess lipids that are taken up from outside the cell (Villanueva et al. 2009). As such, DGAT1 may be important for preventing activation of ER stress pathways due to lipotoxic lipids (discussed below). DGAT2, a member of the DGAT2 gene family (Yen et al. 2008), in contrast, is localized to both the ER and LDs (Kuerschner et al. 2008, Stone et al. 2009, Wilfling et al. 2013). Unlike DGAT1, DGAT2 does not have multiple transmembrane domains but instead has a hydrophobic sequence, possibly a hairpin, that is embedded in the ER bilayer (Stone et al. 2006, Wilfling et al. 2013). DGAT2 appears to be a major enzyme for TG synthesis from fatty acid substrates derived from de novo lipogenesis (Irshad et al. 2017, Li et al. 2015, Qi et al. 2012, Villanueva et al. 2009, Wurie et al. 2012). In agreement with the different biochemical and cell biological properties of the two enzymes, mice lacking either DGAT1 or DGAT2 have very different phenotypes. DGAT1 knockout mice are viable and have body fat but are lean and resistant to diet-induced obesity, diabetes, and liver steatosis (Chen et al. 2002, Smith et al. 2000). In addition, these mice live on average ∼25% longer than control mice (Streeper et al. 2012). Mice lacking DGAT2 die shortly after birth in large part due to a lipid defect in the skin impairing its barrier function (Stone et al. 2004). These mice also nearly completely lack TGs, implicating DGAT2 as a major contributor to mammalian TG synthesis.
Both DGAT enzymes localize to the ER in cells without LDs (Shockey et al. 2006, Stone et al. 2009). The relationship of the localization of these enzymes to TG synthesis within the ER is uncertain. A current model suggests that TG synthesis occurs throughout the ER wherever enzyme substrates are found in excess and that the product of the reaction, TGs, is deposited within the ER bilayer. Either DGAT enzyme localized within the ER appears to generate TGs for initial LD formation (Harris et al. 2011, Li et al. 2015, Wilfling et al. 2013). Studies in plant cells suggested that DGAT1 and DGAT2 may reside in distinct domains of the ER (Shockey et al. 2006). Whether this property is evolutionarily conserved and how this might impact LD formation remain to be determined.
Of note, SEs are also formed in the ER, for example, by the enzymes Are1 and Are2 in yeast (Yang et al. 1996) and by the acyl CoA:cholesterol acyltransferase enzymes in mammals (Anderson et al. 1998, Cases et al. 1998a, Chang et al. 1993, Oelkers et al. 1998). Most LDs contain a mixture of TGs and SEs. However, genetic manipulations in yeast allow for the generation of LDs that contain primarily one or the other neutral lipid (Fu et al. 2014). Compared with studies of TG-containing LDs, there has been relatively less investigation into the mechanisms of formation of SE-containing LDs.
Step 2: Formation of an Oil Lens in the ER Membrane
The second step of LD formation is the formation of a lens of neutral lipids within the ER bilayer. As TG is synthesized and accumulates within the bilayer of the ER, biophysical processes drive lens formation. One model is that TG will nucleate an oil phase to minimize the entropy costs of disrupting the ER bilayer leaflets. The transition to an oil lens in the ER will accommodate newly synthesized TG and will lead to the relative depletion of TG within other spaces of the bilayer leaflet. In agreement with this model, only a limited amount of TG (∼3 mol%) can be accommodated in a dispersed form in model bilayers before oil droplets appear (Hamilton et al. 1983).
Whether cells have specific sites for TG lens nucleation and how such sites are determined are unclear. The nucleation model predicts, for instance, that the lipid composition of the ER membrane, its local curvature, and possibly proteins restricting the diffusion of lipids may have a role in lens formation. However, this prediction has not yet been tested comprehensively.
Several studies using biophysical techniques reported data that are consistent with a model of TG lens formation. As noted above, TGs can reach maximally ∼3 mol% of lipids in small vesicle membranes before forming a distinct droplet phase (Hamilton & Small 1981, Hamilton et al. 1983). However, another recent study reports that TGs can accumulate in higher concentrations in multilamellar vesicles, and when TGs are found at 5–10 mol%, they tend to form oil domains (Duelund et al. 2013).
ER structure may have an important role in modulating LD formation, although how it might exert effects remains to be determined. The ER is a vast membranous network that is spread throughout the cell and consists of membrane sheets and tubules (Nixon-Abell et al. 2016; Shibata et al. 2006, 2010; Voeltz et al. 2006). These different domains are characterized by different membrane curvatures. Whereas membrane sheets are flat with essentially no curvature, tubules have positive curvature in one principal direction (but not in the other). Moreover, tips of tubules have positive curvature in both principal directions, and junctions between tubules have positive curvature in one principal direction but negative curvature in the other principal direction (Schweitzer et al. 2015). Examples for the different ER structures include the outer nuclear membrane, which is a vast ER membrane sheet, and the dense interconnected network of cytoplasmic ER tubules. Proteins such as reticulons and atlastin (Hu et al. 2009, Voeltz et al. 2006) help to maintain ER tubules and sheets and to form and build junctions of tubules, respectively. For reviews of these proteins, please see Chen et al. (2013), Park & Blackstone (2010), and Shibata et al. (2009).
TG lens formation and subsequent LD formation occur at ER tubules (Jacquier et al. 2011, Kassan et al. 2013), where local curvature may aid TG lens formation by locally disturbing the bilayer order. The relationship of TG lens formation with ER sheets, which lack curvature, is less clear, as is whether lens formation occurs at tubular tips or junctions. What is clear, however, is that perturbations of ER structure can affect LD formation. For example, depletion of proteins that affect ER shape and the balance between sheets and tubules, or the formation of tubular junctions, results in alterations of LD morphologies. For example, depletion of atlastin dramatically affects LD size (Klemm et al. 2013), as does deletion of the protein REEP1 (Falk et al. 2014, Klemm et al. 2013, Renvoise ˊ et al. 2016), a protein involved in generating ER tubules. Intriguingly, many ER proteins whose genetic mutations give rise to hereditary spastic paraplegia also result in changes in LD morphology (Blackstone 2012, Eastman et al. 2009, Ito & Suzuki 2009, Renvoise ˊ et al. 2016). Hereditary spastic paraplegia appears to be due to ER structural deficiencies that are particularly manifested in large motor neurons. It is unclear whether the LD abnormalities found in cells with some of the mutant proteins are involved in the pathogenesis of hereditary spastic paraplegia or, instead, are markers for the disrupted ER.
Proteins are likely involved in TG lens formation, although this remains to be confirmed. A candidate for such a function is the protein FIT2. FIT2 is an ER protein with multiple trans- membrane domains that has been linked to LD formation (Choudhary et al. 2015, Kadereit et al. 2008). FIT2 binds lipids, such as diacylglycerol and TGs, and FIT2 may be involved in partitioning neutral lipids within the ER for LD formation (Gross et al. 2011). Overexpression of FIT2 in cells results in larger LDs, although levels of TG synthesis are unchanged, suggesting altered partitioning of newly synthesized TGs into LDs (Kadereit et al. 2008). Loss of FIT2 function in cells results in smaller LDs, possibly with a reduction in TG synthesis (Kadereit et al. 2008). In mice, deletion of FIT2 in intestinal enterocytes results in severe intestinal pathology, including a near absence of LDs and massive intestinal dysfunction (Goh et al. 2015), highlighting the importance of FIT2 in neutral lipid biology of dietary fat intake. These data suggest that FIT2 plays a role in LD formation, possibly at the step of lens formation and/or a TG partitioning step. However, the functional role of FIT2 in LD formation remains to be elucidated.
In addition to FIT2, other proteins may be involved in regulating TG lens formation in the ER. For example, perilipin3 (or Tip47) has been suggested to bind nascent lenses, thus stabilizing them in the earliest steps of LD formation (Bulankina et al. 2009). Other proteins may be involved in determining lens nucleation sites or, alternatively, in preventing TG lenses from forming in specific regions of the ER (Kassan et al. 2013, Wilfling et al. 2014a).
