

SUMMARY
Voltage-gated sodium channel Nav1.5 generates cardiac action potentials and initiates the heartbeat. Here we report structures of NaV1.5 at 3.2–3.5 Å resolution. NaV1.5 is distinguished from other sodium channels by a unique glycosyl moiety and loss of disulfide-bonding capability at the NaVβ subunit-interaction sites. The antiarrhythmic drug flecainide specifically targets the central cavity of the pore. The voltage sensors are partially activated, and the fast-inactivation gate is partially closed. Activation of the voltage sensor of Domain III allows binding of the IFM motif to the inactivation-gate receptor. Asp and Ala in the selectivity motif DEKA line the walls of the ion-selectivity filter, whereas Glu and Lys are in positions to accept and release Na+ ions via a charge-delocalization network. Arrhythmia mutation sites undergo large translocations during gating, providing a potential mechanism for pathogenic effects. Our results provide detailed insights into Nav1.5 structure, pharmacology, activation, inactivation, ion selectivity, and arrhythmias.
In Brief
The high-resolution structure of the cardiac sodium channel gives detailed insights into voltage-dependent activation, ion selectivity, arrhythmia mechanisms, and antiarrhythmic drug action at the atomic level.
Graphical Abstract
INTRODUCTION
Cardiac sodium channels generate the up-stroke of the cardiac action potential by activating rapidly in response to membrane depolarization and conducting Na+ inward across the membrane (Catterall and Maier, 2015). The rate of rise of the action potential, which depends on rapid activation of voltage-gated sodium channels, controls the rate and homogeneity of propagation of the action potential through cardiac tissue (Catterall and Maier, 2015). Voltage-gated sodium channel 1.5 (Nav1.5) is the predominant sodium channel expressed in myocardial cells (Fozzard and Hanck, 1996; Rogart et al., 1989). Dysfunction of NaV1.5 owing to mutations or pathophysiological conditions causes life-threatening cardiac arrhythmias (Liu et al., 2003b), which are terminated and prevented by treatment with antiarrhythmic drugs (Sampson and Kass, 2011). Although other voltage-gated sodium channels are expressed in the heart, they are unable to compensate for dysfunction of NaV1.5, which highlights the importance of this sodium channel in cardiac physiology and pathophysiology.
Voltage-gated sodium channels in eukaryotes consist of a high molecular weight pore-forming α-subunit of approximately 2000 amino acid residues containing four homologous domains (DI-DIV) (Catterall, 2000; Noda et al., 1984). Each domain of the α-subunit is composed of six transmembrane helices organized in two functional modules. The first four segments (S1-S4) form the voltage-sensing module (VS), whereas the last 2 segments (S5-S6) and a P-loop between them form the pore module (PM). The VS and PM are connected by the α-helical S4-S5 linker. The intracellular linker connecting DIII and DIV serves as the fast inactivation gate, which closes the pore within 1–2 msec after opening. In contrast, prokaryotic sodium channels are composed of four identical subunits, which are similar in structure to one domain of the eukaryotic α-subunits, and there is no structural equivalent of the fast inactivation gate (Payandeh et al., 2011; Ren et al., 2001).
Most eukaryotic sodium channels contain one or two auxiliary β-subunits (Isom et al., 1994; O’Malley and Isom, 2015). β1 and β3 bind to the α-subunit by noncovalent interactions, whereas β2 and β4 are linked by a disulfide bond. The β-subunits are composed of an extracellular Ig-like domain and a single transmembrane segment. They regulate channel voltage dependence, gating kinetics, cell-surface localization, and density.
High-resolution crystal structures of bacterial sodium channels and cryo-electron microscopic (cryo-EM) structures of eukaryotic sodium channels from nerve and skeletal muscle reveal similar architectures (Catterall et al., 2017; Pan et al., 2019; Pan et al., 2018; Payandeh et al., 2011; Shen et al., 2019). The central pore is formed by the PMs of the four subunits or domains. The four VS are positioned on the periphery of the PM in approximately square symmetry, and they are activated in these structures, placing the S4 segment containing the gating charges in an outward position. We used voltage-shifting mutations and disulfide crosslinking to trap the resting state of the bacterial sodium channel NaVAb at 0 mV and determined its structure by cryo-EM (Wisedchaisri et al., 2019). Major changes in the conformation of the VS, the S4-S5 linker, and the intracellular activation gate were observed. These studies have given important insights into the structure and function of sodium channels
To discover the key structural features of NaV1.5 and relate them to the structural basis for its physiological function and its dysfunction in cardiac arrhythmias, we have determined the cryo-EM structure of the rat Nav1.5 α-subunit at 3.2–3.5 Å resolution. Our experiments provide a blueprint for understanding cardiac sodium channel structure, pharmacology, activation, inactivation, sodium selectivity, and disease mutations.
RESULTS AND DISCUSSION
Structure Determination of Nav1.5 by CryoEM
NaV1.5 is composed of approximately 2000 amino acid residues with large intracellular N- and C-termini and large intracellular segments that connect the four domains (Catterall et al., 2017; Fozzard and Hanck, 1996; Rogart et al., 1989). These intracellular segments, which are predicted to be structurally flexible, were not observed in previous cryo-EM structures of sodium channels (Pan et al., 2018; Shen et al., 2019; Shen et al., 2017). In order to optimize expression and homogeneity of rat Nav1.5, constructs with truncated N- and C-termini and shortened interdomain linkers were screened by expression as fusion proteins with GFP and Fluorescence-detection Size Exclusion Chromatography (FSEC) (Goehring et al., 2014). Constructs were selected that have combined truncations and deletions, improved homogeneity and expression, and high voltage-activated Na+ conductance. One of these NaV1.5 constructs with deletions in DI-DII (Leu462-Gly658), DII-DIII (Ser1069-Pro1189), and the C-terminus (Leu1898-Val2019) was selected for further electrophysiological and structural studies.
This well-structured core transmembrane construct of rat NaV1.5 (rNav1.5C) was expressed in human embryonic kidney (HEK) cells and analyzed by whole-cell voltage clamp (see Star Methods and (Yarov-Yarovoy et al., 2012)). These recordings revealed typical inward sodium currents in response to depolarizing stimuli (Figure S1A). Proteolytic cleavage of cytoplasmic loops shifts activation curves of mammalian sodium channels negatively (Bennett, 2001). Likewise, we found that rNav1.5C has a substantial negative shift in the voltage dependence of activation (−50 mV) compared to native rat Nav1.5, suggesting that activated/inactivated states are favored (Figure S1A).
rNav1.5C was expressed at high levels in the cell line HEK293S GnTI−, which limits N-linked glycosylation (Reeves et al., 2002). After solubilization, affinity purification, and two rounds of size-exclusion chromatography, a peak of protein containing only the 230-kDa rNaV1.5C α subunit was collected for structural studies (Figure S1D, E). Detailed descriptions of protein expression, purification, cryo-EM imaging, and data processing are presented in Star Methods.
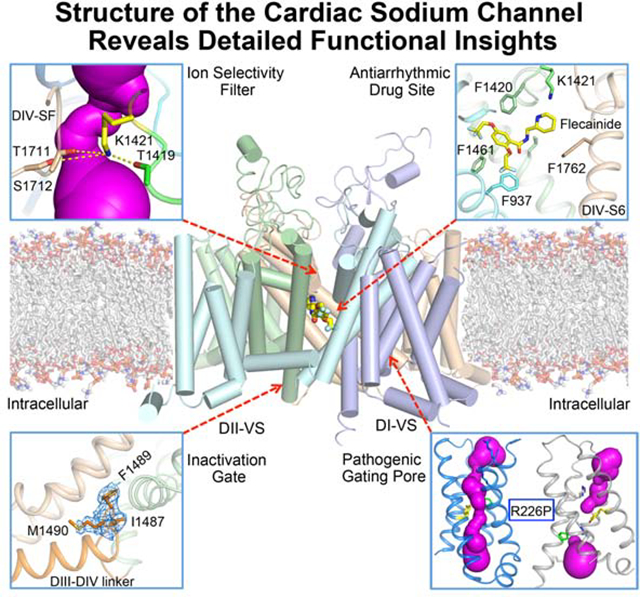
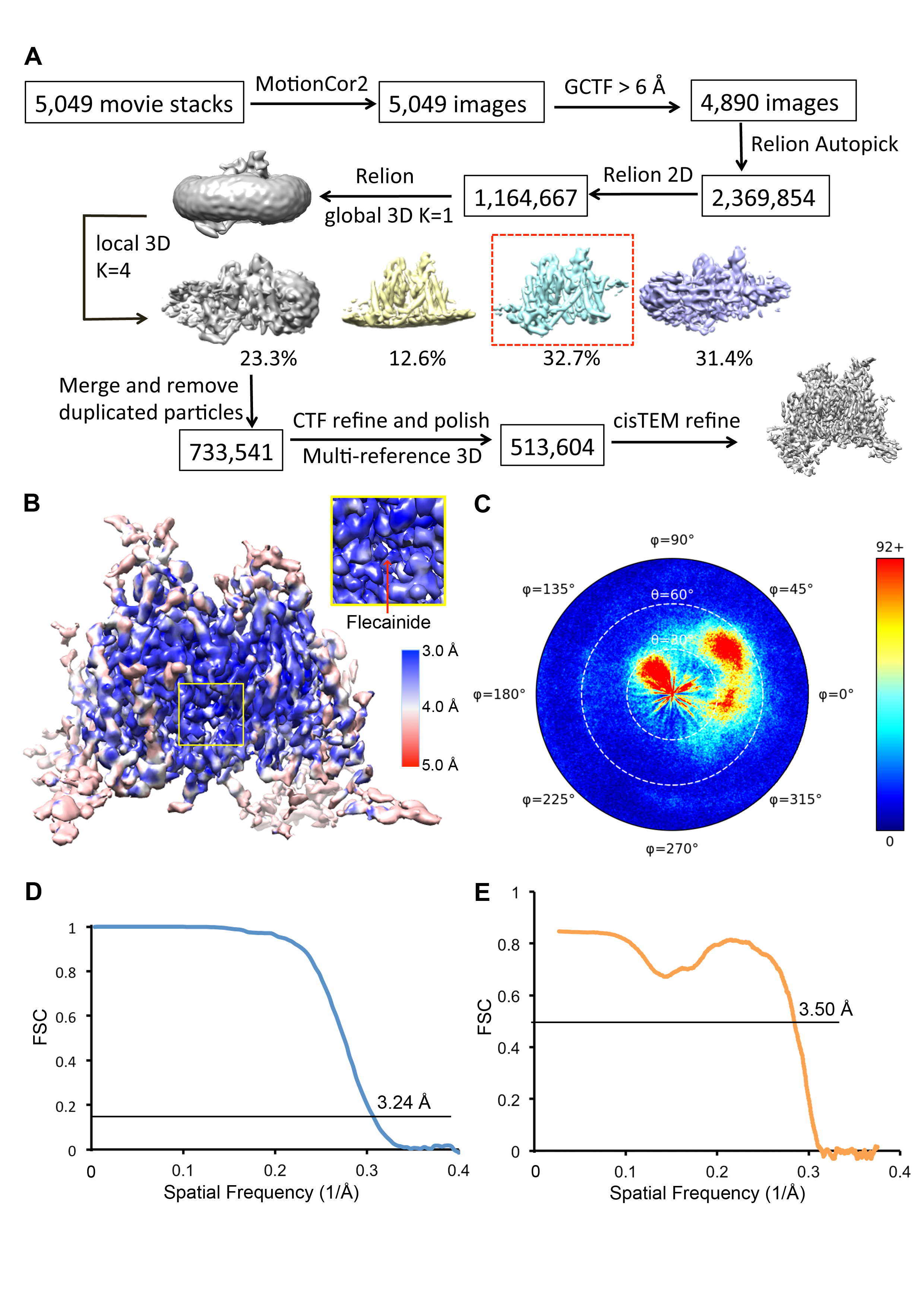
We imaged rNav1.5C by cryo-EM (Figure S1F, G; Figure S2A) and constructed a 3D map to an overall resolution of 3.5 Å (Figure 1A), based on the gold standard Fourier shell correlation criterion (Figure S2B–D) (Henderson et al., 2012). The PM in the center of the structure has higher resolution, approaching 3.3 Å, while the four VS have slightly lower resolution (Figure S2B). The high-quality electron density map allowed us to build an atomic model for rNav1.5C containing DI (missing a short extracellular loop Ala298-Asn303), DII (missing the sequence Leu799-Leu805 between S3 and S4), DIII, DIV, and the DIII-DIV linker (Figure 1A–D, Figure S3). We observed weak electron density on the intracellular side of DI-VS representing the N-terminus (Figure 1A). In a different protein sample prepared with 0.5 mM flecainide, the resolution of the rNav1.5C-flecainide complex was improved to 3.24 Å (Figure 2A; Figures S4–S6). No electron density was found for loops between DI and DII, DII and DIII, or the CTD containing the EF-hand like domain, suggesting that these segments are disordered or mobile.
