

Abstract
Soft tissue tumours of the elbow are mostly benign. Malignant tumours in this area, although uncommon, often present unique clinical and histopathological characteristics that are helpful for diagnosis.
Management of soft tissue tumours around the elbow may be challenging because of their rarity and the proximity to neurovascular structures. Careful staging, histological diagnosis and treatment are essential to optimize clinical outcome. A missed or delayed diagnosis or an improperly executed biopsy may have devastating consequences for the patient.
This article reviews the most common benign and malignant soft tissue tumours of the elbow and discusses the clinicopathological findings, imaging features and current therapeutic concepts.
Cite this article: EFORT Open Rev 2019;4:668-677. DOI: 10.1302/2058-5241.4.190002
Keywords: benign, elbow, malignant, soft tissue, tumours
Introduction
Soft tissue tumours around the elbow are rare, with an incidence of around 3.8% of all soft tissue tumours.1 Benign soft tissue tumours occur approximately 10 times more frequently than malignant ones.2 Nevertheless, although the clinical presentation of the most frequent lesions might be straightforward, it can often be difficult to differentiate benign and reactive lesions from malignant and aggressive ones on purely clinical grounds. Thus, it is important for the clinician to be aware of the wide variety of these lesions and treat appropriately or refer to a specialist centre. When a lesion raises suspicions, it should not be only treated with excisional biopsy, as this approach might lead to errors which are difficult to remedy. Where there is doubt after initial assessment, this should prompt referral to a specialized tumour centre, where an appropriate biopsy and pre-operative and reconstructive planning should take place prior to undertaking any treatment. This article reviews the most common benign and malignant soft tissue tumours of the elbow and discusses the clinicopathological findings, imaging characteristics and current concepts of treatment.
Benign soft tissue tumours of the elbow
Lipomata (Lipomas)
Lipomas are palpable, mobile, painless masses that are either superficial or deep to the fascia and in the elbow and represent 5.2% of all lipomas.1 Deep lesions are difficult to evaluate with ultrasound and a magnetic resonance imaging (MRI) scan is required. Although lipomas are benign in nature, lipomatous lesions that are deep to the fascia could be intra or inter-muscular lipomas, or atypical lipomatous tumours such as well-differentiated lipomas like liposarcomas with amplification of the MDM2 gene.3 On MRI, a lipoma presents as a homogeneous non-enhancing fatty mass.4 The T1 and T2-weighted MRI images show high signal intensity, whereas low signal intensity is seen on Short Tau Inversion Recovery (STIR) images or fat saturated sequences (Fig. 1A–C). Biopsy is not necessary in most cases. ‘Watchful waiting’ may be satisfactory for small or asymptomatic lipomas but large tumours which manifest with pain and/or limitation of function justify marginal excision.
Fig. 1.

Forty-five-year-old female complaining of a painless mass at the front of the elbow area which proved to be an intramuscular lipoma. (AB) Anteroposterior and lateral radiograph of the elbow demonstrating a well circumscribed mass at the anterior surface of the elbow. (C) Magnetic resonance imaging contrast-enhanced sagittal T1 sequences of the mass measuring 4.5 x 2.7 cm.
Synovial osteochondromatosis
Synovial osteochondromatosis is a monoarticular metaplastic proliferative disorder of the synovium characterized by the formation of multiple cartilaginous nodules in the synovium, many of which detach creating loose bodies. When the lesion occurs in the upper limb it has a predilection for the elbow followed by the shoulder.5–8 Symptoms include diffuse joint discomfort and decreased elbow range of motion with a sensation of joint locking or catching. Large intra or extra-articular calcified cartilaginous masses, which are formed by the fusion of multiple synovial chondromas or due to the growth of a solitary synovial chondroma, have been described as ‘giant solitary synovial osteochondromatosis’.9 The last may cause ulnar nerve neuropathy due to nerve compression.6,10,11 The diagnosis is based on plain radiographs of the elbow which show multiple oval, well defined, intra-articular calcified loose bodies that are present in up to 66% of cases.12,13 If radiolucent, these lesions may be detected via ultrasonography, arthrography, CT, arthro-CT or MRI.12,13 Differential diagnosis includes chronic articular infection, osteoarthritis, tenosynovial giant cell tumour (TGCT), monoarticular inflammatory arthritis and synovial sarcoma.14 The treatment consists of open or arthroscopic synovectomy with removal of loose bodies.15,16 Open radical synovectomy of the elbow joint requires a circumferential approach through medial/lateral or anterior/posterior surgical procedures.7 Currently, arthroscopic synovectomy with loose-body removal via two anterior (medial/lateral) and two posterior (posterior/posterolateral) portals is a safe and effective option, resulting in low disease recurrence, low morbidity and early return to activities.17,18 The mainstay of treatment should be complete removal of the synovium, otherwise recurrences may occur in up to 22% of cases and in these cases synovectomy must be repeated.17 Although this is a benign tumour, transformation to chondrosarcoma has been reported in up to 5% of cases, especially when periosteal reaction and cortical erosion are present.19
Tenosynovial giant cell tumour
Another monoarticular benign but locally aggressive synovial neoplastic process of the elbow is the diffuse type of tenosynovial giant cell tumour (TGCT) or pigmented villonodular synovitis (PVNS). As a term, PVNS is no longer used by the World Health Organization.20 The tumour can be localized or diffuse and is rarely malignant.21 TGCT is associated with characteristic cytogenetic abnormalities resulting in the overexpression of the CSF1 gene.22 It commonly affects adults in their third or fourth decade of life.23 Although the knee is most frequently involved, the shoulder, followed by the elbow are the most common sites of occurrence in cases of upper limb involvement.24 Approximately 0.8% of all TGCT are located in the elbow.1,25–31 Pain, stiffness, recurrent effusion, functional impairment and posterior interosseous nerve palsy are the most common clinical presentations.32 MRI with signal attenuation by haemosiderin, results in low signal intensity on both T1 and T2-weighted sequences and has a positive predictive value of almost 85% (Fig. 2).33,34 A biopsy is always required for definitive diagnosis. Optimal treatment of TGCT of the elbow remains controversial.31 Complete open or arthroscopic synovectomy is the currently recommended.35 Unfortunately, the incidence of local recurrence after synovectomy ranges from 9% to 44%36 and destruction of the elbow joint37,38 with secondary arthritis remains a common outcome. Because of the anatomical complexity of the elbow joint, complete resection of the synovium is often challenging and, in order to minimize the risk of recurrence, adjuvant external beam radiation therapy (RT)39,40 is sometimes recommended as an adjuvant therapy, mainly after incomplete resection or in case of renewed progression after failure of multiple previous procedures. In select cases, instillation of intra-articular radioactive colloid such as 90-yttrium41 can be a safe and potentially effective local adjuvant treatment; however, further studies are required to assess long-term outcomes.31,42 Neo-adjuvant systemic targeted therapy targeting the CSF1/CSF1R axis (imatinib)43 or other tyrosine kinase inhibitors such as nilotinib, emactuzumab44 and pexidartinib (PLX3397) have been tested in patients with locally advanced or relapsed disease, particularly when RT is contraindicated.45
Fig. 2.