Step 3: Budding and Nascent Lipid Droplet Formation
In a prevailing model of LD formation, when sufficient TG accumulates within the ER bilayer, budding of the droplet into the cytosol occurs. However, it is unclear whether this happens for all LDs or whether some (or indeed all) of them remain connected to the ER. At least in yeast, a substantial fraction of LDs remains physically connected with the ER (Jacquier et al. 2011), whereas in mammalian cells at least some LDs appear to be completely budded from the ER (Wilfling et al. 2013). It is currently unclear what determines the directionality of the budding reaction toward the cytosol and away from the ER lumen, although FIT2 may be important in regulating this directionality (Choudhary et al. 2015). In addition, specific lipids may have a role in LD budding. For instance, dephosphorylation of phosphatidic acid by the yeast enzyme Pah1 to generate diacylglycerol appears to be important for packaging of TGs into LDs (Adeyo et al. 2011).
The process of budding itself can be thought of as a dewetting process, in which a TG lens is formed and converted to a bud on the ER. A gradual increase in the contact angle between the droplet and ER bilayer eventually leads to fission of the bud from the ER when a sufficient angle is achieved. Whether proteins are involved in the scission of LDs from the ER remains to be determined. For a more in-depth discussion of the biophysics of dewetting as it pertains to LD formation, see recent reviews (Thiam & Foret 2016, Thiam et al. 2013b).
Although budding of LDs from the ER is a biophysical process of emulsion formation, proteins are likely involved in mediating or regulating this step. One possibility is that specific ER proteins mark and modulate the sites of LD budding in the ER. Such proteins may act as gatekeepers for the formation of domains with specific phospholipid or protein compositions that facilitate the budding of nascent LDs. Alternatively, these proteins may affect the ER bilayer and emerging LD structure in a way that facilitates the dewetting and budding process.
The protein seipin appears to mark sites that correlate with the initial growth of nascent budded LDs and, therefore, may be involved at sites of initial LD formation. Seipin was originally identified as deficient in a particular form of congenital lipodystrophy (Magre et al. 2001). Subsequently, several screens in yeast revealed that deficiency of the yeast ortholog for seipin results in abnormal LDs characterized by both giant LDs and numerous very small LDs, depending on the growth conditions (Fei et al. 2008, Szymanski et al. 2007). Seipin is an ER protein with two transmembrane domains and an evolutionarily conserved ER luminal loop (Lundin et al. 2006). Biochemical studies showed that purified seipin appears to form multimers that may organize into a toroid- shaped structure (Binns et al. 2010, Sim et al. 2014). Several recent studies indicated that seipin is involved in the early steps of LD formation. For example, in yeast, seipin is required for normal LD formation in a model system in which LD biogenesis can be induced (Cartwright et al. 2015). In fly cells, endogenous seipin forms patches in the ER, and these patches move rapidly along ER tubules (Wang et al. 2016). During LD formation, some seipin foci encounter sites of TG accumulation, subsequently become colocalized with these TG collections, and enable the initial growth of nascent LDs and their conversion to larger initial LDs (Wang et al. 2016). In the absence of seipin, in Drosophila and mammalian cells, a multitude of small TG collections associated with the ER accumulate. Ultrastructural studies, including electron tomography, showed that these TG collections are nascent, budded LDs of 100–200-nm diameter that remain tightly associated with the ER (Binns et al. 2010, Choudhary et al. 2015). These regions of association are characterized by the absence of ribosomes and by regions of increased electron density, suggesting that they represent areas of LD-ER contact sites.
These data indicate that seipin plays a functional role in the initial steps of LD formation either by helping to generate nascent LDs or by facilitating their growth and expansion (Salo et al. 2016, Wang et al. 2016). Supporting a model for seipin regulating these early steps, several groups showed that seipin regulates the protein composition of LDs (Grippa et al. 2015, Wang et al. 2016) and that aberrant proteins can access LDs at early times when seipin is deficient (Wang et al. 2016). Among these proteins are the enzymes of TG synthesis that can stimulate LD expansion (discussed below), contributing to both the formation of LDs that are prone to fusion and the formation of giant LDs (Wang et al. 2016). Seipin deficiency is associated with alterations of phospholipid composition of LDs at late times during formation (Fei et al. 2011, Pagac et al. 2016, Wang et al. 2016, Wolinski et al. 2015), which might explain, in part, the observed abnormalities of phospholipid composition of yeast LDs in seipin deficiency (Fei et al. 2011, Wolinski et al. 2015). Because LDs and the ER stay in contact even when seipin is absent (Salo et al. 2016, Wang et al. 2016), other proteins may be involved in defining the initial budding and growth step of LD formation.
During LD budding, some proteins may gain immediate access to the nascent LDs, but others may be excluded. We recently described a protein marker for nascent LDs that we named LiveDrop (Wang et al. 2016). The LiveDrop marker consists of a membrane-embedded hairpin motif from the GPAT4 protein linked to a fluorescent marker. In our experience, LiveDrop localizes to all nascent LDs at the time of their formation and is more sensitive to detecting nascent LDs than are neutral lipid dyes, such as BODIFY 493/503 (Wang et al. 2016). A similar protein that includes a hydrophobic domain from the protein ALDI also appears to mark nascent LDs (Kassan et al. 2013). Interestingly, although LiveDrop localizes to all nascent LDs, the GPAT4 protein from which LiveDrop is derived does not localize to nascent LDs (Wilfling et al. 2013). This finding suggests that mechanisms exist to exclude specific proteins from nascent LDs. How this exclusion occurs molecularly is unknown.
Step 4: Lipid Droplet Growth and Expansion via Acquisition of Specific Proteins
Within minutes to hours after initial LD formation, a subset of these newly formed LDs can be converted to a subpopulation of LDs termed expanding LDs (eLDs) (Wilfling et al. 2013, 2014a). This phenomenon is prominent in and was best described in Drosophila cells but can also occur in cultured mammalian cells (Wilfling et al. 2013). The formation of eLDs within cells leads to the existence of two LD populations in cultured cells: initial LDs (iLDs) and eLDs. iLDs are those LDs of 400–800-nm diameter that are formed from the ER. eLDs are iLDs that are converted to a specialized LD, and eLDs are characterized by localized TG synthesis. This TG synthesis is catalyzed by specific isoforms of the TG synthesis pathway, such as GPAT4 and DGAT2, that appear to relocalize from the ER to and around the surfaces of LDs (Kuerschner et al. 2008, Stone et al. 2009, Wilfling et al. 2013). This relocalization occurs through membrane bridges, where the LD surface is continuous with the ER membrane (Wilfling et al. 2013, 2014b).
What triggers the conversion of an iLD to an eLD is unknown. However, under normal cell physiology conditions, the actions of the ARF1 and COP-I/coatamer protein machinery are clearly required for this process (Wilfling et al. 2014b). Initial screens in Drosophila cells showed that deficiency of ARF1 or COP-I/coatamer proteins resulted in a striking phenotype of intermediate- sized, dispersed LDs (Beller et al. 2008, Guo et al. 2008). Subsequently, ARF1/COP-I proteins mediate the budding of 60–80-nm nano-LDs from LDs in an in vitro system (Thiam et al. 2013a). Furthermore, a portion of the ARF1/COP-I protein localizes specifically to the surfaces of LDs in cells (Wilfling et al. 2014b). A current model is that the LD expansion pathway is triggered by activation of ARF1/COP-I proteins at the surfaces of LDs to form nano-LDs and to remove predominantly phospholipids from the LD surface, thereby increasing surface tension and enabling the modified iLD to interact with the ER. This process then allows for the formation of ER-LD membrane bridges (Wilfling et al. 2014b) and for the migration of TG synthesis enzymes to the surface for LD expansion. Consistent with this model, a recent study implicates TRAPPII (transport protein particle II) and Rab18 as factors in the process downstream of the COP-I proteins (Salo et al. 2016). An alternative model, proposed from studying the requirement of the ARF1/COP-I proteins for targeting of ATGL to LDs, suggests that these proteins are involved in vesicle formation from the ERGIC region of the ER for vesicular targeting of specific proteins to the LD surface (Soni et al. 2009).