Figure 1. Cryo-EM Structure of rNaV1.5C.

(A) Side view of the overall cryo-EM reconstruction of the rNaV1.5C.
(B) Cartoon representation of the overall structure of the rNaV1.5C. DI, DII, DIII, DIII-DIV linker, and DIV were colored blue, cyan, green, orange and wheat, respectively. The same color scheme is applied hereafter in this manuscript unless specified otherwise. The glycosyl moieties, lipids, and detergents shown in sticks are colored in magenta and yellow, respectively.
(C) Side view of rNaV1.5C with surface colored in grey.
(D) Side view of human NaV1.7-β1-β2 complex shown in grey surface for the α-subunit and in cartoons for β1 and β2 subunits colored in magenta and gold, respectively.
(E) Close-up view of β1 (Top panel) and β2 (Bottom panel) binding sites.
(F) SF alignment between rNaV1.5C and NaV1.7 (Grey). The side chains of the DEKA motif (DEKA: Asp373-Glu901-Lys1421-Ala1713) are shown in sticks.
(G) Comparison of the SF of the TTX-resistant rNaV1.5c (colored) and the TTX-sensitive human NaV1.7 (grey) with TTX bound. Cys374 and Arg377 are shown with electron density mesh presented at 3 a. TTX is shown in yellow. DEKA: Asp373-Glu901-Lys1421-Ala1713.
Figure 2. The Cryo-EM Structure of rNaV1.5C and Flecainide Complex.

(A) Side view of the overall cryo-EM reconstruction of the rNaV1.5C and flecainide complex.
(B) Chemical structure of flecainide.
(C) EM density for flecainide binding pocket with key residues for flecainide binding shown in yellow sticks. The densities for the residues and flecainide were countered at 3 σ and colored in blue and green, respectively.
(D) Top and side view of rNaV1.5C structure with flecainide shown in yellow sticks.
(E) Side view of DII-DIII fenestration.
Similar to other sodium channel structures, the four domains are assembled in a domain- swapped arrangement (Figure 1B). The structure of rNav1.5C can be superimposed well with the human Nav1.7 α-subunit (Shen et al., 2019) with R.M.S.D of 1.3 Å over 1153 Ca atoms in the core region, consistent with high sequence similarity (75.5% identity). The transmembrane region also overlays well with the bacterial sodium channel NaVAb (Cα R.M.S.D of 3.2 Å for DI) (Payandeh et al., 2011), illustrating the strong structural conservation of the VS and PM in sodium channels from bacteria to man. Strong electron density is observed for five Asn-linked glycosylation sites and 16 lipid or detergent molecules (Figure 1B, Figure S3E, F). Three of these specifically bound lipid molecules are also observed in NaV1.4 (Pan et al., 2018), and two are conserved in NaVAb as well (Payandeh et al., 2011), highlighting the potential functional significance of these conserved lipid moieties and their binding sites.
Altered Interaction Sites for NaVβ Subunits
Despite high overall similarity with other sodium channels, there are key differences in the binding sites for the auxiliary NaVβ-subunits in rNav1.5C. In contrast to sodium channels from brain and skeletal muscle, no β subunits were observed in biochemical studies of cardiac sodium channels (Gordon et al., 1988; Lombet and Lazdunski, 1984), and co-expression of the β-subunit increases expression but does not modulate the kinetics or voltage dependence of NaV1.5 (Makita et al., 1994; Qu et al., 1995). In our hands, co-expressed NaVβ1 and NaVβ2 do not stably associate with NaV1.5. These studies indicate that NaVβ-subunits do not interact with NaV1.5 as strongly as with sodium channels in nerve and skeletal muscle. In comparison to NaV1.7, (Figure 1C, D, magenta), we found unexpectedly that Asn319 has unambiguous density for an N-linked carbohydrate moiety, which would clash with β-sheet 2 and β-sheet 3 of the Ig-like domain of the p1-subunit for its binding to the Nav1.7 (Figure 1E, upper panel; Figure S3F). The amino acid sequence near Asn319 (Asn-Gly-Thr) encodes a typical core N-linked glycosylation site. Sequence alignment of Asn319 indicates that it is a unique glycosylation site among Nav channels (Nav1.1- Nav1.9) (Figure S7A, blue arrow). Moreover, a hydrogen-bond between Tyr304 (α-subunit) and Glu48 (β1-subunit) that supports binding of the β1 subunit in Nav1.7 is also missing in Nav1.5, where the equivalent position is occupied by Leu316 (Figure S7A, red arrow). These structural changes suggest why Nav1.5 cannot form the same regulatory interface with NaVβ subunits as other NaV α subunits.
Unlike β1, the NaVβ2 subunit forms a disulfide bond with the α-subunit of neuronal sodium channels (Isom et al., 1995; O’Malley and Isom, 2015). Surprisingly, we found that the disulfide bond-forming Cys895 from Nav1.7 (Shen et al., 2019) is replaced by Leu869 in Nav1.5 (Figure 1D, tan; Figure 1E, lower panel; Figure S7B, cyan arrow). This Cys residue is conserved in all other NaV channels except Nav1.8, which has Ala822 at the same position (Figure S7B, cyan arrow). This finding indicates that the β2 subunit does not form a disulfide linkage at this position with either the Nav1.5 or Nav1.8 α subunit. Together with the placement of a blocking glycosyl moiety in the binding site for the β1 subunit, these results establish a structural basis for reduced binding and modulation of NaV1.5 by β subunits.
Structural Basis for Tetrodotoxin Selectivity at Its Receptor Site in the Outer Pore
The classic sodium channel blocker tetrodotoxin (TTX) binds in the outer mouth of the ion selectivity filter (SF) and blocks the pore (Hille, 1975b; Terlau et al., 1991). Its characteristic low affinity for NaV1.5 channels (2 μM) compared to sodium channels in nerve and skeletal muscle (4–10 nM) is a hallmark of sodium channel pharmacology, determined primarily by substitution of a single amino acid residue ((Figure S7C); (Fozzard and Hanck, 1996; Sivilotti et al., 1997)). The overall structure of the SF of rNav1.5C is nearly identical to that of Nav1.7, which has a high affinity for TTX (Figure 1F; Figure S7C). However, rNav1.5C shows clear density for Cys374 and Arg377 on the extracellular side of Asp373 (Figure 1G), in positions where Nav1.7 has Tyr362 and Asn365 (Shen et al., 2019). Tyr362 helps TTX bind tightly to Nav1.7 through a n-n stacking interaction, which is missing in NaV1.5 (Figure 1G). The lack of this key interaction reduces the binding affinity of rNav1.5C for TTX by up to 500-fold (Sunami et al., 2000; Walker et al., 2012). Therefore, this residue is the structural determinant that differentiates the TTX-insensitive sodium channels (Nav1.5 and Nav1.8–9) with a Cys or Ser from the TTX-sensitive channels (Nav1.1–1.4, Nav1.6 and Nav1.7) with a Tyr or Phe. Our rNav1.5C structure shows that this amino acid substitution does not alter local protein conformation but eliminates a key side chain interaction with bound TTX by removing the aromatic side chain.
Structural Basis for Antiarrhythmic Drug Block of the Inner Pore
Class I antiarrhythmic drugs terminate and prevent cardiac arrhythmias by blocking cardiac sodium channels in a complex state-dependent manner (Sampson and Kass, 2011). They are divided into three classes (IA, IB, and IC) based on their rates of binding and unbinding, which determine their clinical uses (Sampson and Kass, 2011). The Class IC antiarrhythmic drug flecainide (Figure 2B) blocks cardiac sodium channels and is widely used to treat atrial arrhythmias, including the increasingly common problem of atrial fibrillation (Anno and Hondeghem, 1990; Konzen et al., 1990; Sampson and Kass, 2011). Flecainide blocks rNaV1.5C with IC50=16.6 μM when tested with depolarizing stimuli at 100 Hz (Figure S1B). The EC50 was increased to 140 μM when single stimuli were applied (Figure S1C), consistent with state- dependent block of open and inactivated rNaV1.5C, as observed for native NaV1.5. Flecainide binds in the central cavity of rNaV1.5C, just on the intracellular side of the SF (Figure 2C, 2D). Its piperidine ring lies across the top of the central cavity, hindering exit of Na+ from the SF (Figure 2C, 2D). The positively charged piperidine nitrogen points upward toward the inner exit from the SF, and the hydrophobic edge of the piperidine ring extends toward the phenyl sidechain of Phe1762 in the IVS6 segment (Figure 2C). This binding pose for flecainide is consistent with previous structure-function studies of NaV1.5 (Liu et al., 2003a; Ragsdale et al., 1996; Wang et al., 2003) and with the less well-resolved binding pose of flecainide in NaVAb (Gamal El-Din et al., 2018). Phe1762 may interact more strongly with the bound drug in the fully inactivated state observed in NaVAb, in which two S6 segments move toward the pore axis (Payandeh et al., 2012).
The wall of the central cavity of NaV channels is penetrated by four fenestrations that lead inward from the lipid bilayer between two PM (Pan et al., 2018; Payandeh et al., 2011; Shen et al., 2019). The two hydrophobic trifluoroethyoxy tails of flecainide latch onto the inner end of the fenestration between the PM in DII and DIII (Figure 2C, 2E). Flecainide can enter the central cavity through the fenestration and bind to its receptor site in NaVAb (Gamal El-Din et al., 2018). Flecainide may access its binding site by entering through the fenestration between DII and DIII in the PM of NaV1.5 (Figure 2D, 2E). Entry is also possible through other fenestrations, including DIII-DIV as suggested by molecular modeling studies based on NaV1.4 (Nguyen et al., 2019), but the DII-DIII fenestration is the largest and DIII-DIV is significantly smaller in rNaV1.5C.
Arrhythmia Mutations in NaV1.5
NaV1.5 and the NaVβ subunits are molecular targets for mutations that cause cardiac arrhythmias (Kapplinger et al., 2010; Li et al., 2018; Liu et al., 2003b; O’Malley and Isom, 2015). There are over 400 disease mutations of human Nav1.5 (UniProt database), including 230 mutations in Brugada Syndrome Type-1 (BS1), 153 in Long QT Syndrome Type-3 (LQT3), nine in Progressive Familial Heart Block Type-1A (PFHA1A), five in Sick Sinus Syndrome 1 (SSS1), and ten in Atrial Fibrillation Type-10 (ATFB10) (Liu et al., 2003b). Our high-resolution structure of rNav1.5C provides a structural template for these disease mutations (Figure 3). The mutations are distributed in every structural component, but they are especially common in conserved regions of the VS, SF, activation gate, and fast inactivation gate. The structural basis for pathogenic effects of these mutations is considered in the sections below.
Figure 3. Molecular Mapping of Arrhythmia Mutations.

(A-D). Arrhythmia related mutations are located on DI, DII, DIII and DIV. Mutation sites are shown in sticks for BS1, LQT3, PFHB1A and ATFB10 colored in red, yellow, magenta and blue, respectively.
State Dependent Conformations of the VS
The S4 segment in the VS contains 4 to 8 repeats of a positively charged amino acid residue flanked by two hydrophobic residues (Catterall et al., 2017; Numa and Noda, 1986). It is exposed to aqueous clefts on its extracellular and intracellular ends, and it is sealed to prevent transmembrane movement of water and ions at the narrow (~ 5 Å) hydrophobic constriction site (HCS; Figure 1B; Figure 4A–D, yellow). The steep voltage dependence of activation results from outward movement of the positive gating charges (Figure 4A–D, blue) in the S4 segment across membrane electric field through the HCS. This charge movement was first measured as a small capacitive gating current in the squid giant axon (Armstrong and Bezanilla, 1973). Chemical labeling, disulfide crosslinking, and gating pore current studies directly detected the outward movement of the S4 segments and their gating charges during activation (DeCaen et al., 2011; DeCaen et al., 2009; DeCaen et al., 2008; Gamal El-Din et al., 2014; Yang et al., 1996; Yang and Horn, 1995). Previous bacterial and mammalian sodium channel structures have captured activated states of the VS, which are most stable at 0 mV (Pan et al., 2018; Payandeh et al., 2011; Shen et al., 2019). However, recent determination of the structure of the NavAb, captured in the resting state, revealed the structural basis for gating charge movement, and supported the “sliding helix” model for VS activation, in which the gating charges move through the HCS under the influence of the electric field and exchange ion pair interactions with the intracellular negative cluster (INC) and extracellular negative cluster (ENC) of negatively charged amino acid residues (Figure 4A–D, red; (Wisedchaisri et al., 2019; Yarov-Yarovoy et al., 2012).