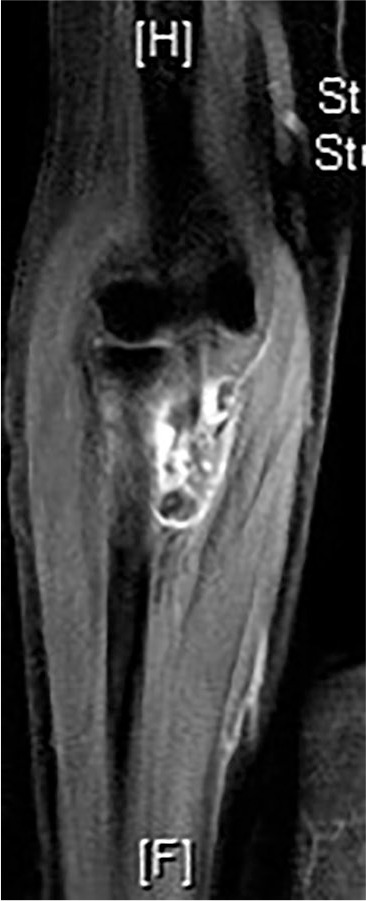
Tenosynovial giant cell tumour (TGCT) diffuse type in a 74-year-old female. Coronal sequence magnetic resonance imaging of the elbow/upper forearm.
Desmoid tumours
Desmoid tumours are benign but locally aggressive fibroblastic neoplasms that arise sporadically due to mutations leading to increased beta-catenin protein level and activity.46,47 These tumours mostly occur in the proximal part of the limbs. In the elbow, the incidence of desmoid tumours is approximately 3%.1 The progress of these tumours is not fully understood48 and while some spontaneously regress, others can growth quickly but do not metastasize. Little is known about the prognostic factors that can differentiate between indolent and aggressive cases; however, mutations in the beta-catenin gene have been shown to have prognostic value and may predict the risk of recurrence.47 The recommended treatment varies depending on tumour aggressiveness and location. Treatment consists of surgery, cryo-ablation,49 isolated limb perfusion,50,51 RT52 and pharmacological therapy: anti-inflammatory medication such as sulindac or other non-steroidal anti-inflammatory drugs such as celecoxib,53 tamoxifen,53 interferon-alpha,54 cytotoxic chemotherapy such as methotrexate,55, vinblastine,55 doxorubicin,56 and targeted therapies with tyrosine kinase inhibitors such as imatinib,57 sunitinib, pazopanib and sorafenib.58,59 The infiltrative nature of desmoid tumours and the fact that they frequently lack a pseudocapsule makes it difficult to determine the true extent of disease at the time of excision. In the upper extremity, the ability to achieve adequate surgical margins can lead to patient morbidity due to the removal of nerves, tendons, and other vital structures. Factors associated with increased risk of local recurrence (LR) after surgery are younger age, positive margins,60 tumour located in the extremities and a large tumour size.61 Regarding positive margins though, they do not lead inevitably to LR, since many tumours recur after wide resection whereas after incomplete resection disease may remain stable for many years.62 If the tumour relapses or the surgical margins are positive, RT may be utilized and reduce the risk of further recurrence.52 However, a consensus regarding the optimal treatment approach is lacking.63 Surgical resection is nowadays less frequently used; instead, an initial period of observation is recommended when the patient is asymptomatic and the tumour is not progressing.59,62,64
Schwannomas
Schwannomas also called neurilemmomas or neurinomas, are benign nerve sheath tumours originating from Schwann cells. Most schwannomas are single sporadic benign neoplasms; however, sometimes they can be part of multiple schwannomatosis, regarded as the third type of neurofibromatosis.65 They are the commonest tumours of peripheral nerves and their incidence in the upper limb is 19% of all schwannomas, with approximately 5.2% occurring in the elbow.(1,66) They present as painless swellings and usually remain asymptomatic for a few years. They occur more frequently in mixed nerves rather than pure sensory or motor nerves. Most schwannomas can be diagnosed clinically. Schwannomas are mobile firm masses in the longitudinal plane, along the course of the involved nerve.67 They may cause symptoms with or without a Tinel’s sign upon percussion of the tumour, if the affected nerve is a sensory or a mixed nerve.68 On imaging, the tumour may cause bone scalloping and appears longitudinal along the course of a peripheral nerve (Fig. 3A-C).69 A specific finding in the MRI is the ‘target’ sign due to the difference in signal intensity between the periphery and central portion of the mass which corresponds to the biphasic histological pattern (Antoni A and B areas).70 Surgery, as a ‘shell out’ procedure/marginal excision is all that is required in symptomatic schwannomas. Despite the classical description that schwannomas are well encapsulated and can be completely enucleated during excision without producing a neurological deficit, a percentage of them have fascicular involvement and could not be completely shelled out.71,72 Even with meticulous dissection, removal of a tumour without damaging any fascicles can be technically difficult and increases the risk of transient or permanent neurological damage. However, the threshold for iatrogenic injury during surgical dissection of a peripheral nerve tends to be lower in the upper limb than in the trunk or lower limbs.72
Fig. 3.

Elbow schwannoma in a 58-year-old female. Magnetic resonance imaging showing the 1.3 x 1.3 x 3.6 cm mass abutting the proximal ulna. (A) Axial T1. (B) Axial T1 with contrast. (C) Sagittal T2 fat saturated sequence of the mass.