Studies of the eLD pathway have been instrumental in our understanding of the mechanisms of protein targeting to LD surfaces. The targeting of proteins to the LD surface provides an unusual circumstance that is different from other membrane targeting events within the cell in that the LD surface exists as a monolayer that covers a neutral lipid oil phase. As such, the biophysical properties of LDs do not allow for targeting of ER proteins with membrane domains that completely span the bilayer, as such targeting would be energetically extremely unfavorable. From the study of enzymes that target eLDs in Drosophila cells, we proposed that proteins that target the LD surface fall in one of two major classes (see Figure 3, and for review, see Kory et al. 2016). Class I proteins target the LD surface from the ER bilayer (Kory et al. 2016). This class is particularly well illustrated by the enzyme GPAT4, which specifically marks eLDs in Drosophila cells (Wilfling et al. 2013). GPAT4 and other class I proteins are typically characterized by a membrane-embedded domain that is hydrophobic and possibly adopts a hairpin motif. This hydrophobic domain is embedded in the ER but does not completely span the ER, which thereby allows it to be localized to the ER in the basal condition and to be translocated to the monolayer surface of LDs that are expanding, presumably via ER-LD membrane bridges (Wilfling et al. 2014b). Other likely examples of class I proteins are DGAT2, ATGL, UBXD8, and ACSL3. For UBXD8, the insertion of the hairpin-containing protein initially into the ER bilayer is dependent on the proteins PEX9 and PEX3 (Schrul & Kopito 2016). It remains to be determined whether this is a general membrane targeting pathway for this class of proteins.
Figure 3.
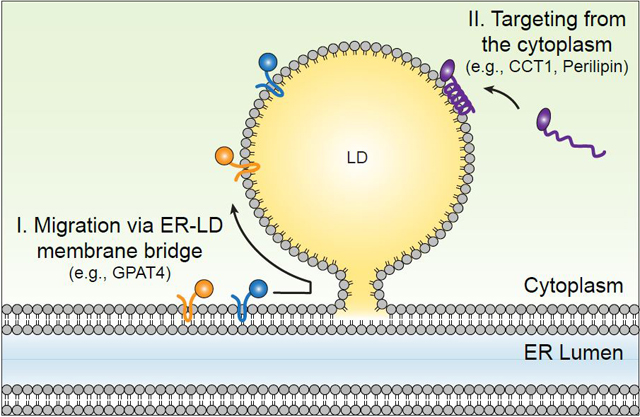
Mechanisms for targeting of proteins to lipid droplet (LD) surfaces during LD formation and growth. Class I proteins, such as GPAT4, are inserted into the ER and from there translocate to the surfaces of LDs either during LD formation or after formation via membrane bridges. Class II proteins, such as CCT1, target from the cytosol via amphipathic helices or other short hydrophobic domains.
A second category, termed class II proteins, target LD surfaces from the cytosol typically through hydrophobic domains, such as amphipathic helices or short, hydrophobic-rich sequences (Kory et al. 2016). An example of this second category is the CCT1 protein, which targets the surfaces of Drosophila eLDs via an amphipathic helix in its membrane-binding domain (Krahmer et al. 2011). This amphipathic helix is both necessary and sufficient for targeting of CCT1 to LDs (Krahmer et al. 2011). Studies of CCT1 during eLD formation indicate that CCT1 apparently recognizes PC deficiency at the surfaces of eLDs. Binding of CCT1 to these surfaces activates the enzyme (Cornell & Vance 1987, Krahmer et al. 2011), thereby catalyzing a rate-limiting step of PC synthesis. How CCT1 or other amphipathic helix–containing proteins recognize the surface of eLDs remains to be determined. Another example of class II proteins that bind LD surfaces is the perilipin family proteins (Li et al. 2017). The amount of class II proteins on LD surfaces in particular is determined by binding affinity and macromolecular crowding (Rowe et al. 2016). During lipolysis, when LD surfaces shrink, class II proteins with relatively lower affinity are displaced from the monolayer surface. In contrast, class I proteins, which are likely more deeply embedded in the neutral lipid core of LDs, generally remain bound to LDs.
In addition to these pathways used by the two major classes of proteins targeting LDs, other routes may exist. For instance, lipid anchors could mediate targeting of proteins to LDs. Such a mechanism could be responsible for the recruitment of small Rab GTPases such as Rab18 (Martin et al. 2005) and ELMOD2, a noncanonical Arf-GAP (Suzuki et al. 2015). However, evidence for the biological function of these proteins on LDs is currently sparse. In addition, protein-protein interactions can mediate the recruitment of proteins to LDs. For instance, during Drosophila development, the adaptor protein Jabba recruits specific histones for temporary storage on LDs (Z. Li et al. 2012).
THE FORMATION OF GIANT LIPID DROPLETS
Some cells have LDs whose diameters far exceed the 1–2-μm diameter of eLDs; such LDs are often referred to as giant LDs. Such cells include hepatocytes or adipocytes, in which LD diameters can exceed tens of micrometers. These giant LDs are formed by one of two processes (discussed above). The first process is coalescence, in which two LDs almost instantaneously fuse to form a larger LD. An example in which LD coalescence is prominent is PC deficiency. A number of gene depletions in a screen of Drosophila or yeast cells that result in giant LDs appear to have PC deficiency in common (Guo et al. 2008), and genetic depletion of CCT1 or culture conditions that result in PC deficiency also specifically lead to LD coalescence (Krahmer et al. 2011). Coalescence can also be induced by adding agents that raise surface tension and act as poor surfactants. For example, propranolol and some LD dyes induce LD coalescence (Murphy et al. 2010).
A second process that results in the formation of giant LDs is diffusion mediated by Ostwald ripening. This process is much slower than coalescence and is characterized by the gradual enlargement of one LD and the depletion of oil in the adjacent second LD. Although this process would be quite slow for TG exchange between LDs in the absence of proteins, there is evidence that specific proteins facilitate this process in cells such as adipocytes. For example, the protein FSP27/CIDE-C localizes to contact zones between two LDs and facilitates the slow transfer of TGs from a smaller droplet to LDs (Gong et al. 2011), suggesting that FSP27 combines LDs by facilitating TG diffusion. A related protein, CIDE-B, is found in the liver, where it has been implicated in TG secretion (Ye et al. 2009), and may be involved in an analogous diffusion process of TGs from LDs to the ER, where it is used for very low density lipoprotein assembly.
INTERACTIONS OF LIPID DROPLETS WITH OTHER CELLULAR ORGANELLES
As knowledge of LD biology increases, we are beginning to understand the interactions of LDs with other cellular organelles. Far and away the most prominent organelle that interacts with LDs is the ER. As described above, LDs are formed from the ER and often remain in close association with the ER after their budding and formation. Ultrastructural studies from mammalian cells indicate that LDs may sometimes be associated with the ER in a structure that resembles an egg cup (Robenek et al. 2006), with the LD being cradled by a region of ER membrane. In yeast, LDs may be associated with the ER and may remain connected to it after formation as part of specialized ER domains (Jacquier et al. 2011). In nonyeast cells, budded iLDs appear to reconnect with the ER during eLD formation (discussed above; see Wilfling et al. 2013). This reconnection has been shown by electron microscopy to consist of a membrane bridge that spans the cytosolic leaflet of the ER via a continuous bridge with the surface monolayers of LDs (Wilfling et al. 2013). Proteins may be involved in establishing or maintaining these bridges, but the identity of such proteins is unknown. One function of ER-LD bridges appears to be in allowing enzymes of TG synthesis to migrate to LD surfaces (Wilfling et al. 2013). Because DGAT2 is one of these enzymes and DGAT2 has been linked functionally to de novo lipogenesis (discussed above), one function of these ER-LD bridges may be to enable partitioning of de novo lipogenesis to a region of the ER that is linked to TG storage.