Figure 4. Structural Comparison of the DI-DIV VS in Activated and Resting States.

(A-D) Structures of each of the four VS and the corresponding resting state models. Resting models are colored in grey. Key residues are shown with sidechains in sticks. Arg and Lys gating charges are colored blue, Glu residues in the ENC and INC are colored red, and the key Phe in the HCS is colored yellow. Extracellular Negative Cluster (ENC): DI, Asn145, Glu162; DII, Glu738, Glu747, Asn754; DIII, Glu1227, Glu1242, Asp1245; DIV, Glu1550, Asp1556. Intracellular Negative Cluster (INC): DI, Glu172, Asp198; DII, Glu764, Asp786; DIII, Glu1255, Asp1277; DIV, Asp1533, Glu1576, Asp1597.
(E) The VS of rNaV1.5C DIII aligned with the VS of the open-state structure of NavAb-Δ40 (PDB code: 5VB8) shown in grey.
(F) The VS of rNaV1.5C DIII aligned with the VS of DIII of NavPas in grey (PDB code: 6A90).
(G) Structure of the activated state of the VS of DI with the R226P mutation in green. Brugada Syndrome target residue Y169 is nearby in yellow. Water-accessible space calculated by MOLE2 is shown in magenta.
(H) Structure of the resting state of the VS of DI with the R226P mutation in green. Brugada Syndrome target residue Y169 is nearby in yellow. Water-accessible space calculated by MOLE2 is shown in magenta.
As expected from its negative voltage dependence of activation (Figure S1A), all four VS in rNav1.5C are in activated states (Figure 4A–D, left panels). The VS of DI closely resembles the activated state of NaVAb (Figure S7D) (Payandeh et al., 2011) and other mammalian sodium channels (Pan et al., 2018; Shen et al., 2019). The DI gating charges R1-R3 point up toward the extracellular side above the HCS, and both R2 and R3 interact with the ENC. K4 is on the intracellular side of the HCS, stabilized by interaction with the INC (Figure 4A, left panel). Similar to DI, gating charges R1-R3 of the DII VS are also on the extracellular side of the HCS, but their outward translocation is less than in DI. R1 and R2 interact with the ENC, but R3 is just above the HCS, and K4 is still on the intracellular side of the HCS (Figure 4B, left panel). In contrast, the VS of DIII is more activated. Four gating charges (K1-R4) are on the extracellular side of the HCS and both R3 and R4 have moved far enough outward to interact with the ENC. R5 is located in the HCS forming a n-cation interaction with Phe1252 (Figure 4C, left panel). Four gating charges in DIV (R1-R4) are on the extracellular side of the HCS, while R5 and R6 are on the intracellular side interacting with the INC (Figure 4D, left panel). In addition, two more conserved positively charged residues (Lys1643 and Arg1646) that continue the S4 helix on the intracellular side of the HCS and could potentially serve as gating charges K7 and R8 (Figure S7E). In our structure, R8 serves as the C-cap, directed toward S4 helix dipole to stabilize the helical conformation (Figure S7E). These additional potential gating charges indicate that S4 of DIV VS could undergo larger outward movement during depolarization.
Recent structure determination of the resting state of NaVAb (Wisedchaisri et al., 2019) allows us to view the conformational change associated with activation of the four VS of NaV1.5. We generated a resting state model for each VS of Nav1.5C by aligning the amino acid sequence of each individual VS with the VS of NaVAb and using MODELLER to determine the best fit structure of the VS of Nav1.5C (Figure 4A–D, right panels). We found that two gating charges from each VS of Nav1.5C moved inward through the HCS, consistent with the NavAb resting state structure. These results suggest that the gating charge movements observed in the transition to the resting state in NaVAb also apply to NaV1.5.
In contrast to bacterial sodium channels, whose VS are identical and activate stochastically, the four VS in eukaryotic sodium channels are nonequivalent and activate sequentially (Chanda and Bezanilla, 2002). Our rNav1.5C structure reveals the asymmetric conformation of the four VS that leads to sequential voltage-dependent activation. Fluorescent labeling and gating pore current studies showed that the S4 segment in the VS of DIV moves five-fold slower than the other three S4 segments (Capes et al., 2013; Chanda and Bezanilla, 2002; Goldschen-Ohm et al., 2013). During depolarization, the VS of DI, DII and DIII move S4 quickly outward to activate the channel, and subsequently full activation of the DIV VS induces a conformational change that leads to fast inactivation. DI VS and DII VS also recover quickly from inactivation, whereas DIII VS and DIV VS recover more slowly. Because DIII VS and DIV VS have more gating charges than DI VS and DII VS, translocating these additional gating charges across the HCS may be the rate limiting step for their S4 movement.
Arrhythmia Mutations in the VS
To understand better how mutations in the VS cause arrhythmias, we compared the positions of the affected amino acid residues in our activated state structure of NaV1.5C with their positions in the modeled resting state of the VS of NaV1.5C. We found that 18 of 24 mutations in the S3 and S4 segments that we examined moved greater than 4 Å between the resting and activated state conformations (Table 1). Mutations of these amino acid residues that move substantially during activation and deactivation of the voltage sensor likely cause dysfunction by altering the normal conformational changes that are required for VS function. Moreover, the six residues with arrhythmia mutations that moved smaller distances might perturb movement of the VS by altered interactions of their mutant sidechains with neighboring amino acid residues that do undergo substantial movements during VS function.
Table 1.
Arrhythmia Mutations in the Conserved Transmembrane Domains
| VS location | Position in rNav1.5c | Human Mutation | Disease | Shift (Å) |
|---|---|---|---|---|
| DI-S3 | A205 | A204V | BS-1 | 5.8 |
| DI-S4 | T221 | T220I | BS-1/SSS1 | 10.3 |
| DI-S4 | V224 | V223L | BS-1 | 9.9 |
| DI-S4 | A227 | A226V | BS-1 | 9.7 |
| DI-S4 | I231 | I230V | BS-1 | 9.8 |
| DI-S4 | V233 | V232I | BS-1 | 11.7 |
| DI-S4 | R223 | R222Q | BS-1/LQT-3 | 9.9 |
| DI-S4 | R226 | R225W | BS-1/LQT-3/PFHB1A | 10.0 |
| DII-S3 | V790 | V789I | BS-1 | 1.8 |
| DII-S4 | R809 | R808P | BS-1 | 7.0 |
| DII-S4 | L813 | L812Q | BS-1 | 6.1 |
| DII-S4 | R815 | R814Q | BS-1 | 5.3 |
| DII-S4 | K818 | K817E | BS-1 | 4.7 |
| DII-S4 | F817 | F816Y | LQT-3 | 5.0 |
| DIII-S3 | L1285 | L1283M | LQT-3 | 1.7 |
| DIII-S4 | T1306 | T1304M | LQT-3 | 8.8 |
| DIII-S4 | L1313 | L1311P | BS-1 | 6.9 |
| DIV-S3 | I1595 | I1593M | LQT-3 | 1.7 |
| DIV-S3 | F1596 | F1594S | LQT-3 | 1.2 |
| DIV-S3 | D1597 | D1595N | PFHB1A | 1.2 |
| DIV-S3 | V1606 | V1604M | BS-1 | 1.8 |
| DIV-S4 | R1625 | R1623Q | LQT-3/ BS-1 | 9.6 |
| DIV-S4 | R1628 | R1626H/P | LQT-3 | 9.7 |
| DIV-S4 | R1631 | R1629Q | BS-1 | 8.0 |
The human arrhythmia mutations located in each domain S3 and S4 of rNav1.5C are listed. Diseases caused by those mutations are indicated individually. The shift of each mutation represents the distance of Cα between rNav1.5C VS and the resting state model.
Mutations of Arg gating charges in NaV1.4 cause pathogenic gating pore current in Hypokalemic and Normokalemic Periodic Paralysis owing to leak of ions through the mutant VS (Sokolov et al., 2007, 2008; Struyk and Cannon, 2007). The hole in the center of the VS that conducts pathogenic gating pore current in the mutants was visualized by X-ray crystallography of NaVAb, revealing an aqueous conducting pathway 2–3 Å in diameter (Jiang et al., 2018). Nine VS mutations in NaV1.5 convert a gating charge Arg to another uncharged residue and therefore are candidates to conduct gating pore current (Moreau and Chahine, 2018). Two of these mutant channels have been shown to conduct substantial gating pore current by direct electrophysiological measurements (Moreau et al., 2015). We introduced the mutation R225P at the position of R3 in our rNaV1.5C structure (R226P in rat) and examined the resulting model with MOLE2 to detect water-filled space. As for HypoPP mutations, we found that the pathogenic arrhythmia mutation R226P does indeed make a conductive hole 2.2 Å in diameter through the VS in Oil of rNaV1.5C (Figure 4G, H). As expected from electrophysiological studies, this conduction pathway through the mutant VS is open in the activated state but not in the resting state (Figure 4G, H). These results define the structural basis for pathogenic gating pore current in cardiac arrhythmias.
Intracellular Activation Gate
The intracellular activation gate formed by the intracellular ends of the S6 transmembrane segments controls sodium channel opening and closing, which are coupled to voltage-dependent conformational changes in the VS through the S4-S5 linker, as we observed by comparing the resting and open states of NaVAb (Wisedchaisri et al., 2019). Our rNav1.5C structure has a partially open pore, with a water-accessible diameter inside the activation gate of 5.6 Å calculated by MOLE2 (Figure 5A–B). When compared with the open-state structure of NavAb, which has a water-accessible diameter of 8.0 Å, the 5.6 Å opening of the activation gate of rNav1.5C would not be wide enough to allow uninterrupted diffusion of hydrated Na+. Movements of the S4-S5 linkers of DII, DIII, and DIV toward the center of the activation gate also indicate that the gate is not fully open (Figure 5B).
Figure 5. Activation and Fast Inactivation Gates.

(A) Bottom view of the rNaV1.5C intracellular activation gate. The IFM motif (Ile1485-Phe1486-Met1487) and the sidechains of amino acid residues that partially close the activation gate are shown in sticks with half-transparent van der Waals surface.
(B) Close-up view of the rNaV1.5C intracellular activation gate aligned with the open-state structure of NavAb-Δ40 (PDB code: 5VB8) colored in magenta.
(C) Close-up view of the NaV1.5C intracellular activation gate aligned with the resting state model of rNaV1.5C activation gate calculated by MODELLER based on the resting state structure of NavAb (PDB code: 6P6W). The resting model is colored in orange with key residues sealing the activation gate shown in sticks and half-transparent van der Waals surface.
(D) Side view of the IFM motif shown in sticks with electron density mesh contoured at 3 σ.
(E) Close-up view of IFM motif bound to the hydrophobic pocket shown in yellow. Dotted line in red, hydrogen bond.
(F) Interaction of DIII-DIV linker with DIII and DIV S4-S5 linker. “(O)” indicates an important interaction with a backbone carbonyl group. Dotted line in red, hydrogen bond.
(G) Interaction of DIII S6 with DIII S4-S5 linker. “(O)” indicates an important interaction with a backbone carbonyl group. Dotted lines in red, hydrogen bonds.
In order to investigate the closed pore structure of the rNav1.5C, a resting state pore model was generated by MODELLER based on the resting state structure of NavAb. Ths the activation gate. As a consequence, the gate is completely sealed by the sidechains of a ring of hydrophobic residues (Val413, Leu941, Ile1471 and Ile1773) (Figure 5C). This closed and sealed model of the resting state has no aqueous space for water or ions to penetrate the closed activation gate.