Haemangiomata (Haemangiomas)
Haemangiomas are unusual benign soft tissue tumours originating from vascular tissues, that can occur in the elbow. The most common type of haemangioma of the limbs is the intramuscular variant which affects mainly adolescents and young adults. They may cause pain and fluctuate in size over time.73 On imaging, calcifications may be observed (phleboliths) whereas on an MRI scan they appear homogeneous and lobulated (‘bunch of grapes’).74 Symptoms can be aggravated following excision and a conservative approach is usually advocated. However, in cases of outsized tumours that may cause symptoms, excision or embolization is recommended.75
Malignant soft tissue tumours of the elbow
The incidence of soft tissue sarcomas (STS) in the upper extremity is approximately 15–30%,2,76,77 and the elbow is involved in less than 1% of cases.78–83 The most common STS subtypes encountered in the elbow are the synovial sarcoma, myxofibrosarcoma and undifferentiated pleomorphic sarcoma (formerly known as malignant fibrous histiocytoma).76,84,85 The clinical behaviour of STS is frequently misleading and the diagnosis may be delayed due to the characteristic slow growth of the tumours, which also remain asymptomatic for a long time.86 However, when these tumours are located in the elbow, symptoms arise earlier due to the anatomical proximity of the tumours to neurovascular structures which are compressed.80 Contrast-enhanced MRI of the elbow is the diagnostic method of choice for STS but biopsy is also required as a confirmatory test. Because STS often metastasize to the lung, a spiral computerized tomography (CT) scan of the thorax is included in the diagnostic work-up. The positron emission tomography (18Fluoro-D-Glucose Positron Emission Tomography (PET)) has not been established as standard imaging modality59,78 but it is useful to evaluate the response of the tumour to neo-adjuvant treatment.87
The treatment of STS of the elbow is challenging since it is difficult to balance the necessity for resection in ‘safe’ oncological margins while preserving the native anatomical relationships of the structures as well as their function. STS of the upper extremities are twice as likely to undergo unplanned excision compared to those occurring in the lower extremities, probably because of their smaller size and their superficial location.76,88 Consequently, residual disease and local recurrence is also encountered more often.89 Although in the past high-grade sarcomas of the elbow were treated with amputation due to high rates of LR, currently limb-sparing surgery can be performed in more than 90% of patients.90
The most established adjuvant treatments for STS are chemotherapy and radiation therapy (RT). Whether chemotherapy is beneficial for the treatment of STS is controversial.91 Newer agents such as Trabectedin,92 Eribulin,93 and targeted therapies such as Oralatumab that blocks PDGFR-a,94 Pazobanib a tyrosine kinase inhibitor,95 Sunitinib/Sorafenib,96 other mTOR inhibitors such as Sirolimus/Terserolimus97 and Palbociclib a cyclin-dependent kinase inhibitor,98 are promising. In contrast, RT is an established treatment modality in combination with en bloc tumour excision. Radiation therapy significantly reduces the likelihood of LR, except in cases with small, low grade, superficial tumours. It may be given pre or post-operatively with similar oncological results but different toxicities: pre-operative RT that utilizes a smaller radiation field and smaller dose (around 50 Gy) has a higher wound complication rate (around 35%); post-operative RT that utilizes larger fields (to accommodate for the surgical manoeuvres) and higher doses (60–66 Gy) is associated with reduced wound complications (around 17%) but higher late toxicity (limb oedema and fibrosis).99,100 Newer cutting-edge modalities, such as intensity modulated RT (IMRT) or particle RT (proton or carbon ion) maximize efficacy while minimizing toxicity to the surrounding tissues, and may gradually replace the traditional techniques.101
Synovial sarcoma
Synovial sarcoma is one of the most common soft tissue sarcomas in the elbow. In the upper extremity these sarcomas have and incidence rate of 16–25%.76,102 They may be encountered either as an extra-articular or intra-articular tumour and typically affect adolescents and young adults.102 The cytogenetic abnormality t(X:18)(p11;q11) is found in 95% of patients with synovial sarcoma.103 On imaging studies, findings such as a slow calcified growing soft tissue mass near but not in the elbow joint in a young patient suggest the presence of this tumour. The tumour size, location, patient age, and presence of poorly differentiated areas determine the prognosis in patients with these lesions. Tumours located in the upper extremity have more favourable prognosis compared with those in the head and neck, axial or lower extremity lesions,104 along with those in patients younger than 15–20 years.105 The presence of extensive calcification suggests improved long-term survival.106 There is considerable controversy regarding the prognostic significance of tumour cell type (monophasic or biphasic). The gene fusion type SYT-SSX2 (more common in monophasic lesions) has been associated with a better prognosis and an 89% metastasis-free survival compared with that for SYT-SSX1.(107–109) The gold standard treatment for synovial sarcoma is wide tumour resection (Fig. 4A-H) plus chemoradiation because it is a chemosensitive tumour.110,111 Newer agents have been successfully administered such as trabectedin112 and some centres advocate neo-adjuvant treatment.62,113 The clinical course of synovial sarcoma is characterized by a high LR rate of 30–50% and marginal excision is associated with even higher rates.114 Metastatic disease is observed in approximately 41% of patients115 with the majority of metastases occurring within the first two to five years after treatment. The most frequent metastatic site is the lung, followed by lymph nodes (4–18%) and bone (8–11%).107,116,117 The five-year survival rate of patients with synovial sarcoma ranges from 36% to 76%. At 10 years, the survival rate has been reported to range from 20% to 63%.115
Fig. 4.

A 64-year-old man presented with a small painful mass of his left elbow. (A) Sagittal T1-weighted magnetic resonance imaging (MRI) showing a well circumscribed mass with homogenous intensity. (B) Sagittal T2 Short Tau Inversion Recovery (STIR) MRI. (C) Axial T1 fat saturated contrast MRI. (D) Intra-operative image following wide excision of the tumour, including the proximal part of the ulna attached to the tumour. The biopsy confirmed the diagnosis of synovial sarcoma. (E) Post-operative radiographs after tumour resection. However, a pathological fracture of the proximal ulna occurred secondary to radiation therapy. (F) Anteroposterior and (G) lateral elbow radiographs following open reduction and internal fixation of the pathological fracture of the ulna with two plates. (H) Post-operative axial T1 MRI with contrast showing no signs of recurrence three years post-operatively.