LDs may also interact with other organelles. For example, in yeast, LDs associate under specific circumstances with peroxisomes (Binns et al. 2006), possibly to facilitate the β-oxidation of fatty acids. In mammalian cells, LDs sometimes appear to interact closely with mitochondria. This interaction may be regulated metabolically, as activation of the master metabolic kinase AMPK results in dispersion of LDs on stable microtubules, possibly to increase interactions with mitochondria (Herms et al. 2015). The LD-mitochondria interaction is particularly prominent in skeletal or heart muscle, as demonstrated by electron micrographs (Wang et al. 2013, Zhang et al. 2011). LDs may interact with mitochondria in mammalian cells to facilitate the transfer of fatty acids to mitochondria for β-oxidation under starvation conditions. In support of this possibility, LDs in cultured mammalian cells that are preloaded with labeled fatty acids transfer these fatty acids to mitochondria, apparently for β-oxidation (Rambold et al. 2015), although whether this process requires LD-mitochondria contacts is unclear.
IMPLICATIONS OF LIPID DROPLET FORMATION FOR CELL PHYSIOLOGY
The formation of LDs has many implications for cell physiology. The storage of fatty acids and other lipids in LDs provides important stores of metabolic energy and lipids and lipid precursors for membrane lipid synthesis during cell proliferation or cell remodeling. Illustrating this function, yeast that are cultured to stationary phase with energy substrate depletion store lipids in LDs. When yeast from stationary phase are transferred to fresh medium and proliferation is induced, these cells consume LDs and utilize the lipids from LDs for cell proliferation (Kurat et al. 2009). Intriguingly, some cancer cells accumulate massive amounts of LDs. For example, clear cell carcinoma of the kidney is characterized by vast amounts of LDs in the cytoplasm (Saito et al. 2016, Yu et al. 2013), although it remains to be tested whether these LDs are important for enabling the proliferation of such cells. More investigation is needed to better understand whether LDs provide stores of energy or membrane lipids during cancer cell proliferation.
LD formation also appears to be central to cell physiology in preventing the accumulation of toxic lipids within the ER. The ER is a large cellular organelle that is involved in a multitude of processes, including lipid synthesis and the generation of membrane and secreted proteins. Glycosylation of membrane and secreted proteins also occurs in the ER. For these processes to occur normally, the proper composition of lipids in the ER must be maintained. Thus, when excess simple lipids, such as fatty acids or sterols, accumulate in the ER, mechanisms exist to convert these lipids to more inert neutral lipids for storage in LDs. For example, the ACAT enzymes generate SEs, and the DGAT enzymes generate TGs and other neutral lipids to be stored in LDs. The absence of these enzymes may thus result in abnormal accumulation of lipids in the ER bilayer, which can trigger ER stress and the unfolded protein response. Indeed, deficiency of the ACAT enzymes results in ER stress when cells are loaded with excess cholesterol (Devries-Seimon et al. 2005, Kedi et al. 2009). Similarly, DGAT1 may play such a role in protecting the ER from lipotoxic ER stress. Interestingly, triggering of ER stress in the liver results in upregulation of IRE1 and downstream lipid metabolism targets, including DGAT2, as part of a homeostatic response (Lee et al. 2008). Related to this finding, LDs could protect phospholipid pools by preventing their oxidation, serving as a sink for radical oxygen species (Bailey et al. 2015, Liu et al. 2015).
LDs have also been implicated in modulating pathways of oxidative stress in cells. For example, studies in Drosophila show that LDs can limit reactive oxygen species in glial cells and can inhibit oxidation of polyunsaturated fatty acids (Bailey et al. 2015). In contrast, glial LDs have been implicated in the propagation of oxidative stress in Drosophila models of neurodegeneration induced by mitochondrial defects (Liu et al. 2015).
Studies in mice and humans highlight the importance of LD formation in normal metabolism and physiology. For example, humans lacking DGAT1 are characterized by a severe congenital diarrhea syndrome that is exacerbated by exogenous fat (Lee et al. 2008). Also, humans treated with inhibitors of DGAT1 develop diarrhea in a dose-related fashion (DeVita & Pinto 2013, Lee et al. 2008). Humans appear to lack expression of DGAT2 in their intestine (Haas et al. 2012), which may make them particularly susceptible to the effects of DGAT1 deficiency.
Additionally, a large number of LD proteins are linked to metabolic disease. As mentioned above, deficiency of FIT2 in the murine small intestine results in severe pathology and intestinal failure that are associated with impaired LD formation, and deficiency of FIT2 in murine adipose tissue results in lipodystrophy (Goh et al. 2015, Miranda et al. 2014). Deletion of seipin in mice results in postnatal lipodystrophy and numerous metabolic abnormalities (Cui et al. 2011, Dollet et al. 2014, Gao et al. 2015, L. Liu et al. 2014). Similarly, homozygous or compound heterozygous mutations of CCTα lead to a lipodystrophy syndrome (Payne et al. 2014). FSP27/CIDE-C deletion results in adipose tissue characterized by many small LDs, rather than one large unilocular LD, and in protection from the insulin resistance that accompanies these morphological changes (Nishino et al. 2008). Another LD protein, PNPLA3, is the major genetic risk factor for liver steatosis (Anstee & Day 2013, Romeo et al. 2008). A variant of PNPLA3 is found in increased concentrations in LD fractions, where it may alter cell physiology in a manner that promotes TG and LD accumulation (He et al. 2010, J.Z. Li et al. 2012, Smagris et al. 2015). The evidence is growing that proteins involved in the formation or maintenance of LDs are of tremendous importance for normal cell physiology and that mutations of specific LD proteins can lead to cell dysfunction, severe metabolic abnormalities, and disease (for reviews, see Carr & Ahima 2016, Greenberg et al. 2011, Krahmer et al. 2013a).
CURRENT QUESTIONS IN LIPID DROPLET FORMATION RESEARCH
In this review, we propose a model of LD formation and describe progress that has been made to understand the molecular aspects of this process. Relative to other cellular organelles, however, an understanding of LD biology is incomplete, and many questions remain. For example, it is unclear how sites of TG synthesis are localized within the ER and how such localization correlates with the sites of LD formation. The model in which TG initially forms lenses within the ER membrane is attractive but is largely untested. How and which proteins participate in TG lens formation and organization are mostly unknown. Additionally, whether proteins are required for the LD budding process is also unknown. How some LDs are selected for the expansion pathway is similarly mostly unknown. The coming years of investigation into LD formation will likely yield many insights into these processes. Advances will come from clues from unbiased screening approaches, such as genetic screens or proteomics, combined with mechanistic studies of cell biology and the biochemistry of candidate proteins. A molecular understanding of how cells utilize proteins to control and regulate the biophysical process of emulsion formation will likely emerge. These advances will provide a multitude of molecular insights affecting our understanding of cell physiology, metabolic diseases, and the basic science underlying oil droplet formation that is central to industrial processes such as seed oil production and biofuels.