Fast Inactivation and Its Coupling to Voltage Sensing and Activation
Cardiac sodium channels are inactivated within 1–2 ms after opening. Fast inactivation of sodium channels decreases Na+ influx and allows potassium channels to activate and repolarize the membrane potential. This mechanism is very important for generating repeated action potentials to drive the heartbeat at a consistent frequency. Mutagenesis studies indicated that an isoleucine-phenylalanine-methionine (IFM) motif located in the DIII-DIV linker is responsible for fast inactivation of eukaryotic sodium channels (West et al., 1992), through interactions with amino acid residues in the S4-S5 linkers in DIII and DIV and the intracellular ends of the S5 and S6 segments of DIV (Kellenberger et al., 1996; McPhee et al., 1998; McPhee et al., 1994, 1995; Smith and Goldin, 1997). Site-directed fluorescent labeling and gating-pore current studies suggested that fast inactivation is coupled with the movement of DIII and DIV gating charges and causes immobilization of those gating charges (Capes et al., 2013; Chanda and Bezanilla, 2002).
In our rNav1.5C structure, the DIII-DIV linker, which is responsible for fast inactivation, packs in parallel with DIV S4-S5 linker, and the IFM motif dips into a pocket formed by DIII S6 and DIV S5 and S6 (Figure 5A). The IFM motif is well resolved with unambiguous electron density (Figure 5D). Remarkably, its structure closely resembles the structure of the IFM motif determined by NMR analysis in solution (Rohl et al., 1999), indicating that its fold is highly stable. IFM penetrates into a compact hydrophobic pocket ~8 Å deep formed by the S4-S5 linker from DIV, S5 from DIII, and S5 and S6 from DIV (Figure 5E), in close agreement with mutagenesis studies (Kellenberger et al., 1996; McPhee et al., 1998; McPhee et al., 1994, 1995; Smith and Goldin, 1997). Besides the hydrophobic interaction inside this pocket, additional interactions from both ends stabilize binding of the IFM motif. At its C-terminus, Asn1661, the carbonyl oxygens of Phe1488 and Thr1490, and the carbonyl oxygen of Gln1320 form two hydrogen bonds (Figure 5F and G). Interactions with a cluster of hydrophobic residues further stabilizes binding of the DIII-DIV linker to the DIV S4-S5 linker (Figure 5F). At its N-terminus, Asn1474 and Gln1478 from DIII S6 interact with carbonyl oxygens of Gly1338 and Ala1328, respectively (Figure 5G). Mutations of those interacting residues disrupt binding of the IFM motif, which in turn affect sodium channel inactivation. For instance, the single mutation Asn1661Ala or the double mutations Phe1650Ala and Leu1659Ala almost completely abolish fast inactivation (McPhee et al., 1998; McPhee et al., 1994, 1995; Smith and Goldin, 1997).
The sequential activation of the four VS in mammalian sodium channels finishes with activation of DIV VS, which is thought to be the main driving force for fast inactivation (Capes et al., 2013; Goldschen-Ohm et al., 2013). DIII VS, which is activated third in sequence, is also involved in fast inactivation, and its gating charges are stabilized in their outward positions by fast inactivation. By comparing the VS of rNav1.5C DIII with the activated VS of NavAb, we found that the gating charges of rNav1.5C DIII VS have moved further outward than those of NavAb, thereby lifting S4-S5 linker of DIII more outward with respect to the intracellular membrane surface (Figure 4E) and opening a tight binding pocket for the IFM motif. Thus, DIII VS must be activated to move its S4-S5 linker into the proper position for binding the IFM motif. The binding energy of the IFM motif would stabilize this activated state of the DIII VS, revealing why movement of the gating charges of DIII VS is slower than DI and DII during recovery from inactivation (Capes et al., 2013; Goldschen-Ohm et al., 2013).
Our rNav1.5C structure shows the IFM motif binding to the pocket on the periphery of the activation gate, which is consistent with the structures of human Nav1.2, Nav1.4 and Nav1.7 (Pan et al., 2019; Pan et al., 2018; Shen et al., 2019). Those similar structures demonstrate a conserved mechanism for fast inactivation. Moreover, the close interactions of the IFM motif detailed here for rNav1.5C (Figure 5D–G) suggest that the inactivation gate is in an inactivating conformation because it interacts directly with several amino acid residues in the “inactivation gate receptor” whose mutation disrupts fast inactivation (McPhee et al., 1998; McPhee et al., 1994, 1995; Smith and Goldin, 1997). Inactivation is evidently triggered by outward movement of the gating charges of DIII and DIV VS, which move the IFM motif into the binding pocket (Kellenberger et al., 1996) and decouple the DIII S4-S5 linker from DIV S6. In rNav1.5C, we observe that the DIII S4-S5 linker shifts significantly toward the center of the activation gate, compared with the other three S4-S5 linkers (Figure 5A, B). In this position, the activation gate cannot open wide enough for free conductance of hydrated Na+ (Chakrabarti et al., 2013). Thus, our structure suggests that NaV1.5C is in an intermediate inactivated state because the fast inactivation gate is not as far open as observed in the NaVPas channel from cockroach (Shen et al., 2017), and the pore is not yet as completely closed as in the inactivated state of NaVAb (Payandeh et al., 2012).
Arrhythmia Mutations in the Fast Inactivation Gate and Its Receptor
LQT3 and other cardiac arrhythmias are often caused by mutations that slow or prevent fast inactivation (Li et al., 2018). LQT3 mutants are most often located in the inactivation gate or its receptor (Figure 3). Given their short lengths, it is surprising that 11 arrhythmia mutations are located in the S4-S5 linkers in DI, DIII, and DIV (Figure 3). Nine of these mutations cause LQT3 by impairing fast inactivation. Our structure of NaV1.5 gives clear insight into the mechanism of action of these mutations (Figure 5E–G). These amino acid substitutions would impair the close fit of the IFM motif into its receptor site by altering the collar of S4-S5 segments that form the receptor. The LQT3 mutant Ile1662Val (Ile1660Val in hNav1.5) may cause reduced amplitude of sodium current because the IFM motif binds deeper and makes recovery from fast inactivation more difficult.
Mutations in the inactivation gate itself also cause LQT3. For example, Phe1475Cys (Phe1473Cys in hNav1.5) alters the key Phe in the IFM motif (Figure 5D and E) and causes slowed and incomplete inactivation and increased persistent sodium current (Bankston et al., 2007). The nearby ATFB10 mutant Gln1478Lys (Gln1476Lys in hNav1.5; Figure 5G) also accelerates recovery from fast inactivation, due to decreased affinity for IFM (Dichgans et al., 2005). The original LQT3 triple mutation, ΔKPQ(1507–1509), causes a three-residue in-frame deletion downstream from the inactivation gate and dramatically impairs fast inactivation (Bennett et al., 1995). This deletion mutation of rNav1.5C is located in the flexible loop following the highly conserved a-helix in the DIII-DIV linker (Figures 1B and 3C), which would shorten the loop by 13 Å and significantly limit the movement of IFM during cycles of activation followed by fast inactivation in the repeated cardiac action potentials necessary to trigger the heartbeat.
Structure of the Ion Selectivity Filter
The location of the SF was first revealed by mapping the binding site for TTX (Noda et al., 1989; Terlau et al., 1991). Mutagenesis studies identified amino acid residues from equivalent positions in the four domains, Asp373-Glu901-Lys1421-Ala1713 (DEKA), as the key determinants of Na+ selectivity (Favre et al., 1996; Heinemann et al., 1992; Sun et al., 1997). In NaVAb, this highly charged site at the extracellular entry to the SF, termed the high field strength site (HFS), is formed by four Glu177 sidechains. This site interacts first with permeating Na+ and catalyzes partial dehydration of the conducted ion (Chakrabarti et al., 2013). In the rNav1.5C structure, the DEKA motif stands in the center of the pore, forming a similar HFS (Figure 6A, B). The conformations of the four Ca atoms form a nearly four-fold symmetrical square, similar to the conformation of the HFS in NaVAb (Payandeh et al., 2011), but the difference in sidechains makes the site in rNav1.5C asymmetric. The high local resolution for the SF structure allows us to build a reliable atomic model of these sidechains (Figure 6B). Unambiguous density for Lys1421 shows the sidechain pointing inward, deep into the SF as a pinch point (Figure 6B–D). This position for the sidechain of Lys1421 is surprising, as it seems likely that cation permeation would be blocked if a full positive charge were located in this position. The electron density for the distal end of the sidechain of Glu901 is not as well resolved, which suggests sidechain flexibility. This conformational flexibility of Glu901 may allow it to function via the “sidechain-dunking” mechanism of Na+ permeation observed in molecular dynamics studies of NaVAb, in which rotation of the corresponding Glu177 side chains at a single torsion angle allowed their negative charges to “dunk” in concert with movement of Na+ through the SF (Chakrabarti et al., 2013).
Figure 6. Structure of the Pore and the Ion Selectivity Filter.

(A) The open ion permeation path shown in magenta surface calculated by MOLE2. DI and DIII PM with Asp373 and Lys1421 shown in sticks (Left panel), DII and DIV PM with Glu901 and Ala1713 shown in sticks (Right panel). DEKA: Asp373-Glu901-Lys1421-Ala1713.
(B) Top view of the rNaV1.5C SF with electron density shown in grey mesh contoured at 3 σ.
(C) SF comparison of NaV1.5c and activated NavAb (PDB,3RVY) shown in grey.
(D) Side view comparing the SF of rNaV1.5c and activated NavAb (PDB,3RVY).
(E) The charge delocalization complex of Lys1421. Lys1421 side chain interacts with adjacent backbone carbonyl group of Thr1419, Thr1711 and Thr1712, which are indicated by “(O)”. Magenta surface represents the ion permeation pathway inside the SF.
Compared to the approximately square orifice of the SF of NaVAb (Payandeh et al., 2011), which has van der Walls dimensions of 4.6 × 4.6 Å, rNav1.5C has a rectangular SF with van der Walls dimensions of 3.0 × 4.6 Å (Figure 6B–C). These dimensions are remarkably close to the model developed from classic studies of selective permeation of inorganic and organic cations, which suggested 3.1 × 5.1 Å (Hille, 1971, 1972, 1975a). In contrast to the four Glu177 residues that form the HFS in NaVAb, the Asp373 and Glu901 sidechain positions are more inward in NaV1.5, and the other half of the negatively charged tetrad is replaced by Lys1421 and Ala1713 on the opposite side of the SF. However, the HFS of Nav1.5 is extended inward by the sidechains of Glu376, Asp904 and Asp1716 (Figure 6D), which may act together with Asp373 and Glu901 to form the asymmetric HFS of NaV1.5.
Arrhythmia Mutations in the Pore and Selectivity Filter
A remarkable panel of 71 arrhythmia mutations target the PM and SF (Figure 3). Most of these mutations cause Brugada Syndrome (Figure 3, red), which is thought to arise from heterozygous loss of function of NaV1.5 by impairing its folding and functional expression (Baroudi et al., 2002; Baroudi et al., 2001). However, mutations in key amino acid residues in the PM may also cause loss of function by impairing ion conductance through an otherwise structurally normal NaV1.5 channel protein. We found eight Brugada Syndrome mutations in the SF itself, and our structure gives clear insights into the likely pathogenic mechanism for five of them.
Two Brugada Syndrome mutations are likely to have local structural effects that impair ion conductance. The mutation Trp375Gly removes the large indole sidechain from this highly conserved Trp residue, whose backbone carbonyl is crucial in stabilizing the SF structure by forming a network of hydrogen bonds around the HFS (Payandeh et al., 2011). It is likely that this mutation alters the square shape of the four Cα atoms that form the backbone corners of the SF. The nearby mutation Met370Lys replaces a hydrophobic sidechain with a hydrophilic one in a buried hydrophobic pocket. This mutation may also alter the precise local folding of the SF.
Even more strikingly, we found that the key SF DEKA signature residue Lys1421 in DIII is actually a target for Brugada Syndrome mutation (Lys1421Glu; (Kapplinger et al., 2010)). As our structure reveals, this residue plays a key role in forming the HFS in the SF of NaV1.5, and functional studies show that it is essential for selective sodium conductance (Favre et al., 1996; Heinemann et al., 1992). It occupies a unique dunked position deep in the SF in our structure, and we propose that it has a unique function as a coordinating ligand for Na+ through delocalization of its positive charge by formation of a charge transfer complex, as developed in more detail in the following section. Our structure predicts that mutation of Lys1421 to Glu, as in Brugada Syndrome, would create an ion coordination site for Ca2+ in the SF and thereby cause a loss of function by competing with sodium conductance, as was shown in mutagenesis studies of NaV1.4 (Heinemann et al., 1992). Thus, our structure defines the molecular basis for NaV1.5 dysfunction caused by this Brugada Syndrome mutation at the atomic level. The adjacent Gly is the target of the Brugada Syndrome mutation Gly1422Arg. This mutation in DIII places the large positively charged Arg sidechain squarely in the aqueous ion conduction pathway and would surely reduce or prevent sodium conductance. Finally, the Brugada Syndrome mutation Thr1711Arg in DIV has a similar effect to Gly1422Arg in DIII by placing a positively charged Arg sidechain in the aqueous ion conduction pathway in the SF, thereby reducing or blocking sodium conductance.