Myxofibrosarcoma
Another STS frequent in the elbow area is the myxofibrosarcoma. These sarcomas are malignant fibroblastic tumours with myxoid stroma. They appear more frequently in the sixth to eight decades with a slight male predominance. There are encountered more frequently in the upper extremity than other STSs (such as leiomyosarcoma) with a frequency of between 22–32%118,119 and the elbow is involved in approximately 3% of cases.118,120 Half of them arise subcutaneously, often associated with unplanned excision.121,122 The most striking feature is high incidence of LR: between 30–60% in many series (around 15% in some modern series).118,119 This can probably be attributed to a highly infiltrative pattern and growth along fascial planes, seen on MRI scans as a high signal ‘tail’.121,123 Indeed, high incidence of positive margins (43% with microscopic spread of up to 29 mm beyond macroscopic tumour) has been reported.119 Therefore wide excision with further re-excision if needed is of utmost importance. All of the high signal area in the MRI should be resected if possible, leading to significant soft tissue defects.119,124 Reconstruction/flap coverage should probably be carried out in a delayed fashion as a second-stage procedure, after ensuring that margins are negative.119,125 Peri-operative RT is also very important. The radiation field should include the initial tumour plus adjacent suspicious tissues; reducing field size may not be advisable in this particular tumour.118,119,121
Undifferentiated pleomorphic sarcoma
Undifferentiated pleomorphic sarcoma, formerly known as Malignant Fibrous Histiocytoma (MFH) mainly occurs in the upper extremities in a frequency of 19%, with the region of the elbow most commonly affected.117 It is the second most common high-grade STS among adults between 60 and 70 years old, and frequently presents as a slow growing painless and enlarging mass.126 Wide resection with negative margins plus peri-operative chemo and RT achieves optimal results for local control and overall survival.127
Conclusions
Soft tissue tumours around the elbow are rare. Although the characteristics and treatment of benign lesions such as lipoma are relatively straightforward, synovial disorders such as synovial chondromatosis and TGCT remain difficult, with high incidence of local recurrence even after complete synovectomy. Soft tissue sarcomas in the elbow are sometimes misdiagnosed and the treatment is delayed. Limb salvage surgery is the treatment of choice for malignant soft tissue tumours of the elbow. Unfortunately, in many cases, resection in ‘safe’ oncological margins results in significant compromise of the function of the forearm and hand. Following wide tumour resection, adjuvant therapies may lessen local recurrence, but the effect on overall survival rate remains unclear. The role of chemotherapy in the treatment of malignant soft tissue tumours of the elbow is controversial. By contrast, RT is an established therapeutic modality in the managements of these lesions.
Footnotes
OA licence text: This article is distributed under the terms of the Creative Commons Attribution-Non Commercial 4.0 International (CC BY-NC 4.0) licence (https://creativecommons.org/licenses/by-nc/4.0/) which permits non-commercial use, reproduction and distribution of the work without further permission provided the original work is attributed.
ICMJE Conflict of interest statement: The authors declare no conflict of interest relevant to this work.
Funding statement
No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
References
- 1. Picci P, Vanel D, Gambarotti M, Ruggieri P, Ferrari S. Epidemiology. In: Picci P, Vanel D, Gambarotti M, Fabbri N, Manfrini M, eds. Atlas of musculoskeletal tumors and tumorlike lesions: the Rizzoli case archive. Cham: Springer, 2014:259–269. [Google Scholar]
- 2. Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Altekruse SF, et al. SEER Cancer Statistics Review, 1975–2013. Bethesda: National Cancer Institute, MD2016, updated 3/5/2017, http://seer.cancer.gov/csr/1975_2013/ (date last accessed April 2016).
- 3. Thway K, Wang J, Swansbury J, Min T, Fisher C. Fluorescence in situ hybridization for MDM2 amplification as a routine ancillary diagnostic tool for suspected well-differentiated and dedifferentiated liposarcomas: experience at a tertiary center. Sarcoma 2015;2015:812089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gaskin CM, Helms CA. Lipomas, lipoma variants, and well-differentiated liposarcomas (atypical lipomas): results of MRI evaluations of 126 consecutive fatty masses. AJR Am J Roentgenol 2004;182:733–739. [DOI] [PubMed] [Google Scholar]
- 5. Terra BB, Moraes EW, de Souza AC, Cavatte JM, Teixeira JC, De Nadai A. Arthroscopic treatment of synovial osteochondromatosis of the elbow. Case report and literature review. Rev Bras Ortop 2015;50:607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Al-Najjim M, Mustafa A, Fenton C, Morapudi S, Waseem M. Giant solitary synovial osteochondromatosis of the elbow causing ulnar nerve neuropathy: a case report and review of literature. J Brachial Plex Peripher Nerve Inj 2013;8:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Giannetti S, Santucci A, Patricola A, Stancati A, Di Sanzo V. Neglected synovial osteochondromatosis of the elbow: a rare case. World J Surg Oncol 2013;11:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kamineni S, O’Driscoll SW, Morrey BF. Synovial osteochondromatosis of the elbow. J Bone Joint Surg Br 2002;84:961–966. [DOI] [PubMed] [Google Scholar]
- 9. Edeiken J, Edeiken BS, Ayala AG, Raymond AK, Murray JA, Guo SQ. Giant solitary synovial chondromatosis. Skeletal Radiol 1994;23:23–29. [DOI] [PubMed] [Google Scholar]
- 10. Kim CH, Kim SH, Kim MS, Chang CH. Cubital tunnel syndrome, associated with synovial chondromatosis. J Korean Neurosurg Soc 2008;43:109–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ruth RM, Groves RJ. Synovial osteochondromatosis of the elbow presenting with ulnar nerve neuropathy. Am J Orthop (Belle Mead NJ) 1996;25:843–844. [PubMed] [Google Scholar]