ACKNOWLEDGMENTS
We thank Michel Becuwe, Nora Kory, Florian Wilfling, and Huajin Wang for a critical reading of the manuscript and G. Howard for editorial assistance. The authors are supported by the National Institutes of Health [R01 DK101579 (R.V.F.), R01 DK056084 (R.V.F.), R01 GM097194 (T.C.W.)], the Mathers Foundation (T.C.W.), and the Howard Hughes Medical Institute (T.C.W.). J.C. is a fellow of the Damon Runyon Cancer Research Foundation (DRG-2296-17).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Adeyo O, Horn PJ, Lee S, Binns DD, Chandrahas A, et al. 2011. The yeast lipin orthologue Pah1p is important for biogenesis of lipid droplets. J. Cell Biol. 192:1043–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmann R 1894. Die Elementarorganismen und ihre Beziehungen zu den Zellen. Leipzig: Viet & Co; 271 pp. [Google Scholar]
- Anderson RA, Joyce C, Davis M, Reagan JW, Clark M, et al. 1998. Identification of a form of acyl- CoA:cholesterol acyltransferase specific to liver and intestine in nonhuman primates. J. Biol. Chem. 273:26747–54 [DOI] [PubMed] [Google Scholar]
- Anstee QM, Day CP. 2013. The genetics of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 10:645–55 [DOI] [PubMed] [Google Scholar]
- Bailey AP, Koster G, Guillermier C, Hirst EM, MacRae JI, et al. 2015. Antioxidant role for lipid droplets in a stem cell niche of Drosophila. Cell 163:340–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barneda D, Christian M. 2017. Lipid droplet growth: regulation of a dynamic organelle. Curr. Opin. Cell Biol. 47:9–15 [DOI] [PubMed] [Google Scholar]
- Beller M, Riedel D, Jansch L, Dieterich G, Wehland J, et al. 2006. Characterization of the Drosophila lipid droplet subproteome. Mol. Cell. Proteom. 5:1082–94 [DOI] [PubMed] [Google Scholar]
- Beller M, Sztalryd C, Southall N, Bell M, Jackle H, et al. 2008. COPI complex is a regulator of lipid homeostasis.PLOS Biol. 6:e292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binns D, Januszewski T, Chen Y, Hill J, Markin VS, et al. 2006. An intimate collaboration between peroxisomes and lipid bodies. J. Cell Biol. 173:719–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binns D, Lee S, Hilton CL, Jiang QX, Goodman JM. 2010. Seipin is a discrete homooligomer. Biochemistry 49:10747–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstone C 2012. Cellular pathways of hereditary spastic paraplegia. Annu. Rev. Neurosci. 35:25–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasaemle DL, Dolios G, Shapiro L, Wang R. 2004. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J. Biol. Chem. 279:46835–42 [DOI] [PubMed] [Google Scholar]
- Brasaemle DL, Rubin B, Harten IA, Gruia-Gray J, Kimmel AR, Londos C. 2000. Perilipin A increases triacylglycerol storage by decreasing the rate of triacylglycerol hydrolysis. J. Biol. Chem. 275:38486–93 [DOI] [PubMed] [Google Scholar]
- Buhman KK, Chen HC, Farese RV Jr. 2001. The enzymes of neutral lipid synthesis. J. Biol. Chem. 276:40369–72 [DOI] [PubMed] [Google Scholar]
- Bulankina AV, Deggerich A, Wenzel D, Mutenda K, Wittmann JG, et al. 2009. TIP47 functions in the biogenesis of lipid droplets. J. Cell Biol. 185:641–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr RM, Ahima RS. 2016. Pathophysiology of lipid droplet proteins in liver diseases. Exp. Cell Res. 340:187–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright BR, Binns DD, Hilton CL, Han S, Gao Q, Goodman JM. 2015. Seipin performs dissectible functions in promoting lipid droplet biogenesis and regulating droplet morphology. Mol. Biol. Cell 26:726–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cases S, Novak S, Zheng YW, Myers HM, Lear SR, et al. 1998a. ACAT-2, a second mammalian acyl-CoA:cholesterol acyltransferase: its cloning, expression, and characterization. J. Biol. Chem. 273:26755–64 [DOI] [PubMed] [Google Scholar]
- Cases S, Smith SJ, Zheng YW, Myers HM, Lear SR, et al. 1998b. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. PNAS 95:13018–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cases S, Stone SJ, Zhou P, Yen E, Tow B, et al. 2001. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J. Biol. Chem. 276:38870–76 [DOI] [PubMed] [Google Scholar]
- Cermelli S, Guo Y, Gross SP, Welte MA. 2006. The lipid-droplet proteome reveals that droplets are a protein-storage depot. Curr. Biol. 16:1783–95 [DOI] [PubMed] [Google Scholar]
- Chang CC, Huh HY, Cadigan KM, Chang TY. 1993. Molecular cloning and functional expression of human acyl-coenzyme A:cholesterol acyltransferase cDNA in mutant Chinese hamster ovary cells. J. Biol. Chem. 268:20747–55 [PubMed] [Google Scholar]
- Chang CCY, Sun J, Chang T-Y. 2011. Membrane-bound O-acyltransferases (MBOATs). Front. Biol. 6:177 [Google Scholar]
- Chen HC, Smith SJ, Ladha Z, Jensen DR, Ferreira LD, et al. 2002. Increased insulin and leptin sensitivity in mice lacking acyl CoA:diacylglycerol acyltransferase 1. J. Clin. Investig. 109:1049–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Novick P, Ferro-Novick S. 2013. ER structure and function. Curr. Opin. Cell Biol. 25:428–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D, Meegalla RL, He B, Cromley DA, Billheimer JT, Young PR. 2001. Human acyl-CoA:diacylglycerol acyltransferase is a tetrameric protein. Biochem. J. 359:707–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D, Nelson TC, Chen J, Walker SG, Wardwell-Swanson J, et al. 2003. Identification of acyl coenzyme A:monoacylglycerol acyltransferase 3, an intestinal specific enzyme implicated in dietary fat absorption. J. Biol. Chem. 278:13611–14 [DOI] [PubMed] [Google Scholar]
- Choudhary V, Jacquier N, Schneiter R. 2011. The topology of the triacylglycerol synthesizing enzyme Lro1 indicates that neutral lipids can be produced within the luminal compartment of the endoplasmatic reticulum: implications for the biogenesis of lipid droplets. Commun. Integr. Biol. 4:781–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary V, Ojha N, Golden A, Prinz WA. 2015. A conserved family of proteins facilitates nascent lipid droplet budding from the ER. J. Cell Biol. 211:261–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RA, Haynes EB. 1985. Subcellular location and topography of rat hepatic monoacylglycerol acyl-transferase activity. Biochim. Biophys. Acta 834:180–87 [DOI] [PubMed] [Google Scholar]
- Cornell R, Vance DE. 1987. Binding of CTP: phosphocholine cytidylyltransferase to large unilamellar vesicles. Biochim. Biophys. Acta 919:37–48 [DOI] [PubMed] [Google Scholar]
- Cui X, Wang Y, Tang Y, Liu Y, Zhao L, et al. 2011. Seipin ablation in mice results in severe generalized lipodystrophy. Hum. Mol. Genet. 20:3022–30 [DOI] [PubMed] [Google Scholar]
- Currie E, Guo X, Christiano R, Chitraju C, Kory N, et al. 2014. High confidence proteomic analysis of yeast LDs identifies additional droplet proteins and reveals connections to dolichol synthesis and sterol acetylation. J. Lipid Res. 55:1465–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVita RJ, Pinto S. 2013. Current status of the research and development of diacylglycerol O-acyltransferase 1 (DGAT1) inhibitors. J. Med. Chem. 56:9820–25 [DOI] [PubMed] [Google Scholar]
- Devries-Seimon T, Li Y, Yao PM, Stone E, Wang Y, et al. 2005. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J. Cell Biol. 171:61–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Yang L, Zhang S, Wang Y, Du Y, et al. 2012. Identification of the major functional proteins of prokaryotic lipid droplets. J. Lipid Res. 53:399–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollet L, Magre J, Cariou B, Prieur X. 2014. Function of seipin: new insights from Bscl2/seipin knockout mouse models. Biochimie 96:166–72 [DOI] [PubMed] [Google Scholar]