Sidechain Dunking and Interaction with Lys1421 During Sodium Permeation
It is surprising that bacterial channels have a HFS formed by four negatively charged Glu sidechains, whereas vertebrate sodium channels have a signature sequence of DEKA at the HFS. Is there a common mechanism that mediates conduction in both cases? Our previous crystal structure of NavAb and correlated molecular dynamics simulations suggested that hydrated Na+ is partially dehydrated by the HFS formed by four Glu177 residues and further that Glu177 sidechains rotate around a single torsion angle and “dunk” with permeating Na+ (Chakrabarti et al., 2013). The most common modes of Na+ conductance have two dunking Glu177 carboxyl groups (Chakrabarti et al., 2013). In the structure of the SF in NaV1.5C, Lys1421 is in a dunked conformation, as if it interacts with permeating Na+. How is that possible? Detailed analysis of the pKa’s of Lys residues in different positions in a known protein structure demonstrated large decreases in pKa of the Lys −NH2 group to values as low at pH 5.3 in nonaqueous environments and in environments with nearby hydrogen bond acceptors, comparable to the pKa values of carboxyl groups (Isom et al., 2011; Wu et al., 2017). In the SF itself, there are no free water molecules and many surrounding hydrogen bond acceptors (Figure 6B–E). Therefore, we propose that Lys1421 sidechain dunked deep in the SF forms a complex in which its positive charge is delocalized by interaction of all three NH3+ protons with nearby backbone carbonyl groups of Thr1419, Thr1711, and Ser1712 (Figure 6E). Remarkably, despite the presence of Lys1421, the surface of the SF in this region is uniformly electronegative (Figure S7F, red). This delocalization of the −NH3+ charge may allow the unpaired electrons of the N atom to serve as a specific coordinating ligand of Na+, consistent with the unique requirement for Lys at this position for Na+ selectivity (Favre et al., 1996; Heinemann et al., 1992; Sun et al., 1997). Based on these considerations, we propose a unified model for SF function in prokaryotes and eukaryotes in which Na+ is conducted in concert with dunking of the sidechain of Glu901 followed by formation of a unique charge-delocalization complex with Lys1421 in eukaryotes. This model retains the sidechain dunking mechanism of prokaryotic sodium channels and explains the extraordinary importance for Lys1421 in determining the Na+ selectivity of eukaryotic sodium channels, because only charged Na+ would have small enough size and sufficiently high field strength to join the charge delocalization complex formed by Lys1421 and engage in an energetically favorable coordination with it. Consistent with this model, Li+ is also highly permeant and has a similar high field strength surrounding its positively charged nucleus, whereas the larger alkali metal cations K+, Rb+, and Cs+ are not highly permeant and do not have a high-field-strength positive charge. Thus, this model for sodium selectivity would bring together several unexpected aspects of sodium permeation, the sidechain-dunking mechanism of the prokaryotic SF, and the unique structure of the eukaryotic SF.
CONCLUSIONS
The NaV1.5 channel has unique physiological roles and stands alone on a branch of the dendrogram of related sodium channel proteins (Goldin et al., 2000). Even heterozygous disease mutations in NaV1.5 can be lethal, indicating that other sodium channels cannot compensate for loss of its unique functions. Despite these specific functional requirements, our structure of NaV1.5C is highly similar to other sodium channels in the transmembrane core. Based on this detailed structure, we have elucidated the molecular basis for several specific functions of NaV1.5.
The interaction sites for NaVβ subunits are strikingly altered in NaV1.5C. An Asn residue with N-linked carbohydrate chain attached is substituted for a key amino acid residue in the interaction site for the β1 subunit. Differential glycosylation of this site has the potential for controlling modulation of sodium channel expression and function by β1 and β3 subunits. Similarly, the highly conserved Cys that forms a disulfide bond with the β2 and β4 subunits is replaced by Leu869. The lack of disulfide-bond formation is likely to make binding of β2 and β4 much less stable.
The characteristic low affinity for block of NaV1.5 by TTX is a hallmark of sodium channel pharmacology. The NaV1.5C structure shows that the Cys374 residue that is substituted for aromatic residues in NaV1.5 does not change the local conformation but fails to make a productive molecular interaction with bound TTX because of steric constraints. The loss of interactions between TTX and the aromatic side chains at this position is likely to be responsible for the 500-fold reduction of TTX-binding affinity in NaV1.5.
Our results give a detailed, high-resolution view of the receptor site for antiarrhythmic drugs. Flecainide binds to a platform at one side of the central cavity and blocks sodium permeation directly. Binding of its piperidine nitrogen to the Lys1421 charge delocalization complex at the inner end of the SF would prevent conductance of Na+. This platform may mediate binding of other Class IC antiarrhythmic drugs, most of which have a large hydrophobic ring structure similar to flecainide. The Class IA antiarrhythmic drugs (eg., procainamide) and the Class IB antiarrhythmic drugs (eg. lidocaine) are smaller and less hydrophobic; therefore, it is likely that they occupy only a portion of the flecainide-binding surface. The comparatively high affinity and slow binding kinetics that are the hallmark of block by flecainide and other Class IC drugs likely reflect their interaction with this larger binding surface.
The four VS in the rNaV1.5C are all in partially or fully activated conformations, because each of them has moved at least two gating charges outward with respect to their positions in our resting state model of NaV1.5. The outward position of the gating charges is most prominent for the VS in DIII because four gating charges are located outside the HCS. This outward movement of the S4 segment in DIII is coupled to conformational changes in the S4-S5 linkers that form the receptor site for the IFM motif in the fast inactivation gate. In the resting state model of NaV1.5, the interaction site for the inactivation gate is blocked by the intracellular end of the S4 segment in DIII and the S4-S5 linker connecting DIII and DIV, but it is available and occupied by the IFM motif of the inactivation gate in our structure of NaV1.5C. These structural results reveal the molecular basis for coupling of activation to fast inactivation in atomic detail.
The structure of the SF in rNaV1.5C reveals crucial insights into the mechanism of Na+ conductance and selectivity. The image of the sidechain of Lys1421 in a dunked conformation deep in the SF leads directly to a model of cation permeation that combines two key elements: dunking of the sidechain of Glu901, in analogy to the model proposed for bacterial sodium channels from molecular dynamics analysis (Chakrabarti et al., 2013), plus interaction with the unique charge-delocalization complex of the amino group of Lys1421 and its neighboring hydrogen bond acceptors. As illustrated in Figure S7F, the positive charge of Lys1421 is completely transferred to the three backbone carbonyls with which it forms hydrogen bonds, leaving a negative surface for interaction with conducted cations. Among alkali metal cations, only Li+ and Na+ have sufficient charge density to replace a proton as a ligand of a primary NH2 group. Thus, this model explains the unique requirement for Lys1421 in order to achieve high Na+ selectivity and fits well with the high conductance of Li+ in sodium channels.
Finally, the structure of rNaV1.5C provides key insights into the mechanism of action of arrhythmia mutations. We find that many mutations located in the VS affect amino acid residues that move substantially during activation. Changing these amino acid residues is likely to disrupt activation and/or deactivation by altering structural interactions that are essential for normal VS movement. Arrhythmia mutations that cause gating pore currents generate a transmembrane hole in the VS, like corresponding mutations in NaV1.4 that are pathogenic in periodic paralysis (Jiang et al., 2018). This finding elucidates the mechanism of arrhythmogenesis by these mutations at the atomic level. We find that mutations that alter amino acid residues in the fast inactivation gate and its receptor disrupt close interactions between the IFM motif and its binding partners in the S4-S5 linkers in DIII and DIV and the intracellular end of the S6 segment in DI. Similarly, mutation of the key SF residue Lys1421 to Glu may cause loss of function by inducing a calcium binding site in a blocking position in the SF. Future experiments may allow structure-based diagnosis and drug development for life-threatening cardiac arrhythmias based on this structural information.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, William A. Catterall (wcatt@uw.edu). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbe strains
E. coli GC10 was cultured at 37°C in LB medium supplemented with 100 μg/mL of ampicillin for plasmid DNA extraction. E. coli DH10Bac was cultured at 37°C in LB medium supplemented with 50 μg/mL kanamycin sulfate, 7 μg/mL gentamicin and 10 μg/mL tetracycline for bacmid production.
Cell lines
Sf9 (Spodoptera frugiperda) insect cells were maintained in Grace’s Insect Medium and supplemented with 10% FBS and penicillin/streptomycin at 27°C and passaged at 80–95% confluence for baculovirus production. HEK293S GnTI− (Homo sapiens) mammalian cells were maintained and infected on cell culture plates in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS and glutamine/penicillin/streptomycin at 37°C and 5% CO2 for electrophysiology.
METHOD DETAILS
Electrophysiological Recordings
All experiments were performed at room temperature (21–24°C). HEK293S GnTI− cells were held at −140 mV and 50-ms pulses were applied in 10 mV increments. Holding leak potential was set to −140 mV. Extracellular solution contains in mM: 140 NaCl, 2 CaCl2, 2 MgCl2, 10 HEPES, pH 7.4. Intracellular solution: 35 NaCl, 105 CsF, 10 EGTA, 10 HEPES, pH 7.4. Glass electrodes had a resistance 2–3 MW. Currents resulted from applied pulses were filtered at 5 kHz with a low-pass Bessel filter, and then digitized at 20 kHz. Voltage commands were generated using Pulse 8.5 software (HEKA, Germany) and ITC18 analog-to-digital interface (Instrutech, Port Washington, NY).
Rat Nav1.5 Expression and Purification
The full-length rat Nav1.5 gene was cloned into pEG_BacMam vector, with a recombinant human rhinovirus (RHV 3C) protease site, eGFP and a DYKDDDK FLAG peptide fused at its C-terminus. Non-structured loop deleted constructs were screened by fluorescence-detection size- exclusion chromatography (FSEC)(Kawate and Gouaux, 2006) for improved expression level and thermostability. The plasmid was transfected into DH10Bac competent cells to make baculovirus for transduction of HEK293S GnTI− cells (ATCC). HEK293S GnTI− cells were cultured in FreeStyle 293 expression medium (ThermoFisher Scientific) within 8% CO2 37 °C incubator shaking at 130 rpm. 30 mL of β3 baculovirus was added to 800 mL medium when cell density reached 2.5–3.0 × 106 cells per mL. 10 mM sodium butyrate was added to cell culture 8 hours after and cells were cultured for another 48 h at 30 with 8% CO 2 before harvesting.
For each purification, 6 L 0of transfected cells were harvested and washed with buffer containing 25 mM HEPES pH = 7.4, 150 mM NaCl and 10% glycerol (buffer A). Washed cells were collected by ultra-centrifugation at 100,000xg for 1 h, the cell pellet was resuspended in buffer A supplemented with 1% (w/v) n-dodecyl-p-D-maltopyranoside (DDM, Anatrace), 0.2% (w/v) cholesteryl hemisuccinate Tris salt (CHS, Anatrace), 5 mM lidocaine, and protease inhibitor cocktail (Sigma), and agitated for 1.5 h at 4°C. The supernatant was separated by ultra-centrifugation at 100,000×g for 35 min, then incubated with anti-Flag M2 affinity gel (Sigma) and agitated for 1 h at 4°C. The resin was re-loaded to a column and washed 5 times with buffer A supplemented with 0.06% glycol-diosgenin (GDN, Anatrace) and 2 mM lidocaine in a total volume of 100 ml. The rNaV1.5C protein was eluted with buffer A supplemented with 0.06% GDN, 2 mM lidocaine and 100 μg/mL Flag peptide (Bio Basic). The eluted protein was concentrated and loaded onto a Superose-6 column (GE Healthcare) equilibrated with buffer containing 25 mM imidazole pH=6.0, 150 mM NaCI, 1 mM lidocaine and 0.006% GDN. Peak fractions were collected and concentrated to ~ 500 μgl at 1 mg/ml and mixed with 50 μgM ProTx II overnight. The mixture was then re-loaded to Superose-6 column within the same buffer. Finally, peak fractions were concentrated to 50 μgl at 4.5 mg/ml.