- 12. Campeau NG, Lewis BD. Ultrasound appearance of synovial osteochondromatosis of the shoulder. Mayo Clin Proc 1998;73:1079–1081. [DOI] [PubMed] [Google Scholar]
- 13. Rao JP, Spingola C, Mastromonaco E, Villacin A. Synovial osteochondromatosis. Computerized axial tomography, frozen section and arthrography in diagnosis and management. Orthop Rev 1986;15:245–248. [PubMed] [Google Scholar]
- 14. Shanbhag A, Balakrishnan A, Bhaduri R, Chinoy R. Primary synovial osteochondromatosis. J Indian Rheumatol Assoc 2004;12:29–30. [Google Scholar]
- 15. Ranalletta M, Bongiovanni S, Calvo JM, Gallucci G, Maignon G. Arthroscopic treatment of synovial chondromatosis of the shoulder: report of three patients. J Shoulder Elbow Surg 2009;18:e4–e8. [DOI] [PubMed] [Google Scholar]
- 16. Shpitzer T, Ganel A, Engelberg S. Surgery for synovial chondromatosis: 26 cases followed up for 6 years. Acta Orthop Scand 1990;61:567–569. [DOI] [PubMed] [Google Scholar]
- 17. Byrd JW. Arthroscopy of the elbow for synovial chondromatosis. J South Orthop Assoc 2000;9:119–124. [PubMed] [Google Scholar]
- 18. Griesser MJ, Harris JD, Likes RL, Jones GL. Synovial chondromatosis of the elbow causing a mechanical block to range of motion: a case report and review of the literature. Am J Orthop (Belle Mead NJ) 2011;40:253–256. [PubMed] [Google Scholar]
- 19. Wittkop B, Davies AM, Mangham DC. Primary synovial chondromatosis and synovial chondrosarcoma: a pictorial review. Eur Radiol 2002;12:2112–2119. [DOI] [PubMed] [Google Scholar]
- 20. Fletcher CDM, Unni KK, Mertens F. Editorial, Consensus C. In: Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press, 2006. [Google Scholar]
- 21. Sistla R, Vidyasagar JVS, Afroz T. Malignant pigmented villonodular synovitis: a rare entity. J Orthop Case Rep 2014;4:9–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Molena B, Sfriso P, Oliviero F, et al. Synovial colony-stimulating factor-1 mRNA expression in diffuse pigmented villonodular synovitis. Clin Exp Rheumatol 2011;29:547–550. [PubMed] [Google Scholar]
- 23. DiCaprio MR, Damron TA, Stadnick M, Fuller C. Pigmented villonodular synovitis of the elbow: a case report and literature review. J Hand Surg Am 1999;24:386–391. [DOI] [PubMed] [Google Scholar]
- 24. Abdelwahab IF, Kenan S, Steiner GC, Abdul-Quader M. True bursal pigmented villonodular synovitis. Skeletal Radiol 2002;31:354–358. [DOI] [PubMed] [Google Scholar]
- 25. Aydingöz U, Leblebicioglu G, Gedikoglu G, Atay OA. Pigmented villonodular synovitis of the elbow in a 6-year-old girl. J Shoulder Elbow Surg 2002;11:274–277. [DOI] [PubMed] [Google Scholar]
- 26. Geiger EV, Reize P, Wehrmann M, Wülker N. Radial and ulnar neuropathy due to pigmented villonodular synovitis of the elbow. J Shoulder Elbow Surg 2006;15:e8–e10. [DOI] [PubMed] [Google Scholar]
- 27. Koto K, Murata H, Sakabe T, et al. Magnetic resonance imaging and thallium-201 scintigraphy for the diagnosis of localized pigmented villonodular synovitis arising from the elbow: a case report and review of the literature. Exp Ther Med 2013;5:1277–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miller WE. Villonodular synovitis: pigmented and nonpigmented variations. South Med J 1982;75:1084–1086, 92. [PubMed] [Google Scholar]
- 29. Schwartz HS, Unni KK, Pritchard DJ. Pigmented villonodular synovitis: a retrospective review of affected large joints. Clin Orthop Relat Res 1989;247:243–255. [PubMed] [Google Scholar]
- 30. Sekiya H, Ozawa H, Sugimoto N, Kariya Y, Hoshino Y. Pigmented villonodular synovitis of the elbow in a 6-year-old girl: a case report. J Orthop Surg (Hong Kong) 2007;15:106–108. [DOI] [PubMed] [Google Scholar]
- 31. Wyatt MC, Rolton N, Veale GA. Pigmented villonodular synovitis of the elbow with a fenestrated fossa: a case report. J Orthop Surg (Hong Kong) 2009;17:127–129. [DOI] [PubMed] [Google Scholar]
- 32. Kohyama K, Sugiura H, Yamada K, Hyodo I, Kato H, Kamei Y. Posterior interosseous nerve palsy secondary to pigmented villonodular synovitis of the elbow: case report and review of literature. Orthop Traumatol Surg Res 2013;99:247–251. [DOI] [PubMed] [Google Scholar]
- 33. Barile A, Sabatini M, Iannessi F, et al. Pigmented villonodular synovitis (PVNS) of the knee joint: magnetic resonance imaging (MRI) using standard and dynamic paramagnetic contrast media. Report of 52 cases surgically and histologically controlled. Radiol Med 2004;107:356–366. [PubMed] [Google Scholar]
- 34. Masih S, Antebi A. Imaging of pigmented villonodular synovitis. Semin Musculoskelet Radiol 2003;7:205–216. [DOI] [PubMed] [Google Scholar]
- 35. Dürr HR, Stäbler A, Maier M, Refior HJ. Pigmented villonodular synovitis: review of 20 cases. J Rheumatol 2001;28:1620–1630. [PubMed] [Google Scholar]
- 36. Sharma V, Cheng EY. Outcomes after excision of pigmented villonodular synovitis of the knee. Clin Orthop Relat Res 2009;467:2852–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nishida Y, Tsukushi S, Nakashima H, et al. Osteochondral destruction in pigmented villonodular synovitis during the clinical course. J Rheumatol 2012;39:345–351. [DOI] [PubMed] [Google Scholar]
- 38. Ogilvie-Harris DJ, McLean J, Zarnett ME. Pigmented villonodular synovitis of the knee: the results of total arthroscopic synovectomy, partial, arthroscopic synovectomy, and arthroscopic local excision. J Bone Joint Surg Am 1992;74:119–123. [PubMed] [Google Scholar]
- 39. O’Sullivan B, Cummings B, Catton C, et al. Outcome following radiation treatment for high-risk pigmented villonodular synovitis. Int J Radiat Oncol Biol Phys 1995;32:777–786. [DOI] [PubMed] [Google Scholar]
- 40. Nassar WA, Bassiony AA, Elghazaly HA. Treatment of diffuse pigmented villonodular synovitis of the knee with combined surgical and radiosynovectomy. HSS J 2009;5:19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pimpalnerkar A, Barton E, Sibly TF. Pigmented villonodular synovitis of the elbow. J Shoulder Elbow Surg 1998;7:71–75. [DOI] [PubMed] [Google Scholar]