- Duelund L, Jensen GV, Hannibal-Bach HK, Ejsing CS, Pedersen JS, et al. 2013. Composition, structure and properties of POPC-triolein mixtures. Evidence of triglyceride domains in phospholipid bilayers. Biochim. Biophys. Acta 1828:1909–17 [DOI] [PubMed] [Google Scholar]
- Eastman SW, Yassaee M, Bieniasz PD. 2009. A role for ubiquitin ligases and Spartin/SPG20 in lipid droplet turnover. J. Cell Biol. 184:881–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk J, Rohde M, Bekhite MM, Neugebauer S, Hemmerich P, et al. 2014. Functional mutation analysis provides evidence for a role of REEP1 in lipid droplet biology. Hum. Mutat. 35:497–504 [DOI] [PubMed] [Google Scholar]
- Fei W, Shui G, Gaeta B, Du X, Kuerschner L, et al. 2008. Fld1p, a functional homologue of human seipin, regulates the size of lipid droplets in yeast. J. Cell Biol. 180:473–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei W, Shui G, Zhang Y, Krahmer N, Ferguson C, et al. 2011. A role for phosphatidic acid in the formation of “supersized” lipid droplets. PLOS Genet. 7:e1002201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu D, Yu Y, Folick A, Currie E, Farese RV Jr., et al. 2014. In vivo metabolic fingerprinting of neutral lipids with hyperspectral stimulated Raman scattering microscopy. J. Am. Chem. Soc. 136:8820–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Wang M, Guo X, Qiu X, Liu L, et al. 2015. Expression of seipin in adipose tissue rescues lipodystrophy, hepatic steatosis and insulin resistance in seipin null mice. Biochem. Biophys. Res. Commun. 460:143–50 [DOI] [PubMed] [Google Scholar]
- Gao Q, Goodman JM. 2015. The lipid droplet—a well-connected organelle. Front. Cell Dev. Biol. 3:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh VJ, Tan JS, Tan BC, Seow C, Ong WY, et al. 2015. Postnatal deletion of fat storage–inducing transmembrane protein 2 (FIT2/FITM2) causes lethal enteropathy. J. Biol. Chem. 290:25686–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J, Sun Z, Wu L, Xu W, Schieber N, et al. 2011. Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. J. Cell Biol. 195:953–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg AS, Coleman RA, Kraemer FB, McManaman JL, Obin MS, et al. 2011. The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Investig. 121:2102–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grippa A, Buxo L, Mora G, Funaya C, Idrissi FZ, et al. 2015. The seipin complex Fld1/Ldb16 stabilizes ER–lipid droplet contact sites. J. Cell Biol. 211:829–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronke S, Mildner A, Fellert S, Tennagels N, Petry S, et al. 2005. Brummer lipase is an evolutionary conserved fat storage regulator in Drosophila. Cell Metab. 1:323–30 [DOI] [PubMed] [Google Scholar]
- Gross DA, Zhan C, Silver DL. 2011. Direct binding of triglyceride to fat storage–inducing transmembrane proteins 1 and 2 is important for lipid droplet formation. PNAS 108:19581–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Walther TC, Rao M, Stuurman N, Goshima G, et al. 2008. Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature 453:657–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas JT, Winter HS, Lim E, Kirby A, Blumenstiel B, et al. 2012. DGAT1 mutation is linked to a congenital diarrheal disorder. J. Clin. Investig. 122:4680–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JA, Miller KW, Small DM. 1983. Solubilization of triolein and cholesteryl oleate in egg phosphatidylcholine vesicles. J. Biol. Chem. 258:12821–26 [PubMed] [Google Scholar]
- Hamilton JA, Small DM. 1981. Solubilization and localization of triolein in phosphatidylcholine bilayers: a 13C NMR study. PNAS 78:6878–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CA, Haas JT, Streeper RS, Stone SJ, Kumari M, et al. 2011. DGAT enzymes are required for triacylglycerol synthesis and lipid droplets in adipocytes. J. Lipid Res. 52:657–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, McPhaul C, Li JZ, Garuti R, Kinch L, et al. 2010. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem. 285:6706–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms A, Bosch M, Reddy BJ, Schieber NL, Fajardo A, et al. 2015. AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat. Commun. 6:7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Shibata Y, Zhu PP, Voss C, Rismanchi N, et al. 2009. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell 138:549–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irshad Z, Dimitri F, Christian M, Zammit VA. 2017. Diacylglycerol acyltransferase 2 links glucose utilization to fatty acid oxidation in the brown adipocytes. J. Lipid Res. 58:15–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D, Suzuki N. 2009. Seipinopathy: a novel endoplasmic reticulum stress-associated disease. Brain 132:8–15 [DOI] [PubMed] [Google Scholar]
- Jacquier N, Choudhary V, Mari M, Toulmay A, Reggiori F, Schneiter R. 2011. Lipid droplets are functionally connected to the endoplasmic reticulum in Saccharomyces cerevisiae. J. Cell Sci. 124:2424–37 [DOI] [PubMed] [Google Scholar]
- Kadereit B, Kumar P, Wang WJ, Miranda D, Snapp EL, et al. 2008. Evolutionarily conserved gene family important for fat storage. PNAS 105:94–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassan A, Herms A, Fernandez-Vidal A, Bosch M, Schieber NL, et al. 2013. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J. Cell Biol. 203:985–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedi X, Ming Y, Yongping W, Yi Y, Xiaoxiang Z. 2009. Free cholesterol overloading induced smooth muscle cells death and activated both ER- and mitochondrial-dependent death pathway. Atherosclerosis 207:123–30 [DOI] [PubMed] [Google Scholar]
- Khor VK, Ahrends R, Lin Y, Shen WJ, Adams CM, et al. 2014. The proteome of cholesteryl-ester-enriched versus triacylglycerol-enriched lipid droplets. PLOS ONE 9:e105047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel AR, Sztalryd C. 2016. The perilipins: major cytosolic lipid droplet–associated proteins and their roles in cellular lipid storage, mobilization, and systemic homeostasis. Annu. Rev. Nutr. 36:471–509 [DOI] [PubMed] [Google Scholar]
- Klemm RW, Norton JP, Cole RA, Li CS, Park SH, et al. 2013. A conserved role for atlastin GTPases in regulating lipid droplet size. Cell Rep. 3:1465–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliwad SK, Streeper RS, Monetti M, Cornelissen I, Chan L, et al. 2010. DGAT1-dependent triacylglycerol storage by macrophages protects mice from diet-induced insulin resistance and inflammation. J. Clin. Investig. 120:756–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kory N, Farese RV Jr., Walther TC. 2016. Targeting fat: mechanisms of protein localization to lipid droplets. Trends Cell Biol. 26:535–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahmer N, Farese RV Jr., Walther TC. 2013a. Balancing the fat: lipid droplets and human disease. EMBO Mol. Med. 5:973–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahmer N, Guo Y, Wilfling F, Hilger M, Lingrell S, et al. 2011. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholine cytidylyltransferase. Cell Metab. 14:504–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahmer N, Hilger M, Kory N, Wilfling F, Stoehr G, et al. 2013b. Protein correlation profiles identify lipid droplet proteins with high confidence. Mol. Cell. Proteom. 12:1115–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuerschner L, Moessinger C, Thiele C. 2008. Imaging of lipid biosynthesis: how a neutral lipid enters lipid droplets. Traffic 9:338–52 [DOI] [PubMed] [Google Scholar]
- Kurat CF, Wolinski H, Petschnigg J, Kaluarachchi S, Andrews B, et al. 2009. Cdk1/Cdc28-dependent activation of the major triacylglycerol lipase Tgl4 in yeast links lipolysis to cell-cycle progression. Mol. Cell 33:53–63 [DOI] [PubMed] [Google Scholar]