For rNaV1.5C-flecainide complex purification, the sample was purified similarly with some modifications. First, no lidocaine was used throughout the purification. Second, the sample was mixed with purified human FGF12b and incubated overnight at 4 °C. Third, the solutions for the second gel filtration contained 0.5 mM flecainide. Peak fractions were collected and concentrated to 50 μgl at 5.5 mg/ml.
CryoEM Grid Preparation and Data Collection
Three microliters of purified sample was applied to glow-discharged holey gold grids (UltraAuFoil, 300 mesh, R1.2/1.3), and blotted for 3.0 – 3.5 s at 100% humidity and 4°C before being plunge-frozen in liquid ethane cooled by liquid nitrogen using an FEI Mark IV Vitrobot. All data were acquired using a Titan Krios transmission electron microscope operated at 300 kV, a Gatan K2 Summit direct detector and Gatan Quantum GIF energy filter with a slit width of 20 eV. A total of 7,626 movie stacks for rNaV1.5C-ProTxII and 5,049 movie stacks for rNaV1.5C-flecainide were automatically collected using Leginon (Suloway et al., 2005) at a nominal magnification of 130,000x with a pixel size of 0.528 Å (super-resolution mode). Defocus range was set between −1.0 and −2.5 μm. The dose rate was adjusted to 8 counts/pixel/s, and each stack was exposed for 8.4 s with 42 frames with a total dose of 60 e−/Å2.
Cryo-EM Data Processing
The movie stacks were motion corrected with MotionCorr2 (Zheng et al., 2017), binned 2- fold, and dose weighted, yielding a pixel size of 1.056 Å. Defocus value of each aligned sum were estimated with Gctf (Zhang, 2016). For rNaV1.5C-ProTxII and rNaV1.5C-FGF12b-flecainide respectively, 7,427/4,890 micrographs with CTF fits better than 6 Å were used for particle picking. A total of 2,188,972/2,369,854 particles were automatically picked in RELION2.0 (Scheres, 2012). After several rounds of 2D classification, 590,667/1,164,667 good particles were selected and subjected to one class global angular search 3D classification with an angular search step at 7.5°, using the low-pass filtered cryoEM map of NaVPas as an initial model. Each of the last five iterations was further subjected to four-class local angular search and 3D classification with an angular search step at 3.75°. After combining particles from the best 3D classes and removing duplicated particles, 293,629/733,541 particles were subjected to per-particle CTF estimation by Gctf followed by Bayesian polishing. The polished particles were subjected to a final three-class multi-reference 3D classification. The best class containing 106,104/513,604 particles was imported into cisTEM (Grant et al., 2018) for refinement. A protein-only mask was used to avoid over-fitting of the detergent micelles. Several iterative global and local refinement improved protein density of the map. For the rNaV1.5C-ProTxII map, only weak density for ProTxII was observed on DI-VS, which was not sufficient to build a model. For the rNaV1.5C-FGF12b-flecainide map, weak density at the C-terminus was observed, but it was not sufficient to build a model for the CTD or FGF12b. The final map was sharpened in cisTEM by applying a B-factor of −60. Local resolution was estimated by MonoRes (Vilas et al., 2018). A diagram of data processing is presented in Figure S2 and Figure S5.
Model Building and Refinement
The structure of human NaV1.4 (PDB code: 6AGF) α-subunit was fitted into the cryo-EM density map of rNaV1.5C in Chimera (Pettersen et al., 2004). Then the model was manually rebuilt in COOT (Emsley et al., 2010) and subsequently refined in Phenix (Adams et al., 2010). The model vs map FSC curve was calculated by Phenix.mtriage. Statistics for cryo-EM data collection and model refinement are summarized in Supplementary Table S1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of electrophysiological data
Current–voltage (I–V) relationships were recorded in response to voltage steps ranging from − 150 to + 50 mV in 10-mV increments from a holding potential of− 150 mV. Conductance-voltage (G-V) curves were calculated from the corresponding (I–V) curves. Pulses were generated and currents were recorded using Pulse software controlling an InstruTECH ITC18 interface (HEKA). Data were analyzed using Igor Pro 6.37 (WaveMetrics). G-V curves were fit with a simple two-state Boltzmann Equation 1/(1+exp(V1/2-Vp)/k) in which Vp is the stimulus potential, V1/2 is the half-activation voltage, and k is the slope factor. Dose-Response curve was fit with a Hill equation (base+(max-base)/(1+KA/[L])n), where KA is flecainide concentration producing 50% block and [L] is the free unbound flecainide, and n is the Hill coefficient. The best fit gives a Hill coefficient n= 0.75.
Estimation
Protein concentrations were calculated by an A280 extinction coefficient of 258,015 M−1 cm−1 from a spectrophotometer. Global resolution estimation of the cryo-EM density maps were based on the Fourier Shell Correlation criterion.
DATA AND CODE AVAILABILITY
The cryo-EM map of the rNav1.5C and rNav1.5C/flecainide complex have been deposited in the Electron Microscopy Data Bank (EMDB) under accession code EMD-20951 and EMD-20949, respectively. The coordinates of the rNav1.5C and rNav1.5C/flecainide complex atomic model have been deposited in the Protein Data Bank (PDB) under accession code 6UZ3 and 6UZ0, respectively.
Supplementary Material
Figure S1. Purification and Cryo-EM Images of rNav1.5C, Related to Figures 1 and 2.
(A) Electrophysiological characterization of the rNav1.5C construct. Conductance–voltage relationships (green squares) and steady-state inactivation curves (red squares) for rNaV1.5C. The lines through the curves are fits of Boltzmann relationships with values of Va = −73.3 ± 3.5 mV and k = 6 ± 2.8 mV for the conductance-voltage curve and Vh = −110 ± 0.6 mV and k = 6.9 ± 0.58 mV for the steady-state inactivation curve. Inset. Example sodium currents in response to 40-ms depolarizing pulses in 10 mV increments from a holding potential of −150 mV (green).
(B) Concentration-response curve for state-dependent block of NaV1.5C by flecainide. Use-dependent block was measured after perfusing the indicated drug concentration and applying 40-ms pulses at 100 Hz from − 150 mV to −40 mV. Each point represents the average of 5-8 cells. Data were fit by the Hill equation with nH = 1. IC50 = 16 ± 1 μM. Inset. Use-dependent block of sodium currents by different concentrations of flecainide.
(C) A histogram showing the differential affinity for flecainide block of activated (UDB) versus resting (TB) states. IC50 = 16 ± 1 μM for activated state block and IC50 = 108 ± 7 μM for tonic block.
(D) Representative size-exclusion chromatography profile of purified rNav1.5C. Peak fractions collected for cryo-EM grid preparation are shown in blue.
(E) SDS-PAGE of the SEC peak fractions stained by Coomassie blue.
(F) Representative cryo-EM micrograph of rNav1.5C sample.
(G) Selected reference-free 2D classification averages.
Figure S2. Cryo-EM Data Processing of the rNav1.5C, Related to Figure 1
(A) The flowchart of the rNav1.5C data processing.
(B) Local resolution of the EM map generated by MonoRes colored from blue to red representing resolution from high to low (side bar).
(C) Particle angular distribution of the final reconstruction.
(D) The gold-standard Fourier Shell Correlation curve for rNav1.5C reconstruction.
(E) The Fourier Shell Correlation curve of the model versus the map used for model refinement.
{kind=link}
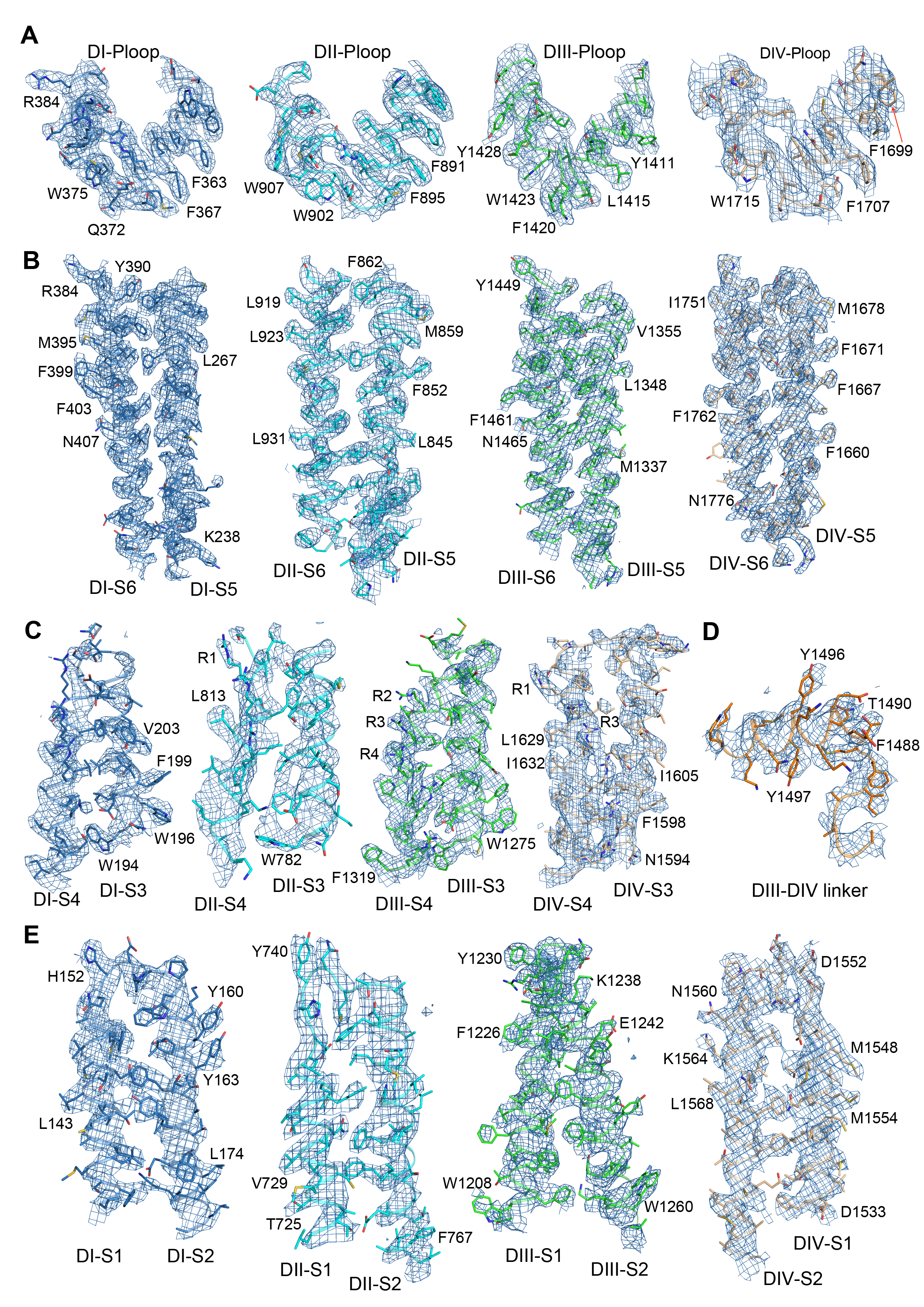
Figure S3. Representative EM Map for Key Components of rNav1.5C, Related to Figure 1
(A) The EM density for the P-loop of each domain.
(B) The EM density for the S5 and S6 helices of each domain.
(C) The EM density for the S3 and S4 helices of each domain.
(D) The EM density for the DIII-DIV linker.
(E) The EM density for the lipids and detergents bound to the channel.
(F) The EM density for the glycosylation sites of the channel.
{kind=link}
Figure S4. Purification and Cryo-EM Images of rNav1.5C/Flecainide Complex, Related to Figure 2
(A) Representative size-exclusion chromatography profile of purified rNav1.5C and flecainide complex. Peak fractions collected for cryo-EM grid preparation are shown in blue.
(B) SDS-PAGE of the SEC peak fractions stained by Coomassie blue.
(C) Representative cryo-EM micrograph of rNav1.5C and flecainide complex sample.
(D) Selected reference-free 2D classification averages.
Figure S5. Cryo-EM data Processing of rNav1.5C/Flecainide Complex, Related to Figure 2
(A) The flowchart of the rNav1.5C and flecainide complex data processing.
(B) Local resolution of the EM map generated by MonoRes colored from blue to red representing resolution from high to low (side bar). Yellow square indicates high local resolution for DII-DIII fenestration.
(C) Particle angular distribution of the final reconstruction.
(D) The gold-standard Fourier Shell Correlation curve for rNav1.5C and flecainide complex reconstruction.
(E) The Fourier Shell Correlation curve of the model versus the map used for model refinement.