- 42. Koca G, Ozsoy H, Atilgan HI, Demirel K, Dincel VE, Korkmaz M. Application of rhenium-186 radiosynovectomy in elbow diffuse pigmented villonodular synovitis: a case report with multiple joint involvement. Nucl Med Mol Imaging 2012;46:215–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cassier PA, Gelderblom H, Stacchiotti S, et al. Efficacy of imatinib mesylate for the treatment of locally advanced and/or metastatic tenosynovial giant cell tumor/pigmented villonodular synovitis. Cancer 2012;118:1649–1655. [DOI] [PubMed] [Google Scholar]
- 44. Cassier PA, Italiano A, Gomez-Roca CA, et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: a dose-escalation and dose-expansion phase 1 study. Lancet Oncol 2015;16:949–956. [DOI] [PubMed] [Google Scholar]
- 45. van der Heijden L, Gibbons CL, Dijkstra PD, et al. The management of diffuse-type giant cell tumour (pigmented villonodular synovitis) and giant cell tumour of tendon sheath (nodular tenosynovitis). J Bone Joint Surg Br 2012;94:882–888. [DOI] [PubMed] [Google Scholar]
- 46. Inoue Y, Ishida H, Ueno H, et al. The treatment of desmoid tumors associated with familial adenomatous polyposis: the results of a Japanese multicenter observational study. Surg Today 2017;47:1259–1267. [DOI] [PubMed] [Google Scholar]
- 47. Mullen JT, DeLaney TF, Rosenberg AE, et al. β-Catenin mutation status and outcomes in sporadic desmoid tumors. Oncologist 2013;18:1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van Broekhoven DL, Verhoef C, Elias SG, et al. Local recurrence after surgery for primary extra-abdominal desmoid-type fibromatosis. Br J Surg 2013;100:1214–1219. [DOI] [PubMed] [Google Scholar]
- 49. Schmitz JJ, Schmit GD, Atwell TD, et al. Percutaneous cryoablation of extraabdominal desmoid tumors: a 10-year experience. AJR Am J Roentgenol 2016;207:190–195. [DOI] [PubMed] [Google Scholar]
- 50. Bonvalot S, Rimareix F, Causeret S, et al. Hyperthermic isolated limb perfusion in locally advanced soft tissue sarcoma and progressive desmoid-type fibromatosis with TNF 1 mg and melphalan (T1-M HILP) is safe and efficient. Ann Surg Oncol 2009;16:3350–3357. [DOI] [PubMed] [Google Scholar]
- 51. Grunhagen DJ, de Wilt JH, Verhoef C, van Geel AN, Eggermont AM. TNF-based isolated limb perfusion in unresectable extremity desmoid tumours. Eur J Surg Oncol 2005;31:912–916. [DOI] [PubMed] [Google Scholar]
- 52. Janssen ML, van Broekhoven DL, Cates JM, et al. Meta-analysis of the influence of surgical margin and adjuvant radiotherapy on local recurrence after resection of sporadic desmoid-type fibromatosis. Br J Surg 2017;104:347–357. [DOI] [PubMed] [Google Scholar]
- 53. Hansmann A, Adolph C, Vogel T, Unger A, Moeslein G. High-dose tamoxifen and sulindac as first-line treatment for desmoid tumors. Cancer 2004;100:612–620. [DOI] [PubMed] [Google Scholar]
- 54. Leithner A, Schnack B, Katterschafka T, et al. Treatment of extra-abdominal desmoid tumors with interferon-alpha with or without tretinoin. J Surg Oncol 2000;73:21–25. [DOI] [PubMed] [Google Scholar]
- 55. Azzarelli A, Gronchi A, Bertulli R, et al. Low-dose chemotherapy with methotrexate and vinblastine for patients with advanced aggressive fibromatosis. Cancer 2001;92:1259–1264. [DOI] [PubMed] [Google Scholar]
- 56. de Camargo VP, Keohan ML, D’Adamo DR, et al. Clinical outcomes of systemic therapy for patients with deep fibromatosis (desmoid tumor). Cancer 2010;116:2258–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chugh R, Wathen JK, Patel SR, et al. Efficacy of imatinib in aggressive fibromatosis: results of a phase II multicenter Sarcoma Alliance for Research through Collaboration (SARC) trial. Clin Cancer Res 2010;16:4884–4891. [DOI] [PubMed] [Google Scholar]
- 58. Gounder MM, Lefkowitz RA, Keohan ML, et al. Activity of Sorafenib against desmoid tumor/deep fibromatosis. Clin Cancer Res 2011;17:4082–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. vonMehren M Randall RL Benjamin RS et al. Soft tissue sarcoma, version 2.2016, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw 2016;14:758–786. [DOI] [PubMed] [Google Scholar]
- 60. Leithner A, Gapp M, Leithner K, et al. Margins in extra-abdominal desmoid tumors: a comparative analysis. J Surg Oncol 2004;86:152–156. [DOI] [PubMed] [Google Scholar]
- 61. Houdek MT, Rose PS, Kakar S. Desmoid tumors of the upper extremity. J Hand Surg Am 2014;39:1761–1765. [DOI] [PubMed] [Google Scholar]
- 62. Dangoor A, Seddon B, Gerrand C, Grimer R, Whelan J, Judson I. UK guidelines for the management of soft tissue sarcomas. Clin Sarcoma Res 2016;6:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Al-Jazrawe M, Au M, Alman B. Optimal therapy for desmoid tumors: current options and challenges for the future. Expert Rev Anticancer Ther 2015;15:1443–1458. [DOI] [PubMed] [Google Scholar]
- 64. Casali PG, Blay JY, ESMO/CONTICANET/EUROBONET Consensus Panel of experts. Soft tissue sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2010;21:v198–v203. [DOI] [PubMed] [Google Scholar]
- 65. Seppälä MT, Sainio MA, Haltia MJ, Kinnunen JJ, Setälä KH, Jääskeläinen JE. Multiple schwannomas: schwannomatosis or neurofibromatosis type 2? J Neurosurg 1998;89:36–41. [DOI] [PubMed] [Google Scholar]
- 66. Adani R, Baccarani A, Guidi E, Tarallo L. Schwannomas of the upper extremity: diagnosis and treatment. Chir Organi Mov 2008;92:85–88. [DOI] [PubMed] [Google Scholar]
- 67. White NB. Neurilemomas of the extremities. J Bone Joint Surg Am 1967;49:1605–1610. [PubMed] [Google Scholar]
- 68. Donner TR, Voorhies RM, Kline DG. Neural sheath tumors of major nerves. J Neurosurg 1994;81:362–373. [DOI] [PubMed] [Google Scholar]
- 69. Gambarotti M. Schwannoma (Neurilemoma, Neurinoma). In: Picci P, Vanel D, Gambarotti M, Fabbri N, Manfrini M, eds. Atlas of musculoskeletal tumors and tumorlike lesions: the Rizzoli case archive. Cham: Springer, 2014:337–339. [Google Scholar]