- Lardizabal KD, Mai JT, Wagner NW, Wyrick A, Voelker T, Hawkins DJ. 2001. DGAT2 is a new diacylglycerol acyltransferase gene family: purification, cloning, and expression in insect cells of two polypeptides from Mortierella ramanniana with diacylglycerol acyltransferase activity. J. Biol. Chem. 276:38862–69 [DOI] [PubMed] [Google Scholar]
- Lee AH, Scapa EF, Cohen DE, Glimcher LH. 2008. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320:1492–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Li L, Lian J, Watts R, Nelson R, et al. 2015. Roles of acyl-CoA:diacylglycerol acyltransferases 1 and 2 in triacylglycerol synthesis and secretion in primary hepatocytes. Arterioscler. Thromb. Vasc. Biol. 35:1080–91 [DOI] [PubMed] [Google Scholar]
- Li C, Luo X, Zhao S, Siu GK, Liang Y, et al. 2017. COPI-TRAPPII activates Rab18 and regulates its lipid droplet association. EMBO J. 36:441–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, et al. 2012. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J. Clin. Investig. 122:4130–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Thiel K, Thul PJ, Beller M, Kuhnlein RP, Welte MA. 2012. Lipid droplets control the maternal histone supply of Drosophila embryos. Curr. Biol. 22:2104–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr., et al. 2003. Triglyceride accumulation protects against fatty acid–induced lipotoxicity. PNAS 100:3077–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Jiang Q, Wang X, Zhang Y, Lin RC, et al. 2014. Adipose-specific knockout of SEIPIN/BSCL2 results in progressive lipodystrophy. Diabetes 63:2320–31 [DOI] [PubMed] [Google Scholar]
- Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, et al. 2015. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160:177–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Ying Y, Zhao Y, Mundy DI, Zhu M, Anderson RG. 2004. Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J. Biol. Chem. 279:3787–92 [DOI] [PubMed] [Google Scholar]
- Liu Z, Li X, Ge Q, Ding M, Huang X. 2014. A lipid droplet–associated GFP reporter–based screen identifies new fat storage regulators in C. elegans. J. Genet. Genom. 41:305–13 [DOI] [PubMed] [Google Scholar]
- Lundin C, Nordstrom R, Wagner K, Windpassinger C, Andersson H, et al. 2006. Membrane topology of the human seipin protein. FEBS Lett. 580:2281–84 [DOI] [PubMed] [Google Scholar]
- Magre J, Delepine M, Khallouf E, Gedde-Dahl T Jr., Van Maldergem L, et al. 2001. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat. Genet. 28:365–70 [DOI] [PubMed] [Google Scholar]
- Martin S, Driessen K, Nixon SJ, Zerial M, Parton RG. 2005. Regulated localization of Rab18 to lipid droplets: effects of lipolytic stimulation and inhibition of lipid droplet catabolism. J. Biol. Chem. 280:42325–35 [DOI] [PubMed] [Google Scholar]
- Miranda DA, Kim JH, Nguyen LN, Cheng W, Tan BC, et al. 2014. Fat storage–inducing transmembrane protein 2 is required for normal fat storage in adipose tissue. J. Biol. Chem. 289:9560–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moessinger C, Klizaite K, Steinhagen A, Philippou-Massier J, Shevchenko A, et al. 2014. Two different pathways of phosphatidylcholine synthesis, the Kennedy Pathway and the Lands Cycle, differentially regulate cellular triacylglycerol storage. BMC Cell Biol. 15:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy S, Martin S, Parton RG. 2010. Quantitative analysis of lipid droplet fusion: inefficient steady state fusion but rapid stimulation by chemical fusogens. PLOS ONE 5:e15030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino N, Tamori Y, Tateya S, Kawaguchi T, Shibakusa T, et al. 2008. FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J. Clin. Investig. 118:2808–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon-Abell J, Obara CJ, Weigel AV, Li D, Legant WR, et al. 2016. Increased spatiotemporal resolution reveals highly dynamic dense tubular matrices in the peripheral ER. Science 354(6311):aaf3928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelkers P, Behari A, Cromley D, Billheimer JT, Sturley SL. 1998. Characterization of two human genes encoding acyl coenzyme A:cholesterol acyltransferase–related enzymes. J. Biol. Chem. 273:26765–71 [DOI] [PubMed] [Google Scholar]
- Ohsaki Y, Kawai T, Yoshikawa Y, Cheng J, Jokitalo E, Fujimoto T. 2016. PML isoform II plays a critical role in nuclear lipid droplet formation. J. Cell Biol. 212:29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagac M, Cooper DE, Qi Y, Lukmantara IE, Mak HY, et al. 2016. SEIPIN regulates lipid droplet expansion and adipocyte development by modulating the activity of glycerol-3-phosphate acyltransferase. Cell Rep.17:1546–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Blackstone C. 2010. Further assembly required: construction and dynamics of the endoplasmic reticulum network. EMBO Rep. 11:515–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul A, Chan L, Bickel PE. 2008. The PAT family of lipid droplet proteins in heart and vascular cells. Curr. Hypertens. Rep. 10:461–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne F, Lim K, Girousse A, Brown RJ, Kory N, et al. 2014. Mutations disrupting the Kennedy phosphatidyl-choline pathway in humans with congenital lipodystrophy and fatty liver disease. PNAS 111:8901–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploegh HL. 2007. A lipid-based model for the creation of an escape hatch from the endoplasmic reticulum. Nature 448:435–38 [DOI] [PubMed] [Google Scholar]
- Qi J, Lang W, Geisler JG, Wang P, Petrounia I, et al. 2012. The use of stable isotope-labeled glycerol and oleic acid to differentiate the hepatic functions of DGAT1 and −2. J. Lipid Res. 53:1106–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Cohen S, Lippincott-Schwartz J. 2015. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 32:678–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renvoise ˊ B, Malone B, Falgairolle M, Munasinghe J, Stadler J, et al. 2016. Reep1 null mice reveal a converging role for hereditary spastic paraplegia proteins in lipid droplet regulation. Hum. Mol. Genet. 25:5111–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robenek H, Hofnagel O, Buers I, Robenek MJ, Troyer D, Severs NJ. 2006. Adipophilin-enriched domains in the ER membrane are sites of lipid droplet biogenesis. J. Cell Sci. 119:4215–24 [DOI] [PubMed] [Google Scholar]
- Robenek MJ, Severs NJ, Schlattmann K, Plenz G, Zimmer KP, et al. 2004. Lipids partition caveolin-1 from ER membranes into lipid droplets: updating the model of lipid droplet biogenesis. FASEB J. 18:866–68 [DOI] [PubMed] [Google Scholar]
- Roingeard P, Melo RC. 2017. Lipid droplet hijacking by intracellular pathogens. Cell Microbiol. 19:e12688 [DOI] [PubMed] [Google Scholar]
- Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, et al. 2008. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 40:1461–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe ER, Mimmack ML, Barbosa AD, Haider A, Isaac I, et al. 2016. Conserved amphipathic helices mediate lipid droplet targeting of perilipins 1–3. J. Biol. Chem. 291:6664–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Arai E, Maekawa K, Ishikawa M, Fujimoto H, et al. 2016. Lipidomic signatures and associated transcriptomic profiles of clear cell renal cell carcinoma. Sci. Rep. 6:28932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salo VT, Belevich I, Li S, Karhinen L, Vihinen H, et al. 2016. Seipin regulates ER–lipid droplet contacts and cargo delivery. EMBO J. 35:2699–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Ploier B, Koch B, Daum G. 2013. Analysis of yeast lipid droplet proteome and lipidome. Methods Cell Biol. 116:15–37 [DOI] [PubMed] [Google Scholar]