{kind=link}
Figure S6. Representative EM Map for Key Components of rNav1.5C/Flecainide Complex, Related to Figure 2
(A) The EM density for the P-loop of each domain.
(B) The EM density for the S5 and S6 helices of each domain.
(C) The EM density for the S3 and S4 helices of each domain.
(D) The EM density for the DIII-DIV linker.
(E) The EM density for the S1 and S2 helices of each domain.
{kind=link}
Figure S7. Sequence Alignment of Key Regions Among Sodium Channels, Related to Figures 1, 4 and 6
(A) Sequence alignment of the region interacting with β1 subunit.
(B) Sequence alignment of the region forming disulfide bound with β2 subunit.
(C) Sequence alignment in the region of the TTX binding site.
(D) Comparison of rNaV1.5C DI-VS and NaVAb VS colored in blue and grey, respectively. Gating charges are shown in sticks.
(E) Bottom view of DIV-VS with gating charges shown in sticks illustrating the C-cap for the S4 segment in DIV. K7, Lys1643; R8, Arg1646.
(F) Orthogonal views are shown with the calculated electrostatic potential displayed over the solvent accessible surface of rat NaV1.5C. The electrostatic potential was calculated and visualized with the APBS plugin in Pymol (Jurrus et al., 2018), with positive potentials shown in blue and negative potentials shown in red (scale below the model is in units of kT/ec). The highly conserved lysine K1421 shows a negative electrostatic potential, suggesting that deprotonated lysine may be involved in selectivity (Jurrus et al., 2018).
Highlights.
The structure of the cardiac sodium channel reveals key functional features
The antiarrhythmic drug flecainide blocks the pore below the selectivity filter
The ion selectivity filter and inactivation gate are revealed in atomic detail
An arrhythmia mutation creates a pathogenic gating pore ~2Å in diameter
Acknowledgements
We are grateful to Dr. Eric Gouaux (Vollum Institute, Oregon Health & Science University) and the staff at the Cryo-EM Facility of Oregon Health & Science University for their advice and help in the early phase of this work. We thank J. P. Quispe of the University of Washington Cryo-EM Facility for advice and technical assistance during the main phase of data collection and Dr. Jin Li, Department of Pharmacology, University of Washington, for technical and editorial support. This research was supported by National Institutes of Health Research Grant R01 HL112808 (to W.A.C. and N.Z.) and by the Howard Hughes Medical Institute (N.Z.).
Footnotes
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anno T, and Hondeghem LM (1990). Interactions of flecainide with guinea pig cardiac sodium channels. Importance of activation unblocking to the voltage dependence of recovery. Circ Res 66, 789–803. [DOI] [PubMed] [Google Scholar]
- Armstrong CM, and Bezanilla F (1973). Currents related to movement of the gating particles of the sodium channels. Nature 242, 459–461. [DOI] [PubMed] [Google Scholar]
- Bankston JR, Yue M, Chung W, Spyres M, Pass RH, Silver E, Sampson KJ, and Kass RS (2007). A novel and lethal de novo LQT-3 mutation in a newborn with distinct molecular pharmacology and therapeutic response. PLoS One 2, e1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroudi G, Acharfi S, Larouche C, and Chahine M (2002). Expression and intracellular localization of an SCN5A double mutant R1232W/T1620M implicated in Brugada syndrome. Circ Res 90, E11–16. [PubMed] [Google Scholar]
- Baroudi G, Pouliot V, Denjoy I, Guicheney P, Shrier A, and Chahine M (2001). Novel mechanism for Brugada syndrome: defective surface localization of an SCN5A mutant (R1432G). Circ Res 88, E78–83. [DOI] [PubMed] [Google Scholar]
- Bennett ES (2001). Channel cytoplasmic loops alter voltage-dependent sodium channel activation in an isoform-specific manner. J Physiol 535, 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett PB, Yazawa K, Makita N, and George AL Jr. (1995). Molecular mechanism for an inherited cardiac arrhythmia. Nature 376, 683–685. [DOI] [PubMed] [Google Scholar]
- Capes DL, Goldschen-Ohm MP, Arcisio-Miranda M, Bezanilla F, and Chanda B (2013). Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. J Gen Physiol 142, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA (2000). From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 26, 13–25. [DOI] [PubMed] [Google Scholar]
- Catterall WA, and Maier SKG (2015). Voltage-gated sodium channels and the electrical excitability of the heart. In Cardiac Electrophysiology: From Bench to Bedside, Zipes DJ, and Jalife J, eds. (Saunders/Elsevier; ). [Google Scholar]
- Catterall WA, Wisedchaisri G, and Zheng N (2017). The chemical basis for electrical signaling. Nature Chem Biol 13, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti N, Ing C, Payandeh J, Zheng N, Catterall WA, and Pomes R (2013). Catalysis of Na+ permeation in the bacterial sodium channel NaVAb. Proc Natl Acad Sci U S A 110, 11331–11336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda B, and Bezanilla F (2002). Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol 120, 629–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, Yarov-Yarovoy V, Scheuer T, and Catterall WA (2011). Gating charge interactions with the S1 segment during activation of a Na+ channel voltage sensor. Proc Natl Acad Sci U S A 108, 18825–18830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, Yarov-Yarovoy V, Sharp EM, Scheuer T, and Catterall WA (2009). Sequential formation of ion pairs during activation of a sodium channel voltage sensor. Proc Natl Acad Sci U S A 106, 22498–22503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, Yarov-Yarovoy V, Zhao Y, Scheuer T, and Catterall WA (2008). Disulfide locking a sodium channel voltage sensor reveals ion pair formation during activation. Proc Natl Acad Sci U S A 105, 15142–15147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, Ferrari MD, Herzog J, van den Maagdenberg AM, Pusch Mv et al. (2005). Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366, 371–377. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre I, Moczydlowski E, and Schild L (1996). On the structural basis for ionic selectivity among Na+, K+, and Ca2+ in the voltage-gated sodium channel. Biophys J 71, 3110–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fozzard HA, and Hanck DA (1996). Structure and function of voltage-dependent sodium channels: Comparison of brain II and cardiac isoforms. Physiol Rev 76, 887–926. [DOI] [PubMed] [Google Scholar]
- Gamal El-Din TM, Lenaeus MJ, Zheng N, and Catterall WA (2018). Fenestrations control resting-state block of a voltage-gated sodium channel. Proc Natl Acad Sci U S A 115, 13111–13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamal El-Din TM, Scheuer T, and Catterall WA (2014). Tracking S4 movement by gating pore currents in the bacterial sodium channel NaChBac. J Gen Physiol 144, 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring A, Lee CH, Wang KH, Michel JC, Claxton DP, Baconguis I, Althoff T, Fischer S, Garcia KC, and Gouaux E (2014). Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat Protoc 9, 2574–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Berwald Netter Y, et al. (2000). Nomenclature of voltage-gated sodium channels. Neuron 28, 365–368. [DOI] [PubMed] [Google Scholar]
- Goldschen-Ohm MP, Capes DL, Oelstrom KM, and Chanda B (2013). Multiple pore conformations driven by asynchronous movements of voltage sensors in a eukaryotic sodium channel. Nature communications 4, 1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Merrick D, Wollner DA, and Catterall WA (1988). Biochemical properties of sodium channels in a wide range of excitable tissues studied with site-directed antibodies. Biochemistry 27, 7032–7038. [DOI] [PubMed] [Google Scholar]
- Grant T, Rohou A, and Grigorieff N (2018). cisTEM, user-friendly software for single-particle image processing. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann SH, Terlau H, Stuhmer W, Imoto K, and Numa S (1992). Calcium channel characteristics conferred on the sodium channel by single mutations. Nature 356, 441–443. [DOI] [PubMed] [Google Scholar]
- Henderson R, Sali A, Baker ML, Carragher B, Devkota B, Downing KH, Egelman EH, Feng Z, Frank J, Grigorieff N, et al. (2012). Outcome of the first electron microscopy validation task force meeting. Structure 20, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (1971). The permeability of the sodium channel to organic cations in myelinated nerve. J Gen Physiol 59, 599–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (1972). The permeability of the sodium channel to metal cations in myelinated nerve. J Gen Physiol 59, 637–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (1975a). Ionic selectivity, saturation, and block in sodium channels. A four-barrier model. J Gen Physiol 66, 535–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (1975b). The receptor for tetrodotoxin and saxitoxin: a structural hypothesis. BiophysJ 15, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom DG, Castaneda CA, Cannon BR, and Garcia-Moreno B (2011). Large shifts in pKa values of lysine residues buried inside a protein. Proc Natl Acad Sci U S A 108, 5260–5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom LL, De Jongh KS, and Catterall WA (1994). Auxiliary subunits of voltage-gated ion channels. Neuron 12, 1183–1194. [DOI] [PubMed] [Google Scholar]
- Isom LL, Ragsdale DS, De Jongh KS, Westenbroek RE, Reber BF, Scheuer T, and Catterall WA (1995). Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 83, 433–442. [DOI] [PubMed] [Google Scholar]
- Jiang D, Gamal El-Din TM, Ing C, Lu P, Pomes R, Zheng N, and Catterall WA (2018). Structural basis for gating pore current in periodic paralysis. Nature 557, 590–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurrus E, Engel D, Star K, Monson K, Brandi J, Felberg LE, Brookes DH, Wilson L, Chen J, Liles K, et al. (2018). Improvements to the APBS biomolecular solvation software suite. Protein Sci 27, 112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A, Harris-Kerr C, et al. (2010). An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 7, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawate T, and Gouaux E (2006). Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 14, 673–681. [DOI] [PubMed] [Google Scholar]
- Kellenberger S, Scheuer T, and Catterall WA (1996). Movement of the Na+ channel inactivation gate during inactivation. J Biol Chem 271, 30971–30979. [DOI] [PubMed] [Google Scholar]
- Konzen G, Reichardt B, and Hauswirth O (1990). Fast and slow blockade of sodium channels by flecainide in rabbit cardiac Purkinje fibres. Naunyn Schmiedebergs ArchPharmacol 341, 565–576. [DOI] [PubMed] [Google Scholar]
- Li W, Yin L, Shen C, Hu K, Ge J, and Sun A (2018). SCN5A Variants: Association With Cardiac Disorders. Front Physiol 9, 1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Atkins J, and Kass RS (2003a). Common molecular determinants of flecainide and lidocaine block of heart Na+ channels: evidence from experiments with neutral and quaternary flecainide analogues. J Gen Physiol 121, 199–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Clancy C, Cormier J, and Kass R (2003b). Mutations in cardiac sodium channels: clinical implications. Am J Pharmacogenomics 3, 173–179. [DOI] [PubMed] [Google Scholar]
- Lombet A, and Lazdunski M (1984). Characterization, solubilization, affinity labeling, and purification of the cardiac sodium channel using Tityus toxin. Eur J Biochem 141, 651–660. [DOI] [PubMed] [Google Scholar]
- Makita N, Bennett PB Jr., and George AL Jr. (1994). Voltage-gated Na+ channel beta-1 subunit mRNA expressed in adult human skeletal muscle, heart, and brain is encoded by a single gene. J Biol Chem 269, 7571–7578. [PubMed] [Google Scholar]
- McPhee JC, Ragsdale D, Scheuer T, and Catterall WA (1998). A critical role for the S4-S5 intracellular loop in domain IV of the sodium channel alpha subunit in fast inactivation. J Biol Chem 273, 1121–1129. [DOI] [PubMed] [Google Scholar]
- McPhee JC, Ragsdale DS, Scheuer T, and Catterall WA (1994). A mutation in segment IVS6 disrupts fast inactivation of sodium channels. Proc Natl Acad Sci U S A 91, 12346–12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhee JC, Ragsdale DS, Scheuer T, and Catterall WA (1995). A critical role for transmembrane segment IVS6 of the sodium channel alpha subunit in fast inactivation. J Biol Chem 270, 12025–12034. [DOI] [PubMed] [Google Scholar]