- 70. Koga H, Matsumoto S, Manabe J, Tanizawa T, Kawaguchi N. Definition of the target sign and its use for the diagnosis of schwannomas. Clin Orthop Relat Res 2007;464:224–229. [DOI] [PubMed] [Google Scholar]
- 71. Tang CY, Fung B, Fok M, Zhu J. Schwannoma in the upper limbs. Biomed Res Int 2013;2013:167196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Park MJ, Seo KN, Kang HJ. Neurological deficit after surgical enucleation of schwannomas of the upper limb. J Bone Joint Surg Br 2009;91:1482–1486. [DOI] [PubMed] [Google Scholar]
- 73. Gambarotti M. Vascular tumors: hemangioma, epithelioid hemangioendothelioma, and angiosarcoma. In: Picci P, Vanel D, Gambarotti M, Fabbri N, Manfrini M, eds. Atlas of musculoskeletal tumors and tumorlike lesions: the Rizzoli case archive. Cham: Springer, 2014:321–328. [Google Scholar]
- 74. Griffin N, Khan N, Thomas JM, Fisher C, Moskovic EC. The radiological manifestations of intramuscular haemangiomas in adults: magnetic resonance imaging, computed tomography and ultrasound appearances. Skeletal Radiol 2007;36:1051–1059. [DOI] [PubMed] [Google Scholar]
- 75. Nazzi V, Messina G, Dones I, Ferroli P, Broggi G. Surgical removal of intramuscular arteriovenous hemangioma of the upper left forearm compressing radial nerve branches. J Neurosurg 2008;108:808–811. [DOI] [PubMed] [Google Scholar]
- 76. Gerrand CH, Bell RS, Wunder JS, et al. The influence of anatomic location on outcome in patients with soft tissue sarcoma of the extremity. Cancer 2003;97:485–492. [DOI] [PubMed] [Google Scholar]
- 77. Pisters PW, Leung DH, Woodruff J, Shi W, Brennan MF. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14:1679–1689. [DOI] [PubMed] [Google Scholar]
- 78. Wong JC, Abraham JA. Upper extremity considerations for oncologic surgery. Orthop Clin North Am 2014;45:541–564. [DOI] [PubMed] [Google Scholar]
- 79. Baroudi MR, Ferguson PC, Wunder JS, et al. Forearm soft tissue sarcoma: tumors characteristics and oncologic outcomes following limb salvage surgery. J Surg Oncol 2014;110:676–681. [DOI] [PubMed] [Google Scholar]
- 80. Koulaxouzidis G, Simunovic F, Bannasch H. Soft tissue sarcomas of the arm: oncosurgical and reconstructive principles within a multimodal, interdisciplinary setting. Front Surg 2016;3:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lehnhardt M, Hirche C, Daigeler A, et al. [Soft tissue sarcoma of the upper extremities: analysis of factors relevant for prognosis in 160 patients]. Chirurg 2012;83:143–152. [DOI] [PubMed] [Google Scholar]
- 82. Müller M, Bickert B, Germann G, Sauerbier M. [Soft-tissue sarcoma of the forearm and hand: plastic surgical management]. Chirurg 2008;79:682–688. [DOI] [PubMed] [Google Scholar]
- 83. Muramatsu K, Ihara K, Doi K, Hashimoto T, Taguchi T. Sarcoma in the forearm and hand: clinical outcomes and microsurgical reconstruction for limb salvage. Ann Plast Surg 2009;62:28–33. [DOI] [PubMed] [Google Scholar]
- 84. Morrey BF. The elbow and its disorders. London: Elsevier Health Sciences, 2008. [Google Scholar]
- 85. Murray PM. Soft tissue sarcoma of the upper extremity. Hand Clin 2004;20:325–333, vii. [DOI] [PubMed] [Google Scholar]
- 86. Berger F, Winkler EC, Ruderer C, Reiser MF. [Imaging of soft tissue sarcomas: standard approaches and new strategies]. Chirurg 2009;80:175–185. [DOI] [PubMed] [Google Scholar]
- 87. Schuetze SM, Rubin BP, Vernon C, et al. Use of positron emission tomography in localized extremity soft tissue sarcoma treated with neoadjuvant chemotherapy. Cancer 2005;103:339–348. [DOI] [PubMed] [Google Scholar]
- 88. Korah MP, Deyrup AT, Monson DK, et al. Anatomic tumor location influences the success of contemporary limb-sparing surgery and radiation among adults with soft tissue sarcomas of the extremities. Int J Radiat Oncol Biol Phys 2012;82:933–939. [DOI] [PubMed] [Google Scholar]
- 89. Collin C, Hajdu SI, Godbold J, Friedrich C, Brennan MF. Localized operable soft tissue sarcoma of the upper extremity: presentation, management, and factors affecting local recurrence in 108 patients. Ann Surg 1987;205:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hoekstra HJ, Thijssens K, van Ginkel RJ. Role of surgery as primary treatment and as intervention in the multidisciplinary treatment of soft tissue sarcoma. Ann Oncol 2004;15:iv181–iv186. [DOI] [PubMed] [Google Scholar]
- 91. Pervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008;113:573–581. [DOI] [PubMed] [Google Scholar]
- 92. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol 2016;34:786–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Schöffski P, Ray-Coquard IL, Cioffi A, et al. European Organisation for Research and Treatment of Cancer (EORTC) Soft Tissue and Bone Sarcoma Group (STBSG). Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histological subtypes. Lancet Oncol 2011;12:1045–1052. [DOI] [PubMed] [Google Scholar]
- 94. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 2016;388:488–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kasper B, Sleijfer S, Litière S, et al. Long-term responders and survivors on pazopanib for advanced soft tissue sarcomas: subanalysis of two European Organisation for Research and Treatment of Cancer (EORTC) clinical trials 62043 and 62072. Ann Oncol 2014;25:719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Santoro A, Comandone A, Basso U, et al. Phase II prospective study with sorafenib in advanced soft tissue sarcomas after anthracycline-based therapy. Ann Oncol 2013;24:1093–1098. [DOI] [PubMed] [Google Scholar]
- 97. Benson C, Vitfell-Rasmussen J, Maruzzo M, et al. A retrospective study of patients with malignant PEComa receiving treatment with sirolimus or temsirolimus: the Royal Marsden Hospital experience. Anticancer Res 2014;34:3663–3668. [PubMed] [Google Scholar]
- 98. Dickson MA, Tap WD, Keohan ML, et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J Clin Oncol 2013;31:2024–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 2002;359:2235–2241. [DOI] [PubMed] [Google Scholar]