- Schrul B, Kopito RR. 2016. Peroxin-dependent targeting of a lipid-droplet-destined membrane protein to ER subdomains. Nat. Cell Biol. 18:740–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer Y, Shemesh T, Kozlov MM. 2015. A model for shaping membrane sheets by protein scaffolds. Biophys. J. 109:564–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y, Hu J, Kozlov MM, Rapoport TA. 2009. Mechanisms shaping the membranes of cellular organelles. Annu. Rev. Cell Dev. Biol. 25:329–54 [DOI] [PubMed] [Google Scholar]
- Shibata Y, Shemesh T, Prinz WA, Palazzo AF, Kozlov MM, Rapoport TA. 2010. Mechanisms determining the morphology of the peripheral ER. Cell 143:774–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y, Voeltz GK, Rapoport TA. 2006. Rough sheets and smooth tubules. Cell 126:435–39 [DOI] [PubMed] [Google Scholar]
- Shockey JM, Gidda SK, Chapital DC, Kuan JC, Dhanoa PK, et al. 2006. Tung tree DGAT1 and DGAT2 have nonredundant functions in triacylglycerol biosynthesis and are localized to different subdomains of the endoplasmic reticulum. Plant Cell 18:2294–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim MF, Talukder MU, Dennis RJ, Edwardson JM, Rochford JJ. 2014. Analyzing the functions and structure of the human lipodystrophy protein seipin. Methods Enzymol. 537:161–75 [DOI] [PubMed] [Google Scholar]
- Smagris E, BasuRay S, Li J, Huang Y, Lai KM, et al. 2015. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 61:108–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SJ, Cases S, Jensen DR, Chen HC, Sande E, et al. 2000. Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat. Nat. Genet. 25:87–90 [DOI] [PubMed] [Google Scholar]
- Soni KG, Mardones GA, Sougrat R, Smirnova E, Jackson CL, Bonifacino JS. 2009. Coatomer-dependent protein delivery to lipid droplets. J. Cell Sci. 122:1834–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone SJ, Levin MC, Farese RV Jr. 2006. Membrane topology and identification of key functional amino acid residues of murine acyl-CoA:diacylglycerol acyltransferase-2. J. Biol. Chem. 281:40273–82 [DOI] [PubMed] [Google Scholar]
- Stone SJ, Levin MC, Zhou P, Han J, Walther TC, Farese RV Jr. 2009. The endoplasmic reticulum enzyme DGAT2 is found in mitochondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. J. Biol. Chem. 284:5352–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone SJ, Myers HM, Watkins SM, Brown BE, Feingold KR, et al. 2004. Lipopenia and skin barrier abnormalities in DGAT2-deficient mice. J. Biol. Chem. 279:11767–76 [DOI] [PubMed] [Google Scholar]
- Streeper RS, Grueter CA, Salomonis N, Cases S, Levin MC, et al. 2012. Deficiency of the lipid synthesis enzyme, DGAT1, extends longevity in mice. Aging 4:13–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Murakami T, Cheng J, Kano H, Fukata M, Fujimoto T. 2015. ELMOD2 is anchored to lipid droplets by palmitoylation and regulates adipocyte triglyceride lipase recruitment. Mol. Biol. Cell 26:2333–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski KM, Binns D, Bartz R, Grishin NV, Li WP, et al. 2007. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. PNAS 104:20890–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiam AR, Antonny B, Wang J, Delacotte J, Wilfling F, et al. 2013a. COPI buds 60-nm lipid droplets from reconstituted water-phospholipid-triacylglyceride interfaces, suggesting a tension clamp function. PNAS 110:13244–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiam AR, Farese RV Jr., Walther TC. 2013b. The biophysics and cell biology of lipid droplets. Nat. Rev. Mol. Cell Biol. 14:775–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiam AR, Foret L. 2016. The physics of lipid droplet nucleation, growth and budding. Biochim. Biophys. Acta 1861:715–22 [DOI] [PubMed] [Google Scholar]
- Villanueva CJ, Monetti M, Shih M, Zhou P, Watkins SM, et al. 2009. Specific role for acyl CoA:diacylglycerol acyltransferase 1 (Dgat1) in hepatic steatosis due to exogenous fatty acids. Hepatology 50:434–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. 2006. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 124:573–86 [DOI] [PubMed] [Google Scholar]
- Walther TC, Farese RV Jr. 2012. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 81:687–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Becuwe M, Housden BE, Chitraju C, Porras AJ, et al. 2016. Seipin is required for converting nascent to mature lipid droplets. eLife 5:e16582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Lei M, Hsia RC, Sztalryd C. 2013. Analysis of lipid droplets in cardiac muscle. Methods Cell Biol. 116:129–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss SB, Kennedy EP, Kiyasu JY. 1960. The enzymatic synthesis of triglycerides. J. Biol. Chem. 235:40–44 [PubMed] [Google Scholar]
- Welte MA. 2015. Expanding roles for lipid droplets. Curr. Biol. 25:R470–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker R, Loy PA, Sisman E, Suyama E, Aza-Blanc P, et al. 2010. Identification of microRNAs that control lipid droplet formation and growth in hepatocytes via high-content screening. J. Biomol. Screen.15:798–805 [DOI] [PubMed] [Google Scholar]
- Wilfling F, Haas JT, Walther TC, Farese RV Jr. 2014a. Lipid droplet biogenesis. Curr. Opin. Cell Biol. 29:39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfling F, Thiam AR, Olarte MJ, Wang J, Beck R, et al. 2014b. Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. eLife 3:e01607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfling F, Wang H, Haas JT, Krahmer N, Gould TJ, et al. 2013. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev. Cell 24:384–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolinski H, Hofbauer HF, Hellauer K, Cristobal-Sarramian A, Kolb D, et al. 2015. Seipin is involved in the regulation of phosphatidic acid metabolism at a subdomain of the nuclear envelope in yeast. Biochim. Biophys. Acta 1851:1450–64 [DOI] [PubMed] [Google Scholar]
- Wurie HR, Buckett L, Zammit VA. 2012. Diacylglycerol acyltransferase 2 acts upstream of diacylglycerol acyltransferase 1 and utilizes nascent diglycerides and de novo synthesized fatty acids in HepG2 cells. FEBS J. 279:3033–47 [DOI] [PubMed] [Google Scholar]
- Yang H, Bard M, Bruner DA, Gleeson A, Deckelbaum RJ, et al. 1996. Sterol esterification in yeast: a two-gene process. Science 272:1353–56 [DOI] [PubMed] [Google Scholar]
- Ye J, Li JZ, Liu Y, Li X, Yang T, et al. 2009. Cideb, an ER- and lipid droplet–associated protein, mediates VLDL lipidation and maturation by interacting with apolipoprotein B. Cell Metab. 9:177–90 [DOI] [PubMed] [Google Scholar]
- Yen CL, Farese RV Jr. 2003. MGAT2, a monoacylglycerol acyltransferase expressed in the small intestine. J. Biol. Chem. 278:18532–37 [DOI] [PubMed] [Google Scholar]
- Yen CL, Monetti M, Burri BJ, Farese RV Jr. 2005. The triacylglycerol synthesis enzyme DGAT1 also catalyzes the synthesis of diacylglycerols, waxes, and retinyl esters. J. Lipid Res. 46:1502–11 [DOI] [PubMed] [Google Scholar]
- Yen CL, Stone SJ, Cases S, Zhou P, Farese RV Jr. 2002. Identification of a gene encoding MGAT1, a monoacylglycerol acyltransferase. PNAS 99:8512–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen CL, Stone SJ, Koliwad S, Harris C, Farese RV Jr. 2008. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 49:2283–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Wang H, Zhao J, Yuan Y, Wang C, et al. 2013. Expression of CIDE proteins in clear cell renal cell carcinoma and their prognostic significance. Mol. Cell. Biochem. 378:145–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wang Y, Li J, Yu J, Pu J, et al. 2011. Proteome of skeletal muscle lipid droplet reveals association with mitochondria and apolipoprotein A-I. J. Proteome Res. 10:4757–68 [DOI] [PubMed] [Google Scholar]
- Zhang P, Na H, Liu Z, Zhang S, Xue P, et al. 2012. Proteomic study and marker protein identification of Caenorhabditis elegans lipid droplets. Mol. Cell. Proteom. 11:317–28 [DOI] [PMC free article] [PubMed] [Google Scholar]