- Moreau A, and Chahine M (2018). A new cardiac channelopathy: from clinical phenotypes to molecular mechanisms associated with Nav1.5 gating pores. Front Cardiovasc Med 5, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau A, Gosselin-Badaroudine P, Boutjdir M, and Chahine M (2015). Mutations in the voltage sensors of domains I and II of Nav1.5 that are associated with arrhythmias and dilated cardiomyopathy generate gating pore currents. Front Pharmacol 6, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PT, DeMarco KR, Vorobyov I, Clancy CE, and Yarov-Yarovoy V (2019). Structural basis for antiarrhythmic drug interactions with the human cardiac sodium channel. Proc Natl Acad Sci U S A 116, 2945–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M, Shimizu S, Tanabe T, Takai T, Kayano T, Ikeda T, Takahashi H, Nakayama H, Kanaoka Y, Minamino N, et al. (1984). Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature 312, 121–127. [DOI] [PubMed] [Google Scholar]
- Noda M, Suzuki H, Numa S, and Stuhmer W (1989). A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett 259, 213–216. [DOI] [PubMed] [Google Scholar]
- Numa S, and Noda M (1986). Molecular structure of sodium channels. Ann N Y Acad Sci 479, 338–355. [DOI] [PubMed] [Google Scholar]
- O’Malley HA, and Isom LL (2015). Sodium channel beta subunits: emerging targets in channelopathies. Annu Rev Physiol 77, 481–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Li Z, Huang X, Huang G, Gao S, Shen H, Liu L, Lei J, and Yan N (2019). Molecular basis for pore blockade of human Na+ channel NaV1.2 by the mu-conotoxin KIIIA. Science 363, 1309–1313. [DOI] [PubMed] [Google Scholar]
- Pan X, Li Z, Zhou Q, Shen H, Wu K, Huang X, Chen J, Zhang J, Zhu X, Lei J, et al. (2018). Structure of the human voltage-gated sodium channel NaV1.4 in complex with beta1. Science 362, pii: eaau2486. [DOI] [PubMed] [Google Scholar]
- Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, and Catterall WA (2012). Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J, Scheuer T, Zheng N, and Catterall WA (2011). The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Qu Y, Isom LL, Westenbroek RE, Rogers JC, Tanada TN, McCormick KA, Scheuer T, and Catterall WA (1995). Modulation of cardiac Na+ channel expression in Xenopus oocytes by beta-1 subunits. J Biol Chem 270, 25696–25701. [DOI] [PubMed] [Google Scholar]
- Ragsdale DS, McPhee JC, Scheuer T, and Catterall WA (1996). Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc Natl Acad Sci U S A 93, 9270–9275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves PJ, Callewaert N, Contreras R, and Khorana HG (2002). Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc Natl Acad Sci U S A 99, 13419–13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren D, Navarro B, Xu H, Yue L, Shi Q, and Clapham DE (2001). A prokaryotic voltage-gated sodium channel. Science 294, 2372–2375. [DOI] [PubMed] [Google Scholar]
- Rogart RB, Cribbs LL, Muglia LK, Kephart DD, and Kaiser MW (1989). Molecular cloning of a putative tetrodotoxin-resistant rat heart Na+ channel isoform. Proc Natl Acad Sci USA 86, 8170–8174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohl CA, Boeckman FA, Baker C, Scheuer T, Catterall WA, and Klevit RE (1999). Solution structure of the sodium channel inactivation gate. Biochemistry 38, 855–861. [DOI] [PubMed] [Google Scholar]
- Sampson KJ, and Kass RK (2011). Antiarrhythmic Drugs. In Goodman & Gilman’s Pharmacological Basis of Therapeutics, pp. 815–848. [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Liu D, Wu K, Lei J, and Yan N (2019). Structures of human NaV1.7 channel in complex with auxiliary subunits and animal toxins. Science. [DOI] [PubMed] [Google Scholar]
- Shen H, Zhou Q, Pan X, Li Z, Wu J, and Yan N (2017). Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science, eaal4326. [DOI] [PubMed] [Google Scholar]
- Sivilotti L, Okuse K, Akopian AN, Moss S, and Wood JN (1997). A single serine residue confers tetrodotoxin insensitivity on the rat sensory-neuron-specific sodium channel SNS. FEBS Lett 409, 49–52. [DOI] [PubMed] [Google Scholar]
- Smith MR, and Goldin AL (1997). Interaction between the sodium channel inactivation linker and domain III S4-S5. BiophysJ 73, 1885–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov S, Scheuer T, and Catterall WA (2007). Gating pore current in an inherited ion channelopathy. Nature 446, 76–78. [DOI] [PubMed] [Google Scholar]
- Sokolov S, Scheuer T, and Catterall WA (2008). Depolarization-activated gating pore current conducted by mutant sodium channels in potassium-sensitive normokalemic periodic paralysis. Proc Natl Acad Sci U S A 105, 19980–19985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struyk AF, and Cannon SC (2007). A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J Gen Physiol 130, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, and Carragher B (2005). Automated molecular microscopy: the new Leginon system. J Struct Biol 151, 41–60. [DOI] [PubMed] [Google Scholar]
- Sun YM, Favre I, Schild L, and Moczydlowski E (1997). On the structural basis for size-selective permeation of organic cations through the voltage-gated sodium channel - Effect of alanine mutations at the DEKA locus on selectivity, inhibition by Ca2+ and H+, and molecular sieving. J Gen Physiol 110, 693–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunami A, Glaaser IW, and Fozzard HA (2000). A critical residue for isoform difference in tetrodotoxin affinity is a molecular determinant of the external access path for local anesthetics in the cardiac sodium channel. Proc Natl Acad Sci U S A 97, 2326–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlau H, Heinemann SH, Stuhmer W, Pusch M, Conti F, Imoto K, and Numa S (1991). Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett 293, 93–96. [DOI] [PubMed] [Google Scholar]
- Vilas JL, Gomez-Blanco J, Conesa P, Melero R, Miguel de la Rosa-Trevin J, Oton J, Cuenca J, Marabini R, Carazo JM, Vargas J, et al. (2018). MonoRes: Automatic and accurate estimation of local resolution for electron microscopy maps. Structure 26, 337–344 e334. [DOI] [PubMed] [Google Scholar]
- Walker JR, Novick PA, Parsons WH, McGregor M, Zablocki J, Pande VS, and Du Bois J (2012). Marked difference in saxitoxin and tetrodotoxin affinity for the human nociceptive voltage-gated sodium channel (NaV1.7) Proc Natl Acad Sci U S A 109, 18102–18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GK, Russell C, and Wang SY (2003). State-dependent block of wild-type and inactivation-deficient Na+ channels by flecainide. J Gen Physiol 122, 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, and Catterall WA (1992). A cluster of hydrophobic amino acid residues required for fast Na+ channel inactivation. Proc Natl Acad Sci U S A 89, 10910–10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisedchaisri G, Tonggu L, McCord E, Gamal El-Din TM, Wang L, Zheng N, and Catterall WA (2019). Resting-state structure and gating mechanism of a voltage-gated sodium channel. Cell 178, 993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Lee J, and Brooks BR (2017). Origin of pKa Shifts of Internal Lysine Residues in SNase Studied Via Equal-Molar VMMS Simulations in Explicit Water. J Phys Chem B 121, 3318–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, George AL Jr., and Horn R (1996). Molecular basis of charge movement in voltage-gated sodium channels. Neuron 16, 113–122. [DOI] [PubMed] [Google Scholar]
- Yang N, and Horn R (1995). Evidence for voltage-dependent S4 movement in sodium channels. Neuron 15, 213–218. [DOI] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, Decaen PG, Westenbroek RE, Pan CY, Scheuer T, Baker D, and Catterall WA (2012). Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci USA 109 E93–E102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Purification and Cryo-EM Images of rNav1.5C, Related to Figures 1 and 2.
(A) Electrophysiological characterization of the rNav1.5C construct. Conductance–voltage relationships (green squares) and steady-state inactivation curves (red squares) for rNaV1.5C. The lines through the curves are fits of Boltzmann relationships with values of Va = −73.3 ± 3.5 mV and k = 6 ± 2.8 mV for the conductance-voltage curve and Vh = −110 ± 0.6 mV and k = 6.9 ± 0.58 mV for the steady-state inactivation curve. Inset. Example sodium currents in response to 40-ms depolarizing pulses in 10 mV increments from a holding potential of −150 mV (green).
(B) Concentration-response curve for state-dependent block of NaV1.5C by flecainide. Use-dependent block was measured after perfusing the indicated drug concentration and applying 40-ms pulses at 100 Hz from − 150 mV to −40 mV. Each point represents the average of 5-8 cells. Data were fit by the Hill equation with nH = 1. IC50 = 16 ± 1 μM. Inset. Use-dependent block of sodium currents by different concentrations of flecainide.
(C) A histogram showing the differential affinity for flecainide block of activated (UDB) versus resting (TB) states. IC50 = 16 ± 1 μM for activated state block and IC50 = 108 ± 7 μM for tonic block.
(D) Representative size-exclusion chromatography profile of purified rNav1.5C. Peak fractions collected for cryo-EM grid preparation are shown in blue.
(E) SDS-PAGE of the SEC peak fractions stained by Coomassie blue.
(F) Representative cryo-EM micrograph of rNav1.5C sample.
(G) Selected reference-free 2D classification averages.
Figure S2. Cryo-EM Data Processing of the rNav1.5C, Related to Figure 1
(A) The flowchart of the rNav1.5C data processing.
(B) Local resolution of the EM map generated by MonoRes colored from blue to red representing resolution from high to low (side bar).
(C) Particle angular distribution of the final reconstruction.
(D) The gold-standard Fourier Shell Correlation curve for rNav1.5C reconstruction.
(E) The Fourier Shell Correlation curve of the model versus the map used for model refinement.
Figure S3. Representative EM Map for Key Components of rNav1.5C, Related to Figure 1
(A) The EM density for the P-loop of each domain.
(B) The EM density for the S5 and S6 helices of each domain.
(C) The EM density for the S3 and S4 helices of each domain.
(D) The EM density for the DIII-DIV linker.
(E) The EM density for the lipids and detergents bound to the channel.
(F) The EM density for the glycosylation sites of the channel.
Figure S4. Purification and Cryo-EM Images of rNav1.5C/Flecainide Complex, Related to Figure 2
(A) Representative size-exclusion chromatography profile of purified rNav1.5C and flecainide complex. Peak fractions collected for cryo-EM grid preparation are shown in blue.
(B) SDS-PAGE of the SEC peak fractions stained by Coomassie blue.
(C) Representative cryo-EM micrograph of rNav1.5C and flecainide complex sample.
(D) Selected reference-free 2D classification averages.
Figure S5. Cryo-EM data Processing of rNav1.5C/Flecainide Complex, Related to Figure 2
(A) The flowchart of the rNav1.5C and flecainide complex data processing.
(B) Local resolution of the EM map generated by MonoRes colored from blue to red representing resolution from high to low (side bar). Yellow square indicates high local resolution for DII-DIII fenestration.
(C) Particle angular distribution of the final reconstruction.
(D) The gold-standard Fourier Shell Correlation curve for rNav1.5C and flecainide complex reconstruction.
(E) The Fourier Shell Correlation curve of the model versus the map used for model refinement.
Figure S6. Representative EM Map for Key Components of rNav1.5C/Flecainide Complex, Related to Figure 2
(A) The EM density for the P-loop of each domain.
(B) The EM density for the S5 and S6 helices of each domain.
(C) The EM density for the S3 and S4 helices of each domain.
(D) The EM density for the DIII-DIV linker.
(E) The EM density for the S1 and S2 helices of each domain.
Figure S7. Sequence Alignment of Key Regions Among Sodium Channels, Related to Figures 1, 4 and 6
(A) Sequence alignment of the region interacting with β1 subunit.
(B) Sequence alignment of the region forming disulfide bound with β2 subunit.
(C) Sequence alignment in the region of the TTX binding site.
(D) Comparison of rNaV1.5C DI-VS and NaVAb VS colored in blue and grey, respectively. Gating charges are shown in sticks.
(E) Bottom view of DIV-VS with gating charges shown in sticks illustrating the C-cap for the S4 segment in DIV. K7, Lys1643; R8, Arg1646.
(F) Orthogonal views are shown with the calculated electrostatic potential displayed over the solvent accessible surface of rat NaV1.5C. The electrostatic potential was calculated and visualized with the APBS plugin in Pymol (Jurrus et al., 2018), with positive potentials shown in blue and negative potentials shown in red (scale below the model is in units of kT/ec). The highly conserved lysine K1421 shows a negative electrostatic potential, suggesting that deprotonated lysine may be involved in selectivity (Jurrus et al., 2018).
Data Availability Statement
The cryo-EM map of the rNav1.5C and rNav1.5C/flecainide complex have been deposited in the Electron Microscopy Data Bank (EMDB) under accession code EMD-20951 and EMD-20949, respectively. The coordinates of the rNav1.5C and rNav1.5C/flecainide complex atomic model have been deposited in the Protein Data Bank (PDB) under accession code 6UZ3 and 6UZ0, respectively.