- 100. McGee L, Indelicato DJ, Dagan R, et al. Long-term results following postoperative radiotherapy for soft tissue sarcomas of the extremity. Int J Radiat Oncol Biol Phys 2012;84:1003–1009. [DOI] [PubMed] [Google Scholar]
- 101. Tiong SS, Dickie C, Haas RL, O’Sullivan B. The role of radiotherapy in the management of localized soft tissue sarcomas. Cancer Biol Med 2016;13:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kransdorf MJ. Malignant soft-tissue tumors in a large referral population: distribution of diagnoses by age, sex, and location. AJR Am J Roentgenol 1995;164:129–134. [DOI] [PubMed] [Google Scholar]
- 103. Panagopoulos I, Mertens F, Isaksson M, et al. Clinical impact of molecular and cytogenetic findings in synovial sarcoma. Genes Chromosomes Cancer 2001;31:362–372. [DOI] [PubMed] [Google Scholar]
- 104. Machen SK, Easley KA, Goldblum JR. Synovial sarcoma of the extremities: a clinicopathologic study of 34 cases, including semi-quantitative analysis of spindled, epithelial, and poorly differentiated areas. Am J Surg Pathol 1999;23:268–275. [DOI] [PubMed] [Google Scholar]
- 105. Raney RB. Synovial sarcoma in young people: background, prognostic factors, and therapeutic questions. J Pediatr Hematol Oncol 2005;27:207–211. [DOI] [PubMed] [Google Scholar]
- 106. Varela-Duran J, Enzinger FM. Calcifying synovial sarcoma. Cancer 1982;50:345–352. [DOI] [PubMed] [Google Scholar]
- 107. Miettinen MM. Soft tissue tumors with epithelial differentiation. In: Miettinen MM, ed. Diagnostic soft tissue pathology. New York: Churchill Livingstone, 2003:463–468. [Google Scholar]
- 108. Murphey MD, Gibson MS, Jennings BT, Crespo-Rodríguez AM, Fanburg-Smith J, Gajewski DA. From the archives of the AFIP: imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics 2006;26:1543–1565. [DOI] [PubMed] [Google Scholar]
- 109. Weiss SW, Goldblum JR. Malignant soft tissue tumors of uncertain type. In: Weiss SW, Goldblum JR, Enzinger FM, eds. Enzinger and Weiss’s soft tissue tumors. St. Louis: Mosby, 2001:1483–1565. [Google Scholar]
- 110. Vlenterie M, Litière S, Rizzo E, et al. Outcome of chemotherapy in advanced synovial sarcoma patients: review of 15 clinical trials from the European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group; setting a new landmark for studies in this entity. Eur J Cancer 2016;58:62–72. [DOI] [PubMed] [Google Scholar]
- 111. Spurrell EL, Fisher C, Thomas JM, Judson IR. Prognostic factors in advanced synovial sarcoma: an analysis of 104 patients treated at the Royal Marsden Hospital. Ann Oncol 2005;16:437–444. [DOI] [PubMed] [Google Scholar]
- 112. Kawai A, Araki N, Sugiura H, et al. Trabectedin monotherapy after standard chemotherapy versus best supportive care in patients with advanced, translocation-related sarcoma: a randomised, open-label, phase 2 study. Lancet Oncol 2015;16:406–416. [DOI] [PubMed] [Google Scholar]
- 113. Payne CE, Hofer SO, Zhong T, Griffin AC, Ferguson PC, Wunder JS. Functional outcome following upper limb soft tissue sarcoma resection with flap reconstruction. J Plast Reconstr Aesthet Surg 2013;66:601–607. [DOI] [PubMed] [Google Scholar]
- 114. de Silva MV, McMahon AD, Reid R. Prognostic factors associated with local recurrence, metastases, and tumor-related death in patients with synovial sarcoma. Am J Clin Oncol 2004;27:113–121. [DOI] [PubMed] [Google Scholar]
- 115. Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer 2004;101:627–634. [DOI] [PubMed] [Google Scholar]
- 116. Kransdorf MJ, Murphey MD. Imaging of soft tissue tumors. Philadelphia, PA: Lippincott Williams & Wilkins, 2006. [Google Scholar]
- 117. Weiss SW, Enzinger FM. Malignant fibrous histiocytoma: an analysis of 200 cases. Cancer 1978;41:2250–2266. [DOI] [PubMed] [Google Scholar]
- 118. Mutter RW, Singer S, Zhang Z, Brennan MF, Alektiar KM. The enigma of myxofibrosarcoma of the extremity. Cancer 2012;118:518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ghazala CG, Agni NR, Ragbir M, et al. Myxofibrosarcoma of the extremity and trunk: a multidisciplinary approach leads to good local rates of LOCAL control. Bone Joint J 2016;98-B:1682–1688. [DOI] [PubMed] [Google Scholar]
- 120. Gambarotti M. Myxofibrosarcoma. In: Picci P, Vanel D, Gambarotti M, Fabbri N, Manfrini M, eds. Atlas of musculoskeletal tumors and tumorlike lesions: the Rizzoli case archive. Cham: Springer, 2014:355–357. [Google Scholar]
- 121. Manoso MW, Pratt J, Healey JH, Boland PJ, Athanasian EA. Infiltrative MRI pattern and incomplete initial surgery compromise local control of myxofibrosarcoma. Clin Orthop Relat Res 2006;450:89–94. [DOI] [PubMed] [Google Scholar]
- 122. Dewan V, Darbyshire A, Sumathi V, Jeys L, Grimer R. Prognostic and survival factors in myxofibrosarcomas. Sarcoma 2012;2012:830879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lefkowitz RA, Landa J, Hwang S, et al. Myxofibrosarcoma: prevalence and diagnostic value of the ‘tail sign’ on magnetic resonance imaging. Skeletal Radiol 2013;42:809–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kaya M, Wada T, Nagoya S, et al. MRI and histological evaluation of the infiltrative growth pattern of myxofibrosarcoma. Skeletal Radiol 2008;37:1085–1090. [DOI] [PubMed] [Google Scholar]
- 125. Kaya M, Wada T, Nagoya S, Yamashita T. Bone and/or joint attachment is a risk factor for local recurrence of myxofibrosarcoma. J Orthop Sci 2011;16:413–417. [DOI] [PubMed] [Google Scholar]
- 126. Murphey MD, Gross TM, Rosenthal HG. From the archives of the AFIP: musculoskeletal malignant fibrous histiocytoma: radiologic-pathologic correlation. Radiographics 1994;14:807–826. [DOI] [PubMed] [Google Scholar]
- 127. Vasileios KA, Eward WC, Brigman BE. Surgical treatment and prognosis in patients with high-grade soft tissue malignant fibrous histiocytoma of the extremities. Arch Orthop Trauma Surg 2012;132:955–961. [DOI] [PubMed] [Google Scholar]